
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTranslocation of Ti2CO2 MXene monolayer through the cell membranes†
Hamed Ahmadi,
Rouhollah Abdolvahab and
Mahdi Esmaeilzadeh*
and
Mahdi Esmaeilzadeh*
Department of Physics, Iran University of Science and Technology, Narmak, Tehran 16844, Iran. E-mail: mahdi@iust.ac.ir
First published on 4th October 2024
Abstract
Nanoparticle-based therapies represent a cutting-edge direction in medical research. Ti2CO2 MXene is a novel two-dimensional transition metal carbide with a high surface area and reactivity, making it suitable for biomedical applications due to its biocompatibility. In biomedicine, Ti2CO2 MXene is particularly used in photothermal therapy, where its ability to absorb light and convert it into heat can be utilized to target and destroy cancer cells. The study of how temperature influences the interaction between nanoparticles and cell membranes is a critical aspect of this field. Our study conducts a thorough coarse-grained molecular dynamics analysis of a Ti2CO2 MXene nanosheet interacting with a phosphatidylcholine (POPC) membrane under various thermal conditions and nanosheet orientations. We show that the hydrophilic nature of the nanosheet presents a substantial barrier to membrane penetration and an increase in temperature significantly enhances the permeability of the membrane, thereby facilitating the migration of the MXene nanoparticles across it. The peak force required to translocate the nanosheet through the membrane decreases e.g., from 2150 pN at 300 kelvin to 1450 pN at 370 kelvin indicating significant reduction in resistance at higher temperatures. The study also highlights the critical role of the nanosheets' spatial orientation in cellular uptake. Our research underscores the importance of the application of MXenes for nanomedical and photothermal therapy purposes.
Introduction
Biological membranes comprise various structures, including protein and ion channels that regulate interactions between cells and solutes. The type of phospholipids in the cell membrane influences its physical properties.1 Variances in lipid head group and electric charges can lead to distinct elastic properties, head group area, membrane fluctuations, bending characteristics, and compressibility. For example phosphatidylserine (PS) has a negative charge and phosphatidylethanolamine (PE) has a phosphate group combined with ethanolamine and phosphatidylcholine (PC) has a phosphate group combined with choline.2By increasing our knowledge of the role of membranes in cell function and designing novel nanoparticles, nanoparticle-based therapies are being developed. Many nanoparticles with unique physical and chemical properties and ability of specialization to various applications in nanomedicine have been developed. From imaging to drug delivery and photothermal therapy.3 All these methods are based on the interaction between nanoparticles and membranes. Molecular dynamics simulation has made it possible to investigate and study this interaction. MXenes are generally comprised of transition metal carbides, nitrides, or carbonitrides and are distinguished by their remarkable electrical conductivity,4 structural robustness, and superior optical attributes.5 The general formula for MXenes is Mn+1XnTx, where M represents an early transition metal (e.g., titanium, zirconium, hafnium, vanadium, or niobium), X represents carbon or nitrogen, n can vary from 1 to 4, T represents surface terminations, which can include groups like oxygen (O), hydroxyl (OH), or fluorine (F). This formula allows for a wide range of compositions and properties, making MXenes highly versatile for various applications. The high surface area inherent to MXenes is instrumental in enhancing drug encapsulation and release mechanisms, while their inherent hydrophilicity contributes to improved biological compatibility.6 The efficacy of MXenes in converting light to heat is leveraged in photothermal therapy applications, and their unique composition renders them effective as contrast agents in various imaging techniques, including computed tomography (CT) and magnetic resonance imaging (MRI).7 Additionally, strong tissue penetration capabilities, enabling targeted treatment with rapid and effective results, minimal invasiveness, and swift recovery. These advantages underscore the potential of MXenes in the field of tumor treatment.8,9 The broad applicability of MXenes extends to their use in biosensing devices, theranostic strategies for cancer treatment, and antibacterial interventions, underscoring their transformative potential in biomedical applications. Ti2CO2 MXene is a highly promising material for biomedical applications due to its high biocompatibility, abundant reactive surface groups, good conductivity, and excellent photothermal properties.10,11
There are some nanoparticle properties that affect the nanoparticle–membrane interaction. Shape, size, surface charge and surface modification of nanoparticles12–15 which can be changed to suit the application of nanoparticles. The interaction between the nanoparticle and the membrane has been investigated by studying the impact of charge, shape, surface modifications, as well as the hydrophilicity and hydrophobicity of the nanoparticles.16 Solutes with size of smaller than 1 nm enter membrane via permeation and larger size solutes enter membrane via endocytosis.17 During the penetration of Polyethylenimine (PEI) and Polyethylenimine–Lactic Acid (PEI–LA) nanoparticles, membrane deformation, and pore formation investigated using a series of steered molecular dynamics simulations. Three membrane models were created based on 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphocholine (POPC), Dipalmitoylphosphatidylcholine (DPPC), and 1,2-Dilauroyl-sn-Glycero-3-Phosphocholine (DLPC) lipids. The study revealed that the acyl chains of POPC and DLPC were less aligned compared to DPPC. The addition of long-tailed LA substitutions enabled them to embed themselves into the less aligned tails of the hydrophobic part of the membranes, thereby reducing the force necessary for the nanoparticle penetration.18 In a study,19 to explore the interaction between a model lung membrane and three different nanoparticles (Ag, SiO2, TiO2) of varying sizes (1 nm, 3 nm, 5 nm) using an all-atom simulation, the results indicate that the 3 nm and 5 nm Ag nanoparticles were spontaneously taken up by the membrane, suggesting a potential affinity for cellular uptake. On the other hand, the SiO2 nanoparticle showed a preference for residing at the interface between the membrane and water. The TiO2 nanoparticle did not exhibit a favorable interaction with the membrane. The hydrophobic coating on gold nanoparticles aids in the uptake process. Research indicates that these nanoparticles are unable to fully penetrate a completely flat membrane, but they can easily insert into highly curved ribbon edges.20 A study investigated how multiple nanoparticles penetrate lipid membranes. The research highlights the importance of particle quantity in the penetration process.
However, increasing the number of particles does not always improve nanoparticle penetration. This is due to changes in the interaction mechanism between the nanoparticles and the membrane.21
It is a well-established fact that hydrophobic nanoparticles have a greater ease of penetration into the membrane22 and nanoparticles with the size of 1 nm and with 50% hydrophobicity have high translocation rates.23 A study investigated how the size of nanoparticles relates to their aggregation behavior. When exposed to low concentrations and placed on membranes with different curvatures, nanoparticles of medium size quickly moved towards areas of high curvature. This suggests that these nanoparticles can sense changes in membrane curvature. On the other hand, at high concentrations, the nanoparticles caused noticeable deformations in the membrane while still maintaining its mechanical stability. This indicates that these nanoparticles can induce changes in membrane curvature.24 Nanoparticles with different charges exhibit enhanced adhesion to the membrane surface, depending on the membrane's charge, the presence of opposite electric charges on a nanoparticle facilitates its adhesion and the presence of an asymmetric charge density enables the nanoparticle to reorient itself in the vicinity of the membrane surface.25 Furthermore, PEGylated nanoparticles have a higher energy barrier because of polyethylene glycol (PEG) aggregation process, leading to a greater free energy expense.26 Nanoparticles possessing a greater aspect ratio exhibit enhanced ease of penetration into the membrane.27 Using of an electric field in molecular dynamics simulation28 has resulted in the observation of direct translocation29 of nanoparticles across the membrane. The impact of temperature on the interaction between nanoparticle and membrane has been investigated through a coarse-grained simulation with implicit solvent. The results indicate that as the temperature rises, the fraction of nanoparticle wrapped by the membrane also increases. Furthermore, there exists a critical temperature at which a sudden surge in nanoparticle wrapping is observed.30
Soft nanoparticles, owing to their deformability, have a greater penetration energy barrier in comparison to rigid nanoparticles.31 Nanoparticles coated with cell membranes exhibit improved biocompatibility, prolonged lifetime, and enhanced cellular uptake.32 A study revealed that nanoparticles possessing a high hydrophobic coating degree (>50%) are capable of entering the cells independently, while nanoparticles with a low coating degree (<50%) necessitate aggregation before internalization.33 The examination of the cellular uptake of gold nanoparticles with mobile ligands discovered that the endocytic pathway is greatly impacted by the mobility of the ligands.34
A coarse-grained SMD simulation,35 provides a detailed molecular-level analysis of how MXenes, particularly the Ti3C2 variant, interact with cell membranes, a process metaphorically described as a “nano thermal blade”. The study employs molecular dynamics simulations to explore the mechanism by which MXenes penetrate cell membranes. The results indicate that the penetration is a non-spontaneous process requiring significant external force, suggesting that MXenes are more force-dependent compared to other nanomaterials for membrane disruption. These insights are crucial for understanding the therapeutic potential of MXenes in cancer treatment and antibacterial applications, offering a foundation for future biomedical innovations.
