DOI:
10.1039/D4RA05799F
(Paper)
RSC Adv., 2024,
14, 33235-33246
Novel pyrimidine linked acyl thiourea derivatives as potent α-amylase and proteinase K inhibitors: design, synthesis, molecular docking and ADME studies†
Received
10th August 2024
, Accepted 16th October 2024
First published on 21st October 2024
Abstract
To discover promising therapeutic agents, novel diaryl pyrimidine linked acyl thiourea derivatives (6a–j) were designed and synthesized via straightforward and multistep synthesis. The structure of these derivatives (6a–j) was confirmed by FTIR, 1H, and 13C NMR spectroscopic techniques. The in vitro biological screening of these compounds was carried out to assess their bacterial, α-amylase, and proteinase K inhibition potential. The results manifested that the developed molecules (6a–j) possessed a remarkable inhibition potential against targeted α-amylase and proteinase K enzymes. The compounds 6j and 6g were found to be the most potent α-amylase inhibitors with IC50 values of 1.478 ± 0.051 and 1.509 ± 0.039 μM, respectively. Meanwhile, the compounds 6a, 6f, and 6e having IC50 values of 1.790 ± 0.079, 1.794 ± 0.080, and 1.795 ± 0.080 μM, respectively, showed high proteinase K inhibitory activity. A moderate antibacterial activity is also displayed by these compounds (6a–j). The different substitution on the framework of pyrimidine linked acyl thiourea pharmacophore, provided the valuable basis for structure–activity relationship studies. Additionally, to identify the binding affinities of our desired compounds, molecular docking study was used. ADME analysis was also conducted to explore the physicochemical properties. Hence, these studies shed light on the significance of pyrimidine-based acyl thiourea to attain potent efficacy in drug discovery.
1. Introduction
Nitrogen-containing six-membered heterocyclic rings play an immense role in medicinal chemistry due to their effective correlation with diverse therapeutic features.1 The pyrimidine based heterocycles are prominent, as they comprise a well-known class of molecules with remarkable biological and therapeutic potential.2 The simplest explanation for their numerous medicinal properties is the presence of pyrimidine bases in uracil, cytosine, and thymine, which are fundamental moieties of nucleic acids.3 Their biological activities include anticancer, anti-inflammatory, antiviral, antimalarial antifungal, antibacterial and enzyme inhibition.4 Different marketed drugs with 2-aminopyrimidine scaffolds are Imatinib (antitumor), Glipizide (antidiabetic), Nilotinib (anticancer), and Iclaprim (antibiotic).5
Diabetes mellitus, a metabolic disorder, is a global health concern caused by the inability to either produce sufficient levels of insulin or effectively respond to the insulin being produced.6 Consequently, due to the abnormally excessive blood glucose level, there is always a high risk of cardiovascular events, cognitive impairment, fractures, and mortality.7 α-Amylase, a digestive calcium-ion metalloenzyme involved in the breakdown of starch and oligosaccharides, is the main trigger of diabetes and is frequently released by the pancreas and salivary glands. These enzymes can be easily obtained from various sources including plants, animals and microorganisms.8 Therefore, targeting the α-amylase is one of the methods for finding new antidiabetic drugs.
Protease enzymes cleave peptide linkage in proteins and are involved in many biological processes, like blood coagulation, protein degradation, and digestion.9 The serine protease enzyme namely, proteinase K produced by the fungus Tritirachium album Limber, is widely used to digest proteins (especially native keratin) and expose the nucleic acids that have been isolated from various cells or tissues.10 As proteases play a key role in all biological processes, anomalies and mis regulation in proteolytic activities lead to adverse pathological conditions, including neurodegenerative, cardiovascular, cancer, and inflammatory diseases.11 Further, the numerous inhibitors of proteinase are efficient regulators for the normal proteolytic activity in various diseases. They successfully bind to the targeted proteinase to yield a complex that maintains the substrate's peptide bond.12 Protease inhibitors are deeply investigated as potentially effective drugs for various diseases by targeting different proteases responsible for neurodegenerative, cardiovascular, and autoimmune-related disorders.13
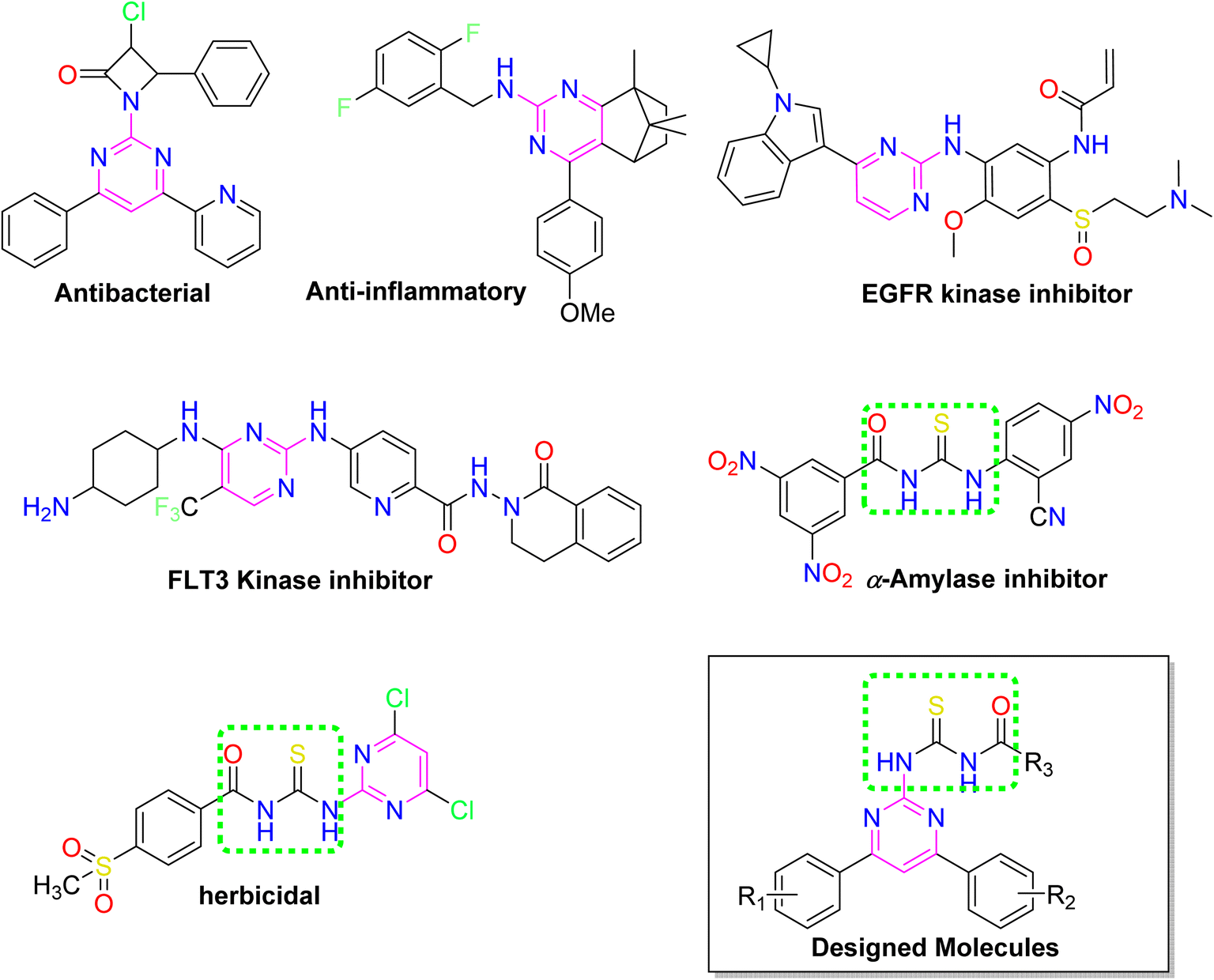
Over the years, acyl thiourea moiety has been noted to have a diverse range of pharmaceutical applications like anticancer, antiviral, antimalarial, antidiabetic, anti-inflammatory, antimicrobial, herbicidal, insecticidal,14 and various enzyme inhibitors.15–17 Extensive research shows that the donor atoms N, O, and S of acyl thiourea afford effective binding interaction as well as their derivatives can be easily cross-linked with peptides due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond in the farmwork.18 Moreover, they appear as essential components for developing various heterocycles like triazole, thiazole, tetrazole, and thiazolidinone via cyclization.19 The acyl thiourea scaffold is employed as a basic precursor for corrosion inhibitors, non-ionic surfactants, and organo-catalysts.20 Some selected pyrimidine and acyl thiourea based derivatives have been reported (Fig. 1) with significant biological activities.21–26
S bond in the farmwork.18 Moreover, they appear as essential components for developing various heterocycles like triazole, thiazole, tetrazole, and thiazolidinone via cyclization.19 The acyl thiourea scaffold is employed as a basic precursor for corrosion inhibitors, non-ionic surfactants, and organo-catalysts.20 Some selected pyrimidine and acyl thiourea based derivatives have been reported (Fig. 1) with significant biological activities.21–26
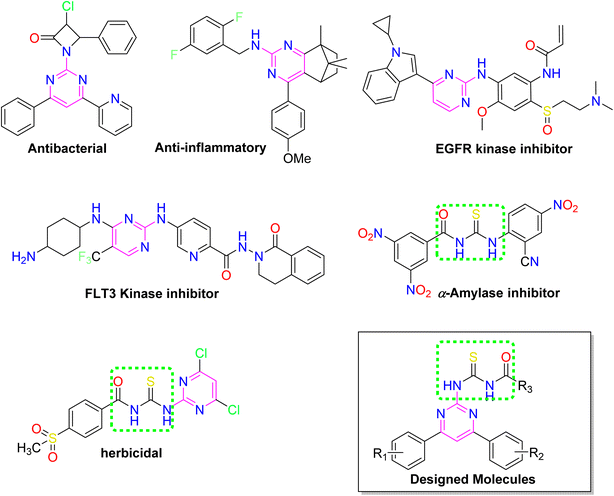 |
| | Fig. 1 Some biologically active compounds incorporating pyrimidine and acyl thiourea scaffolds and architecture of currently studies molecules. | |
Therefore, stimulated by the therapeutic potential of pyrimidine and acyl thiourea scaffolds, an effective and straightforward approach was established to develop pyrimidine linked acyl thioureas to address the global health challenges. The present research work includes synthesis, characterization, molecular docking, ADME prediction, and biological evaluation of 2-amino-4,6-diarylpyrimidine linked acyl thioureas against bacteria, α-amylase, and proteinase K.
