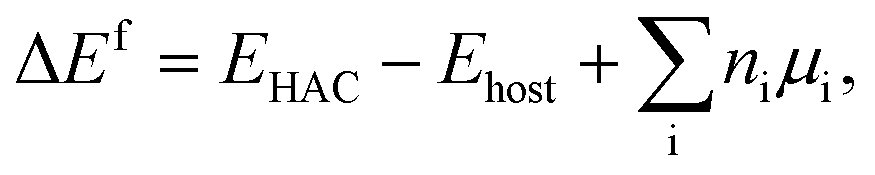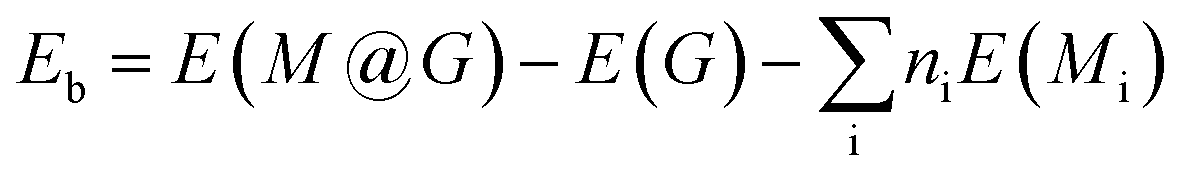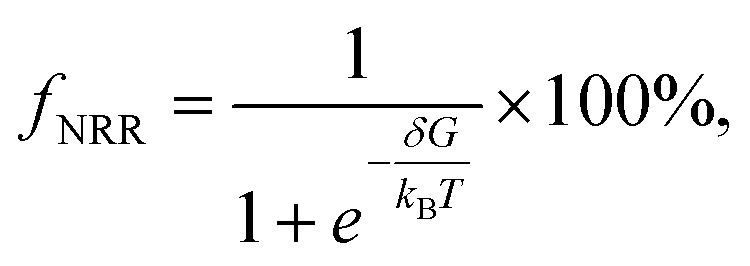DOI:
10.1039/D4RA06077F
(Paper)
RSC Adv., 2024,
14, 34893-34903
Theoretical study of a CuCo dual-atom catalyst for nitrogen fixation†
Received
22nd August 2024
, Accepted 25th October 2024
First published on 31st October 2024
Abstract
Developing low-cost catalysts for highly efficient nitrogen reduction reaction (NRR) in industrial applications is a great challenge. Dual-atom catalysts (DACs) have also aroused scientific interest as potential NRR catalysts due to their possible higher activity and atom utilization than single atom catalysts. Using density functional theory, we have investigated the NRR performances of heteronuclear CuCo DACs with different coordination configurations. Comparisons with the possible Cu or Co SACs and homonuclear dual-atom catalysts for the NRR performance were also made. We find that O-CuCo-N DAC has superior NRR performance, where the NRR easily takes place through the alternating pathway from the side-on N2 adsorption mode and the potential limiting step is the first hydrogenation step (*NN* → *NHN*) with a Gibbs free energy change of 0.55 eV. The good activity of O-CuCo-N DAC benefits from the regulation provided by Cu and the coordination environment. In addition, O-CuCo-N DAC also exhibits good selectivity and durability.
1 Introduction
Reduction of dinitrogen (N2) to ammonia (NH3) is of critical importance to sustain life on earth, since ammonia is an essential commodity chemical for agriculture and the chemical industry. Currently, ammonia production is still dominated by the traditional Haber–Bosch process,1,2 which is emission and energy intensive due to the high working temperature and pressure. The electrochemical nitrogen reduction reaction (NRR, N2 + 6H+ + 6e− → 2NH3), as performed by the nitrogenase enzymes in bacteria, is a promising alternative because it can be powered by clean energy and operated under ambient conditions, and does not cause CO2 emissions.3–6 The main challenge of the NRR lies in finding an electrocatalyst with high stability, activity and selectivity. To date, different types of NRR catalysts such as noble metals,7–11 two dimensional materials,12–14 oxides,15,16 nitrides17,18 and sulfides19–21 have been explored. However, their selectivity is generally low because of competition with the hydrogen evolution reaction (HER) and thus they cannot achieve a high faradaic efficiency. Recently, single-atom catalysts (SACs) with atomically distributed metal active centers have emerged as a new frontier in catalysis and aroused widespread attention since they can theoretically realize the largest metal atom utilization, a higher catalytic activity and selectivity.22,23 Various single metal atoms anchored on different systems have been extensively investigated for their catalytic performance.24–29 SACs also exhibit excellent potential for N2 fixation.30–34 Nitrogen or oxygen-doped carbon materials, which are earth-abundant and cost-effective, have a great advantage as substrates to support single atoms. High faradaic efficiency and NH3 yield rate have been reported in experiments from single atom dispersed, N- or O-doped carbon materials for ambient ammonia synthesis.35,36 Also, other kinds of single atoms, anchored on doped graphene were proposed for the NRR from theoretical computations.37 In those models, the single atoms were normally coordinated purely by either N or O atoms.38 However, based on experiments, assuming nearby O and N dopants in a carbon material is also reasonable.
Inspired by enzymes containing metal pair, dual metal atoms catalysts (DACs), including both homonuclear and heteronuclear DACs, have also been developed to boost the activity of SACs by providing diverse active sites and synergistic interactions between adjacent atoms for the oxygen reduction reaction (ORR)39,40 and NRR.41–44 Fe and Mo metal dimers situ-anchored on defect-rich graphene layer (FeMoNxC), and isolated Fe, Mo atoms anchored by hierarchical N doped carbon nanotubes (FeMo/NC) were reported that they could achieve selective electroreduction of N2 to NH3 and a stronger catalytic activity than its single-metal counterparts.41,42 Wang et al. reported that the synergy of Fe and Cu multi-atom clusters on graphitic carbon nitride provides greatly improved NRR performance, with nearly doubled NH3 yield and Faradaic efficiency of up to 34% when compared to the single-metal counterparts.43 Effects of adjacent single-TM atoms on Fe anchored C2N SACs,44 as well as dual metal atoms anchored on graphite carbon nitride were theoretically investigated.45 Nevertheless, a deeper understanding of the synergistic mechanism is imperative to bridge the gap between theory and experiments. It is noted that Cu is an earth-abundant non-noble metal element, Cu-based catalysts have attracted much attention due to their facile preparation in NRR and Co-based catalysts have also been widely investigated to be excellent NRR catalysts.46–48 We speculate that the coexistence of dual metal atoms Cu and Co, as well as the O and N coordination, could lead to a synergistic regulation on the NRR intermediates. Also, carbon-based catalyst with diatomic Fe–Co sites in which the Fe and Co atoms are respectively coordinated to N and O atoms (as witnessed by HAADF-STEM and XAS) have been recently successfully synthesized for electrocatalytic oxygen reduction and evolution in experiments,49 which proves that DACs with N and O coordination are experimentally feasible.
Herein, we chose Cu and Co as the two hetero metal atoms of our dual-atom catalysts,50–53 as well as N and O doped graphene as the support, to study its NRR performance. In our work, the NRR activities of heteronuclear CuCo DACs with various N and O coordination structures were investigated and compared with the Cu(Co) single atom catalysts and homonuclear CuCu(CoCo) dual atom catalysts. Our results show that an O-CuCo-N DAC has the lowest NRR reaction energy barrier (0.55 eV) and exhibits the best NRR performance. We also reveal that the Cu–O3 moiety efficiently regulates the d orbital energy levels of Co in the Co–N3 moiety and optimizes the adsorption energy of N containing intermediates on CuCo DAC.
2 Computational methods
All the computations were performed within spin-polarized density functional theory (DFT) methods using the Vienna Ab initio Simulation Package (VASP).54–56 Though, transition metals with partially filled d orbitals were involved as active center, negligible differences (<0.05 eV) between DFT and DFT+U method were reported for the free energies.57 Therefore, the electron–electron exchange–correlation interaction was described within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof functional through out.58 The van der Waals interactions were accounted for by using the Grimme custom method for DFT-D3.59 The projector augmented wave (PAW) method has been used to describe the inert core electrons.60,61 A cut-off energy of 450 eV was employed for the expansion of the wave functions. A vacuum gap of 15 Å was imposed to avoid interactions between periodic images. Since the solvation-induced stabilization of reaction intermediates in the NRR is within 0.2 eV (affecting the limiting potential for the NRR by about 0.1 eV),62 the effects of solvation were not taken into account. The different configurations were constructed in a 72-atom graphene layer, where 4 carbon atoms were removed to achieve a cavity and anchor the Cu and Co atoms. The coordination of Cu and Co was realized by substitution of C atoms with O and N atoms. Therefore, the basic model was a periodic graphene structure containing 62C atoms, one Cu atom, one Co atom, three O and three N atoms. We relaxed the structures until the total energy changes within 1 × 10−4 eV per atom and the Hellmann–Feynman force on each atomic site was less than 0.02 eV Å−1. A 2 × 2 × 1 Monkhorst–Pack k-point sampling was used for Brillouin-zone integration throughout.63 The formation energy of the anchored dual atom system was calculated according to the equation| |
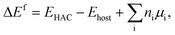 | (1) |
where EHAC is the total energy of the supercell with the hetero atoms anchored, Ehost is the total energy of the supercell without the metal atoms, ni is the number of atoms type i, introduced into the supercell when forming the configuration, and μi is the chemical potential of that atom in its standard metallic states.
