
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses of optically active monapterin, 7,8-dihydromonapterin, and 5,6,7,8-tetrahydromonapterin from L-xylose†
Arun K. Ghosh *ab,
Ashish Sharmaa,
Satish Nagama and
Clay Fuquac
*ab,
Ashish Sharmaa,
Satish Nagama and
Clay Fuquac
aDepartment of Chemistry, Purdue University, West Lafayette, IN 47907, USA. E-mail: akghosh@purdue.edu
bDepartment of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, IN 47907, USA
cDepartment of Biology, Indiana University, Bloomington, IN 47405, USA
First published on 8th November 2024
Abstract
We describe the syntheses of monapterin, dihydromonapterin and tetrahydromonapterin in optically active forms. The syntheses involved the condensation of L-xylose with phenylhydrazine, providing a hydrazone derivative. The reaction of the resulting hydrazone with triamino-pyrimidinone followed by oxidation of the resulting pteridinone with molecular oxygen furnished pterin containing a hydroxylated side chain. Hydrogenation of the pterin derivatives over RANEY® Ni catalyst afforded dihydromonapterin and tetrahydromonapterin in optically active forms. We also investigated an alternative route involving an Amadori rearrangement, followed by the Polonovski–Boon reaction as the key step to make these monapterin derivatives.
Introduction
Pterins are nitrogen-rich fused bicyclic biomolecules widely produced across all domains of life.1,2 They play critical roles in diverse biological processes, including cell signaling, modulation of the immune system, pigmentation and metabolism.3–5 Xanthopterin (1, Fig. 1) was first discovered in the late nineteenth century from the yellow and blue fluorescent pigments of butterfly wings. Later, xanthopterin was found in gastrointestinal endocrine cells.6,7 Pteridine rings are biosynthesized from guanosine triphosphate (GTP), and they are key building blocks for many biomolecules, including folic acid 2.8,9 Pterins with hydroxylated side chains exist in the fully oxidized form as well as partially reduced and fully reduced forms. In the human immune system, oxidized pterins have been demonstrated to modulate the oxidative burst of reactive oxygen and nitrogen species.10,11 Biopterin 3, a 6-(D-erythro-1,2-dihydroxypropyl) pterin, is a representative member of the family of biopterins. Biopterins exist in many different reduction states. They are endogenous enzyme cofactors for a family of aromatic amino acid hydroxylases involved in the synthesis of neurotransmitters, including dopamine, epinephrine, norepinephrine, and serotonin.12,13 Biopterin derivatives are also utilized as cofactors in the synthesis of nitric oxide.14 Monapterin, also known as neopterin 4, a 6-(D-erythro-1,2,3-trihydroxypropyl) pterin, is ubiquitous in insects, bacteria, and mammals. It is found in high levels, particularly in cerebrospinal fluid and plasma. Also, the level surges upon infection and during immune system activation.15,16 Neopterin is viewed as an early biomarker for diseases associated with the activation of cell-mediated immunity.17,18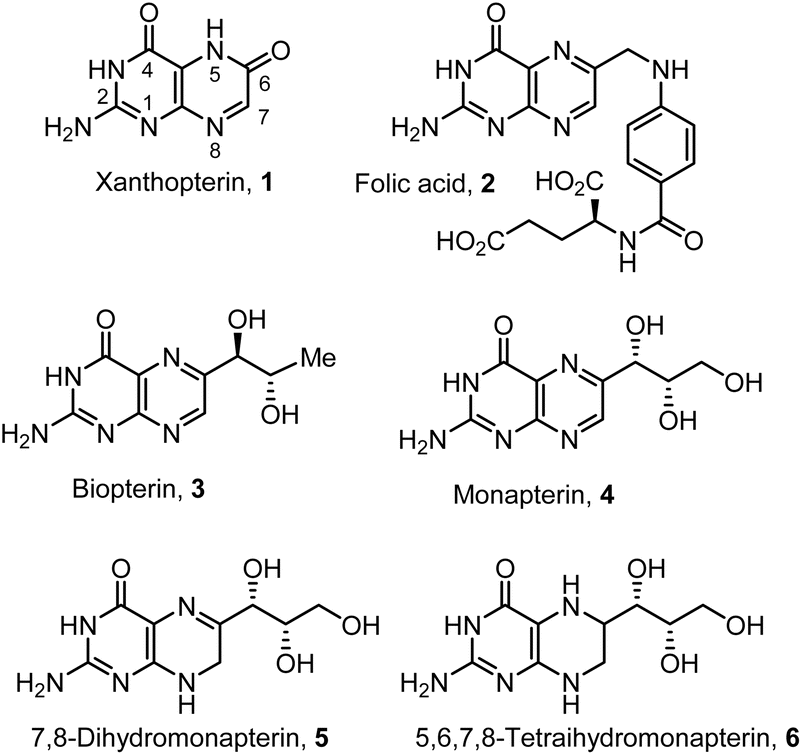 | ||
| Fig. 1 Structures of bioactive pterin derivatives 1–6. | ||