2. Results and discussion
2.1. Chemistry
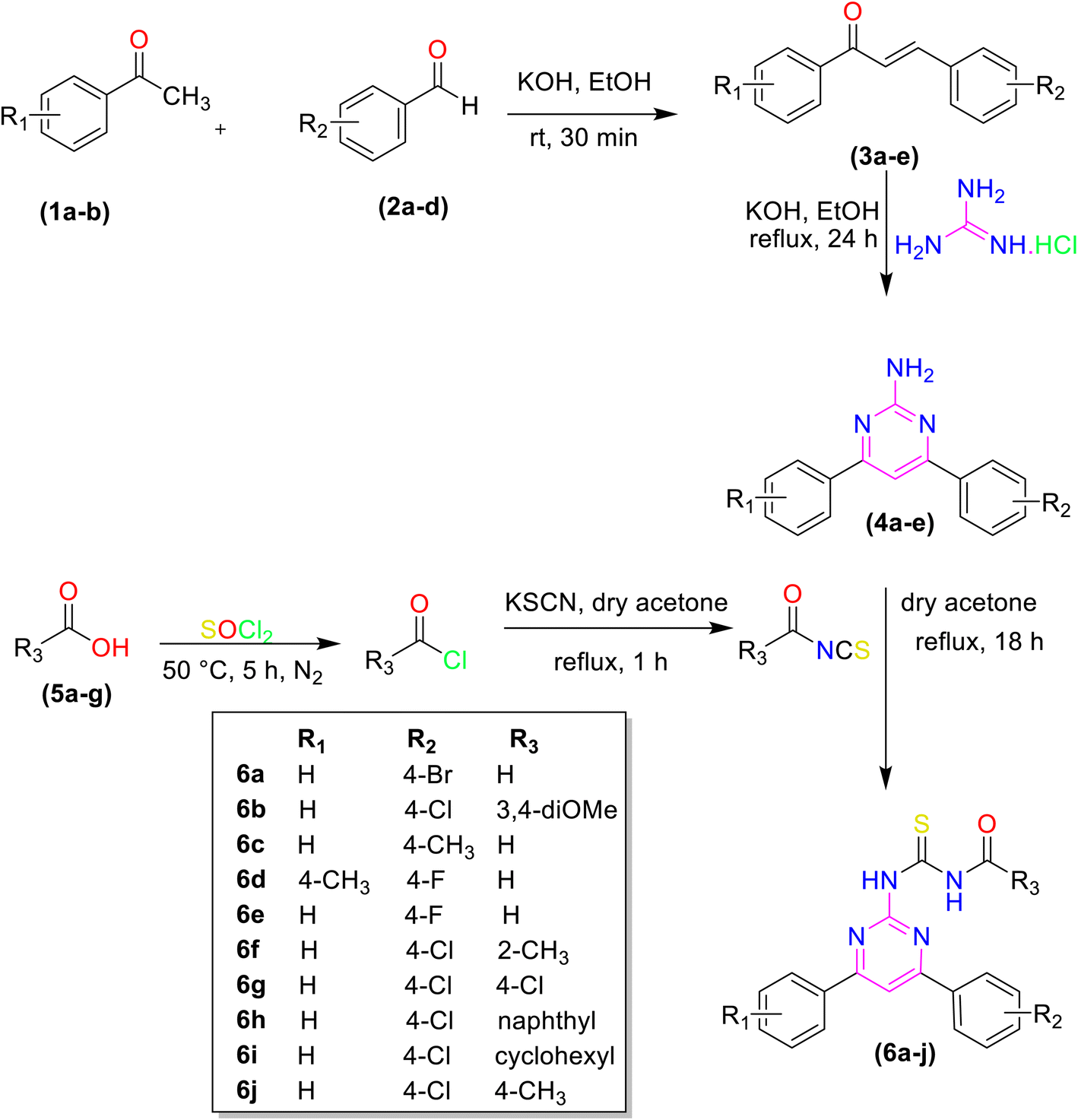
The pyrimidine linked acyl thiourea derivatives (6a–j) were synthesized according to the reported procedure.27 First, different chalcones were synthesized using different aldehydes and acetophenones via aldol condensation. These chalcones were modified into 2-amino-4,6-disubstituted pyrimidines (4a–e) with guanidine hydrochloride. Different acids (5a–f) were converted into acyl chlorides and acyl isothiocyanates by using thionyl chloride and potassium thiocyanate respectively. Finally, different acyl isothiocyanates were treated with amino pyrimidines (4a–e) through a nucleophilic addition to afford desired pyrimidine linked acyl thiourea derivatives (6a–j) with an appropriate yield. The synthetic pathway for the synthesis of desired molecules is given in (Scheme 1).
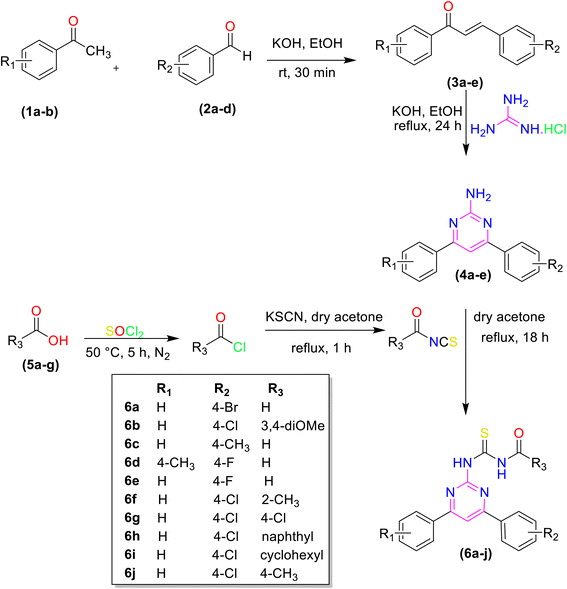 |
| | Scheme 1 Synthetic trail of pyrimidine linked acyl thiourea derivatives. | |
The synthesized acyl thiourea derivatives (6a–j) were analysed via FTIR, 1H-, and 13C NMR spectroscopy. The IR spectrum of compounds revealed the characteristic functional group regions. For compound (6f) the two medium-intensity broad absorption bands corresponding to N–H were found at 3260 and 3130 cm−1. The sp2 and sp3 C–H stretching appeared at 2970 and 2940 cm−1, respectively. The carbonyl group of acyl thiourea appeared as a strong absorption peak at 1721 cm−1. The CN and CC stretching's are confirmed by the presence of two strong peaks at 1590 and 1509 cm−1, respectively. While the peak at 1230 cm−1 was assigned for thiocarbonyl. In 1H NMR spectrum of compound (6f), two characteristic signals of NH in the most deshielded region (12.74 and 12.62 ppm) confirmed the formation of acyl thiourea. The proton attached to sp2 hybridized carbon of the pyrimidine ring gave singlet at 8.40 ppm. All the aromatic protons of phenyl rings appeared in their respective regions ranging from 8.34 to 7.23 ppm. The singlet in the shielded region at 2.42 ppm is a clear indication of the presence of methyl group in the structure. In 13C NMR, the two signals at 178.39 and 169.51 ppm correspond to thiocarbonyl and carbonyl carbons respectively. These two signals of carbon are characteristic signals of acyl thiourea. The carbons of the pyrimidine ring appeared at 165.85, 164.46, 157.97, and 109.03 ppm. While the rest of the signals at 136.83–126.26 ppm corresponds to phenyl carbons, and the signal at 19.9 ppm represents methyl carbon.
2. 2. In vitro α-amylase inhibition and structure–activity relationship
All compounds (6a–j) were evaluated against α-amylase to assess their inhibitory potential via in vitro inhibition assay. The results expressed as IC50 values are given in Table 1. These results revealed that all the derivatives (6a–j) exhibited significant inhibition activity, as their IC50 values are nearly equal to the IC50 values of positive control acarbose 1.063 ± 0.013 μM. The compound (6j) with an IC50 value of 1.478 ± 0.051 μM appears to be the most potent inhibitor among all. Further, the compound (6g) (IC50 1.509 ± 0.039 μM) also showed a quite good inhibition potential against α-amylase. While the rest of the compounds have moderate α-amylase inhibition activity.
Table 1 Results of biological activities of synthesized compoundsa
| Compd |
α-Amylase inhibition IC50 ± SEM (μM) |
