
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular design and functional outcomes of RTP and TADF traits in isomers†
Meiling Chen‡
ab,
Yuzhuo Chen‡cd,
Ting Zhange,
Hua Zhange,
Zhiwen Xiaof,
Zhongzhen Su *cd and
Yunan Wu*gh
*cd and
Yunan Wu*gh
aState Key Laboratory of Oncology in South China, Guangdong Provincial Clinical Research Center for Cancer, Sun Yat-sen University Cancer Center, Guangzhou, 510060, China
bDepartment of Nuclear Medicine, Sun Yat-sen University Cancer Center, Guangzhou, 510060, China
cDepartment of Ultrasound, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai, 519000, China
dDepartment of Interventional Medicine, Guangdong Provincial Key Laboratory of Biomedical Imaging, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai, 519000, China
eShenzhen Key Laboratory of Systems Medicine for inflammatory diseases, School of Medicine, Shenzhen Campus of Sun Yat-Sen University, Sun Yat-sen University, Shenzhen, 518107, China
fDepartment of Otolaryngology Head and Neck Surgery, The Sixth Affiliated Hospital, Sun Yat-sen University, Guangzhou, 510655, China
gGuangzhou Institute of Chemistry, Chinese Academy of Sciences, Guangzhou, 510650, China. E-mail: wuynan@foxmail.com
hHengyang Aijie Technology Co, Ltd., Hengyang, 421006, China
First published on 11th October 2024
Abstract
This study reports the synthesis and photophysical analysis of three isomeric compounds, namely 3Fmo, 3Fmm, and 3Fmp, which were engineered using carbazole as the electron donor, phthalimide as the electron acceptor, and a benzene ring as the bridging moiety. Among these, 3Fmm was distinguished by its ability to exhibit immediate room-temperature white phosphorescence following the cessation of UV illumination, whereas 3Fmo and 3Fmp demonstrated TADF properties. Crystallographic analysis revealed unique intermolecular π–π stacking interactions within 3Fmm, absent in the other two isomers. Advanced TD-DFT computations indicated that such π–π stacking in 3Fmm not only facilitates intersystem crossing but also effectively reduces the free volume within the crystal, leading to a decrease in non-radiative transitions. These molecular interactions promote the manifestation of room-temperature phosphorescence. Furthermore, leveraging the superior luminescent properties of 3Fmo, the compound was successfully utilized in cellular imaging, where it achieved excellent imaging results, showcasing its potential for biomedical applications.
Introduction
Triplet excitons are pivotal due to their extensive applications in sensors,1–3 biological imaging,4–6 and organic light-emitting diodes (OLEDs).7–10 Various methodologies have been investigated for the effective harvesting of triplet excitons within purely organic luminophores.11–13 One prevalent method involves the conversion of triplet excitons into fluorescent singlets via thermally activated delayed fluorescence (TADF),14–17 necessitating a molecular architecture that minimizes orbital overlap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Alternatively, phosphorescence can be facilitated through radiative decay of triplet excitons.18,19 This approach mandates the integration of aromatic aldehyde groups,20 carbonyl groups,21,22 or heavy halogens23–26 to enhance spin–orbit coupling (SOC), thereby promoting efficient intersystem crossing (ISC). Organic room temperature phosphorescence (RTP) refers to phosphorescence that can be observed at ambient conditions, often continuing to emit light even after the excitation source is turned off. RTP materials stabilize triplet excited states by optimizing molecular structures and microenvironments, enabling long-lived phosphorescence without the need for heavy metals. The unique feature of RTP is its long emission delay, which lends itself to important applications in anti-counterfeiting, data encryption, and bioimaging. To mitigate non-radiative relaxation of excited states, strategies such as crystallization,27–29 incorporation within rigid matrices,30–32 leveraging effective intermolecular interactions,33,34 or utilizing deuterated carbon are employed,35,36 enhancing the photoluminescent quantum efficiency and longevity of the emitted light.However, the relationship between the properties of TADF and phosphorescence and their aggregation states has not been thoroughly explored. Recently, Yang and colleagues demonstrated the coexistence of dual emission pathways for TADF and RTP in crystalline forms, attributed to separate monomeric and dimeric states.37 Tang et al. reported on the RTP properties of three carbazolyl benzoate isomers, finding that the meta isomer exhibited longer RTP lifetimes.38 An and colleagues developed an isomeric strategy to enhance RTP efficiency through the formation of intermolecular π-type halogen bonds, achieving lifetimes of approximately 150 ms.39 Bryce et al. achieved a switch between RTP and thermally activated delayed fluorescence by controlling methyl isomerization. The optoelectronic properties of organic luminescent materials hinge both on their molecular chemistry and their physical packing configurations.40 Thus, exploring the influence of molecular aggregation on controlling RTP and TADF emissions is crucial.
The aromatic-imide group, characterized by its rigid π-conjugated structures and potent electron-withdrawing properties, is extensively utilized in the development of organic luminescent materials.41–45 In this study, we utilized phthalimide as the electron acceptor, carbazole as the electron donor, and a benzene ring as the conjugated π-bridge. Carbazole was strategically positioned at different sites on the benzene ring to design and synthesize three isomeric compounds: 3Fmo, 3Fmm, and 3Fmp. Our findings indicate that 3Fmo and 3Fmp exhibited the properties associated with TADF, whereas the meta-substituted compound, 3Fmm, was characterized by pronounced RTP traits. Remarkably, an immediate white afterglow was observed upon deactivation of the UV light. Alterations in the substitution sites not only modulated the excited-state electronic structure of the monomers but also influenced the molecular packing within the crystals, thereby facilitating the fine-tuning of both TADF and RTP phenomena.