Six coupled proton and electron (H+/e−) transfer steps are involved in the NRR process. In each step, the coupled H+/e− pair transfers from solution to the adsorbed species on the surface of the catalyst. The Gibbs free energy change (ΔG) of each step was calculated by employing the standard hydrogen electrode (SHE) model proposed by Norskov et al., where the chemical potential of the H+/e− pair was considered as half of the energy of H2 gas molecule.64 This way, the ΔG can be calculated as
| | |
ΔG = ΔE + ΔEZPE − TΔS + ΔGU + ΔGpH
| (2) |
where Δ
E is the reaction energy which can be obtained directly from the DFT calculations, Δ
EZPE and Δ
S represent the difference in zero point energy and entropy,
65 which were computed by considering only the vibrational frequencies. The frequencies and entropies of the molecules in the gas phase were taken from the NIST database. The temperature
T was set to be 298.15 K. Δ
GU is the free energy contribution related to the applied electrode potential. Δ
GpH is the concentration dependent correction of the H
+ free energy and can be obtained by the formula Δ
GpH =
kBT × pH × ln10, where the Boltzmann constant is denoted as
kB and the pH is set to zero. The potential-limiting step in the NRR processes possess the most positive Δ
G (expressed as Δ
Gmax) and the overpotential (
h) is calculated by the definition:
| | |
η = Uequilibrium − (−ΔGmax/e),
| (3) |
where the
Uequilibrium is the equilibrium potential of NRR and set to be −0.16 V.
66
3 Results and discussion
3.1 Heteronuclear CuCo DACs
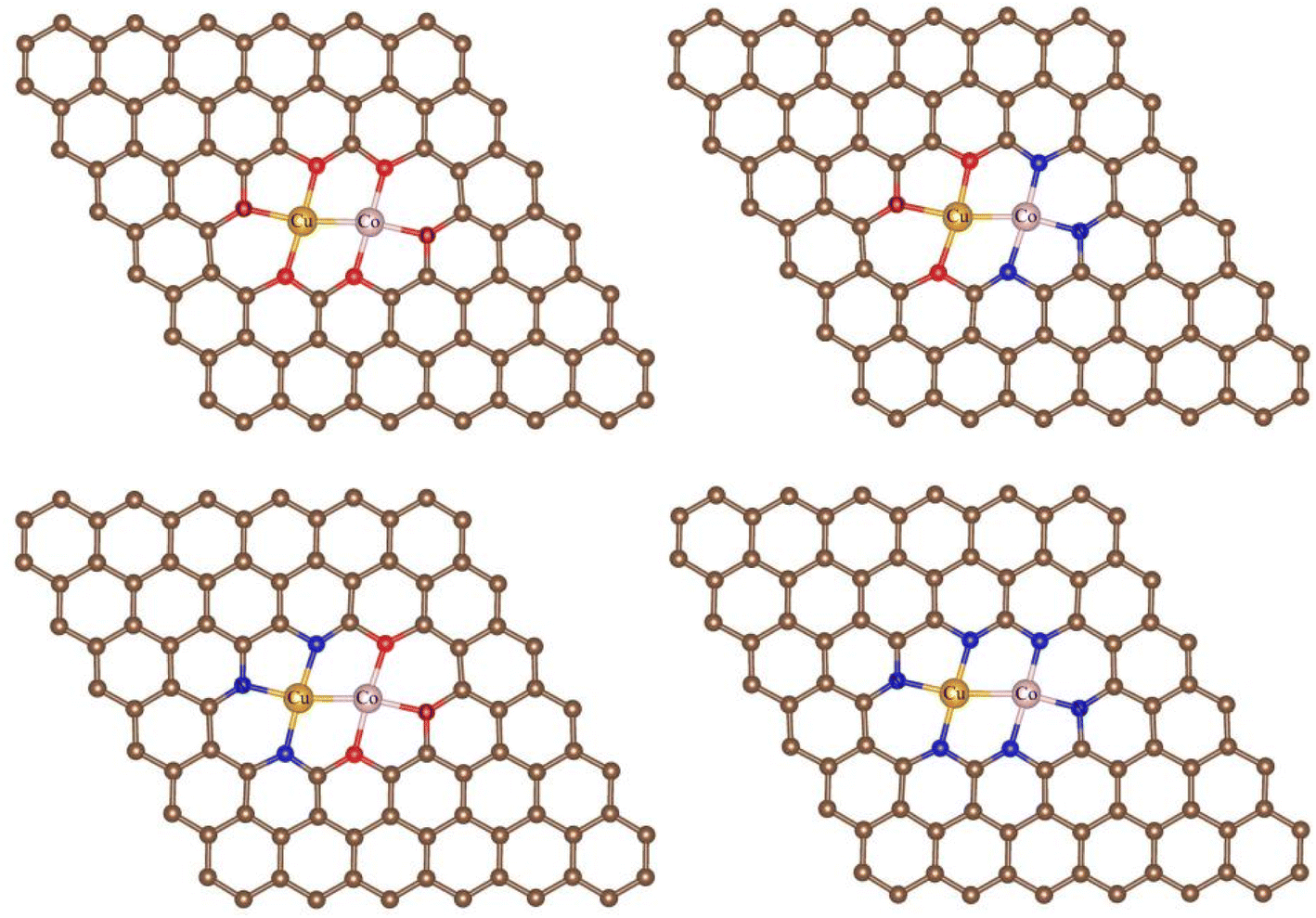
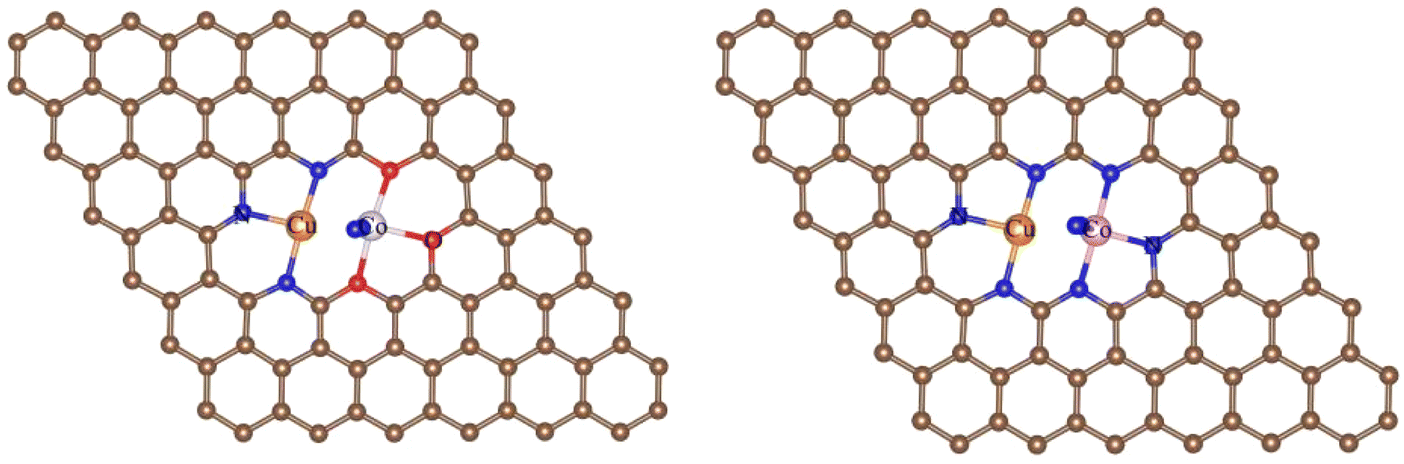
3.1.1 Stabilities. In the NRR process, besides the metal species, the coordination of the metal center also has a great impact on the adsorption, activation, and conversion of N2. According to experimental observation, Cu and Co are likely to be coordinated with N or O. While, theoretically, there should be many configurations (C36 = 120 in total), we can not consider all of them and just chose those typical as the experimental indicated.42 Therefore, four different coordination structures of heteronuclear DACs were constructed, as shown in Fig. 1. Table 1 shows the formation energies of these, with N-CuCo-N having the lowest value, while for O-CuCo-O it is so high that this configuration is unlikely to form. Then we investigated the stability of DACs by calculating the binding energies (Eb) of the hetero atoms with doped graphene, and compared with their corresponding cohesive energies (Ec) of Cu and Co metals. For a stable catalyst, it is vital that the metal atoms should not diffuse and aggregate. The definitions of Eb and Ec are:| |
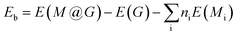 | (4) |
| | |
EC = E(Mbulk)/N − E(M)
| (5) |
where the E(M@G) is the energy of DAC, E(M), E(Mbulk) and N represent the energy of the single atom in vacuum, the energy of the bulk crystal unit cell of the corresponding metal, and the number of atoms in the unit cell, respectively.
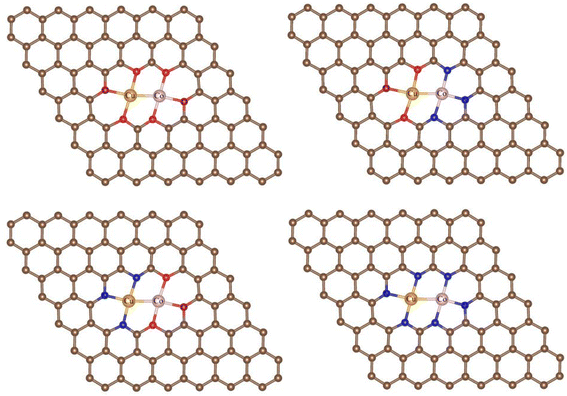 |
| | Fig. 1 Coordination structures of O-CuCo-O, O-CuCo-N, N-CuCo-O, N-CuCo-N. Brown spheres: C, red spheres: O, orange spheres: Cu, pink spheres: Co, blue spheres: N. | |
Table 1 Cohesive energy (Ec), binding energy (Eb) and formation energy (Ef) of HACs, all in eV
| |
Ec (cal.) |
Ec (exp.) |
| Cu |
−3.70 |
−3.5 |
| Co |
−5.27 |
−4.4 |
| |
Eb |
Ef |
| O-CuCo-O |
−0.25 |
4.72 |
| O-CuCo-N |
−5.59 |
3.10 |
| N-CuCo-O |
−5.37 |
3.33 |
| N-CuCo-N |
−10.89 |
2.96 |
The binding of metal atoms with N and O doped graphene is thermodynamically more favorable if Eb is more negative than Ec. Our results show that except for O-CuCo-O, all the other 3 configurations have a much more negative binding energy than the cohesive energy of Cu and Co in bulk metal, which means that Cu and Co atoms can anchor on doped graphene stably without aggregation. O-CuCo-O, with Eb being −0.25 eV, was proved to be thermodynamically unstable under 500 K. Therefore, further investigations were made only on the other three configurations in following.