Pterins serve as critical cofactors in eukaryotic redox enzymes and in many bacterial systems.1,19 In the model pathogen Agrobacterium tumefaciens, the pteridine reductase PruA catalyzes the nicotinamide adenine dinucleotide phosphate hydrogen (NADPH)-dependent reduction of 7,8-dihydromonapterin 5 to 5,6,7,8-tetrahydromonapterin 6.20,21 Recently, we reported mechanistic insights into a complex regulatory system discovered in A. tumefaciens that controls biofilm formation depending upon excreted pterins.22 This pathway is conserved among a wide range of Gram-negative bacteria, including pathogens of animals and plants. These studies may lead to effective treatment modalities for biofilms.23,24 As part of our studies on the mechanism for biofilm regulation in A. tumefaciens, we required both dihydromonapterin and tetrahydromonapterin for our studies. Commercial supplies of specific hydroxylated pterins are very limited. Also, the practical syntheses as well as purification protocols of these very polar biomolecules have not been reported.25–27 Herein, we report the syntheses of monapterin, 7,8-dihydromonapterin, and 5,6,7,8-tetrahydromonapterin in optically active forms using commercially available L-xylose.
Results and discussion
The synthesis of monapterin is shown in Scheme 1. First, L-xylose phenyl hydrazone was prepared by the condensation of phenyl hydrazine and L-xylose in ethanol at 20 °C in the presence of a catalytic amount of acetic acid to provide 8 in a 99% yield. Interestingly, the treatment of L-xylose with phenyl hydrazine hydrochloride salt in ethanol at 23 °C for 3 h furnished (phenylhydrazinyl)pentan-2-one 9 in a 69% yield. Pterins are generally synthesized using the Viscontini reaction with amino pyrimidinone and sugar hydrazone.1,28 Therefore, commercially available 2,5,6-triaminopyrimidine-4(1H)-one sulfate 10 was treated with L-xylose phenyl hydrazone 8 in MeOH in the presence of 5 N HCI solution and a catalytic amount of mercaptoethanol at 23 °C. The resulting reaction mixture was heated at reflux for 3 h to provide a mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of tetrahydropyrano pteridine 11 and tetrahydrofurano pteridine 12 derivatives.28,29 Similarly, the reaction of (phenylhydrazinyl)pentan-2-one 9 with 2,5,6-triaminopyrimidine-4(1H)-one sulfate 10 provided the pteridine derivatives 11 and 12. These pteridine derivatives 11 and 12 are quite unstable as they are prone to autoxidation. Therefore, these compounds were not separated, and the mixture was dissolved in a pH 10 buffer and the resulting solution was stirred at 23 °C under a balloon filled with oxygen for 20 h. Acidification followed by evaporation of the solvents provided a crude product, which was recrystallized using a mixture (1:10) of methanol and ether to furnish monapterin 4 in a 66% isolated yield.
:1) of tetrahydropyrano pteridine 11 and tetrahydrofurano pteridine 12 derivatives.28,29 Similarly, the reaction of (phenylhydrazinyl)pentan-2-one 9 with 2,5,6-triaminopyrimidine-4(1H)-one sulfate 10 provided the pteridine derivatives 11 and 12. These pteridine derivatives 11 and 12 are quite unstable as they are prone to autoxidation. Therefore, these compounds were not separated, and the mixture was dissolved in a pH 10 buffer and the resulting solution was stirred at 23 °C under a balloon filled with oxygen for 20 h. Acidification followed by evaporation of the solvents provided a crude product, which was recrystallized using a mixture (1:10) of methanol and ether to furnish monapterin 4 in a 66% isolated yield.
