
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a multivalent α-1,2-mannobiose ligand for targeting C-type lectins†
Jannis Langer ab,
Laura Hartmann*ab and
Nicole L. Snyder*c
ab,
Laura Hartmann*ab and
Nicole L. Snyder*c
aInstitute for Organic Chemistry and Macromolecular Chemistry, Heinrich Heine University Düsseldorf, Universitätsstraße 1, Düsseldorf 40225, Germany
bInstitute for Macromolecular Chemistry, University Freiburg, Stefan-Meier-Str. 31, 79104 Freiburg i.Br., Germany. E-mail: laura.hartmann@makro.uni-freiburg.de
cDepartment of Chemistry, Davidson College, Davidson, NC 28035, USA. E-mail: nisnyder@davidson.edu
First published on 28th November 2024
Abstract
The importance of lectins in biological processes such as pathogen recognition, cell adhesion, and cell recognition is well documented. C-Type lectins, which require calcium for binding, play an important role in the innate immune response by engaging carbohydrates presented as part of the human and pathogen glycocalyx. For example, lectins such as MBL, Dectin-2, langerin and DC-SIGN selectively recognize mannose rich (high-mannose) structures presented as part of the glycocalyx. One common sugar binding motif that is recognized by these lectins on the pathogen glycocalyx is α-1,2-mannobiose, a disaccharide that consists of two mannose units connected via a α-1,2-linkage. To study the binding of these motifs in different contexts, synthetic replicas of α-1,2-mannobiose that can be presented in a multivalent fashion mimicking their presentation on the glycocalyx are required. Here we present the synthesis of a novel α-1,2-mannobiose analog bearing an azido linker from known precursors using a split and combine approach guided by neighboring group participation. Our approach makes it possible to achieve comparatively high yields and stereoselectivities while reducing the number of steps required to prepare such structures. We also introduce, for the first time, the trivalent presentation of our α-1,2-mannobiose ligand on a precision glycomacromolecule using copper-catalyzed azide–alkyne cycloaddition (CuAAC) to create high-mannose mimetics. Such structures have the potential to serve as probes for unlocking the rules of engagement between high-mannose glycans and C-type lectins like langerin and DC-SIGN.
1 Introduction
α-1,2-Mannobiose, where the two mannose saccharides are connected via an α-1,2-glycosidic linkage (Manα(1–2)Manα), is a common motif found in the terminal regions of glycosylated chains on the pathogens glycocalyx1 where it serves as an important recognition element for several C-type lectins involved in innate immunity such as Dectin-2, MBL (mannose binding lectin), langerin and DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin).2–7 For this reason, there is significant interest in understanding how the presentation of α-1,2-mannobiose influences it's engagement with these lectins and, in turn, the corresponding immune response. This has led to the desire for increased access to synthetic α-1,2-mannobiose ligands that can be used for such studies. Notably, such structures are neither readily nor commercially available.The few examples in literature that build up a selective α-1,2-linkage between two mannose units often include the use of highly toxic chemicals such as mercury(II) bromide and/or involve complex synthetic approaches.8–10 In addition, most synthetic routes lack modularity limiting further application e.g. for multivalent presentation or further chemical modification.10,11 In an effort to overcome these limitation we sought to develop a more convenient method based on the split and combine approach by Gronenborn et al.12 that would provide us with both a linker for multivalent presentation and options for further structural exploration including extensions of the glycan chain.
Our efforts in this area are aligned with our goals to develop precision glycomacromolecules as glycomimetics.13–16 Our approach, which is based on known precursors, provides for a modular synthetic route to a novel α-1,2-mannobiose ligand with an anomeric azidoethanol linker for multivalent presentation via click chemistry, and a temporary 2′-O-protecting group for possible further extension via glycosylation. Furthermore, as proof of principle, we demonstrate, for the first time, the utility of this structure through its subsequent conjugation to a sequence defined oligo(amido)amine scaffold. The resulting glycooligo(amidoamine), which serves as a high-mannose glycan mimetic, will be used in the future as a tool to explore how the presentation of α-1,2-mannobiose influences C-type lectin binding and immune function.
2 Material and methods
2.1 Chemicals
All chemicals used were purchased from commercial suppliers and used without further purification. D(+)-Mannose (99%) was purchased from Abcr GmbH. Acetonitrile (Reag. Ph. Eur., for UHPLC) and triethylamine (ultrapure) were purchased from AppliChem. p-Toluenesulfonic acid (98%), acetic acid (99.8%), 2,4,6-collidine (99%), methanol (≥99.8%), sodium hydride (60% dispersion in mineral oil), tin(II) triflate (97%), Celite 535 coarse, sodium ascorbate (99.0%), TIPS (98%) and diethyl ether (≥99.8%) were purchased from Sigma-Aldrich. Acetic anhydride (99%) was purchased from Grüssing GmbH. Ethyl acetate (analytical reagent grade), sodium hydrogen carbonate (analytical reagent grade), dichloromethane (99.99%), and toluene (analytical reagent grade) were purchased from Fischer Scientific. Hydrogen bromide 33% (w/w) in acetic acid was purchased from Thermo Scientific. Magnesium sulphate was acquired from VWR BDH Prolabo Chemicals. n-Hexane (97%) was purchased from HiPerSolv CHROMANORM. Ethanol (99%) was purchased from ChemSolute. Sodium methoxide (98%) and boron trifluoride diethyl etherate were purchased from Alfa Aesar. DMF (99.8%, for peptide synthesis), benzyl bromide, ethanethiol (99+ %), sodium azide, piperidine (99%), and copper(II) sulphate (98%) were purchased from Acros Organics. N-Iodosuccinimide (98%) was purchased from BLDPharm. N,N-Diisopropylethylamine (≥99%) was purchased from Carl Roth. PyBOP and trifluoroacetic acid (99%) were purchased from Fluorochem. Tentagel S RAM resin (loading: 0.23 mmol g−1) was purchased from RAPP Polymers.2.2 Analytical methods
1H-NMR measurements were recorded using a Bruker Avance III 600 (600 MHz) at room temperature. 13C-NMR measurements were recorded using a Bruker Avance Neo 400 (100 MHz) at room temperature. All 1H spectra are referenced to their solvent peaks (CDCl3: δ = 7.26 ppm, DMSO-d6: δ = 2.50 ppm, D2O: δ = 4.79 ppm, MeOH-d4: δ = 4.87 ppm). The multiplicities are given as follows s = singlet, d = doublet, t = triplet, q = quartet and m = multiplet. The coupling constant J was given in Hz. All 13C spectra are referenced to their solvent peaks (CDCl3: δ = 77.16 ± 0.06 ppm).MALDI-TOF MS measurements were recorded on the Ultraflex I instrument from Bruker Daltonics. 2,5-Dihydroxybenzoic acid (DHB) and α-cyano-4-hydroxycinnamic acid (CHCA) were used as matrices. The ratio of the compound to be analysed to the matrix was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
The HRMS spectra were recorded on the UHR QTOF maXis 4G instrument from Bruker Daltonics.