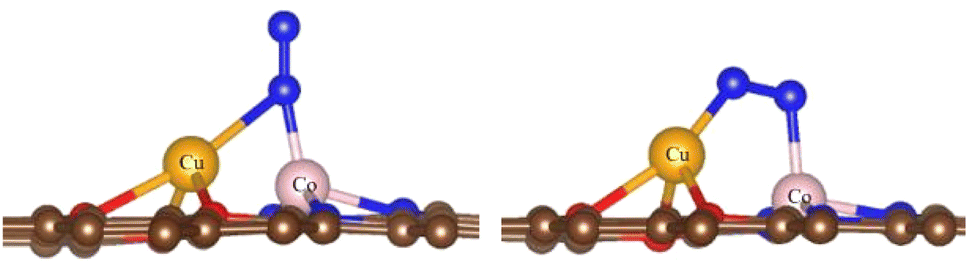
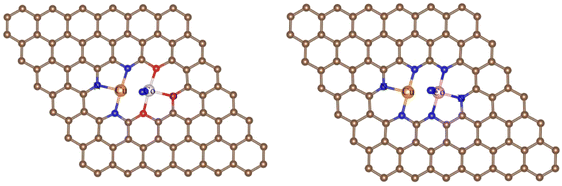
3.1.2 N2 conversion into NH3. The adsorption of N2 is an essential prerequisite of NRR, so we first calculated the adsorption properties of N2.For the O-CuCo-N configuration, we considered three possibilities: adsorption on top of the Cu site, on top of the Co site and on the CuCo site. The structural optimization showed that, upon adsorption of N2 on the Cu site, the structure will break down due to the weak bonding between Cu and O, but N2 can be easily adsorbed on the CuCo site with end-on and side-on adsorption mode, as shown in Fig. 2. The adsorption energy in the side-on mode is −0.57 eV and in the end-on mode −0.50 eV, the difference being negligible.
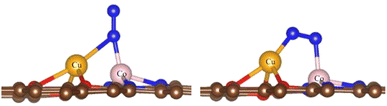 |
| | Fig. 2 Adsorption modes of N2 on the Cu-Co site with end-on (left) and side-on mode (right). Brown spheres: C, red spheres: O, orange spheres: Cu, pink spheres: Co, blue spheres: N. | |
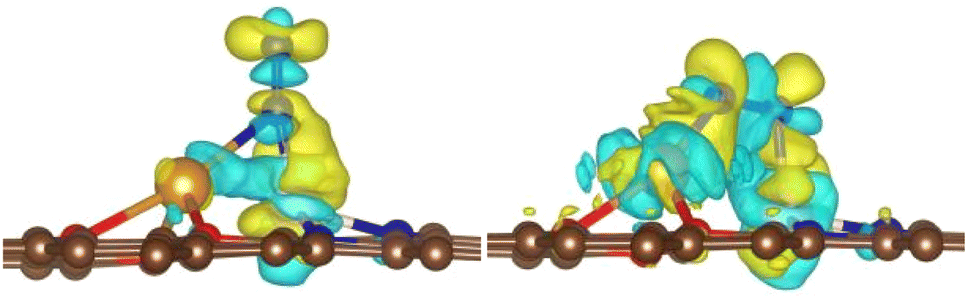
In addition, the charge density difference in Fig. 3 clearly show that Cu and Co donate electrons to the empty π* orbitals of N2, while, the loss of electrons in the N–N σ bond indicates that the N2 is indeed activated. The Bader charge analysis also shows that, after adsorption, N2 gains 0.41 e and 0.53 e from the substrate with end-on and side-on adsorption mode, respectively. Consequently, the bond lengths of N2 are elongated from 1.12 Å in the free gas state to 1.15 Å and 1.18 Å for end-on and side-on adsorption mode, respectively. These results suggest that the side-on adsorbed N2 is better activated.
 |
| | Fig. 3 Charge density difference (δρ = ρA+B − ρA − ρB) of adsorbed N2 with end-on (left) and side-on mode (right) on O-CuCo-N DAC. Yellow stands for electron accumulation and cyan for electron loss, the isosurface value is 0.004 e/a02. | |
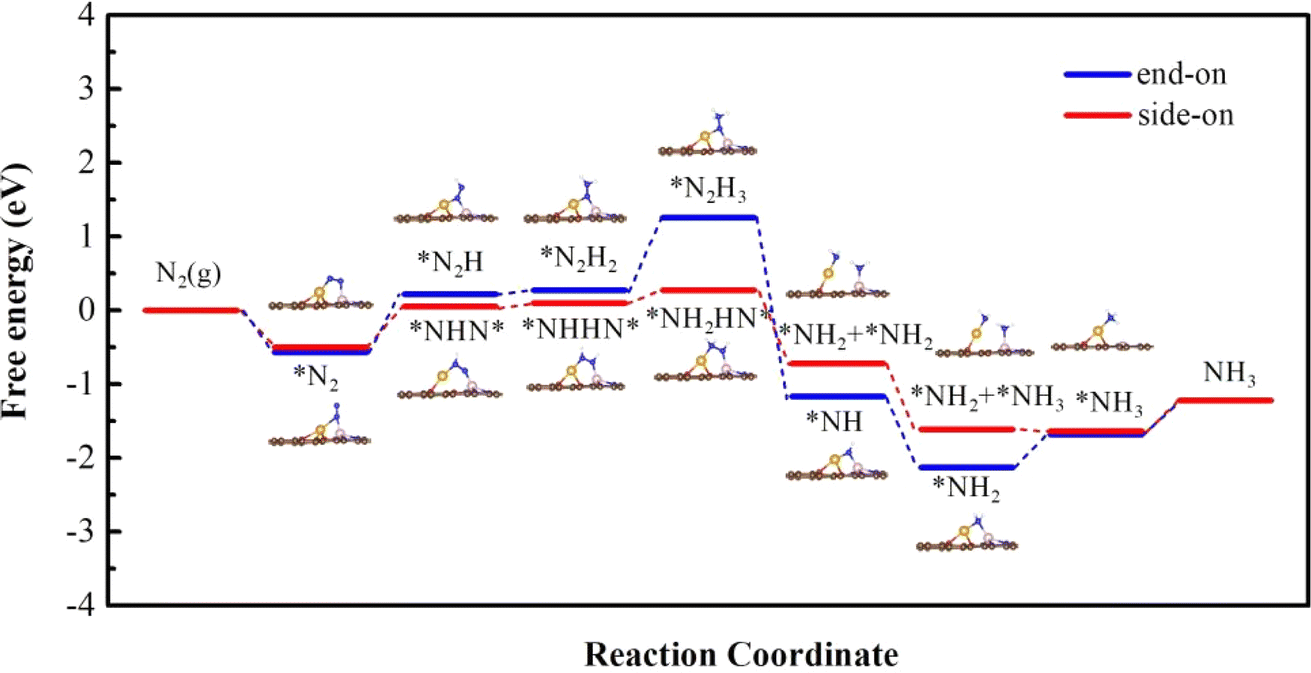
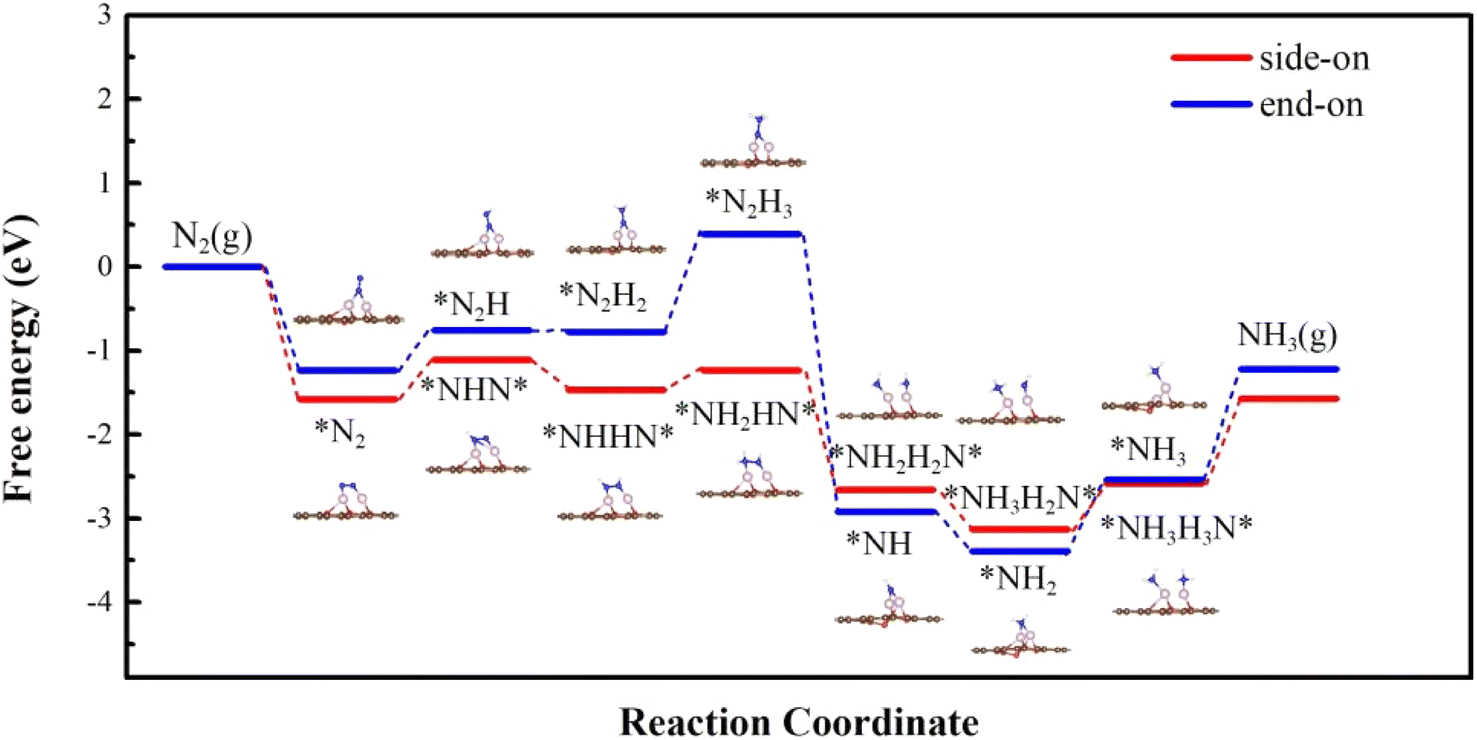
We calculated first the possible nitrogen reduction reaction pathway starting from the side-on mode (red lines in Fig. 4.) As can be seen, the first H+/e− pair prefers to attack the N atom that is connected to Cu. The energy change of this step is 0.55 eV, and induces a N–N bond length elongation from 1.18 Å to 1.27 Å. In the second step, *NHHN* or *NNH2 can form through the enzymatic, by H+/e− alternatively attacking the N atom connected with Co or through an alternative pathway by H+/e− attacking the same N atom. The results show that to form *NHHN* and *NNH2, it needs 0.05 eV and 0.95 eV respectively. Therefore, the enzymatic pathway from *NHNH* is further considered. In the third step, *NH2HN* is formed with an uphill energy of 0.17 eV, and the bond length is correspondingly further elongated from 1.33 Å in *NHHN* to 1.46 Å. In the fourth step, the N–N bond breaks and forms *NH2 + *NH2 on the Cu and Co atoms with an energy release of 0.99 eV. The next two steps to form*NH3 are still downhill in energy by 0.90 eV and 0.02 eV. Desorption free energy of NH3 is calculated to be 0.42 eV, indicating the excellent durability of O-CuCo-N DAC.
