
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into ESIPT-induced multicolor fluorescence emission in 2-(2′-hydroxy-5′-bromo)phenylbenzimidazole: a spectroscopic and TDDFT study†
Yumeng Wang‡
a,
Mingxia Hu‡a,
Qianqian Yuea,
Feng Xiaoqingb and
Yanying Zhao *a
*a
aSchool of Chemistry and Chemical Engineering, School of Materials Science and Engineering, Zhejiang Sci-Tech University, Hangzhou 310018, China. E-mail: dameng0902@163.com; H_mx469@outlook.com; 1326443499@qq.com; yyzhao@zstu.edu.cn
bSchool of Pharmacy, School of Medicine, Changzhou University, Changzhou 213164, China. E-mail: fxq@163.com
First published on 17th December 2024
Abstract
Although multicolor luminescent materials are widely used in information encryption and decryption based on the excited-state intramolecular proton transfer (ESIPT) reaction, there remains a significant gap in the mechanistic understanding of how solvent and pH conditions influence the ESIPT process. Owing to their ability to avoid self-absorption as well as provide large Stokes' shift and strong emission properties, ESIPT-generated molecules (ESIPT gens) have recently emerged as highly potential fluorophores. Herein, the ESIPT mechanism of bromine-based (2′-hydroxy-5′-bromo)phenylbenzimidazole (HBI-pBr) was investigated in solvents using spectroscopic measurements and time-dependent density functional theory (TD-DFT) calculations. The results indicated that multi-color fluorescence emissions were observed at 470, 458 and 416 nm in CH3OH doped with a base and acid. The potential energy profile rationalized the fluorescence mechanistic insights into the ESIPT reaction and pH-dependent dual response. Notably, nucleus-independent chemical shift (NICS_ZZ) values were applied to reveal the ESIPT process. We leveraged the bromine atom as an electron withdrawing group to manipulate ground and excited-state proton transfer, thereby offering a strategic approach for designing and developing an ESIPT fluorescence sensor for the detection of H+ and OH−. By studying the effect of solvent and pH conditions on HBI-pBr, the multicolor fluorescence mechanism of ESIPT was elucidated, thus laying a solid foundation for the design and synthesis of luminescent materials based on the ESIPT reaction.
Introduction
Excited-state intramolecular proton transfer (ESIPT) routinely results in a large Stokes' shift and even strong fluorescence emission, effectively avoiding interference from self-absorption and inner-filter effects.1–3 Intramolecular hydrogen bonds play a crucial role in ESIPT reactions and contribute to enhancing photostability as the typical photoacid.4–6 Based on the characteristic four-level-energy photochemical cycle (enol → enol* → keto* → keto), the ESIPT molecules are utilized as fluorescent probes for biomarker detection,7–9 optoelectronics for lasers,10 OLEDs11,12 and anticounterfeiting encryption materials,13,14 including Förster resonance energy transfer (FRET),15 intramolecular charge transfer (ICT),16,17 multi-proton transfer,18–20 and aggregated-induced emission (AIE)21 coupled with ESIPT. More