
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anti-hepatocellular carcinoma activities of novel hydrazone derivatives via downregulation of interleukin-6†
Ahmed Nabil *abc,
Marwa Abdel-Motaal*d,
Ayman Hassanc,
Mohamed M. Elshemye,
Medhat Asemf,
Mariam Elwang,
Mitsuhiro Ebaraahi,
Mohammed Abdelmageedjk,
Gamal Shihacl and
Hassan M. E. Azzazy*m
*abc,
Marwa Abdel-Motaal*d,
Ayman Hassanc,
Mohamed M. Elshemye,
Medhat Asemf,
Mariam Elwang,
Mitsuhiro Ebaraahi,
Mohammed Abdelmageedjk,
Gamal Shihacl and
Hassan M. E. Azzazy*m
aResearch Center for Macromolecules and Biomaterials, National Institute for Materials Science (NIMS), Tsukuba, 305-0044, Japan. E-mail: TOLBA.AhmedNabil@nims.go.jp; Tel: +201000618349
bBiotechnology and Life Sciences Department, Faculty of Postgraduate Studies for Advanced Sciences (PSAS), Beni-Suef University, Beni-Suef, Egypt
cEgyptian Liver Research Institute and Hospital (ELRIAH), Sherbin, El Mansoura, Egypt
dDepartment of Chemistry, College of Science, Qassim University, Qassim, Buraydah, 51452, Saudi Arabia. E-mail: ma.mohammed@qu.edu.sa; Tel: +966569909737
eFaculty of Science, Menoufia University, Menoufia, Egypt
fDepartment of Civil Engineering, College of Engineering and Information Technology, Onaizah Colleges, Qassim, Saudi Arabia
gEgyptian Ministry of Health, El Mansoura, Dakahlia, Egypt
hGraduate School of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki, 305-8577, Japan
iGraduate School of Industrial Science and Technology, Tokyo University of Science, 6-3-1 Niijuku, Katsushika-ku, Tokyo 125-8585, Japan
jDepartment of Pharmacology and Toxicology, Faculty of Pharmacy, Buraydah Colleges, Qassim, Saudi Arabia
kHot Laboratory Center, Atomic Energy Authority, Cairo, Egypt
lHepatology and Gastroenterology Unit, Internal Medicine Department, Faculty of Medicine, Mansoura University, Egypt
mDepartment of Chemistry, School of Sciences & Engineering, The American University in Cairo, New Cairo, 11835, Egypt. E-mail: hazzazy@aucegypt.edu; Tel: +201000565727
First published on 28th November 2024
Abstract
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related morbidity worldwide. Sorafenib is a first-line drug for the treatment of HCC, however, it is reported to cause serious adverse effects and may lead to resistance in many patients. In this study, 20 hydrazone derivatives incorporating triazoles, pyrazolone, pyrrole, pyrrolidine, imidazoline, quinazoline, and oxadiazine moieties were designed, synthesized, and characterized. In addition to molecular docking and in silico ADME study, the cytotoxic activity of the synthesized compounds was evaluated against the human hepatocellular cancer cell line (HepG2) and liver mesenchymal stem cells as a normal cell line. The antitumor activities of the derivatives against sorafenib were compared. Of the 20 synthesized compounds, compound 16 demonstrated potential as a potent anti-HCC drug candidate through downregulation of interleukin 6 which reduces inflammation and tumorigenesis with a strong binding interaction and bioavailability.
1. Introduction
HCC is considered the sixth most identified neoplasm and the second most common cause of cancer-related death worldwide.1,2 It usually occurs in patients with liver cirrhosis.3 The most recognized risk factors for HCC include chronic viral infections of hepatitis B and C, autoimmune hepatitis, alcohol, aflatoxin B1, non-alcoholic steatohepatitis, obesity, and diabetes mellitus.4Sorafenib is a first-line drug for patients with advanced HCC. It reduces tumor cell growth and progression through inhibition of multiple serine/threonine kinases involved in tumor progression and angiogenesis.5 These include the vascular endothelial growth factor receptor (VEGFR-2/3), platelet-derived growth factor receptor (PDGF-R), Flt3, c-Kit, and Raf kinase.5,6
Although sorafenib has demonstrated clinical advantages in patients with HCC, it is reported to exert multiple side effects primarily caused by its suppression of kinases in healthy cells. These include diarrhea and dermatological effects such as hand-foot skin reactions, alopecia, stomatitis, and multiforme erythema.7 Other side effects include fatigue, hypertension, cardiovascular problems, hemorrhage, renal toxicity, pancreatitis, mouth ulcers, and weight loss.8 Furthermore, resistance to sorafenib represents a major challenge in the treatment of advanced/recurrent HCC.9 Therefore, there is a need to develop other agents to overcome the previous drawbacks.
Interleukin 6 (IL-6) is a key molecule of the immune response that is produced in response to infections and tissue damage.10 Elevated blood levels of IL-6, independent of other risk factors for HCC, have been associated with a higher risk of developing HCC.11 Furthermore, by promoting the repair and stimulation of countersignalling (antioxidant and anti-apoptotic/pro-survival) pathways, IL-6 protects tumor cells against DNA damage, oxidative stress, and/or apoptosis caused by anticancer treatments. Therefore, a potential therapeutic approach for cancer treatment could involve suppressing IL-6 or its signalling pathway alone or in conjunction with conventional anticancer strategies.12
Hydrazone derivatives represent a class of compounds containing a biologically active pharmacophore (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NH–R) with various biological effects13–15 such as antibacterial,16–18 anti-inflammatory,19 antimalarial,20 anticonvulsant,21 antidepressant, and antiproliferative22 activities. Different heterocycle-containing hydrazones have been reported to exert powerful cytotoxic and antitumor actions.23,24 Importantly, various classes of hydrazones based on coumarin, triazoles, pyridine, indole, quinolone, caffeine, pyrimidine have been evaluated for their anticancer potential against various cell lines,25–27 as depicted in Fig. 1. Additionally, hydrazone derivatives have shown promising therapeutic potential for the treatment of neurodegenerative diseases such as Alzheimer's disease.28 Hydrazone links are also used as pH-responsive drug delivery systems29 and in nifuroxazide, an antibiotic used for the treatment of colitis and dehydration. Consequently, these pharmacophores were used as intermediates for the synthesis of heterocycles with numerous biological activities.30–32
N–NH–R) with various biological effects13–15 such as antibacterial,16–18 anti-inflammatory,19 antimalarial,20 anticonvulsant,21 antidepressant, and antiproliferative22 activities. Different heterocycle-containing hydrazones have been reported to exert powerful cytotoxic and antitumor actions.23,24 Importantly, various classes of hydrazones based on coumarin, triazoles, pyridine, indole, quinolone, caffeine, pyrimidine have been evaluated for their anticancer potential against various cell lines,25–27 as depicted in Fig. 1. Additionally, hydrazone derivatives have shown promising therapeutic potential for the treatment of neurodegenerative diseases such as Alzheimer's disease.28 Hydrazone links are also used as pH-responsive drug delivery systems29 and in nifuroxazide, an antibiotic used for the treatment of colitis and dehydration. Consequently, these pharmacophores were used as intermediates for the synthesis of heterocycles with numerous biological activities.30–32
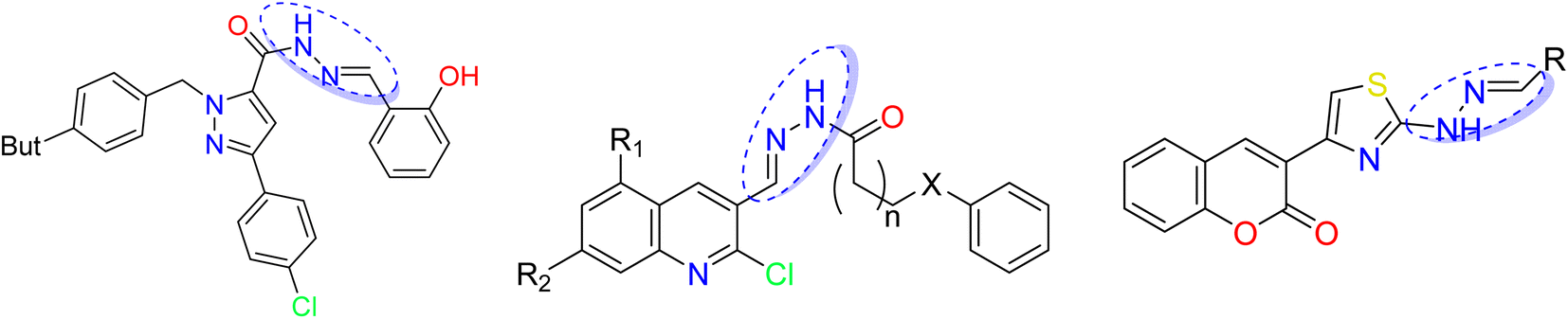 | ||
| Fig. 1 Hydrazones based on heterocycles reported showing antitumor activity. | ||
Synthesis of broad-spectrum biologically active hydrazone-based heterocycles was reported.33 Some of these compounds exhibited high in vitro anticancer activity against breast cancer cell lines (MCF7). Therefore, in this study, 20 hydrazone derivatives were investigated using structure–activity relationships (SAR) and molecular docking to select those with better anticancer activity and higher selectivity. Furthermore, the cytotoxicity of the hydrazone derivatives was assessed against the human hepatocellular carcinoma cell line (HepG2) and liver mesenchymal stem cells using MTT assay and flow cytometry. Their effects on IL-6 and cytochrome C levels and tumor cell migration were investigated.
2. Materials and methods
2.1 Synthesis and characterization of hydrazone derivatives
All chemicals, which are of the highest purity grade, were acquired from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). The synthesis of hydrazone derivatives was performed as previously reported.33 The melting points were measured using a Gallenkamp electric melting point apparatus (Gemini Lab, Apeldoorn, The Netherlands), and the results were uncorrected. Using KBr discs and a FT-IR spectrophotometer, the IR spectra of different compounds were obtained and analyzed. 1H-NMR and 13C-NMR spectra were assessed in DMSO-d6 on a JNM-ECA500II at 500 MHz NMR spectrometer (JEOL Ltd., Peabody, MA) reported as δ ppm and using tetramethyl silane (TMS) as internal reference. With the Kratos MS apparatus (Kratos Analytical Ltd, Manchester, UK), mass spectra (EI) were recorded at 70 eV.2.2 Reagents and kits
Dulbecco's modified Eagles culture medium (DMEM), fetal bovine serum (FBS), trypsin/EDTA 0.25%, MTT assay kit, trypan blue 0.4%, and penicillin–streptomycin were purchased from (Lonza, Belgium), while Kreps ringer bicarbonate buffer was purchased from (sigma Aldrich, USA). Bovine serum albumin (BSA) and 1X phosphate buffer saline (PBS) were acquired from (Hyclone, USA). Sorafenib was purchased from (Bayer, Germany).The high-capacity cDNA reverse transcription kit, TRIzol reagent, and RT-PCR grade water were purchased from (Thermo Fisher Scientific), and the Sso-Fast EvaGreen supermix was from (BIO-RAD). Propidium iodide was purchased from (Miltenyi Biotec), the cytochrome C ELISA kit was supplied by (Abcam, Cambridge, UK), and other chemicals and reagents used were of the highest purity grade.