Results and discussion
The three specified molecules, designated as 3Fmo, 3Fmm, and 3Fmp, possess a donor–acceptor (D–A) configuration, with carbazole serving as the donor and phthalimide as the acceptor, linked via a benzene ring forming the conjugated π-bridge (refer to Fig. 1). These molecules were synthesized through straightforward procedures involving amide formation and Suzuki coupling reactions (depicted in Scheme 1). Comprehensive structural confirmation was obtained through various spectroscopic methods including 1H NMR, 13C NMR, and high-resolution mass spectrometry (HR-MS), along with electron ionization (EI) techniques (illustrated in Fig. S1–S16†). The crystallographic structures were definitively established via single-crystal X-ray diffraction analysis (CCDC 2353412–2353414†).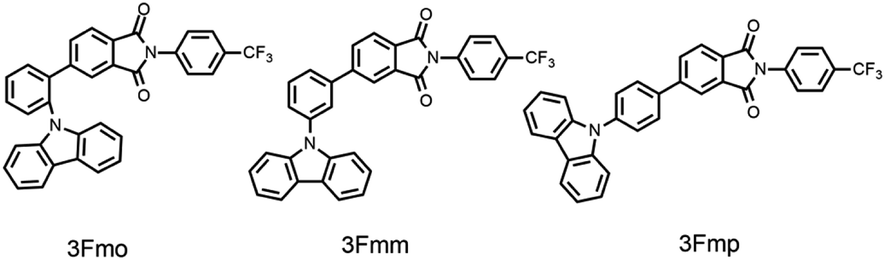 | ||
| Fig. 1 Chemical structure of 3Fmo, 3Fmm and 3Fmp. | ||
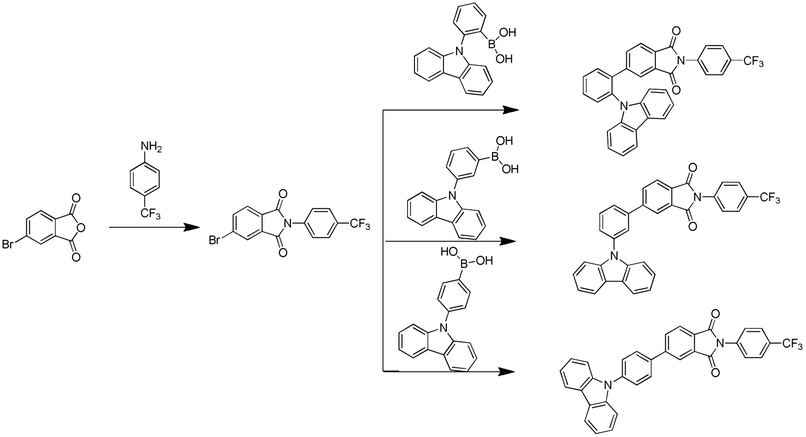 | ||
| Scheme 1 Synthetic route of products. | ||
To gain a deeper understanding of the photophysical properties of these three newly synthesized compounds, their absorption spectra, photoluminescence spectra, and excited-state lifetimes were characterized in dilute tetrahydrofuran solutions (concentration of 10−5 M), as shown in Fig. 2. At 292 nm, all three compounds exhibited absorption peaks corresponding to the π–π* transitions of carbazole; 3Fmp displayed significant CT absorption peaks within the 300–400 nm range, while 3Fmo and 3Fmm exhibited relatively weaker CT peaks between 300 and 350 nm. In tetrahydrofuran, the emission wavelengths of 3Fmo, 3Fmm, and 3Fmp were respectively 517 nm, 528 nm, and 542 nm, with corresponding lifetimes of 19.32 ns, 9.73 ns, and 3.38 ns. Although the positions of these absorption peaks remained nearly constant with changes in solvent polarity, indicating negligible electronic interaction between the ground state and the solvent, the emission bands significantly red-shifted with changes in solvent polarity, demonstrating a typical solvatochromic effect attributed to ICT transitions (Fig. S17–S19†). For example, the emission band of 3Fmo shifted from 424 nm to 560 nm when the solvent was changed from cyclohexane to dichloromethane. To further elucidate the ICT process, quantum mechanical computations were performed using Gaussian 09 software to analyze the electron cloud distribution of the three isomers. As shown in Fig. S24,† the analysis revealed that the electron cloud of the HOMO was predominantly located on the carbazole units, while that of the LUMO was concentrated on the phthalimide units, and the effective separation between HOMO and LUMO facilitated the ICT transition.
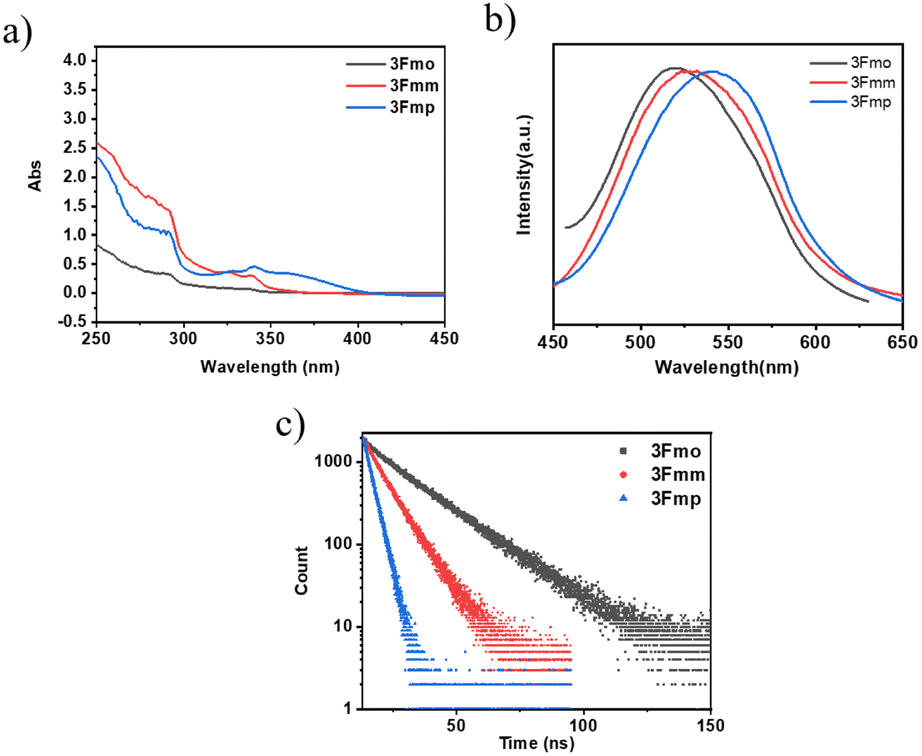 | ||
| Fig. 2 The compound in dilute tetrahydrofuran solution (c = 10−5 M) for: (a) UV-visible absorption spectrum; (b) fluorescence spectrum; (c) time-resolved PL-decay profiles of 3Fmo (at 517 nm), 3Fmm (at 528 nm) and 3Fmp (at 542 nm). All of these were obtained under ambient conditions. | ||
We conducted an advanced examination of the photophysical characteristics of the target compounds under conditions of molecular aggregation. As delineated in Fig. 3, the peak emissions of the powdered spectra for 3Fmo, 3Fmm, and 3Fmp occur at 494 nm, 456 nm, and 468 nm, respectively. These findings highlight a marked divergence in the PL profiles of these compounds in the solid state compared to their behavior in dilute solutions, which is likely attributable to their aggregated states. The aggregation of molecules facilitates the coupling and splitting of initially degenerate molecular orbitals, leading to the formation of novel molecular orbitals. This differential aggregation alters the luminescence characteristics significantly, as evidenced by varying emission behaviors in aggregated versus solution states. These discrepancies likely stem from distinct aggregation modes that result in heterogeneous orbital reorganizations among the compounds. Such reorganizations profoundly influence the optical properties, manifesting in distinct absorption and emission profiles in the aggregated state. Confirmation of these effects can be obtained through the analysis of UV absorption and excitation spectra in the solid state. In these solid-state spectra, 3Fmo and 3Fmp exhibit maximum absorption peaks at 429 nm and 421 nm, respectively, whereas 3Fmm presents its peak at 378 nm, signifying the highest energy state among them when aggregated. The excitation spectra further corroborate these findings, with 3Fmm displaying the shortest excitation wavelength at 385 nm. In contrast, the excitation wavelengths for 3Fmo and 3Fmp are observed at 486 nm and 454 nm, respectively. As depicted in Fig. 3d, all three compounds demonstrate fluorescence lifetimes at the microsecond scale, with 3Fmo at 2.55 μs, 3Fmm at 59.6 μs, and 3Fmp at 11.7 μs. This sustained emission duration may implicate the involvement of triplet excitons in their luminescent processes.