Proteinase K inhibition IC50 ± SEM (μM) |
Antibacterial zone of inhibition (mm) |
| Escherichia coli |
Bacillus subtilis |
| SEM = standard error of the mean. |
| 6a |
1.676 ± 0.014 |
1.790 ± 0.079 |
7 |
8 |
| 6b |
1.612 ± 0.004 |
1.814 ± 0.086 |
6 |
9 |
| 6c |
1.551 ± 0.024 |
1.829 ± 0.062 |
7 |
8 |
| 6d |
1.553 ± 0.023 |
1.828 ± 0.062 |
8 |
9 |
| 6e |
1.662 ± 0.010 |
1.795 ± 0.080 |
6 |
7 |
| 6f |
1.664 ± 0.010 |
1.794 ± 0.080 |
7 |
6 |
| 6g |
1.509 ± 0.039 |
1.847 ± 0.067 |
6 |
8 |
| 6h |
1.601 ± 0.008 |
1.818 ± 0.087 |
6 |
8 |
| 6i |
1.521 ± 0.034 |
1.841 ± 0.066 |
7 |
11 |
| 6j |
1.478 ± 0.051 |
1.861 ± 0.071 |
6 |
7 |
| Acarbose |
1.063 ± 0.013 |
— |
— |
— |
| Phenylmethyl sulfonyl fluoride |
— |
0.1196 ± 0.014 |
— |
— |
| Rifampin |
— |
— |
17 |
26 |
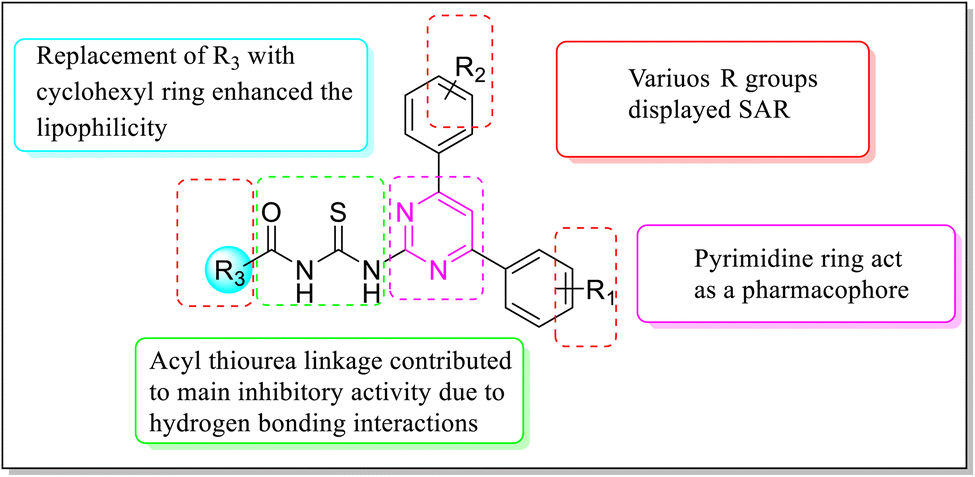
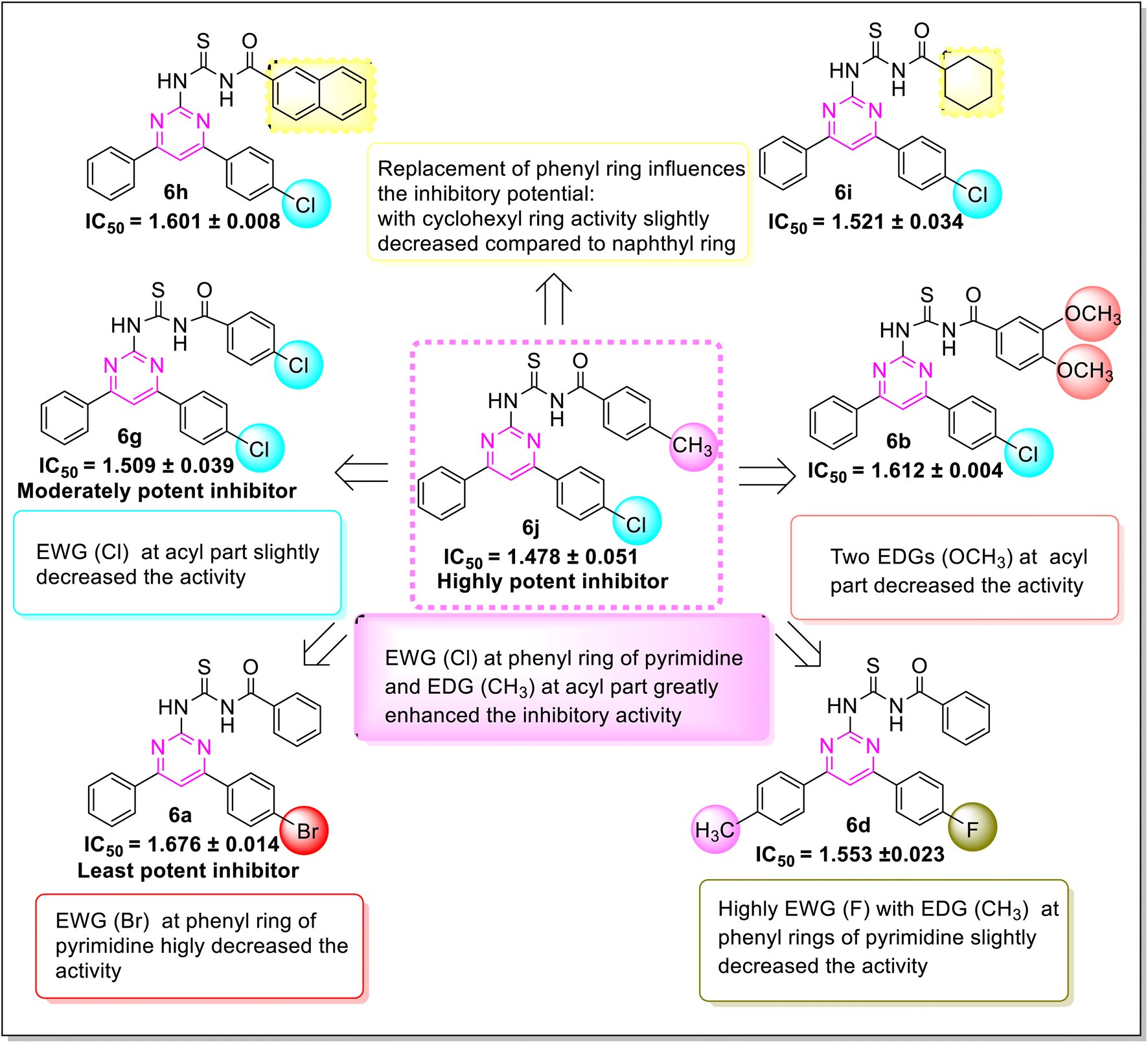
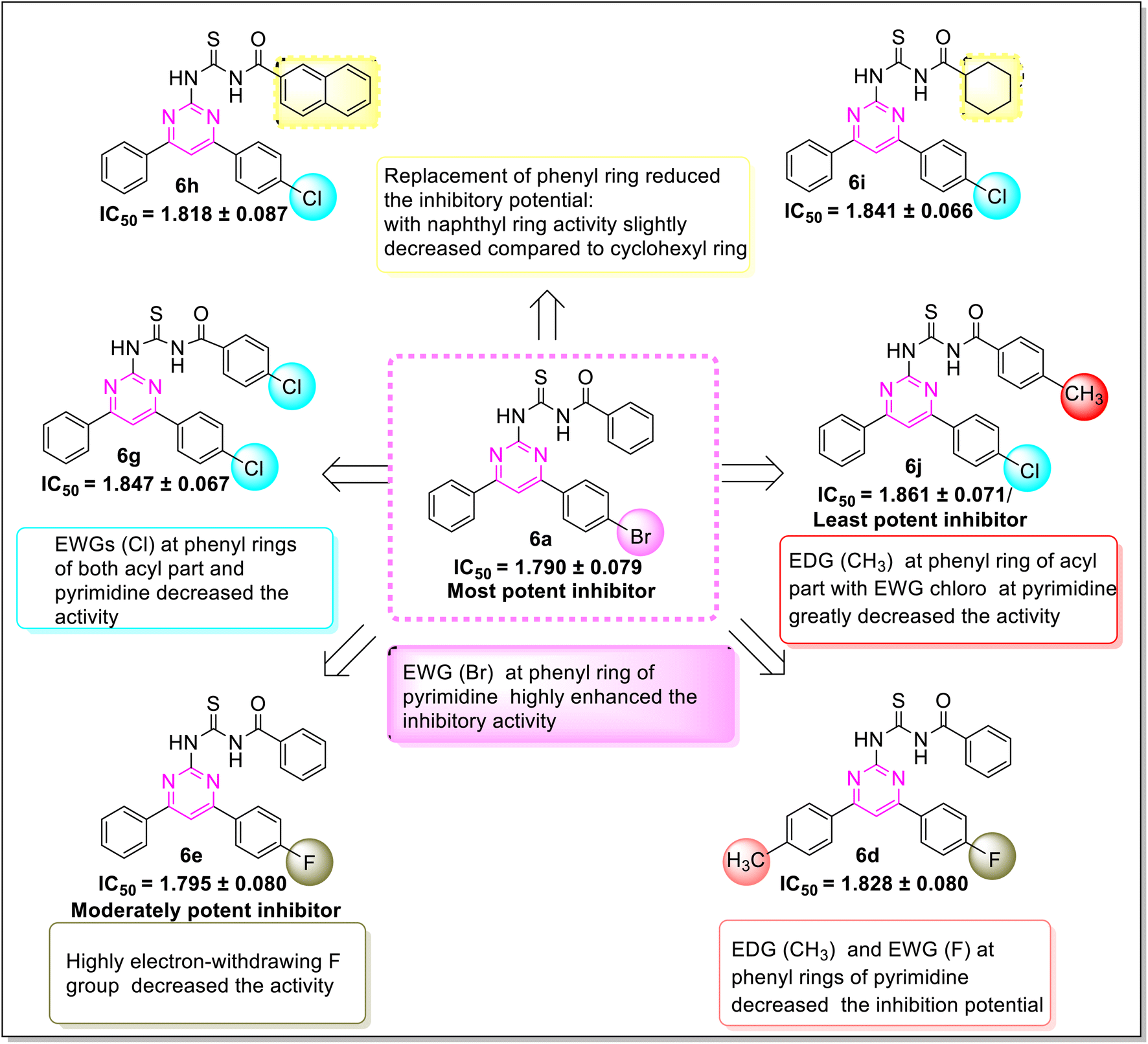
Various structure–activity relationships (SAR) unveiled that the position and nature of the substituents on the peripheral phenyl moiety of the pyrimidine pharmacophore and acyl part has a strong influence on the inhibition activity (Fig. 2). From the synthesized library (6a–j), compound (6j) incorporating methyl (electron-donating group, EWG) and chloro (electron-withdrawing group, EWG) substituents at the para position of phenyl rings provided the strongest activity against the targeted α-amylase enzyme. Compound (6i) (IC50 1.521 ± 0.034 μM) incorporating cyclohexyl group instead of phenyl ring exhibited a significant improvement in α-amylase inhibition. The addition of EDG methyl at the para position of the phenyl ring of pyrimidine pharmacophore (6d) (IC501.553 ± 0.023 μM) enhanced the inhibition activity compared to (6e) (IC50 1.662 ± 0.010 μM). The replacement of methyl with chloro substituent of compound (6g) (IC50 1.509 ± 0.039 μM) slightly reduced the activity. The introduction of two methoxy substituents at the phenyl ring of acyl part (6b) yielded diminished activity with an IC50 value of 1.612 ± 0.004 μM. Furthermore, EDG at para position of phenyl ring of acyl part (6j) showed a more prominent potency than compound (6f) (IC50 1.664 ± 0.010 μM) with a methyl group at ortho position. The presence of a large size bromo group at para position of compound (6a) exhibited a drastic decline in α-amylase inhibition activity (Fig. 3).
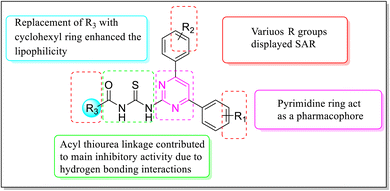 |
| | Fig. 2 General illustration of structural requirements for inhibition activity. | |
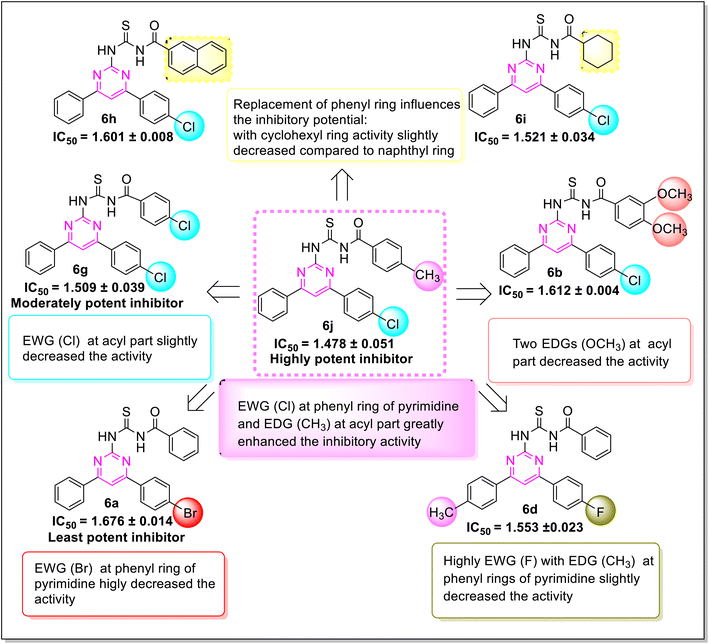 |
| | Fig. 3 SAR of synthesized compounds against α-amylase inhibition. | |
2.3. In vitro proteinase K inhibition and structure–activity relationship
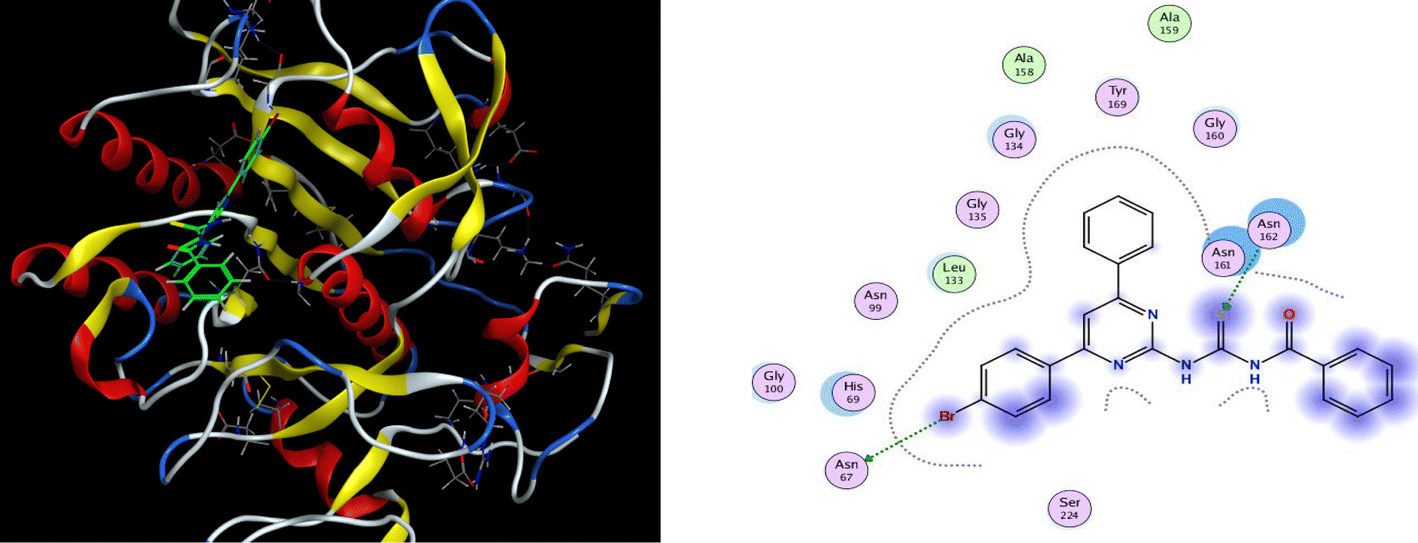
Proteinase K inhibitory activity of the compounds (6a–j) was assessed using a reported in vitro assay and the results are described as IC50 values in Table 1. Phenylmethyl sulfonyl fluoride was employed as a positive control having noteworthy enzyme inhibition with IC50 value of 0.119 ± 0.014 μM. The results revealed that the highest inhibition potential is exhibited by compound (6a) with IC50 value of 1.790 ± 0.079 μM. Additionally, compounds (6f) (IC50 1.794 ± 0.080 μM) and (6e) (IC50 1.795 ± 0.080 μM) also showed significant activity. The other derivatives have moderate inhibitory activity with the range of IC50 values (1.81–1.86 μM). Hence, all the compounds showed prominent proteinase K inhibition activity.
