
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of carbonyl functionalized organotellurium(IV) derivatives†
Puspendra Singh *a,
Mariya Khanb,
Andrew Duthiec and
Ray J. Butcherd
*a,
Mariya Khanb,
Andrew Duthiec and
Ray J. Butcherd
aDepartment of Chemistry, Dr Shakuntala Misra National Rehabilitation University, Lucknow, 226017, India. E-mail: pushpendrasingh0612@gmail.com
bDepartment of Chemistry, University of Lucknow, Lucknow, 226007, India
cSchool of Life and Environmental Sciences, Deakin University, Geelong, 3217, Australia
dDepartment of Chemistry, Howard University, Washington DC20059, USA
First published on 8th November 2024
Abstract
The current study focuses on synthesis and characterization of carbonyl functionalized unsymmetrical diorganotellurium(IV) dichlorides (1–5), dibromide (6), and their characterization by elemental analysis, 1H, 13C{1H}, and 125Te NMR spectroscopy. In addition to this, compounds 1, 4 and 5 were further confirmed via single-crystal X-ray studies. Reduction of all the dichlorides with Na2S2O5 affords labile tellurides, which decompose quickly even at room temperature into the more stable symmetric ditellurides, Ar2Te2. Mesityl fragments bearing organotellurium(IV) derivatives show separate 1H and 13C{1H} NMR signals for the ortho methyls and meta protons of the mesityl ring. Among the Te(IV) dichlorides, the observed O–H⋯O, C–H⋯O, C–H⋯Cl, Te⋯O and Te⋯Cl hydrogen and secondary bonding interactions are longer than Σrcov (sum of the covalent bond radii) and significantly shorter than Σrvdw (sum of the van der Waal radii) respectively. The linearity of the C–Te⋯O, C–H⋯O and C–H⋯Cl makes n → σ* orbital interaction possible.
1 Introduction
The structure and reactivity of acetylacetone and benzoylacetone molecules is important in the creation of inter- and intramolecular hydrogen bonds in many fields of science,1–7 with the bonds playing an essential role in areas of chemistry ranging from biochemistry8 to crystal engineering,9 self-assembly of large pore zeolites10 and supramolecular chemistry11–14 to catalysis.15–19 The acetylacetone and benzoylacetone molecules can exist in keto and enol forms, generally the keto form is more thermodynamically stable than the enol form. Interestingly, it is observed that the enol form is predominant in acetylacetone and benzoylacetone, with percentages of 80.0 and 89.4% respectively.20 The following three main types of the more stable enol tautomer are present: (i) in which the enolic double bond is in conjugation with another double bond, (ii) a molecule bearing two or three bulky aryl groups, and (iii) a molecule bearing highly fluorinated enols (Chart 1).20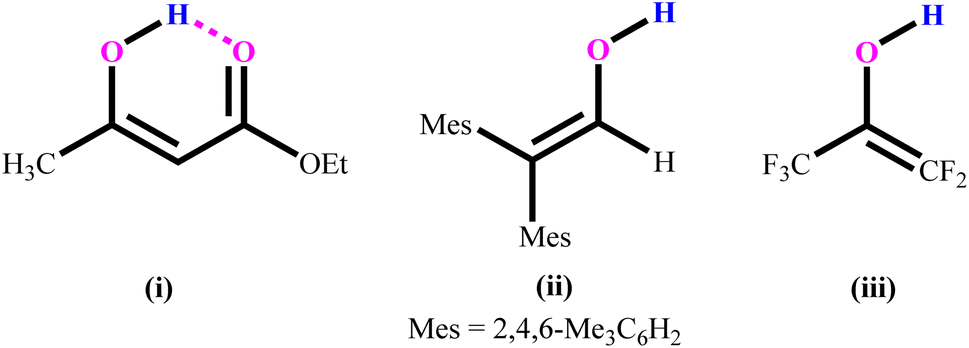 | ||
| Chart 1 Three main types of the more stable enols. | ||
Morgan and co-workers have reported chalcogen derivatives (A–G) of acetylacetone through the treatment of acetylacetone with SeCl4 and TeCl4 (Chart 2).21 Reaction of acetylacetone with SeCl4 can afford [(MeCO)2CSe]2 (A) as the major product. Reduction of A with hydroiodic acid can afford diselenide (B). Similarly, reaction of acetylacetone with TeCl4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in chloroform affords the labile acetylacetonyltellurium trichloride [CH3C(O)CH2C(O)CH2]TeCl3 (C), with further addition of one equivalent of acetylacetone affording a mixture of the three derivatives: [CH3C(O)CH2C(O)CH2]2TeCl2 (D),
:1) in chloroform affords the labile acetylacetonyltellurium trichloride [CH3C(O)CH2C(O)CH2]TeCl3 (C), with further addition of one equivalent of acetylacetone affording a mixture of the three derivatives: [CH3C(O)CH2C(O)CH2]2TeCl2 (D),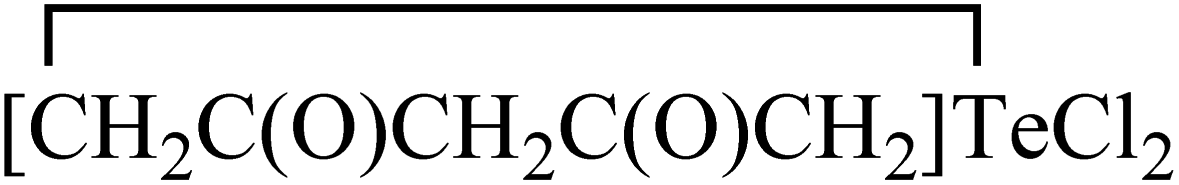 (E) and
(E) and  (F).
(F).