2.3 Cytotoxicity
The HepG2 cell line was obtained from ATCC (Rockville, MD). Liver mesenchymal stem cells were harvested from liver biopsy according to previously published protocols.34 Cells were grown in DMEM media with 10% fetal bovine serum (FBS), 100 U per mL penicillin, and 100 g per mL streptomycin at 37 °C, 5% CO2. The MTT assay kit, which relies on succinate dehydrogenase in living cells' mitochondria to convert the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye to violet formazan crystals, was used to calculate the vitality of individual cells. 6000 cells per well were seeded in the 96-well plate containing, which were then incubated for 24 h. Different concentrations of drug were dissolved in DMEM and then added to the wells to replace the culture medium. The medium was discarded after incubation for 24 h under the same conditions, and 100 μL of MTT (2 mg mL−1) were added. After 3 h of incubation at 37 °C, the generated formazan crystals generated were dissolved in 50 μL of DMSO. The optical density was then measured using a Stat Fax ELISA plate reader (Romer Labs, Getzersdorf, Austria) at 570 nm with a reference wavelength of 630 nm after incubating the plate at 37 °C for 15 min. As a positive control, the tyrosine kinase inhibitor sorafenib (Bayer, Germany) was employed. DMSO was used as a solvent and its final concentration was less than 0.2%. SI and IC50 were calculated. Triplicates of each in vitro test were performed.332.4 Flow cytometry analysis
Cells were harvested, aliquoted up to 1 × 106 cells/100 μL in FACS tubes, washed twice with 2 mL of PBS, centrifuged at 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ×g for five minutes, and the buffer was decanted. Reconstituted cells in 100 μL of staining buffer for flow cytometry. HepG2 cells were stained with propidium iodide according to the technique offered via the kit and subsequently performed on the cytometer. The distribution of the cell cycles was estimated and analyzed using a FACScan TM device (BD, Franklin Lakes, NJ). Furthermore, the caspase-3 apoptotic marker levels were investigated by flow cytometry. During the flow cytometry assay, we investigated a population of HepG2 cells to obtain enough cells for statistically significant detection.
×g for five minutes, and the buffer was decanted. Reconstituted cells in 100 μL of staining buffer for flow cytometry. HepG2 cells were stained with propidium iodide according to the technique offered via the kit and subsequently performed on the cytometer. The distribution of the cell cycles was estimated and analyzed using a FACScan TM device (BD, Franklin Lakes, NJ). Furthermore, the caspase-3 apoptotic marker levels were investigated by flow cytometry. During the flow cytometry assay, we investigated a population of HepG2 cells to obtain enough cells for statistically significant detection.
FITC active caspase-3 apoptosis kit (BD Biosciences, Franklin Lakes, NJ, USA) was used for detection of active caspase 3 followed by flow cytometry analysis according to the instructions of the kit manufacturer.
2.5 IL-6 gene expression
After two rounds of PBS washing, total RNA was extracted using the TRIzol Reagent, and cDNA synthesis was performed using the high-capacity cDNA reverse transcription kit in accordance with the manufacturer's instructions. The qPCR reaction mixture contained so-fast EvaGreen supermix (10 μL), cDNA (2 μL), invitrogen RT-PCR grade water (6 μL), and the primer pair (500 nM; forward primer 5′-CAAATTCGGTACATCCTC-3′, reverse primer 5′-CTGGCTTGTTCCTCACTA-3′). The amplification started with heating 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C, 20 s at 55 °C and 30 s at 72 °C. The amplification data was then analyzed as previously described previously with normalization to β-actin.352.6 Cell migration assay
The wound healing assay was used to investigate the anti-cell migration/antiproliferation effects of synthesized hydrazone derivatives on the HepG2 cell line. In DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 U per mL penicillin, and 100 g per mL streptomycin, cells were cultured at 37 °C and 5% CO2. After reaching 80% density, the monolayer cells were scratched using a sterile tip and then washed with phosphate buffered saline, pH 7.4. Different hydrazone derivatives were applied to cells, which were then incubated for 24 h at 37 °C and 5% CO2. Finally, an Olympus microscope (Olympus Europa, Hamburg, Germany) was used to take pictures of the wound areas at 0 and 24 h. The relative cell migration was calculated using the following equation:| Relative migration = (width0h − width24h)/width0h |
2.7 Cytochrome C assay
Cytochrome C was quantified using a sandwich ELISA kit (Abcam, Cambridge, UK) according to the manufacturer's procedures. After the addition of the TMB substrate and the development of the color, the optical density was measured using a Stat fax microplate reader, USA, at 450 nm.2.8 Docking studies
Molecular Operating Environment (MOE) 2015.10 software was used to perform the docking studies. The 3D structures and conformations of the fibroblast growth factor receptor 4 (FGFR4) complex with N-(3,5-dichloro-2-((5-((2,6-dichloro-3,5-dimethoxybenzyl)oxy)pyrimidin-2-yl)amino)phenyl)acrylamide were downloaded from the PDB website (http://www.rcsb.org/; ID: 6NVG). The structures of compound 16 and the standard drug, 5-fluorouracil (5-FU), were drawn using Chem Draw Ultra 16.0 (ChemOffice). Before docking, preparatory steps were performed for the ligand (including protonation, partial charge, and energy minimization in the database) and protein (including elimination of water molecules and repeating chains, addition of hydrogens, calculation of the partial charges, and determination of the active site). The performance of the docking method was assessed by redocking the co-crystalline ligand in the detected active pocket site of FGFR4. The validation of the docking process was affirmed by determining the scoring energy (lower binding energy), the root means standard deviation (RMSD) values, and detecting amino acid interactions for the best pose by free rotation of the rotatable bonds into the rigid receptor binding site. The validation process was carried out by redocking the co-crystal within its binding pocket. A valid performance was verified when an RMSD value less than 2 Å was obtained.2.9 In silico ADME simulation
The prediction of the ADME properties of the synthesized compounds was determined from https://www.swissadme.ch/online toolkit according to Lipinski's molecular rules. Lipinski's ‘Rule of Five’ was used to establish the druglikeness of the synthesized hydrazone derivatives compared to the reference drug 5-fluorouracil.2.10 Statistical analysis
Data were expressed as mean ± standard error (SE) of the mean. Statistical significance was determined by one-way ANOVA and Tukey post hoc test using data analyzed using SPSS software version 22 (SPSS Inc., Chicago, IL, United States). A level of p < 0.05 was defined as statistically significant.3. Results and discussion
3.1 Synthesis of hydrazone derivatives
Dicyclopropyl ketone 1 was condensed with hydrazine hydrate to generate dicyclopropylmethylene hydrazone (2). Compound 2 was used as the lead compound for the synthesis of 3–20 derivatives through the synthetic routes depicted in Schemes 1–3.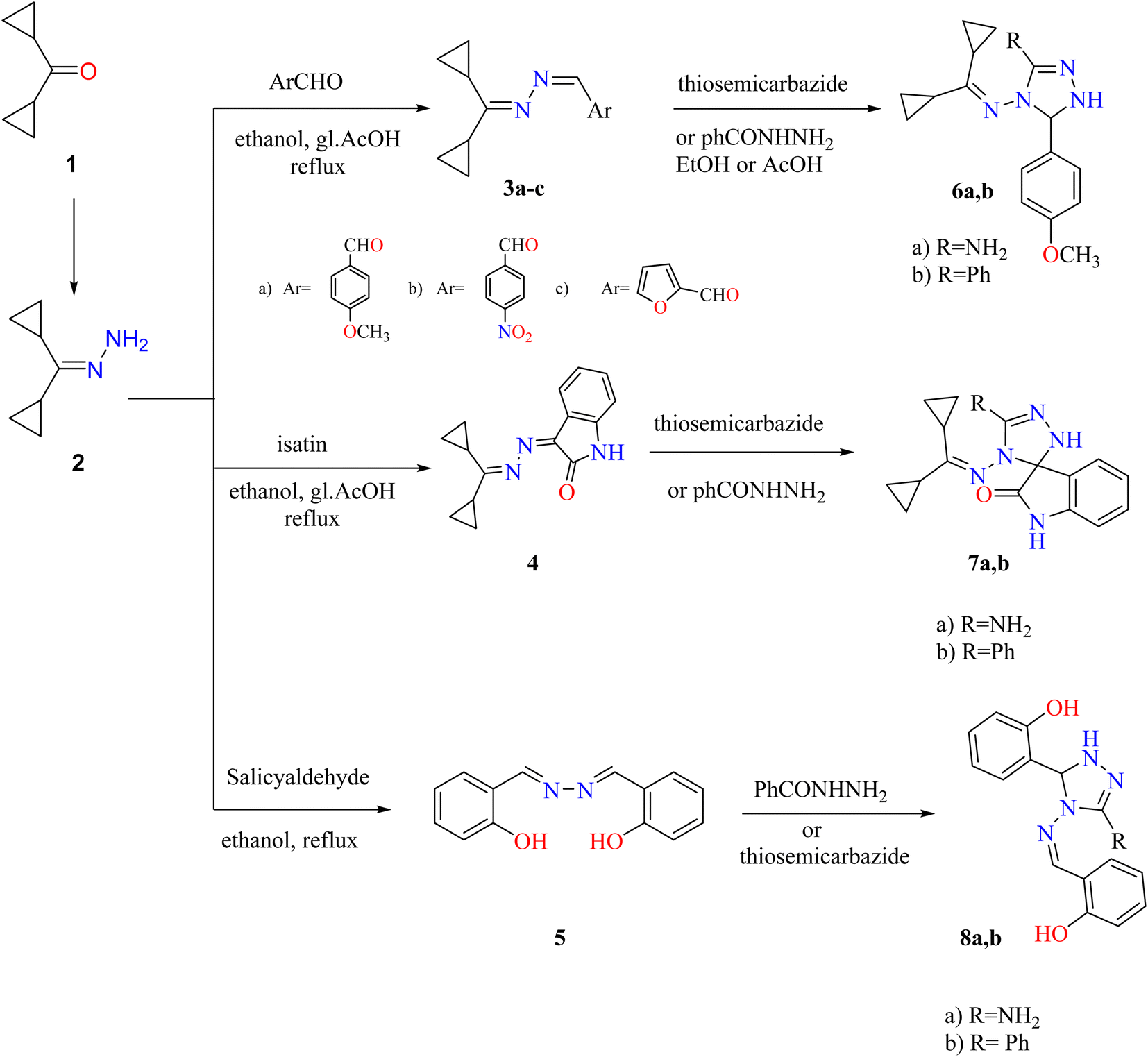 | ||
| Scheme 1 Synthetic route to obtain triazoles 3–8. Reproduced from ref. 33. Copyright (2019), Deuton-X Ltd. | ||
Treatment of hydrazone 2 with suitable aromatic aldehydes (such as p-anisaldehyde, p-nitrobenzaldehyde, furfural, and salicylaldehyde) in refluxed ethanol furnished the corresponding Schiff bases 3a–c and the bisazene derivative 5. Similarly, condensation of hydrazone 2 with isatin (1H-indole-2,3-dione) in EtOH and reflux with a few drops of glacial acetic acid (gl. AcOH) gave a high yield of 3-(dicyclopropylmethylene)hydrazone indolin-2-one (4). Triazoles 6a,b, 7a,b, and 8a,b were generated by cycloaddition on the azine derivatives 3a, 4, and 5 with thiosemicarbazide or benzohydrazide (Scheme 1).