Extensive researches have been conducted that underscores the profound influence of nanoparticles' physical and chemical characteristics on their interactions with biological membranes.36 However, the role of temperature in this interaction remains insufficiently explored.
This study aims to examine the effect of temperature on the interactions potentials between the Ti2CO2 MXene and the POPC membrane. This research demonstrates that higher temperatures enhance the uptake of Ti2CO2 MXene nanosheet by increasing membrane permeability. We show that the nanosheet orientation affects uptake and certain alignments reduces the barrier and thus penetration becomes easier. Van der Waals forces dominate the interaction, aiding in photothermal therapy by causing membrane damage. This highlights the potential of MXenes in nanomedicine and photothermal treatments, emphasizing the importance of temperature and orientation.
The rest of this paper is structured into three sections: method, results, and conclusion, detailing the classical molecular dynamics simulation of a nanosheet and membrane interaction across various temperatures. The method section outlines the simulation procedures, including research design, parameters, and computational techniques utilized. Following this, the results section presents the outcomes of the simulations systematically, analyzing the behavior of the nanosheet and membrane interaction at different temperature settings. Lastly, we summarize the key findings and their implications in the conclusion section.
Method
Steered Molecular Dynamics (SMD) is a technique used to study the mechanical properties and interactions of molecules by applying external forces to specific atoms or groups of atoms within a molecular system. The basic idea is to simulate the effect of an external force, such as that applied by an atomic force microscope (AFM), to induce conformational changes or unbinding events in biomolecules. In constant velocity pulling, a molecule or a part of a molecule is pulled at a constant speed, allowing researchers to observe how it interacts with its environment. There are key components of constant velocity pulling. The SMD atom(s) is the atom or group of atoms that we want to pull. The dummy atom is a virtual atom connected to the SMD atom(s) via a virtual spring. This dummy atom is moved at a constant velocity. The SMD atom(s) is attached to the dummy atom through a virtual spring. The force exerted on the SMD atom(s) is proportional to the displacement of the spring, following Hooke's Law, F(x) = −K(x − x0). Where F is the force, K is the spring constant, x is the current position, and x0 is the initial position. The dummy atom is moved at a constant velocity, v. The position of the dummy atom at any time t is given by xdummy(t) = xdummy(0) + vt. The force exerted on the SMD atom(s) is measured throughout the simulation. This force provides insights into the interactions and mechanical properties of the system. The work done by the external force during the SMD simulation is given by W(t) = v∫F(t)dt (W = ∫Fdx = v∫Fdt, dx = vdt) where F is the applied force and dt is the differential time step and v is the constant velocity of the dummy atom. Steps in constant velocity pulling simulation:• Defining the SMD atom(s) and dummy atom.
• Setting the initial positions and velocities.
• Moving the dummy atom at a constant velocity.
• Recording the position and force on the SMD atom(s) at each time step.
In this study, pulling a MXene nanosheet toward a POPC membrane at constant velocity, involves the following
• Selecting atoms of the MXene nanosheet that will be pulled.
• Attaching a virtual spring to the SMD atom(s) and defining the dummy atom that will move at a constant velocity.
• Choosing an appropriate pulling velocity that balances computational efficiency and accuracy.
• Moving the dummy atom at the chosen constant velocity.
• Recording the force exerted on the MXene and the position of the nanosheet relative to the membrane.
• Examining the force–time or force–distance curve to understand the interaction between the MXene nanosheet and the POPC membrane.
• Identifying any changes in the structure of the membrane and interaction between the MXene and the membrane during the pulling process.
The Martini force field37 consists of four main bead types: polar (P), apolar (C), nonpolar (N), and charged (Q). Each of the main bead types is further divided into subtypes to capture a range of chemical properties. There is a distinction between apolar and nonpolar beads. Apolar beads represent hydrophobic interactions and are typically used to model regions of molecules that do not interact favorably with water. They are characterized by their lack of polarity and their tendency to avoid aqueous environments. Nonpolar beads are used to represent regions of molecules that are neutral and do not have a significant dipole moment. They interact weakly with both polar and apolar environments. The distinction lies in the specific interactions and the context in which these beads are used within the force field. Apolar beads are more strictly hydrophobic, whereas nonpolar beads are neutral and can interact with a broader range of environments. The Martini force field is particularly advantageous for some reasons. It uses a coarse-grained (CG) approach, which allows for the simulation of larger systems and longer timescales compared to all-atom models. This is crucial for studying complex biological processes that require extensive computational resources. The Martini force field is systematically parameterized to reproduce experimental partitioning free energies between polar and apolar phases, ensuring accurate representation of molecular interactions. It has been successfully applied to a variety of systems, including lipids, proteins, nucleotides, and polymers, demonstrating its robustness and flexibility.
In our study, we employ the Martini force field and SMD method to explore the impact of temperature and MXene nanosheet orientations on the interaction between the MXene nanosheet and the biological membrane.
We obtain the atomistic structure of a monolayer of Ti2CO2 MXene from38,39 and consider the oxygen and titanium atom as a single bead. The Ti2C structure is then considered as the Coarse-Grained (CG) structure of Ti2CO2. The decision to treat the oxygen and titanium atoms as a single unit in our coarse-grained model was based on some considerations. Combining these atoms reduces the complexity of the model, allowing for more efficient simulations without significantly compromising the accuracy of the results. The coarse-grained model retains the essential structural characteristics and interactions of the (Ti2CO2) MXene, ensuring that the simulations remain representative of the real material. Similar approaches have been successfully employed in previous studies to model Ti2CO2 MXene, demonstrating the validity of this simplification.35 The all-atom and coarse-grained structure of the Ti2CO2 is represented in Fig. S1.† We assigned Q (charged) type parameter to the merged beads of titanium and oxygen atoms because the MXene surface is negatively charged in a water solution. This choice accurately represents the electrostatic interactions of the negatively charged surface. For the carbon atoms, we assigned C type parameter to reflect their specific properties within the simulation.
The nanosheet took on a rhombus shape with a large diameter of 36 Å, a small diameter of 22 Å, a thickness of 3.4 Å, and surface area of 207.81 Å2.
The CG structure of the POPC and water molecules, along with the ions, are generated using charmm-gui.40 The membrane at the center of the box consisted of a total of 660 lipids. The box, measuring 157 × 157 × 162 Å3, contains 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 101 water beads and 290 ions. The MXene, membrane, and water box are merged using VMD software,41 and a periodic boundary condition is set. Three sets of steered molecular dynamics (SMD) simulations are carried out, investigating the behavior of a nanosheet in relation to a membrane at five different temperatures: 300, 320, 340, 360, and 370 kelvin. The temperature and pressure of the system are kept constant using Langevin dynamics. Non-bond interactions are limited to a cutoff distance of 20 Å. Three distinct configurations are selected for analysis: CF1, where the normal vector of the nanosheet is aligned parallel to the normal vector of the membrane, CF2, where the normal vector of the nanosheet is perpendicular to the normal vector of the membrane, CF3, where the normal vector of the nanosheet forms a 45-degree angle with the normal vector of the membrane (Fig. 1). To maintain the orientation of the nanosheet, harmonic constraints are applied to the x and y coordinates of its atoms. However, the atoms are allowed to freely move along the z direction. This constraint ensures that the selected atoms remain at their equilibrium positions and only exhibited vibrations within the x − y plane. Furthermore, bonds are introduced between the atoms of the nanosheet to ensure the preservation of its shape and orientation. Our primary goal is to investigate the effect of temperature on the interaction between the MXene and the membrane. Although we considered three orientations, our main focus was on understanding how temperature influences the interactions in these specific orientations. In the CF1 configuration, the nanosheet faces the membrane with its surface, requiring a high force to pass through the membrane. In the CF2 configuration, the nanosheet faces the membrane with its vertices, requiring a lower force to penetrate the membrane. In the CF3, nanosheet touches the membrane with vertex of the major diagonal of the rhombus. To consider the effect of temperature on these three states, we fixed the orientation of the nanosheet. So we concentrated our analysis on these three configurations to investigate effect of the temperature rise on the interactions between the nanosheet and membrane.