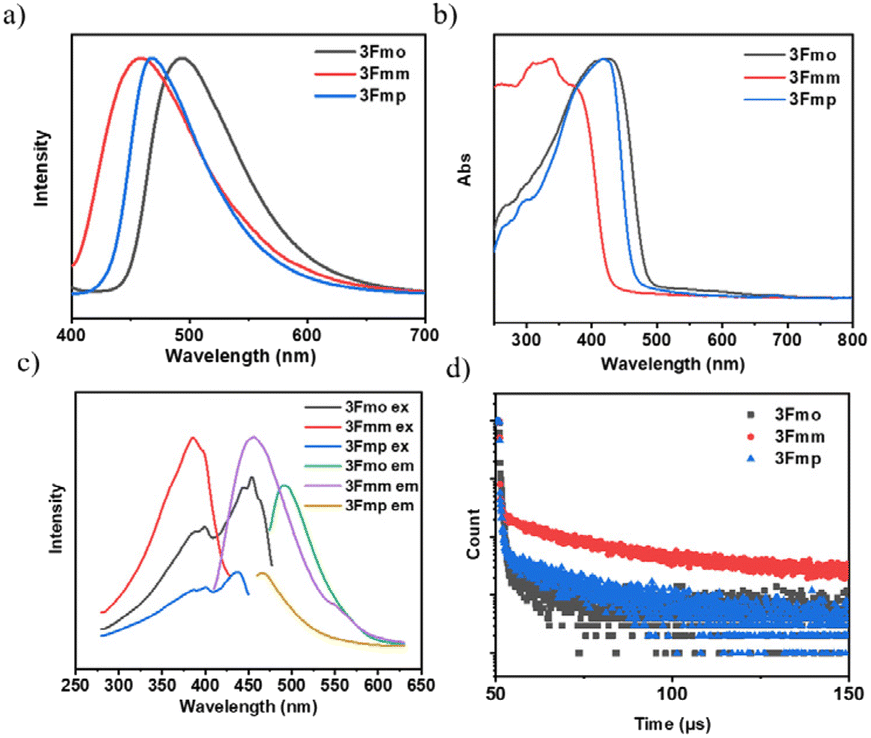 | ||
| Fig. 3 Photophysical properties of isomers in the aggregated state: (a) fluorescence spectra; (b) solid-state ultraviolet-visible absorption spectra; (c) excitation spectra and emission spectra; (d) transient decay curves. | ||
Upon deactivation of the UV lamp, both 3Fmo and 3Fmp cease luminescence, whereas 3Fmm maintains its glow for several seconds (refer to Fig. 4a). Under UV irradiation, 3Fmm exhibits a bright blue emission. When the UV source is removed, it initially displays a white emission, which progressively transitions to yellow before gradually diminishing and ultimately extinguishing. This behavior suggests that the phosphorescence of 3Fmm may involve diverse luminescent centers.
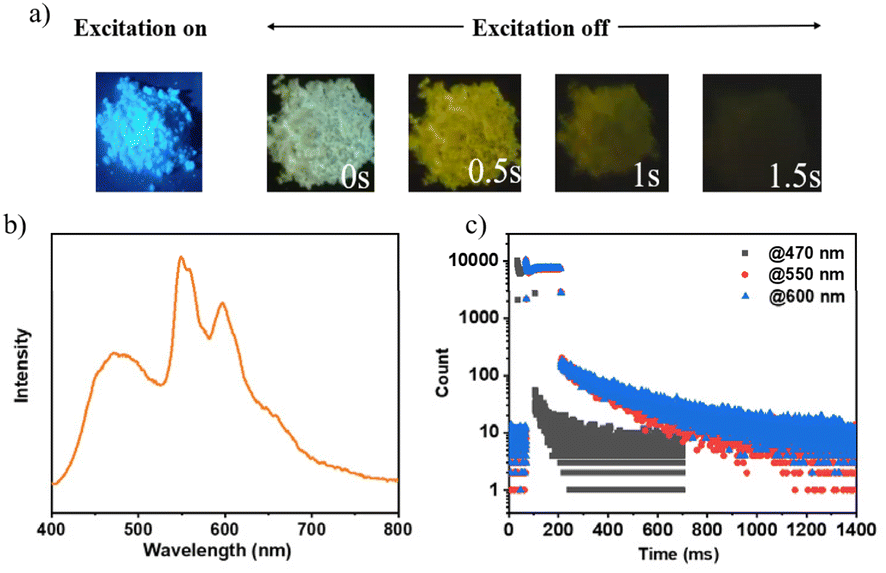 | ||
| Fig. 4 (a) Photographic documentation of 3Fmm exposed to ultraviolet light and in the absence of ultraviolet illumination; the phosphorescent characteristics of 3Fmm powder are delineated as follows: (b) phosphorescence spectrum at ambient temperature; (c) transient decay profiles for distinct emission peaks. | ||
The phosphorescence spectrum of the compound 3Fmm was investigated and the results are presented in Fig. 4b. The spectrum exhibited multiple emission peaks at wavelengths of 470 nm, 550 nm, and 600 nm. Detailed analysis through time-resolved spectroscopy revealed disparate decay kinetics across these peaks. Measurements conducted using a Horiba integrated fluorescence spectrometer ascertained the lifetimes associated with these emission peaks. Notably, the lifetime at 470 nm was significantly shorter, registered at 99 milliseconds, whereas the emissions at 550 nm and 600 nm displayed nearly identical lifetimes of approximately 0.39 seconds, indicative of a common luminescent origin. The more rapid decay at 470 nm led initially to a white light phosphorescence emission at ambient temperature following the deactivation of the UV light source. As the emission progressed, the relative intensity of the 470 nm peak diminished, resulting in a gradual transition of the emitted light from white to yellow before ultimately ceasing. This observation underscores the presence of dual luminescent centers within 3Fmm, which are responsible for the dynamic shifts observed in the phosphorescence color spectrum.
The emission spectra and lifetimes of 3Fmm were measured at various temperatures, as depicted in Fig. 5. At 100 K and 200 K, phosphorescence peaks at 550 nm and 600 nm were observed; however, these peaks were not detected at 300 K. This absence is likely due to an increase in non-radiative transitions with rising temperature, which significantly reduces the phosphorescence intensity relative to fluorescence, rendering the phosphorescence peaks indiscernible in the steady-state spectra. Additionally, the intensity of the steady-state spectra was found to be consistent across different temperatures, indicating that the triplet excitons in 3Fmm are predominantly involved in phosphorescence rather than TADF. Lifetimes measured at 456 nm, which remained nearly constant across temperatures, further confirm the absence of TADF characteristics in 3Fmm. Furthermore, when comparing lifetimes at 470 nm and 550 nm, a significant reduction was observed with increasing temperature, demonstrating that the triplet excitons are highly sensitive to temperature and are rapidly quenched at higher temperatures.