Also, synthesis of imidazolones 10a,b by treating hydrazone 2 with oxazolones 9a,b in acetic acid under reflux. Furthermore, the reaction of hydrazone 2 with several anhydrides such as phthalic, succinic, maleic anhydrides proceeded by the same methodology and generated amido and pyrrole derivatives 14–16, respectively. Although 3-((dicyclopropylmethylene) amino)-2-methylquinazolin-4(3H)-one (11) was achieved by reaction of hydrazone 2 with an equimolar amount of 2-methylbenzoxazinone in boiling acetic anhydride. Heating of hydrazone (2) (dicyclopropylmethylene) with 2,3-epoxy-1,4-naphthoquinone 12 in CH3CN produced naphthoxadiazine-5,6-dione 13 (Scheme 2).
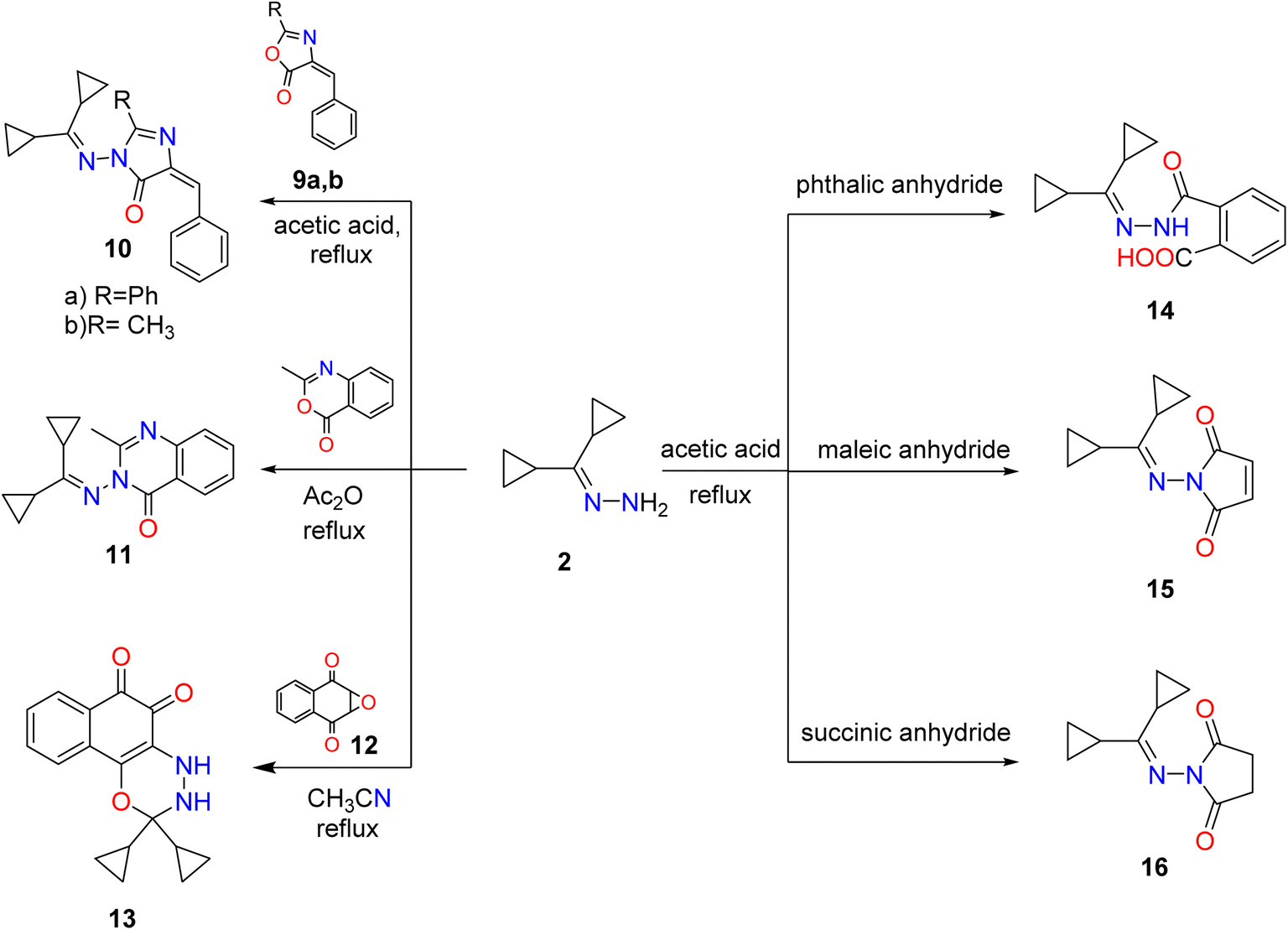 | ||
| Scheme 2 Synthesis of imidazole, cyclic imides, fused diazine, and oxadiazine targets. Reproduced from ref. 33. Copyright (2019), Deuton-X Ltd. | ||
Additionally, a mixture of cyano acetohydrazone and pyridazinone derivatives 17 and 18 was acquired by condensation of 1 with cyano acetic acid hydrazide in boiling ethanol and the two products were easily separated. Finally, dicyclopropyl ketone was treated with the thiosemicarbazide derivative 19 in refluxing ethyl alcohol, providing the corresponding quinazoline derivative 20 (Scheme 3).
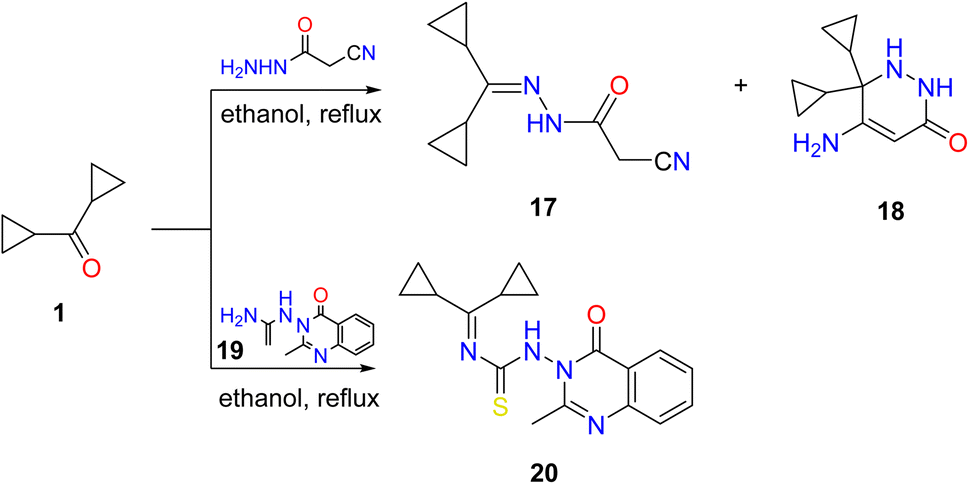 | ||
| Scheme 3 Condensation of dicyclopropyl ketone 1 with hydrazide, and thiosemicarbazide derivatives. Reproduced from ref. 33. Copyright (2019), Deuton-X Ltd. | ||
The structures of these targets were identified and confirmed based on IR, NMR, and MS spectral and analytical data as previously reported.33 FTIR, 1H-NMR, 13C-NMR, and MS spectra of most derivatives are provided in ESI (Fig. S1 and Table S1†).
3.2. Cytotoxicity assay
The cytotoxic effects of the synthetic hydrazone derivatives were compared to those of sorafenib using the HepG2 cell line using the MTT assay. The mean IC50 values ranged from 23.6 to 94.7 μM and the SI values are presented in Table 1. The results demonstrated that sorafenib and compounds 2, 4, 8a, 13, 16, 18, and 20 had excellent cytotoxic action against HepG2. Compound 16 had IC50 and SI values similar to those of sorafenib, indicating a strong potency and selectivity for activated HepG2 cell lines with little cytotoxicity and the potential to be a successful anticancer agent.| MTT assay IC50 24 h (μM) | SI | ||
|---|---|---|---|
| Compounds | HepG2 | Liver mesenchymal stem cells | |
| a IC50: drug concentration that inhibits cell growth by 50%.b SI: calculated by dividing the IC50 value against liver mesenchymal stem cells for each compound by the IC50 value of that compound against the cancer cell line HepG2. | |||
| Sorafenib | 25.6 | 100 | 3.9 |
| 2 | 94.7 | >100 | 1.58 |
| 3a | 48.2 | >100 | — |
| 3b | >100 | 90.2 | — |
| 4 | 53.7 | 100 | 1.86 |
| 5 | 75.8 | >100 | — |
| 7a | >100 | >100 | — |
| 7b | >100 | 100 | — |
| 8a | 62.2 | 100 | 1.6 |
| 8b | >100 | 75.5 | — |
| 11 | >100 | >100 | — |
| 13 | 61.4 | 85.2 | 1.38 |
| 16 | 23.6 | 76.5 | 3.2 |
| 18 | 89.5 | 100 | 1.11 |
| 19 | >100 | >100 | — |
| 20 | 48.6 | 61.5 | 1.26 |
3.3 Cell migration assay
The ability of sorafenib and the synthesized hydrazone derivatives to attenuate the migration of HepG2 cells was evaluated using the wound healing assay. Cell migration increased after 24 h for untreated cells (control) but was substantially reduced in the presence of sorafenib and hydrazone derivative compounds 2, 4, 8a, 13, 16, 18, and 20 (Fig. 2). These results suggest that compounds 2, 4, 8a, 13, 16, 18, and 20 delay wound closure and significantly inhibit migration/invasion of HepG2 cells similar to or better than sorafenib. Quantified relative migration calculations showed that compounds 16, 13, and 20 had a significant antimigration and antiproliferative effects on HepG2 cells compared to the commercially available anticancer drug sorafenib. | ||
| Fig. 2 In vitro wound healing assay of HepG2 cells treated with sorafenib and synthesized hydrazone derivatives (A) cell morphology before and after drug screening, (B) quantified relative migration summarized as a bar graph. Data are mean ±SD (n = 3). (a) significant with DMEM Medium, (b) significant with sorafenib, (c) significant with 16, (d) significant with 13, (e) significant with 20, (f) significant with 8a, (g) significant with 4, (h) significant with 18. Cell migration was dramatically suppressed after treatment with compounds 2, 4, 8a, 13, 16, 18, and 20 for 24 h. Wound healing assay of HepG2 cells treated with treatment with sorafenib and hydrazone derivatives. Compared to the commercially available anticancer drug, sorafenib, the data showed that compounds 16, 13, and 20 had a significant antimigration effect. | ||
3.4 Apoptosis and cell cycle analyses
The effects of treatment with sorafenib and hydrazone derivatives on HepG2 cells were investigated. Apoptotic and necrotic effects of the tested compounds on HepG2 cells were evaluated using caspase 3 assay kit which employs an FITC-antibody reported to specifically recognize the active form of caspase 3 (not its proenzyme). On the other hand, cell cycle arrests at different stages, caused by treatment with hydrazone derivatives were assessed using propidium iodide staining (Fig. 3, ESI Fig. S2 and S3†).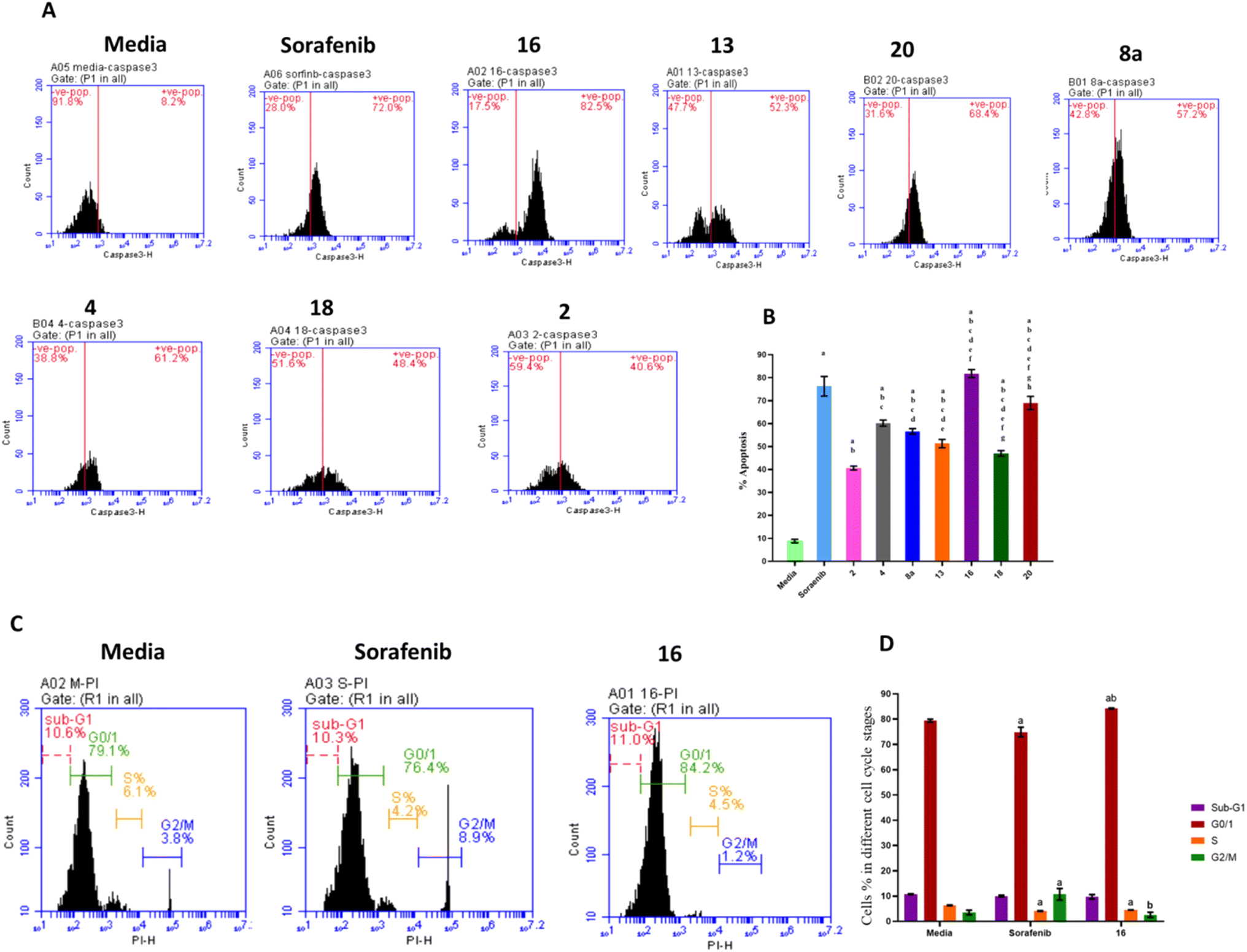 | ||