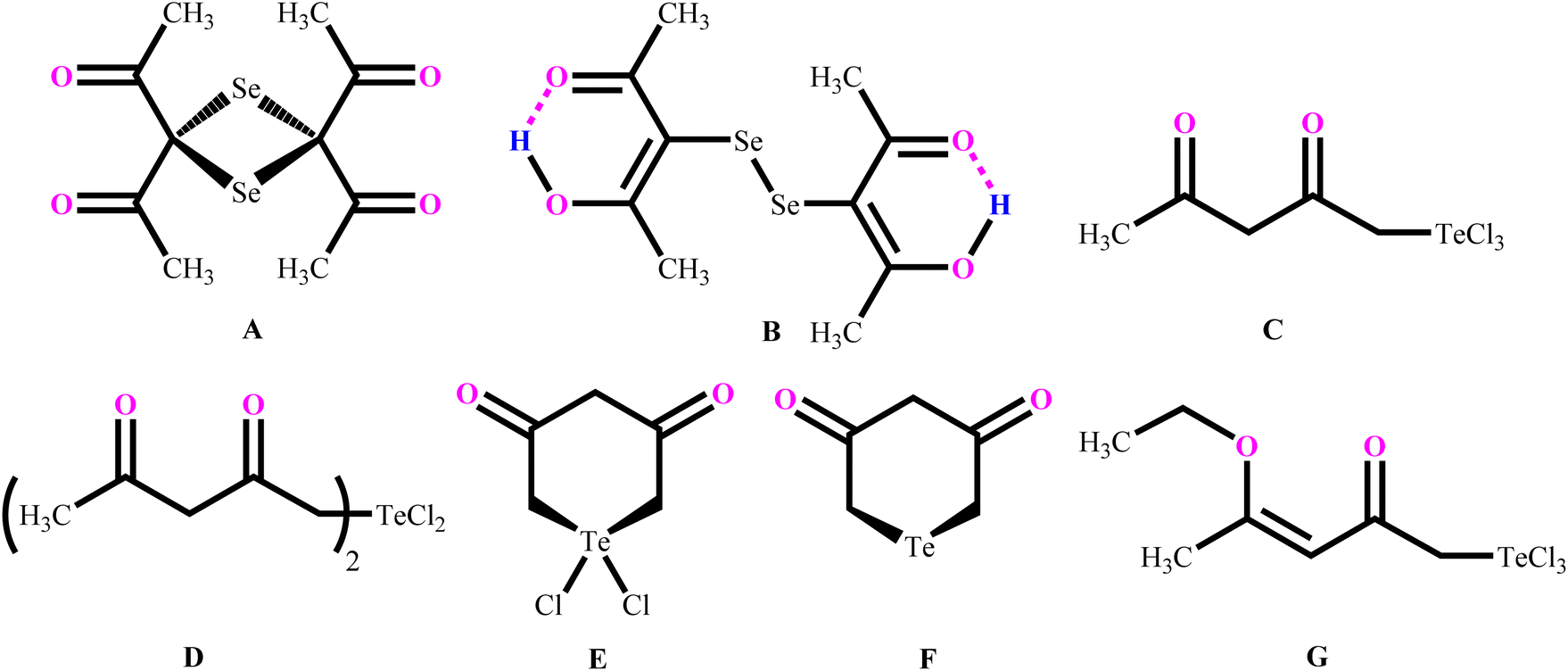 | ||
| Chart 2 Reported organoselenium and organotellurium derivatives of acetylacetone. | ||
Reaction of in situ generated molecule C with ethyl chloride can afford [CH3C(OEt)CHC(O)CH2]TeCl3 (G) in quantitative yield. Reaction of phenylstibonic acid with acetylacetone in the presence of HCl can afford trichloro(acetylacetonato)phenylantimony(V) as transparent needle-shaped crystals. Investigations with 1H-NMR and IR spectra revealed that only oxygen atoms of acetylacetone are covalently bonded with Sb atoms in the trichloro(acetylacetonato)phenylantimony(V).22
Similarly, methyl fragments bearing ketones can undergo electrophilic substitution reactions with aryltellurium trichlorides under mild conditions to obtain the aryl(acylmethyl)tellurium dichlorides Ar[RC(O)CH2]TeCl2 (Ar = phenyl, p-tolyl, p-anisyl, 1-naphthyl, mesityl; R = Me, i-Pr, t-Bu, mesityl).23 In the present study, we create the carbonyl functionalized organotellurium(IV) derivatives, Ar[RC(OH)CHC(O)CH2]TeCl2 (1–5) [Ar = phenyl, (Ph); p-tolyl, (p-tol) 1-napthyl, (1-Nap); and mesityl (Mes); R = methyl, phenyl] through the treatment of acetylacetone or benzoylacetone with aryltellurium trichlorides under mild conditions. In addition, Mes[CH3C(OH)CHC(O)CH2]TeBr2 (6) are also prepared through the metathetical reaction of 5 with NaBr in chloroform at room temperature.