101 water beads and 290 ions. The MXene, membrane, and water box are merged using VMD software,41 and a periodic boundary condition is set. Three sets of steered molecular dynamics (SMD) simulations are carried out, investigating the behavior of a nanosheet in relation to a membrane at five different temperatures: 300, 320, 340, 360, and 370 kelvin. The temperature and pressure of the system are kept constant using Langevin dynamics. Non-bond interactions are limited to a cutoff distance of 20 Å. Three distinct configurations are selected for analysis: CF1, where the normal vector of the nanosheet is aligned parallel to the normal vector of the membrane, CF2, where the normal vector of the nanosheet is perpendicular to the normal vector of the membrane, CF3, where the normal vector of the nanosheet forms a 45-degree angle with the normal vector of the membrane (Fig. 1). To maintain the orientation of the nanosheet, harmonic constraints are applied to the x and y coordinates of its atoms. However, the atoms are allowed to freely move along the z direction. This constraint ensures that the selected atoms remain at their equilibrium positions and only exhibited vibrations within the x − y plane. Furthermore, bonds are introduced between the atoms of the nanosheet to ensure the preservation of its shape and orientation. Our primary goal is to investigate the effect of temperature on the interaction between the MXene and the membrane. Although we considered three orientations, our main focus was on understanding how temperature influences the interactions in these specific orientations. In the CF1 configuration, the nanosheet faces the membrane with its surface, requiring a high force to pass through the membrane. In the CF2 configuration, the nanosheet faces the membrane with its vertices, requiring a lower force to penetrate the membrane. In the CF3, nanosheet touches the membrane with vertex of the major diagonal of the rhombus. To consider the effect of temperature on these three states, we fixed the orientation of the nanosheet. So we concentrated our analysis on these three configurations to investigate effect of the temperature rise on the interactions between the nanosheet and membrane.
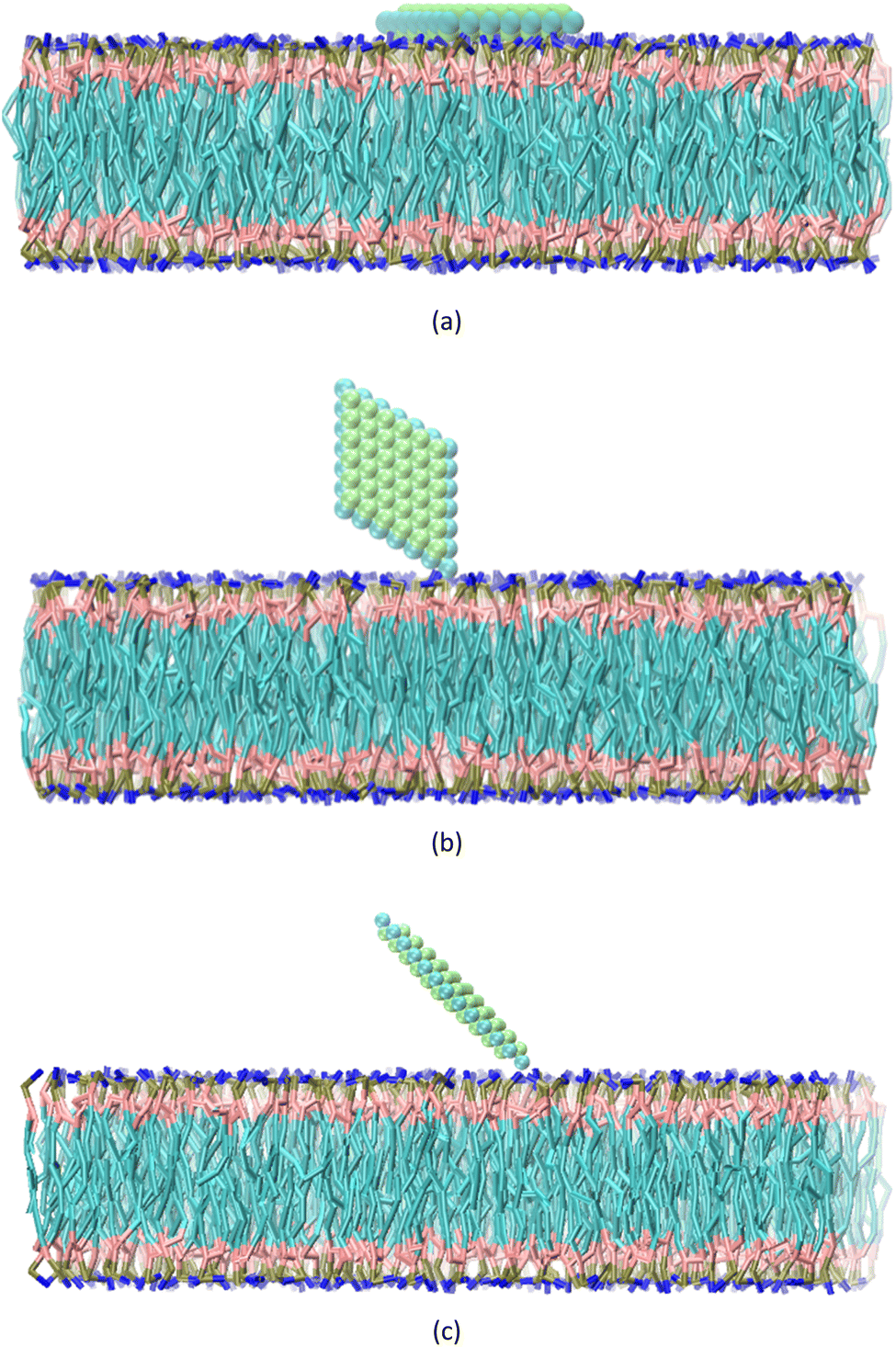 | ||
| Fig. 1 Schematic view of initial state of (a) CF1, (b) CF2 and (c) CF3 configurations. | ||
The nanosheet is initially positioned directly above the upper layer of the membrane in CF1, CF2 and CF3 configurations. Subsequently, a 2 nano-second (ns) minimization process is conducted for each temperature setting. After completing the minimization process, a 5 ns NPT (constant pressure, constant temperature) simulation, with the nanosheet atoms stationary, is performed. This is followed by a 5 ns constant velocity SMD simulation within the NPT ensemble at each specified temperature. In order to execute the SMD simulation with constant velocity, a spring constant of 5 kcal mol−1 Å2 and a pull rate of 22 × 10−6 Å fs−1 is utilized. To ensure that the fast-pulling speed and the size of the system do not affect the results, we have performed simulations with a larger system size and a slower pulling speed. As shown in Fig. S2,† the force converges to the same value in these simulations. This indicates that the results are not significantly dependent on either the pulling speed or the size of the system. This consistency suggests that our findings are robust and reliable, regardless of these variables.
The SMD simulations are sustained until the nanosheet had completely traversed the membrane. All simulations are conducted using NAMD.42
Results
In this section, we investigate how the hydrophilic properties of Ti2CO2 MXene can hinder its spontaneous uptake by the membrane, which leads to the presence of an energy barrier. Nevertheless, by increasing the temperature, this energy barrier can be modified, thereby promoting the uptake process. Furthermore, we also analyze the influence of the orientation of the nanosheet on the penetration process.The correlation between the rise in temperature and the interaction between the nanosheet and the membrane can be evaluated by analyzing the pulling force–time graph and the subsequent work–time graph. We analyze various factors, including the van der Waals interaction between the nanosheet and the membrane, the role of the water molecules within the pore, the membrane thickness, as well as the electrostatic interaction between the nanosheet and the membrane. Fig. 2 illustrates the force versus the stimulation time for the CF1, CF2 and CF3 configurations. In CF1, the nanosheet is positioned with its surface facing the membrane, while in CF2, the nanosheet faces the membrane with its edge. In CF3, nanosheet touches the membrane with vertex of the major diagonal of the rhombus. The CF1 configuration allows for a greater contact area between the nanosheet and the lipid head groups. In this configuration, a larger pore is required for the nanosheet to penetrate the membrane, resulting in an anticipated increase in the force barrier for penetration.