interestingly, ESIPT probes exhibit high sensitivity for detecting and visualizing metal ions, SO2, H2O2 and other molecules.22–26 Furthermore, sensitivity to the environment, such as solvent and pH, can promote multicolor fluorescence emission.27–30Excited-state proton transfer in 2-(2′-hydroxy)phenylbenzimidazoles (HBIs), a fascinating and typical ESIPT-generated molecule (ESIPT gen), is of utmost importance in chemistry and materials. The intricate process is understood through a combination of spectroscopic methods and time-dependent density functional theory (TDDFT).31–34 The proton transfer (PT) requires a proton-donating entity (–OH, >NH and –NH2) and a proton-accepting entity (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– and –CO), which effectively enhance the fluorescence quantum yield.35,36 Furthermore, HBI derivatives display the nature of fluorescent probes owing to the smart sensing of Zn2+ and Al3+ based on the ESIPT reaction mechanism.37,38 Additionally, HBI derivatives demonstrate multi-color fluorescent and pH-dependent fluorescence properties due to special substituents.39 Thus, substituted functional groups in HBIs enable the fine-tuning of their photophysical and photochemical properties, providing a strategy for designing and developing luminescent materials while improving and even mediating efficient fluorescent quantum yields.40 In particular, heavy atoms in HBIs, such as Br and S, can shift emission pathways from fluorescence emission to intersystem crossing (ISC).41,42
N– and –CO), which effectively enhance the fluorescence quantum yield.35,36 Furthermore, HBI derivatives display the nature of fluorescent probes owing to the smart sensing of Zn2+ and Al3+ based on the ESIPT reaction mechanism.37,38 Additionally, HBI derivatives demonstrate multi-color fluorescent and pH-dependent fluorescence properties due to special substituents.39 Thus, substituted functional groups in HBIs enable the fine-tuning of their photophysical and photochemical properties, providing a strategy for designing and developing luminescent materials while improving and even mediating efficient fluorescent quantum yields.40 In particular, heavy atoms in HBIs, such as Br and S, can shift emission pathways from fluorescence emission to intersystem crossing (ISC).41,42
Herein, an ESIPT-based multicolor fluorescence molecule was initially synthesized via the introduction of a bromine atom on the p-substituted position of hydroxyl group 2-(2′-hydroxy)phenylbenzimidazole (HBI), as shown in Scheme 1(a). Combined with density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations, we revealed the multicolor fluorescence mechanism through the spectroscopic experiments, potential energy profiles and nucleus-independent chemical shift (NICS(1)_ZZ) values, which are key indicators of aromaticity (negative values) and antiaromaticity (positive values).
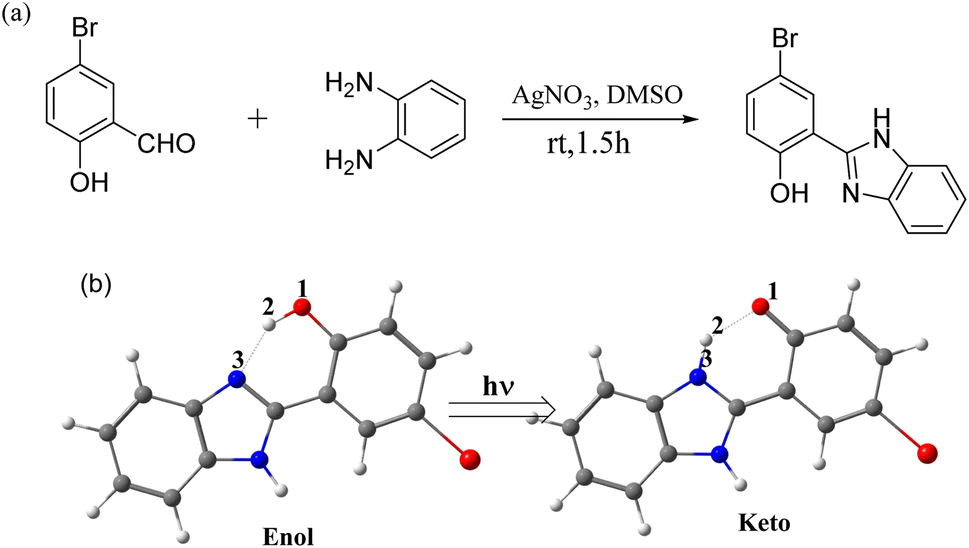 | ||
| Scheme 1 Synthesis route (a) and structural tautomers (b) of HBI-pBr. | ||
Experimental and computational methods