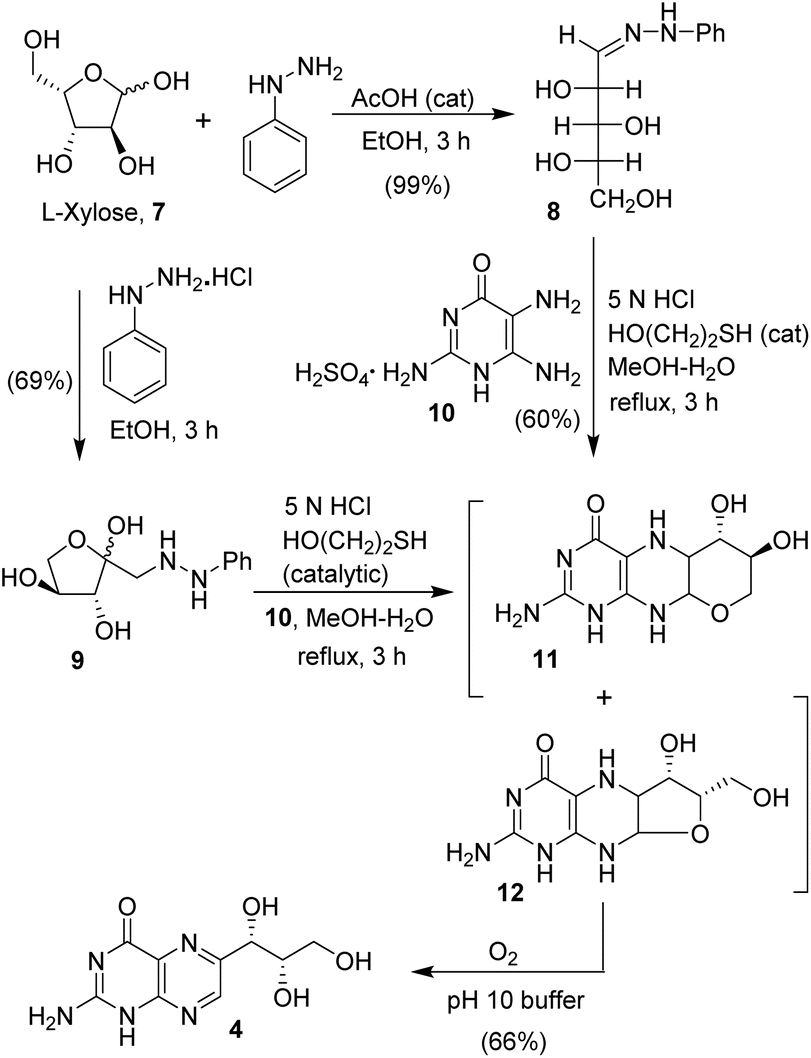 | ||
| Scheme 1 Synthesis of monapterin 4. | ||
Monapterin 4 was converted to dihydromonapterin 5 and tetrahydromonapterin 6 as shown in Scheme 2. We investigated the selective reduction of monapterin to dihydromonapterin under a variety of hydrogenation conditions, as summarized in Table 1. This hydrogenation requires careful monitoring otherwise the overreduction product tetrahydromonapterin 6 becomes a major by-product. The catalytic hydrogenation of monapterin 4 with Pearlman's catalyst (100% by weight) in NaH2PO4 buffer (pH 5.7) under a hydrogen-filled balloon at 23 °C for 2 h resulted in a mixture (30:70 by HPLC analysis) of dihydromonapterin 5 and fully reduced tetrahydromonapterin 6 in a 56% yield (entry 1). The hydrogenation of 4 with 10% Pd–C (100% by weight) in pH 5.7 buffer for 4 h improved the selectivity and the reaction mixture (70:30) showed more of the desired dihydromonapterin 5 in a combined yield of 63% (entry 2). Reducing the time to 1.3 h led to improvements in the yield and selectivity, providing a 95:5 mixture of 5 and 6 in a 79% yield (entry 3). We then examined hydrogenation over the RANEY® Ni catalyst. The hydrogenation of 4 over RANEY® Ni (W2, 100% by weight) in pH 5.7 buffer under a hydrogen-filled balloon at 23 °C for 2 h furnished dihydromonapterin 5 in a highly selective manner (98:2 ratio) in an 88% yield. Only a trace amount of the fully reduced tetrahydromonapterin 6 was formed during this reaction (entry 4). This optimized procedure was utilized for the synthesis of dihydromonapterin for our biological studies.22 The hydrogenation of 4 in EtOH after 2 h resulted in only a trace amount of product, and mainly unreacted monapterin remained (entry 5). We also investigated hydrogenation using PtO2 and 5% Rh/Al2O3 as the catalysts. In both cases, there were hardly any hydrogenation products, mostly the starting monapterin remained after the work up (entries 6 and 7). For the synthesis of tetrahydromonapterin 6, monapterin 4 was hydrogenated over Pd on C (100% by weight) in NaH2PO4 buffer (pH 5.7) at 23 °C for 8 h to furnish tetrahydromonapterin 6 in a 75% yield.