2 Result and discussion
Aryltellurium trichlorides react under mild conditions with acetylacetone/benzoylacetone to give proton responsive unsymmetrical diorganotellurium(IV) dichlorides, Ph[PhC(OH)CHC(O)CH2]TeCl2, (1); p-Tol[PhC(OH)CHC(O)CH2]TeCl2, (2); 1-Nap[PhC(OH)CHC(O)CH2]TeCl2, (3); Mes[PhC(OH)CHC(O)CH2]TeCl2, (4); and Mes[MeC(OH)CHC(O)CH2]TeCl2, (5) (Scheme 1). The aryltellurium trichlorides employed have two axial and one equatorial chlorine atoms along with the aryl fragment. Only the equatorial chlorine atom of the aryltellurium trichlorides participates in the reactions.23 We can conclude that aryl fragment have more trans directing effect than the chlorine atoms in the aryltellurium trichlorides. However, the reaction of 1-napthyltellurium triiodide with benzoylacetone did not take place, even at temperatures up to 60 °C. Similarly, decomposition takes place during reaction of 1-napthyltellurium tribromide, affording the bi-naphthyl product through aryl–aryl coupling.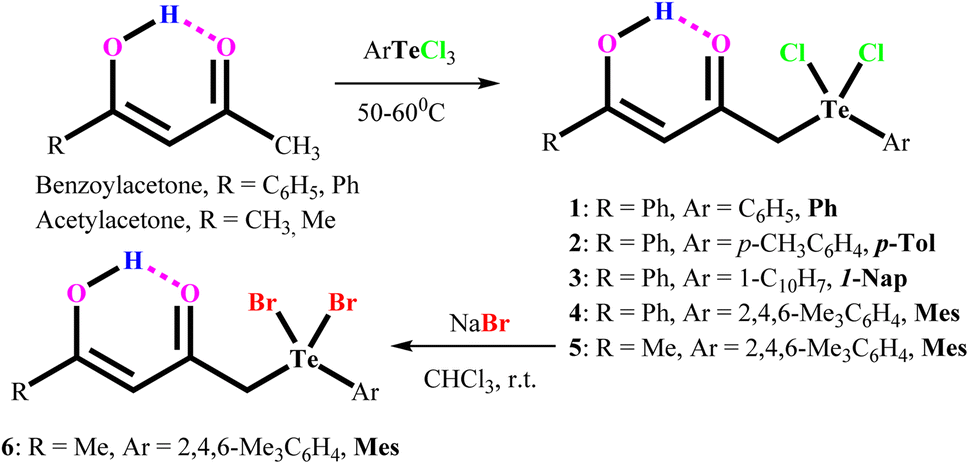 | ||
| Scheme 1 Development of carbonyl functionalized organotellurium(IV) derivatives. | ||
Plausible mechanism for aryl–aryl coupling are shown in Scheme 2. The first two steps are the formation of a precursor complex and the formation of bridged binuclear intermediate. Subsequently electron transfer through the bridging ligand to give the successor complex, followed by dissociation to 1-napthyl cation, 1-napthyl anion, TeBr4 and TeBr2. Finally, 1-napthyl cation and anion undergo aryl–aryl coupling to give bi-naphthyl product. Simultaneously, during dissociation process also afforded two equivalent of elemental Te and three equivalent of elemental Br2 through decomposition of in sittu generated TeBr4 and TeBr2.
 | ||
| Scheme 2 Plausible mechanism for aryl–aryl coupling. | ||
The dibromide Mes[PhC(OH)CHC(O)CH2]TeBr2, (6) can also be obtained with 95% yield from the corresponding dichlorides by metathesis with NaBr in chloroform at room temperature. Biphasic (H2O/CH2Cl2) reduction of dichlorides 1–5 with Na2S2O5 affords respective labile tellurides, which decompose quickly even at room temperature into the more stable symmetric ditellurides, Ar2Te2. The filtrate showed the presence of parent ketone (1H NMR).
All the proton responsive unsymmetrical diorganotellurium(IV) dihalides are crystalline solids that are soluble in common organic solvents. The 1H and 13C{1H} NMR spectra of the mesityltellurim(IV) derivatives are quite interesting. The restricted rotation of the mesityl fragment about the Te–C(mesityl) bond in 4, 5 and 6 is evidenced from their 1 H NMR spectra, which show separate signals for each of the meta ring protons and those of the two ortho methyl groups.23,24 All of the corresponding 13C{1H} NMR spectra consist of separate signals for each of the six ring and the two ortho methyl carbon atoms. The 1H chemical shifts for the CH2, CH and OH protons of the ketone fragments in 1–6 show singlets at ∼5, ∼6.22 and ∼15.12 ppm respectively.
The 125Te NMR of all the isolated diorganotellurium(IV) derivatives (1–6) show the presence of only one tellurium containing species in solution, as well as in the solid state. A single resonance signal suggests they are stabile in solution state. The 125Te chemical shifts for 5 and 6 in CDCl3 move 78 ppm upfield from Cl to Br as expected in terms of increasing shielding of the tellurium atom.
3 Crystal structure of carbonyl functionalized organotellurium(IV) derivatives 1, 4 and 5
Crystal data and structure refinement details for compounds 1, 4 and 5 are given in Table S1.† ORTEPS diagrams of 1, 4 and 5 are shown in Fig. 1, 2 and 3 respectively, each captioned with the selected interatomic distances and angles. Table S1† and packing diagrams of 1, 4 and 5 are presented in the ESI (Fig. S1–S10).† Compounds 1 and 4 crystallize in a monoclinic crystal system with the P21/c space group, while 5 crystallizes in a monoclinic system with the P21 space group. The primary geometry around the Te(IV) atom in these diorganotellurium dichlorides is ψ-trigonal bipyramidal with one equatorial position occupied by a stereochemically active lone pair. Interatomic Te⋯O(carbonyl) distances (d(Te⋯O) 2.861(1) Å in 1, 2.847(1) in 4 and 2.926(3) in 5) are longer than the Σrcov (Te,O) of 2.03 Å, and significantly shorter than Σrvdw (Te,O) of 3.58 Å, enough to imply the presence of attractive intramolecular 1,4-Te⋯O secondary bonding interactions.25 Moreover, the smaller Te–C–C(carbonyl) angle (104.66(9)° in 1, 105.37(9)° in 4 and 104.29(16)° in 5) compared to the tetrahedral angle indicate appreciable bending, consequentially Te(IV) and O atoms are attracted closer to each other. There are possible rotations about the Te–CH2 bonds, with the acetylacetone/benzoylacetone fragments in each case being oriented so that the carbonyl oxygen atoms are almost in the equatorial C–Te–C plane. Along with planarity, adjacent linearity of the C(trans)–Te⋯O triad(s) (155.05(5)° 1, 158.51(5)° in 4 and 156.82(8)° in 5) make n → σ* orbital interactions feasible. The observed intramolecular hydrogen bonding interaction in compound 5 (1.744(3) Å) is shorter than compounds 1 (1.824(28) Å) and 4 (1.854(25) Å), probably due to presence of an electron-donating methyl group on the hydroxyl carbon atom. The outstanding feature in all cases is a very short intramolecular O2–H⋯O1 hydrogen bond [d(O⋯O) in the range 2.498(4)–2.559(2) Å]. The downfield shifts of the δ(O–H) and δ(C–H) NMR signals are spectroscopic evidences of such strength. In the crystal structures the OH proton is observed to occupy a slightly asymmetric position, supporting all known solid state and solution spectroscopic data.26 Due to the presence of short strong intramolecular hydrogen bonding interactions, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC bonds in the adopted six membered rings, together with the carbonyl oxygen and hydroxyl atoms, almost lie in a plane.