A significant structure–activity relationship is also observed in the case of proteinase K inhibition. The inhibition potential of the compound (6a) revealed that the (EWG) bromo substituted derivative was the most active inhibitor of the library. As the electronegativity of substituents on phenyl moiety of compounds (6a, 6f, and 6e) increased (Br > Cl > > F), a gradual decline in inhibitory activity was observed. The compound (6b) (IC50 1.814 ± 0.086 μM) with an EDG methoxy on phenyl ring of the acyl part exhibited a decreased potency. The IC50 values of compounds (6i) (IC50 1.841 ± 0.066 μM) and (6h) (IC50 1.818 ± 0.087 μM) ascertained that the exchange of the cyclohexyl with naphthyl group on the acyl part slightly improved the inhibition potency. The compound (6j) (IC50 1.861 ± 0.071 μM) bearing methyl and chloro substituents at para position showed a minimum activity as compared to another derivative (6f) (IC50 1.794 ± 0.080 μM) having a methyl group at ortho position (Fig. 4).
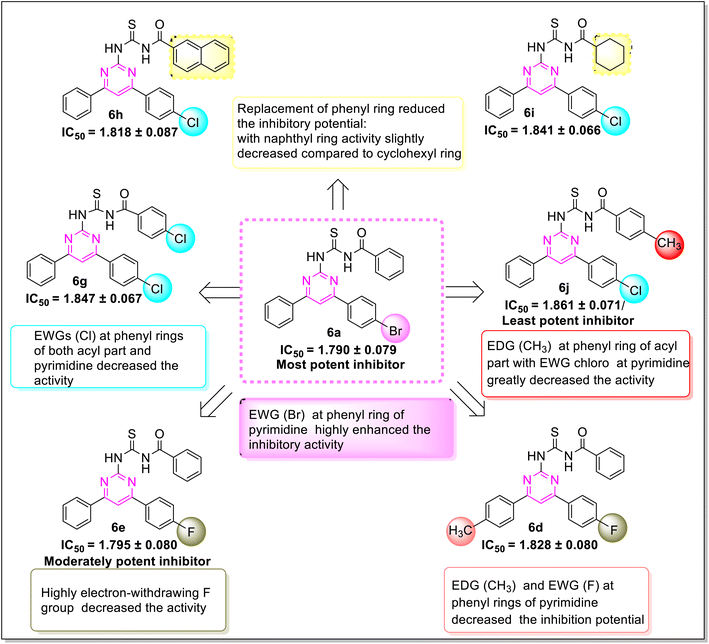 |
| | Fig. 4 SAR of synthesized compounds against proteinase K Inhibition. | |
2.4. Antibacterial activity and structure–activity relationship
The antibacterial potential of the synthesized series (6a–j) was estimated through a disc diffusion method. The bacterial strains, namely Escherichia coli and Bacillus subtilis were employed in this assay and Rifampin was utilized as a positive control. Table 1 summarizes all the results, which were expressed in terms of inhibition zones. These results illustrated that all compounds have antibacterial potential. Amongst all, the compound (6d) exhibited significant inhibition activity for E. coli with 8 mm zone of inhibition. Similarly compound (6i) showed highest activity for B. subtilis with 11 mm zone of inhibition. All other compounds showed moderate antibacterial activity with a range of zone of inhibition (6–9 mm) against both bacterial strains. SAR showed that the simultaneous presence of both EDGs, as well as EWGs at phenyl rings of pyrimidine scaffold (6d) enhanced the activity against E. coli. In the case of another bacterial strain B. subtilis, compound (6i) with more lipophilic cyclohexyl ring as compared to other derivatives displayed high antibacterial potential.
2.5. Molecular docking studies
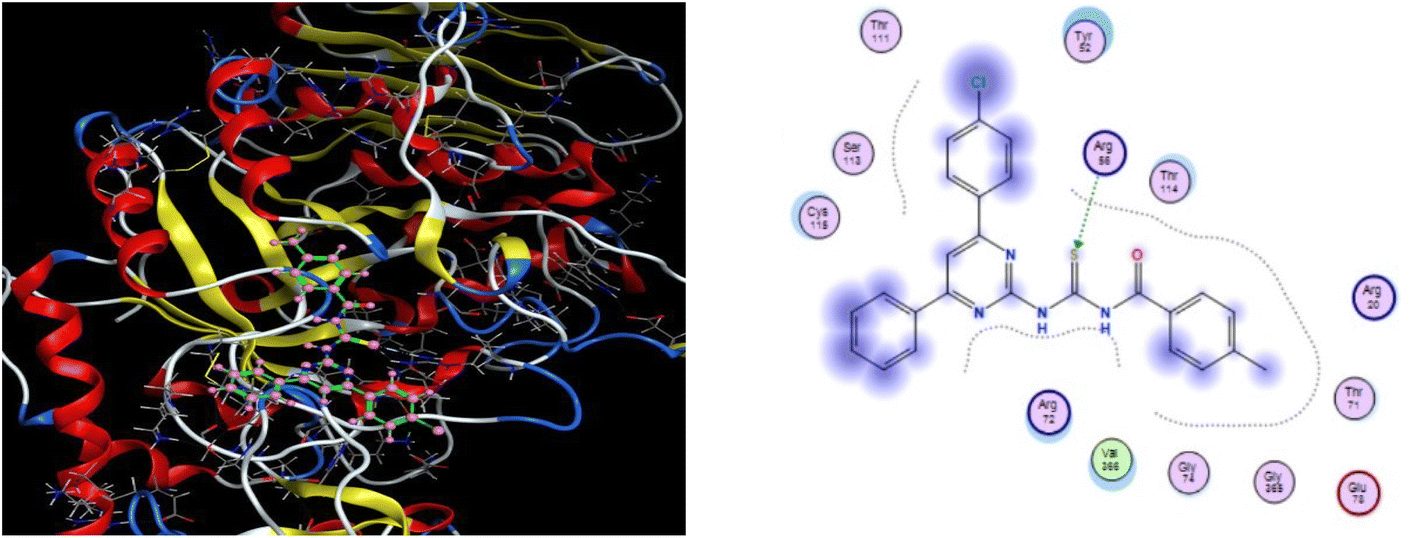
Molecular docking of all the synthesized ligands (6a–j) was conducted using MOE software to evaluate the binding affinities between the prepared ligand molecules (6a–j) and targeted enzymes (α-amylase and proteinase K). The results of docking studies are illustrated in Table 2. Interestingly, the ligand (6b) showed the highest binding interaction with a −7.18 kcal mol−1 docking score in the active site of α-amylase. The S atom and N–H of thiourea formed a hydrogen bonding interaction with the residue GLN-63 and HIS-305 in the active pocket of the α-amylase receptor protein. Further, the phenyl ring of the pyrimidine scaffold showed arene–H interaction with GLU-233. For the most potent compound (6j), the S atom interacted with ARG-56 residue in the active pocket of α-amylase at the distance of 3.2 Å and showed significant binding score of −6.15 kcal mol−1 (Fig. 5). The other remaining compounds presented good binding interaction with optimum binding score (−6.14 to −6.84 kcal mol−1) at different residues with a range of bond distance (2.1–4.3 Å).
Table 2 Docking results of synthesized ligands(6a–j) against targeted enzymesa
| Compd |
α-Amylase inhibition |
Proteinase K inhibition |
| Binding energy (kcal mol−1 |
Distance (Å) |
Receptor residues |
| a |
b |
a |
b |
a |
b |
| a: α-amylase inhibition; b: proteinase K inhibition. |
| 6a |
−6.190 |
−6.11 |
3.83 |
3.89 |
ASN-64, 162 |
ASN-67, 162 |
| 6b |
−7.1822 |
−6.5684 |
3.21 |
2.97 |
GLU-233, HIS-305, GLN-63 |
ASN-161 |
| 6c |
−6.6531 |
−6.2101 |
4.09 |
4.06 |
VAL-366, ILE-367, ARG-56 |
ASN161 |
| 6d |
−6.1470 |
−6.5185 |
3.02 |
3.52 |
ARG-185, ASP-5 |
ARG-56, 72, GLY-365 |
| 6e |
−6.5302 |
−5.9442 |
2.15 |
2.56 |
TRP-59, HIS-305 |
TRY-82, PRO-7 |
| 6f |
−6.8378 |
−6.3726 |
2.93 |
3.25 |
TRP-59, HIS-305 |
PRO-7, ARG-19, GLY-185 |
| 6g |
−6.1474 |
−5.7494 |
4.3 |
2.36 |
GLY-163, THR-63 |
TRY-8, PRO-7 ALA-6, LEU-209 |
| 6h |
−6.7454 |
−6.2833 |
3.57 |
4.32 |
ARG-56 |
ASN-161 |
| 6i |
−6.4653 |
−6.4931 |
2.89 |
2.02 |
THR-59, GLN-63 |
ASN-162 |
| 6j |
−6.1592 |
−6.4836 |
3.26 |
3.70 |
ARG-56 |
HIS-69, ASN-161 |
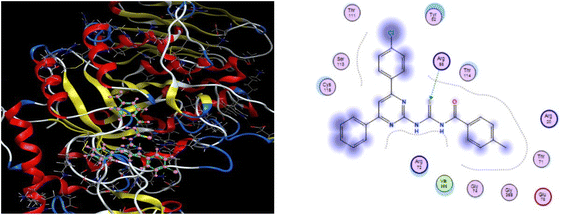 |
| | Fig. 5 3D docking pose and 2D binding interaction of the most potent compound (6j) in the binding pocket of α-amylase. | |
Similarly, all the ligands were also docked with proteinase K receptor protein. The strongest binding interaction with binding energy −6.56 kcal mol−1 was observed for compound (6b) which unveiled the hydrogen bonding interaction of N–H with ASN-161 at the distance of 2.9 Å. For the most active compound (6a), S atom and Br showed interaction with ASN-67 and ASN-162 residue in the active pocket of receptor protein, respectively and exhibited the docking score of −6.11 kcal mol−1 (Fig. 6). Moreover, the other remaining compounds also presented good binding interactions with the optimum binding energy range −5.7 to −6.5 kcal mol−1 and showed various interactions at different residues with a range of bond distance (2.0–4.3 Å).