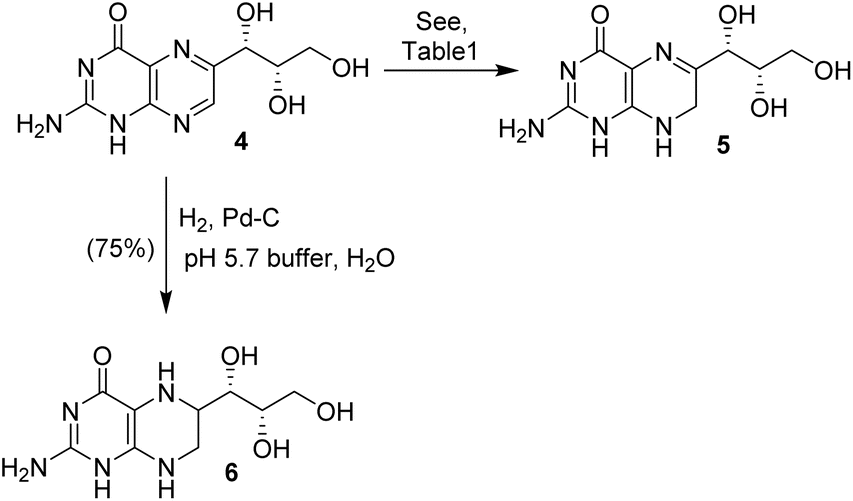 | ||
| Scheme 2 Syntheses of dihydromonapterin and tetrahydromonapterin. | ||
| Entry | Catalysts (wt%) | Solvent | Time (h) | Yieldb (%) | Ratioc (5:6) |
|---|---|---|---|---|---|
| a Reactions were carried out in a 0.1 mmol scale at 23 °C.b Combined yields after purification by HPLC.c Ratios determined by HPLC and LCMS analysis of the crude products. | |||||
| 1 | 20% Pd(OH)2 | pH 5.7 buffer | 2 | 56 | 30:70 |
| 2 | 10% Pd/C | pH 5.7 buffer | 4 | 63 | 70:30 |
| 3 | 10% Pd/C | pH 5.7 buffer | 1.3 | 79 | 95:5 |
| 4 | Ra–Ni (W2) (25%) | pH 5.7 buffer | 2 | 88 | 98:2 |
| 5 | Ra–Ni (W2) (125%) | EtOH | 2 | Trace | — |
| 6 | PtO2 (25%) | THF | 6 | Trace | — |
| 7 | 5% Rh/Al2O3 | EtOH | 9 | Trace | — |
| 8 | 10% Pd/C | pH 5.7 buffer | 8 | 75 | 1:99 |
We also explored an alternative route to monapterin and dihydromonapterin due to issues of overreduction. Andrews and co-workers reported the synthesis of biopterin using L-arabinose as the starting sugar.30 In a modified synthesis, as shown in Scheme 3, L-xylose was reacted with dibenzylamine in ethanol in the presence of an equivalent amount of acetic acid at 70 °C for 6 h. This condition resulted in the Amadori rearrangement product,31 α-ketobenzylamine derivative 13 in a 70% yield. The catalytic hydrogenolysis of ketoamine 13 with 10% Pd–C under a hydrogen-filled balloon in a mixture (4:1) of ethanol and 1 N HCl at 23 °C for 48 h furnished the corresponding amine in an excellent yield. The resulting amino ketone was condensed with 2-amino-6-chloro-5-nitropyrimidinone 14 in ethanol at 50 °C for 30 min. Then, the addition of NaHCO3 followed by heating the resulting mixture in a (3:1) mixture of ethanol and water at 60 °C for 1 h resulted in the nitro derivative 15 in a 42% yield over 2 steps. Exposure of the nitro derivative 15 to RANEY® Ni under a hydrogen-filled balloon in water at 23 °C for 2 h furnished 7,8-dihydromonapterin 4 in an 80% yield. The oxidation of dihydromonapterin with MnO2 in water for 3 h afforded monapterin 5 in a 60% yield. Overall, the route is straight forward and provided convenient access to dihydromonapterin 5 and monapterin 4 in good yields.