RP-HPLC MS measurements were performed on an Aglient 1260 Infinity instrument coupled to a wavelength detector (VWD, set to 214 nm) and a 6120 quadrupole LC/MS electron spray ionisation (ESI) source (positive mode, m/z = 200 to 2000). The column used was an MZ-AquaPerfekt C18 (3.0 × 50 mm, 3 μm) RP (reverse phase) column from Mz-Analysentechnik with a flow rate of 0.4 mL min−1 at 25 °C. A water/acetonitrile mixture mixed with 0.1 vol% formic acid was used as eluent. All purities were determined by integration of the RP-HPLC spectrum.
The ESI MS measurements were performed on the aforementioned instrument.
The FTIR measurements were recorded on a Fourier-Transform IR spectrometer FTIR 5SXB from Nicolet.
Lyophilisation was performed with an Alpha 1-4 LD plus instrument from Martin Christ Freeze Dryers GmbH. The parameters chosen during the process were −52 °C and 0.1 mbar.
The thin-layer chromatography was carried out on TLC plates from E. Merck Silica Gel 60 F252 (0.25 mm thickness) and the spots were stained using an anisaldehyde/sulphuric acid solution and subsequent heating of the DC plates.
Flash chromatography was carried out using the CombiFlash Rf device from Teledyne ISCO.
2.3 α-1,2-Mannobioseazide synthesis
:1 ethyl acetate:n-hexane, v/v, Rf = 0.6). Upon completion, the solvent was removed by rotary evaporation. The remaining oil was dissolved in 250 mL ethyl acetate and washed three times each with 250 mL sodium hydrogen carbonate solution and 300 mL water. The collected organic phases were dried over magnesium sulphate and the solvent was removed on the rotary evaporator under reduced pressure to provide the desired product 2 in 98% yield. 1H-NMR: (600 MHz, chloroform-d) δ (ppm) 6.07 (d, 3JH1α–H2α = 1.89 Hz, 1H, H1α), 5.85 (d, 3JH1β–H2β = 1.20 Hz, 0.5H, H1β), 5.47 (dd, 3JH2β–H1β = 1.19 Hz, 3JH2β–H3β = 3.32, 0.5H, H-2β), 5.34–5.32 (m, 2H, H2α, H3α), 5.30–5.24 (m, 1.5H, H3β, H3α), 5.12 (dd, 3JH4β–H3β = 3.30 Hz, 3JH4β–H5β = 9.97 Hz, 0.5H, H4β), 4.31–4.25 (m, H6α, H6β, 1.5H), 4.15–4.05 (m, 1.5H, H6′α, H6′β), 4.07–4.00 (m, 1H, H5α) 3.81–3.77 (m, 0.5H, H5β), 2.20 (s, 1.5H, β –C(O)CH3), 2.16 (s, 3H, α –C(O)CH3), 2.15 (s, 3H, α –C(O)CH3), 2.09 (s, 1.5H, β –C(O)CH3), 2.08 (s, 3H, α –C(O)CH3), 2.04 (s, 3H, α –C(O)CH3), 2.03 (s, 1.5H, β –C(O)CH3), 1.99 (s, 3H, α –C(O)CH3), 1.98 (s, 1.5H, β –C(O)CH3). HRMS calculated for C16H22NaO11 [M + Na]+ 413.1054; found 413.1057, calculated for the fragmentation product C14H19O9 [M + H]+ 331.1024 found 331.1018. RP HPLC: (linear gradient from 0 to 50 vol% MeCN in H2O in 30 min at 25 °C) tR = 20.86 min and tR = 21.87 min, respectively.:1 n-hexane:ethyl acetate v/v, Rf = 0.5), 400 mL chloroform was added to the solution, and the organic phase was washed three times each with sodium hydrogen carbonate solution and water (100 mL per washing). Subsequently, the organic phase was dried over magnesium sulphate and concentrated on the rotary evaporator to provide the desired product 3 in 97% yield. 1H-NMR: (600 MHz, chloroform-d) δ (ppm) 6.28 (d, 3JH1–H2 = 0.95 Hz, 1H, H1), 5.70 (dd, 3JH4–H5 = 10.17 Hz, 3JH4–H3 = 3.42 Hz, 1H, H4), 5.43 (dd, 3JH2–H1 = 1.64 Hz, 3JH2–H3 = 3.45 Hz, 1H, H2), 5.35 (t, 3JH3–H4=H3–H2 = 10.18 Hz, 1H, H3), 4.31 (dd, 2JH6–H6′ = 12.55 Hz, 3JH6–H5 = 4.93 Hz, 1H, H6), 4.20 (m, 1H, H5), 4.12 (dd, 2JH6′–H6 = 12.53 Hz, 3JH6′–H5 = 2.23 Hz, 1H, H6′), 2.16 (s, 3H, –C(O)CH3), 2.09 (s, 3H, –C(O)CH3), 2.06 (s, 3H, –C(O)CH3), 1.99 (s, 3H, –C(O)CH3). HRMS: calculated for the fragmentation product C14H19O9 [M + H]+ 331.1024 found 331.1018. RP HPLC: (linear gradient from 0 to 50 vol% MeCN in H2O in 30 min at 25 °C) tR = 10.34 min.:1 n-hexane:ethyl acetate, v/v, Rf = 0.6). Upon completion, the solution was diluted with 350 mL chloroform and washed three times with water (300 mL of water per washing). The aqueous phase was also extracted once with 200 mL chloroform. The combined organic phases were dried over magnesium sulphate and concentrated by rotary evaporation. The residue was recrystallized from an ethanol/n-hexane mixture, precipitating a white solid to provide the desired product 4 in 57% yield. 