O and CC bonds in the adopted six membered rings, together with the carbonyl oxygen and hydroxyl atoms, almost lie in a plane.
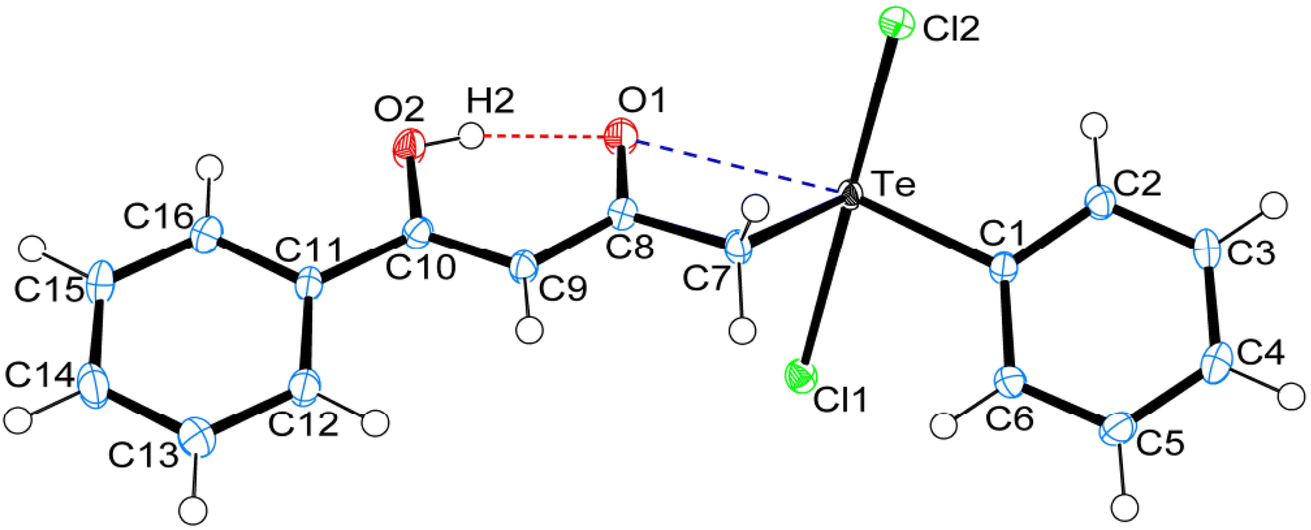 | ||
| Fig. 1 ORTEPs diagram showing 50% probability displacement ellipsoids and crystallographic numbering scheme for 1. Selected bond distances (Å) and angles (°): Te–C1 2.126(2), Te–C7 2.137(2), Te–Cl1 2.555(1), Te–Cl2 2.463(1), C8–O1 1.258(2), C10–O2 1.328(2), Te⋯O1, 2.861(1), O1⋯H2, 1.824(28), O2–H2, 0.783(29), O1⋯O2 2.559(2); C1–Te–C7 100.77(6), Te–C7–C8 104.66(9), Cl1–Te–Cl2 171.80(2), Te⋯O1⋯H2 165.45(85), O1⋯H2–O2 153.56(27), C7–Te⋯O1 54.34(4), C1–Te⋯O1 155.05(5), C10–O2⋯H2 104.46(2). | ||
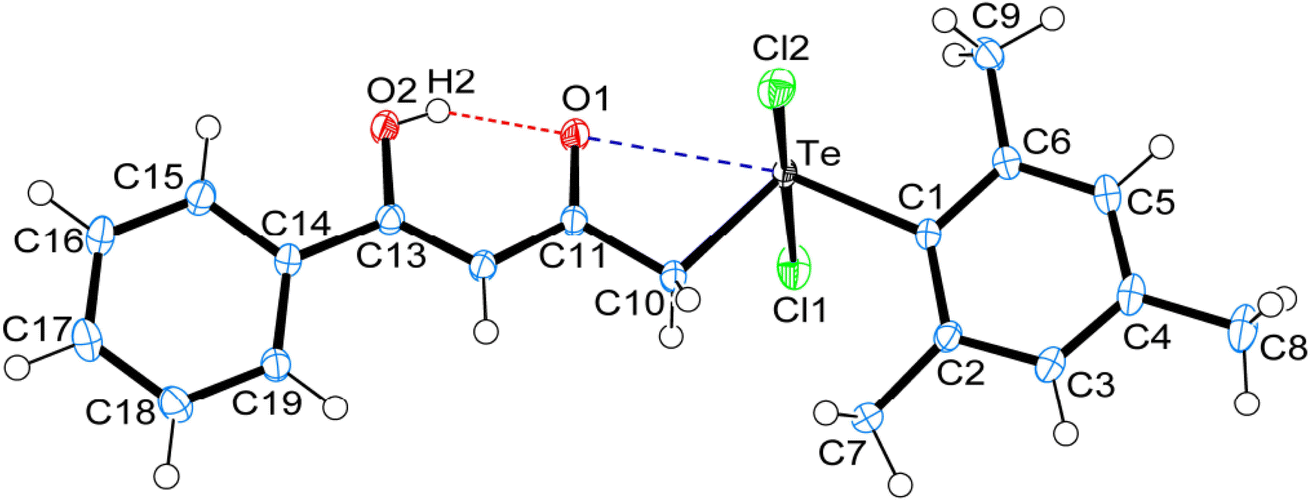 | ||
| Fig. 2 ORTEPs diagram showing 50% probability displacement ellipsoids and crystallographic numbering scheme for 4. Selected bond distances (Å) and angles (°): Te–C1 2.125(1), Te–C10 2.148(1), Te–Cl1 2.521(1), Te–Cl2 2.498(1), C11–O1 1.254(2), C13–O2 1.335(2), Te⋯O1, 2.847(1), O1⋯H2, 1.854(25), O2–H2, 0.756(25), O1⋯O2 2.541(2); C1–Te–C10 106.89(54), Te–C10–C11 105.37(9), Cl1–Te–Cl2 173.71(8), Te⋯O1⋯H2 174.65(78), O1⋯H2–O2 159.92(26), C10–Te⋯O1 54.62(4), C1–Te⋯O1 158.51(5), C13–O2⋯H2 107.92(2). | ||
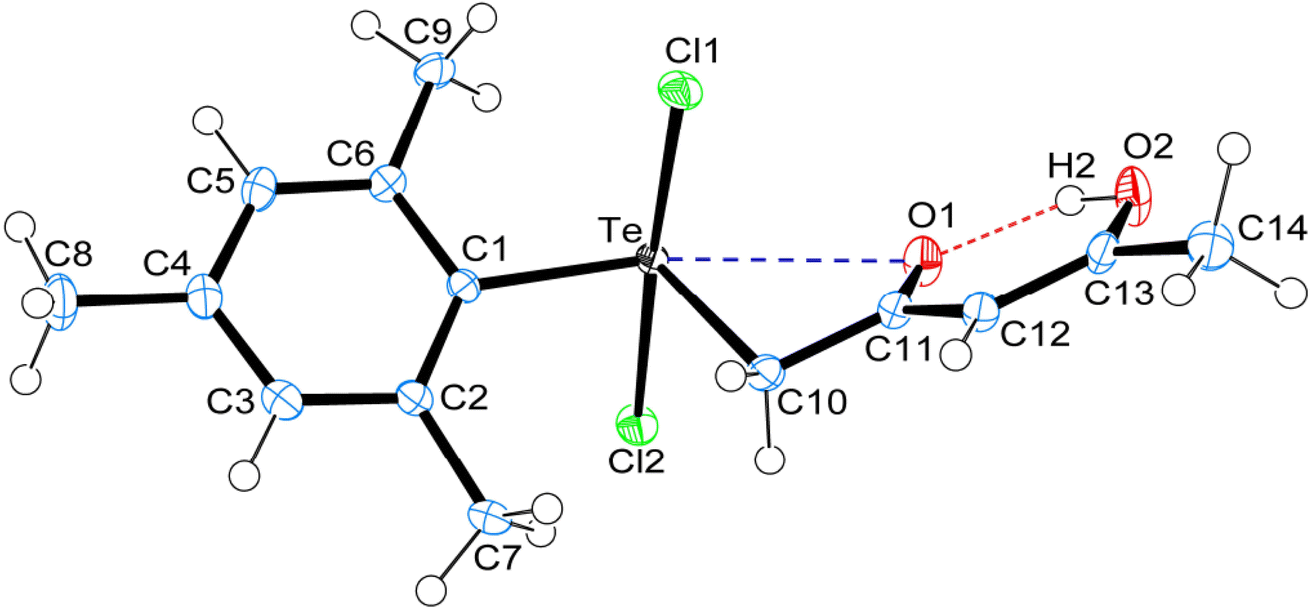 | ||
| Fig. 3 ORTEPs diagram showing 50% probability displacement ellipsoids and crystallographic numbering scheme for 5. Selected bond distances (Å) and angles (°): Te–C1 2.127(3), Te–C10 2.141(3), Te–Cl1 2.534(2), Te–Cl2 2.479(2), C11–O1 1.262(3), C13–O2 1.321(3), Te⋯O1, 2.926(3), O1⋯H2, 1.744(3), O2–H2, 0.840(3), O1⋯O2 2.498(4); C1–Te–C10 105.24(10), Te–C10–C11 104.29(16), Cl1–Te–Cl2 173.66(3), Te⋯O1⋯H2 153.18(12), O1⋯H2–O2 148.42(19), C10–Te⋯O1 52.97(8), C1–Te⋯O1 156.82(8), C13–O2⋯H2 109.44(2). | ||
3.1 Supramolecular architectures in the crystal lattices of compound 1, 4 and 5