The synthesis route of 2-(2′-hydroxy-5′-bromo)phenylbenzimidazole (HBI-pBr) is shown in Scheme 1(a) and detailed in the ESI.†43 Fig. S1† shows the 1H NMR spectrum (400 MHz, DMSO-d6) δ 13.32 (s, 2H), 8.32 (d, J = 2.5 Hz, 1H), 7.71 (s, 2H), 7.55 (dd, J = 8.8, 2.5 Hz, 1H), 7.37–7.30 (m, 2H), and 7.05 (d, J = 8.8 Hz, 1H). Various concentrations of HBI-pBr were prepared using high-performance liquid chromatography grade acetonitrile (99.9%, Tedium Company, Inc., USA), methanol, cyclohexane, dichloromethane, tetrahydrofuran, and dimethyl sulfoxide (both 99.9%, Spectrum Chemical Mfg. Corp., USA). UV absorption spectra were measured in cyclohexane, dichloromethane, tetrahydrofuran, methanol, acetonitrile and dimethyl sulfoxide at concentrations of ∼10−5 mol L−1 using a UV-visible spectrometer (UV-2501PC, Shimadzu Corp., Japan). Fluorescence emission spectra were recorded using an F-4600 FL spectrophotometer at the same concentration.Density functional theory (DFT) calculations were performed to acquire the ground geometries.44–46 The vibrational analysis was carried out at the same level of theory as geometry optimization to identify the characteristics of all the obtained stationary points (minima and transition states). To obtain the minima of the excited state, a time-dependent DFT (TD-DFT) calculation was performed at the B3LYP functional.47–55 The 6-311+G(d, p) basis was applied for all elements for both the DFT and TD-DFT calculations.56 The solvation energies were evaluated using the polarized continuum model (PCM).57 The intrinsic reaction coordinate (IRC)58 calculations were analyzed to confirm the transition states connecting the corresponding reactant and product in Scheme 1(b). The NICS values were computed as 1 Å above the ring center along the ZZ axis based on the ZZ component of the nuclear shielding tensor at that point.59,60 Notably, Baird's rule61 elucidates the molecular behavior in the triplet excited state (T1), where molecules that are aromatic in the ground state (S0) become antiaromatic in T1, and vice versa. This principle has been extended to encompass the singlet excited state (S1). Schleyer et al.62 proposed that negative NICS(1)_ZZ values indicate S0 aromaticity, with more negative values correlating to stronger aromaticity, while positive values suggest anti aromaticity. Recent studies have shown that changes in NICS(1)_ZZ during the ESIPT signal reduce aromaticity, driving the ESIPT process.63,64
All DFT and TD-DFT calculations were carried out using the Gaussian 09 program package.65
Results and discussion
Fig. 1 shows the UV-Vis absorption and fluorescence emission spectra in five solvents. In the 300–360 nm region, two shoulder absorption peaks were observed at ∼340 and 330 nm, displaying the solvent-dependent shift. As shown in Table 1, the absorption band was observed at 344 nm in c-C6H12, while in CH3CN and CH3OH, it shifted to 339.6 and 338 nm, respectively. Simultaneously, similar absorptions were observed at 342.4 and 342 nm in CH2Cl2 and THF, respectively. The above-mentioned results indicated that the maximum absorption peaks are blue-shifted with increasing solvent polarity.66,67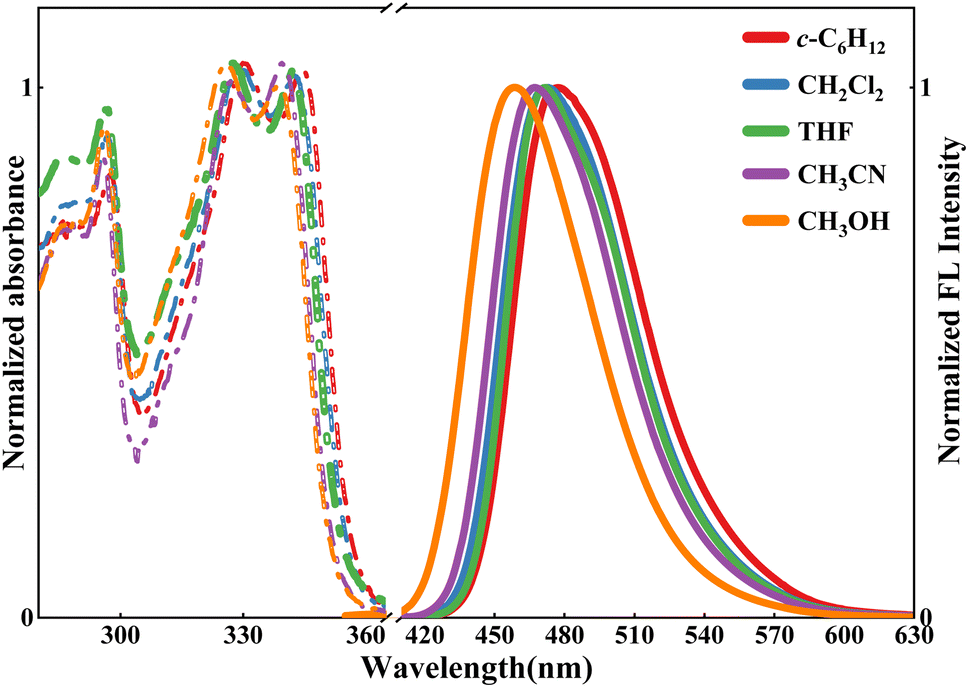 | ||