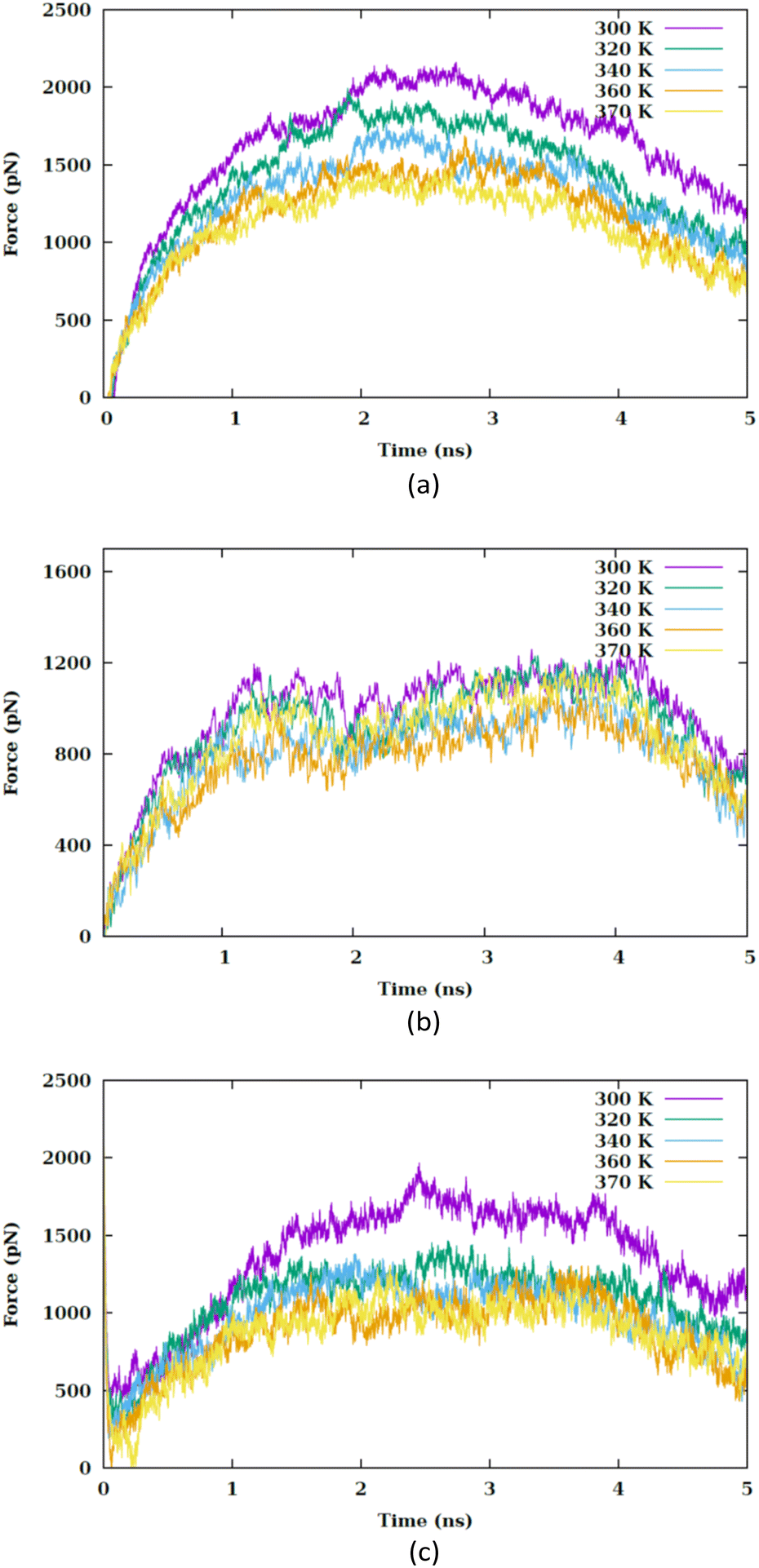 | ||
| Fig. 2 Pulling forces versus the simulation time at different values of temperature (i.e., 300, 320, 340, 360 and 370 K) for (a) CF1, (b) CF2 and (c) CF3 configurations. | ||
In each temperature for the three configurations, there exists a peak in the force graph representing the penetration of the nanosheet into the membrane. This peak indicates the minimum force required for the nanosheet to surpass the energy barrier that hinders its penetration into the membrane. The energy barrier encompasses the energy necessary to disrupt the membrane's structure, create a pore, and overcome the hydrophobic interaction between the hydrophilic nanosheet and the carbon chains of the lipids within the membrane. In the CF1 configuration, the force increases and decreases in a parabolic manner, reaching a maximum before dropping. In the CF2 configuration, the force gradually increases, peaks remain nearly constant for a period, and then drops. In the CF3 configuration, the force variation is similar to CF1 but with smoother transitions. In the three configurations, as the temperature increases, the maximum force reduces. Fig. 3(a) compares the maximums in the force graph for three configurations.
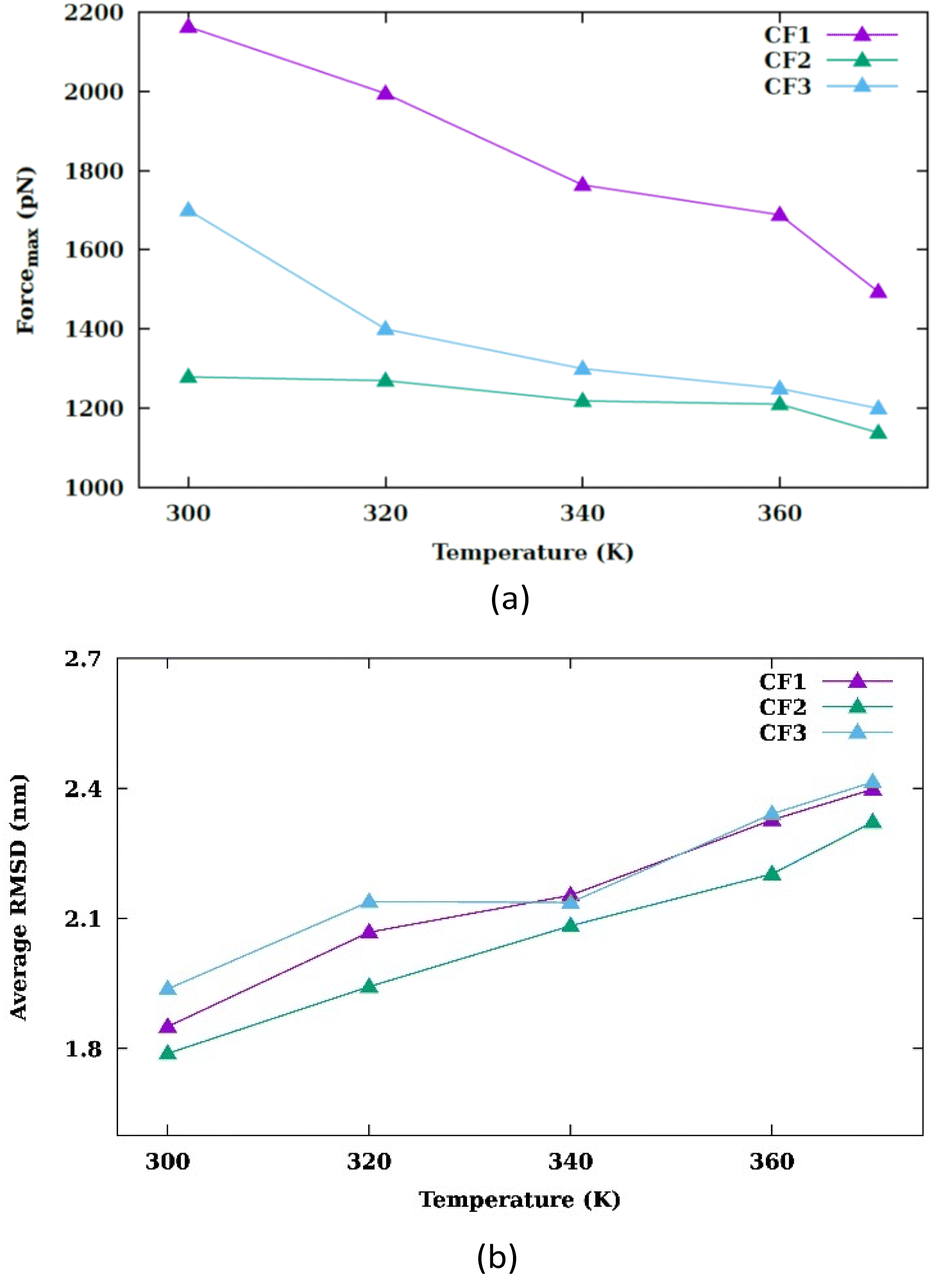 | ||
| Fig. 3 (a) Maximum pulling force at different values of temperature for CF1, CF2 and CF3 configurations. (b) Average Root Mean Square Deviation of CF1, CF2 and CF3 configurations at different values of temperature. | ||
The CF2 configuration shows no notable variation in maximum force across different temperatures, whereas the CF1 and CF3 configurations exhibits a substantial alteration in maximum forces under varying temperature conditions. When the nanosheet infiltrates the membrane, a pore begins to develop.
During the pore formation process, water molecules, lipid head groups and ions enter the pore, leading to the creation of a hydrophilic pore (refer to Fig. S5†).
To explore the impact of nanosheet size on the variation of pulling force, we utilize a scaled-down variant of the primary nanosheet, designated as n2 and a reduced surface area of 138.5 Å2 at 340 kelvin. This is compared against the original nanosheet, referred to as n1, within the CF1 configuration. As depicted in Fig. 4, the force–time graph is influenced by the nanosheet's dimensions. A reduction in nanosheet size leads to a decreased interaction area with the membrane, thereby facilitating the nanosheet's traversal through the membrane. The force–time profile nearly returns to its initial value at the commencement of the simulation. This phenomenon can be attributed to the reduced surface area of n2, resulting in a lesser degree of interaction with the membrane.