In addition to the short strong intramolecular hydrogen bonding interactions (O⋯H) and Te⋯O secondary bonding interactions, the other intermolecular hydrogen bonding interactions (C–H⋯O, C–H⋯Cl) and secondary bonding interactions Te⋯Cl and π⋯π that have been recognized to play a vital role in the self-assembly of carbonyl functionalized organotellurium(IV) dichlorides. Parametric details of such interactions are depicted in Table S2.†The structure of 1 consists of a 2D helical structure running along the b-axis (Fig. S1†), with C–H⋯Cl interactions along the c-axis. Each Te(IV) atom is covalently bonded with two axial Cl atoms and two equatorial C-atoms. Simultaneously each Te(IV) atom is also interconnected with two SBIs [Te⋯O 2.861(1) Å and Te⋯Cl 3.477(1) Å] (Fig. S2†). The phenyl ring of benzylacetone fragment within each helical structure in 1 is glide such that the para-carbon atom of one molecule forms a π⋯π stacking interaction with the phenyl ring of benzylacetone fragment of a neighbouring molecule. The para-carbon⋯centroid distance between the phenyl ring of a neighbouring molecule in the structure of 1 is 3.652(2) Å (Fig. S3†). The crystal packing of 1 also consists of a centrosymmetric dimeric unit through C–H⋯Cl [2.861(1) Å] interactions repetition of these units gives rise to three-dimensional supramolecular architectures via self-assembly (Fig. S4†).27,28 The crystal structure of 4 consist of a centrosymmetric dimeric unit through reciprocal O–H⋯O [2.419(24) Å] intermolecular hydrogen bonding interaction (Fig. S5†). These dimeric units further connected with another dimeric unit through reciprocal C–H⋯Cl [2.902(0) Å] intermolecular hydrogen bonding interaction give rise to a 2D supramolecular architecture along c-axis (Fig. S6†). In the dimeric unit of 4 also consist of two strong intramolecular hydrogen bonding interaction C9–H9B⋯Cl2 [2.881(0) Å] and C7–H7B⋯Cl1 [2.956(0) Å]. Probably due to these interactions and steric effect of mesityl ring in compound 4 exhibits separate signals for both ortho methyl proton (2.74 & 2.79 ppm) and for both meta proton (6.43 & 6.97 ppm).
