
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive Zn(II) complexes showing the structural conversion and on/off switching of catalytic properties†
So Hyeon Kwona,
Sunwoo Leeb,
Jacopo Tessarolo *b and
Haeri Lee*a
*b and
Haeri Lee*a
aDepartment of Chemistry, Hannam University, 34054, Republic of Korea. E-mail: haeri.lee@hnu.ac.kr
bDepartment of Chemistry, Chonnam National University, Gwangju, 61186, Republic of Korea. E-mail: jacopo@jnu.ac.kr
First published on 16th October 2024
Abstract
In this work, we report a series of dinuclear Zn(II) complexes and their corresponding catalytic properties for a transesterification reaction. We show that the structures and catalytic activity of the complexes are strongly dependent on their molecular structures surrounding the metal centres. The use of halides yields a series of [Zn2X4L] (X = Cl, Br, and I) complexes with low catalytic activity because of the fully saturated coordination environment, whereas Zn(ClO4)2 results in two isomeric [ZnL]n 1D coordination polymers with efficient catalytic properties, despite being susceptible to structural rearrangement and consequent changes in catalytic activity over time. The response to chemical stimuli to trigger anion exchange allows for switching on/off the systems' catalytic activity, simultaneously recovering the catalytic effect upon degradation and thus reconstructing the coordination environment of the 1D polymer.
Research studies on catalytic effects of coordination-based materials have piqued the interest of molecular engineers. Controlling the coordination environment can modulate the properties of such compounds.1,2 Among the several reactions, transesterification is often used as a benchmark to study such structure–property relationships.3,4 This is because biodiesel plays a pivotal role in fostering sustainable energy practices by offering a renewable and biodegradable alternative to traditional fossil fuels while remaining compatible with existing infrastructures.5–9 Its use contributes to reducing greenhouse gas emissions, mitigating climate change, and enhancing energy security by diversifying energy supply. Biodiesel production entails the transesterification of feedstock oil with alcohols to yield alkyl esters,10–14 often in the presence of a metal complex catalyst.15,16 In this context, the design of efficient, cheap and sustainable catalysts plays a key role in improving the transesterification reaction.17–20 Generally, homogeneous catalysts demonstrate superior activity, while heterogeneous catalysts offer advantages in terms of separation from reaction solution and recyclability.8,21–24 In the latter case, access to metal centres is challenging and a critical step towards compound activity. The lability of ligands or coordinating anions needs to be tuned to gain access to coordination sites without sacrificing complex stability.25–27 Control of the anions can also be used to tune complex charge, favouring an interaction with reaction intermediates when charged.1 At the same time, solid state packing can provide void spaces around metal ions in heterogeneous systems, thereby tuning substrate access to the catalytic centre.28,29 The packing structure can be controlled by the template, ligand bulkiness, coordination environment of metal ions, and intrinsic anion effects.1,2,25,30,31 The structure of metal catalysts for transesterification reactions can vary from complexes,32–34 clusters,35–37 coordination polymers38–40 and metal organic frameworks,41–43 with the latter two being particularly interesting for heterogeneous catalysis.44,45 In particular, Zn(II) metal ions play a pivotal role in esterification and transesterification reactions owing to their cheap, abundant, and non-toxic nature.1–3,46 While a plethora of Zn(II)-based systems have been reported and applied to substrates, ranging from simple commercially available esters to vegetable oils,46,47 the possibility to exploit coordination chemistry to develop stimuli-responsive systems has been overlooked.24,29,42 For instance, the addition or removal of coordinating anions can be exploited to tune catalyst reactivity and generate a catalytic on/off switch.4
To study these aspects, we synthesized a series of complexes based on Zn(II) metal ions (Fig. 1). Owing to the coordination geometry versatility of its d10 electronic configuration and the stable, yet dynamic, character of the coordination bonds, this allows for structural versatility and control. These complexes are based on a chelating ligand, (E)-N′,N′,N′′,N′′-tetra(2-pyridyl)-2-butene-1,2-diamine (L) (details in the ESI, Fig. S1–S6 and S8a†). The ligand design couples flexibility from the butene backbone and tertiary amines with the thermodynamic stability from the chelating pyridines.