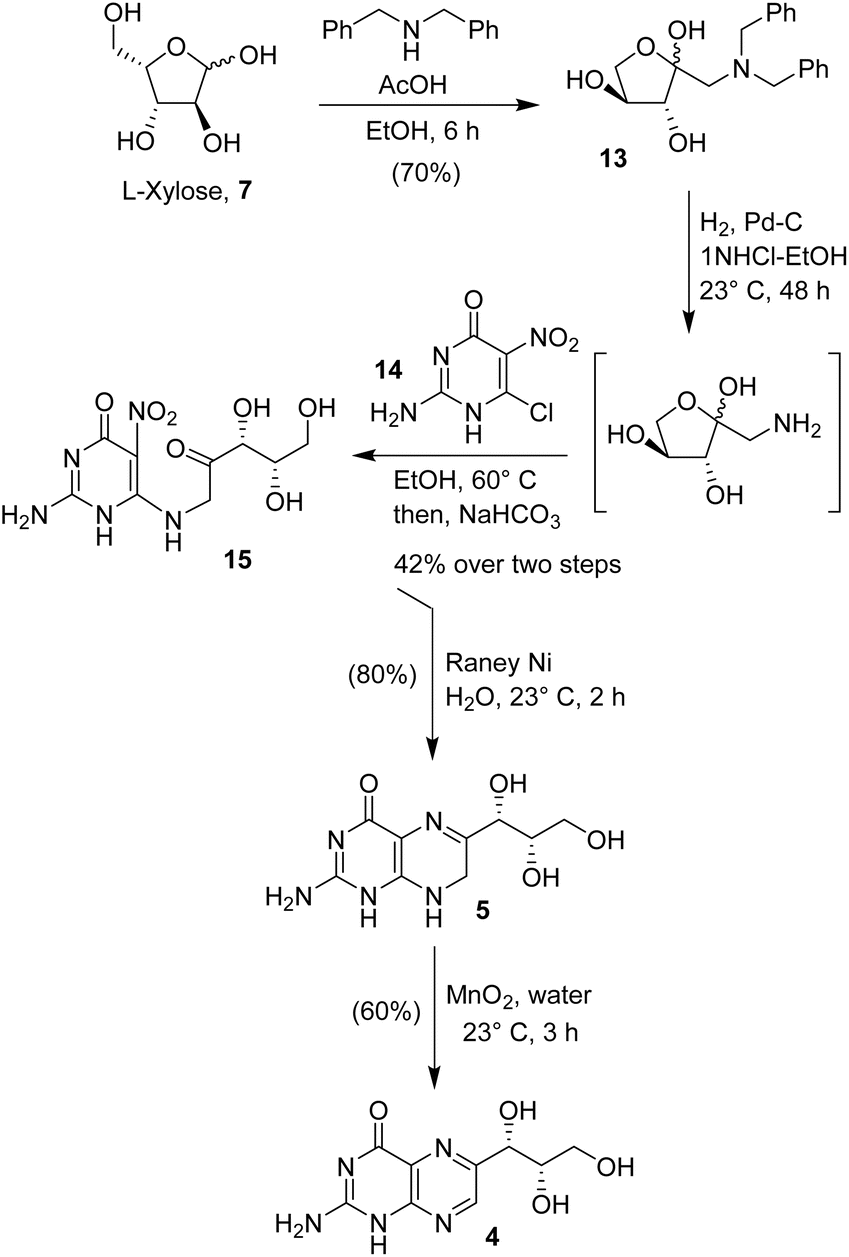 | ||
| Scheme 3 Syntheses of dihydromonapterin and monapterin. | ||
Conclusions
In summary, we describe the practical syntheses of monapterin, 7,8-dihydromonapterin, and 5,6,7,8-tetrahydromonapterin in optically active forms using commercially available L-xylose as the starting material. The synthesis of dihydromonapterin involved the selective hydrogenation of monapterin. We investigated a number of catalytic systems and solvents to obtain optimum results. Catalytic hydrogenation with RANEY® Ni in pH 5.7 buffer under a hydrogen-filled balloon provided the best results. The product dihydromonapterin is highly sensitive to air and it underwent spontaneous autooxidation to provide monapterin along with a small amount of decomposition product. We investigated the stability of dihydromonapterin in D2O under atmospheric conditions. It turns out that dihydromonapterin completely converted to monapterin within 24 h at 23 °C in D2O. The current synthetic route provides access to both dihydro- and tetrahydro monapterin for biological studies. We also developed an alternate route to avoid overreduction, as shown in Scheme 3, in which L-xylose was converted to the α-ketobenzylamine derivative 13 by using Amadori rearrangement followed by Polonovski–Boon cyclization32 to afford the nitro derivative 15. RANEY® Ni reduction of the nitro compound afforded dihydromonapterin, and then further oxidation with MnO2 gave monapterin in a more efficient manner. Further work involving monapterin is ongoing in our laboratories.Experimental section
All the reactions were carried out under an inert atmosphere, in either nitrogen or argon, using a magnetic stirrer and oven-dried glassware. All the solvents were anhydrous and distilled prior to use. Dichloromethane and triethylamine were distilled from calcium hydride. Tetrahydrofuran, diethyl ether, and benzene were distilled from sodium and benzophenone. All the other solvents were HPLC grade or better. Flash column chromatography was performed using EM Science 60–200 mesh silica gel. Thin-layer chromatography was performed using 60 F-254 E. Merck silica gel plates. 1H- and 13C-NMR were recorded using Bruker AV-500, Avance DRX-500, Varian Mercury-Vx-300, and Gemini-2300 spectrometers with Me4Si as an internal standard. Optical rotations were recorded on a PerkinElmer 341 polarimeter. A Thermo Finnigan LCQ Classic mass spectrometer was used for the MS analyses. The purity of test compounds was determined by HRMS and HPLC analyses. All the test compounds showed ≥95% purity.Experimental procedures