| Fig. 3 Effects of the selected derivatives of hydrazones on HepG2 cell apoptosis, necrosis, and cell cycle. The treated cells were stained using propidium iodide cell cycle staining and caspase 3 and analyzed by flow cytometry to determine percentages of necrotic and apoptotic cells characterizing cells in different cell cycle stages. (A) Caspase 3 flow cytometry histograms for all derivatives of hydrazone compared to Sorafinib. (B) The quantified percentages are summarized as a bar graph. Data are mean ±SD (n = 3). (a) significant with DMEM Medium, (b) significant with Sorafenib, (c) significant with 2, (d) significant with 4, (e) significant with 8a, (f) significant with 13, (g) significant with 16, (h) significant with 18. Treatment with sorafenib and hydrazone derivatives (compounds 2, 4, 8a, 13, 16, 18, and 20) significantly increased the apoptotic percentage of HepG2 cells compared to the control group. Compound 16 caused a higher percentage of apoptosis compared to sorafenib and the other derivatives. (C) Histograms showing different cell cycle stages for compound 16 compared to sorafenib and media. (D) The quantified percentages are summarized as a bar graph. | ||
Caspase 3 is synthesized as a proenzyme that is cleaved by other proteases into small and large subunits which then associate to form the active enzyme. Active caspase 3 proteolytically cleaves other caspases and cellular targets leading to execution of apoptosis. Caspase 3 flow cytometry analysis of HepG2 cells treated with the synthesized hydrazone derivatives or sorafenib was conducted and the results showed that, compared to the control group, the percentage of apoptotic cells increased significantly after treatment with sorafenib and the derivatives of hydrazone (2, 4, 8a, 13, 16, 18, and 20). Treatment with compound 16 showed a higher increase in the percentage of apoptotic cells (80%) than sorafenib (75%) (Fig. 3A and B). This explains the key effect, of the synthesized hydrazone derivatives on caspase-3 activation that leads to cellular apoptosis upregulation that supports the anticancer potential of the synthesized compounds especially compound 16.
Additionally, the anticancer potential of the most potent hydrazone derivatives and sorafenib on the cell cycle progression, crucial for proliferation of cancer cells, was determined. Flow cytometry cell cycle analysis (Fig. 3C and D) was used to investigate the effect of compound 16 on HepG2 cell cycle stained with propidium iodide. Approximately 79.38% of untreated HepG2 cells were in the G0/G1 phase, 6.23% in the S-phase, and 3.5% in the G2/M phase. For cells treated with sorafenib, a positive control, 74.93% of cells were in the G0/G1 phase, 4.17% in S-phase, and 10.68% in G2/M. The most potent anticancer hydrazone derivative, compound 16, reduced the number of cells in S-phase to 4.5% while 2.5% of the cell population was in the G2/M phase which suggest a significant inhibitory effect of compound 16 on the HepG2 cells. These data confirm that compound 16 has a greater impact on HepG2 cell cycle when compared to sorafenib. This cellular proliferation inhibitory effect of the synthesized hydrazones, especially compound 16, could enable their application as anticancer agents following further assessments. Additional flow cytometry graphs illustrating the effects of different treatments on caspase 3 levels and cell cycle of HepG2 cells can be found in ESI.†
3.5 Effects of sorafenib and compound 16 treatment on IL-6 gene expression and cytochrome C concentration
IL-6 plays a key role in the regulation, inflammation, and oncogenesis of the immune system.36 When IL-6 binds to its receptor, glycoprotein 130, JAK phosphorylates the receptor, activating the JAK/STAT3 signalling pathway. Through the reduction of oxidative damage and inhibition of the apoptotic cascade, these pathways play a crucial role in liver regeneration.37 However, continuing activation of the IL-6 signalling pathway is detrimental to the liver and can eventually lead to HCC progression or recurrence.12 High serum levels of IL-6 have been suggested as a tumor marker of HCC. Furthermore, IL-6 has been reported to protect cancer cells against DNA damage, oxidative stress, and apoptosis caused by cancer therapy.38 Inhibition of IL-6 was proposed as a possible therapeutic strategy to make HCC cells more susceptible to sorafenib.39 Because the JAK/STAT3 cascade is critical for the development of HCC, and various intracellular signalling pathways are activated by the IL-6 cytokine, a potential therapeutic approach for the treatment of malignancies could involve blocking IL-6 or interfering with its signalling pathways alone or in conjunction with traditional anticancer medicines. Cellular death is significantly influenced by cytochrome C.40 The expression of the mitochondrial respiratory chain proteins cytochrome C, cytochrome C oxidase subunits I and IV, and the manganese superoxide dismutase, which scavenges free radicals, increases in response to chemotherapy. An apoptotic signal is produced when Bax encourages the release of cytochrome C from mitochondria, which in turn triggers the caspase 9 apoptotic pathway. Caspase 9 activates downstream caspases, including caspase 3, which execute apoptosis and cell death.12,41 Cytochrome C is released from injured mitochondria into plasma after induction of apoptosis and its serum levels could be used to assess the effect of chemotherapy. In this study, the administration of sorafenib and compound 16 caused a significant (P < 0.001) decrease (P < 0.001) in the expression of IL-6 (Fig. 4A) and a significant increase in cytochrome C concentration (Fig. 4B) compared to the control group. Treatment with compound 16 showed a significant decrease in the level of IL-6 level and an increase in cytochrome C greater than sorafenib. Therefore, the proposed anticancer mechanism of compound 16 may include a marked reduction in IL-6 gene expression that causes suppression of the JAK/STAT3 pathway and ultimately decreases inflammation and tumorigenesis, as shown in Fig. 5.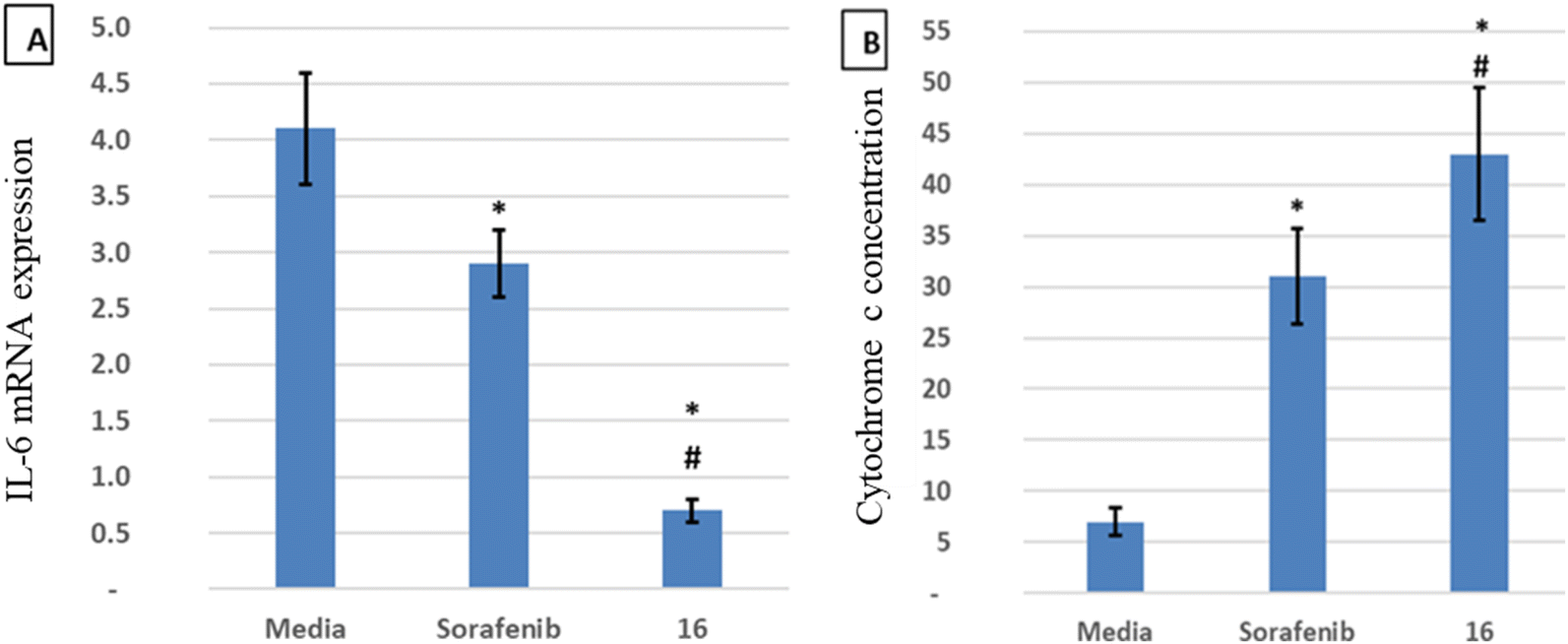 | ||