| Fig. 1 Normalized absorption (dotted) and fluorescence spectra (solid, λex = 340.0 nm) in cyclohexane (c-C6H12), dichloromethane (CH2Cl2), tetrahydrofuran (THF), acetonitrile (CH3CN) and methanol (CH3OH). | ||
| Solvent | λmaxabs/nm a | λmaxem/nm b | Δν (cm−1) c | ||
|---|---|---|---|---|---|
| Exp | Cal | Exp | Cal | ||
| a Calculated at TD-B3LYP/6-311+G(d, p) on optimized S0 at the B3LYP/6-311+G(d, p) level using the corresponding PCM solvent model.b Calculated at TD-B3LYP/6-311+G(d, p) on optimized S1 at the TD-B3LYP/6-311+G(d, p) level using the corresponding PCM solvent model.c Data in parentheses calculated Stokes shift. | |||||
| Cyclohexane | 344 | 333 | 477 | 454 | 8105(8004) |
| Dichloromethane | 342 | 329 | 473 | 432 | 8098(7247) |
| Tetrahydrofuran | 342 | 329 | 472 | 433 | 8053 (7300) |
| Acetonitrile | 340 | 327 | 468 | 428 | 8044 (7217) |
| Methanol | 338 | 327 | 458 | 428 | 7752(7217) |
To further interpret the experimental results, four ground configurations, cis-phenol, trans-phenol, cis-phenol-1, and keto in CH3OH were optimized at B3LYP-TD/6-311+G(d, p) level. As shown in Fig. S2,† the cis-phenol isomer is the most stable, while both keto and trans-phenol are about 6.5 kcal mol−1 higher energy. Table S2† indicates the vertical transition energies calculated using the corresponding solvent model. The transition energy at 332 nm for the cis-phenol aligns well with the experimentally observed shoulder absorptions at 344 and 330 nm, which mainly corresponds to the π → π* transition. The additional maximum absorption bands observed at 298 and 288 nm also matched the calculated values at 287 and 283 nm. As shown in Table S2,† the absorption band at 330 nm seems close to the calculated electronic transition of the trans-phenol at 323 nm. However, the potential energy profile scan indicates that the torsional process from cis-phenol to trans-phenol requires a barrier of 10.4 kcal mol−1, while the reverse barrier is only 5.4 kcal mol−1 along the dihedral angle of D15-13-7-21, as shown in Fig. S3.† Thus, cis-phenol is the dominant isomer, both thermodynamically and kinetically.68,69
From Fig. 1, when excited at 340 nm, HBI-pBr exhibits the solvent-dependent emission observed at 477 nm in c-C6H12, 473 nm in CH2Cl2, 472 nm in THF, 468 nm in CH3CN and 458 nm in CH3OH. As illustrated in Tables 1 and S1,† the Stokes' shift exhibits a negative correlation with the solvent polarity.70 Table S1† indicates that para-bromine substitution modulates the proton transfer dynamics and charge transfer efficiency likely through its influence on electronic distribution and steric factors. The lowest unoccupied molecular orbital (LUMO) is more localized than the highest unoccupied molecular orbital (HOMO). With a bromine (Br) atom at the tip apex, there is a pronounced localization of electron density within the phenol group (fragment 2), particularly evident in the LUMO. As an electron-attracting group, the bromine atom causes the electron density to concentrate more within the phenol group, as opposed to the more balanced charge transfer in HBI. The reduction in the HOMO–LUMO charge distribution gap from 6.0% in HBI to 1.5% in HBI-pBr further validates that bromine substitution enhances electron localization and diminishes intramolecular charge transfer (ICT), inhibiting charge transfer with minimal impact on the emission spectrum.71,72