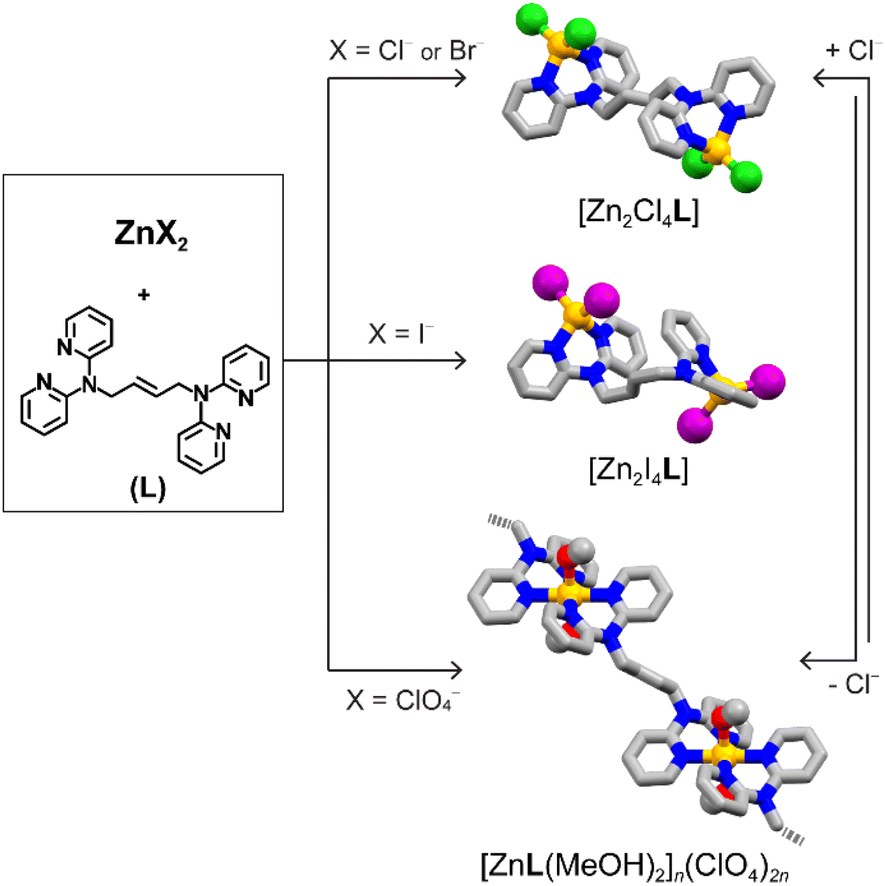 | ||
| Fig. 1 Synthetic diagram for zinc(II) complexes [Zn2X4L] (X = Cl, Br, and I) and [ZnL]n, and their anion triggered structural conversion (colour code: yellow, Zn2+; green, Cl−; purple, I−; grey, C; blue, N; red, O). | ||
The ligand has been combined with a variety of Zn(II) metal precursors (ZnX2, X = Cl−, Br−, I−, ClO4−). In all cases, this has resulted in colourless crystalline materials in high yields, as confirmed by single crystal and powder X-ray diffraction (SCXRD and PXRD).
Using halide precursors results in a series of dinuclear complexes with the general formula [Zn2X4L]. Such complexes are insoluble in organic solvents, such as methanol, chloroform, tetrahydrofuran, and toluene, while the use of DMSO or DMF dissolves the crystalline material upon complex dissociation. Thermogravimetric analyses (TGA) confirmed the increased stability of the complex, showing decomposition of [Zn2X4L] at 316 °C, 320 °C and 320 °C for X = Cl−, Br−, and I−, respectively, compared to the ligand which melts at 117 °C and decomposes at 190 °C (Fig. S5 and S7†). Interestingly, TGA data show a direct decomposition to ZnO without loss of the crystallization solvent, suggesting the absence of crystalline solvent-accessible voids (vide infra). The reaction was carried in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (L:M) stoichiometric ratio. However, the dinuclear compound can also be obtained in high yields when using a 1:1 molar ratio, proving the system stability and preference for such dinuclear complexes. Single crystals of [Zn2X4L] suitable for SCXRD were obtained upon assembly of the ligand and ZnX2 (X = Cl−, Br−, I−) in a methanolic solution (Tables S1 and S2†). In all three structures, the molecular structure consists of two ZnX2 moieties at the side of the molecule, connected by one bridging ligand (Fig. 2a and b). Free rotation of the pyridyl groups and flexibility of the tertiary amine result in a chelating binding mode. Each neutral Zn(II) node shows a typical tetrahedral geometry with an X–Zn–X angle of 118.91(8)° for [Zn2Cl4L], 118.31(3)° for [Zn2Br4L], and 119.55(2)° for [Zn2I4L]. Structural analysis shows no solvent-accessible voids in the structure, in agreement with the TGA data (Fig. S7a–c†). The use of different anions impacts the intermolecular interactions, influencing their packing structures along with anions. [Zn2Cl4L] and [Zn2Br4L] are isostructural with an intermolecular π–π stacking of 3.45 Å (Fig. 2a). However, when using a larger iodide anion, the intramolecular π–π stacking occurs between the pyridyl moieties of the same ligand, resulting in folding of the dinuclear complex and shortening the Zn⋯Zn distance.
:2 (L:M) stoichiometric ratio. However, the dinuclear compound can also be obtained in high yields when using a 1:1 molar ratio, proving the system stability and preference for such dinuclear complexes. Single crystals of [Zn2X4L] suitable for SCXRD were obtained upon assembly of the ligand and ZnX2 (X = Cl−, Br−, I−) in a methanolic solution (Tables S1 and S2†). In all three structures, the molecular structure consists of two ZnX2 moieties at the side of the molecule, connected by one bridging ligand (Fig. 2a and b). Free rotation of the pyridyl groups and flexibility of the tertiary amine result in a chelating binding mode. Each neutral Zn(II) node shows a typical tetrahedral geometry with an X–Zn–X angle of 118.91(8)° for [Zn2Cl4L], 118.31(3)° for [Zn2Br4L], and 119.55(2)° for [Zn2I4L]. Structural analysis shows no solvent-accessible voids in the structure, in agreement with the TGA data (Fig. S7a–c†). The use of different anions impacts the intermolecular interactions, influencing their packing structures along with anions. [Zn2Cl4L] and [Zn2Br4L] are isostructural with an intermolecular π–π stacking of 3.45 Å (Fig. 2a). However, when using a larger iodide anion, the intramolecular π–π stacking occurs between the pyridyl moieties of the same ligand, resulting in folding of the dinuclear complex and shortening the Zn⋯Zn distance.