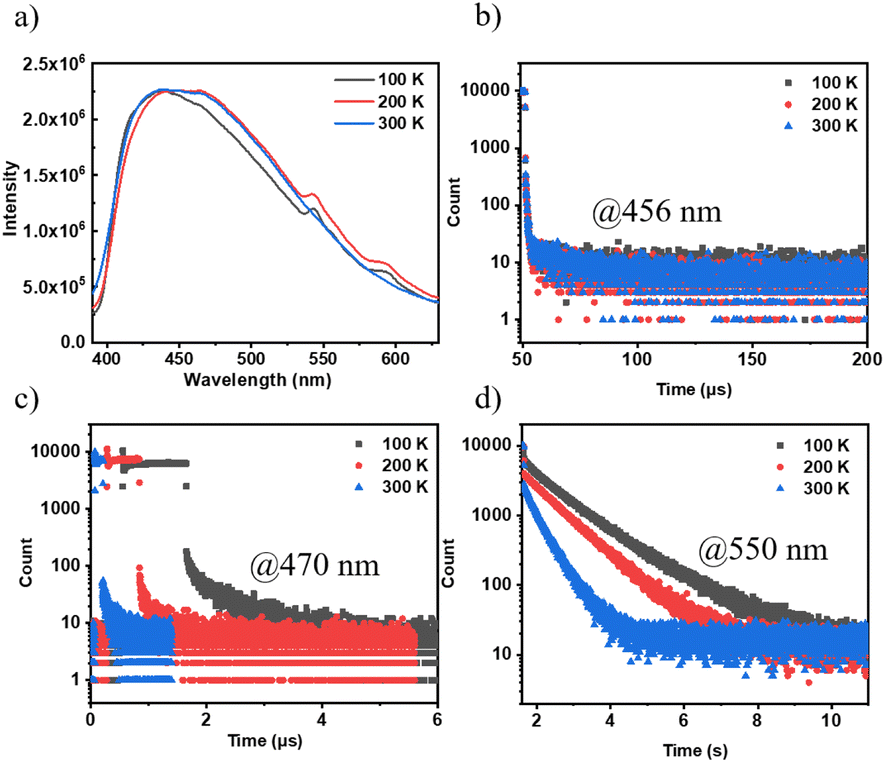 | ||
| Fig. 5 The properties of 3Fmm powder are described as follows: (a) temperature-variable spectroscopic profiles; (b) temperature-dependent lifetimes of the excited state corresponding to the 456 nm emission peak; (c) temperature-dependent lifetimes of the excited state corresponding to the 470 nm emission peak; (d) temperature-dependent lifetimes of the excited state corresponding to the 550 nm emission peak. | ||
To further elucidate the origin of this ultra-long-lived phosphorescence, we initially examined 3Fmm in dilute solution and observed that RTP was absent under these conditions. This observation suggests that 3Fmm requires an aggregated state to manifest RTP. Upon further cooling of the dilute solution to 77 K using liquid nitrogen, pronounced phosphorescence was detected. The corresponding phosphorescence spectra were recorded and compared with those obtained from the powdered sample at 77 K and at room temperature. The comparison revealed notable discrepancies between the spectra of the powder and the dilute solution at 77 K. In general, the phosphorescence spectra of a dilute solution at 77 K are considered indicative of single-molecule phosphorescence. This is because, in dilute solutions, fluorescent molecules are surrounded by solvent molecules, which significantly reduces the likelihood of close molecular interactions. Consequently, the observed emission predominantly originates from single molecules. As illustrated in Fig. 6, the phosphorescence emission peak of 3Fmm in dilute solution is centered at 528 nm. In the aggregated state, the phosphorescence spectrum of 3Fmm is characterized by multiple emission centers. At 77 K, the emission peak differs markedly from that observed at room temperature. Specifically, the 530 nm peak present at 77 K is absent at room temperature, indicating that the 530 nm phosphorescence at 77 K likely arises from single-molecule phosphorescence. In contrast, the phosphorescence peaks at 550 nm and 600 nm, which occur at lower energy levels compared to the single-molecule phosphorescence, are likely due to intermolecular electronic coupling in the aggregated state. These lower energy phosphorescence emissions are consistent with the yellow RTP observed at room temperature.
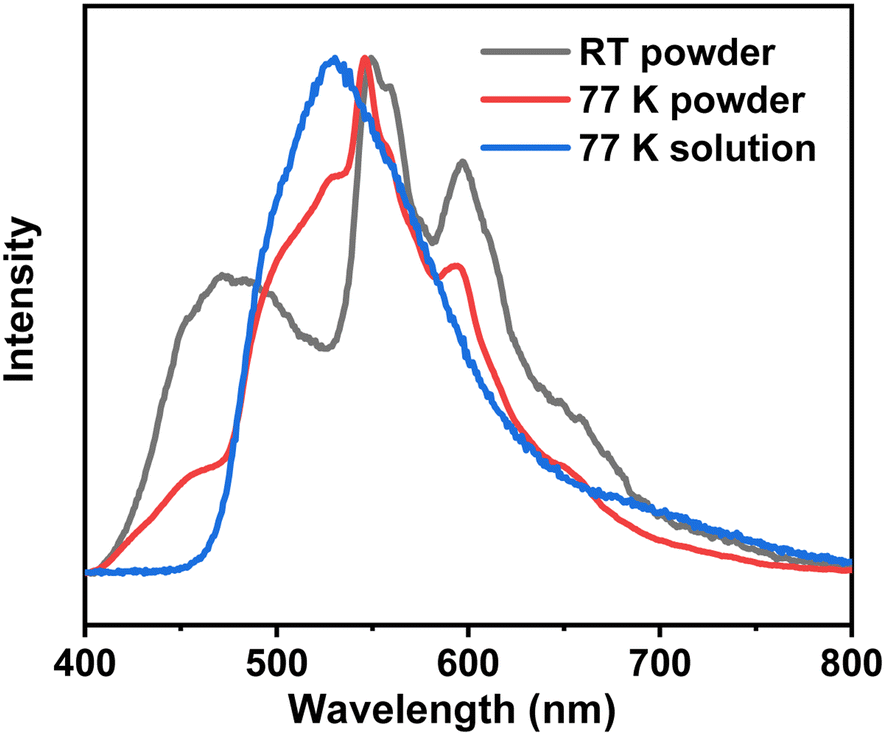 | ||
| Fig. 6 The phosphorescence spectra of the 3Fmm compound in powder form at room temperature and 77 K, as well as in dilute solution at 77 K. | ||
To investigate the effect of aggregation on the RTP of the 3Fmm compound, we heated the 3Fmm powder to melt it, then rapidly cooled it using liquid nitrogen. It was observed that the quenched sample did not exhibit RTP. X-ray powder diffraction was employed to analyze the crystallinity of the samples before and after quenching, as shown in Fig. 7. The original sample was crystalline, while the quenched sample was amorphous. The fluorescence spectra of the samples before and after quenching were collected, revealing a significant red shift in the emission peak of 3Fmm in the amorphous state to 530 nm. The absence of RTP in the amorphous sample obtained after quenching suggests that, in addition to requiring molecular packing, 3Fmm also necessitates a specific ordered structure to exhibit room-temperature phosphorescence.