1H-NMR: (600 MHz, chloroform-d) (ppm) δ 5.48 (d, 3JH1–H2 = 1.01 Hz, 1H, H1exo), 5.38 (t, 3JH2–H3 = 9.99 Hz, 0.33H, H2endo), 5.28 (t, 3JH2–H3 = 9.79 Hz, 1H, H2exo), 5.25 (d, 3JH1–H2 = 1.08 Hz, 0.33H, H1endo), 5.19 (dd, 2JH6–H6′ = 10.07 Hz, 3JH6–H5 = 4.15 Hz, 0.33H, H6endo), 5.13 (dd, 2JH6–H6′ = 9.98 Hz, 3JH6–H5 = 4.03 Hz, 1H, H6exo), 4.62–4.57 (m, 1H, H6′exo), 4.39–4.35 (m, 0.33H, H6′endo), 4.28–4.19 (m, 1.33H, H5endo + exo), 4.16–4.08 (m, 1.33H, H4 endo + exo), 3.74–3.64 (m, 1.33H, H3endo + exo), 3.48 (s, 1H, –O–CH3(endo)), 3.27 (s, 3H, –O–CH3(exo)), 2.11 (s, 4H, –C(O)CH3 (endo + exo)), 2.06 (s, 3H, –C(O)CH3 (exo)), 2.04 (s, 4H, –C(O)CH3 (endo + exo)), 2.01 (s, 1H, –C(O)CH3 (endo)), 1.73 (s, 3H, –CH3(exo)), 1.51 (s, 1H, CH3(endo)). HR-MS: calculated for C15H22NaO10 [M + Na]+ 385.1105; found 385.1106.:1, n-hexane:ethyl acetate v/v, Rf = 0.8). Upon completion, the solvent was removed under reduced pressure and the product was used without further purification. 1H-NMR (600 MHz, methanol-d4) δ (ppm) 5.46 (d, 3JH1–H2 = 2.41 Hz, 1H, H1exo), 5.23 (d, 3JH1–H2 = 2.41 Hz, 0.33H, H1endo), 4.47 (dd, 3JH2–H1 = 2.45 Hz, 3JH2–H3 = 4.14 Hz, 1H, H2(exo)), 4.27 (dd, 3JH2–H1 = 2.47 Hz, 3JH2–H3 = 4.39 Hz, 0.33H, H2endo), 3.89–3.83 (two dd overlapping, 1.33H, H6(endo + exo)), 3.77 (dd, 2JH3–H4 = 9.46 Hz, 3JH3–H2 = 4.3 Hz, 0.33H, H3endo), 3.73–3.63 (m, 3H, H3(exo + endo), H6′exo, H4endo), 3.57 (t, 3JH4–H5 = 9.5 Hz, 1H, H4exo), 3.42 (s, 1H, –O–CH3(endo)), 3.28 (s overlapped with m, 3H, –O–CH3(exo), 0.3H, H5endo), 3.24 (m, 1H, H5exo), 1.63 (s, 3H, –CH3(exo)), 1.48 (s, 1H, –CH3(endo)). HR-MS: (not measured here).:1, n-hexane:ethyl acetate v/v, Rf = 0.4), the reaction was stopped by the addition of 80 mL methanol. The solution was extracted twice with 250 mL of ethyl acetate, and the collected organic phases were dried over magnesium sulphate. After removing the solvents under reduced pressure, the crude product was purified by column chromatography using an automated flash chromatography system with a running mixture of n-hexane and ethyl acetate (3:1, v/v). This method was also partially successful in separating the resulting endo:exo isomeric mixture. A running mixture of toluene and ethyl acetate (3:1, v/v) was also used as a substitute in a different trial, which provided a similar separation. The final yield was 73%. 1H-NMR: (600 MHz, chloroform-d) δ (ppm) 7.42–7.14 (m, overlapping with the singlet from CHCl3, 15H, Ar-H), 5.35 (d, 3JH1–H2 = 2.58 Hz, 1H, H1), 4.90 (d, 3JH9–H10/11 = 10.70 Hz, 1H, –O–CH2), 4.82–4.75 (m, 2H, –O–CH2), 4.63–4.58 (m, 2H, –O–CH2), 4.55 (d, 3JH9–H10/11 = 12.09 Hz, 1H, –O–CH2), 4.40 (dd, 3JH2–H3 = 4.02 Hz, 3JH2–H1 = 2.56 Hz, 1H, H2), 3.92 (t, 3JH4–H5 = 9.31 Hz, 1H, H4), 3.78–3.68 (m, 3H, H6, H6′ H3), 3.45–3.40 (m, 1H, H5), 3.29 (s, 3H, –O–CH3), 1.74 (s, 3H, –CH3). HR-MS: calculated for C30H34NaO7 [M + Na]+ 529.2197; found 529.2198. RP-HPLC: (not measured here).:ethyl acetate (2:1, v/v) to provide the desired product 7 in 47% yield. 1H-NMR: (600 MHz, chloroform-d) δ (ppm) 7.37–7.13 (m, overlapping with the singlet from CHCl3, 15H, Ar-H), 5.43 (dd, 3JH2–H3 = 2.91 Hz, 3JH2–H1 = 1.64 Hz, 1H, H2), 5.32 (d, 3JH1–H2 = 1.62 Hz, 1H, H1), 4.85 (d, 3JH10–H11/12 = 10.7 Hz, 1H, –O–CH2), 4.68 (dd, 3JH10–H11/12 = 11.6, 2.0 Hz, 2H, –O–CH2), 4.56–4.46 (m, 3H, O–CH2), 4.18–4.14 (m, 1H, H4), 3.96–3.89 (m, 2H, H6, H3), 3.84 (dd, 2JH6′–H6 = 10.82 Hz, 3JH6′–H5 = 4.29 Hz, 1H, H6′), 3.69 (dd, 3JH5–H6 = 10.83, 3JH5–H4 = 1.97 Hz, 1H, H5), 2.70–2.54 (m, 2H, –S–CH2–CH3), 2.16 (s, 3H, –C(O)–CH3), 1.30–1.26 (m, 3H, –S–CH2–CH3). HR-MS: calculated for C31H40NO6S [M + NH4]+ 554.2571; found 554.2568. RP-HPLC: (linear gradient from 0 to 50 vol% MeCN in H2O in 30 min at 25 °C) tR = 16.74 min.:1, n-hexane:ethyl acetate, v/v, Rf = 0.5), the orange solution was washed three times each with ice water and then cold sodium hydrogen carbonate solution (30 mL per washing). The organic phase was dried over magnesium sulphate and the solvent was removed under reduced pressure on a rotary evaporator. The crude product in a yield of 91% was used in the next reaction without further purification. 