Subsequently, TD-DFT calculations were applied to identify the electronic spectrum. The calculated emission band at 454 nm for keto HBI-pBr corresponds well with the experimental emission band at 477 nm in c-C6H12, as shown in Table 1. Similarly, the calculated emission bands in the other four solvents also align well with the experimental data illustrated in Table 1. Relatively large Stokes' shifts were observed in c-C6H12 compared to the other solvents. Table 2 compares the bond distances of O1–H2 and H2–N3, and the bond angles of O1–H2–N3 in both the ground and first singlet excited states. For phenol, the O1–H2 bond length was elongated from 0.998 Å in S0 to 1.015 Å in S1, while the distance of H2–N3 was shortened from 1.690 Å in S0 to 1.639 Å in S1. The above-mentioned results indicate that HBI-pBr is a characteristic of photoacid, similar to HBO.73 For keto, the H2–N3 bond length further decreased from 1.036 Å in S0 to 1.016 Å in S1, while the O1–H2 bond increased by 0.232 Å in S1. δ(O1–H2–N3) angle was largest in S1 for phenol, while a compact S1 of keto structure perhaps is attributed to the smallest δ(O1–H2–N3). Photoisomerization between cis-phenol and keto is calculated along the O1–H2 bond distances, and the potential energy curves of the ground and first singlet excited states are also shown in Fig. 2. The energy barrier of PT is 6.1 kcal mol−1 in S0 along the O1–H2 bond, which, however, reduces to only 1.5 kcal mol−1 in S1. More importantly, the energy barrier on S1 reaches 10.4 kcal mol−1 from keto to cis-phenol, contrary to S0.74–76 Thus, the ESIPT reaction is validated both thermodynamically and kinetically. Fig. S4† shows the natural atom charge on S1 and S0 of the phenol and keto forms. For phenol, the charges on O1 and N4 are −0.710 and −0.531 in S0 and -0.707 and −0.524 in S1, respectively, indicating a slight decrease in the negative charge excited state. Meanwhile, the positive charges on H2 and H5 increase by 0.001 and 0.022, respectively, from S0 to S1 states, with the H2 proton showing greater photoacidity than the H5 proton. Thus, H2 has perhaps stronger photoacidity than H5. However, for keto, the positive charges on H5 are consistently 0.433 larger than those in the phenolic structure, making H5 protons more likely to be deprotonated by OH−, which facilitates the formation of an anion. These findings are in accordance with the significant charge redistribution upon excitation, enhancing the molecule's polarity. Consequently, we can comprehensively investigate the photophysical process of HBI-pBr in relation to base concentration, enabling us to conduct analogous spectral measurements with enhanced precision.