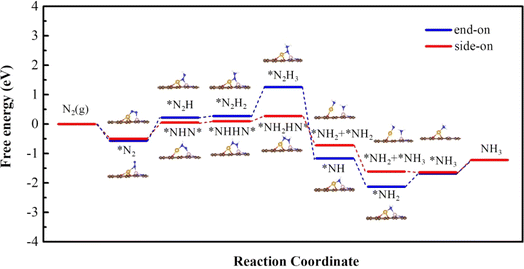 |
| | Fig. 4 Free energy diagram along the reaction path of NRR proceeded on O-CuCo-N starting with side-on and end-on configuration. | |
Next, we considered the distal reaction pathway from the end-on mode. As shown in Fig. 4 (blue lines), the Gibbs free energy change of the first hydrogenation step is 0.79 eV, which is 0.24 eV higher than that from the side-on mode. From *N2H to *N2H2, it only needs an energy input of 0.06 eV. The potential limiting step is from *N2H2 to *N2H3 with an energy change of 0.98 eV. The following two steps are downhill in energy, whereas, the last step to form *NH3 and the final desorption of NH3 are endothermic processes where an energy investment of 0.44 eV and 0.46 eV is needed, respectively.
Therefore, for O-CuCo-N DAC the NRR prefers to take place along the enzymatic pathway from the initial side-on N2 adsorption mode and the potential-limiting step is the first hydrogenation step of N2 with overpotential η being 0.39 V.
For the N-CuCo-O and N-CuCo-N configuration, we found that after relaxation N2 will finally bond to the O/N coordinated Co atom with end-on adsorption mode (see Fig. 5). The adsorption of N2 is strong in the N-CuCo-O configuration, while much weaker in N-CuCo-N, with the adsorption energy calculated to be -0.94 eV and −0.13 eV, respectively. For N-CuCo-O, the first hydrogenation step of N2, which is also the potential determining step, needs an input energy of 1.41 eV (Fig. S1†). This is much higher than 0.79 eV/0.55 eV in O-CuCo-N configuration. Also in N-CuCo-N, it takes 1.50 eV to be first hydrogenated. Therefore, we may safely draw the conclusion that for the heteronuclear CuCo DACs considered herein, the O-CuCo-N configuration is the best one given the formation energy, stability, and NRR activities.
 |
| | Fig. 5 Adsorption mode of N2 on Co site of N-CuCo-O and N-CuCo-N. | |
3.2 Homonuclear DACs
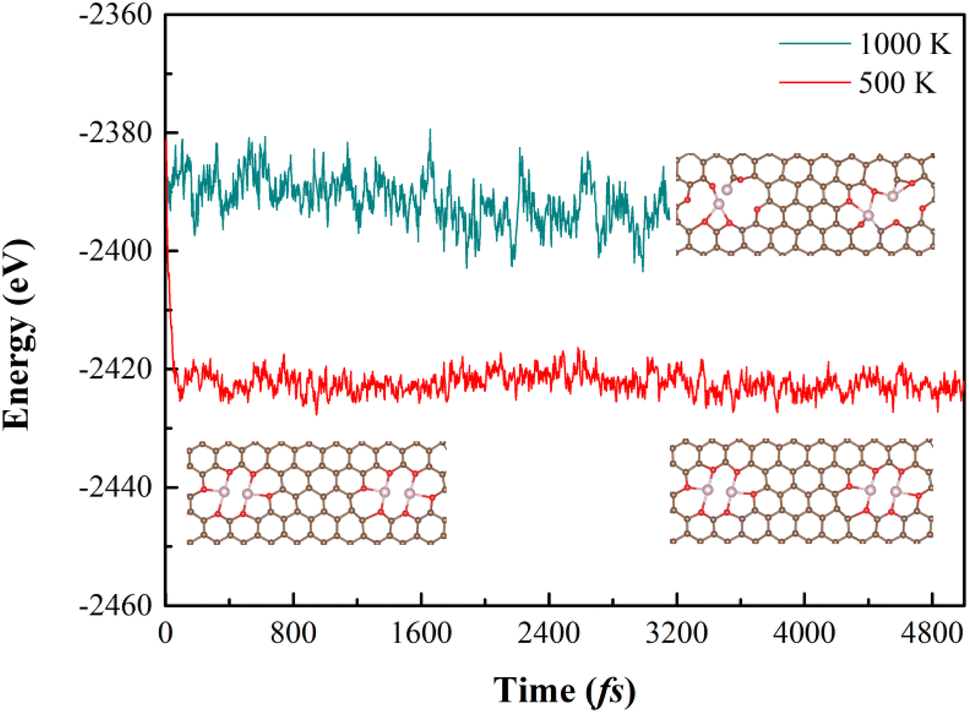
3.2.1 Stabilities. Since homonuclear DACs also has a big chance to form during the experimental fabrication, we investigated the NRR properties of the following possible DACs: O-CoCo-O, O-CoCo-N, N-CoCo-N, O-CuCu-O, O-CuCu-N, N-CuCu-N. Results of geometry optimization suggest that O-CuCu-O is structurally not stable because of the weak bonding between Cu and O. The formation and binding energies listed in Table 2 indicate that O-CoCo-O has a relatively weak binding (Eb being −1.34 eV) between CoCo and the carbon support, as well as a high formation energy. All the other configurations are strongly bonded and have a formation energy less than 4 eV. Considering that O-CoCo-O is most likely not stable, we carried out the molecule dynamics simulation. The results show that the energy fluctuates and the structure also breaks down in 3000 fs under 1000 K, but under 500 K, the structure is thermodynamically stable (see Fig. 6). We subsequently studied their NRR properties.
Table 2 Cohesive energy (Ec), binding energy (Eb) and formation energy (Ef) of DACs, all in eV
| |
Ec (cal.) |
Ec (exp.) |
| Cu |
−3.70 |
−3.5 |
| Co |
−5.27 |
−4.4 |
| |
Eb |
Ef |
| O-CoCo-O |
−1.34 |
5.26 |
| O-CoCo-N |
−6.65 |
3.67 |
| N-CoCo-N |
−12.12 |
3.36 |
| O-CuCu-N |
−4.73 |
2.34 |
| N-CuCu-N |
−9.36 |
2.86 |
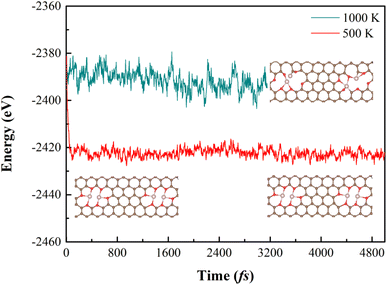 |
| | Fig. 6 The energy and structure of O-CoCo-O vs. number of steps in 5000 fs. | |
3.2.2 N2 conversion into NH3. For O-CoCo-O, our results show that N2 can be strongly adsorbed with both side-on and end-on mode, and the first hydrogenation step takes 0.47 and 0.48 eV, respectively, which are even lower than that in O-CuCo-N. Therefore, we continue to study the following reaction steps through the advantageous distal pathway from end-on mode and enzymatic pathway from the side-on mode. As shown in Fig. 7, it needs a high energy input of 1.17 eV for *N2H2 to form *N2H3 and it takes 1.32 eV for the *NH3 desorption through the distal pathway. While, through the enzymatic pathway, the highest hydrogenation barrier takes places in forming *NH3NH3* with an uphill energy of 0.54 eV, which is comparable with the limiting potential in O-CuCo-N. Whereas, the desorption of *NH3 requires a high energy of 1.01 eV, indicating a relatively poor durability.