1H-NMR (600 MHz, chloroform-d) δ (ppm) 7.39–7.09 (m, overlapping with the singlet from CHCl3, 15H, Ar-H), 5.39 (dd, 3JH2–H3 = 3.40 Hz, 3JH2–H1 = 1.85 Hz, 1H, H2), 4.90 (d, 3JH1–H2 = 1.87 Hz, 1H, H1), 4.86 (d, 3JH10–H11/12 = 10.73 Hz, 1H, –O–CH2), 4.76–4.62 (m, 3H, –O–CH2), 4.60–4.45 (m, 5H, –O–CH2), 4.01–3.93 (m, 3H, H4, –O–CH2–CH2), 3.89–3.87 (m, 2H, H6, H3), 3.83–3.78 (m, 1H, H6′), 3.74–3.68 (m, 1H, H5), 3.50–3.44 (m, 2H, –O–CH2–CH2), 2.15 (s, 3H, –C(O)–CH3). HR-MS: calculated for C31H39BrNO7 [M + NH4]+616.1904; found 616.1903. RP-HPLC: (linear gradient from 0 to 50 vol% MeCN in H2O in 30 min at 25 °C) tR = 28.90 min.:1, n-hexane:ethyl acetate, v/v, Rf = 0.4) and the reaction was diluted with 50 mL toluene after complete conversion in about 6 h. The solvents were removed under reduced pressure by rotary evaporation and dried at high vacuum. The resulting product 7b-II was used without purification in the next step. 1H-NMR (600 MHz, chloroform-d) δ (ppm) 7.37–7.09 (m, overlapping with the singlet from CHCl3, 15H, Ar-H), 5.39 (dd, 3JH2–H3 = 3.40 Hz, 3JH2–H1 = 1.85 Hz, 1H, H2), 4.90 (d, 3JH1–H2 = 1.87 Hz, 1H, H1), 4.86 (d, 3JH10–H11/12 = 10.73 Hz, 1H, –O–CH2), 4.72–4.65 (m, 3H, –O–CH2), 4.56–4.45 (m, 4H, –O–CH2), 4.03–3.99 (m, 1H, –O–CH2–CH3), 3.91–3.76 (m, 5H, H4, H6, H6′, H3), 3.73–3.69 (m, 1H, H6′), 3.64–3.59 (m, 1H, –O–CH2–CH2), 3.41–3.35 (m, 2H, –O–CH2–CH2), 2.15 (s, 3H, –C(O)–CH3). HR-MS: calculated for C31H39N4O7 [M + NH4]+ 579.2813; found 579.2808. RP-HPLC: (linear gradient from 0 to 50 vol% MeCN in H2O in 30 min at 25 °C) tR = 28.34 min.:1, n-hexane:ethyl acetate, v/v, Rf = 0.3). Upon completion, the solvent was removed under reduced pressure by rotary evaporation. For purification, the crude product was separated by column chromatography with a running mixture of n-hexane:ethyl acetate (3:1, v/v) to provide the desired product 8 in 42% yield. 1H-NMR (600 MHz, chloroform-d) δ (ppm) 7.39–7.14 (m, overlapping with the singlet from CHCl3, 15H, Ar-H), 4.95 (d, 3JH1–H2 = 1.69 Hz, 1H, H1), 4.82 (d, 3JH10–H11/12 = 10.8 Hz, 1H, –O–CH2), 4.71–4.67 (m, 2H, –O–CH2), 4.64 (d, 3JH10–H11/12 =12.2 Hz, 1H, –O–CH2), 4.55–4.49 (m, 2H, –O–CH2), 4.07 (dd, 3JH2–H3 = 3.33 Hz, 3JH2–H1 = 1.74 Hz, 1H, H2), 3.93–3.87 (m, 2H, H3, H4), 3.86–3.82 (m, 1H, H5), 3.82–3.78 (m, 1H, –O–CH2–CH2), 3.76–3.68 (m, 2H, H6, H6′), 3.65–3.60 (m, 1H, –O–CH2–CH2), 3.43–3.32 (m, 2H, –O–CH2–CH2). HR-MS: calculated for C29H37N4O6 [M + NH4]+ 537.2708; found 537.2704. RP-HPLC: (linear gradient from 0 to 50 vol% MeCN in H2O in 30 min at 25 °C) tR = 14.76.:ethyl acetate (3:1, v/v) to provide the desired product 9 in 58% yield and 91% relative purity (determined by HPLC). 1H-NMR (600 MHz, chloroform-d) δ (ppm) 7.36–7.15 (m, overlapping with the singlet from CHCl3, 30H, Ar-H), 5.53 (dd, 3JH2–H3 = 3.33 Hz, 3JH2–H1 = 1.86 Hz, 1H, H2), 5.03 (d, 3JH1–H2 = 1.81 Hz, 1H, H1), 4.94 (d, 3JH1′–H2′ = 1.92 Hz, 1H, H1′), 4.87–4.83 (m, 2H, –O–CH2), 4.70–4.62 (m, 5H, –O–CH2), 4.54 (t, 3JH10–H11/12 = 11.02 Hz, 2H, –O–CH2), 4.48 (t, 3JH10–H11/12 = 11.23 Hz, 2H, –O–CH2), 4.41 (d, 3JH10–H11/12 = 10.93 Hz, 1H, –O–CH2), 4.01–3.91 (m, 4H), 3.84–3.68 (m, 7H), 3.41–3.35 (m, 1H, –O–CH2–CH3), 3.30–3.18 (m, 2H, –O–CH2–CH3), 2.13 (s, 3H, –C(O)–CH3). An exact assignment of the carbohydrate signals of both mannose units in the range of 4.01 to 3.68 ppm has not been made due to the very similar chemical environment. 13C-NMR (100 MHz, chloroform-d) δ (ppm) 170.22, 138.47, 138.44, 138.42, 138.37, 138.23, 138.04, 128.83–127.34 (m), 99.78, 98.86, 79.54, 78.14, 75.17, 75.10, 74.50, 74.47, 73.53, 73.36, 72.23, 72.14, 71.97, 71.90, 69.36, 69.29, 68.97, 68.78, 66.53, 50.46, 21.20. HR-MS: calculated for C58H67N4O12 [M + NH4]+ 1011.4750; found 1011.4743. RP-HPLC: (linear gradient from 0 to 50 vol% MeCN in H2O in 30 min at 25 °C) tR = 15.77 min, relative purity 91%2.4 Solid phase glycomacromolecule synthesis