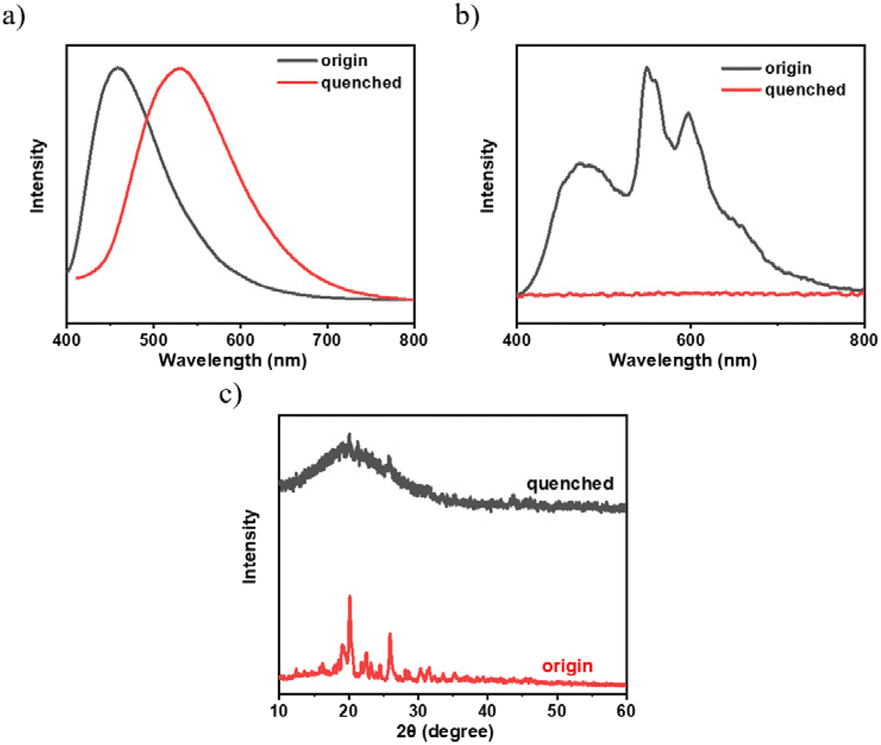 | ||
| Fig. 7 (a) Steady-state spectra; (b) spectra after the removal of the UV lamp; (c) X-ray powder diffraction patterns of 3Fmm powder and the quenched sample. | ||
Based on the experimental results, the room-temperature phosphorescence (RTP) process of 3Fmm can be illustrated as shown in the schematic diagram (Fig. S20†): upon excitation, 3Fmm generates singlet excitons (S1), which then undergo intersystem crossing to form triplet excitons (T1). The triplet excitons of the individual molecules further stabilize, transitioning to an electronically coupled multi-molecular state (Tn*), ultimately returning to the ground state via radiative transition, thereby producing phosphorescence. The phosphorescence emission peak of the single molecules is at 528 nm, while the 470 nm peak in 3Fmm has higher energy and is close to the fluorescence peak at 456 nm. Therefore, it can be inferred that the emission at 470 nm originates from fluorescence generated by the annihilation of triplet excitons, as illustrated in Fig. S21.†
As isomeric derivatives of 3Fmm, 3Fmo and 3Fmp did not exhibit the characteristics of long-lived room temperature phosphorescence. Consequently, we investigated whether the triplet excitons in these compounds exist in the form of TADF. By examining the temperature-dependent lifetimes of these compounds in their powdered state (refer to Fig. S22 and S23†), we observed an increase in the proportion of long-lived excitons at higher temperatures, which aligns with the typical characteristics of TADF. Therefore, the triplet excitons in 3Fmo and 3Fmp predominantly convert into delayed fluorescence via reverse intersystem crossing.
Despite sharing the same electron donors and acceptors, room-temperature phosphorescence is predominantly exhibited by 3Fmm, while TADF characteristics are displayed by 3Fmo and 3Fmp. To further investigate the underlying factors influencing these properties, the solution evaporation technique was employed to prepare and analyze the single crystals of these compounds. By examining the crystal structures and intermolecular interactions, their luminescent mechanisms were explored. Using Gaussian 09 software and time-dependent density functional theory (TD-DFT), the electronic structures of the molecules within the crystals were calculated, as depicted in Fig. S22.† The HOMO of the compounds are mainly located on the carbazole units, whereas the LUMO are primarily situated on the imine rings, facilitating effective separation between HOMO and LUMO. The molecule of 3Fmo is characterized by a “U”-shaped and considerably distorted structure; in contrast, the structure of 3Fmp is linear. Variations in substituent positions are found to alter the extent of ICT. Additionally, the degree of molecular distortion varies among these compounds. Charge distribution in these crystals was also analyzed, with results presented in Table S5.† According to the data, the largest energy gap is possessed by 3Fmm, resulting in the highest levels of fluorescence and phosphorescence energy, which correlate with the shortest fluorescence emission wavelength, consistent with experimental results. By measuring the fluorescence spectra of the three compounds and their phosphorescence spectra in dilute solutions at 77 K, the S1 and T1 energy levels of the compounds were determined, as illustrated in Fig. S26–28.† It is evident that 3Fmo and 3Fmp exhibit relatively small ΔEST values of 0.12 eV and 0.23 eV, respectively, which favor the conversion of T1 excitons to S1 via reverse intersystem crossing, thereby leading to delayed fluorescence. Conversely, 3Fmm exhibits a larger ΔEST of 0.46 eV, which hinders the conversion of T1 excitons to S1. However, S1 excitons can convert to T1 excitons, resulting in room-temperature phosphorescence.
Based on earlier findings, room-temperature phosphorescence is exhibited exclusively in the crystalline state of 3Fmm, requiring a particular ordered arrangement of its structure. Various stacking configurations of these three compounds in their crystal forms were investigated (refer to Fig. 8a–c), and it was noted that π–π interactions are present only in 3Fmm, with no such interactions observed in 3Fmo and 3Fmp. This observation prompts an investigation into whether π–π stacking interactions are essential for enabling room-temperature phosphorescence. Consequently, detailed theoretical calculations on the dimer structures of these compounds were performed using the TD-DFT approach in Gaussian 09 software. As depicted in Fig. 8d–i, while limited pathways for intersystem crossing are exhibited by the monomeric models of these compounds, a notable increase in these pathways is evident in the dimeric models, particularly in that of 3Fmm. This increase suggests that electronic coupling is more likely to occur among adjacent molecules in the dimeric models, significantly influencing intersystem crossing. In the dimer model of 3Fmm, the efficiency of intersystem crossing is greatly enhanced by the π–π interactions. Moreover, these π–π interactions effectively mitigate the vibrational motion of the excited-state molecules, thereby curbing non-radiative transitions. Hence, it is proposed that intermolecular π–π interactions play a crucial role in facilitating room-temperature phosphorescence.