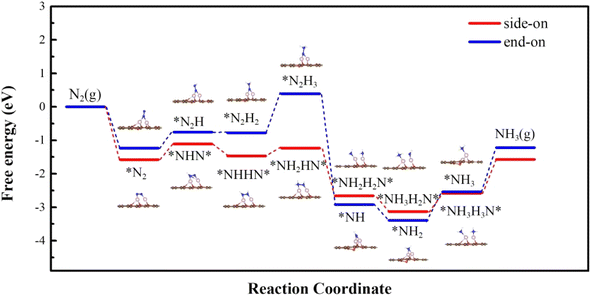 |
| | Fig. 7 Free energy diagram along the reaction path of NRR proceeded on O-CoCo-O starting with side-on and end-on configuration. | |
As can be seen from Table 3, for O-CoCo-N, ΔG(N2→N2H) is lower from the side-on N2 adsorption mode (0.75 eV) than from the end-on N2 adsorption mode (1.29 eV). For N-CoCo-N, ΔG(N2→N2H) is 0.61 eV from side-on N2 adsorption mode. While, for O-CuCu-N, N2 prefers to adsorb with end-on mode on Cu site connected to O, but with a much higher ΔG(N2→N2H) of 1.48 eV. As for N-CuCu-N, N2 cannot be effectively activated as the bond length (1.116 Å) elongation is negligible, also the first hydrogenation step needs an energy of more than 2.00 eV.
Table 3 Calculated adsorption energy of N2 , N–N bond length, Gibbs free energy change for the first hydrogenation step and maximum Gibbs free energy change (corresponds to potential determining step, PDS) of NRR in different DACs. All energy in eV
| Heteronuclear DACs |
ΔG(N2) |
N–N bond length |
ΔG(N2 → N2H) |
ΔGmax (PDS) |
| O-CuCo-N (side-on) |
−0.50 |
1.185 |
0.55 |
0.55 |
| O-CuCo-N (end-on) |
−0.57 |
1.147 |
0.79 |
0.98 |
| N-CuCo-O (end-on Co site) |
−0.94 |
1.142 |
1.41 |
1.41 |
| N-CuCo-N (end-on Co site) |
−0.13 |
1.136 |
1.50 |
— |
| Homonuclear DACs |
ΔG(N2) |
N–N bond length |
ΔG(N2 → N2H) |
ΔGmax (PDS) |
| O-CoCo-O (side-on) |
−1.58 |
1.210 |
0.47 |
1.01 |
| O-CoCo-O (end-on) |
−1.24 |
1.170 |
0.48 |
1.32 |
| O-CoCo-N (side-on) |
−0.21 |
1.186 |
0.75 |
— |
| O-CoCo-N (end-on) |
−0.67 |
1.143 |
1.29 |
— |
| N-CoCo-N (side-on) |
−0.22 |
1.182 |
0.61 |
— |
| O-CuCu-N (end-on) |
−0.22 |
1.129 |
1.48 |
— |
| N-CuCu-N (side-on) |
−0.23 |
1.116 |
2.03 |
— |
As summarized in Table 3, while O-CoCo-O is the only homonuclear DAC that has comparable ΔG(N2→N2H) with O-CuCo-N, it is not as good as O-CuCo-N DAC since the structure is relatively not stable enough and desorption of NH3 is difficult to achieve. Heteronuclear O-CuCo-N DAC exhibits the best NNR activity due to the lowest ΔGmax (PDS) of 0.55 eV, which is also lower than the reported results in different references, as shown in Table 4.
Table 4 Comparisons of the maximum Gibbs free energy change of our work with data reported in difference references
| |
O-CuCo-Nour work |
FeMoNxC41 |
Fe2Cu@C3N443 |
Fe–Fe@C2N44 |
Fe–Mo@C2N44 |
| ΔGmax (eV) |
0.55 |
0.91 |
0.58 |
0.76 |
0.68 |
3.3 Comparisons with SACs
Earlier, N coordinated single atom catalysts have been widely investigated. In reality, dual-atom and single-atom active sites will coexist to a large extent. Therefore, we compared the NRR activities of DACs and possible SACs: CuN4, CoN4, CuO4 and CoO4.
We analyzed the stabilities of SACs and found that both Cu and Co have strong binding with N coordinations, which are more negative than the cohesive energies of Cu and Co in bulk. In contrast, Cu and Co have a pretty weak binding with O coordinations, as can be seen from Table 5. Though both CuO4 and CoO4 can strongly bond with N2, as the adsorption energies being −0.69 eV and −1.08 eV, respectively, the weak binding indicates that Cu and Co may not stay disperse as single atoms but prefer to get aggregated. Inversely, though CuN4 and CoN4 are stable in binding, the adsorption of N2 is very weak in both the end-on and side-on mode. Most importantly, the corresponding Gibbs free energy changes from *N2 to *N2H are all very large in both O and N coordinated SACs, e.g. the lowest ΔG(N2→N2H) is 0.93 eV in CoO4 from the side-on adsorption mode. Therefore, Cu SACs and Co SACs are not in the same league as DACs in NRR catalytic performance.
Table 5 Adsorption energies (s denotes side-on configuration), binding energies and corresponding cohesive energies of N and O coordinated SACs (energy in eV)
| SACs |
Eb |
Gads(N2) |
ΔG(N2→N2H) |
| CuN4 |
−5.28 |
0.37/0.37(s) |
2.13/2.16 |
| CoN4 |
−7.65 |
0.29/0.38(s) |
1.17/1.17 |
| CuO4 |
−0.28 |
−0.69/−0.28(s) |
1.47/1.08 |
| CoO4 |
−0.92 |
−1.08/−0.66(s) |
1.18/0.93 |
3.4 Competition with HER
As has been discussed above, the heteronuclear O-CuCo-N is the most promising highly efficient NRR electrocatalyst. To assure that a good NRR performance can indeed be achieved in O-CuCo-N DAC, we have also investigated the hydrogen evolution reaction (HER), as it is the major competing reaction that affects the faradaic efficiency of the nitrogen reduction reaction. The ΔG of HER was calculated to be 0.92 eV in O-CuCo-N, which is much higher than that of NRR. Theoretically, the faradaic efficiency of NRR can be estimated according to the Boltzmann distribution:67
where δ is the Gibbs free energy difference between HER and NRR limiting-potential step, kB is the Boltzmann constant, and T is the room temperature which was set to be 298.15 K. The O-CuCo-N achieves a high theoretical faradaic efficiency of 100%, demonstrating a high NRR selectivity over HER.
3.5 Activity mechanisms
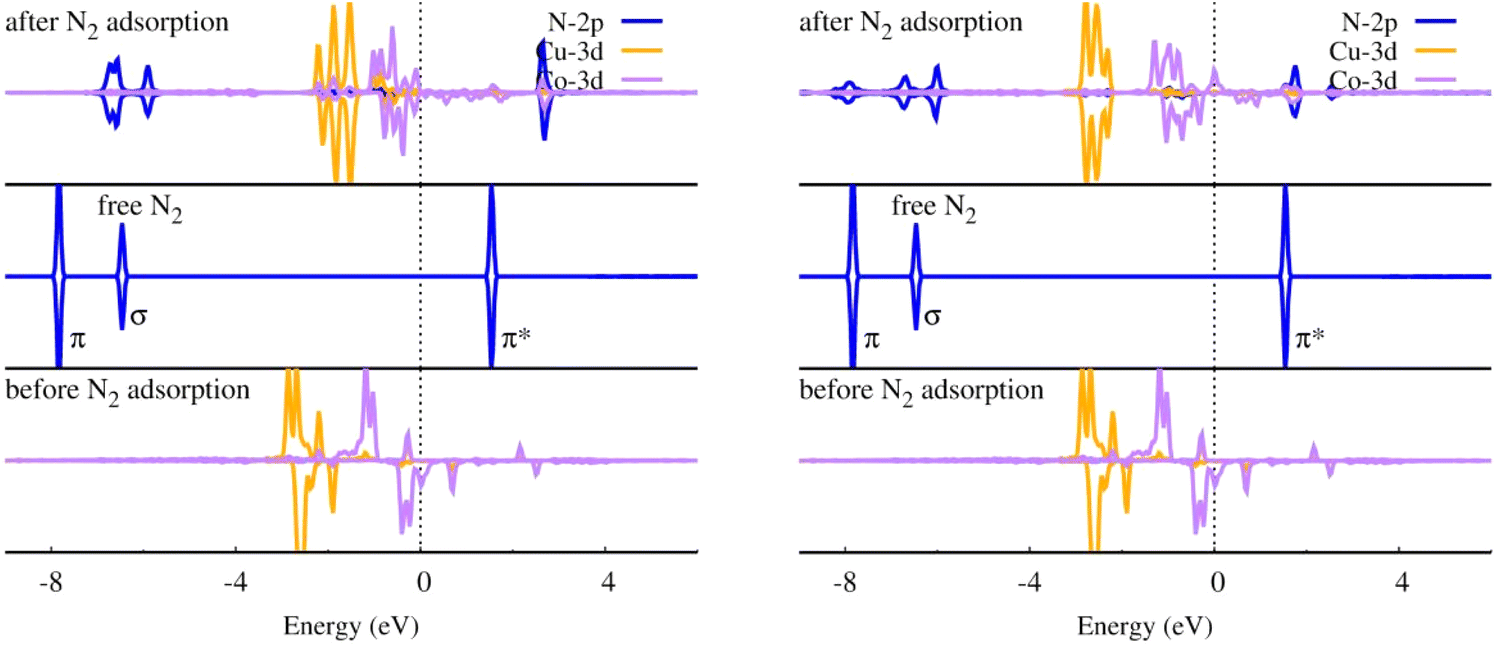
For NRR catalysts, transition metal atoms are always chosen as the active center. N2 adsorption and activation are usually manipulated by the mechanism of electron acceptance-donation between d orbitals of the transition atoms and the frontier molecular orbital of N2.68 From the projected density of states (PDOS) in Fig. 8, it can be seen that before N2 adsorption, the O-CuCo-N configuration has a large spin polarization mainly contributed by Co and the Co spin-down d orbital is much closer to the N2 empty π* orbital than Cu, which means that Co will contribute most to the N2 activation, i.e., the Cu atom may take the role as electrons bank. After N2 adsorption, the spin polarization of Co is weakened, and some d–π* orbitals are partially occupied due to the electrons transferred from Co and Cu d states.