The oligo(amidoamine) scaffold was built up step by step using the general coupling and deprotection protocol with building blocks EDS and TT, which were synthesized according to literature.19,20 The preparation size/scale of the oligo(amidoamine) scaffold was 0.2 mmol. The resin used was a TentaGel® S RAM resin preloaded with ethylenediamine. Before each reaction on the resin, the resin was allowed to swell in DCM for approx. 20 min and subsequently washed five times with the solvent of the following reaction.:1, v/v), ten times with dimethylformamide and ten times with dichloromethane. These steps were repeated until no discoloration of the sodium diethyldithiocarbamate was visible.3 Results and discussion
3.1 α-1,2-Mannobioseazide synthesis
Our synthetic approach, inspired by Groneborn et al., is shown in Fig. 1.12 The key requirement of the approach is the generation of a regio- and stereoselective α-1,2-connection between the two mannose units to obtain desired product 9 with both a linker in place for conjugation, and a temporary protecting group in the 2′-O-position for possible extension. For this purpose, we envisioned generating two different glycosides from known orthoester 6 (ref. 9): (i) known glycosyl donor 8 (ref. 18) bearing a shelf stable thioglycoside which could be readily activated for glycosylation, and (ii) glycosyl acceptor 7a (ref. 17) bearing an azido linker for further conjugation e.g., for presentation on a multivalent scaffold. To ensure the regio- and stereoselective formation of the desired α-1,2-linkage, an orthogonal protecting group strategy was employed, and a 2-O-acetate protecting group was initially installed to drive α-selectivity during glycosylation. This group could then be selectively deprotected for subsequent glycosylation. Meanwhile, the installation of O-benzyl groups more distant from the anomeric site could serve to arm each monosaccharide for glycosylation.21 Notably, orthoester 6 can be prepared in high yields from readily available D-mannose over four steps.9 Overall, the resulting split and combine approach consisted of ten steps from commercially available D-mannose to the desired novel α-1,2-mannobioside 9.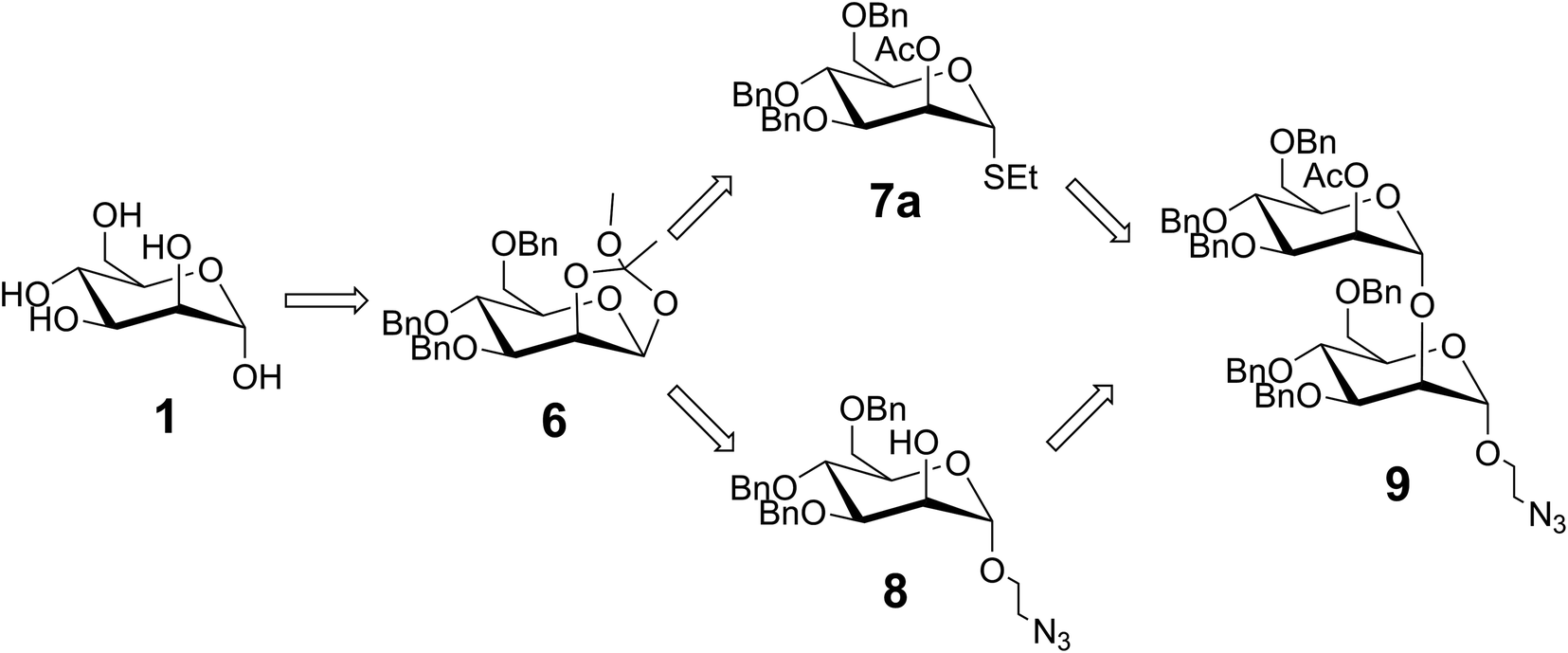 | ||
| Fig. 1 Synthetic pathway of final α-1,2-mannobiose disaccharide 9 starting from D-(+)-mannose. | ||
The synthesis of compound 9 is shown in Fig. 2. Known orthoester 6 was prepared based on work by Ahadi et al.9 Acetylation of α-D-mannose 1 under acidic conditions gave 2 in near quantitative yield. Bromination of 2 with hydrogen bromide in acetic acid gave 3 almost exclusively as the α-anomer in 97% yield, with the stereochemistry of this reaction being driven by the neighboring group effect of the acetylated 2-O-position which creates an intermediate oxonium ion that blocks the top face of the sugar. In the next step, the C2-acetyl group, aided by the assistance of 2,4,6-trimethylpyridine, underwent an intramolecular substitution reaction at the anomeric position. The resulting acetonide was then captured by methanol to generate orthoester 4 in 57% yield after purification. The acetyl protecting groups of 4 were hydrolyzed with sodium methoxide in methanol to yield 5, which was subsequently benzylated with benzyl chloride in sodium hydroxide to afford 6 in a yield of 73% over two steps and after purification via chromatography. This key intermediate was then split and used in the second part of the synthesis.
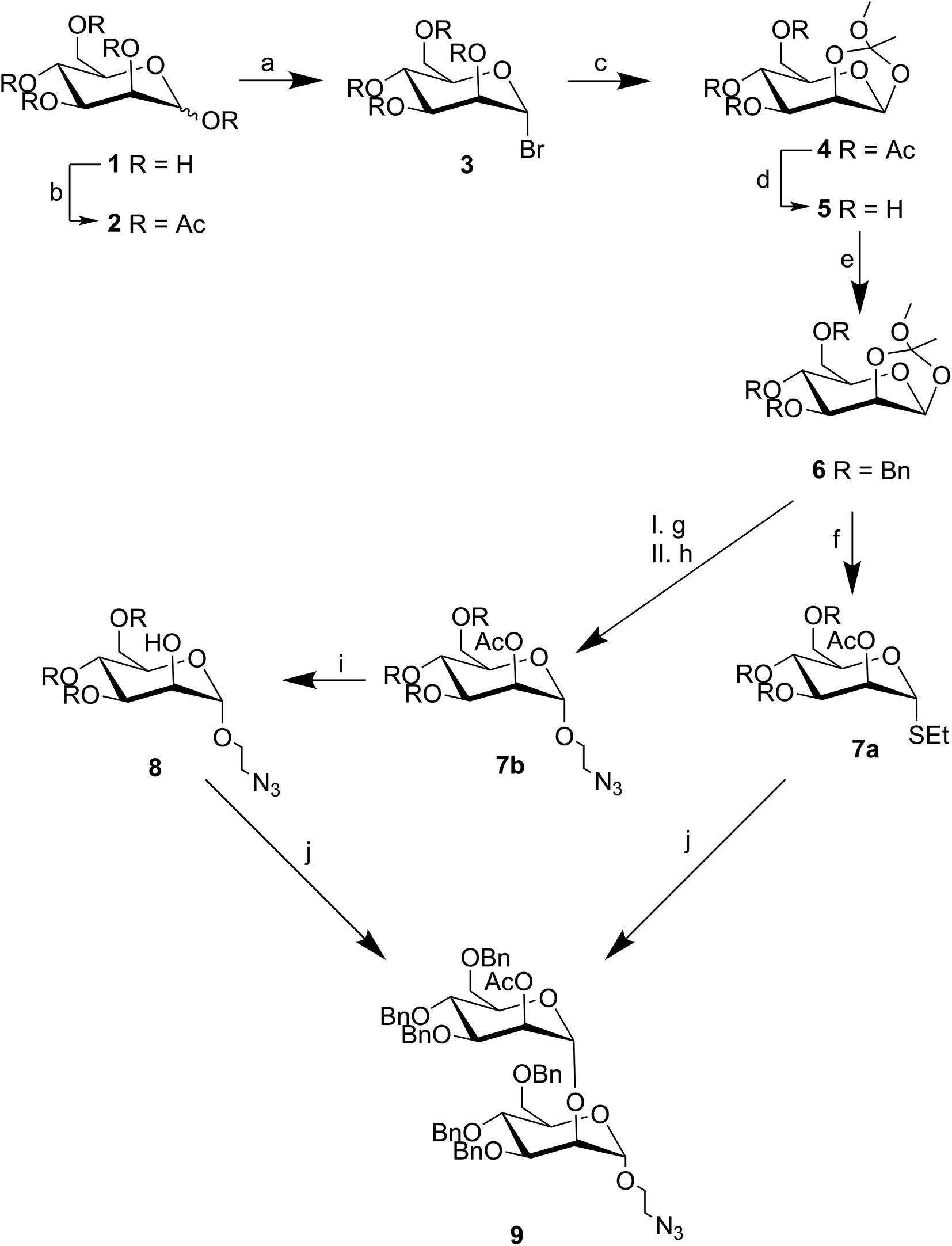 | ||
| Fig. 2 Synthesis of α-1,2-mannobioseazide starting from D-(+)-mannose 1 via orthoester 6. (a) Ac2O, MeCN, 24 h. (b) HBr, AcOH, 16 h. (c) 2,4,6-trimethylpyidine, MeOH, 16 h. (d) NaOMe, DCM, MeOH, 2 h. (e) BnBr, NaH, DMF, 16 h. (f) EtSH, BF3*OEt2, 30 min. (g) BrEtOH, BF3*OEt2, DCM, 16 h. (h) NaN3, DMF, 55 °C. (i) NaOMe, DCM, MeOH, 2 h. (j) NIS, Sn(OTf)2, DCM, −20 °C, 30 min. | ||
The second part of the synthesis required the introduction of the azide linker to 6 to generate known compound 8 for multivalent presentation, and the addition of the thiol group to 6 to generate 7a for subsequent glycosylation with 8; both reactions required nucleophilic ring opening of the orthoester. The synthesis of compound 8 was based on work by Neralkar et al.18 Anomeric substitution of 6 with 2-bromoethanol via Lewis acid catalyzed activation, followed by nucleophilic substitution at the resulting primary bromide with sodium azide afforded crude compound 7b in 91% yield over two steps. Subsequent deprotection of the temporary acetyl protecting group at the C2 position of 7b via sodium methanolate yielded glycosyl acceptor 8 as a single anomer in 42% after purification. The synthesis of glycosyl donor 7a was based on work by Barresi et al.17 Orthoester 6 was activated under Lewis acid catalyzed conditions, but this time in the presence of ethanethiol. This provided a 47% yield of the desired product 7a after purification via chromatography.