| Fig. 4 Sorafenib and compound 16 effects on (A) IL-6 mRNA expression (RT-PCR) and (B) cytochrome C concentration (ELISA). All samples were measured separately in duplicate, and the data were provided as mean standard deviation. *Statistical significance compared to the control group (p < 0.001); # statistical significance compared to the sorafenib group (p < 0.001). Treatment with compound 16 showed a significant decrease in the level of IL-6 and an increase in cytochrome C greater than sorafenib. Therefore, the proposed anticancer mechanism of compound 16 may include a marked reduction in the expression of the IL-6 gene. | ||
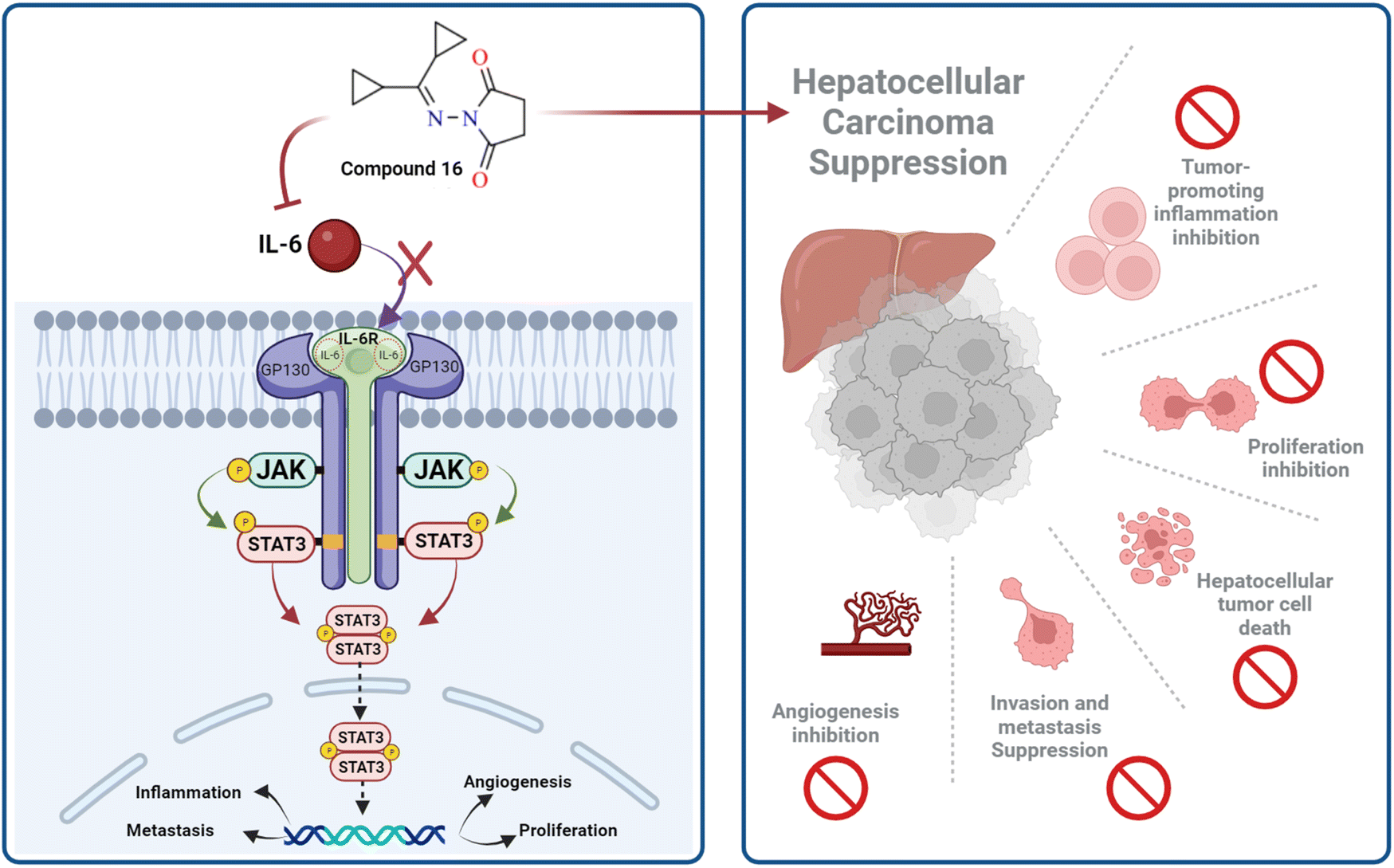 | ||
| Fig. 5 Proposed mechanism by which compound 16 may exert its anticancer effect. | ||
3.6. Molecular docking studies
The fibroblast growth factor receptor (FGFR) family of receptor tyrosine kinases consists of 4 members (FGRF 1–4) encoded by different genes (50–70% sequence homology). FGFRs are key to activation of mitogen-activated protein kinase; phosphatidylinositol 3-kinase, phospholipase Cγ, and signal transducer and activator of transcription (STAT) which lead to activation of target genes responsible for cellular proliferation, metastasis, angiogenesis, and development of HCC.42 The fibroblast growth factor receptor 4 (FGFR4) is involved in cell proliferation, differentiation, and migration, and its abnormal signalling was linked to development and progression of HCC. Similar to other tyrosine kinases, FGFR consists of an extracellular receptor domain and a transmembrane helix connected to a cytoplasmic kinase domain.36,43–45 Therefore, compound 16 and a standard drug, 5-fluorouracil, were docked to the active site of the FGFR4 kinase domain (PDB ID: 6NVG) to predict their ability to inhibit the kinase function using Molecular Operating Environment 2015.10 (MOE) software to analyze all docking poses and binding energies (Fig. 6 and 7). RMSD was employed to measure the overall stability of the protein and ligand in relation to the initial protein backbone structure. Therefore, validation of the docking protocol indicates that the ligand is confirmed by the active site pocket with a score = 4.6292 kcal mol−1 and rsmd = 1.1392 Å. Compound 16 presented better binding interactions compared to 5-fluorouracil (score = −3.8501 kcal mol−1, rsmd = 1.2706 Å). The imino and carbonyl groups of compound 16 showed hydrogen bond interactions with His 704 and Arg 701 amino acids. On the other hand, when the same pose was docked with a different pocket on the same protein, compound 16 also formed a hydrogen bond with Lys 503. These interactions suggest a strong polar connection between compound 16 and the binding pocket of the protein, potentially stabilizing the complex (Table 2 and Fig. 7). In addition to hydrogen bonds, hydrophobic interactions (typically enable the molecule to fit more tightly into the binding site and increase its affinity for the protein) are achieved by the orientation of compound 16 within the protein pocket which contribute to the binding stability of compound 16. The ability of compound 16 to display better binding interactions to EGFR4 as compared to 5-FU, a common chemotherapeutic drug used for treatment of several cancers, supports its potential as an effective anticancer agent.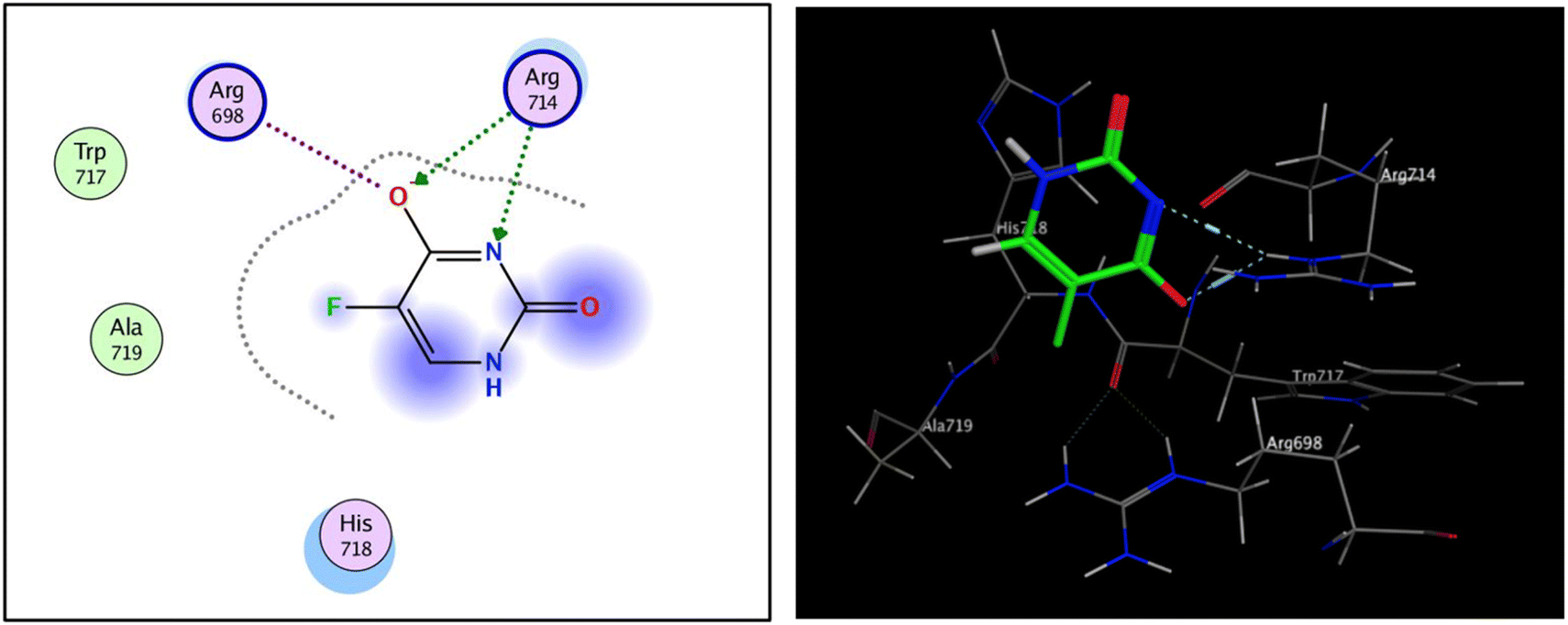 | ||
| Fig. 6 2D and 3D interaction analysis of structural redocked with 5-fluorouracil with FGFR4 showing clear active site interactions. | ||
| Ligand | Receptor | Interaction Distance (oA) | E (kcal mol−1) | |
|---|---|---|---|---|
| Compound 16 | N 5 | N HIS 704 | H-acceptor 3.49 | −1.0 |
| O 15 | NH1 ARG 701 | H-acceptor 2.96 | −4.8 | |
| O 5 | N Lys 503 | H-acceptor 3.28 | −0.7 | |
|
||||
| 5-Fluorouracil | O 5 | NE ARG 714 | H-acceptor 2.99 | −5.5 |
| N 6 | NE ARG 714 | H-acceptor 3.43 | −1.7 | |
| O 5 | NE ARG 698 | Ionic 3.80 | −0.9 | |
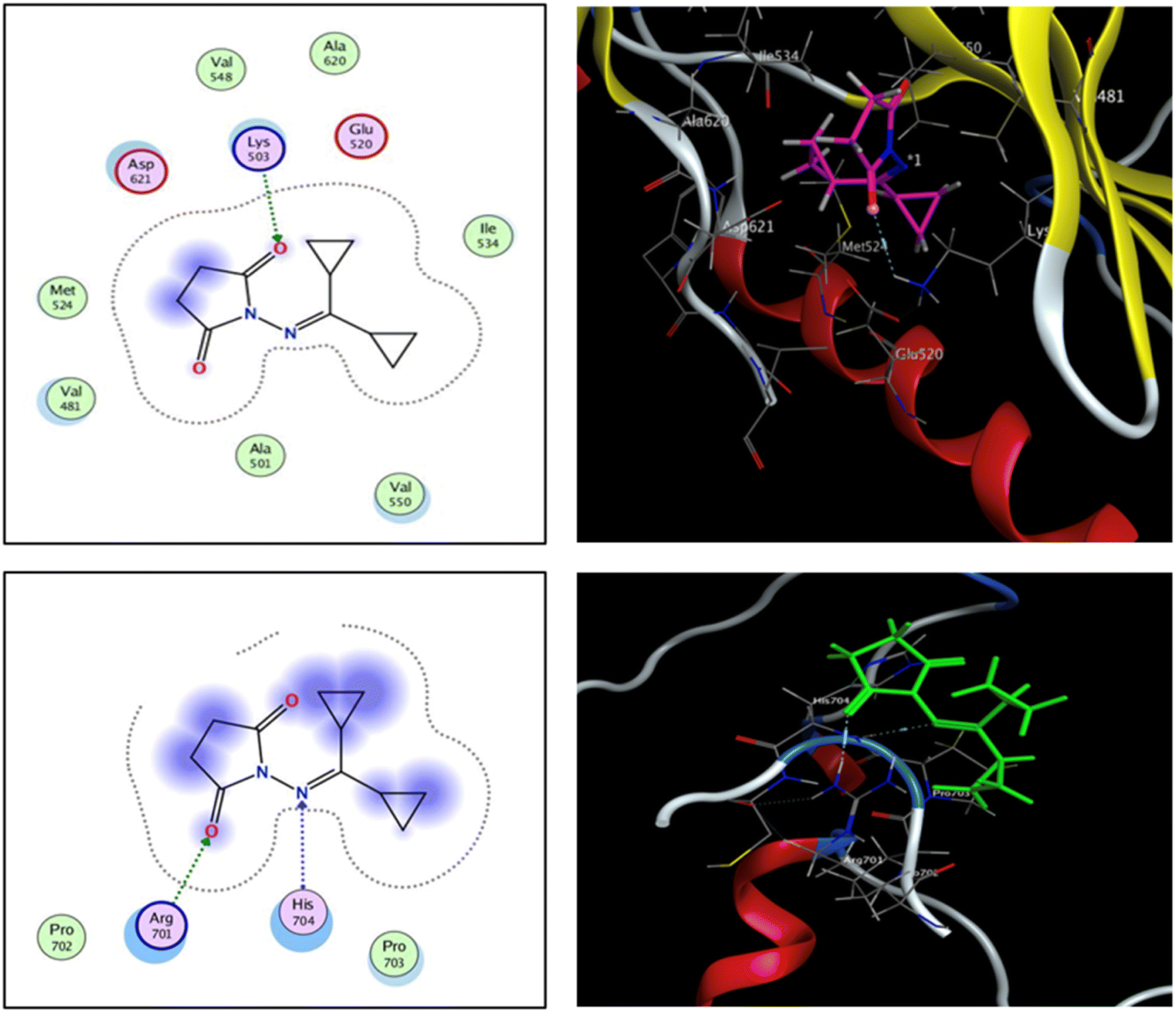 | ||
| Fig. 7 2D and 3D interaction analysis of 16 newly redocked compounds with two different pockets of FGFR4 showing significant active site interactions. | ||
3.7 In silico ADME predictions