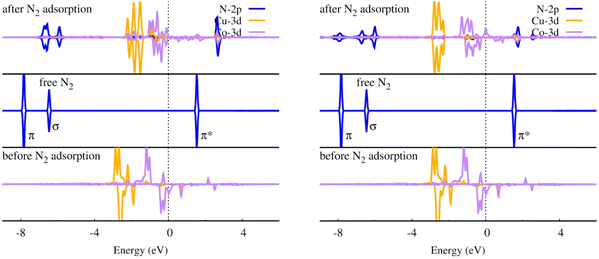 |
| | Fig. 8 Density of states of N2 with end-on (left) and side-on adsorption mode (right) on O-CuCo-N DAC. | |
To deepen our insight into the origin of the adsorption behaviour of N2, we made a calculation on the spin moment of Fe and Co. We found that N2 prefers to adsorb with end-on mode on the metal site which is more highly spin-polarized, namely the transition metal Co. The side-on N2 adsorption mode can be achieved as the two metal atoms are both spin polarized.
Compared with the homonuclear DACs, we found that the existence of Cu can regulate the spin polarization of Co. The magnetic moment of Co in O-CoCo-N is 1.208 μB (O–Co) and 0.552 μB (Co–N), respectively, leading to N2 end-on adsorption on O coordinated Co site. Whereas, the magnetic moment of Co in O-CuCo-N changes to 1.326 μB (Co–N) due to regulation of Cu (0.202 μB), leading to the N2 end-on adsorption site change to N coordinated Co in O-CuCo-N. Since both Cu and Co are spin polarized in O-CoCo-N and O-CuCo-N, N2 side-on adsorption can also be realized. Also, the coordination structure of metal atom has an effect on its spin polarization. Compared with O-CuCo-N (Co: 1.326 μB, Cu: 0.202 μB), the spin polarization of N coordinated Cu in N-CuCo-N is negligible (Co: 0.275 μB, Cu: 0.009 μB), resulting in only an end-on N2 adsorption mode.
In addition, the d orbital energy level center (εd) of the metal atom is of vital role in the charge transfer between N2 and the catalysts. We found that εd of Cu is much lower in energy than that of Co, Co is the main electron donor, as proved in Fig. 8, PDOS of O-CuCo-N. Coexistence with Cu could alter the d orbital energy level of Co. As shown in Table 6, the spin-down εd of Co in O-CoCo-N lies at −0.779 eV, while the presence of Cu pulls the εd of Co in O-CuCo-N up to −0.356 eV, which will facilitate the charge transfer between N2 and DACs. On the other hand, the coordination environment has an effect on the εd too. The εd of Co in O-CuCo-N is 0.438 eV higher than εd of Co in N-CuCo-O. The PDOS of Co-3d in O-CuCo-N, O-CoCo-N, N-CuCo-O and N-CuCo-N as displayed in Fig. S2,† also indicates the effects of coordination on Co.
Table 6 d-Orbital energy level centers of Co in various configurations (all referenced to Fermi level EF = 0 eV)
| Configuration |
O-CuCo-N |
O-CoCo-N |
N-CuCo-O |
| εd (eV) |
Spin-up |
−1.635 |
−1.596 |
−0.901 |
| Spin-dw |
−0.356 |
−0.779 |
−0.794 |
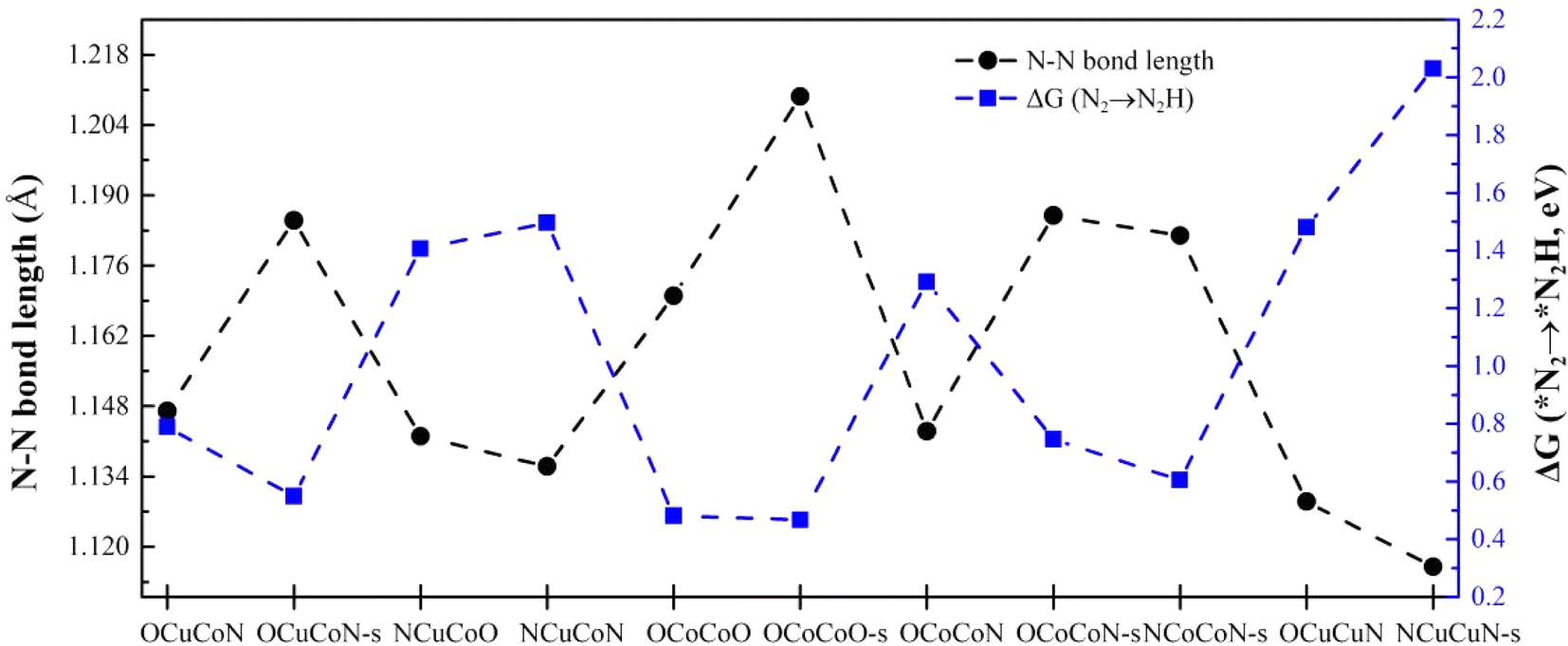
We summarized the adsorption energies, N–N bond length after adsorption and Gibbs free energy change of the first hydrogenation step ΔG(*N2→*N2H) (as shown in Table 4) and found that there exists an inverse relationship between the N–N bond length of *N2 and ΔG(N2→N2H). The longer the N–N bond length is, the lower ΔG(N2→N2H) will be. This phenomenon was observed in both heteronuclear, homonuclear DACs and SACs, as shown in Fig. 9 and S3.† The activation of the inert N2 can be reflected by the bond length elongation, which is determined by the charge transfer between N2 and metal active sites. Therefore, the more charge is transferred to N2, the better will the inert N2 be activated, and the easier will it be hydrogenated. However, such relationship does not exist between the adsorption energy of *N2 and the N–N bond length.
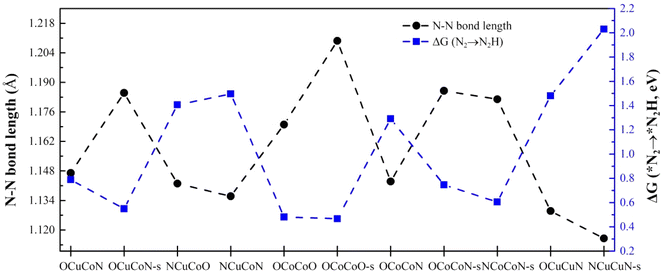 |
| | Fig. 9 The relationship between N–N bond length of adsorbed *N2 and Gibbs free energy change ΔG from *N2 to *N2H on different DACs and SACs. | |
3.6 Practical application of O-CuCo-N as NRR electrocatalyst
For practical application, O-CuCo-N should have good structural stability, electric conductivity and durability. Structural stability has been discussed when screening out the possible candidates. As shown in Fig. 8 and S2,† O-CuCo-N is metallic, endowing it excellent electrical conductivity. Since nitrogen atoms are contained in O-CuCo-N electrocatalyst, under the drive of certain potential the electrocatalyst may decompose and these nitrogen atoms could become the source of the formation of NH4+. Therefore, thermodynamics calculations were performed to evaluate the possibility of substrate decomposition, which can be expressed as O3CuCoN3 + 3(H+ + e−) → O3CuCoN2 + NH3(g). Accordingly, the free energy change for the decomposition reaction can be calculated following ΔGd = E(O3CuCoN2) + G(NH3) − E(O3CuCoN3) − 3G(H+ + e−). The required potential to drive the decomposition and then NH3 release can be obtained by Ud = −ΔGd/3e. Our calculated decomposition potential is −0.85 V, which is lower than the limiting potential of NRR (−0.39 V). Therefore, O-CuCo-N could serve as efficient NRR with high stability and durability.