With both donor 7a and acceptor 8 in hand, the next step involved a stereoselective glycosylation reaction to generate the novel compound 9. To achieve this, N-iodosuccinimide and tin(II) trifluoromethanesulfonate were added to a mixture containing 7a and 8 under argon atmosphere at −20 °C. The reaction was monitored by TLC until completion. The desired α-1,2-mannobioseazide product 9 was achieved in 58% yield after column chromatography. The final, overall yield of the ten step sequence required to produce α-1,2-mannobioseazide was 8.77%.
3.2 Multivalent α-1,2-mannobiose glycomacromolecule synthesis
After successful synthesis and isolation of the α-1,2-mannobioseazide ligand, a trivalent precision glycomacromolecule based on an oligo(amidoamine) scaffold F2 was constructed using solid-phase synthesis as shown in Fig. 3. This structure was inspired by previous efforts in our group to target C-type lectins with precision glycomacromolecules designed to mimic high-mannose/mannose rich glycans.22,23 In this work, we chose to focus on a unique and novel ligand consisting of a hydrophilic tail group conjugated to a functionalizable trivalent head group for optimal presentation of the α-1,2-mannobiose ligand. The EDS (ethylene glycol diamine succinic acid) building block was selected as a spacer unit to enhance the flexibility and hydrophilicity of the ligand. Notably the choice of resin ensured a terminal amine at the end of the EDS spacer which is of special interest to our work as we endeavor to present these ligands on surfaces and membranes for further investigation. The use of the TT (trivalent triple bond) building block was selected for presentation of the α-1,2-mannobiose ligand, and was chosen for its ability to mimic the way high mannose oligosaccharides are presented to C-type lectins found in nature, e.g. Man9GlcNAc2.24,25 Furthermore, the close distance between the alkyne groups in the TT building block and the resulting increase in coupling difficulty acted as a rigorous evaluation for on-resin functionalization with the large benzyl protected α-1,2-mannobioseazide ligand.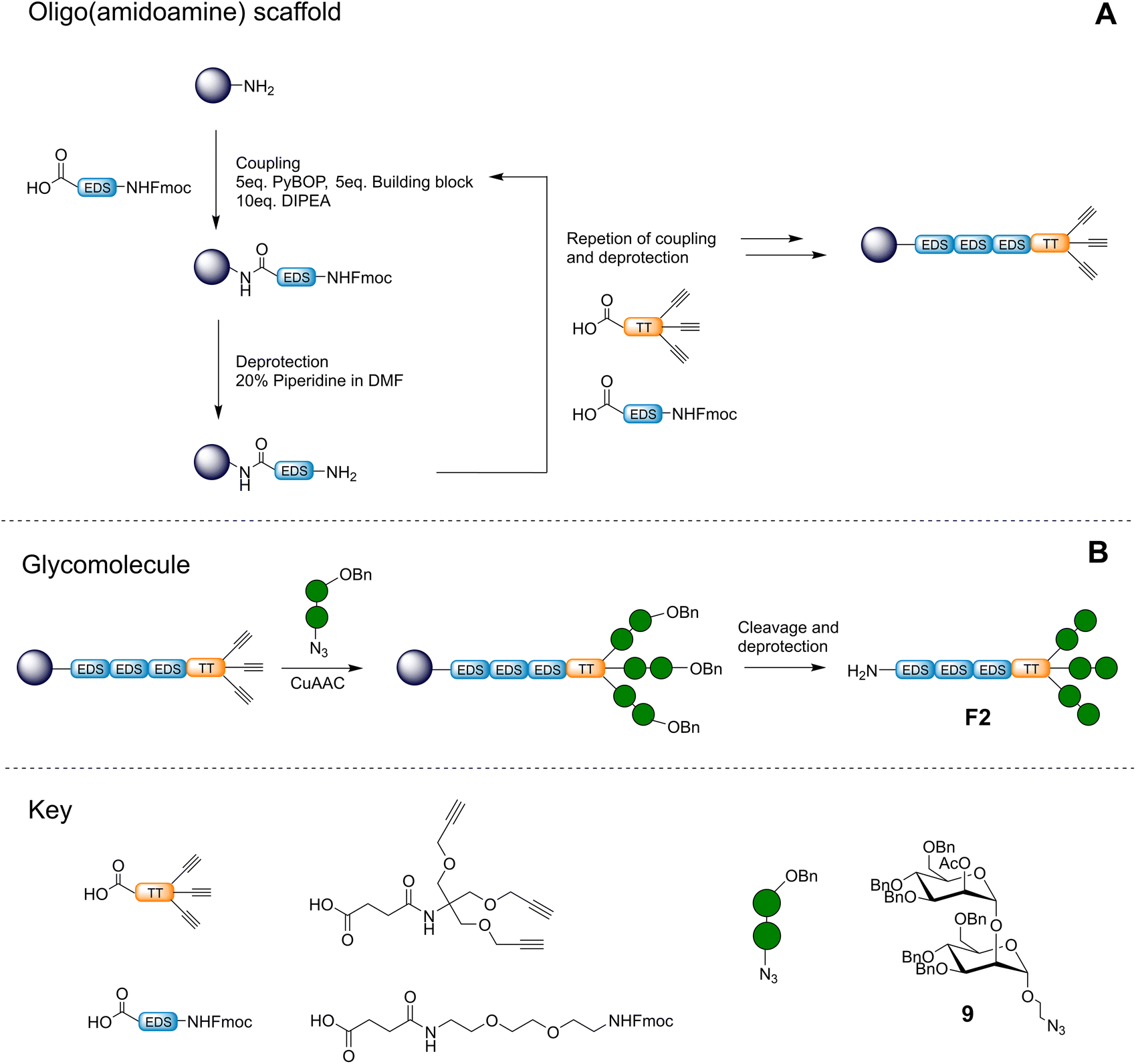 | ||
| Fig. 3 Overview of construction of trivalent scaffold via solid phase polymer synthesis (A) and conjugation of the ligand to the scaffold via CuAAC followed by cleavage and deprotection (B). | ||
The selected precision glycomacromolecule scaffold was synthesized by iterative coupling of building blocks on solid support as shown in Fig. 3A. Couplings were achieved with high efficiency using an excess of building blocks, which were recoverable.26 Post construction, the α-1,2-mannobioseazide ligand was introduced to the scaffold directly on solid support using an excess (7.5 equivalents) of ligand via CuAAC. Notably, we were also able to recover and recycle unreacted α-1,2-mannobioseazide thus improving the overall efficiency of the approach.
Following conjugation of the α-1,2-mannobioseazide ligand, the acetyl protecting groups were cleaved on resin with 1 M sodium methoxide solution in methanol in 1 h. The oligomer was removed from solid support, and the remaining benzyl protecting groups on the oligomer were cleaved by hydrogenolysis with Pd/C in methanol in the presence of hydrogen gas for 72 h; shorter deprotection times led to mixtures of partially benzylated products. The final trivalent glycomacromolecule F2 was characterized with 1H NMR and HRMS. While the NMR showed the spectrum of F2 in high purity, mass spectroscopy results showed a variety of masses which were attributed to fragmentation of the α-1,2-mannobiose ligands (see ESI† for further information).