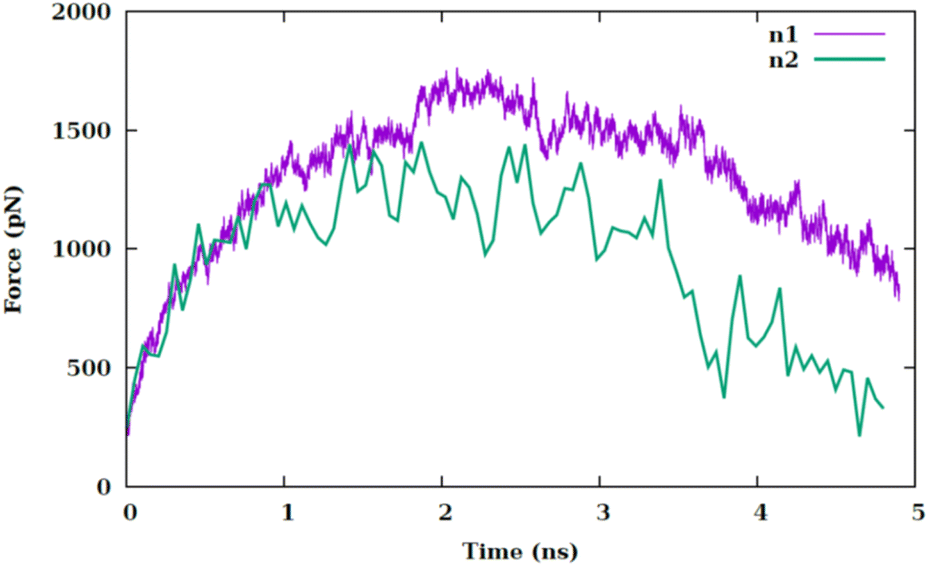 | ||
| Fig. 4 Pulling force versus the simulation time at temperature of 340 K for two different sizes of nanosheet (i.e., n1 and n2) for CF1 configuration. | ||
The peak in the force graph corresponds to the energy barrier required to insert the hydrophilic nanoparticle into the membrane. Upon exceeding the barrier, there is a subsequent diminution in the magnitude of the force. The Root Mean Square Deviation (RMSD) is illustrated in Fig. S3.† Fig. 3(b) displays the average RMSD of the membrane atoms in the three different configurations. The phospholipid tail order parameter and the average area per lipid are common indicators of membrane properties, reflecting local structural changes. To evaluate overall structural changes and the conformational dynamics of the membrane as a whole, we utilized RMSD. An increase in temperature results in an increase in the mobility of the membrane atoms.
This rise in temperature leads to enhanced fluidity and permeability of the membrane, facilitating the penetration of the nanosheet into the membrane. As depicted in Fig. 3(a), the peak force decreases with the increase in temperature. The work graph in Fig. S4† demonstrates a reduction in work as the temperature rises.
The work required to translocate the nanosheet across the membrane is significantly high (of order 108 pN nm), hence spontaneous uptake does not occur. The increase in temperature alters the interaction potentials within the system. Generally, a rise in temperature diminishes the attractive part of the Lennard-Jones potential and accentuates the repulsive part. The increase in temperature elevates the kinetic energy of the particles, enabling them to escape the attractive potential well. Fig. 5 exhibits the variation in the Lennard-Jones potential between the nanosheet and the membrane. Consequently, fewer water molecules are situated between the nanosheet and the membrane atoms, allowing for a closer proximity between the nanosheet and the membrane atoms.
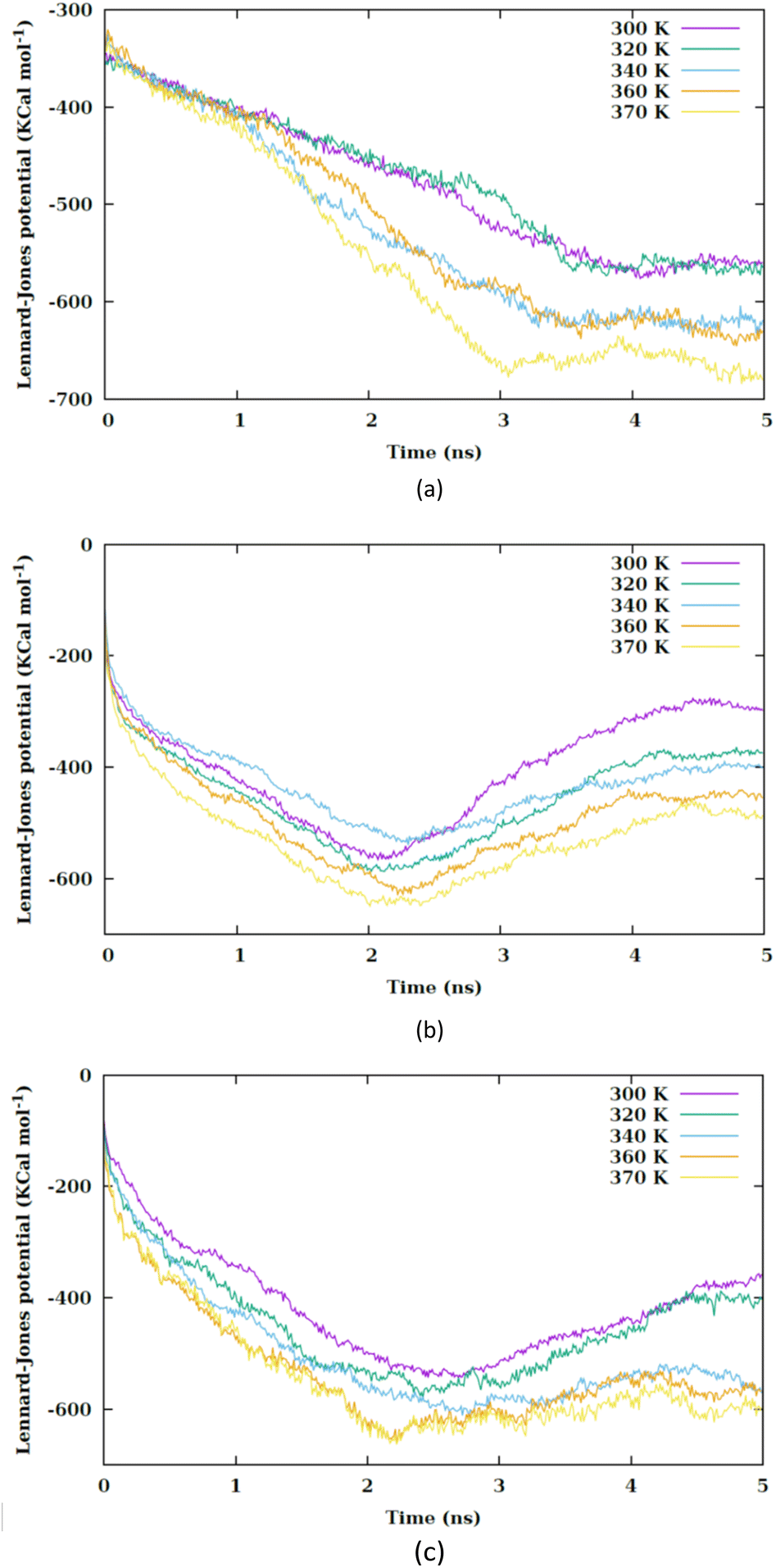 | ||
| Fig. 5 Lennard-Jones potential between the nanosheet and membrane versus the simulation time at different values of temperature for (a) CF1, (b) CF2 and (c) CF3 configurations. | ||
By increasing the temperature, the Lennard-Jones potential is observed to become increasingly attractive. Fig. S5† illustrates the quantity of water molecules present within the cylindrical region with a radius of 4 nm located in the interior of the membrane, specifically centered at the nanoparticle's center of mass. It is evident that with a rise in temperature, the count of water molecules decreases. The movement of the membrane's atoms is notably enhanced at elevated temperatures, leading to a potential reduction in the radius of the pore. Consequently, the pore on the membrane's surface may even close, thereby restricting the entry of water molecules into the membrane. This proximity results in a heightened attraction of the Lennard-Jones potential between the nanosheet and the water molecules. Prior to the nanosheet entering the interior of the membrane, a greater concentration of water molecules surrounds it. Consequently, the Lennard-Jones potential between the water molecules and the membrane exhibits a higher value. As the temperature increases and the quantity of water molecules decreases. There is a corresponding reduction in the Lennard-Jones potential between the nanosheet and the water molecules, as shown in Fig. 6. When comparing Fig. 5 with Fig. 6, it becomes clear that the interactions between the nanosheet and the membrane are primarily governed by van der Waals forces, while the electrostatic potential has a secondary influence. Therefore, in the context of thermal therapy applications involving nanoparticles, van der Waals interaction is deemed more crucial than electrostatic interaction. Nevertheless, long-range electrostatic interaction is more significant in the adhesion of charged nanoparticles. The water molecules and carbon chains are considered electrically inert in the martini force field, and the electrostatic interaction between the nanosheet and the system occurs via the lipids' head groups and ions. As the temperature increases, the electrostatic interaction between the nanosheet and the charged head groups of the lipids becomes more negative in the three configurations, as shown in Fig. 7. This phenomenon is attributed to the reduction in the number of water molecules within the pore at higher temperatures, allowing the charged head groups of the lipids to approach the nanosheet more closely, resulting in a more negative electrostatic potential. In the martini force field, the head group of POPC lipid comprises an NC3 bead with a +1 charge unit and a PO4 bead with a −1 charge unit. The penetration process of the nanosheet is significantly influenced by the van der Waals interaction.