4 Conclusions
In this paper, the potential for N2 fixation of the heteronuclear CuCo dual-atom electrocatalyst anchored on doped graphene, was investigated by DFT calculations. Our computations on various coordination structures revealed that the heteronuclear O-CuCo-N DAC is a promising candidate due to its relatively low formation energy, strong structural stability and high NRR activity, where NRR easily take place though the alternating pathway from the side-on N2 adsorption mode, since the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond can easily break under the pulling force of two metal atoms. The potential limiting step is the first hydrogenation (*NN*→*NHN*) step with a pretty low Gibbs free energy change of 0.55 eV. We also made comparisons with the possible Cu or Co single atom catalysts and homonuclear (CuCu and CoCo) dual-atom catalysts, which were found not good as heteronuclear O-CuCo-N DAC in NRR activity. In addition, O-CuCo-N also exhibits good selectivity and durability. The mechanism study reveals that Cu and the coordination structure could regulate the spin polarization and the d orbital energy level center of Co, adjusting the adsorption of intermediates, leading to the superior NRR performance of O-CuCo-N DAC. The discovery of the distinct interaction between dual metal atom sites that can regulate their electronic spin states provides a powerful strategy for guiding the design of highly efficient DACs or SACs with high loading in the future. We hope that our studies could motivate more experimental research on CuCo as an NRR electrocatalyst.
N bond can easily break under the pulling force of two metal atoms. The potential limiting step is the first hydrogenation (*NN*→*NHN*) step with a pretty low Gibbs free energy change of 0.55 eV. We also made comparisons with the possible Cu or Co single atom catalysts and homonuclear (CuCu and CoCo) dual-atom catalysts, which were found not good as heteronuclear O-CuCo-N DAC in NRR activity. In addition, O-CuCo-N also exhibits good selectivity and durability. The mechanism study reveals that Cu and the coordination structure could regulate the spin polarization and the d orbital energy level center of Co, adjusting the adsorption of intermediates, leading to the superior NRR performance of O-CuCo-N DAC. The discovery of the distinct interaction between dual metal atom sites that can regulate their electronic spin states provides a powerful strategy for guiding the design of highly efficient DACs or SACs with high loading in the future. We hope that our studies could motivate more experimental research on CuCo as an NRR electrocatalyst.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support from the Zhejiang Provincial Natural Science Foundation (ZCLY24A0401) and National Natural Science Foundation of China under Grant No. 61804154. The support from the Hefei advanced computing center for theoretical calculations is acknowledged.
References
- V. Smil, Global Population and the Nitrogen Cycle, Sci. Am., 1997, 277, 76–81 CrossRef CAS.
- J. R. Jennings. Catalytic Ammonia Synthesis: Fundamentals and Practice, Plenum Press, New York, 1st edn, 1991 Search PubMed.
- D. J. Little, M. R. S. III and T. W. Hamann, Electrolysis of liquid ammonia for hydrogen generation, Energy Environ. Sci., 2015, 8, 2775–2781 RSC.
- A. Klerke, C. H. Christensen, J. K. Nfrskov and T. Vegge, Ammonia for hydrogen storage: challenges and opportunities, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- Y. Nishibayashi, Recent progress in transition-metal-catalyzed reduction of molecular dinitrogen under ambient reaction conditions, Inorg. Chem., 2015, 54, 9234–9247 CrossRef CAS PubMed.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Challenges in reduction of dinitrogen by proton and electron transfer, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- S. Back and Y. Jung, On the mechanism of electrochemical ammonia synthesis on the Ru catalyst, Phys. Chem. Chem. Phys., 2016, 18, 9161–9166 RSC.
- M. M. Shi, D. Bao, B. R. Wulan, Y. H. Li, Y. F. Zhang, J. M. Yan and Q. Jiang, Au Sub-Nanoclusters on TiO2 toward Highly Efficient and Selective Electrocatalyst for N2 Conversion to NH3 at Ambient Conditions, Adv. Mater., 2017, 29, 1606550 CrossRef PubMed.
- D. Bao, Q. Zhang, F.-L. Meng, H.-X. Zhong, M.-M. Shi, Y. Zhang, J.-M. Yan, Q. Jiang and X.-B. Zhang, Electrochemical Reduction of N2 under Ambient Conditions for Artificial N2 Fixation and Renewable Energy Storage Using N2/NH3 Cycle, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, A Spectroscopic Study on the Nitrogen Electrochemical Reduction Reaction on Gold and Platinum Surfaces, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS PubMed.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Ambient ammonia synthesis via palladium-catalyzed electrohydrogenation of dinitrogen at low overpotential, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- I.-Y. Jeon, H.-J. Choi, M. J. Ju, I. T. Choi, K. Lim, J. Ko, H. K. Kim, J. C. Kim, J.-J. Lee, D. Shin, S.-M. Jung, J.-M. Seo, M.-J. Kim, N. Park, L. Dai and J.-B. Baek, Direct nitrogen fixation at the edges of graphene nanoplatelets as efficient electrocatalysts for energy conversion, Sci. Rep., 2013, 3, 2260 CrossRef PubMed.
- X. Yu, P. Han, Z. Wei, L. Huang, Z. Gu, S. Peng, J. Ma and G. Zheng, Boron-Doped Graphene for Electrocatalytic N2 Reduction, Joule, 2018, 2, 1610–1622 CrossRef CAS.
- J. Zhao and Z. Chen, Single Mo Atom Supported on Defective Boron Nitride Monolayer as an Efficient Electrocatalyst for Nitrogen Fixation: A Computational Study, J. Am. Chem. Soc., 2017, 139, 12480–12487 CrossRef CAS PubMed.
- R. Zhang, X. Ren, X. Shi, F. Xie, B. Zheng, X. Guo and X. Sun, Enabling Effective Electrocatalytic N2 Conversion to NH3 by the TiO2 Nanosheets Array under Ambient Conditions, ACS Appl. Mater. Interfaces, 2018, 10, 28251–28255 CrossRef CAS PubMed.
- X. Wu, L. Xia, Y. Wang, W. Lu, Q. Liu, X. Shi and X. Sun, Mn3O4 Nanocube: An Efficient Electrocatalyst Toward Artificial N2 Fixation to NH3, Small, 2018, 14, e1803111 CrossRef PubMed.
- X. Ren, G. Cui, L. Chen, F. Xie, Q. Wei, Z. Tian and X. Sun, Electrochemical N2 fixation to NH3 under ambient conditions: Mo2N nanorod as a highly efficient and selective catalyst, Chem. Commun., 2018, 54, 8474–8477 RSC.
- L. Zhang, X. Ji, X. Ren, Y. Luo, X. Shi, A. M. Asiri, B. Zheng and X. Sun, Efficient Electrochemical N2 Reduction to NH3 on MoN Nanosheets Array under Ambient Conditions, ACS Sustainable Chem. Eng., 2018, 6, 9550–9554 CrossRef CAS.
- L. Zhang, X. Ji, X. Ren, Y. Ma, X. Shi, Z. Tian, A. M. Asiri, L. Chen, B. Tang and X. Sun, Electrochemical Ammonia Synthesis via Nitrogen Reduction Reaction on a MoS2 Catalyst: Theoretical and Experimental Studies, Adv. Mater., 2018, 30, 1800191 CrossRef PubMed.
- X. Li, T. Li, Y. Ma, Q. Wei, W. Qiu, H. Guo, X. Shi, P. Zhang, A. M. Asiri, L. Chen, B. Tang and X. Sun, Boosted Electrocatalytic N2 Reduction to NH3 by Defect-Rich MoS2 Nanoflower, Adv. Energy Mater., 2018, 1801357 CrossRef.
- X. Zhao, X. Lan, D. Yua, H. Fu, Z. Liu and T. Mu, Deep eutectic-solvothermal synthesis of nanostructured Fe3S4 for electrochemical N2 fixation under ambient conditions, Chem. Commun., 2018, 54, 13010–13013 RSC.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Single-Atom Electrocatalysts, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Single-Atom Catalysts: Synthetic Strategies and Electrochemical Applications, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- C. Choi, S. Back, N.-Y. Kim, J. Lim, Y.-H. Kim and Y. Jung, Suppression of Hydrogen Evolution Reaction in Electrochemical N2 Reduction Using Single-Atom Catalysts: A Computational Guideline, ACS Catal., 2018, 8, 7517–7525 CrossRef CAS.
- C. Ling, L. Shi, Y. Ouyang, X. C. Zeng and J. Wang, Nanosheet Supported Single-Metal Atom Bifunctional Catalyst for Overall Water Splitting, Nano Lett., 2017, 17, 5133–5139 CrossRef CAS PubMed.
- C. Zhu, S. Fu, J. Song, Q. Shi, D. Su, H. E. Mark, X. Li, D. Xiao, D. Li, L. Estevez, D. Du and Y. Lin, Self-Assembled Fe-N-Doped Carbon Nanotube Aerogels with Single-Atom Catalyst Feature as High-Efficiency Oxygen Reduction Electrocatalysts, Small, 2017, 13, 1603407 CrossRef PubMed.
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Heterogeneous Single-Atom Catalyst for Visible-Light-Driven High-Turnover CO2 Reduction: The Role of Electron Transfer, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed.