4 Conclusion
Here we adopt an efficient, split and mix approach for the synthesis of a novel α-1,2-mannobiose analog bearing an azide linker for conjugation (9) from known precursors. The synthetic route described here has several advantages. First, compound 9 can be synthesized from an easily accessible and inexpensive orthoester precursor (6) which can be prepared on gram scales. Second, the glycosylation conditions provide for the installation of a wide variety of possible linkers. Finally, the temporary 2-O-acetyl protecting that is generated on nucleophilic ring opening of 6 provides the possibility of selectively elongating/functionalizing the disaccharide with a wide range of substrates, which in turn allows for the possibility of further customization and adaptivity.The multivalent presentation of α-1,2-mannobiose on a precision glycomacromolecule (F2) was also achieved using a solid support approach. Notably, the disadvantage of having to use higher equivalents of building blocks during solid-supported synthesis steps and glycoconjugation via CuAAC was mitigated by their collection, recycling, and reuse, making the overall process highly efficient. Furthermore, the terminal amine group and long flexible linker consisting of the EDS units make the compound well suited for presentation on surfaces to better mimic the native presentation of these types of structures.
The final precision glycomacromolecule presenting the α-1,2-mannobiose in a trivalent fashion represents an interesting example for the investigation into the affinity of C-type lectins towards the multivalent presentation of α-1,2-mannobiose ligands. We believe its presentation is likely to mimic high-mannose containing natural glycans that are known to engage C-type lectins like langerin and DC-SIGN. Indeed, our current efforts involve expanding the methodology reported here to generate a library of similar structures like the one presented here to evaluate their binding towards langerin and DC-SIGN to examine possible trends for selectivity and affinity.
Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
NLS would like to thank Davidson College for a Faculty Study and Research Grant, and the National Science Foundation for partial support through an International Research Experience for Scientists grant (#1854028). We would also like to thank the analytical facilities at Heinrich-Heine-Universität, Duesseldorf for support with 1H-NMR and MS analysis.References
- H. Feinberg, S. A. F. Jégouzo, M. J. Rex, K. Drickamer, W. I. Weis and M. E. Taylor, Mechanism of pathogen recognition by human dectin-2, J. Biol. Chem., 2017, 292, 13402–13414 CrossRef CAS PubMed.
- Y. Guo, H. Feinberg, E. Conroy, D. A. Mitchell, R. Alvarez, O. Blixt, M. E. Taylor, W. I. Weis and K. Drickamer, Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR, Nat. Struct. Mol. Biol., 2004, 11, 591–598 CrossRef CAS PubMed.
- J. M. Irache, H. H. Salman, C. Gamazo and S. Espuelas, Mannose-targeted systems for the delivery of therapeutics, Expert Opin. Drug Delivery, 2008, 5, 703–724 CrossRef CAS PubMed.
- A. M. Kerrigan and G. D. Brown, C-type lectins and phagocytosis, Immunobiology, 2009, 214, 562–575 CrossRef CAS PubMed.
- V. Porkolab, E. Chabrol, N. Varga, S. Ordanini, I. Sutkeviči
![[u with combining overline]](https://www.rsc.org/images/entities/char_0075_0305.gif) tė, M. Thépaut, M. J. García-Jiménez, E. Girard, P. M. Nieto, A. Bernardi and F. Fieschi, Rational-Differential Design of Highly Specific Glycomimetic Ligands: Targeting DC-SIGN and Excluding Langerin Recognition, ACS Chem. Biol., 2018, 13, 600–608 CrossRef CAS PubMed.
tė, M. Thépaut, M. J. García-Jiménez, E. Girard, P. M. Nieto, A. Bernardi and F. Fieschi, Rational-Differential Design of Highly Specific Glycomimetic Ligands: Targeting DC-SIGN and Excluding Langerin Recognition, ACS Chem. Biol., 2018, 13, 600–608 CrossRef CAS PubMed. - P. Valverde, J. D. Martínez, F. J. Cañada, A. Ardá and J. Jiménez-Barbero, Molecular Recognition in C-Type Lectins: The Cases of DC-SIGN, Langerin, MGL, and L-Sectin, ChemBioChem, 2020, 21, 2999–3025 CrossRef CAS PubMed.