The crystal structure of 5 is devoid of Te based intermolecular SBIs. Steric demand of the mesityl group, acetylacetone fragment and small inter-atomic distance between Te and the axial Cl ligands make the Te atom inaccessible for intermolecular bonding. As a result, chlorine atoms are interconnected through the bifurcated and trifurcated C–H⋯Cl interactions, along with trifurcated C–H⋯O inter- and intra-connected hydrogen bonding interaction, giving rise to three-dimensional supramolecular packing (Fig. S7–S9†). Among all three Te(IV) dichlorides, the observed (O–H⋯O, ∼1.95 Å; C–H⋯O, ∼2.47 Å; C–H⋯Cl, ∼2.80 Å and Te⋯Cl, 3.45 Å) hydrogen bonding and SBIs are longer than Σrcov [(H,O), 0.97 Å; (H,Cl), 1.33 Å; (Te,Cl), 2.40 Å] and significantly shorter than [Σrvdw (H,O), 2.48 Å; (H,Cl), 2.81 Å; (Te,Cl), 3.83 Å] respectively.25 Linearity of the C–H⋯O and C–H⋯Cl make n → σ* orbital interaction possible. The four electron-three centre covalent bonding interaction and SBIs play a major role in the formation of supramolecular synthons.
4 Experimental
4.1. General
Preparative work was performed under dry nitrogen. Phenyl, p-tolyl, 1-naphthyl, and mesityl tellurium trichlorides were prepared by chlorination of their corresponding ditellurides. Commercial acetylacetone and benzoylacetone were dried with anhydrous Na2SO4 and freshly distilled under inert atmosphere. Melting points were recorded in capillary tubes and are uncorrected. The 1H, 13C{1H}, and 125Te NMR spectra were recorded on Bruker AMX 400, JEOL 400 or JEOL 300 spectrometers. Chemical shifts cited were referenced to TMS (1H, 13C{1H}) as internal and Me2Te (125Te) as external standard. Microanalyses were carried out using a Carlo Erba 1108 elemental analyzer. Tellurium was estimated volumetrically.4.2. Syntheses
(2) Prepared from p-tolyltellurium trichloride (0.65 g, 2.0 mmol) and benzoylacetone (0.34 g, 2.1 mmol) in a way similar to 1. Yield (0.34 g, 38%); mp 128 °C (from C6H6). (Found: C, 45.2; H, 3.6; Te, 28.1. C17H16Cl2O2Te requires C, 45.3; H, 3.6; Te, 28.3%); δH (300.1 MHz; CDCl3; Me4Si) 2.44 (3H, s, Me), 4.76 (2H, s, CH2), 6.34 (1H, s, CH), 7.38 (2H, d J 2.7 Hz, aryl), 7.41–7.50 (2H, m, aryl), 7.55–7.60 (1H, m, aryl), 7.87–7.91 (2H, m, aryl), 8.08–8.14 (2H, m, aryl), 15.31 (1H, br, OH); δC (100.5 MHz; CDCl3; Me4Si) 21.34 (Me), 61.85 (CH2), 96.61 (CH), 126.42, 127.24, 128.77, 130.78, 133.11, 133.14, 133.53, 142.66 (C–Ph), 181.70 (COH), 187.96 (CO); δTe (126.2 MHz; CDCl3; Me2Te) 863.8 (s).
(3) Prepared from 1-naphthyltellurium trichloride (0.72 g, 2.0 mmol) and benzoylacetone (0.34 g, 2.1 mmol) in a way similar to 1. Yield (0.81 g, 83%); mp 145 °C (from C6H6). (Found: C, 49.3; H, 3.4; Te, 26.1. C20H16Cl2O2Te requires C, 49.3; H, 3.3; Te, 26.2%); δH (300.1 MHz; CDCl3; Me4Si) 5.10 (2H, s, CH2), 6.46 (1H, s, CH), 7.49 (2H, t, J 1.9 Hz, aryl), 7.57–7.62 (1H, m, aryl), 7.66 (2H, t, J 1.9 Hz, aryl), 7.73 (1H, t, J 1.9 Hz, aryl), 7.93 (2H, d, J 2.0 Hz, aryl), 7.97 (1H, d, J 2.0 Hz, aryl), 8.08 (1H, d, J 2.1 Hz, aryl), 8.15 (1H, d, J 2.1 Hz, aryl), 8.21 (1H, d, J 1.8 Hz, aryl), 15.32 (1H, br, OH); δC (100.5 MHz; CDCl3; Me4Si) 62.57 (CH2), 96.23 (CH), 126.37, 126.70, 127.25, 128.17, 128.79, 130.70, 132.22, 132.69, 132.77, 133.23, 133.93, 134.22 (C-aryl), 181.05 (COH), 189.08 (CO); δTe (126.2 MHz; CDCl3; Me2Te) 779.5 (s).