The preferable oral bioavailability of the compounds examined was confirmed by in silico ADME predictions (logP < 5, Mw < 500, HBA 10 and HBD <5). The number of values of rotatable bonds (n-ROTB) (which should be <10), indicated molecular flexibility. The topological polar surface area (TPSA) revealed that most of the calculated values of the target compounds indicated good permeability. According to ADME results (Table 3), the compounds tested did not violate Lipinski's rules, which provides positive values of drug similarity. Skin permeation (logKp; cm s−1) showed that the more negative the logKp, the less skin permeant for each compound.46 The Lipinski qualitative model (mostly known as rule of five) is a widely used approach for prediction of the likelihood of a small compound being orally bioavailable. It is used in early drug development to guide compound selection by using criteria such as molecular weight, lipophilicity (logP), hydrogen bond donors, and acceptors. It helps to streamline the prioritize selection of compounds more likely to be absorbed in the gastrointestinal tract and consequently and potentially succeed in clinical trials. Several studies have proven a strong correlation between its criteria and the pharmacokinetic properties of successful drugs. Further, it assists in evaluating ADME parameters for drug candidates and molecules and offers insights that help address uncertainties early in the drug discovery process. Therefore, the predicted oral bioavailability was summarized using the bioavailability radar, in which the pink zone represents the ideal ranges of different characteristics (lipophilicity: XLOGP3 0.7 to +5.0, size 150–500 g mol−1, polarity: TPSA between 20 and 130, solubility: logarithmicS with maximum value 6, saturation: the fraction of hybridized carbons sp3 with minimum value 0.25, and flexibility: rotatable bonds with maximum value 9). Accordingly, all of the evaluated hydrazones exhibit the probability of being orally active drug-like candidates. Compared to the reference drug (5-fluorouracil), the hydrazone derivatives tested showed greater bioavailability (Table 4). These In silico ADME predictions are matched with the in vitro investigations that showed that compared to the control group, the percentage of apoptotic cells increased significantly after treatment with sorafenib and the derivatives of hydrazone (2, 4, 8a, 13, 16, 18, and 20) and the treatment with compound 16 showed a higher increase in the percentage of cell apoptosis than sorafenib (Fig. 3A and B). Also, these computational studies matched with cell cycle analysis as for cells treated with sorafenib, 74.93% of cells were in the G0/G1 phase, 4.17% in S-phase, and 10.68% in G2/M while the most potent anticancer hydrazone derivative, compound 16, reduced the number of cells in S-phase to 4.5% while 1.2% of the cell population was in the G2/M phase which suggest a significant inhibitory effect of compound 16 on the HepG2 cells (Fig. 3C and D). Moreover, treatment with compound 16 showed a significant decrease in the level of IL-6 level and an increase in cytochrome C greater than sorafenib. Therefore, the proposed anticancer mechanism of compound 16 may include a marked reduction in IL-6 gene expression that causes suppression of the JAK/STAT3 pathway and ultimately decreases inflammation and tumorigenesis, as shown in Fig. 5.
| Compounds | Mw (g mol−1) | logP0/w (MLO GP) |
TPSA (Å) | n-ROTB | HBD | HBA | Drug likeness/violation | logKp (skin permeation) (cm s−1) |
|---|---|---|---|---|---|---|---|---|
| a Molecular weight (Mw) < 500, partition coefficient between n-octanol and water (logP) < 5.0, topological polar surface area (TPSA) < 140 Å:, number of rotatable bonds (n-RB) <10, hydrogen-bond donor (HBD) <5, and hydrogen-bond acceptors (HBA) <10. |
||||||||
| 4 | 253.31 | 2.03 | 53.82 | 3 | 1 | 3 | Yes/0 | −6.35 |
| 8a | 297.32 | 2.03 | 53.82 | 3 | 1 | 3 | Yes/0 | −6.35 |
| 13 | 296.33 | 1.43 | 67.43 | 2 | 2 | 3 | Yes/0 | −6.42 |
| 16 | 206.25 | 1.76 | 49.74 | 3 | 0 | 3 | Yes/0 | −7.38 |
| 18 | 193.25 | 1.69 | 63.81 | 3 | 2 | 1 | Yes/0 | −6.80 |
| 20 | 326.43 | 3.66 | 91.37 | 5 | 1 | 3 | Yes/0 | −6.74 |
| 5-Fluorouracil | 130.08 | −0.42 | 66.24 | 0 | 2 | 5 | Yes/0 | −6.91 |
| a The pink area represents the optimal range for each properties as follows: lipophilicity (LIPO): XLOGP3 between 0.7 and + 5.0, Size: Mw from 150 to 500 g mol−1; polarity (POLAR): TPSA between 20 and 130 2, insolubility (INSOLU): logS not greater than 6; saturation (INSATU): fraction of hybridized carbons of sp3 not less than 0.25; and flexibility (FLEX): no more than 9 rotatable bonds. |
|---|
 |
3.8. Structure–activity relationship (SAR)
The synthesized compounds were characterized and evaluated against anticancer activities that support the described SAR (Fig. 8). In a previous investigation of lead cyclopropyl hydrazone 2 with its structural modifications, the presence of the cyclopropyl ring47,48 and the hydrazone fragment49,50 was predicted to exert broad cytotoxic activities against cancer cells. To further enhance its potential as an anticancer agent, important modifications such as the addition of N-heterocycles were introduced to the lead compound.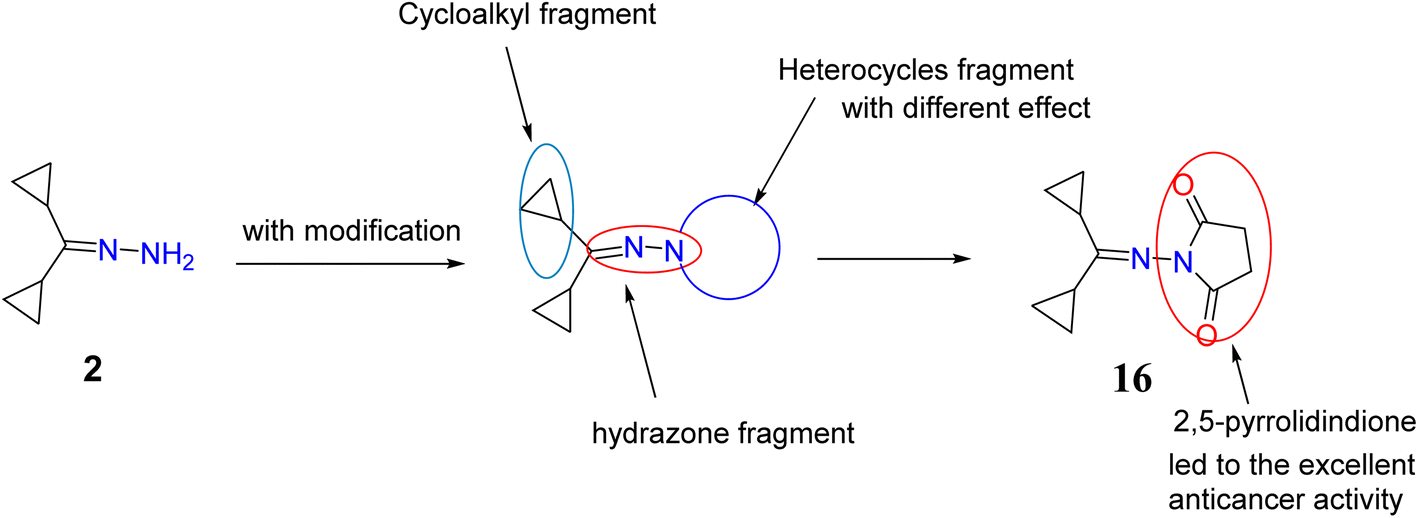 | ||
| Fig. 8 Chemical structure of target compounds with important sites for the structure–activity relationship (SAR). | ||
As evidenced by the effects of the synthesized compounds in the MTT assay, compounds 2, 4, 8a, 13, 16, 18, and 20 possessed the highest cytotoxic activities against HepG2 cells. Although the starting material, compound 2, with the basic structure of cyclopropyl rings, hydrazone, and primary amine exerted moderate cytotoxicity against HepG2 cells, it was found that cyclization of the amino group to a heterocycle moiety or introduction of functional group such as formation of cyanoacetamide fragment (compound 18) increased its potency and selectivity. This may be essential to achieve good binding interactions between the tested compounds and the receptor, possibly through the formation of a hydrogen bond.