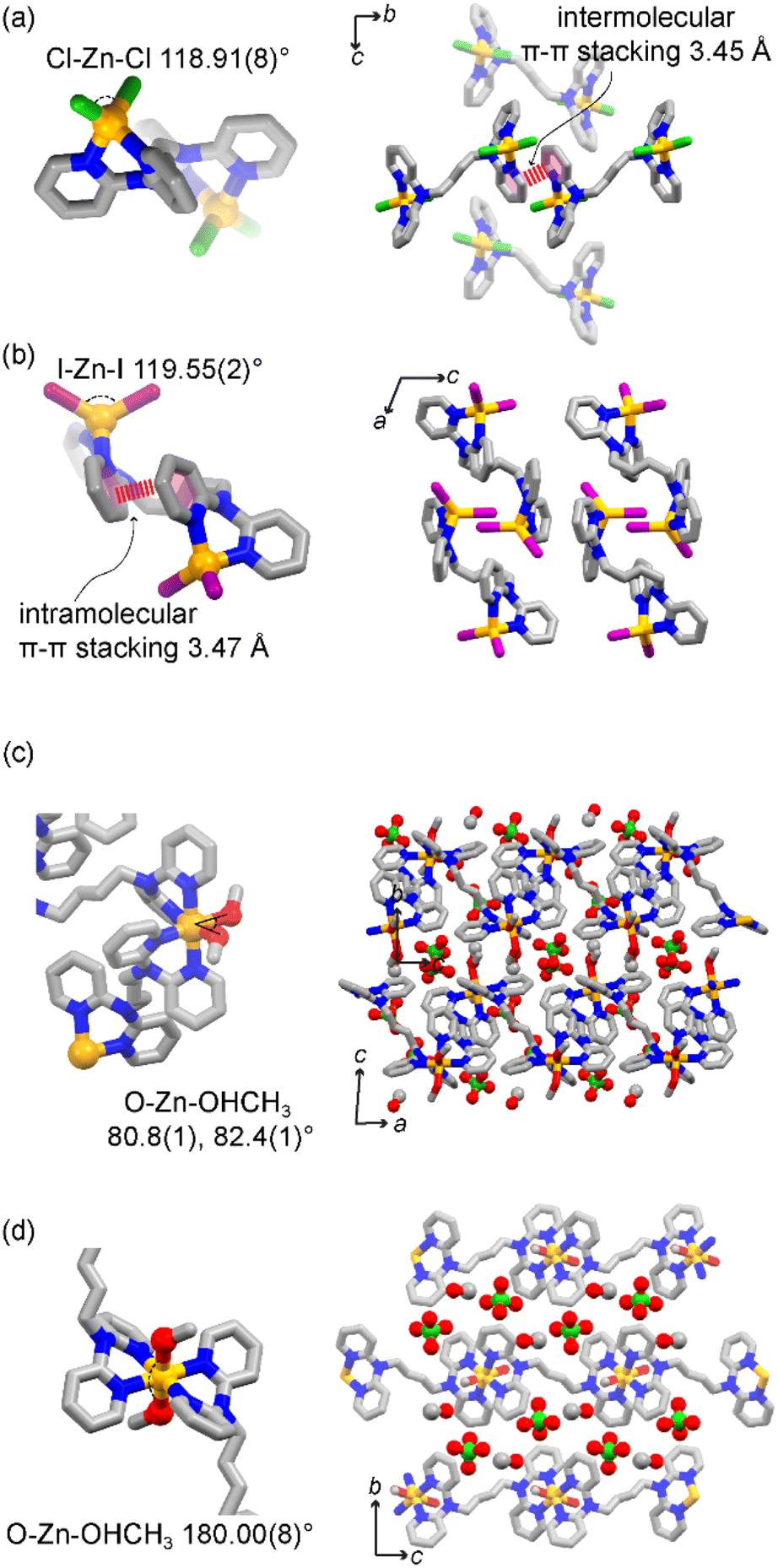 | ||
| Fig. 2 Molecular structures (left) and packing (right) of [Zn2Cl4L] (a), [Zn2I4L] (b), and cis-[ZnL]n (c) and trans-[ZnL]n (d). Colour code: yellow, Zn2+; grey, C; blue, N; red, O; green, Cl; purple, I; H atoms are omitted for clarity. | ||
When starting from Zn(ClO4)2, the resulting compound crystallizes as 1D coordination polymers with the general formula [ZnL(MeOH)2]n(ClO4)2n ([ZnL]n). We obtained single crystals suitable for diffraction studies for two isomeric compounds. In both cases, two ligands and two methanol molecules coordinated with each metal centre, resulting in an octahedral coordination. In one structure, the two coordinated methanol molecules are in the cis-position. Meanwhile, in the other case, they bind in the trans-position, giving cis-[ZnL]n and trans-[ZnL]n 1D coordination polymers, respectively. In cis-[ZnL]n, the O–Zn–O angles are 80.8(1)° and 82.4(1)° (Fig. 2c), resulting in 1D zigzag chains, with perchlorate counter anions and methanol molecules lying between the chains. On the other hand, in trans-[ZnL]n, the two methanol molecules are in the trans-position with a O–Zn–O angle of 180.0(8)° (Fig. 2d), resulting in a more linear 1D polymer. When directly precipitating a crystalline powder for [ZnL]n, the powder XRD pattern shows a mixture with the major presence of characteristic peaks matching the pattern of trans-[ZnL]n. Upon exposing the powder sample to methanol at 50 °C under vigorous stirring, mimicking the conditions for the follow up reaction studies, the pattern gradually shifted to an enriched cis-form over time, showing a structural conversion via crystal-to-crystal transformation (Fig. S10†). TGA analysis of the crystalline materials shows that the compound is thermally stable up to 318 °C upon an initial loss of solvent, compatible with the loss of methanol. Combining a non-coordinating anion and weakly bound solvent molecules results in a positively charged metal complex with easy-to-access metal coordination, both desirable features for catalytical properties.