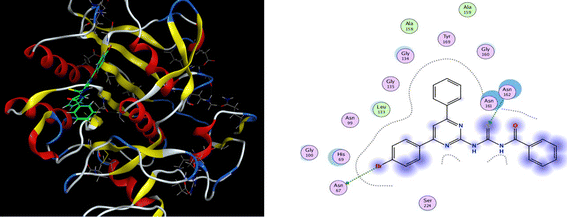 |
| | Fig. 6 3D docking pose and 2D binding interaction of the most potent compound (6a) in the binding pocket of proteinase K. | |
Consequently, all docking results emphasized the prominent role of pyrimidine linked acyl thioureas as amylase and proteinase K inhibitors.
2.6. ADME studies
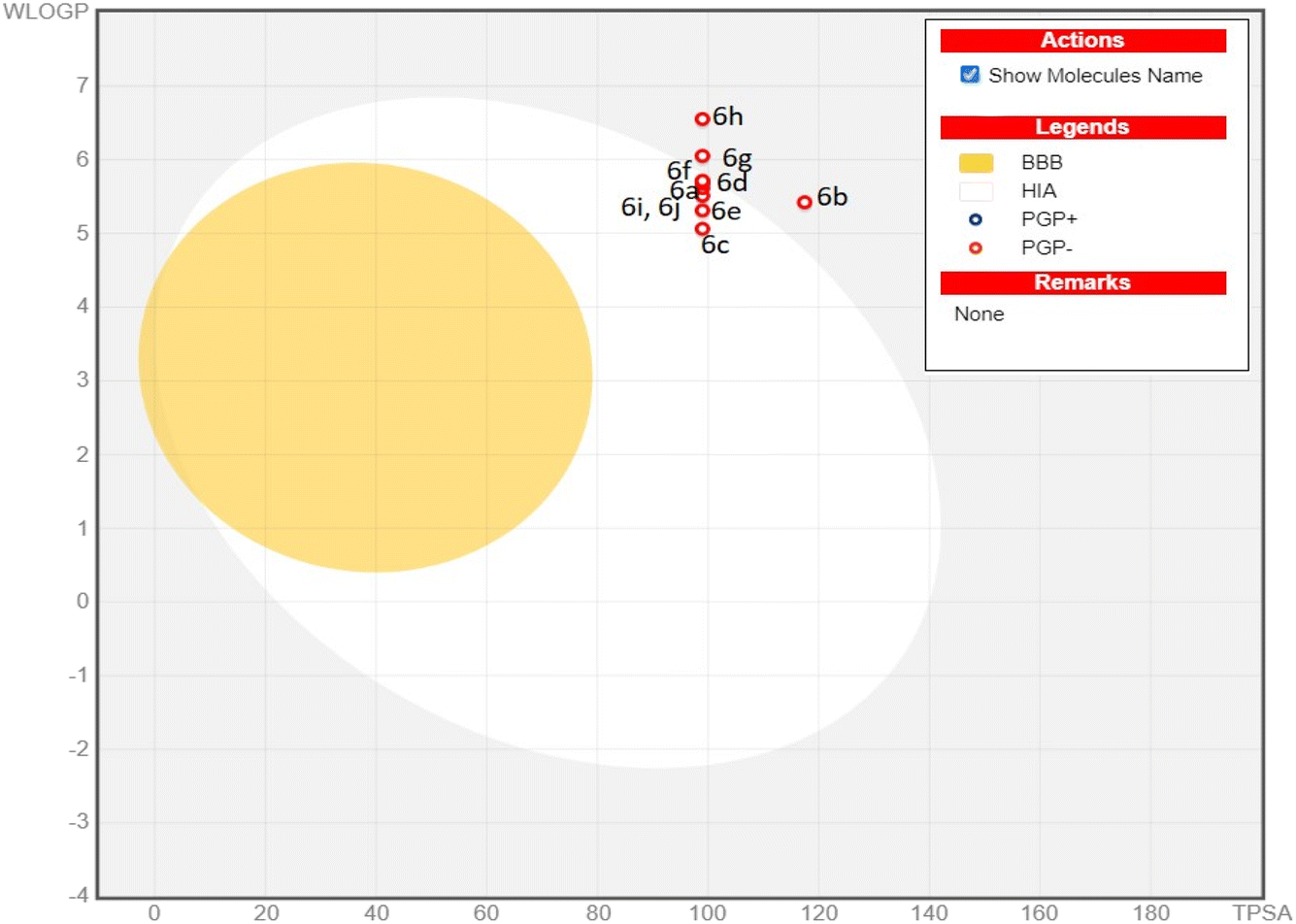
In drug research and development, the chemical compound must have ideal physicochemical properties, pharmacokinetics, and pharmacological activities, to be a drug candidate. Recently, computer-assisted estimations of drug candidates' ADME properties (absorption, distribution, metabolism, and excretion) in drug research and development studies have minimized drug formulation, development, cost, and time.28 Therefore, in this study, the various physicochemical features, pharmacokinetic parameters, and drug-like properties of compounds (6a–j) were determined through the Swiss ADME online web tool, and detailed results are illustrated in Table 3. The results revealed a suitable number of rotatable bonds (7–9), H-bond donors (2), H-bond acceptors (3–5), and value of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/w (iLOGP, lipophilicity 3.1–4.2), indicating that most of the derivatives followed the Lipinski's rule of 5. The compounds (6b) (molecular weight > 500 g mol−1), 6g, and 6h are exceptions due to the low gastrointestinal absorption. Topological polar surface area (TPSA), being a reliable predictor of bioavailability is correlated with the hydrogen bonding of a molecule. The derivatives (6a–j) possessed TPSA in the optimum range of 99–117.4 Å. Therefore, considering the drug-like parameters anticipated by ADME, several compounds exhibited drug-like behaviour. The enzymes, Cytochrome P450 (CYP) are essential for drug metabolism and their inhibition is important in drug discovery, development, and clinical practice.29 All compounds (6a–j) exhibited inhibition of various enzymes, namely CYP1A2, CYP2C9, CYP2D6, CYP2C19, and CYP3A4. The potential for gastrointestinal absorption and blood–brain barrier (BBB) permeability has been analyzed using the BOILED-Egg plot, a predictive model based on the lipophilicity (logP) and polarity (TPSA) of molecules. The plot consists of three distinct regions. As shown in the Fig. 7, the white region (albumin) of the BOILED egg plot represents the molecules with high gastrointestinal (GI) absorption potential, while the yellow region indicates the potential blood–brain barrier (BBB) permeability. The grey area is reserved for molecule with low gastrointestinal absorption and low brain penetration. Some synthesized compounds (6a, 6c, 6d, 6e, 6f, 6i and 6j) located in the white zone, suggesting a high potential for GI absorption and these compounds are more likely to be bioavailable when administered orally. While other compounds (6b, 6g, and 6h) in the grey zone exhibit low GI absorption and low brain penetration. Additionally, the BOILED-Egg plot can predict glycoprotein permeability (PGP). The compounds represented by red dots indicate they are not likely to be effluxed by P-glycoprotein (Pgp), meaning they may have better absorption characteristics and are less likely to be pumped out of cells. All compounds possessed a bioavailability score of 0.55. Consequently, the in silico data supported the potential of all compounds except (6b, 6g and 6h) for oral bioavailability and suggested the pyrimidine-based acyl thiourea scaffold as a promising candidate for developing novel orally active drugs.
Po/w (iLOGP, lipophilicity 3.1–4.2), indicating that most of the derivatives followed the Lipinski's rule of 5. The compounds (6b) (molecular weight > 500 g mol−1), 6g, and 6h are exceptions due to the low gastrointestinal absorption. Topological polar surface area (TPSA), being a reliable predictor of bioavailability is correlated with the hydrogen bonding of a molecule. The derivatives (6a–j) possessed TPSA in the optimum range of 99–117.4 Å. Therefore, considering the drug-like parameters anticipated by ADME, several compounds exhibited drug-like behaviour. The enzymes, Cytochrome P450 (CYP) are essential for drug metabolism and their inhibition is important in drug discovery, development, and clinical practice.29 All compounds (6a–j) exhibited inhibition of various enzymes, namely CYP1A2, CYP2C9, CYP2D6, CYP2C19, and CYP3A4. The potential for gastrointestinal absorption and blood–brain barrier (BBB) permeability has been analyzed using the BOILED-Egg plot, a predictive model based on the lipophilicity (logP) and polarity (TPSA) of molecules. The plot consists of three distinct regions. As shown in the Fig. 7, the white region (albumin) of the BOILED egg plot represents the molecules with high gastrointestinal (GI) absorption potential, while the yellow region indicates the potential blood–brain barrier (BBB) permeability. The grey area is reserved for molecule with low gastrointestinal absorption and low brain penetration. Some synthesized compounds (6a, 6c, 6d, 6e, 6f, 6i and 6j) located in the white zone, suggesting a high potential for GI absorption and these compounds are more likely to be bioavailable when administered orally. While other compounds (6b, 6g, and 6h) in the grey zone exhibit low GI absorption and low brain penetration. Additionally, the BOILED-Egg plot can predict glycoprotein permeability (PGP). The compounds represented by red dots indicate they are not likely to be effluxed by P-glycoprotein (Pgp), meaning they may have better absorption characteristics and are less likely to be pumped out of cells. All compounds possessed a bioavailability score of 0.55. Consequently, the in silico data supported the potential of all compounds except (6b, 6g and 6h) for oral bioavailability and suggested the pyrimidine-based acyl thiourea scaffold as a promising candidate for developing novel orally active drugs.