Similarly, it is suggested that the incorporation of indoline-2-one (compound 4), aminotriazole moiety and imino group (compound 8a) and oxadiazine-5,6-dione (compound 13) is responsible for their biological significance. Introducing a thiourea fragment substituted with a 4-oxoquinazoline moiety remarkably improved the cytotoxic activity. However, compound 16 with the 2,5-pyrrolidinone moiety50 showed an excellent effect compared to sorafenib and the other compounds tested, possibly due to the formation of hydrogen bonds with the receptor (due to the presence of an imide group). Treatment with compound 16 showed a significant increase in cytochrome C greater than that observed upon treatment with sorafenib. Chemotherapeutic agents are reported to significantly increase cytochrome C release from the mitochondrial matrix into the cytosol in addition to the other mitochondrial respiratory chain proteins cytochrome C oxidase subunits I and IV, and manganous superoxide dismutase (which scavenges free radicals). The released cytochrome C indirectly activates caspase-9 and caspase-3 which ultimately execute apoptosis.51–53 This study showed that treatment of HepG2 cells with compound 16 led to high increase in activation of cytochrome C and activated caspase 3 which represent significant markers of apoptosis. This supports the potential role of the synthesized hydrazone derivatives, especially compound 16, as anticancer agents mediated by their ability to trigger cellular apoptosis.
To the best of our knowledge, this report is the first to investigate the potential of the newly synthesized hydrazone derivatives for hepatocellular carcinoma treatment. However, further validations such as assessing the effect of hydrazone derivatives, namely compound 16, on the proteins involved in cell proliferation, apoptosis, JAK/STAT3 pathway, as well as animal studies to confirm the anticancer effects of hydrazone derivatives are required. A further computational molecular dynamics simulation and docking study compared with a co-crystallized ligand are required to better elucidate the interaction between the protein and the ligand.
4. Conclusions
Despite recent improvements in the management of HCC, it remains one of the leading causes of cancer-related mortality. Various molecular targets involved in signaling pathways that regulate tumor proliferation and angiogenesis that characterize HCC were identified. The first HCC medication approved by the FDA is the oral multikinase inhibitor sorafenib. This first-line treatment may not work for all patients for various causes, including drug resistance. Therefore, it is necessary to develop additional effective HCC drugs.In the current study, new hydrazone derivatives with improved activities and selectivity were synthesized and evaluated in silico by molecular docking and structure–activity correlations (SAR) in addition to in vitro studies using HepG2 human hepatocellular carcinoma cell line and liver mesenchymal stem cells as a normal cell line. With mean IC50 values ranging from 23.6 to 94.7 μM, compounds 2, 4, 8a, 13, 16, 18, and 20 exhibited high cytotoxic effects against HepG2 cells. Compound 16 had IC50 and SI of 23.6 μM and 3.2, respectively, against HepG2 as compared to sorafenib (25.6 μM. and SI 3.9) indicating a strong potency and selectivity for activated HepG2 cell lines. Compound 16, however, exhibited low cytotoxicity against liver mesenchymal stem cells (IC50 76.5 μM). In vitro wound scratch assay indicated that several synthesized hydrazone derivatives, including compound 16, attenuated HepG2 cell migration and wound closure by 40–70%. HepG2 cell apoptosis as determined by assay of active caspase 3 enzyme, showed that the most potent hydrazone derivative, compound 16, caused a higher increase in the percentage of cell apoptosis (80%) as compared to sorafenib (75%). Cell cycle analysis showed that compound 16 reduced the number of cells in S-phase to 4.5% while 1.2% of the cell population was in the G2/M phase which suggest a significant inhibitory effect of compound 16 on the HepG2 cells. Furthermore, compound 16 significantly increased cytochrome C release and significantly decreased IL-6 gene expression compared to sorafenib.
These findings suggest that compound 16 might be a potent anticancer candidate for HCC treatment by downregulating the expression of the IL-6 gene that causes suppression of the JAK/STAT pathway, decreases inflammation, and tumorigenesis with favorable binding interaction and bioavailability. Computational analysis including molecular docking and structure–activity correlations also supported the anticancer potential of the synthesized hydrazone derivative 16. Compound 16 displayed better binding interactions to EGFR4 as compared to 5-FU which further supports the potential of compound 16 as an effective chemotherapeutic agent. Furthermore, ADME predictions supported preferable bioavailability of the synthesized hydrazones.
EGFR and IL-6 signaling pathways crosstalk in multiple downstream signaling pathways and both are linked to tumorigenesis. Therefore, co-targeting them could represent a promising strategy for effective cancer treatment.54 Findings of this study indicate that compound 16 targets both signalling pathways by binding to the kinase domain of EGFR4 and decreasing IL-6 expression. Further computational, in vitro, and in vivo evaluations should be considered to support the anticancer potential of the synthesized novel hydrazone derivatives.
Data availability
The data supporting this study are available in the main manuscript and its ESI.†Author contributions
All authors contributed equally to the experimental and writing parts depending on their specialty. All authors read and approved the final version of this manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors of this manuscript express their great gratitude to College of Science, Qassim University for providing laboratory facilities. This work was funded in part by a grant from the American University in Cairo to Prof. Hassan Azzazy.References
- A. Villanueva, Hepatocellular carcinoma, NEJM, 2019, 380(15), 1450–1462 CrossRef CAS PubMed.
- A. M. Attallah, M. El-Far, C. A. A. Malak, M. M. Omran, G. E. Shiha, K. Farid, L. A. Barakat, M. S. Albannan, A. A. Attallah, M. A. Abdelrazek, M. S. Elbendary, R. Sabry, G. A. Hamoda, M. M. Elshemy, A. A. Ragab, B. M. Foda and S. O. Abdallah, HCC-DETECT: a combination of nuclear, cytoplasmic, and oncofetal proteins as biomarkers for hepatocellular carcinoma, Tumor Biol., 2015, 36, 7667–7674 CrossRef CAS PubMed.
- L. A. T orre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Global cancer statistics, 2012, CA Cancer J. Clin., 2015, 65(2), 87–108 CrossRef PubMed.
- J. D. Yang and L. R. Roberts, Hepatocellular carcinoma: a global view, Nat. Rev. Gastroenterol. Hepatol., 2010, 7(8), 448–458 CrossRef PubMed.
- S. M. Rida, S. A. M. El-Hawash, H. T. Y. Fahmy, A. A. Hazzaa and M. M. M. El-Meligy, Synthesis of novel benzofuran and related benzimidazole derivatives for evaluation of in vitro anti-HIV-1, anticancer and antimicrobial activities, Arch. Pharm. Res., 2006, 29, 826–833 CrossRef CAS PubMed.
- M. Cervello, D. Bachvarov, N. Lampiasi, A. Cusimano, A. Azzolina and J. A. McCubrey, et al., Molecular mechanisms of Sorafenib action in liver cancer cells, Cell Cycle, 2012, 11(15), 2843–2855 CrossRef CAS PubMed.
- J. L. MacGregor, D. N. Silvers, M. E. Grossman and W. H. Sherman, Sorafenib-induced erythema multiforme, J. Am. Acad. Dermatol., 2007, 56(3), 527–528 CrossRef PubMed.
- Y. Li, Z. H. Gao and X. J. Qu, The adverse effects of Sorafenib in patients with advanced cancers, Basic Clin. Pharmacol. Toxicol., 2015, 116(3), 216–221 CrossRef CAS PubMed.
- Y.-j. Zhu, B. Zheng, H.-y. Wang and L. Chen, New knowledge of the mechanisms of Sorafenib resistance in liver cancer, Acta Pharmacol. Sin., 2017, 38(5), 614–622 CrossRef CAS PubMed.
- T. Tanaka, M. Narazaki and T. Kishimoto, IL-6 in inflammation, immunity, and disease, Cold Springer Harb. Perspect. Biol., 2014, 6(10), a016295 CrossRef PubMed.
- W. Ohishi, J. B. Cologne, S. Fujiwara, G. Suzuki, T. Hayashi and Y. Niwa, et al., Serum interleukin-6 associated with hepato-cellular carcinoma risk: a nested case–control study, Int. J. Cancer, 2014, 134(1), 154–163 CrossRef PubMed.
- D. Schmidt-Arras and S. Rose-John, IL-6 pathway in the liver: from physiopathology to therapy, J. Hepatol, 2016, 64(6), 1403–1415 CrossRef CAS PubMed.
- S. Rollas and S. Küçükgüzel, Biological activities of hydrazone derivatives, Molecules, 2007, 12(8), 1910–1939 CrossRef CAS PubMed.
- P. Kamur, A. Rai, M. Singh, D. Kumar, A. K. Sahdev and V. Raj, Review on the pharmacological activities of hydrazones derivatives, EC Pharm. Sci., 2016, 2, 278–306 Search PubMed.