Next, we investigated the catalytic activity of the synthesized Zn(II) complexes towards transesterification reactions in methanol. Initially, the four crystalline materials were tested using a 4-fluorophenyl acetate substrate as a case study to then apply optimized conditions to a variety of substrates, as reported in Fig. 3. The aromatic ester reacts with methanol to give the relative aromatic alcohol and methyl acetate. The conditions screened for 4-fluorophenyl acetate are reported in Table 1. The reactions were carried out in 500 μL methanol at 50 °C, with a catalyst loading of 5 mol% or 10 mol%.
 | ||
| Fig. 3 Transesterification reaction scheme and scope of the used substrates. | ||
| Entry | Catalyst | Mol% | Yield at 1 ha |
|---|---|---|---|
| a The catalytic yield after 1 hour of the reaction was determined by integrating NMR spectra corresponding to the ratio of the substrate and products. | |||
| 1 | [Zn2Cl4L] | 5 | 7.4% |
| 2 | [Zn2Cl4L] | 10 | 9.9% |
| 3 | [Zn2Br4L] | 5 | Trace |
| 4 | [Zn2Br4L] | 10 | Trace |
| 5 | [Zn2I4L] | 5 | Trace |
| 6 | [Zn2I4L] | 10 | Trace |
| 7 | [ZnL]n | 5 | 70.0% |
| 8 | [ZnL]n | 10 | 94.2% |
| 9 | Zn(ClO4)2 + L | 5 | 40.8% |
| 10 | Zn(ClO4)2 + L | 10 | 40.5% |
| 11 | Zn(ClO4)2 | 10 | 0 |
| 12 | Zn(ClO4)2 | 20 | 0 |
| 13 | L | 5 | 0 |
| 14 | L | 10 | 0 |
When employing 10 mol% of the [Zn2X4L] species (33 mg for [Zn2Cl4L], 42 mg for [Zn2Br4L], 52 mg for [Zn2I4L], respectively), the amount of methyl acetate and 4-fluorophenol formed after 1 hour is detected only in traces, except for [Zn2Cl4L] yielding ∼10% (entries 1–8). On the other hand, when 10 mol% of the 1D coordination polymer [ZnL]n (19.5 mg) is employed as a catalyst, the reaction reaches a yield of 94.2% after one hour, and reaches full conversion to the desired product after 100 minutes. Interestingly, combining the metal precursor Zn(ClO4)2 with L directly in situ in the presence of the substrate results in the formation of 4-fluorophenol in a 40% yield after 1 hour, presumably due to the partial formation of the catalytic coordination polymer. On the contrary, employing only one of the two building blocks, either the metal salt or L, results in a negligible product formation. When performing the same reaction using [ZnL]n in iso-propanol or tert-butanol, the catalytic activity at 50 °C is hampered. Meanwhile, it yields 11.4% conversion when using iso-propanol at 70 °C (Fig. S11†). Based on these results, we employed [ZnL]n with a catalyst load of 10% in methanol on a larger scope of substrates, varying the electron-withdrawing and electron-donating character of the substituents.
The product formation over time was monitored by NMR spectroscopy (Fig. 4 and S12–S17†), and cross checked by GC analysis at selected time intervals of 50 and 100 minutes (Fig. S18†). Among the selected substrates, 4-nitrophenyl acetate and p-tolyl acetate are readily converted to 4-nitrophenol and p-cresol with a quantitative yield within 1 hour. When pentafluorophenyl acetate is used as a substrate, the reaction is slower and reaches 85% yield after 150 min. The non-substituted phenyl acetate substrate reactivity is even slower, reaching 40% conversion after 100 min, as detected by NMR analysis (Fig. S15 and S16†). The catalysts can be recovered by filtration with 99% yield. After the first cycle, the recovered catalyst shows similar performances as the starting materials with recyclability of the catalyst up to 5 cycles. Meanwhile, with a minimal loss of the compound, it shows a structural change that results in a less efficient catalysis, slowing down the time required to achieve 100% substrate conversion (Table S3†). Catalysts were examined by PXRD (Fig. S19†) before and after the reaction. The PXRD pattern shows some changes, in a similar manner to the structural conversion previously observed while heating and stirring in methanol (Fig. S10†). The significant difference in reactivity between [Zn2X4L] and [ZnL]n can be explained by the difficulties in coordinating the metal centre when bound to halide ions, together with a role of the coordination complex charge, which can help in stabilizing an intermediate species, thus lowering the reaction activation barrier.