:1) MeOH and diluted with diethyl ether (15 mL). The resulting precipitate was collected by filtration and the filter cake was washed with diethyl ether to provide a brown solid. Further purification of the compound was carried out using Pre HPLC-Agilent 1260 infinity (YMC PAK ODS-A C18, 2% ACN/H2O, 10 mL min−1, tr: 6.175, conc. 10 mg/1 mL of H2O) to provide the desired compound monapterin (10 mg, 60%). [α]D20 = +125.8 (c 0.5, DMSO). 1H NMR (500 MHz, DMSO-d6) δ 11.4 (br, 1H), 8.73 (s, 1H), 6.99–6.65 (br, 2H), 5.40 (s, 1H), 4.72 (s, 1H), 4.65–4.53 (m, 2H), 3.64 (m, 1H), 3.55–3.46 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ 164.8, 157.1, 157.0, 151.8, 148.5, 127.7, 74.6, 72.6, 62.9. LRMS ESI (m/z): 254.0 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C9H12N5O4Na, 276.07032; found 276.07062.:44) mixture of diastereomeric compounds based up on integration of the data from 13C NMR (4.5 mg, 75%). 1H NMR (800 MHz, D2O) δ 4.0 (m, 1H), 3.81 (m, 1H), 3.75 (m, 1H), 3.73–3.69 (m, 3H), 3.60–3.56 (m, 7H), 3.49–3.43 (m, 2H). 13C NMR (200 MHz, D2O) δ 165.6, 158.6, 158.3, 154.4, 153.8, 153.6, 85.7, 84.8, 71.2, 71.1, 67.5, 66.4, 61.9, 61.8, 54.3, 53.3, 38.9, 37.8. LRMS-ESI (m/z): 258 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C9H15N5O4H, 258.1202; found 258.1204.The above 2-amino-6-chloro-5-nitropyrimidin-4(3H)-one (100 mg, 0.5 mmol) was dissolved in a mixture of solvents comprising 2.0 mL of EtOH and 0.5 mL of H2O and the reaction mixture was heated at 50 °C for the period of 15 min. After 15 min, the above amine solution was added to the reaction mixture and the reaction mixture was further stirred for 5 min. The reaction mixture was slowly basified by the dropwise addition of aqueous NaHCO3 (300 mg in 3 mL of water) for a period of 45 min and the reaction mixture was further stirred for a period of 1.0 h at 60 °C. The reaction mixture was filtered in a hot condition, and then the filtrate was evaporated off under reduced pressure to afford the crude reside. The crude residues were crystallized in an EtOH:H2O (2:1) mixture of solvents to afford a light-yellow brown solid compound (200 mg, crude solid). Further purification of the compound was carried out using a Pre HPLC-Agilent 1260 infinity