- E.-C. Wamhoff, J. Schulze, L. Bellmann, M. Rentzsch, G. Bachem, F. F. Fuchsberger, J. Rademacher, M. Hermann, B. Del Frari, R. van Dalen, D. Hartmann, N. M. van Sorge, O. Seitz, P. Stoitzner and C. Rademacher, A Specific, Glycomimetic Langerin Ligand for Human Langerhans Cell Targeting, ACS Cent. Sci., 2019, 5, 808–820 CrossRef CAS PubMed.
- M. K. Patel, B. Vijayakrishnan, J. R. Koeppe, J. M. Chalker, K. J. Doores and B. G. Davis, Analysis of the dispersity in carbohydrate loading of synthetic glycoproteins using MALDI-TOF mass spectrometry, Chem. Commun., 2010, 46, 9119–9121 RSC.
- S. Ahadi, S. I. Awan and D. B. Werz, Total Synthesis of Tri-, Hexa- and Heptasaccharidic Substructures of the O-Polysaccharide of Providencia rustigianii O34, Chem.–Eur. J., 2020, 26, 6264–6270 CrossRef CAS PubMed.
- S. Boonyarattanakalin, X. Liu, M. Michieletti, B. Lepenies and P. H. Seeberger, Chemical Synthesis of All Phosphatidylinositol Mannoside (PIM) Glycans from Mycobacterium tuberculosis, J. Am. Chem. Soc., 2008, 130, 16791–16799 CrossRef CAS PubMed.
- A. K. Pathak, V. Pathak, J. M. Riordan, S. S. Gurcha, G. S. Besra and R. C. Reynolds, Synthesis of mannopyranose disaccharides as photoaffinity probes for mannosyltransferases in Mycobacterium tuberculosis, Carbohydr. Res., 2004, 339, 683–691 CrossRef CAS PubMed.
- G. Nestor, T. Anderson, S. Oscarson and A. M. Gronenborn, Exploiting Uniformly 13C-Labeled Carbohydrates for Probing Carbohydrate–Protein Interactions by NMR Spectroscopy, J. Am. Chem. Soc., 2017, 139, 6210–6216 CrossRef CAS PubMed.
- S. A. Hill, C. Gerke and L. Hartmann, Recent Developments in Solid-Phase Strategies towards Synthetic, Sequence-Defined Macromolecules, Chem.–Asian J., 2018, 13, 3611–3622 CrossRef CAS PubMed.
- S. Boden, F. Reise, J. Kania, T. K. Lindhorst and L. Hartmann, Sequence-Defined Introduction of Hydrophobic Motifs and Effects in Lectin Binding of Precision Glycomacromolecules, Macromol. Biosci., 2019, 19, 1800425 CrossRef PubMed.
- D. Ponader, P. Maffre, J. Aretz, D. Pussak, N. M. Ninnemann, S. Schmidt, P. H. Seeberger, C. Rademacher, G. U. Nienhaus and L. Hartmann, Carbohydrate-Lectin Recognition of Sequence-Defined Heteromultivalent Glycooligomers, J. Am. Chem. Soc., 2014, 136, 2008–2016 CrossRef CAS PubMed.
- T. Freichel, D. Laaf, M. Hoffmann, P. B. Konietzny, V. Heine, R. Wawrzinek, C. Rademacher, N. L. Snyder, L. Elling and L. Hartmann, Effects of linker and liposome anchoring on lactose-functionalized glycomacromolecules as multivalent ligands for binding galectin-3, RSC Adv., 2019, 9, 23484–23497 RSC.
- F. Barresi and O. Hindsgaul, The synthesis of β-mannopyranosides by intramolecular aglycon delivery: scope and limitations of the existing methodology, Can. J. Chem., 1994, 72, 1447–1465 CrossRef CAS.
- M. Neralkar, L. Tian, R. L. Redman and I. J. Krauss, Synthesis of Mannosidase-Stable Man3 and Man4 Glycans Containing S-linked Manα1→2Man Termini, Org. Lett., 2021, 23, 3053–3057 CrossRef CAS PubMed.
- D. Ponader, F. Wojcik, F. Beceren-Braun, J. Dernedde and L. Hartmann, Sequence-Defined Glycopolymer Segments Presenting Mannose: Synthesis and Lectin Binding Affinity, Biomacromolecules, 2012, 13, 1845–1852 CrossRef CAS PubMed.
- S. Üclü, C. Marschelke, F. Drees, M. Giesler, D. Wilms, T. Köhler, S. Schmidt, A. Synytska and L. Hartmann, Sweet Janus Particles: Multifunctional Inhibitors of Carbohydrate-Based Bacterial Adhesion, Biomacromolecules, 2024, 25, 2399–2407 CrossRef PubMed.
- D. R. Mootoo, P. Konradsson, U. Udodong and B. Fraser-Reid, Armed and disarmed n-pentenyl glycosides in saccharide couplings leading to oligosaccharides, J. Am. Chem. Soc., 1988, 110, 5583–5584 CrossRef CAS.
- J. Valladeau, V. Duvert-Frances, J.-J. Pin, C. Dezutter-Dambuyant, C. Vincent, C. Massacrier, J. Vincent, K. Yoneda, J. Banchereau, C. Caux, J. Davoust and S. Saeland, The monoclonal antibody DCGM4 recognizes Langerin, a protein specific of Langerhans cells, and is rapidly internalized from the cell surface, Eur. J. Immunol., 1999, 29, 2695–2704 CrossRef CAS PubMed.
- H. Feinberg, M. E. Taylor, N. Razi, R. McBride, Y. A. Knirel, S. A. Graham, K. Drickamer and W. I. Weis, Structural Basis for Langerin Recognition of Diverse Pathogen and Mammalian Glycans through a Single Binding Site, J. Mol. Biol., 2011, 405, 1027–1039 CrossRef CAS PubMed.
- D. A. Mitchell, A. J. Fadden and K. Drickamer, A Novel Mechanism of Carbohydrate Recognition by the C-type Lectins DC-SIGN and DC-SIGNR: Subunit Organization and Binding to Multivalent Ligands, J. Biol. Chem., 2001, 276, 28939–28945 CrossRef CAS PubMed.
- N. S. Stambach and M. E. Taylor, Characterization of carbohydrate recognition by langerin, a C-type lectin of Langerhans cells, Glycobiology, 2003, 13, 401–410 CrossRef CAS PubMed.
- F. Shamout, L. Fischer, N. L. Snyder and L. Hartmann, Recovery, Purification, and Reusability of Building Blocks for Solid Phase Synthesis, Macromol. Rapid Commun., 2020, 41, 1900473 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06526c |
| This journal is © The Royal Society of Chemistry 2024 |