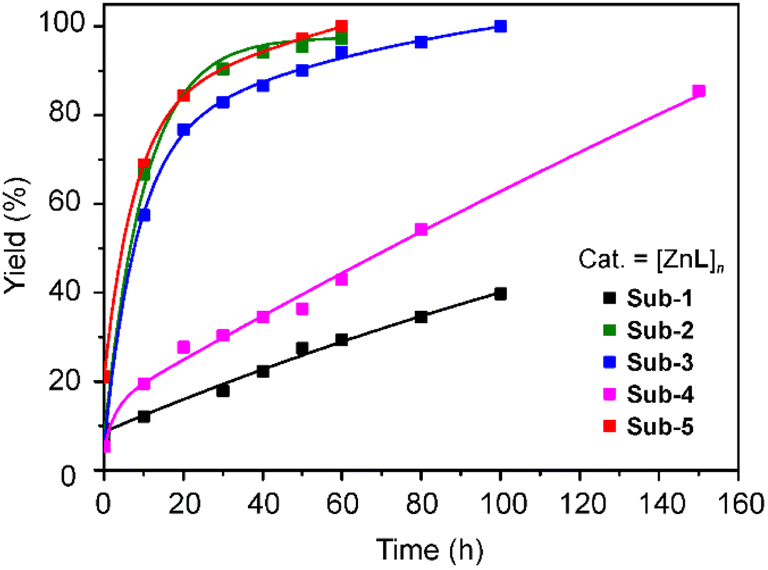 | ||
| Fig. 4 Yield (%) of transesterification conversion over time for various substituted phenyl acetates. | ||
The proposed mechanism (Fig. S22†), by comparison with literature reports, involves three steps. At first, the substrate carbonyl coordinates to Zn(II), followed by a nucleophilic attack from methanol. Then, the elimination and rearrangement of the intermediate result in the formation of phenols and methyl acetate. This mechanism is effective only when metal coordination sites are accessible to the solvent, otherwise rendering the catalytic properties ineffective. Based on this, we investigated the systems' response to the addition or removal of halides to create a stimuli-responsive switch for turning on/off the catalytic performances (Fig. 5). Employing [Zn2Cl4L], the substrate reactivity is not activated. However, the addition of AgClO4 results in an anion exchange, followed by precipitation of AgCl, and an activation of the metal complex catalytic properties (Fig. 5, red). The catalyst can be activated before the addition of the substrate, or by addition of the silver salt in a premixed and preheated reaction mixture. Both cases trigger the phenol formation. Addition of AgClO4 corresponds to the time 0 when the switch is turned on, and the reaction reached 100% yield conversion after 1 h, 30 min (Fig. S20†). To trigger the switch off, NH4Cl is added to a suspension of [ZnL]n in methanol, leading to the opposite anion exchange. Chloride anions coordinate the metal centre, rendering the complex neutral, blocking available coordination sites and partially releasing some ligand to balance the stoichiometry, resulting in no catalytic activity (Fig. 5, blue). Besides a switch of the catalytic properties, this process allows for the regeneration of the initial [ZnL]n structure after running the transesterification reaction for a few cycles.