system (YMC PAK ODS-A C18, 2% ACN/H2O, 10 mL min−1, tr: 19.411, conc. 15 mg/1 mL of H2O) to provide compound 15 (60 mg, 42%, over two steps) as a white amorphous solid, which existed as a mixture of anomers. [α]D20 = +19.3 (c 0.3, H2O): 1H NMR (800 MHz, DMSO) δ 9.80 (s, 1H), 9.71 (s, 1H), 9.59 (s, 1H), 8.55 (s, 1H), 8.00 (s, 1H), 5.96 (d, J = 16.6 Hz, 1H), 5.63 (d, J = 4.8 Hz, 1H), 5.42 (m, 1H), 5.27–5.14 (m, 2H), 4.87 (d, J = 26.0 Hz, 1H), 4.73–4.62 (m, 2H), 4.48 (dd, J = 20.7, 4.5 Hz, 1H), 4.18 (s, 1H), 4.10 (d, J = 5.4 Hz, 1H), 3.98–3.88 (m, 2H), 3.82–3.73 (m, 2H), 3.69 (dd, J = 13.6, 5.5 Hz, 1H), 3.61 (dd, J = 8.5, 5.5 Hz, 1H), 3.59 (d, J = 4.4 Hz, 1H), 3.56–3.49 (m, 2H), 3.45 (t, J = 9.4 Hz, 2H), 3.40 (dd, J = 8.9, 4.7 Hz, 1H); 13C NMR (201 MHz, DMSO) δ 209.9, 161.6, 159.7, 159.7, 159.5, 159.2, 111.3, 111.1, 110.9, 105.3, 102.2, 82.4, 78.7, 76.7, 76.4, 75.4, 73.2, 71.2, 70.7, 61.9, 50.4, 47.4, 45.8; LRMS-ESI (m/z): 304 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C9H13N5O7, 304.0893; found 304.0884.
Data availability
The authors confirm that the data supporting these studies are available within the article and its ESI.†Author contributions
A. K. Ghosh (writing original draft, review, editing, supervision, resources, methodology, funding acquisition, formal analysis); A. Sharma (methodology, investigation, data curation); S. Nagam (methodology, investigation, data curation); C. Fuqua (review, editing, formal analysis).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Institutes of Health (Grants AI150466 and GM120337). The authors would like to thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities.Notes and references
- N. Feirer and C. Fuqua, Pteridine, 2017, 28, 23–36 CrossRef CAS.
- T. Hanaya and H. Yamamoto, IUBMB Life, 2013, 65, 300–309 CrossRef CAS PubMed.
- P. Galland and H. Senger, Photochem. Photobiol., 1988, 48, 811–820 CrossRef CAS.
- C. A. Nichol, G. K. Smith and D. S. Duch, Annu. Rev. Biochem., 1985, 54, 729–764 CrossRef CAS PubMed.
- P. Andrade and M. Carneir, Biol. Lett., 2021, 17, 20210221 CrossRef CAS PubMed.
- J. Ferré, Insects, 2024, 15, 370 CrossRef PubMed.
- B. Wijnen, H. L. Leertouwer, D. G. Stavenga and J. Insect, Physiol, 2007, 53, 1206–1217 CAS.
- A. Kirschning, Angew. Chem., Int. Ed., 2021, 60, 6242–6269 CrossRef CAS PubMed.
- T. D. Ames, D. A. Rodionov, Z. Weinberg and R. R. Breaker, Chem. Biol., 2010, 17, 681–685 CrossRef CAS PubMed.