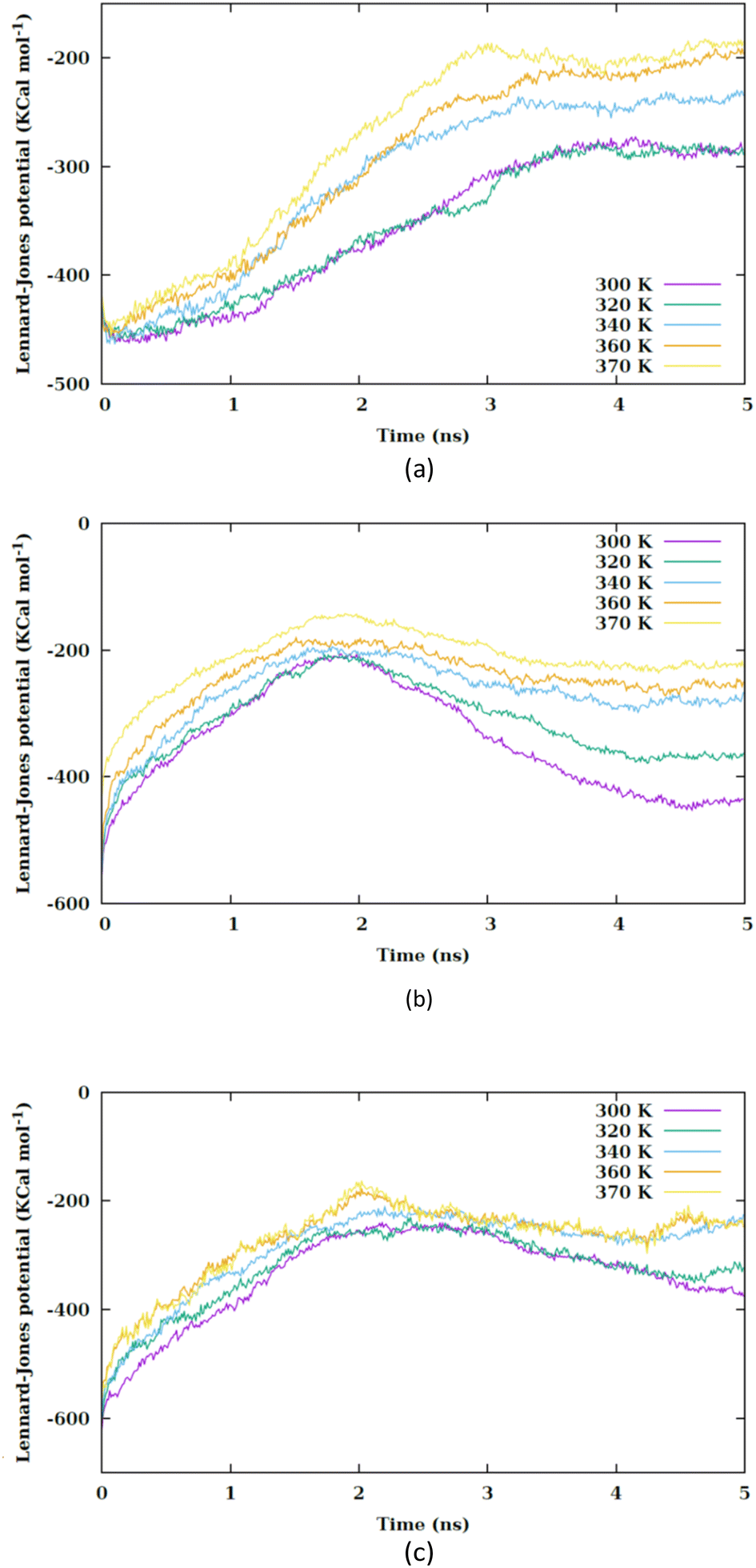 | ||
| Fig. 6 Lennard-Jones potential between the nanosheet and water molecules versus the simulation time at different values of temperature for (a) CF1, (b) CF2 and (c) CF3 configurations. | ||
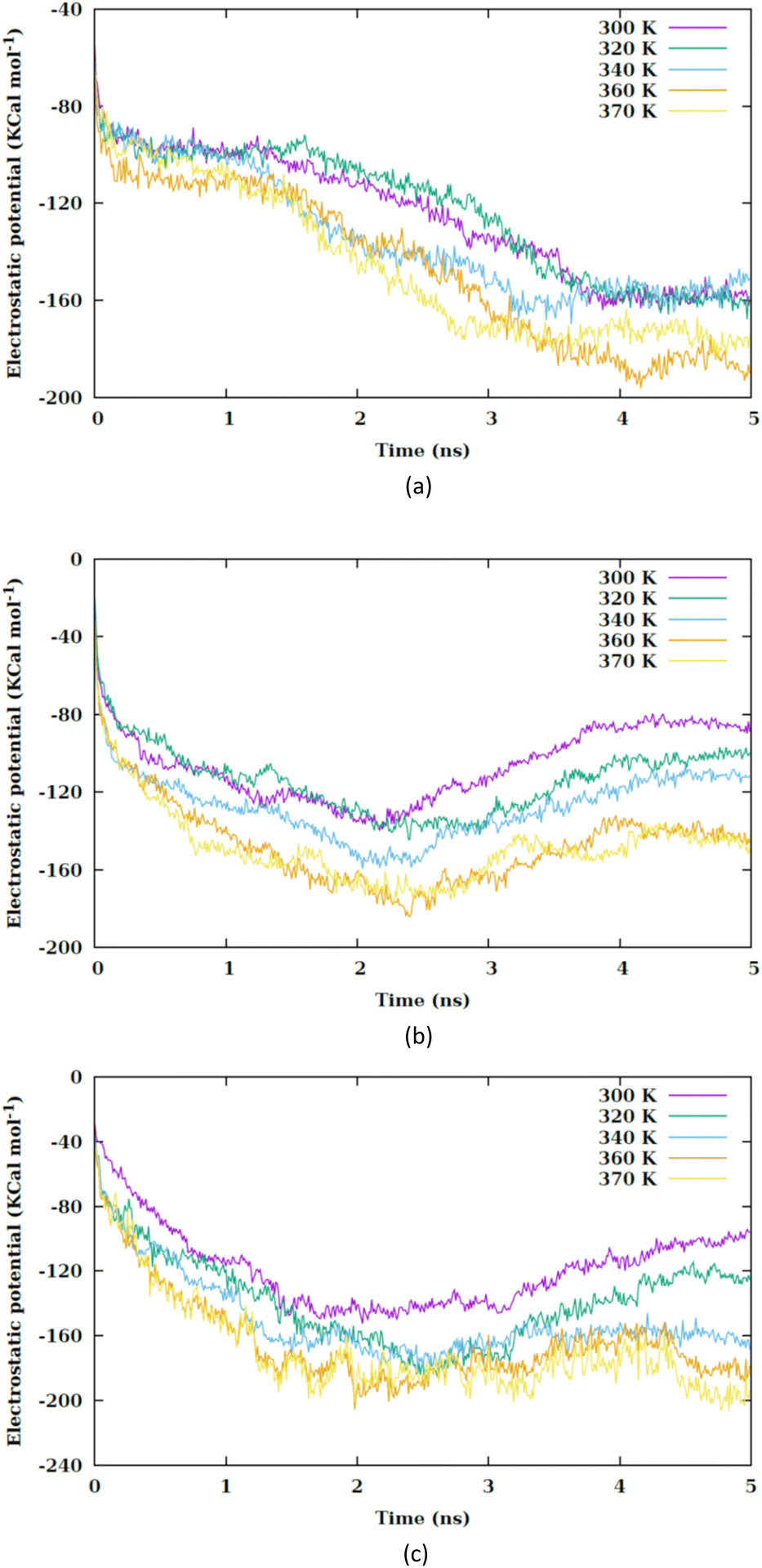 | ||
| Fig. 7 Electrostatic interaction potential between the nanosheet and membrane versus the simulation time at different values of temperature for (a) CF1, (b) CF2 and (c) CF3 configurations. | ||
Hydrophilic nanoparticles tend to remain close to the surface of the membrane, while hydrophobic nanoparticles prefer to be located inside the membrane, where the lipids' carbon chains are present. In Fig. S6(a–c)†, the Lennard-Jones potential illustrates the interaction between the nanosheet and the head groups of the membrane, while Fig. S6(d–f)†, depict the interaction between the nanosheet and the carbon chains of the lipids. It is evident that the van der Waals interaction between the nanosheet and the head groups of the lipids is considerably stronger than the interaction between the nanosheet and the carbon chains. This discrepancy in interaction strength contributes to the existence of a large energy barrier for penetration. Upon passing through the membrane, the nanosheet is observed to extract some lipids from the membrane (Fig. S7–S9†). Fig. S6† demonstrates that even after the nanosheet exits the membrane, there are still interactions between the nanosheet and phospholipids. Notably, this interaction is slightly more pronounced in the CF1 configuration due to its specific penetration pattern. The MEMB-PLUGIN43 is utilized within the VMD environment to determine the thicknesses of lipid bilayer membrane.