Table 3 Physicochemical and ADME properties of synthesized compounds (6a–j)
| Compd |
6a |
6b |
6c |
6d |
6e |
6f |
6g |
6h |
6i |
6j |
| MW |
489.39 |
504.99 |
424.52 |
442.51 |
428.48 |
458.96 |
479.38 |
494.99 |
450.98 |
458.96 |
| Rotatable bonds |
7 |
9 |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
| H-bonds acceptors |
3 |
5 |
3 |
4 |
4 |
3 |
3 |
3 |
3 |
3 |
| H-bond donors |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
| TPSA |
99 |
117.4 |
99 |
99 |
99 |
99 |
99 |
99 |
99 |
99 |
| Lipinski violations |
0 |
1 |
0 |
0 |
0 |
0 |
1 |
1 |
0 |
0 |
| iLOGP |
3.97 |
4.29 |
3.81 |
4 |
3.86 |
4.01 |
4.1 |
3.12 |
4.16 |
4.18 |
| CYP1A2 inhibitor |
— |
— |
— |
— |
— |
— |
— |
— |
Yes |
— |
| CYP2C19 inhibitor |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
| CYP2C9 inhibitor |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
— |
Yes |
Yes |
| CYP2D6 inhibitor |
— |
Yes |
Yes |
— |
Yes |
— |
— |
— |
— |
— |
| CYP3A4 inhibitor |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
Yes |
| Synthetic accessibility |
3.03 |
3.33 |
3.09 |
3.14 |
3.01 |
3.1 |
3 |
3.22 |
3.24 |
3.12 |
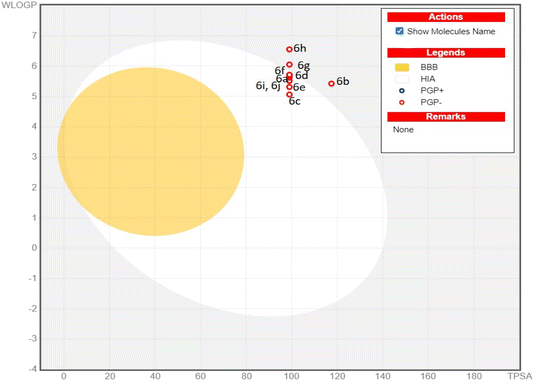 |
| | Fig. 7 Boiled egg plot for the synthesized compounds (6a–j). | |
3. Experimental section
3.1. Materials and methods
StuartSMP3 melting point equipment was utilized to record the melting points of all compounds (uncorrected). 1H and 13C-NMR spectra of the compounds were acquired from Bruker Avance (300 and 75 MHz) in deuterated DMSO-d6 solvent. The ppm-scale (δ) was used to describe the chemical shifts and the J values (coupling constants) were measured in hertz (Hz). To monitor the reaction, (TLC) using aluminum pre-coated plates with silica gel Kiesel 60 F254 was performed in a solvent system (7:3 n-hexane:ethyl acetate), and Rf values were calculated carefully. UV active compounds on the chromatogram were easily visualized with UV-light at the wavelength of 254 and 360 nm. Staining agents were also used for the visualization of compounds.
3.2. General protocol for the synthesis of pyrimidine linked acyl thioureas (6a–j)
Differently substituted chalcones (3a–e) were prepared from various acetophenones (1a–b) (0.004 mol) and aldehydes (2a–d) (0.004 mol) using KOH base (0.004 mol) in dry ethanol at 25 °C. Then these derivatives were further converted into aminopyrimidines (4) with guanidine hydrochloride (0.015 mol) and KOH base (0.001 mol) in dry ethanol at 78 °C. Upon completion, as observed by TLC, the whole mixture was added to chilled water and the solid product was collected from filtration. Different acyl chlorides were synthesized by treating acids (5a–g) (0.004 mol) with SOCl2 (0.004 mol) at 50 °C under N2 for 5–6 hours. Then anhydrous KSCN (0.006 mol) dissolved in dry acetone is added to the acyl chloride and refluxed for 1.5 hours. The solution of aminopyrimidine (4a–e) (0.003 mol) in dry acetone is added to the freshly prepared acyl isothiocyanates and refluxed for 17–18 hours. In the end, after the successful completion of the reaction, the solid product from the aqueous workup was washed with hot ethanol. The pure acyl thioureas (6a–j) were obtained in good yields.
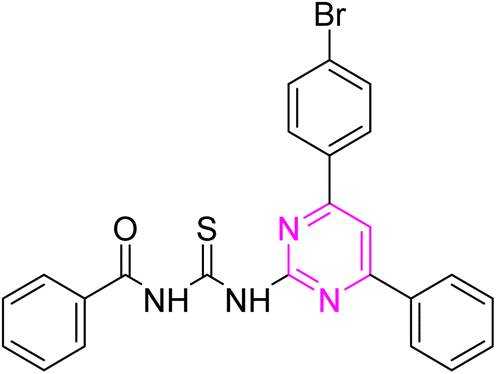
3.2.1. N-((4-(4-Bromophenyl)-6-phenylpyrimidin-2-yl)carbamothioyl) benzamide (6a). Pale yellow solid; mp.: 214– 216 °C; yield: 68%; Rf: 0.64 (n-hexane/ethyl acetate 7:3); FTIR (ATR, cm−1), 3143, 3050 (N–H), 2970 (C–H, Ar), 1723 (CO), 1597 (CN), 1513 (CC), 1238 (CS), 1176 (C–N). 1H NMR (300 MHz, DMSO-d6) δ: 12.70 (s, 1H, NH), 12.64 (s, 1H, NH), 8.41 (s, 1H), 8.31–8.27 (m, 3H), 8.23–8.17 (m, 1H), 7.98–7.93 (m, 2H), 7.79–7.72 (m, 2H), 7.67–7.44 (m, 6H); 13C NMR (75 MHz, DMSO-d6) δ: 178.74 (CS), 167.44 (CO), 165.93, 164.65, 158.00, 136.09, 135.41, 133.53, 133.31, 132.34, 132.01, 130.04, 129.73, 129.34, 129.05, 128.09, 125.81, 109.11. Anal. calcd for C24H17BrN4OS [489.39]: C, 58.90; H, 3.50; N, 11.45; S, 6.55%. Found: C, 58.88; H, 3.53; N, 11.44; S, 6.56%.
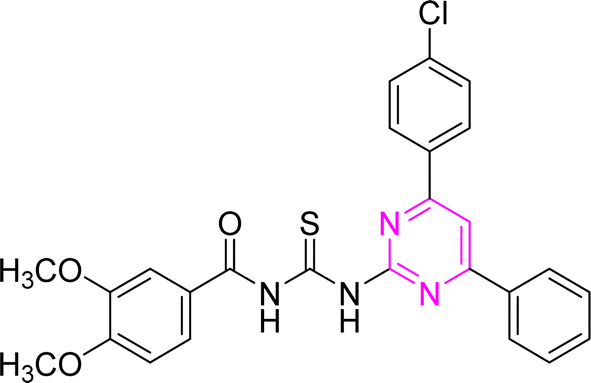
3.2.2. N-((4-(4-Chlorophenyl)-6-phenylpyrimidin-2-yl)carbamothioyl)-3,4-dimethoxybenzamide (6b). Yellow solid; mp.: 254–257 °C; yield: 66%; Rf: 0.52 (n-hexane/ethyl acetate 7:3); FTIR (ATR, cm−1), 3230, 3100 (N–H), 2968 (C–H, Ar), 2941 (C–H, CH3), 1718 (CO), 1590 (CN), 1514 (CC), 1220 (CS), 1154 (C–N). 1H NMR (300 MHz, DMSO-d6) δ: 12.85 (s, 1H, NH), 12.55 (s, 1H, NH), 8.40 (s, 1H), 8.34–8.26 (m, 4H), 7.59–7.49 (m, 5H), 7.07 (s, 2H), 6.73 (s, 1H), 3.71 (s, 6H, OCH3). 13C NMR (75 MHz, DMSO-d6) δ: 178.78 (CS), 166.02 (CO), 164.59, 160.84, 158.02, 136.64, 135.99, 135.01, 134.93, 132.11, 132.05, 131.49, 129.83, 129.36, 129.36, 129.30, 128.04, 122.51, 106.60, 55.92 (CH3). Anal. calcd for C26H21ClN4O3S [504.99]: C, 61.84; H, 4.19; N, 11.09; S, 6.35%. Found: C, 61.83; H, 4.18; N, 11.08; S, 6.33%.
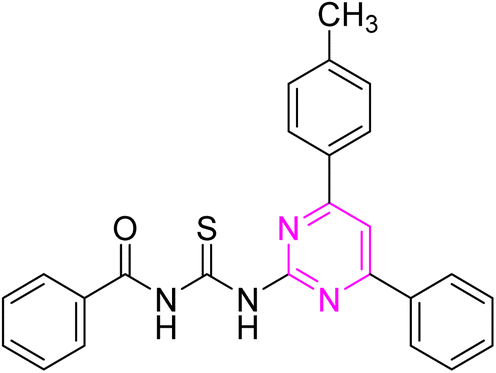
3.2.3. N-((4-Phenyl-6-(p-tolyl)pyrimidin-2-yl)carbamothioyl)benzamide (6c). Yellow solid; mp.: 236–238 °C; yield: 69%; Rf: 0.63 (n-hexane/ethyl acetate 7:3); FTIR (ATR, cm−1), 3328, 3148 (N–H), 3064 (C–H, Ar), 2926 (C–H, CH3), 1736 (CO), 1610 (CN), 1524 (CC), 1250 (CS), 1180 (C–N). 1H NMR (300 MHz, DMSO-d6) δ: 12.78 (s, 1H, NH), 12.55 (s, 1H, NH), 8.33 (s, 1H), 8.28 (d, 2H, J = 7.2 Hz), 8.21 (d, 2H, J = 8.1 Hz), 8.08–8.05 (m, 1H), 7.94 (d, 1H, J = 7.5 Hz), 7.67–7.62 (m, 1H), 7.57–7.43 (m, 4H), 7.32 (d, 1H, J = 8.1 Hz), 7.19 (d, 1H, J = 8.1 Hz), 7.04 (d, 1H, J = 7.8 Hz), 2.39 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ: 178.71 (CS), 167.33 (CO), 165.74, 164.57, 157.98, 142.05, 136.50, 136.25, 133.50, 133.40, 131.85, 130.71, 129.94, 129.31, 129.08, 128.99, 128.04, 126.17, 108. 75, 21.53 (CH3). Anal. calcd for C25H20N4OS: [424.52]: C, 70.73; H, 4.75; N, 13.20; S, 7.55%. Found: C, 70.72; H, 4.76; N, 13.22; S, 7.54%.
3.2.4 N-((4-(4-Fluorophenyl)-6-(p-tolyl)pyrimidin-2-yl)carbamothioyl)benzamide (6d). Yellow solid; mp.: 207–209 °C; yield: 83%; Rf: 0.57 (n-hexane/ethyl acetate 7:3); FTIR (ATR, cm−1), 3250, 3150 (N–H), 3000 (C–H, Ar), 2886 (C–H, CH3), 1716 (CO), 1586 (CN), 1520 (CC), 1228 (CS), 1185 (C–N). 1H NMR (300 MHz, DMSO-d6) δ: 12.61 (s, 2H, NH), 8.39–8.31 (m, 3H), 8.18 (d, J = 7.8 Hz, 2H), 7.94 (d, 2H, J = 7.5 Hz), 7.65 (t, 1H, J = 7.5 Hz), 7.47 (t, 2H, J = 7.8 Hz), 7.36–7.28 (m, 4H), 2.37 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ: 178.65 (CS), 167.43 (CO), 165.73, 164.59 (1JC–F = 245 Hz), 164.42, 157.89, 142.03, 133.34, 133.00, 132.75, 130.54 (3JC–F = 9 Hz), 129.90, 129.06, 129.01,127.97, 116.25 (2JC–F = 21 Hz), 108.52, 21.51 (CH3). Anal. calcd for C25H19FN4OS [442.51]: C, 67.86; H, 4.33; N, 12.66; S, 7.24%. Found: C, 67.87; H, 4.31; N, 12.65; S, 7.28%.