(4) Prepared from mesityltellurium trichloride (0.72 g, 2.0 mmol) and benzoylacetone (0.34 g, 2.1 mmol) in a way similar to 1. Yield (0.33 g, 35%); mp 148 °C (from C6H6). (Found: C, 47.7; H, 4.3; Te, 26.7. C19H20Cl2O2Te requires C, 47.7; H, 4.2; Te, 26.6%); δH (300.1 MHz; CDCl3; Me4Si) 2.32 (3H, s, p-Me), 2.74 (3H, s, o-Me), 2.79 (3H, s, o-Me), 5.09 (2H, s, CH2), 6.43 (1H, s, CH), 6.97 (1H, s, aryl), 6.99 (1H, s, aryl), 7.48 (2H, t, J 1.9 Hz, aryl), 7.58 (1H, t, J 1.9 Hz, aryl), 7.91 (2H, d, J 1.9 Hz, aryl), 15.34 (1H, br, OH); δC (100.5 MHz; CDCl3; Me4Si) 21.01 (p-Me), 23.64 (Me), 23.76 (Me), 60.46 (CH2), 96.48 (CH), 127.30, 128.83, 130.41, 131.59, 133.01, 133.22, 135.47, 139.79, 141.00, 142.34 (C-Mes), 181.38 (COH), 189.40 (CO); δTe (126.2 MHz; CDCl3; Me2Te) 802.9 (s).
(5) Prepared from mesityltellurium trichloride (0.72 g, 2.0 mmol) and acetylacetone (1.5 mL, 15 mmol) in a way similar to 1. Yield (0.40 g, 48%); mp 145 °C (from C6H6). (Found: C, 40.2; H, 4.6; Te, 30.5. C14H18Cl2O2Te requires C, 40.3; H, 4.4; Te, 30.6%); δH (300.1 MHz; CDCl3; Me4Si) 2.14 (3H, s, p-Me), 2.32 (3H, s, Me), 2.70 (3H, s, o-Me), 2.80 (3H, s, o-Me), 4.95 (2H, s, CH2), 5.76 (1H, s, CH), 6.99 (1H, s, aryl), 7.03 (1H, s, aryl), 14.7 (1H, br, OH); δC (100.5 MHz; CDCl3; Me4Si) 21.00 (p-Me), 23.50 (o-Me), 23.60 (o-Me), 23.7 (Me), 60.10 (CH2), 100.10 (CH), 130.30, 131.50, 135.20, 139.80, 141.00, 142.30 (C-Mes), 188.10 (COH), 188.40 (CO); δTe (126.2 MHz; CDCl3; Me2Te) 801.0 (s).
4.3. Crystallography
Single crystals of 1, 4 and 5 suitable for X-ray crystallography were grown by slow evaporation of their C6H6 solutions at room temperature. ORTEPs and packing diagrams were generated with Ortep 3 for windows29 and DIAMOND 3.2 program respectively.30 Intensity data were collected on a Bruker SMART Apex CCD diffractometer at 100(2) K with graphite-monochromated Mo-Kα (0.7107 Å) radiation. The data was integrated with SAINT software.31 An experimental absorption modification was applied to the collected reflections with SADABS.32 The structure was confirmed by direct methods using SHELXTL and was refined on F2 by the full-matrix least-squares procedure using the program SHELXL-2018.33 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms attached to carbon were included in geometrically calculated positions using a riding model and were refined isotropically. Packing diagrams for the molecular structures of 1, 4 and 5 are shown in Fig. S1–S10 of the ESI.† Additionally, the 1H, 13C{1H} and 125Te NMR spectra are also included in the ESI (Fig. S11–S38).†5 Conclusions
Electrophilic substitution reaction of aryltellurium trichlorides, ArTeCl3 (Ar = C6H5, Ph; 4-Me-C6H4, p-Tol; 1-C10H7, 1-Nap; 2,4,6-Me3C6H2, Mes) with acetylacetone/benzoylacetone to give carbonyl functionalized unsymmetrical diorganotellurium(IV) dichlorides, Ph[PhC(OH)CHC(O)CH2]TeCl2 (1), p-Tol[PhC(OH)CHC(O)CH2]TeCl2 (2), 1-Nap[PhC(OH)CHC(O)CH2]TeCl2 (3), Mes[PhC(OH)CHC(O)CH2]TeCl2 (4) and Mes[MeC(OH)CHC(O)CH2]TeCl2 (5). Mes[PhC(OH)CHC(O)CH2]TeBr2 (6) can also be obtained with 95% yield from the corresponding dichlorides by metathesis with NaBr in chloroform at room temperature. Biphasic (H2O/CH2Cl2) reduction of all the dichlorides with Na2S2O5 affords labile tellurides, which decompose quickly even at room temperature into the more stable symmetric ditellurides, Ar2Te2. Mesityl fragments bearing organotellurium(IV) derivatives show separate 1H and 13C{1H} NMR signals for the ortho methyls and meta protons of the mesityl ring.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compounds 1, 4 & 5 has been deposited at the Cambridge Crystallographic Data Centre (CCDC) under accession numbers 2378222, 2378223 and 2378224 and can be obtained from “https://www.ccdc.cam.ac.uk/”.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
PS is heartily thankful to the Science and Engineering Research Board, New Delhi, India, for Teachers Associateship for Research Excellence Grant (Project No. TAR/2021/000075). MK and PS are particularly grateful to Dr R. C. Srivastava and Dr Ashok Kumar Singh Chauhan (Retired Professor of Department of Chemistry, University of Lucknow) for guidance and valuable suggestions. We are also thankful to Central Drug Research Institute Lucknow for recording analytical data.References
- G. S. Hammond, W. G. Borduin and G. A. Guter, J. Am. Chem. Soc., 1959, 81, 4682–4686 CrossRef CAS.
- M. S. Gordon and R. D. Koob, J. Am. Chem. Soc., 1973, 95, 5863–5867 CrossRef CAS.
- R. H. Holm and F. A. Cotton, J. Am. Chem. Soc., 1958, 80, 5658–5663 CrossRef CAS.
- B. Bock, K. Flatau, H. Junge, M. Kuhr and H. Musso, Angew. Chem., Int. Ed. Engl., 1971, 10, 225–235 CrossRef CAS.
- X.-B. Chen, W.-H. Fang and D. L. Phillips, J. Phys. Chem. A, 2006, 110, 4434–4441 CrossRef CAS PubMed.