- C. Murr, B. Widner, B. Wirleitner and D. Fuchs, Curr. Drug Metab., 2002, 3, 175–187 CrossRef CAS PubMed.
- G. Weiss, D. Fuchs, A. Hausen, G. Reibnegger, E. R. Werner, G. Werner-Felmayer, E. Semenitz, M. P. Dierich and H. Wachter, FEBS Lett., 1993, 321, 89–92 CrossRef CAS PubMed.
- H. Fanet, L. Capuron, N. Castanon, F. Calon and S. Vancassel, Curr. Neuropharmacol., 2021, 19, 591–609 CAS.
- E. R. Werner, N. Blau and B. Thöny, Biochem. J., 2011, 438, 397–414 CrossRef CAS PubMed.
- N. S. Kwon, C. F. Nathan and D. J. Stuehr, J. Biol. Chem., 1989, 264, 20496–20501 CrossRef CAS PubMed.
- H. Wachter, D. Fuchs, A. Hausen, G. Reibnegger and E. R. Werner, Adv. Clin. Chem., 1989, 27, 81–141 CAS.
- A. Lindsay, G. Baxter-Parker and S. P. Gieseg, J. Clin. Med., 2019, 8, 1383 CrossRef CAS PubMed.
- D. Fuchs, P. Avanzas, R. Arroyo-Espliguero, M. Jenny, L. Consuegra-Sanchez and J. C. Kaski, Curr. Med. Chem., 2019, 16, 4644–4653 CrossRef PubMed.
- S. K. Pingle, R. G. Tumane and A. A. Jawade, Indian J. Occup. Environ. Med., 2008, 12, 107–111 CrossRef PubMed.
- A. Pribat, I. K. Blaby, A. Lara-Núnez, J. F. Gregory III, V. de Crécy-Lagard and A. D. Hanson, J. Bacteriol., 2010, 192, 475–482 CrossRef CAS PubMed.
- M. Labine, L. DePledge, N. Feirer, J. Greenwich, C. Fuqua and K. D. Allen, J. Bacteriol., 2020, 202, e00098 CrossRef CAS PubMed.
- K. L. Kavanagh, H. Jörnvall, B. Persson and U. Oppermann, Cell. Mol. Life Sci., 2008, 65, 3895–3906 CrossRef CAS PubMed.
- L. Greenwich, J. L. Eagan, N. Feirer, K. Boswinkle, G. Minasov, L. Shuvalova, N. L. Inniss, J. Raghavaiah, A. K. Ghosh, K. J. F. Satchell, K. D. Allen and C. Fuqua, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2319903121 CrossRef PubMed.
- O. Ciofu, C. Moser, P. Ø. Jensenand and N. Høiby, Nat. Rev. Microbiol., 2022, 20, 621–635 CrossRef CAS PubMed.
- L. Hall-Stoodley, J. W. Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95–108 CrossRef CAS PubMed.
- K. J. Colston and P. Basu, Molecules, 2022, 27, 3324 CrossRef CAS PubMed.
- Biochemical and Clinical Aspects of Pteridines. Cancer Immunology Metabolic Diseases, ed. R. Soyka and W. Pfleiderer, Walter de Gruyter, Berlin, 1985, vol. 4, pp. 33–43 Search PubMed.
- W. Pfleiderer, in Folates and Pterins, ed. R. L. Blakley and S. J. Benkovic, Wiley, New York, 1985, vol. 2, pp. 43–114 Search PubMed.
- M. Viscontini, R. Provenzale, S. Ohlgart and J. Mallevialle, Helv. Chim. Acta, 1970, 53, 1202–1207 CrossRef CAS.
- R. Soyka, W. Pfleiderer and R. Prewo, Helv. Chim. Acta, 1990, 73, 808–826 CrossRef CAS.
- K. J. Andrews, W. E. Barber and B. P. Tong, J. Chem. Soc. C, 1969, 6, 928–930 RSC.
- J. E. Hodge, Adv. Carbohydr. Chem., 1955, 10, 169–205 CAS.
- W. R. Boon, W. G. M. Jones and G. R. Ramage, J. Chem. Soc., 1951, 21, 96–102 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07179d |
| This journal is © The Royal Society of Chemistry 2024 |