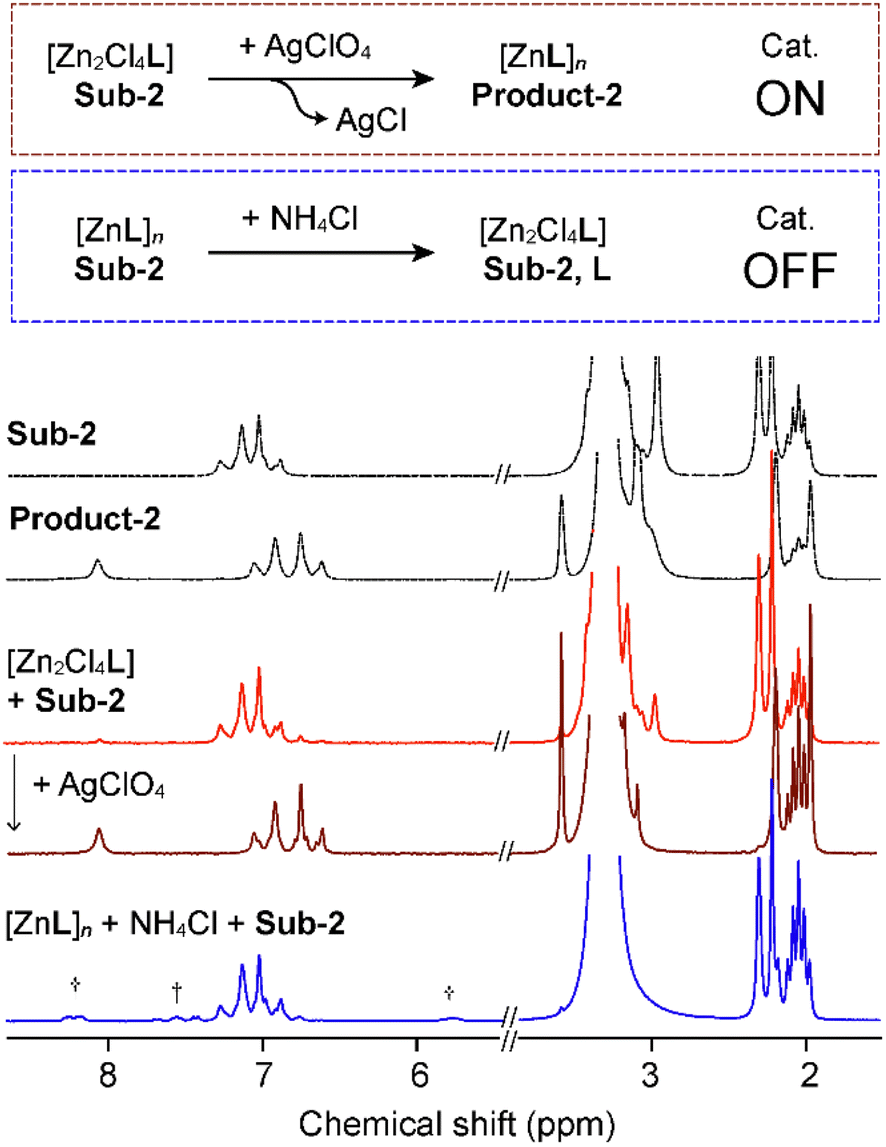 | ||
| Fig. 5 Anion-triggered switching of catalytic properties with Sub-2 († free L). | ||
The composition of the crystalline material upon anion exchange after running the reaction for 5 cycles was confirmed by PXRD patterns (Fig. 6). The possibility to tune the structure by modulation of the counterion offers a possibility to regenerate a catalyst to initial efficiency by undergoing structural change with progressive reduction of the catalytic efficiency.
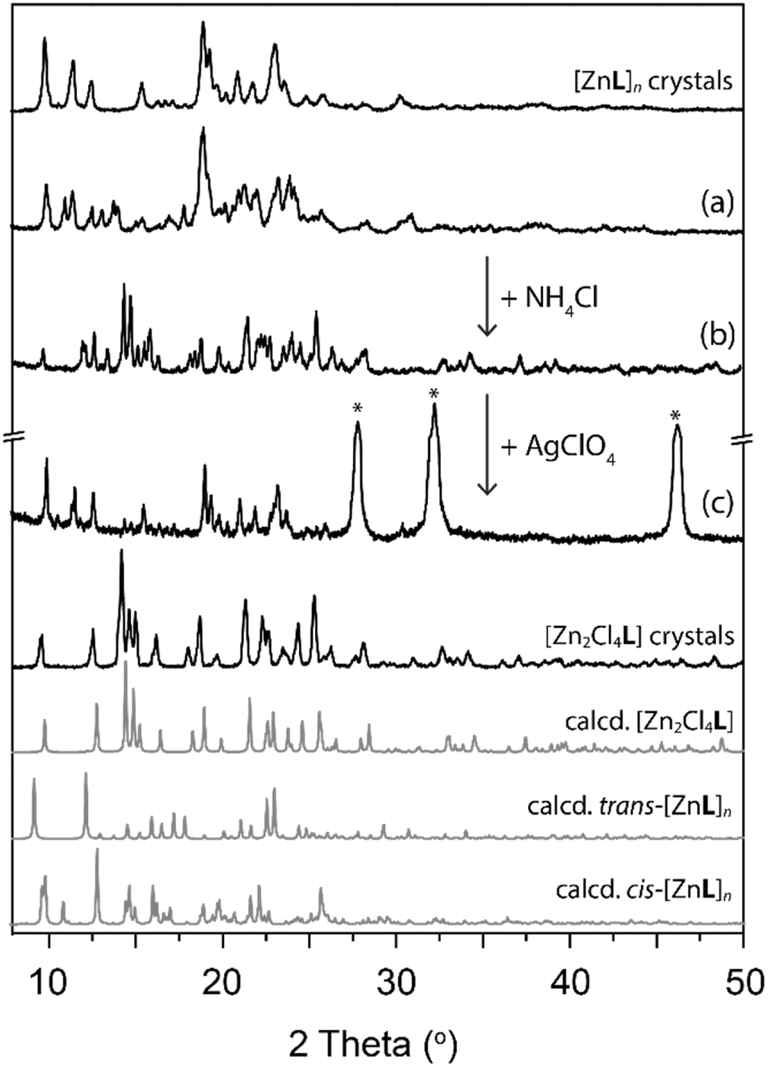 | ||
| Fig. 6 Powder XRD patterns for anion exchange. [ZnL]n crystals (a), used as catalysts in transesterification reaction for 5 cycles, were converted into [Zn2Cl4L] (b) after reaction with NH4Cl. Subsequently, (b) was treated with AgClO4 to obtain [ZnL]n (c) (* AgCl). | ||
In conclusion, we elucidated the significant role of zinc(II) complexes and coordination polymers in catalysing transesterification reactions. Control of the initial building blocks allows us to control the metal coordination geometry, charge of the complex, structure, and solid-state packing. Such features reflect on the catalytic properties towards transesterification reactions on 5 different substrates. When halides are used in [Zn2X4L], the systems' reactivity is hampered. Meanwhile, the use of 1D coordination polymers [ZnL]n with ClO4− anions results in an efficient and recyclable catalyst under mild conditions. Finally, the response to chemical stimuli, as the addition or removal of Cl− ions, acts as an on/off switch to trigger the catalytical properties, which can be applied as a regeneration method after losing catalytic efficiency. The findings underscore the importance of metal precursor selection in influencing catalytic efficiency, and highlight the innovative approach of anion exchange to regulate catalytic activity. This work not only advances our understanding of metal–organic catalysts, but also suggests possible pathways for the development of tunable catalysts for potential green chemistry applications. Our research contributes to the broader objective of enhancing catalytic processes through the strategic design of metal complexes and their structural transformations.