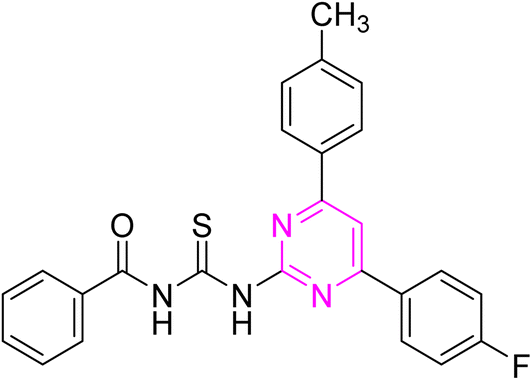
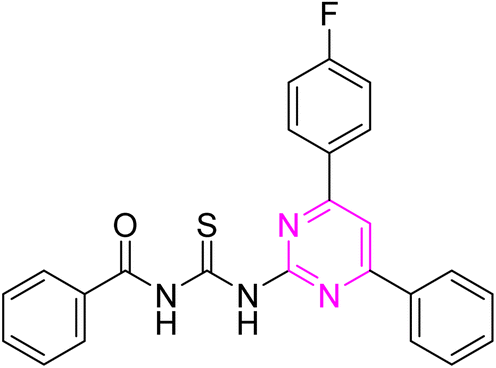
3.2.5. N-((4-(4-Fluorophenyl)-6-phenylpyrimidin-2-yl)carbamothioyl)benzamide (6e). Yellow solid; mp.: 221 –223 °C; yield: 79%; Rf: 0.61 (n-hexane/ethyl acetate 7:3); FTIR (ATR, cm−1), 3144, 3010 (N–H), 2979 (C–H, Ar), 1718 (CO), 1577 (CN), 1506 (CC), 1229 (CS), 1158 (C–N). 1H NMR (300 MHz, DMSO-d6) δ: 12.65 (s, 2H, NH), 8.39–8.27 (m, 5H), 7.95 (m, 2H, J = 6.6 Hz), 7.68–7.36 (m, 8H). 13C NMR (75 MHz, DMSO-d6) δ: 178.71 (CS), 167.40 (CO), 165.82, 164.67, 164.48, (1JC–F = 231 Hz), 157.95, 136.15, 133.53, 133.36, 132.74, 131.94, 130.60 (3JC–F = 8.2 Hz), 129.33, 129.06, 129.01, 128.06, 116.32 (2JC–F = 21 Hz), 108.99. Anal. calcd for C24H17FN4OS [428.49]: C, 67.28; H, 4.00; N, 13.08; S, 7.48%. Found: C, 67.27; H, 3.98; N, 13.09; S, 7.47%.
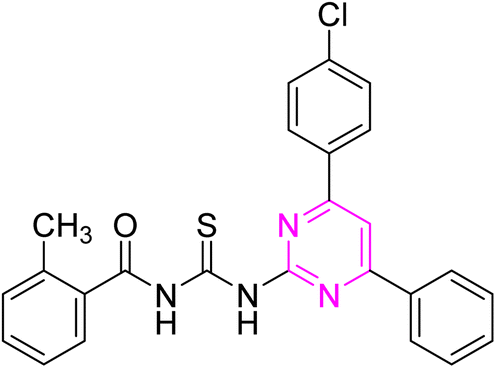
3.2.6. N-((4-(4-Chlorophenyl)-6-phenylpyrimidin-2-yl)carbamothioyl)-2 methylbenzamide (6f). Light yellow solid; mp.: 240–242 °C; yield: 64%; Rf: 0.62 (n-hexane/ethyl acetate 7:3); FTIR (ATR, cm−1), 3260, 3130 (N–H), 2970 (C–H, Ar), 2940 (C–H, CH3), 1721 (CO), 1590 (CN), 1509 (CC), 1230 (CS), 1178 (C–N). 1H NMR (300 MHz, DMSO-d6) δ: 12.74 (s, 1H, NH), 12.62 (s, 1H, NH), 8.40 (s, 1H), 8.34–8.25 (m, 4H), 7.59–7.43 (m, 7H), 7.33 (d, 1H, J = 7.5 Hz), 7.26 (t, 1H, J = 7.2 Hz), 2.42 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ: 178.39 (CS), 169.51 (CO), 165.85, 164.46, 157.97, 136.83, 136.73, 135.98, 134.97, 134.92, 132.03, 131.53, 131.32, 129.76, 129.51, 129.36, 128.43, 127.97, 126.26, 109.03, 19.95 (CH3). Anal. calcd. for C25H19ClN4OS [458.96]: C, 65.42; H, 4.17; N, 12.21; S, 6.99%. Found: C, 65.40; H, 4.16; N, 12.22; S, 6.97%.
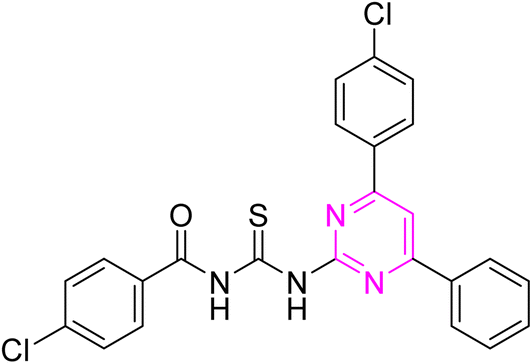
3.2.7. 4-Chloro-N-((4-(4-chlorophenyl)-6-phenylpyrimidin-2-yl)carbamothioyl) benzamide (6g). Light green solid; mp.: 261–263 °C; yield: 74%; Rf: 0.59 (n-hexane/ethyl acetate 7:3); FTIR (ATR, cm−1), 3148, 3114 (N–H), 2926 (C–H, Ar), 1721 (CO), 1593 (CN), 1500 (CC), 1226 (CS), 1176 (C–N). 1H NMR (300 MHz, DMSO-d6) δ: 12.67 (s, 2H, N–H), 8.41 (s, 1H), 8.361–8.287 (m, 5H), 7.94 (d, 2H, J = 8.1 Hz) 7.61–7.50 (m, 6H). 13C NMR (75 MHz, DMSO-d6) δ: 178.42 (CS), 165.98 (CO), 165.64, 164.52, 157.92, 136.84, 136.02, 135.09, 132.26, 131.96, 131.92, 131.00, 129.85, 129.40, 129.32, 129.09, 128.09, 109.09. Anal. calcd for C24H16Cl2N4OS [479.38]: C, 60.13; H, 3.36; N, 11.69; S, 6.69%. Found: C, 60.12; H, 3.35; N, 11.67; S, 6.67%.
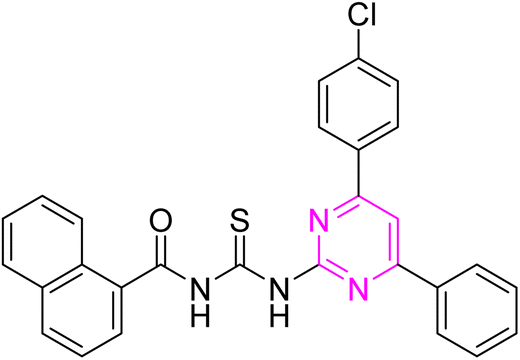
3.2.8. N-((4-(4-Chlorophenyl)-6-phenylpyrimidin-2-yl)carbamothioyl)-1-naphthamide (6h). Yellow solid; mp.: 286–288 °C; yield: 82%; Rf: 0.51 (n-hexane/ethyl acetate 7:3); FTIR (ATR, cm−1), 3358, 3115 (N–H), 3023 (C–H, Ar), 1738 (CO), 1584 (CN), 1557 (CC), 1299 (CS), 1157 (C–N). 1H NMR (300 MHz, DMSO-d6) δ: 13.00 (s, 1H, NH), 12.67 (s, 1H, NH), 8.38 (s, 1H), 8.31–8.22 (m, 5H), 8.14 (d, 1H, J = 8.1 Hz), 8.06–8.03 (m, 1H), 7.89 (d, 1H, J = 7.2 Hz), 7.65–7.43 (m, 8H). 13C NMR (75 MHz, DMSO-d6) δ: 178.53 (CS), 168.86 (CO), 165.84, 164.45, 158.01, 136.81, 135.97, 134.96, 133.60, 132.34, 132.21, 131.99, 130.00, 129.72, 129.37, 129.31, 128.98, 128.21, 127.94, 127.57, 127.08, 125.37, 125.18, 109.03. Anal. calcd for C28H19ClN4OS [495.00]: C, 67.94; H, 3.87; N, 11.32; S, 6.48%. Found: C, 67.95; H, 3.86; N, 11.30; S, 6.47%.
3.2.9. N-((4-(4-Chlorophenyl)-6-phenylpyrimidin-2-yl)carbamothioyl) cyclohexane carboxamide (6i). Pale yellow solid; mp.: 175–177 °C; yield: 71%; Rf: 0.70 (n-hexane/ethyl acetate 7:3); FTIR (ATR, cm−1), 3180, 3028 (N–H), 2927 (C–H, Ar), 2851 (C–H, aliphatic), 1692 (CO), 1584 (CN), 1524 (CC), 1466 (CH2 bending), 1234 (CS), 1142 (C–N). 1H NMR (300 MHz, DMSO-d6) δ: 13.26 (s, 1H, NH), 11.41 (s, 1H, NH), 8.31 (s, 1H), 7.97–7.95 (m, 4H), 7.60–7.49 (m, 5H), 2.38 (q, 1H, J = 7.2 Hz), 1.73–1.48 (m, 6H), 1.46–1.43 (m, 4H). 13C NMR (75 MHz, DMSO-d6) δ: 177.52 (CS), 167.40 (CO), 165.21, 162.05, 158.32, 135.81, 134.33, 133.92, 129.31, 129.27, 128.93, 128.71, 127.59, 108.79, 43.31, 29.76, 25.37, 24.81. Anal. calcd for C24H23ClN4OS [450.99]: C, 63.92; H, 5.14; N, 12.42; S, 7.11%. Found: C, 63.91; H, 5.11; N, 12.40; S, 7.12%.
3.2.10. N-((4-(4-Chlorophenyl)-6-phenylpyrimidin-2-yl)carbamothioyl)-4-methylbenzamide (6j). Light green solid; mp.: 233–235 °C; yield: 67%; Rf: 0.66 (n-hexane/ethyl acetate 7:3); FTIR (ATR, cm−1), 3166, 3060 (N–H), 2971 (C–H Ar), 2950 (C–H, CH3), 1716 (CO), 1599 (CN), 1505 (CC), 1253 (CS), 1159 (C–N). 1H NMR (300 MHz, DMSO-d6) δ: 12.91 (s, 1H, NH), 12.13 (s, 1H, NH), 8.35 (s, 1H), 8.27–8.11 (m, 4H), 7.93 (d, 2H, J = 7.5 Hz), 7.60–7.49 (m, 5H), 7.33 (d, 2H, J = 7.5 Hz), 2.41 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ: 177.51 (CS), 167.46 (CO), 165.21, 162.06, 158.31, 141.82, 135.84, 134.32, 133.94, 130.20, 129.31, 129.24, 129.15, 128.91, 128.76, 127.52, 127.43, 108.39, 21.34 (CH3). Anal. calcd for C25H19ClN4OS [458.96]: C, 65.42; H, 4.17; N, 12.21; S, 6.99%. Found: C, 65.40; H, 4.19; N, 12.20; S, 6.98%.