- I. Hussain and A. Ali, Exploring the pharmacological activities of hydrazone derivatives: a review, J. Phytochem. Biochem., 2017, 1, 1–5 Search PubMed.
- Ł. Popiołek, Hydrazide-hydrazones as potential antimicrobial agents: an overview of the literature since 2010, Med. Chem. Res., 2017, 26(2), 287–301 CrossRef PubMed.
- A. Zakeyah, J. Whitt, C. Duke, D. Gilmore, D. Meeker and M. Smeltzer, et al., Synthesis and antimicrobial studies of hydrazone derivatives of 4-[3-(2,4-difluorophenyl)-4-formyl-1H-pyrazol-1-yl] benzoic acid and 4-[3-(3, 4-difluorophenyl)-4-formyl-1H-pyrazol-1-yl] benzoic acid, Bioorg. Med. Chem. Lett., 2018, 28(17), 2914–2919 CrossRef CAS PubMed.
- M. A. A. Radwan, M. A. Alshubramy, M. Abdel-Motaal, B. A. Hemdan and D. S. El-Kady, Synthesis, molecular docking and antimicrobial activity of new fused pyrimidine and pyridine derivatives, Bioorg. Chem., 2020, 96, 103516 CrossRef CAS PubMed.
- S. M. Sondhi, M. Dinodia and A. Kumar, Synthesis, anti-inflammatory and analgesic activity evaluation of some amidine and hydrazone derivatives, Bioorg. Med. Chem., 2006, 14(13), 4657–4663 CrossRef CAS PubMed.
- P. Melnyk, V. Leroux, C. Sergheraert and P. Grellier, Design, synthesis and in vitro antimalarial activity of an acylhydrazone library, Bioorg. Med. Chem. Lett., 2006, 16(1), 31–35 CrossRef CAS PubMed.
- V. Angelova, V. Karabeliov, P. A. Andreeva-Gateva and J. Tchekalarova, Recent developments of hydra-zide/hydrazone derivatives and their analogs as anticonvulsant agents in animal models, Drug Dev. Res., 2016, 77(7), 379–392 CrossRef CAS PubMed.
- A. Barakat, F. F. El-Senduny, Z. Almarhoon, H. H. Al-Rasheed, F. A. Badria, A. M. Al-Majid, H. A. Ghabbour and A. El-Faham, Synthesis, X-ray crystal structures, and preliminary antiproliferative activities of new s-triazine-hydroxybenzylidene hydrazone derivatives, J. Chem., 2019, 2019, 1–10 CrossRef.
- P. Kumar and B. Narasimhan, Hydrazides/hydrazones as antimicrobial and anticancer agents in the new millennium, Mini Rev. Med. Chem., 2013, 13, 971–987 CrossRef CAS PubMed.
- M. C. Mandewale, U. C. Patil, S. V. Shedge, U. R. Dappadwad and R. S. Yamgar, A review on quinoline hydrazone derivatives as a new class of potent antitubercular and anticancer agents, Beni-Suef Univ. J. Basic Appl. Sci., 2017, 6(4), 354–361 Search PubMed.
- R. M. Mohareb, D. H. Fleita and O. K. Sakka, Novel synthesis of hydrazide-hydrazone derivatives and their utilization in the synthesis of coumarin, pyridine, thiazole, and thiophene derivatives with antitumor activity, Molecules, 2010, 16(1), 16–27 CrossRef PubMed.
- R. Kaplánek, M. Jakubek, J. Rak, Z. Kejík, M. Havlík and B. Dolenský, et al., Caffeine-hydrazones as anticancer agents with pronounced selectivity toward T-lymphoblastic leukaemia cells, Bioorg. Chem., 2015, 60, 19–29 CrossRef PubMed.
- N. M. Ahmed, M. Youns, M. K. Soltan and A. M. Said, Design, synthesis, molecular modelling, and biological evaluation of n ovel substituted pyrimidine derivatives as potential anticancer agents for hepatocellular carcinoma, J. Enzyme Inhib. Med. Chem., 2019, 34(1), 1110–1120 CrossRef CAS PubMed.
- Z. Haghighijoo, O. Firuzi, B. Hemmateenejad, S. Emami, N. Edraki and R. Miri, Synthesis and biological evaluation of quinazolinone-based hydrazones with potential use in Alzheimer's disease, Bioorg. Chem., 2017, 74, 126–133 CrossRef CAS PubMed.
- S. J. Sonawane, R. S. Kalhapure and T. Govender, Hydrazone linkages in pH-responsive drug delivery systems, Eur. J. Pharm. Sci., 2017, 99, 45–65 CrossRef CAS PubMed.
- S. N. Pandeya, H. Manjula and P. N. Singh, Antidepressant activity of some phenylacetic acid hydrazones and 2-chlorophenyl semicarbazones, Indian J. Physiol. Pharmacol., 2000, 44(4), 509–510 CAS.
- N. Gökhan-Kelekçi, S. Koyunoğlu, S. Yabanoğlu, K. Yelekçi, O. Ozgen and G. Uçar, et al., New pyrazoline bearing 4(3H)-quinazolinone inhibitors of monoamine oxidase: synthesis, biological evaluation, and structural determinants of MAO-A and MAO-B selectivity, Bioorg. Med. Chem., 2009, 17(2), 675–689 CrossRef PubMed.
- R. Kulandasamy, A. V. Adhikari and J. P. Stables, A new class of anticonvulsants possessing 6 Hz activity: 3,4-dialkyloxy thiophene bishydrazones, Eur. J. Med. Chem., 2009, 44(11), 4376–4384 CrossRef CAS PubMed.
- M. Abdel-Motaal and A. Nabil, Biological activity of some newly synthesized hydrazone derivatives derived from (dicy-clopropylmethylene) hydrazone, Eur. Chem. Bull., 2018, 7(10), 280–287 CrossRef CAS.
- A. Nabil, K. Uto and F. Zahran, et al., The potential safe antifibrotic effect of stem cell conditioned medium and nilotinib combined therapy by selective elimination of rat activated HSCs, Biomed. Res. Int., 2021, 2021, 6678913 Search PubMed.
- K. J. Livak and T. D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method, Methods, 2001, 25(4), 402–408 CrossRef CAS PubMed.
- T. Kishimoto, Interleukin-6: from basic science to medicine-40 years in immunology, Annu. Rev. Immunol., 2005, 23, 1–21 CrossRef CAS PubMed.
- R. Taub, Hepatoprotection via the IL-6/Stat3 pathway, J. Clin. Invest., 2003, 112(7), 978–980 CrossRef CAS PubMed.
- K. Aleksandrova, H. Boeing, U. Nöthlings, M. Jenab, V. Fedirko and R. Kaaks, et al., Inflammatory and metabolic biomarkers and risk of liver and biliary tract cancer, Hepatology, 2014, 60(3), 858–871 CrossRef CAS PubMed.
- J. Yang, J. Wang and J. Luo, Decreased IL-6 induces sensitivity of hepatocellular carcinoma cells to Sorafenib, Pathol. Res. Pract., 2019, 215(10), 152565 CrossRef CAS PubMed.
- X. Jiang and X. Wang, Cytochrome C-mediated apoptosis, Annu. Rev. Biochem., 2004, 73, 87–106 CrossRef CAS PubMed.
- J. Lokau, V. Schoeder, J. Haybaeck and C. Garbers, Jak-stat signaling induced by interleukin-6 family cytokines in hepatocellular carcinoma, Cancers (Basel), 2019, 11(11), 1704 CrossRef CAS PubMed.
- Y. Wang, D. Liu, T. Zhang and L. Xia, FGF/FGFR signalling in hepatocellular carcinoma: from carcinogenesis to recent therapeutic intervention, Cancers (Basel), 2021, 13(6), 1360 CrossRef CAS PubMed.
- S. Ogasawara, F. Kanai, S. Obi, S. Sato, T. Yamaguchi and R. Azemoto, et al., Safety and tolerance of Sorafenib in Japanese patients with advanced hepatocellular carcinoma, Hepatol. Int., 2011, 5(3), 850–856 CrossRef PubMed.
- M. Hagel, C. Miduturu, M. Sheets, N. Rubin, W. Weng and N. Stransky, et al., First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway, Cancer Discov., 2015, 5(4), 424–437 CrossRef CAS PubMed.
- X. Lin, Y. Yosaatmadja, M. Kalyukina, M. J. Middleditch, Z. Zhang and X. Lu, et al., Rotational freedom, steric hindrance, and protein dynamics explain BLU554 selectivity for the hinge cysteine of FGFR4, ACS Med. Chem. Lett., 2019, 10(8), 1180–1186 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- P. Pevarello, M. G. Brasca, P. Orsini, G. Traquandi, A. Longo and M. Nesi, et al., 3-Aminopyrazole inhibitors of CDK2/cyclin a as antitumor agents. 2. Lead optimization, J. Med. Chem., 2005, 48(8), 2944–2956 CrossRef CAS PubMed.
- P. De, M. Baltas, D. Lamoral-Theys, C. Bruyère, R. Kiss and F. Bedos-Belval, et al., Synthesis and anticancer activity evaluation of 2(4-alkoxyphenyl)cyclopropyl hydrazides and triazolo phthalazines, Bioorg. Med. Chem., 2010, 18(7), 2537–2548 CrossRef CAS PubMed.
- T. I. de Santana, Md. O. Barbosa, P. A. Td. M. Gomes, A. C. N. da Cruz, T. G. da Silva and A. C. L. Leite, Synthesis, anticancer activity and mechanism of action of new thiazole derivatives, Eur. J. Med. Chem., 2018, 144, 874–886 CrossRef PubMed.
- A. A. Helwa, E. M. Gedawy, A. T. Taher, A. K. Ed El-Ansary and S. M. Abou-Seri, Synthesis and biological evaluation of novel pyrimidine-5-carbonitriles featuring morpholine moiety as antitumor agents, Future Med. Chem., 2020, 12(5), 403–421 CrossRef CAS PubMed.
- J. A. Sánchez-Alcázar, A. Khodjakov and E. Schneider, Anticancer drugs induce increased mitochondrial cytochrome C expression that precedes cell death, Cancer Res., 2001, 61(3), 1038–1044 Search PubMed.
- V. Raimondi, F. Ciccarese and V. Ciminale, Oncogenic pathways and the electron transport chain: a dangeROS liaison, Br. J. Cancer, 2020, 122(2), 168–181 CrossRef PubMed.
- C. Garrido, L. Galluzzi, M. Brunet, P. E. Puig, C. Didelot and G. Kroemer, Mechanisms of cytochrome C release from mitochondria, Cell Death Differ., 2006, 13(9), 1423–1433 CrossRef CAS PubMed.
- K. Ray, B. Ujvari, V. Ramana and J. Donald, Cross-talk between EGFR and IL-6 drives oncogenic signaling and offers therapeutic opportunities in cancer, Cytokine Growth Factor Rev., 2018, 41, 18–27 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, and FTIR spectral analysis of different hydrazone derivatives (Fig. S1), physicochemical properties and spectroscopy data of the synthesized compounds (Table S1), caspase 3 assay (Fig. S2), and cell cycle analysis (Fig. S3). See DOI: https://doi.org/10.1039/d4ra05854b |
| This journal is © The Royal Society of Chemistry 2024 |