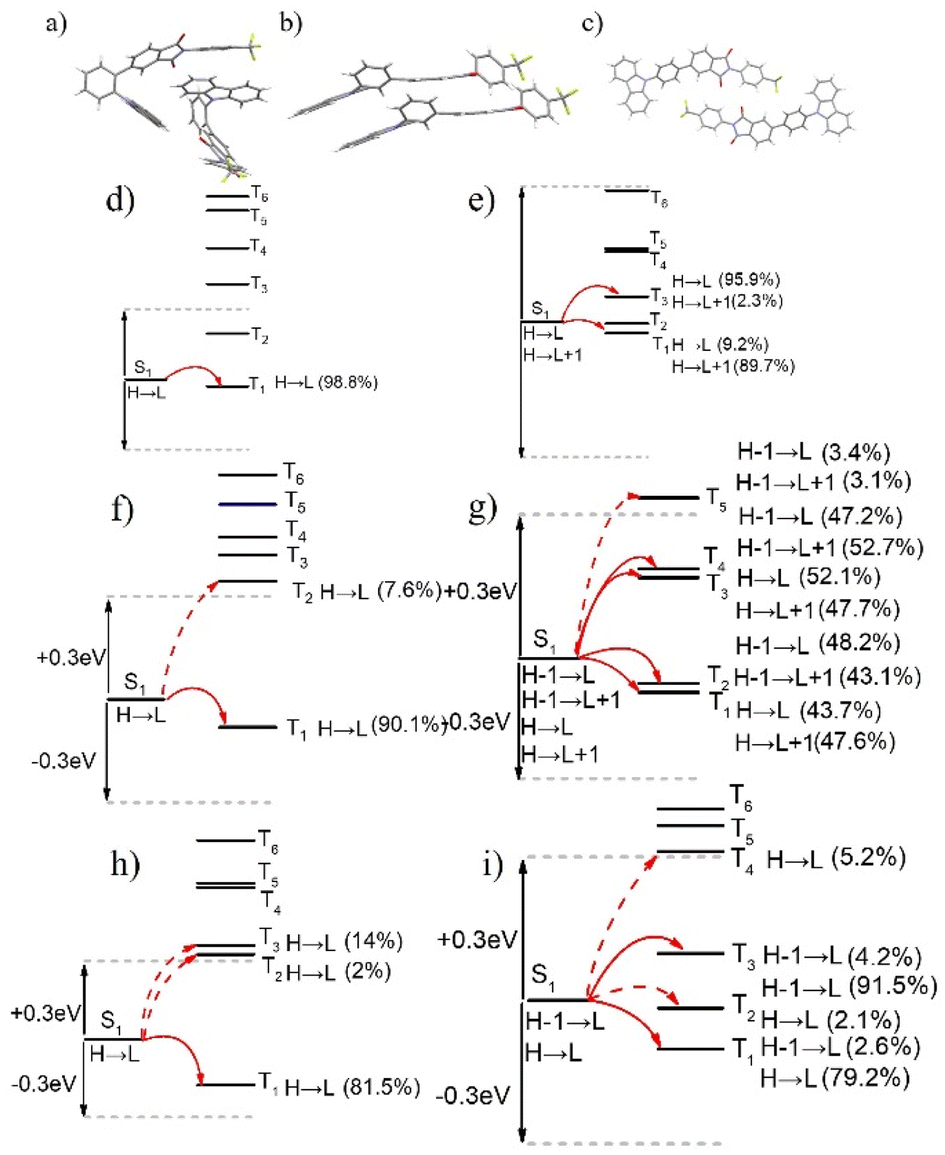 | ||
| Fig. 8 Interactions of dimer molecules in crystals: (a) 3Fmo; (b) 3Fmm; (c) 3Fmp; intersystem crossing pathways in single-molecular model: (d) 3Fmo; (f) 3Fmm; (h) 3Fmp; intersystem crossing pathways in dimer model: (e) 3Fmo; (g) 3Fmm; (i) 3Fmp. | ||
During molecular design, we hypothesized that the degree of molecular distortion increases progressively from 3Fmp to 3Fmm and then to 3Fmo, resulting in a gradual reduction in the ΔEST. A smaller ΔEST is conducive to enhancing the RISC process, thereby favoring the manifestation of TADF characteristics. Thus, we anticipated that molecules with greater structural distortion would be more likely to exhibit TADF properties. However, experimental results revealed that 3Fmp and 3Fmo indeed demonstrated TADF behavior, whereas 3Fmm exhibited RTP characteristics, deviating from our initial expectations. Through single-crystal structural analysis, we observed significant differences in the packing arrangements of 3Fmp, 3Fmm, and 3Fmo, suggesting that the luminescent properties of these compounds are influenced not only by their individual molecular structures but also by their supramolecular packing modes. Specifically, variations in molecular packing can significantly modulate the nature and strength of intermolecular interactions, thereby exerting a profound impact on luminescent behavior. These findings underscore the critical importance of considering molecular packing effects in the design of luminescent materials.
Another approach to enhancing room-temperature phosphorescence involves minimizing non-radiative transitions. Typically, molecular chemical bonds exhibit vibrations and rotations that can dissipate the energy of excited-state molecules non-radiatively. When molecules are densely packed, this movement is curtailed, leading to a reduction in non-radiative transitions. During crystal formation, in addition to the spaces occupied by atoms, there is a considerable amount of unoccupied space within the crystal lattice, known as free space. The reduction in free space correlates with more constrained molecular vibrations and rotations, thereby decreasing non-radiative transition pathways and enhancing luminescence efficiency. The free volume fractions of these three compounds were calculated using a 1 Å diameter probe in Materials Studio, as illustrated in Fig. 9. The findings revealed that the free volume fraction for 3Fmo is 14.2%, while 3Fmm is so compactly packed that gaps measurable by a probe larger than 1 Å in diameter are absent, and 3Fmp has a free volume fraction of 10.5%. Consequently, the low free volume fraction in 3Fmm suggests significant restrictions on the molecular vibrations and rotations, which diminishes non-radiative transition pathways, thereby facilitating the observation of room-temperature phosphorescence.