- J. Zhao and Z. Chen, Single Mo Atom Supported on Defective Boron Nitride Monolayer as an Efficient Electrocatalyst for Nitrogen Fixation: A Computational Study, J. Am. Chem. Soc., 2017, 139, 12480–12487 CrossRef CAS PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong and Y. Wang, Design of Single-Atom Co-N5 Catalytic Site: A Robust Electrocatalyst for CO2 Reduction with Nearly 100% CO Selectivity and Remarkable Stability, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- Y. Q. Le, J. Gu and W. Q. Tian, Nitrogen-fixation catalyst based on graphene: every part counts, Chem. Commun., 2014, 50, 13319–13322 RSC.
- X. F. Li, Q. K. Li, J. Cheng, L. Liu, Q. Yan, Y. Wu, X. H. Zhang, Z. Y. Wang, Q. Qiu and Y. Luo, Conversion of Dinitrogen to Ammonia by FeN3-Embedded Graphene, J. Am. Chem. Soc., 2016, 138, 8706–8709 CrossRef CAS PubMed.
- J. Zhao and Z. Chen, Single Mo Atom Supported on Defective Boron Nitride Monolayer as an Efficient Electrocatalyst for Nitrogen Fixation: A Computational Study, J. Am. Chem. Soc., 2017, 139, 12480–12487 CrossRef CAS PubMed.
- J. Zhao, J. Zhao and Q. Cai, Single transition metal atom embedded into a MoS2 nanosheet as a promising catalyst for electrochemical ammonia synthesis, Phys. Chem. Chem. Phys., 2018, 20, 9248–9255 RSC.
- Z. Wei, Y. Zhang, S. Wang, C. Wang and J. Ma, Fe-doped phosphorene for the nitrogen reduction reaction, J. Mater. Chem. A, 2018, 6, 13790–13796 RSC.
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lu, S. Bak, Z. Liang, S. Zhao, E. Stavitski, J. Luo, R. R. Adzic and H. Xin, Atomically Dispersed Molybdenum Catalysts for Efficient Ambient Nitrogen Fixation, Angew. Chem., Int. Ed., 2019, 58, 2321–2325 CrossRef CAS PubMed.
- M. Wang, S. Liu, T. Qian, J. Liu, J. Zhou, H. Ji, J. Xiong, J. Zhong and C. Yan, Over 56.55% Faradaic efficiency of ambient ammonia synthesis enabled by positively shifting the reaction potential, Nat. Commun., 2019, 10, 341 CrossRef CAS PubMed.
- W. Zhao, L. Zhang, Q. Luo, Z. Hu, W. Zhang, S. Smith and J. Yang, Single Mo1(Cr1) Atom on Nitrogen-Doped Graphene Enables Highly Selective Electroreduction of Nitrogen into Ammonia, ACS Catal., 2019, 9, 3419–3425 CrossRef CAS.
- S. Zhang, M. Jin, T. Shi, M. Han, Q. Sun, Y. Lin, Z. Ding, L. Zheng, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Electrocatalytically Active Fe-(O-C2)4 Single-Atom Sites for Efficient Reduction of Nitrogen to Ammonia, Angew. Chem., Int. Ed., 2020, 59, 13423–13429 CrossRef CAS PubMed.
- T. He, Y. Chen, Q. Liu, B. Lu, X. Song, H. Liu, M. Liu, Y.-N. Liu, Y. Zhang, X. Ouyang and S. Chen, Theory-Guided Regulation of FeN4 Spin State by Neighboring Cu Atoms for Enhanced Oxygen Reduction Electrocatalysis in Flexible Metal-Air Batteries, Angew. Chem., Int. Ed., 2022, 61, e202201007 CrossRef CAS PubMed.
- Z. Xiao, P. Sun, Z. Qiao, K. Qiao, H. Xu, S. Wang and D. Cao, Atomically dispersed Fe-Cu dual-site catalysts synergistically boosting oxygen reduction for hydrogen fuel cells, Chem. Eng. J., 2022, 446, 137112 CrossRef CAS.
- Y. Li, Q. Zhang, C. Li, H.-N. Fan, W.-B. Luo, H.-K. Liu and S.-X. J. J. O. M. C. A. Dou, Atomically dispersed metal dimer species with selective catalytic activity for nitrogen electrochemical reduction, J. Mater. Chem. A, 2019, 7, 22242–22247 RSC.
- W. Cui, B. Geng, X. Chu, J. He, L. Jia, X. Han, X. Wang, S. Song and H. Zhang, Coupling Fe and Mo single atoms on hierarchical N-doped carbon nanotubes enhances electrochemical nitrogen reduction reaction performance, Nano Res., 2022, 16, 5743–5749 CrossRef.
- X. Wang, S. Qiu, J. Feng, Y. Tong, F. Zhou, Q. Li, L. Song, S. Chen, K. H. Wu and P. J. A. M. Su, Confined Fe-Cu Clusters as Sub-Nanometer Reactors for Efficiently Regulating
the Electrochemical Nitrogen Reduction Reaction, Adv. Mater., 2020, 32, e2004382 CrossRef PubMed.
- Y. Zhang, X. Wang, T. Liu, Q. Dang, L. Zhu, Y. Luo, J. Jiang and S. Tang, Charge and spin communication between dual metal single-atom sites on C2N sheets: regulating electronic spin moments of Fe atoms for N2 activation and reduction, J. Mater. Chem. A, 2022, 10, 23704 RSC.
- X. Lv, W. Wei, Y. Huang, Y. Dai and T. Frauenheim, High-throughput screening of synergistic transition metal dual-atom catalysts for efficient nitrogen fixation, Nano Lett., 2022, 22, 4475–4481 CrossRef PubMed.
- F. Wang, Y.-p. Liu, H. Zhang and K. Chu, CuO/Graphene Nanocomposite for Nitrogen Reduction Reaction, ChemCatChem, 2019, 11, 1441–1447 CrossRef CAS.
- S. Liu, M. Wang, H. Ji, X. Shen, C. Yan and T. Qian, Altering the rate-determining step over cobalt single clusters leading to highly efficient ammonia synthesis, Natl. Sci. Rev., 2021, 8, nwaa136 CrossRef CAS PubMed.
- P. Chen, N. Zhang, S. Wang, T. Zhou, Y. Tong, C. Ao, W. Yan, L. Zhang, W. Chu, C. Wu and Y. Xie, Interfacial Engineering of Cobalt Sulfide/Graphene Hybrids for Highly Efficient Ammonia Electrosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2019, 201817881 Search PubMed.
- B. Tang, Y. Zhou, Q. Ji, Z. Zhuang, L. Zhang, C. Wang, H. Hu, H. Wang, B. Mei, F. Song, S. Yang, B. M. Weckhuysen, H. Tan, D. Wang and W. Yan, A Janus dual-atom catalyst for electrocatalytic oxygen reduction and evolution, Nat. Synth., 2024, 3, 878–890 CrossRef.
- P. Chen, N. Zhang, S. Wang, T. Zhou, Y. Tong, C. Ao, W. Yan, W. C. L. Zhang, C. Wu and Y. Xie, Interfacial engineering of cobalt sulfide/graphene hybrids for highly efficient ammonia electrosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6635–6640 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Single Cobalt Atoms with Precise N-Coordination as Superior Oxygen Reduction Reaction Catalysts, Angew. Chem., Int. Ed., 2016, 55, 10800 CrossRef CAS PubMed.
- X. Wang, D. Cullen, Y.-T. Pan, S. Hwang, M. Wang, Z. Feng, J. Wang, M. Engelhard, H. Zhang, Y. He, Y. Shao, D. Su, K. More, J. Spendelow and G. Wu, Nitrogen-Coordinated Single Cobalt Atom Catalysts for Oxygen Reduction in Proton Exchange Membrane Fuel Cells, Adv. Mater., 2018, 30, 1706758 CrossRef PubMed.
- S. Zhang, W. Li, Y. Liu, J. Wang, G. Wang, Y. Zhang, M. Han and H. Zhang, A sulfonate group functionalized active carbon-based Cu catalyst for electrochemical ammonia synthesis under ambient conditions, Inorg. Chem. Front., 2019, 6, 2832–2836 RSC.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium, Phys. Rev. B, 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B, 1996, 54, 11169–11185 CrossRef CAS PubMed.
- W. Guo, S. Wang, H. Wang, Q. Cai and J. Zhao, Cooperation between single atom catalyst and support to promote nitrogen electroreduction to ammonia: a theoretical insight, J. Energy Chem., 2024, 96, 336–344 CrossRef CAS.
- J. P. Perdew, K. Burkeand and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2010, 27, 1787–1799 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B, 1994, 50, 17953 CrossRef PubMed.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Nørskov, The Challenge of Electrochemical Ammonia Synthesis: A New Perspective on the Role of Nitrogen Scaling Relations, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- J. K. Nfrskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, T. B. J. R. Kitchin and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- L. M. Azofra, N. Li, D. R. MacFarlane and C. Sun, Promising prospects for 2D d2-d4 M3C2 transition metal carbides (MXenes) in N2 capture and conversion into ammonia, Energy Environ. Sci., 2016, 9, 2545–2549 RSC.
- C. Ling, X. Bai, Y. Ouyang, A. Du and J. Wang, J. Phys. Chem. C, 2018, 122, 16842–16847 CrossRef CAS.
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S.-Z. Qiao, Building up a Picture of the Electrocatalytic Nitrogen Reduction Activity of Transition Metal Single Atom Catalysts, J. Am. Chem. Soc., 2019, 141, 9664–9672 CrossRef CAS PubMed.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Nitrogen Fixation and Reduction at Boron, Science, 2018, 359, 896–900 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yu Zhang and
Chenyu Zhang
*,
Yu Zhang and
Chenyu Zhang