The mean local thickness of the membranes under conditions CF1, CF2 and CF3 are depicted in Fig. S10–S12,† respectively. An increase in temperature is found to be correlate with an augmentation in the local thickness of the membranes. Specifically, Fig. 8 indicates that in condition CF1, the temperature elevation does not significantly influence the membrane thickness compared to CF2 and CF3.
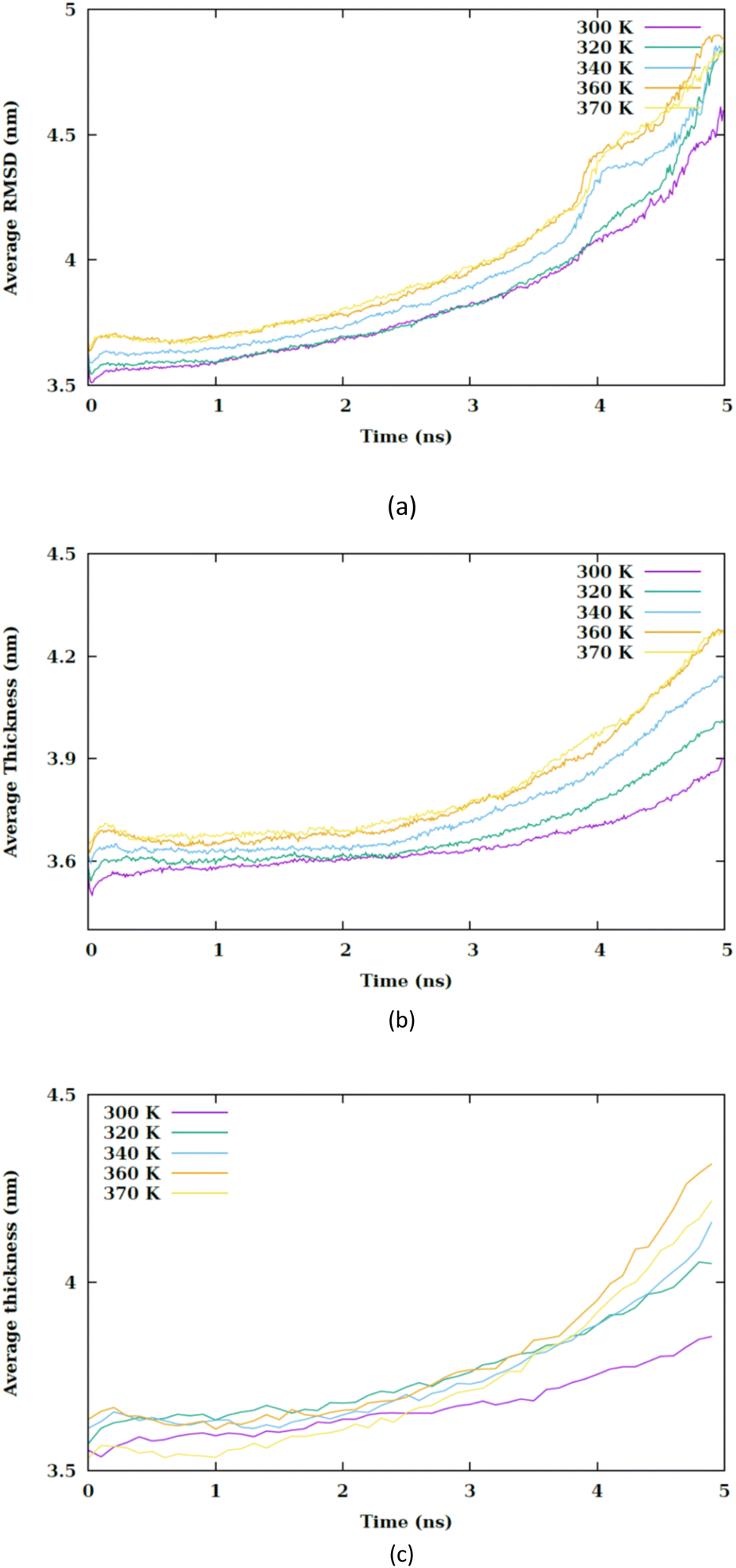 | ||
| Fig. 8 Average thickness of membrane versus the simulation time at different values of temperature for (a) CF1, (b) CF2 and (c) CF3 configurations. | ||
However, an increment in membrane thickness is observed subsequent to the translocation of the nanosheet, indicating that the structural impact of the nanosheet translocation on the membrane is more substantial than that of the temperature increase. Although a slight increase in membrane thickness is associated with temperature rise, it is the translocation of the nanosheet that predominantly affects the membrane structure. In conditions CF2 and CF3 exhibits an alteration in membrane thickness as a result of temperature increase. It is generally observed that an increase in membrane thickness leads to a decrease in the entanglement of phospholipid tails, thereby enhancing their mobility and ultimately contributing to an increase in membrane fluidity.
Conclusions
The impact of temperature and nanosheet orientation on the interaction between a single layer of Ti2CO2 MXene and the POPC membrane has been investigated using coarse-grained molecular dynamics simulation. The results demonstrate that temperature is a critical factor in controlling the cellular uptake of nanoparticles. As increased temperatures improve the membrane's permeability. Hydrophilic nanoparticles, like MXenes, face substantial penetration barriers. However, high temperatures can facilitate the uptake process, particularly under extreme conditions such as unique nanoparticle shapes and orientations or high initial momentum. Significantly, as the temperature rises, there are notable alterations in the interaction potentials between the nanosheet and the membrane, along with changes in the dynamics of the membrane itself. This modification in the interaction potentials leads to a change in the penetration barrier of the nanosheet. Moreover, altering the orientation of the nanosheet and reducing the contact surface area with the membrane have been found to decrease the penetration barrier. This suggests that the alignment of the nanosheet plays a crucial role in its interaction with the membrane. The main mode of interaction between the nanosheet and the membrane seems to be through van der Waals forces, which are responsible for the strong interaction between the hydrophilic MXenes and the polar head groups of lipids in the membrane. This property of MXenes is particularly advantageous in photothermal applications. The nanosheet efficiently transfers heat to the lipids' head groups, causing surface disruption and severe membrane damage. Overall, the study highlights the importance of temperature and nanosheet orientation in understanding the interaction between MXenes and cell membranes, which can have significant implications in various fields, including nanomedicine and photothermal therapy.Data availability
This article contains data generated from a theoretical studies in the field of classical molecular dynamics simulation. The files supporting the findings of this study are available from the corresponding author, Mahdi Esmaeilzadeh, upon reasonable request. The data that support the findings of this study are available within the article and its ESI materials.† Additional files related to this paper may be requested from the authors.Conflicts of interest
There are no conflicts to declare.References
- D. Drabik, G. Chodaczek, S. Kraszewski and M. Langner, Me-chanical properties determination of dmpc, dppc, dspc, and hspc solid-ordered bilayers, Langmuir, 2020, 36(14), 3826–3835 CrossRef CAS PubMed.
- A. Annibal, T. Riemer, O. Jovanovic, D. Westphal, E. Griesser, E. E. Pohl, J. Schiller, R. Hoffmann and M. Fedorova, Structural, biological and biophysical properties of glycated and glycoxidized phosphatidylethanolamines, Free Radical Biol. Med., 2018, 49, 293–307 Search PubMed.
- S. Angioletti-Uberti, Theory, simulations and the design of functionalized nanoparticles for biomedical applications: A soft matter perspective, npj Comput. Mater., 2017, 3(1), 48 CrossRef.
- F. Shahzad, M. Alhabeb, C. B. Hatter, B. Anasori, S. M. Hong, C. M. Koo and Y. Gogotsi, Electromagnetic interference shielding with 2d transition metal carbides (mxenes), Science, 2016, 353(6304), 1137–1140 CrossRef CAS PubMed.
- H. Lin, X. Wang, L. Yu, Y. Chen and J. Shi, Two-dimensional ultrathin mxene ceramic nanosheets for photothermal conversion, Nano Lett., 2017, 17(1), 384–391 CrossRef CAS PubMed.
- J. Liu, H. B. Zhang, R. Sun, Y. Liu, Z. Liu, A. Zhou and Z. Z. Yu, Hydrophilic mxene nanocomposite membranes for highly efficient photothermal conversion under sunlight, Adv. Mater., 2018, 30(15), 1707335 CrossRef PubMed.
- C. Zhang, L. McKeon, M. P. Kremer, S. H. Park, O. Ronan, A. Seral-Ascaso and J. N. Coleman, High capacity silicon anodes enabled by mxene viscous aqueous ink, Nat. Commun., 2019, 10(1), 849 CrossRef PubMed.