- B. Schiùtt, B. B. Iversen, G. K. H. Madsen and T. C. Bruice, J. Am. Chem. Soc., 1998, 120, 12117–12124 CrossRef.
- H. Matsuzawa, T. Nakagaki and M. Iwahashi, J. Oleo Sci., 2007, 56, 653–658 CrossRef CAS PubMed.
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer, Berlin, 1991 Search PubMed.
- (a) C. B. Aakeroy, Acta Crystallogr., 1997, B53, 569–586 CrossRef CAS; (b) G. R. Desiraju, Chem. Commun., 1997, 1475–1482 RSC.
- (a) P. Y. Feng, X. H. Bu and G. D. Stucky, Nature, 1997, 388, 735–741 CrossRef CAS; (b) X. H. Bu, P. Y. Feng and G. D. Stucky, Science, 1997, 278, 2080–2085 CrossRef CAS PubMed.
- F. Huang and E. V. Anslyn, Chem. Rev., 2015, 115, 6999–7000 CrossRef CAS PubMed.
- B. J. Eckstein, L. C. Brown, B. C. Noll, M. P. Moghadasnia, G. J. Balaich and C. M. McGuirk, J. Am. Chem. Soc., 2021, 143, 20207–20215 CrossRef CAS PubMed.
- W. Yang, R. Jiang, C. Liu, B. Yu, X. Cai and H. Wang, Cryst. Growth Des., 2021, 21, 6497–6503 CrossRef CAS.
- E. R. T. Tiekink, Coord. Chem. Rev., 2022, 457, 214397 CrossRef CAS.
- S. Benz, J. Mareda, C. Besnard, N. Sakai and S. Matile, Chem. Sci., 2017, 8, 8164–8169 RSC.
- S. Benz, J. López-Andarias, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 812–815 CrossRef CAS PubMed.
- P. Wonner, L. Vogel, F. Kniep and S. M. Huber, Chem.–Eur. J., 2017, 23, 16972–16975 CrossRef CAS PubMed.
- P. Pale and V. Mamane, Chem.–Eur. J., 2023, 29, e202302755 CrossRef CAS PubMed.
- L. T. Maltz and F. P. Gabbaï, Organometallics, 2024, 43, 1246–1255 CrossRef CAS PubMed.
- M. B. Smith and J. March, Advanced Organic Chemistry Reaction, Mechanism and Structure, 6th edn, 2013, ch. 2, pp. 99–101 Search PubMed.
- (a) G. T. Morgan and H. D. K. Drew, J. Chem. Soc., 1920, 117, 1456–1465 RSC; (b) G. T. Morgan and H. D. K. Drew, J. Chem. Soc., 1921, 119, 610–625 RSC; (c) G. T. Morgan and H. D. K. Drew, J. Chem. Soc., 1922, 121, 922–940 RSC; (d) G. T. Morgan and H. G. Reeve, J. Chem. Soc., Trans., 1923, 123, 444–452 RSC; (e) G. T. Morgan and H. D. K. Drew, J. Chem. Soc., 1924, 125, 731–754 RSC; (f) G. T. Morgan and R. W. Thomason, J. Chem. Soc., Trans., 1924, 125, 754–759 RSC; (g) G. T. Morgan and E. Holmes, J. Chem. Soc., Trans., 1924, 125, 760–765 RSC; (h) G. T. Morgan and C. R. Porter, J. Chem. Soc., 1924, 125, 1269–1277 RSC; (i) G. T. Morgan and H. D. K. Drew, J. Chem. Soc., 1924, 125, 1601–1607 RSC; (j) G. T. Morgan and H. D. K. Drew, J. Chem. Soc., 1925, 127, 531–538 RSC; (k) G. T. Morgan, F. J. Corby, O. C. Elvins, E. Jones, R. E. Kellert and C. J. A. Taylor, J. Chem. Soc., 1925, 127, 2611–2625 RSC; (l) D. H. Dewar, J. E. Fergusson, P. R. Hentschel, C. J. Wilkins and P. P. William, J. Chem. Soc., 1964, 688–691 RSC.
- Y. Kawasaki and R. Okawara, Bull. Chem. Soc. Jpn., 1967, 40, 428 CrossRef CAS.
- (a) A. K. S. Chauhan, P. Singh, A. Kumar, R. C. Srivastava, R. J. Butcher and A. Duthie, Organometallics, 2007, 26, 1955–1959 CrossRef CAS; (b) A. K. S. Chauhan, P. Singh, R. C. Srivastava, A. Duthie and A. Voda, Dalton Trans., 2008, 4023–4028 RSC; (c) A. K. S. Chauhan, P. Singh, R. C. Srivastava, R. J. Butcher and A. Duthie, J. Organomet. Chem., 2010, 695, 2118–2125 CrossRef CAS.
- Y. Takaguchi, H. Fujihara and N. Furukawa, J. Organomet. Chem., 1995, 498, 49–52 CrossRef CAS.
- (a) O. Niyomura, S. Kato and S. Inagaki, J. Am. Chem. Soc., 2000, 122, 2132–2133 CrossRef CAS; (b) A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 1991, 113, 4917–4925 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- D. Braga and F. Grepioni, Acc. Chem. Res., 2000, 33, 601–608 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- K. Brandenburg and H. Putz, DIAMOND (v. 3.2e), Crystal Impact GbR, 1997-2012 Search PubMed.
- Bruker, SAINT, Bruker AXS Inc., Madison, 2016 Search PubMed.
- SADABS, Area Detector Absorption Correction Program, Bruker Analytical X-ray, Madison, 2018 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 1–3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Packing diagrams for the molecular structures of 1, 4 and 5 are shown in Fig. S1–S11 of the ESI. Additionally, the 1H, 13C{1H} and 125Te NMR spectra are also included in the ESI (Fig. S11–S38). The CIF files have also been deposited with the Cambridge Crystallographic Data Centre. CCDC 2378222 (1), 2378223 (4), and 2378224 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra06023g |
| This journal is © The Royal Society of Chemistry 2024 |