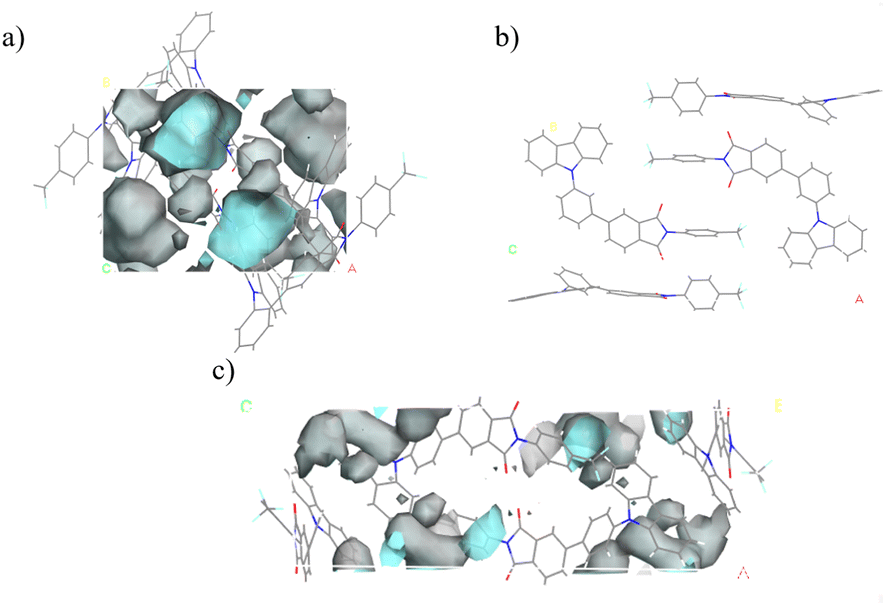 | ||
| Fig. 9 Free volume area in the single crystal cells of: (a) 3Fmo; (b) 3Fmm; (c) 3Fmp. | ||
Given the TADF properties of 3Fmo, its utility in biological imaging was investigated. Utilizing a self-assembly approach, 3Fmo was encapsulated within the amphiphilic block copolymer F127 to form nanoparticles. These nanoparticles were subsequently internalized by cells through the endocytic pathway. As delineated in Fig. 10, under bright field microscopy, the positional context of HeLa cells is clearly discernible. Upon transition to the green fluorescence channel, emission from the cells was observed, indicating that the 3Fmo-containing nanoparticles had effectively penetrated the cells and were functional in situ. These findings highlight the potential of 3Fmo as a fluorescent probe for bioimaging applications, leveraging its TADF capabilities.
 | ||
| Fig. 10 (a) A bright field image of HeLa cells; (b) an image of HeLa cells under the green light channel. | ||
Experimental
Materials
6-Bromobenzo[de]isochromene-1,3-dione, aniline, palladium acetate, potassium carbonate and (3-(9H-carbazol-9-yl)phenyl)boronic acid were purchased from Aldrich. The other reagent were purchased from Guangzhou Jincheng company. All the materials and reagent were used as received.Instrumentation
Nuclear magnetic resonance spectra, including hydrogen (1H NMR) and carbon (13C NMR), were obtained using a Bruker Avance III NMR spectrometer with deuterated dimethyl sulfoxide (DMSO-d6) as the solvent and tetramethylsilane (TMS) as the internal standard. Mass spectrometric evaluations, encompassing both low-resolution and high-resolution electron impact techniques (EI-MS and EI-HRMS), were conducted on Thermo DSQ and Thermo MAT95XP systems, respectively. Ultraviolet-visible (UV-vis) absorption spectra were captured using a Hitachi U-3900 spectrophotometer, and photoluminescence (PL) spectra were gathered with an Ocean Optics QE65 Pro spectrometer. Measurements of fluorescence lifetimes, temperature-dependent lifetimes, time-resolved spectra, and fluorescence quantum efficiencies were performed with a Horiba JY FL-3 combined steady-state/transient fluorescence spectrometer, equipped with a calibrated integrating sphere for quantum efficiency determination. Thermal analyses were conducted using a Shimadzu TGA-50H thermogravimetric analyzer and a NETZSCH DSC 204 F1 differential scanning calorimeter, both under a nitrogen atmosphere, with heating rates of 20 °C min−1 and 10 °C min−1, respectively. Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations were carried out using the Gaussian 09 software with the B3LYP/6-31G(d) method and basis set. X-ray powder diffraction was performed on a Rigaku Miniflex600 instrument, while optical microscopy examinations utilized a Leica Microsystems DM2700P microscope. X-ray single crystal diffraction analysis was executed on a Super Nova single crystal diffractometer. For cellular imaging studies, observations were made using a confocal microscope.Synthesis
The crude product was purified by column chromatography on silica gel with dichloromethane and normal hexane (v/v = 1/2) as eluent. After that 5.39 g white powder 3FmBr were got with a high yield of 90%. 1H NMR (500 MHz, DMSO-d6) δ 8.20 (d, 1H), 8.12 (dd, 1H), 7.92 (dd, 3H), 7.71 (d, 2H). 13C NMR (126 MHz, DMSO) δ 166.40, 165.83, 138.01, 135.90, 134.02, 130.98, 128.83, 128.57, 128.22, 126.91, 126.49, 126.46, 125.88, 125.55, 123.3. 19F NMR (470 MHz, DMSO-d6) δ −61.00. MS (EI, m/z): [M]+ calcd for C15H7BrF3NO2: 368.9612. Found: 368.9604.
Conclusions
In conclusion, three isomeric compounds—3Fmo, 3Fmm, and 3Fmp—were synthesized utilizing carbazole as the electron donor, phthalimide as the electron acceptor, and a benzene ring as the bridging moiety. Of these, 3Fmm exhibited immediate room-temperature white phosphorescence upon the deactivation of UV illumination, whereas 3Fmo and 3Fmp displayed characteristics of TADF. Structural analysis of these isomers' crystals revealed the presence of intermolecular π–π stacking interactions in 3Fmm, which were not observed in 3Fmo and 3Fmp. TD-DFT calculations suggested that π–π stacking in 3Fmm facilitates intersystem crossing. Further analysis indicated that this stacking configuration contributes to a reduction in the free volume within the crystal lattice, thereby decreasing non-radiative transition rates. Such π–π interactions are conducive to the emergence of room-temperature phosphorescence. Additionally, capitalizing on the exceptional luminescent properties of 3Fmo, it has been successfully applied in cellular imaging, achieving favorable outcomes.Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and development program of China (2022YFC2305400), the National Natural Science Foundation of China (82372246, 81872224), Medical Scientific Research Foundation of Guangdong Province of China (A2021082), Guangdong Basic and Applied Basic Research Foundation (2021A1515010734), Shenzhen Science and Technology Program (JCYJ20230807111219040, JCYJ20190807153211194, ZDSYS20220606100803007).References
- D. Li, X. Yang and D. Yan, ACS Appl. Mater. Interfaces, 2018, 10, 34377–34384 CrossRef CAS PubMed.
- S. Qiu, J. Yu, T. Zhou, K. Zhang, Y. Duan, X. Ban, Q. Zhu, L. Shi and D. Zhang, Opt. Mater., 2021, 119, 111301 CrossRef.
- B. Wittmann, T. Biskup, K. Kreger, J. Kohler, H. W. Schmidt and R. Hildner, Nanoscale Horiz., 2021, 6, 998–1005 RSC.
- B. Chang, H. Zhu, Y. Wu, S. Wu, L. Zhang, C. Qu, Y. Ren and Z. Cheng, Adv. Opt. Mater., 2021, 9, 2101382 CrossRef CAS.
- W. Zhang, S. Chen, S. Ye, P. Sun, Q. Fan, J. Song, P. Zeng, J.-L. Qu and W.-Y. Wong, ACS Mater. Lett., 2022, 5, 116–124 CrossRef.
- Y. Zhang, H. Li, M. Yang, W. Dai, J. Shi, B. Tong, Z. Cai, Z. Wang, Y. Dong and X. Yu, Chem. Commun., 2023, 59, 5329–5342 RSC.
- X. Chen, A. Khan, S.-N. Zou, Y. Li, Q.-S. Tian, C. Zhong, M.-K. Fung, Z.-Q. Jiang and L.-S. Liao, J. Mater. Chem. C, 2021, 9, 4792–4798 RSC.