- Y. Wang, W. Yan, J. Zhang, Z. Li and Y. Guo, Ti3C2 mxene nanosheet-based dual-enzyme cascade reaction to facilitate dual-stimulation-mediated breast cancer therapy, ACS Appl. Nano Mater., 2024, 7(7), 7345–7354 CrossRef CAS.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, 2d metal carbides and nitrides (mxenes) for energy storage, Nat. Rev. Mater., 2017, 2(2), 16098 CrossRef CAS.
- I.-C. Lee, Y.-C. Ethan Li, J. L. Thomas, M.-H. Lee and H.-Y. Lin, Recent advances using mxenes in biomedical applications, Mater. Horiz., 2024, 11, 876–902 RSC.
- H. Li, R. Fan, B. Zou, J. Yan, Q. Shi and G. Guo, Roles of mxenes in biomedical applications: recent developments and prospects, J. Nanobiotechnol., 2023, 21(1), 73 CrossRef PubMed.
- H. M. Ding and Y. Q. Ma, Interactions between janus particles and membranes, Nanoscale, 2012, 4, 1116–1122 RSC.
- F. Haque, S. Jamali and M. Moradi, Molecular dynamics simulation analysis of the effects and mechanisms of lipid nanoparticles in drug delivery systems, Biophys. J., 2024, 123(3), 504a CrossRef.
- S. Franco-Ulloa, D. Guarnieri, L. Riccardi, P. P. Pompa and M. De Vivo, Association mechanism of peptide-coated metal nanoparticles with model membranes: A coarse-grained study, J. Chem. Theory Comput., 2021, 17(7), 4512–4523 CrossRef CAS PubMed.
- A. M. Engstrom, R. A. Faase, G. W. Marquart, J. E. Baio, M. R. Mackiewicz and S. L. Harper, Size-dependent interactions of lipid-coated gold nanoparticles: Developing a better mechanistic understanding through model cell membranes and in vivo toxicity, Int. J. Nanomed., 2020, 15, 4091–4104 CrossRef CAS PubMed.
- X. Zhang, G. Ma and W. Wei, Simulation of nanoparticles interacting with a cell membrane: probing the structural basis and potential biomedical application, NPG Asia Mater., 2021, 13(1), 52 CrossRef CAS.
- E. Lavagna, J. Barnoud, G. Rossi and L. Monticelli, Size-dependent aggregation of hydrophobic nanoparticles in lipid membranes, Nanoscale, 2020, 12, 9452–9461 RSC.
- Y. Nademi, T. Tang and H. Uludăg, Nature of bilayer lipids affects membranes deformation and pore resealing during nanoparticle penetration, Mater. Sci. Eng., C, 2022, 132, 112530 CrossRef CAS PubMed.
- D. Drabik, G. Chodaczek, S. Kraszewski and M. Langner, Mechanical properties determination of dmpc, dppc, dspc, and hspc solid-ordered bilayers, Nanoscale Adv., 2021, 3, 6635 RSC.
- R. C. Van Lehn, M. Ricci, P. H. Silva, P. Andreozzi, J. Reguera, K. Voïtchovsky and A. Alexander-Katz, Lipid tail protrusions mediate the insertion of nanoparticles into model cell membranes, Nat. Commun., 2014, 5(1), 4482 CrossRef CAS PubMed.
- H. Zhang, Q. Ji, C. Huang, S. Zhang, B. Yuan, K. Yang and Y. Q. Ma, Cooperative transmembrane penetration of nanoparticles, Sci. Rep., 2015, 5(1), 10525 CrossRef PubMed.
- R. Gupta and B. Rai, In-silico design of nanoparticles for transdermal drug delivery application, Nanoscale, 2018, 10, 4940–4951 RSC.
- C. F. Su, H. Merlitz, H. Rabbel and J. U. Sommer, Nanoparticles of various degrees of hydrophobicity interacting with lipid membranes, J. Phys. Chem. Lett., 2017, 8, 4069–4076 CrossRef CAS PubMed.
- E. Lavagna, J. Barnoud, G. Rossi and L. Monticelli, Size-dependent aggregation of hydrophobic nanoparticles in lipid membranes, Nanoscale, 2020, 12(17), 9452–9461 RSC.
- S. Nangia and R. Sureshkumar, Effects of nanoparticle charge and shape anisotropy on translocation through cell membranes, Langmuir, 2013, 28, 17666–17671 CrossRef PubMed.
- Z. Shen, H. Ye, M. Kröger and Y. Li, Aggregation of polyethylene glycol polymers suppresses receptor-mediated endocytosis of pegylated liposomes, Nanoscale, 2018, 10, 4545–4560 RSC.
- R. Gupta, Y. Badhe, S. Mitragotri and B. Rai, Permeation of nanoparticles across the intestinal lipid membrane: dependence on shape and surface chemistry studied through molecular simulations, Nanoscale, 2020, 12, 6318–6333 RSC.
- H. Nakamura, K. Sezawa, M. Hata, S. Ohsaki and S. Watano, Direct translocation of nanoparticles across a model cell membrane by nanoparticle-induced local enhancement of membrane potential, Phys. Chem. Chem. Phys., 2019, 21(35), 18830–18838 RSC.
- Z. Li, Y. Zhang, D. Zhu, S. Li, X. Yu, Y. Zhao, X. Ouyang, Z. Xie and L. Li, Transporting carriers for intracellular targeting delivery via non-endocytic uptake pathways, Drug Delivery, 2017, 24(sup1), 45–55 CAS.
- H. Cghen, X. Dong, L. Ou, C. Ma, B. Yuan and K. Yang, Thermal-controlled cellular uptake of “hot” nanoparticles, Nanoscale, 2023, 15(30), 12718–12727 RSC.
- Z. Shen, H. Ye and Y. Li, Understanding receptor-mediated endocytosis of elastic nanoparticles through coarse grained molecular dynamic simulation, Phys. Chem. Chem. Phys., 2018, 20, 16372–16385 RSC.
- L. L. Huang, W. Nie, J. Zhang and H. Y. Xie, Cell-membrane-based biomimetic systems with bioorthogonal functionalities, Acc. Chem. Res., 2020, 53, 276–287 CrossRef CAS PubMed.
- L. Liu, X. Bai, M. V. Martikainen, A. Kårlund, M. Roponen, W. Xu and V. P. Lehto, Cell membrane coating integrity affects the internalization mechanism of biomimetic nanoparticles, Nat. Commun., 2021, 12(1), 5726 CrossRef CAS PubMed.
- Y. Q. Chen, M. D. Xue, J. L. Li, D. Huo, H. M. Ding and Y. Ma, Uncovering the importance of ligand mobility on cellular uptake of nanoparticles: Insights from experimental, computational, and theoretical investigations, ACS Nano, 2024, 18(8), 6463–6476 CrossRef CAS PubMed.
- D. Wu, R. Zhao, Y. Chen, Y. Wang, J. Li and Y. Fan, Molecular insights into mxene destructing the cell membrane as a “nano thermal blade”, Phys. Chem. Chem. Phys., 2021, 23(5), 3341–3350 RSC.
- X. Zhang, G. Ma and W. Wei, Simulation of nanoparticles interacting with a cell membrane: probing the structural basis and potential biomedical application, NPG Asia Mater., 2021, 13(1), 52 CrossRef CAS.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. De Vries, The martini force f ield: Coarse grained model for biomolecular simulations, J. Phys. Chem. B, 2007, 27(111), 7812–7824 CrossRef PubMed.
- X. Zhang, X. Zhao, D. Wu, Y. Jing and Z. Zhou, High and anisotropic carrier mobility in experimentally possible ti2co2 (mxene) monolayers and nanoribbons, Nanoscale, 2015, 7(38), 16020–16025 RSC.
- Y.-m. Ding, X. Nie, H. Dong, N. Rujisam-phan and Y. Li, Many-body effects in an mxene Ti2Co2 monolayer modified by tensile strain: Gw-bse calculations, Nanoscale Adv., 2020, 2(6), 2471–2477 RSC.
- Y. Qi, H. I. Ingólfsson, X. Cheng, J. Lee, S. J. Marrink and W. Im, Charmm-gui martini maker for coarse-grained simulations with the martini force field, J. Chem. Theory Comput., 2015, 11(9), 4486–4494 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, Vmd - visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa and K. Schulten, Scalable molecular dynamics with namd, J. Comput. Chem., 2005, 26(16), 1781–1802 CrossRef CAS PubMed.
- R. Guixà-González, I. Rodriguez-Espigares, J. M. Ramírez-Anguita, P. Carrió-Gaspar, H. Martinez Seara, T. Giorgino and J. Selent, Membplugin: studying membrane complexity in vmd, Bioinformatics, 2014, 30(10), 1478–1480 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05821f |
| This journal is © The Royal Society of Chemistry 2024 |