4. Methodologies
4.1. Antibacterial assay
The disc diffusion assay assessed the newly synthesized compounds' antibacterial properties. Escherichia coli and Bacillus subtilis were grown overnight in nutritional broth. The cultures were quickly adjusted to (0.5%) McFarland standard turbidity and uniformly disseminated onto sterile Mueller-Hinton agar plates for bacteria using sterile cotton brushes. Sterile paper discs (4 mm diameter) were impregnated with 10 μL of synthetic compounds and compared to positive and negative controls using standard antibiotics (Rifampin for bacteria) and DMSO. The impregnated discs were then placed on inoculated agar plates and incubated at 37 °C for 24 hours for bacteria. The zone of inhibition around each disc were measured in millimeters after incubation.30
4.2. Proteinase K inhibition assay
The newly prepared compounds (6a–j) were evaluated for their inhibition activity using the in vitro Proteinase K inhibition assay. Proteinase K obtained from Sigma-Aldrich was dissolved in Tris–HCl buffer (pH 8.0) at 25 μg mL−1. The synthesized compounds were dissolved in DMSO to form stock solutions, then diluted in buffer to reach concentrations from 0.1 to 100 μM. For 15 minutes, 45 μL of Proteinase K solution was pre-incubated with 5 μL of each drug in 96-well microtiter plates at 35 °C. Negative controls included buffer with DMSO, while positive controls contained phenyl methyl sulfonyl fluoride (PMSF), a recognized inhibitor. Next, 50 μL of ρ-nitrophenyl acetate substrate solution (0.2–1.25 mM) was added to each well. A UV-visible spectrophotometer recorded absorbance every 10 seconds during the first 10 seconds of the reaction at 405 nm to measure enzyme activity.31
4.3. α-Amylase inhibition assay
The newly prepared compounds (6a–j) were assessed for α-amylase inhibition in vitro. The α-amylase obtained from Sigma-Aldrich was dissolved in phosphate buffer (pH 6.9) to 1 mg mL−1, and starch was the substrate. The synthesized compounds were dissolved in DMSO to form stock solutions and then diluted in buffer to reach concentrations from 0.1 to 100 μM. For the test, 45 μL of α-amylase solution was pre-incubated with 5 μL of each chemical in 96-well microtiter plates at room temperature for 10 minutes. Positive controls included acarbose, and negative controls contained buffer with DMSO. After the addition of 50 μL of substrate solution to each well, the plate was incubated at 50 °C for 30 minutes. To stop the reaction, 100 μL of HCL (1 M) reagent was added. After cooling to room temperature, a microplate reader measured absorbance at 540 nm.32
4.4. Molecular docking study
Molecular Operating Environment (MOE 2015.10) software was used for the molecular docking studies of newly synthesized compounds. Protein 3D structures (.PDB) of Proteinase K and Amylase possessing 2ID8 and 2QMK fetched from the Protein Data Bank for molecular docking simulations with the above-mentioned compounds. First, any hetero atoms, and water molecules from the protein structures were removed. Then, proteins were subjected to a quick preparation method of MOE to make them stable and ready for docking analysis. Then, the active sites of both proteins were located using the Site Finder function of MOE. The most suitable active site was selected and subjected to dummy atoms for docking with the newly synthesized ligands following docking using GBVI/WAS scoring model. The energy of the interaction, distance and amino acid on the proteins with the compounds were evaluated.17
4.5. ADME analysis
Swiss ADME online web tool (http://www.swissadme.ch/) was used to perform ADME analysis of synthesized compounds (6a–j).33 The canonical SMILES of these compounds were generated from ChemDraw and estimation of physicochemical properties of these compounds including lipophilicity, drug likeness, pharmacokinetics, TPSA, number of rotatable bonds, and violations of Lipinski's rule of five was carried out.
5. Conclusions
In summary, a library of novel diaryl pyrimidine linked acyl thioureas (6a–j) was successfully developed and evaluated for bacterial, α-amylase and proteinase K inhibitory activity. The compound (6j) showed excellent α-amylase inhibitory (IC50 1.478 ± 0.051) activity due to the electron-withdrawing chloro and electron-donating methyl substituents among the developed library. While other compounds (6a, 6e, and 6f) possessed promising inhibition potential (IC50 1.790 ± 0.079, 1.795 ± 0.080, 1.794 ± 0.080 μM) against proteinase K due to the electron-withdrawing bromo, fluoro, chloro and electron-donating methyl substituents. The result of antibacterial activity elucidated that the compound (6d and 6i) showed moderate inhibition potential against E. coli and B. subtilis, respectively. The detailed SAR studies ascertained that the type and position of substitution had a dramatic effect on the enzyme inhibition activity. The binding energy (−7.182 and −6.568 kcal mol−1) of most potent compound (6b), highlighted their effective binding interactions within the active site of α-amylase and proteinase K, respectively. Finally, the ADME analysis elicited the drug like properties for most active compounds (6a and 6b). Therefore, our findings from these studies implied that the synthesized molecules (6j and 6a) may be favourable lead compounds for the development of therapeutic agents for diabetes as well as proteinase K-associated diseases.
Data availability
Data available within the article or its ESI.†
Conflicts of interest
There are no potential conflicts to declare.
References
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25(8), 1909 CrossRef CAS.
- V. Sharma, N. Chitranshi and A. K. Agarwal, Int. J. Med. Chem., 2014, 2014(1), 202784 Search PubMed.
- G. Shabir, I. Shafique and A. Saeed, J. Heterocycl. Chem., 2022, 59(10), 1669–1702 CrossRef CAS.
- S. Nadar and T. Khan, Chem. Biol. Drug Des., 2022, 100(6), 818–842 CrossRef CAS.
- B. Nammalwar and R. A. Bunce, Pharmaceuticals, 2024, 17(1), 104 CrossRef CAS PubMed.
- A. Poznyak, A. V. Grechko, P. Poggio, V. A. Myasoedova, V. Alfieri and A. N. Orekhov, Int. J. Mol. Sci., 2020, 21(5), 1835 CrossRef PubMed.
- M. C. Rossi, A. Nicolucci, A. Ozzello, S. Gentile, A. Aglialoro, A. Chiambretti and D. Cucinotta, Nutr. Metabol. Cardiovasc. Dis., 2019, 29(7), 736–743 CrossRef PubMed.
- A. Samrot and A. Vijay, Internet J. Microbiol., 2008, 6(2), 1–6 Search PubMed.
- J. Sabotič and J. Kos, Appl. Microbiol. Biotechnol., 2012, 93, 1351–1375 CrossRef.
- C. J. Silva, E. Vázquez-Fernández, B. Onisko and J. R. Requena, Virus Res., 2015, 207, 120–126 CrossRef CAS.
- C. López-Otín and J. S. Bond, J. Biol. Chem., 2008, 283(45), 30433–30437 CrossRef PubMed.
- A. A. Agbowuro, W. M. Huston, A. B. Gamble and J. D. Tyndall, Med. Res. Rev., 2018, 38(4), 1295–1331 CrossRef CAS PubMed.
- M. Drag and G. S. Salvesen, Nat. Rev. Drug Discovery, 2010, 9(9), 690–701 CrossRef CAS.
- S. A. Ullah, A. Saeed, M. Azeem, M. B. Haider and M. F. Erben, RSC Adv., 2024, 14(25), 18011–18063 RSC.
- A. Saeed, S. U. Rehman, P. A. Channar, F. A. Larik, Q. Abbas, M. Hassan and S. Y. Seo, J. Taiwan Inst. Chem. Eng., 2017, 77, 54–63 CrossRef CAS.
- A. Ahmed, I. Shafique, A. Saeed, G. Shabir, A. Saleem, P. Taslimi, T. T. Tok, M. Kirici, E. M. Üç and M. Z. Hashmi, Eur. J. Med. Chem., 2022, 6, 100082 CAS.
- N. Arshad, M. Shakeel, A. Javed, F. Perveen, A. Saeed, A. Ahmed, H. Ismail, P. A. Channar and F. Naseer, Int. J. Biol. Macromol., 2024, 263, 130231 CrossRef CAS PubMed.
- A. Saeed, F. A. Larik, F. Jabeen, H. Mehfooz, S. A. Ghumro, H. R. El-Seedi, M. Ali, P. A. Channar and H. Ashraf, Russ. J. Gen. Chem., 2018, 88, 541–550 CrossRef CAS.
- W. A. A. Arafa, A. A. Ghoneim and A. K. Mourad, ACS Omega, 2022, 7(7), 6210–6222 CrossRef CAS PubMed.
- A. Saeed, M. N. Mustafa, M. Zain-ul-Abideen, G. Shabir, M. F. Erben and U. Flörke, J. Sulfur Chem., 2019, 40(3), 312–350 CrossRef CAS.
- L. Tong, P. Wang, X. Li, X. Dong, X. Hu, C. Wang, T. Liu, J. Li and Y. Zhou, J. Med. Chem., 2022, 65(4), 3229–3248 CrossRef CAS PubMed.
- R. B. Bakr and N. A. Elkanzi, J. Heterocycl. Chem., 2020, 57(7), 2977–2989 CrossRef CAS.
- J. Li, B. An, X. Song, Q. Zhang, C. Chen, S. Wei, R. Fan, X. Li and Y. Zou, Eur. J. Med. Chem., 2021, 212, 113019 CrossRef CAS PubMed.
- M. Zhang, Y. Wang, S. Wang and H. Wu, Molecules, 2022, 27(22), 8104 CrossRef CAS.
- F. A. Larik, A. Saeed, M. Faisal, P. A. Channar, S. S. Azam, H. Ismail, E. Dilshad and B. Mirza, Comput. Biol. Chem., 2018, 77, 193–198 CrossRef CAS PubMed.
- J. H. Li, Y. Wang, Y. P. Wu, R. H. Li, S. Liang, J. Zhang, Y. G. Zhu and B. Xie, Pestic. Biochem. Physiol., 2021, 172, 104766 CrossRef CAS PubMed.
- U. Zahra, S. Zaib, A. Saeed, M. ur Rehman, G. Shabir, H. O. Alsaab and I. Khan, Int. J. Biol. Macromol., 2022, 198, 157–167 CrossRef CAS PubMed.
- İ. Şahin, M. Çeşme, F. B. Özgeriş and F. Tümer, Chem. Biol. Interact., 2023, 370, 110312 CrossRef PubMed.
- P. Manikandan and S. Nagini, Curr. Drug Targets, 2018, 19(1), 38–54 CAS.
- H. Ismail, B. Mirza, I. U. Haq, M. Shabbir, Z. Akhter and A. Basharat, J. Chem., 2015, 1, 465286 CAS.
- M. Hosseini-Koupaei, B. Shareghi, A. A. Saboury, F. Davar, V. A. Sirotkin, M. H. Hosseini- Koupaei and Z. Enteshari, Int. J. Biol. Macromol., 2019, 122, 732–744 CrossRef CAS.
- H. Ismail, D. Khalid, S. B. Ayub, M. U. Ijaz, S. Akram, M. Z. Bhatti, M. M. Taqi, E. Dilshad, S. Anwaar, G. E. S. Batiha and S. S. Aggad, Biomed Res. Int., 2023, 1, 1725638 CrossRef PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7(1), 42717 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Hammad Ismailb,
Sadaf Anwaarc,
Muhammad Latifd,
Muhammad Zaffar Hashmie and
Hesham R. El-Seedi
*a,
Hammad Ismailb,
Sadaf Anwaarc,
Muhammad Latifd,
Muhammad Zaffar Hashmie and
Hesham R. El-Seedi