- F. Huang, Y.-C. Cheng, H. Wu, X. Xiong, J. Yu, X.-C. Fan, K. Wang and X.-H. Zhang, Chem. Eng. J., 2023, 465, 142900 CrossRef CAS.
- H. Lee, J. H. Park, K. J. Yang, S. J. Hwang, R. Braveenth, T. H. Ha, M. I. Han, C. W. Lee and J. H. Kwon, J. Mater. Chem. C, 2021, 9, 7426–7435 RSC.
- X. Song, S. Shen, S. Zou, Y. Wang, F. Guo, S. Gao and Y. Zhang, Chem. Eng. J., 2024, 481, 148794 CrossRef CAS.
- N. Aizawa, Y.-J. Pu, Y. Harabuchi, A. Nihonyanagi, R. Ibuka, H. Inuzuka, B. Dhara, Y. Koyama, K.-i. Nakayama, S. Maeda, F. Araoka and D. Miyajima, Nature, 2022, 609, 502–506 CrossRef CAS.
- S. Dartar, M. Ucuncu, E. Karakus, Y. Hou, J. Zhao and M. Emrullahoglu, Chem. Commun., 2021, 57, 6039–6042 RSC.
- C. M. Sullivan, J. E. Kuszynski, A. Kovalev, T. Siegrist, R. D. Schaller, G. F. Strouse and L. Nienhaus, Nanoscale, 2023, 15, 18832–18841 RSC.
- J. Gu, Z. Tang, H. Guo, Y. Chen, J. Xiao, Z. Chen and L. Xiao, J. Mater. Chem. C, 2022, 10, 4521–4532 RSC.
- R. K. Konidena, J. Lim and J. Y. Lee, Chem. Eng. J., 2021, 416, 129097 CrossRef CAS.
- H. Yin, Y. Wu, X. Peng and F. Song, Chem. Commun., 2020, 56, 10549–10551 RSC.
- D. Zhang and L. Duan, Nat. Photonics, 2021, 15, 173–174 CrossRef CAS.
- J. Bai, G. Dai, H. Jin, J. Ma, Z. Li, Y. Guan, M. Chen, Z. Ma and Z. Ma, J. Mater. Chem. C, 2023, 11, 16325–16332 RSC.
- Q. Zhou, C. Yang and Y. Zhao, Chem, 2023, 9, 2446–2480 CAS.
- H. Shu, H. Li, J. Rao, L. Chen, X. Wang, X. Wu, H. Tian, H. Tong and L. Wang, J. Mater. Chem. C, 2020, 8, 14360–14364 RSC.
- R. Gao, Y. Cha, H. Fu and Z. Yu, Adv. Opt. Mater., 2023, 11, 2202095 CrossRef CAS.
- Y. Tsukiyama, Y. Yamamoto, D. Koga, L. Cui, Y. Hoshino, Y. Hisaeda and T. Ono, Chem. Asian J., 2024, 19, e202301114 CrossRef CAS PubMed.
- E. Hamzehpoor, C. Ruchlin, Y. Tao, C.-H. Liu, H. M. Titi and D. F. Perepichka, Nat. Chem., 2023, 15, 83–90 CrossRef CAS.
- L. Lai, B. Fang, M. Fan, W. Cheng and M. Yin, J. Phys. Chem. C, 2021, 125, 16350–16357 CrossRef CAS.
- H. Liu, W. Liu, N. Ando, S. Yamaguchi and H. Zhang, J. Mater. Chem. C, 2021, 9, 2738–2743 RSC.
- W. Dai, X. Niu, X. Wu, Y. Ren, Y. Zhang, G. Li, H. Su, Y. Lei, J. Xiao, J. Shi, B. Tong, Z. Cai and Y. Dong, Angew. Chem., Int. Ed., 2022, 61, e202200236 CrossRef CAS.
- S. Jena, J. Eyyathiyil, S. K. Behera, M. Kitahara, Y. Imai and P. Thilagar, Chem. Sci., 2022, 13, 5893–5901 RSC.
- S. H. Lee, M. S. Valverde Paredes, P. M. Forster and D.-C. Lee, RSC Adv., 2024, 14, 6285–6291 RSC.
- K. Liu, S. Li, L. Fu, Y. Lei, Q. Liao and H. Fu, Nanoscale, 2022, 14, 6305–6311 RSC.
- K. Chen, Y. Zhang, Y. Lei, W. Dai, M. Liu, Z. Cai, H. Wu, X. Huang and X. Ma, Nat. Commun., 2024, 15, 1269 CrossRef CAS PubMed.
- X. Meng, Q. Hu, X. Wang, T. Ma, W. Liu, X. Zhu and C. Ye, J. Mater. Chem. C, 2022, 10, 17620–17627 RSC.
- Z. Xie, X. Zhang, H. Wang, C. Huang, H. Sun, M. Dong, L. Ji, Z. An, T. Yu and W. Huang, Nat. Commun., 2021, 12, 3522 CrossRef CAS PubMed.
- F. Liu, H. Yang, D. Sun, F. Gao, X. Zhang, Z. Zhao, X. Han and S. Liu, Chem. Sci., 2022, 13, 7247–7255 RSC.
- S. Cai, X. Yao, H. Ma, H. Shi and Z. An, Aggregate, 2023, 4, e320 CrossRef CAS.
- S. Hirata, K. Totani, T. Yamashita, C. Adachi and M. Vacha, Nat. Mater., 2014, 13, 938–946 CrossRef CAS PubMed.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed.
- Y. Xiong, Z. Zhao, W. Zhao, H. Ma, Q. Peng, Z. He, X. Zhang, Y. Chen, X. He, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 7997–8001 CrossRef CAS PubMed.
- S. Cai, H. Shi, D. Tian, H. Ma, Z. Cheng, Q. Wu, M. Gu, L. Huang, Z. An, Q. Peng and W. Huang, Adv. Funct. Mater., 2018, 28, 1705045 CrossRef.
- C. Chen, R. Huang, A. S. Batsanov, P. Pander, Y.-T. Hsu, Z. Chi, F. B. Dias and M. R. Bryce, Angew. Chem., Int. Ed., 2018, 57, 16407–16411 CrossRef CAS.
- M. Chen, Y. Chen, Y. Li, Y. Lin and Y. Wu, RSC Adv., 2024, 14, 6494–6500 RSC.
- S. Garain, B. C. Garain, M. Eswaramoorthy, S. K. Pati and S. J. George, Angew. Chem., Int. Ed. Engl., 2021, 60, 19720–19724 CrossRef CAS.
- Q. Mu, K. Zhang, H. Liu, Z. Xie, Y. Song, C. K. Wang, L. Lin, Y. Xu and J. Fan, Phys. Chem. Chem. Phys., 2023, 25, 6659–6673 RSC.
- J. Tang, X. Liu, X. Zhang, J. Zhao and Y. Wan, New J. Chem., 2023, 47, 22418–22429 RSC.
- Y. Wu, D. Sun, X. Han, F. Liang and S. Liu, New J. Chem., 2024, 48, 7543–7547 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2353412–2353414. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05807k |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |