
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaleic anhydride derivatives as catalysts for N-oxidation of pyridine using hydrogen peroxide†
Ghellyn Gajeles‡
 *,
Kyung-Koo Lee and
Sang Hee Lee
*,
Kyung-Koo Lee and
Sang Hee Lee
Department of Chemistry, Kunsan National University, Gunsan 573-701, Republic of Korea. E-mail: leesh@kunsan.ac.kr
First published on 7th October 2024
Abstract
Maleic anhydride derivatives were evaluated as catalysts in N-oxidation of various pyridine substrates using hydrogen peroxide (H2O2). Depending on the electronic properties of the pyridine substrates, pyridines with electron-donating groups reacted well with 2,3-dimethylmaleic anhydride (DMMA). In contrast, 1-cyclohexene-1, 2-dicarboxylic anhydride (CHMA) was most effective for electron-deficient pyridines. The different performance of these two anhydrides is attributed to the diacid–anhydride equilibrium, which is crucial for regenerating the peracid oxidant through an anhydride intermediate in the catalytic cycle. This approach using a catalytic amount of anhydride with H2O2 has the potential to replace stoichiometric amounts of percarboxylic acid as an oxidant for a broader range of organic substrates.
1 Introduction
Due to its safe, cheap, clean, and environmentally friendly properties, hydrogen peroxide (H2O2) is a preferred oxidant, producing only molecular oxygen and water as by-products.1 Although it is a powerful oxidant with high potential, H2O2's high activation barriers make it less effective under mild conditions.2–5 To address this limitation and enhance its activity, H2O2 requires activation by an additional reagent that functions as the terminal oxidant.6,7 This has led to the development of numerous catalytic systems utilizing H2O2, with a significant focus on metal-catalyzed oxidations.8–13 Unfortunately, the use of metal catalysts can introduce safety concerns, particularly when producing high-purity pharmaceutical compounds. Metal-free H2O2 oxidation procedures have emerged as a promising solution to address this challenge.14–17Organocatalytic oxidations with H2O2 have recently gained attraction as a powerful and convenient method for synthesizing diverse molecules due to their mild reaction conditions and reduced toxicity.18–20 Various organic catalysts have been explored as mediators for H2O2-based oxidations, including aldehydes,21 hydroxamic acids,22 flavins,23 alloxan,24 heteroarenium salts,25–27 benzoxathiazines,28,29 and aryl trifluoromethyl ketones.30–33 Some organocatalysts have been shown to enable asymmetric transformations such as N-oxidation,34,35 epoxidation,36 and hydroxylation.37
The percarboxylic acids are significantly stronger oxidants compared to H2O2. However, due to their instability, percarboxylic acids are typically generated directly within the reaction mixture (in situ). Even though various anhydrides have been successfully employed as mediators in H2O2 oxidation reactions, unfortunately, mostly stoichiometric amounts of anhydride were used.38–43
Our previous work demonstrated anhydride-containing polymers, featuring repeating succinic anhydride units in the backbone, as effective catalysts for N-oxidation of pyridines using H2O2.44 These anhydride-derived peracids undergo N-oxidation, after which the resulting dicarboxylic acid is converted back to the original anhydride. This regeneration step is essential for a sustained catalytic cycle, allowing the catalyst to be reused for further oxidation reactions. Maleic anhydride is expected to be more readily regenerated from its corresponding diacid compared to succinic anhydride.45 Furthermore, the stronger acidity of maleic acid (pKa = 1.93) compared to succinic acid (pKa = 4.21) suggests that maleic anhydride has the potential to form a stronger peracid oxidant with H2O2. Motivated by this anticipated faster regeneration and stronger peracid formation, this study will investigate maleic anhydride derivatives as replacements for succinic anhydride in our previously reported catalyst system for N-oxidation of pyridine derivatives with H2O2.
2 Experimental
2.1 Materials and instruments
Pyridine derivatives and anhydride derivatives were purchased from Sigma-Aldrich and TCI and used without further purification. Column chromatography was performed using silica gel (230–400 mesh). Hydrogen peroxide (H2O2, 34.5 wt%) was purchased from Samchun chemicals, Korea, and used after titration with KMnO4.1H NMR and 13C NMR spectra were obtained on a 500 MHz Agilent (VARIAN) VNMRS spectrometer. GC experiments were performed on a Shimadzu GC-2010 Gas Chromatograph equipped with a DB-5 column (30 m, 0.25 mm i.d., 0.25 μm film thickness). N2 was used as the carrier gas at a flow rate of 0.87 ml min−1. The oven temperature program began at 60 °C and increased linearly to 300 °C at 15 °C min−1, followed by a hold at 300 °C for 4 min. The total analysis time for each sample was 20 min. The injector and detector (FID) temperatures were both set to 300 °C.
2.2 N-Oxidation reaction
In screening for anhydride catalysts at room temperature, the reaction was conducted with 1 mmol of substrate, 0.1 equivalent of anhydride catalyst, and 1.2 equivalents of 30% H2O2 under vigorous stirring for 24 hours. The reaction mixture is homogeneous in acetonitrile (CH3CN). However, without acetonitrile, all reactions became two-phase systems (water/substrate layer) with the anhydride catalyst being soluble in the substrate layer. The conversion yield was analyzed by GC after the reaction mixture is diluted by acetonitrile.To achieve complete N-oxidation, the reaction was conducted with 0.05 equivalent of catalyst, 2.0 equivalents of 30% H2O2, and vigorous stirring at 80 °C until complete conversion was observed by TLC. The reaction mixture was extracted with dichloromethane and then chromatographed (SiO2, dichloromethane/methanol = 20/1) to isolate and analyze the N-oxide product by 1H NMR and 13C NMR spectroscopy for structure determination. To estimate the conversion percentage, 0.1 ml of the reaction in DMSO-d6 mixture was analyzed by 1H NMR spectroscopy. The relative integrals of the peaks corresponding to the starting material and the product were used to calculate the conversion percentage.
2.3 13C NMR analysis of composition in anhydride-H2O2 solution
For analysis of the composition of CHMA and DMMA in perhydrolysis reaction media, 13C NMR sample solution was prepared by dissolving 1.0 mmol of anhydride and 2.0 mmol of H2O2 (0.20 ml, 30% wt), in 1.0 ml of CD3CN. Similarly, for analysis of the composition of CHMA in N-oxidation media, the NMR sample solution was prepared by dissolving 2.0 mmol of pyridine derivative, 1.0 mmol of CHMA, and 2.0 mmol of H2O2 (0.20 ml, 30% wt) in 1 ml of CD3CN. The 13C NMR spectra were acquired at intervals throughout the incubation at room temperature.3 Results and discussion
3.1 Screening of anhydride catalysts
2-Chloropyridine (CP) and quinoline were chosen as model substrates due to their differing nitrogen atom nucleophilicity in N-oxidation reaction. Readily available maleic anhydride derivatives were evaluated for their catalytic activity in N-oxidation using H2O2 (Fig. 1). The reaction was conducted using 1 mmol of substrate, 0.1 equivalent of anhydride catalyst, 1.2 equivalent of H2O2 (30%) at room temperature for 24 hours. The reaction mixture is homogeneous in acetonitrile (CH3CN), but in its absence, all reactions became two-phase systems (water/substrate layer) with the anhydride catalyst being soluble in the substrate layer.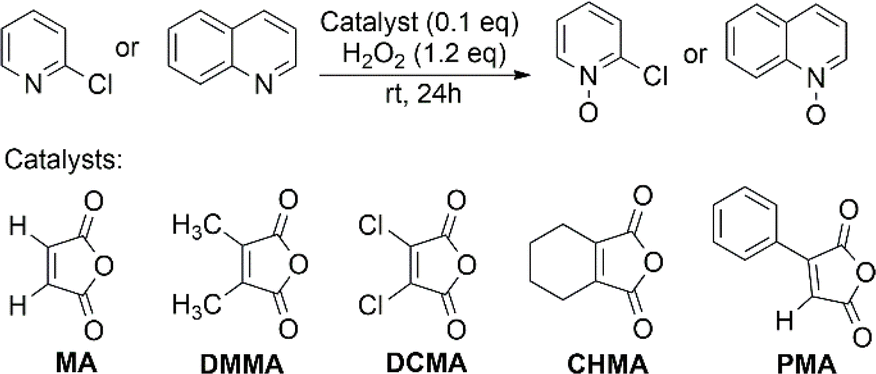 | ||
| Fig. 1 Model reaction for screening the catalytic activity of anhydride catalysts. | ||
The results are shown in Table 1. As expected for electron-rich substrates being more reactive in N-oxidation, 2,3-dimethyl maleic anhydride (DMMA) yielded a high N-oxide conversion (70%) for quinoline, especially in two-phase reactions, but showed no catalytic activity for the electron-deficient pyridine substrate CP. The exceptionally high activity of DMMA toward quinoline in two-phase reactions likely relates to the high partition coefficient of H2O2 favoring quinoline over water.46 This creates a localized environment enriched in H2O2, minimizing the amount of water surrounding the reactants, which is beneficial for the reaction.
2,3-Dichloromaleic acid, derived from dichloromaleic anhydride (DCMA) is a strong acid with pKa1 < 1.72 and pKa2 < 3.86 (compare to pKa1 = 1.72 and pKa2 = 3.86 for chloromaleic acid) due to the electron-withdrawing effect of the two chlorine atoms.47 This suggests its potential effectiveness for less reactive substrates like CP and agrees well with the results (25%). However, intriguingly, DCMA showed no activity towards quinoline, which is a more readily oxidized substrate. This lack of activity for the more reactive quinoline can be attributed to the high basicity of quinoline (pKa = 4.93).48 Quinoline can deprotonate both of the carboxylic acid groups on the diacid form of DCMA, leading to salt formation. This disrupts the anhydride-diacid equilibrium, preventing regeneration of the active anhydride and halting the N-oxidation catalytic cycle (Fig. 2). Conversely, due to its much lower basicity (pKa = 0.54) compared to quinoline, CP cannot deprotonate DCMA. This allows the diacid to readily convert back to the active anhydride species, maintaining the N-oxidation catalytic cycle. In contrast to the substrate-dependent activity observed with DMMA and DCMA, CHMA is effective for both CP and quinoline substrates (24–32%), regardless of whether CH3CN solvent is used. This suggests that CHMA may be less sensitive to the electronic properties of the substrate compared to other anhydride catalysts. To elucidate the contrasting catalytic activities of DMMA and CHMA in N-oxidation reactions, a mechanistic investigation was undertaken.
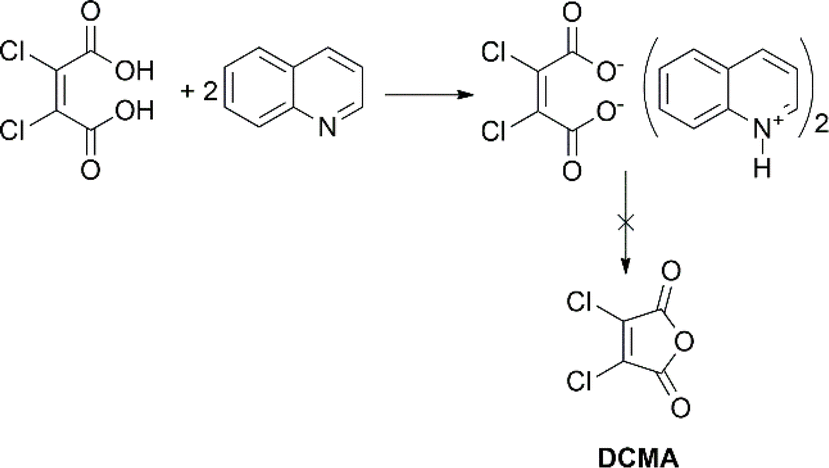 | ||
| Fig. 2 Deactivation of DCMA through salt formation. | ||
3.2 Mechanistic investigation of DMMA and CHMA
Peracid formation, a crucial step in N-oxidation where an organic acid gains oxygen from H2O2, is likely similar to succinic acid's amidation, where anhydride formation is the rate-limiting step.49 We propose that peracid formation from a diacid also proceeds via an anhydride intermediate (Fig. 3). Therefore, the rate of N-oxidation likely depends on the concentration of the active anhydride species, which is related to the equilibrium constant (Keq) between the diacid and the anhydride. In this context, a study by Eberson on cyclic anhydrides investigating their equilibrium behavior in aqueous solution is relevant.50 This study found that the equilibrium constants (Keq) for the hydrolysis of DMMA and CHMA were 5.3 and <0.1, respectively. These significant differences in Keq values for DMMA and CHMA in aqueous H2O2 solution likely explain their contrasting catalytic performance in N-oxidation. The lower Keq value for CHMA indicates a smaller equilibrium concentration of the active anhydride species (precursor to the peracid) compared to DMMA. The difference in their Keq values can be attributed to their structural differences. As shown in Fig. 4, the methyl group in DMMA is more flexible than the methylene group in the cyclohexene ring of CHMA. This increased flexibility may contribute to greater steric hindrance during the hydrolysis of the anhydride, due to the bulkier nature of the methyl group. Additionally, the flexibility of the DMMA methyl group could facilitate the alignment of two carboxyl groups for anhydride formation.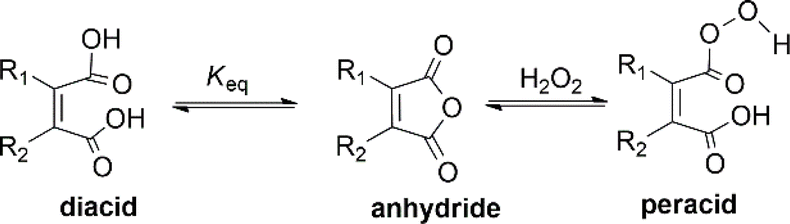 | ||
| Fig. 3 Formation of peracid from diacid via anhydride intermediate. | ||
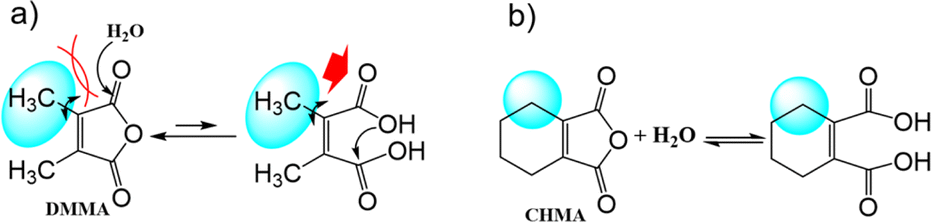 | ||
| Fig. 4 Structural differences of (a) DMMA, and (b) CHMA. | ||
To understand the distribution of species in the CHMA-H2O2 reaction mixture, which is crucial for its catalytic activity, we investigated the molar fractions of anhydride, diacid, and peracid present at equilibrium. While the 1H NMR spectrum of the mixture cannot differentiate the protons of the anhydride, peracid, and diacid, the 13C NMR spectrum (Fig. 5) fortunately allows for clear distinction of the aliphatic carbons ‘a’ and ‘b’ of each species. For similarly connected molecules such as diastereomers, 13C NMR integration with standard broadband decoupling (BBD) closely matches peak areas obtained from 1H NMR, within 3.4%.51 This study proposes that quantification using 13C NMR integration values of carbon “a” and “b” in CHMA derivatives can be correlated well with the actual amounts determined by 1H NMR data. For quantification, we preferentially chose carbon ‘b’ due to its distance from the carbonyl group, which minimizes interference with the integration value caused by relaxation effects.
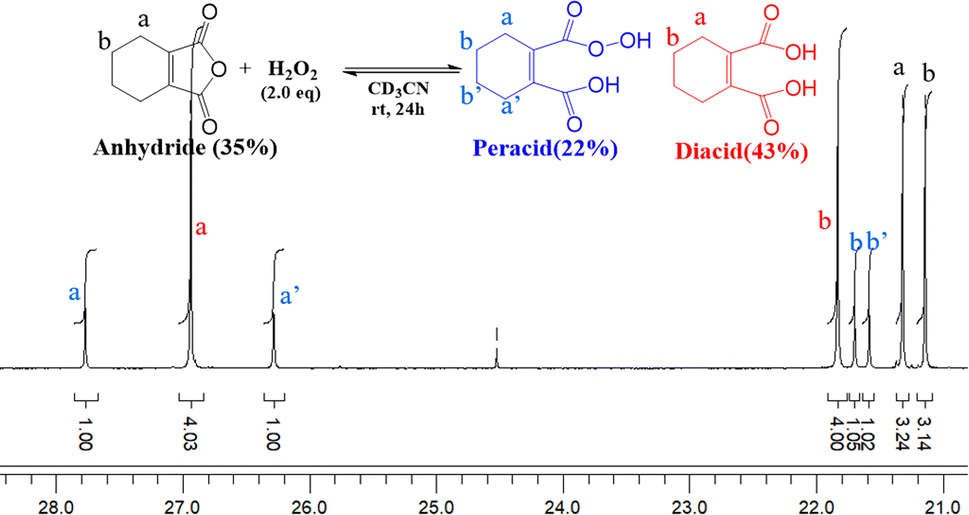 | ||
| Fig. 5 13C NMR spectrum of CHMA in H2O2 and CD3CN after 24 h at room temperature. | ||
A homogeneous solution containing CHMA (1.0 mmol) and H2O2 (2.0 mmol) in deuterated acetonitrile (CD3CN) (1 ml) was analyzed using 13C NMR spectroscopy. 13C NMR analysis of the reaction mixture after incubation for 1 hour at room temperature revealed the formation of approximately 38% peracid and 6% diacid, with the remaining 56% being the starting CHMA (Fig. 6). After 48 hours, the reaction reaches equilibrium, with the relative amounts of anhydride (31%), diacid (49%), and peracid (20%) indicating a dynamic interplay between these species. In the initial stages, CHMA likely undergoes perhydrolysis with H2O2 rather than hydrolysis due to its higher nucleophilicity compared to water. Interestingly, at equilibrium, the diacid concentration is higher than the peracid concentration. This suggests a faster conversion of peracid back to anhydride compared to the conversion rate of diacid back to anhydride. Maintaining the high presence of anhydride at equilibrium (around 31%) likely stems from its continuous regeneration via the equilibrium with the diacid and peracid forms. This continuous regeneration cycle is proposed to be essential for CHMA's effectiveness as an N-oxidation catalyst. It is noteworthy that requiring more than 24 hours to reach complete equilibrium indicates a very slow conversion of diacid to anhydride. Therefore, to achieve fast oxidation, the reaction temperature should be elevated to accelerate the anhydride regeneration rate, independent of the substrate's reactivity.
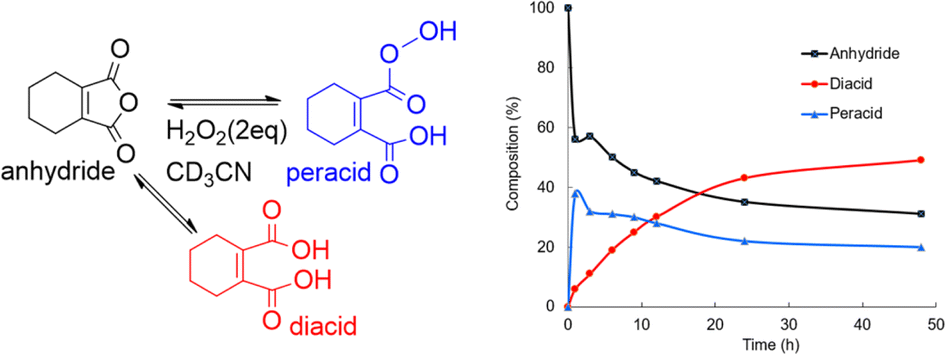 | ||
| Fig. 6 Composition change in the perhydrolysis of CHMA in CD3CN–H2O2 at room temperature. | ||
Perhydrolysis of DMMA yielded only 1.2% of diacid and no detectable peracid according to 13C NMR analysis (Fig. 7, and ESI, SI-3C†). This suggests a strong preference for the anhydride form in equilibrium with H2O2. The equilibrium constant (Keq) of DMMA in a solution of H2O2 (2.0 eq., 30%) and CD3CN is 84, significantly higher compare to DMMA’s value in aqueous solution (Keq = 5.3).50 In addition to the absence of peracid accumulation, the fast regeneration of anhydride can be seen in Fig. 7. The rapid anhydride regeneration favors the reaction with highly reactive substrates such as quinoline because the resulting peracid can react with substrate before it converts back to anhydride. Conversely, for oxidizing less reactive substrate like CP, some amount of peracid accumulation, as seen in CHMA, is likely crucial.
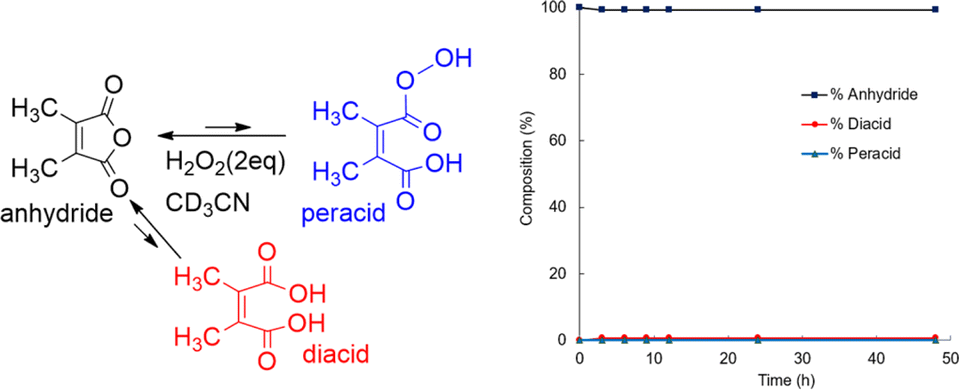 | ||
| Fig. 7 Composition change in the perhydrolysis of DMMA in CD3CN–H2O2 at room temperature. | ||
To understand the mechanistic differences in CHMA's oxidation activity towards quinoline and CP, the reaction mixtures were monitored by 13C NMR spectroscopy at room temperature (Fig. 8). The analysis revealed a clear trend, consistent with quinoline being significantly more reactive than CP. This is evident from the higher N-oxide yield for quinoline (76%) compared to CP (17%). Furthermore, the 13C NMR spectra showed a gradual depletion of the anhydride intermediate in the CP reaction, while it was rapidly consumed in the quinoline reaction. Additionally, the peracid intermediate observed during CP oxidation is not detectable in quinoline oxidation. This suggests that the highly reactive quinoline rapidly consumes the peracid oxidant, preventing its significant accumulation. Based on these observations, the rate-limiting step for quinoline oxidation is likely the formation of anhydride (k3), while for CP, it's the N-oxidation step (k2) (Fig. 9).
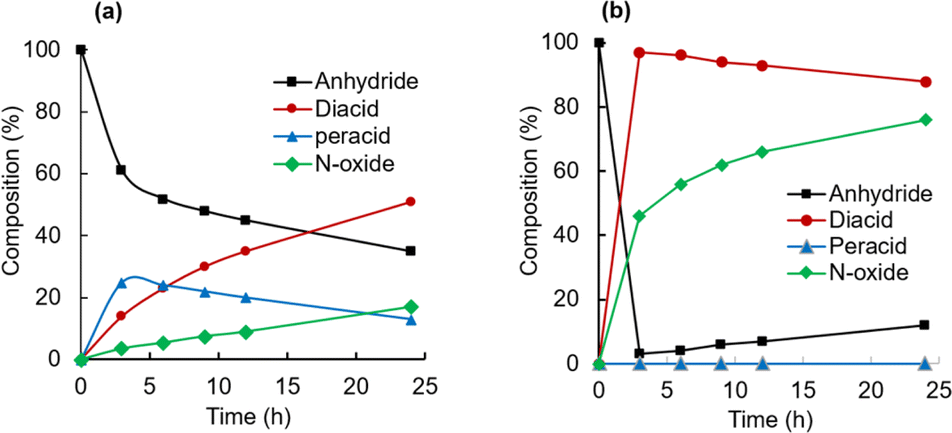 | ||
| Fig. 8 Composition changes during N-oxidation of CP (a) and quinoline (b) by CHMA (1.0 eq.) and H2O2 (2.0 eq.) in CD3CN at room temperature. | ||
 | ||
| Fig. 9 Catalytic cycle of CHMA in N-oxidation. | ||
3.3 Scope of application of DMMA and CHMA
DMMA and CHMA are effective catalysts for the N-oxidation of reactive substrates such as quinoline, even at room temperature, as demonstrated in Table 1 and Fig. 8. However, for less reactive substrates, the reaction temperature must be increased to accelerate the reaction rate. To compare the catalytic performance of DMMA and CHMA in the N-oxidation of various pyridine derivatives, reactions were conducted at 80 °C. The required reaction time for complete N-oxidation and the conversion yield after a 14 hours reaction (for slow reactions) are summarized in Table 2.| Entry | Substrate | Conversionb (%) (reaction time, h) | |
|---|---|---|---|
| DMMA | CHMA | ||
| a Reaction condition: substrate (1 mmol); anhydride (0.05 eq.); H2O2 (2.0 eq., 30%) at 80 °C.b Conversions (%) determined by 1H NMR of the crude reaction mixtures.c Isolated yield (%). | |||
| 1 |  |
100 (3) | 100 (5) |
| 2 |  |
100 (5) | 100 (12) |
| 3 | 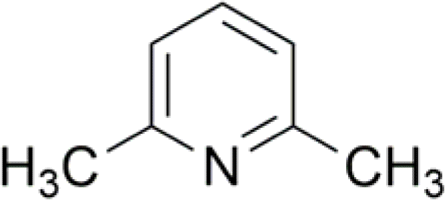 |
97c (14) | 88 (14) |
| 4 | 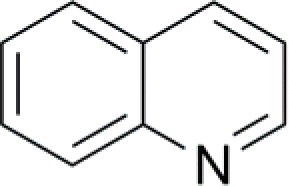 |
100 (3) | 98c (3) |
| 5 | 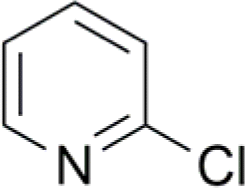 |
37 (14) | 96c (5) |
| 6 | 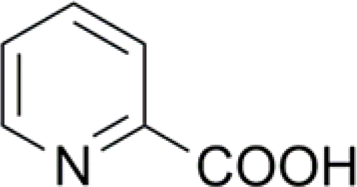 |
98 (14) | 100 (2) |
| 7 | 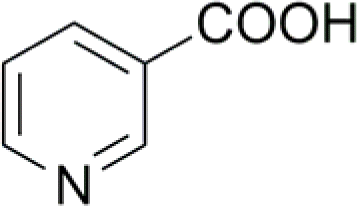 |
80 (14) | 100 (7) |
| 8 | 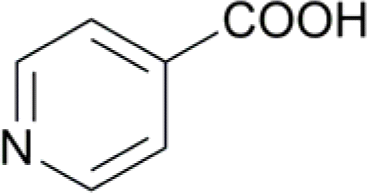 |
13 (14) | 92 (14) |
| 9 | 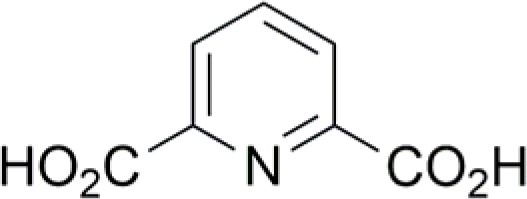 |
0 (14) | 4 (14) |
| 10 | 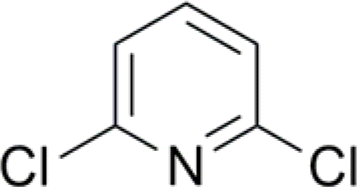 |
0 (14) | 0 (14) |
Both DMMA and CHMA effectively catalyze the N-oxidation of reactive substrates (entries 1–4). Steric hindrance around the nitrogen atom, as observed in entry 3, slows down the reaction rate. For electron-deficient substrates, CHMA outperforms DMMA. CHMA is generally effective for both electron-rich and electron-poor substrates (entries 1–8), except for highly deactivated compounds (entries 9 and 10). Interestingly, 2-picolinic acid (entry 6) exhibits exceptionally high reactivity compared to entries 7 and 8, despite being an electron-poor substrate. This enhanced reactivity is likely attributed to hydrogen bonding between the ortho-carboxyl group of 2-picolinic acid and the peracid. The resulting proximity of the peracid to the nitrogen atom, facilitated by this interaction, promotes the reaction. Based on these results, it is postulated that DMMA is superior to CHMA for reactive substrates, while CHMA is a better choice for less reactive electron-deficient substrates. The catalytic performance of DMMA and CHMA is influenced by both substrate nucleophilicity and steric hindrance.
4 Conclusions
Among the maleic anhydride derivatives, DMMA and CHMA exhibit excellent catalytic activity in pyridine N-oxidation using H2O2. The rapid regeneration of the anhydride from diacid facilitates the catalytic cycle, as anhydride is readily available for conversion to the peracid. DMMA shows good catalytic activity for the N-oxidation of reactive substrates like pyridine, 2-methyl pyridine, and quinoline; whereas CHMA is efficient in oxidizing deactivated pyridine substrates such as CP.This catalytic system, employing a catalytic amount of anhydride with H2O2 as the oxidant, offers a promising alternative to stoichiometric percarboxylic acid oxidants such as m-chloroperoxybenzoic acid (mCPBA). This advantage extends to a broad range of organic substrate oxidations, including olefin epoxidation, sulfide oxidation, Baeyer–Villiger oxidation, etc. A mechanistic study of the vastly different catalytic performances of DMMA and CHMA offered valuable insights for selecting suitable anhydride catalysts in other oxidation reactions.
To achieve optimal catalytic performance and promote green chemistry principles in H2O2 oxidation, further exploration of two promising avenues is recommended: structural modification of anhydride molecules and development of recyclable maleic anhydride catalysts through immobilization.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) under grant number RS-2024-00348663, National Research Foundation of Korea (NRF) grant funded by the Ministry of Education (MOE) (2023RIS-008) and Gunsan City, Korea, under the Human Resources Program for the EV industrial cluster.References
- H. Targhan, P. Evans and K. Bahrami, J. Ind. Eng. Chem., 2021, 104, 295–332 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- J. Kraïem, D. Ghedira and T. Ollevier, Green Chem., 2016, 18, 4859–4864 RSC.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986, 10.1039/b303160h.
- W. R. Sanderson, Pure Appl. Chem., 2000, 72, 1289–1304 CrossRef CAS.
- M. V. Doble, A. C. Ward, P. J. Deuss, A. G. Jarvis and P. C. Kamer, Bioorg. Med. Chem., 2014, 22, 5657–5677 CrossRef CAS.
- B. Martin, J. Sedelmeier, A. Bouisseau, P. Fernandez-Rodriguez, J. Haber, F. Kleinbeck, S. Kamptmann, F. Susanne, P. Hoehn, M. Lanz, L. Pellegatti, F. Venturoni, J. Robertson, M. C. Willis and B. Schenkel, Green Chem., 2017, 19, 1439–1448 RSC.
- M. Alem, S. Kazemi, A. Teimouri and H. Salavati, Asian J. Green Chem., 2019, 3, 366–381 Search PubMed.
- Y. Ding, W. Zhao, W. Song, Z. Zhang and B. Ma, Green Chem., 2011, 13(6), 1486–1489 RSC.
- Y. Kon, J. Jpn. Pet. Inst., 2017, 60, 159–169 CrossRef CAS.
- N. Mizuno and K. Kamata, Coord. Chem. Rev., 2011, 255, 2358–2370 CrossRef.
- F. Sadri, A. Ramazani, A. Massoudi, M. Khoobi, R. Tarasi, A. Shafiee, V. Azizkhani, L. Dolatyari and S. W. Joo, Green Chem. Lett. Rev., 2014, 7, 257–264 CrossRef.
- W. Xie, Y. Zheng, S. Zhao, J. Yang, Y. Liu and P. Wu, Catal. Today, 2010, 157, 114–118 CrossRef.
- A. Rostami, Y. Navasi, D. Moradi and A. Ghorbani-Choghamarani, Catal. Commun., 2014, 43, 16–20 CrossRef.
- S. Rostamnia, B. Gholipour and H. Golchin Hosseini, Process Saf. Environ. Prot., 2016, 100, 74–79 CrossRef.
- F. Secci, A. Frongia and P. P. Piras, Tetrahedron Lett., 2014, 55, 603–605 CrossRef.
- Q. Wei, H. Fan, F. Qin, Q. Ma and W. Shen, Carbon, 2018, 133, 6–13 CrossRef CAS.
- E. T. Poursaitidis, P. L. Gkizis, I. Triandafillidi and C. G. Kokotos, Chem. Sci., 2024, 15, 1177–1203 RSC.
- A. Russo, C. De Fusco and A. Lattanzi, ChemCatChem, 2012, 4, 901–916 CrossRef CAS.
- K. A. Stingl and S. B. Tsogoeva, Tetrahedron: Asymmetry, 2010, 21, 1055–1074 CrossRef CAS.
- I. Triandafillidi, M. G. Kokotou, D. Lotter, C. Sparr and C. G. Kokotos, Chem. Sci., 2021, 12, 10191–10196 RSC.
- E. T. Poursaitidis, F. Trigka, C. Mantzourani, M. G. Kokotou, I. Triandafillidi and C. G. Kokotos, Eur. J. Org Chem., 2024, 27, e202400082 CrossRef CAS.
- K. Bergstad and J.-E. Bäckvall, J. Org. Chem., 1998, 63, 6650–6655 CrossRef CAS.
- S. Zhang, G. Li, L. Li, X. Deng, G. Zhao, X. Cui and Z. Tang, ACS Catal., 2019, 10, 245–252 CrossRef.
- P. Ménová, F. Kafka, H. Dvořáková, S. Gunnoo, M. Šanda and R. Cibulka, Adv. Synth. Catal., 2011, 353, 865–870 CrossRef.
- J. Sturala, S. Bohacova, J. Chudoba, R. Metelkova and R. Cibulka, J. Org. Chem., 2015, 80, 2676–2699 CrossRef CAS PubMed.
- D. Wang, W. G. Shuler, C. J. Pierce and M. K. Hilinski, Org. Lett., 2016, 18, 3826–3829 CrossRef CAS PubMed.
- B. H. Brodsky and J. Du Bois, J. Am. Chem. Soc., 2005, 127, 15391–15393 CrossRef CAS.
- N. D. Litvinas, B. H. Brodsky and J. Du Bois, Angew. Chem., Int. Ed., 2009, 48, 4513–4516 CrossRef CAS PubMed.
- D. Limnios and C. G. Kokotos, J. Org. Chem., 2014, 79, 4270–4276 CrossRef CAS.
- D. Limnios and C. G. Kokotos, Chemistry, 2014, 20, 559–563 CrossRef CAS PubMed.
- C. J. Pierce and M. K. Hilinski, Org. Lett., 2014, 16, 6504–6507 CrossRef CAS PubMed.
- I. Triandafillidi, D. I. Tzaras and C. G. Kokotos, ChemCatChem, 2018, 10, 2521–2535 CrossRef CAS.
- S. Y. Hsieh, Y. Tang, S. Crotti, E. A. Stone and S. J. Miller, J. Am. Chem. Soc., 2019, 141, 18624–18629 CrossRef CAS.
- S. E. Huth, E. A. Stone, S. Crotti and S. J. Miller, J. Org. Chem., 2023, 88, 12857–12862 CrossRef CAS.
- M. Marigo, J. Franzen, T. B. Poulsen, W. Zhuang and K. A. Jorgensen, J. Am. Chem. Soc., 2005, 127, 6964–6965 CrossRef.
- M. Cai, K. Xu, Y. Li, Z. Nie, L. Zhang and S. Luo, J. Am. Chem. Soc., 2021, 143, 1078–1087 CrossRef.
- K. Bahrami, M. Khodaei and A. Karimi, Synthesis, 2008, 2008, 1682–1684 CrossRef.
- S. Caron, N. M. Do and J. E. Sieser, Tetrahedron Lett., 2000, 41, 2299–2302 CrossRef.
- B. Karami, M. Montazerozohori and M. H. Habibi, Molecules, 2005, 10, 1358–1363 CrossRef.
- I. Likhotvorik, M. Lutz and M. Wenzler, Synthesis, 2018, 50, 2231–2234 CrossRef.
- J. Liu, X.-Y. Wei, Y.-G. Wang, D.-D. Zhang, T.-M. Wang, J.-H. Lv, J. Gui, M. Qu and Z.-M. Zong, Fuel, 2015, 142, 268–273 CrossRef.
- P. Pietikäinen, J. Mol. Catal. A: Chem., 2001, 165, 73–79 CrossRef.
- G. Gajeles, S. M. Kim, J.-C. Yoo, K.-K. Lee and S. H. Lee, RSC Adv., 2020, 10, 9165–9171 RSC.
- L. Eberson and H. Welinder, J. Am. Chem. Soc., 1971, 93, 5821–5826 CrossRef.
- J. H. Walton and H. A. Lewis, J. Am. Chem. Soc., 2002, 38, 633–638 CrossRef.
- R. Williams, Bordwell pKa Table, https://organicchemistrydata.org/hansreich/resources/pka/pka_data/pka-compilation-williams.pdf, (accessed August 10, 2024) Search PubMed.
- M. Lõkov, S. Tshepelevitsh, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org Chem., 2017, 2017, 4475–4489 CrossRef.
- T. Higuchi, L. Eberson and J. D. McRae, J. Am. Chem. Soc., 1967, 89, 3001–3004 CrossRef.
- L. Eberson, J. Sundman, I. Toplin, A. Melera and L. Nilsson, Acta Chem. Scand., 1964, 18, 1276–1282 CrossRef.
- D. A. Otte, D. E. Borchmann, C. Lin, M. Weck and K. A. Woerpel, Org. Lett., 2014, 16, 1566–1569 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05962j |
| ‡ Physical Science Department, West Visayas State University, Luna St., La Paz, Iloilo City, 5000 Iloilo, Philippines is the authors present address. |
| This journal is © The Royal Society of Chemistry 2024 |