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for all compounds have been deposited at the CCDC under 2144545–2144548, 2331257, and 2364848 and can be obtained from https://www.ccdc.cam.ac.uk/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT) (grants NRF-2021R1C1C1013037 and RS-2024-00348192), by the Korea Institute for Advancement of Technology (KIAT) grant funded by the Korea Government (MOTIE, RS-2024-00409639), by the Hannam University Research Fund (2022A214), by Chonnam National University (grant number: 2023-0917-01), and by “Regional Innovation Strategy (RIS)” through the NRF funded by the Ministry of Education (MOE) (2021RIS-002). H. L. thanks Dr Dongwook Kim from Institute of Basic Science (IBS) and Jihun Han from Pusan National University for the SCXRD data collection.Notes and references
- A. B. Chakraborty, T. Ganguly and A. Majumdar, Inorg. Chem., 2023, 62, 11095–11111 CrossRef CAS PubMed.
- E. Choi, M. Ryu, H. Lee and O. S. Jung, Dalton Trans., 2017, 46, 4595–4601 RSC.
- D. Nakatake, R. Yazaki and T. Ohshima, Eur. J. Org. Chem., 2016, 2016, 3696–3699 CrossRef CAS.
- M. Melchiorre, M. E. Cucciolito, M. Di Serio, F. Ruffo, O. Tarallo, M. Trifuoggi and R. Esposito, ACS Sustain. Chem. Eng., 2021, 9, 6001–6011 CrossRef CAS.
- A. A. S. Leite, L. V. Weber, J. P. A. Correa, T. L. A. de Castro, C. C. M. da Silva, R. M. F. da Costa e Silva, C. A. L. Cardoso and L. C. Konradt-Moraes, Sci. Rep., 2024, 14, 1586 CrossRef CAS PubMed.
- Y. Deng, X. Hu, L. Cheng, H. Wang, L. Duan and R. Qiu, J. Organomet. Chem., 2018, 870, 116–120 CrossRef CAS.
- F. Moazeni, Y. C. Chen and G. Zhang, J. Cleaner Prod., 2019, 216, 117–128 Search PubMed.
- R. Esquer and J. J. García, J. Organomet. Chem., 2019, 902, 120972 CrossRef CAS.
- J. Aupič, J. Borišek, S. M. Fica, W. P. Galej and A. Magistrato, Nat. Commun., 2023, 14, 8482 CrossRef.
- H. Sun, Y. Ding, J. Duan, Q. Zhang, Z. Wang, H. Lou and X. Zheng, Bioresour. Technol., 2010, 101, 953–958 CrossRef CAS PubMed.
- R. Varghese, J. P. Henry and J. Irudayaraj, Environ. Prog. Sustainable Energy, 2018, 37, 1176–1182 CrossRef CAS.
- X. Liu, H. He, Y. Wang, S. Zhu and X. Piao, Fuel, 2008, 87, 216–221 CrossRef CAS.
- R. Sneha and G. Madhumitha, J. Organomet. Chem., 2023, 1001, 122870 CrossRef CAS.
- K. Li and W. Xie, Fuel, 2024, 364 Search PubMed.
- S. Magens and B. Plietker, J. Org. Chem., 2010, 75, 3715–3721 CrossRef CAS.
- T. Kato, S. Akebi, H. Nagae, K. Yonehara, T. Oku and K. Mashima, Catal. Sci. Technol., 2021, 11, 6975–6986 RSC.
- T. F. Dossin, M. F. Reyniers and G. B. Marin, Appl. Catal., B, 2006, 62, 35–45 CrossRef CAS.
- D. E. López, J. G. Goodwin and D. A. Bruce, J. Catal., 2007, 245, 381–391 CrossRef.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- A. Roy, G. Kedziora, S. Bhusal, C. Oh, Y. Kang, V. Varshney, Y. Ren and D. Nepal, J. Phys. Chem. B, 2021, 125, 2411–2424 Search PubMed.
- X. Ma, F. Liu, Y. Helian, C. Li, Z. Wu, H. Li, H. Chu, Y. Wang, Y. Wang, W. Lu, M. Guo, M. Yu and S. Zhou, Energy Convers. Manage., 2021, 229 CAS.
- K. E. Culley, C. Johnson and D. L. Gin, Chem. Commun., 2023, 59, 11105–11108 RSC.
- M. M. Dell'Anna, V. F. Capodiferro, M. Mali and P. Mastrorilli, J. Organomet. Chem., 2016, 818, 106–114 CrossRef.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS.
- H. W. Zheng, D. D. Yang, Y. S. Shi, T. Xiao, H. W. Tan and X. J. Zheng, Inorg. Chem., 2023, 62, 6323–6331 CrossRef CAS.
- S. Li, Y. Ma, Y. Zhao, R. Liu, Y. Zhao, X. Dai, N. Ma, C. Streb and X. Chen, Angew. Chem., Int. Ed., 2023, 62, e202314999 CrossRef CAS PubMed.
- X. Ma, F. Liu, Y. Helian, C. Li, Z. Wu, H. Li, H. Chu, Y. Wang, Y. Wang, W. Lu, M. Guo, M. Yu and S. Zhou, Energy Convers. Manage., 2021, 229 CAS.
- D. Tian, R. Hao, X. Zhang, H. Shi, Y. Wang, L. Liang, H. Liu and H. Yang, Nat. Commun., 2023, 14, 3226 CrossRef CAS.
- H. Lee, D. Kim, H. Oh and O.-S. Jung, Chem. Commun., 2020, 56, 2841–2844 RSC.
- X. Fan, H. Wang, J. Gu, D. Lv, B. Zhang, J. Xue, M. V. Kirillova and A. M. Kirillov, Inorg. Chem., 2023, 62, 17612–17624 Search PubMed.
- T. Mandal, A. Kumar, J. Panda, T. Kumar Dutta and J. Choudhury, Angew. Chem., Int. Ed., 2023, 62, e202314451 CrossRef CAS PubMed.
- R. R. Behera, R. Ghosh, S. Panda, S. Khamari and B. Bagh, Org. Lett., 2020, 22, 3642–3648 CrossRef CAS PubMed.
- D. Srimani, A. Mukherjee, A. F. G. Goldberg, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben David and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12357–12360 CrossRef CAS PubMed.
- J. C. Lugo-González, P. Gómez-Tagle, M. Flores-Alamo and A. K. Yatsimirsky, Dalton Trans., 2020, 49, 2452–2467 RSC.
- K. Agura, Y. Hayashi, M. Wada, D. Nakatake, K. Mashima and T. Ohshima, Chem.–Asian J., 2016, 11, 1548–1554 CrossRef CAS.
- T. Ohshima, in New Horizons of Process Chemistry, Springer Singapore, Singapore, 2017, pp. 65–87 Search PubMed.
- T. Kato, S. Akebi, H. Nagae, K. Yonehara, T. Oku and K. Mashima, Catal. Sci. Technol., 2021, 11, 6975–6986 RSC.
- D. Nakatake, Y. Yokote, Y. Matsushima, R. Yazaki and T. Ohshima, Green Chem., 2016, 18, 1524–1530 RSC.
- Y. M. Lee, Y. J. Song, J. I. Poong, S. H. Kim, H. G. Koo, J. A. Lee, C. Kim, S.-J. Kim and Y. Kim, Inorg. Chem. Commun., 2010, 13, 101–104 CrossRef CAS.
- M. Y. Hyun, I. H. Hwang, M. M. Lee, H. Kim, K. B. Kim, C. Kim, H.-Y. Kim, Y. Kim and S.-J. Kim, Polyhedron, 2013, 53, 166–171 CrossRef CAS.
- Y.-S. Kang, Y. Lu, K. Chen, Y. Zhao, P. Wang and W.-Y. Sun, Coord. Chem. Rev., 2019, 378, 262–280 CrossRef CAS.
- A. Karmakar, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2014, 43, 7795–7810 RSC.
- J. Won Shin, J. Mi Bae, C. Kim and K. Sik Min, Inorg. Chem., 2013, 52, 2265–2267 CrossRef.
- M. Y. Hyun, I. H. Hwang, M. M. Lee, H. Kim, K. B. Kim, C. Kim, H. Y. Kim, Y. Kim and S. J. Kim, Polyhedron, 2013, 53, 166–171 CrossRef CAS.
- H. Zhang, L. Chen, Y. Li, Y. Hu, H. Li, C. C. Xu and S. Yang, Green Chem., 2022, 24, 7763–7786 Search PubMed.
- R. Esposito, M. Melchiorre, A. Annunziata, M. E. Cucciolito and F. Ruffo, ChemCatChem, 2020, 12, 5858–5879 CrossRef CAS.
- A. Karmakar, M. F. C. Guedes Da Silva and A. J. L. Pombeiro, Dalton Trans., 2014, 43, 7795–7810 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, and crystallographic data. CCDC 2144545–2144548, 2331257 and 2364848. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra06058j |
| This journal is © The Royal Society of Chemistry 2024 |