| Distance | Species | |||
|---|---|---|---|---|
| Phenol | Keto | |||
| S0,min | S1,min | S0,min | S1,min | |
| O1–H2 | 0.998 | 1.015 | 1.742 | 1.974 |
| H2–N3 | 1.690 | 1.639 | 1.036 | 1.016 |
| δ(O1–H2–N3) | 148.3 | 150.2 | 133.4 | 124.0 |
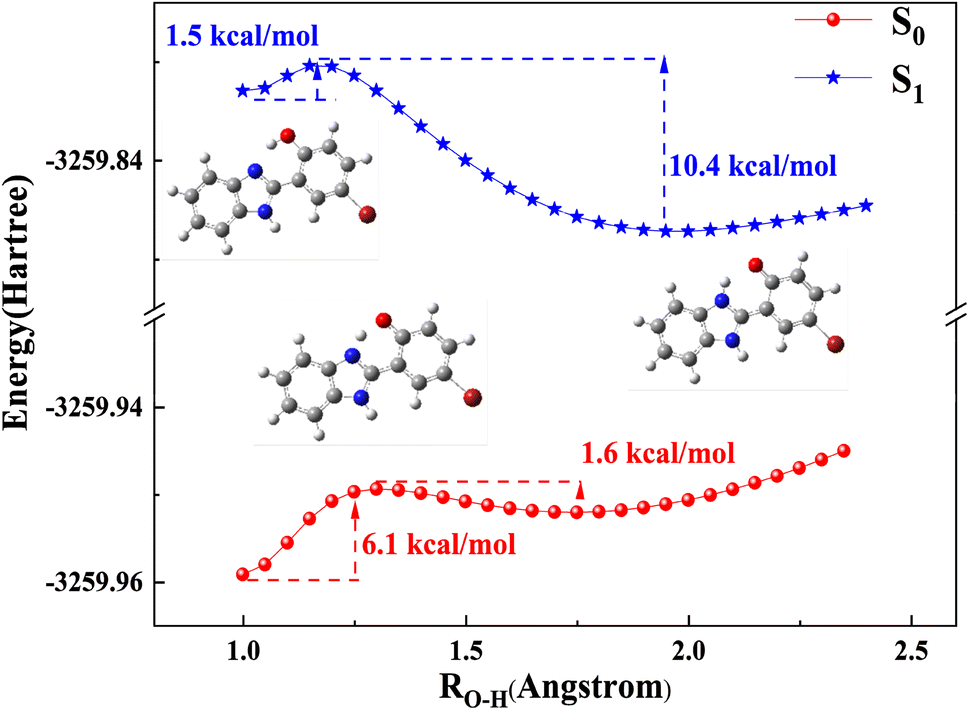 | ||
| Fig. 2 Potential energy curves for the S0 and S1 states of HBI-pBr calculated at the TD-B3LYP/6-311+G(d, p) level (step size 0.05 Å) using PCM (solvent = methanol) model. | ||
When doped with [(Bu)4N]OH in HBI-pBr/CH3OH, as shown in Fig. 3, a series of new absorption bands were generated at 363/360, 298, 289, 253 and 225 nm, displaying significant blue shifts compared to the absorption bands at 338/326, 296, 286, 236 and 217 nm observed in pure CH3OH, respectively. Fig. 4(a) further highlights new bands observed around 360, 300, 260, and 220 nm, close to the absorption bands mentioned. A new fluorescence emission band is observed at 416 nm excited by 340 nm for HBI-pBr/CH3OH solution doped with [(Bu)4N]OH, as shown in Fig. 4(b). This indicates a shift from the emission band at 458 nm observed in pure CH3OH. These newly observed absorption and emission bands are tentatively attributed to the corresponding anions formed in the solution. Given the presence of multiple active hydrogen atoms of the –OH and –NH– groups in HBI-pBr, the mono- and divalent anions were optimized at the B3LYP-TD/6-311+G(d, p) level. As shown in Fig. S2,† four anionic species were identified: DA1 (proton H5 lost), DA2 (proton H2 lost), DA (divalent anion, both H2 and H5 protons lost), and DA3 (proton of H5 loss of phenolic isomer). Among these, DA1 is the most stable. Compared to DA1, a typically shorter C–O bond length of 1.268 Å exists in DA2. In contrast, DA3 has an elongated O–H bond length of 1.014 Å, together with a short N⋯H distance of 1.622 Å. The geometry of the divalent anion DA tends toward non-planarity.
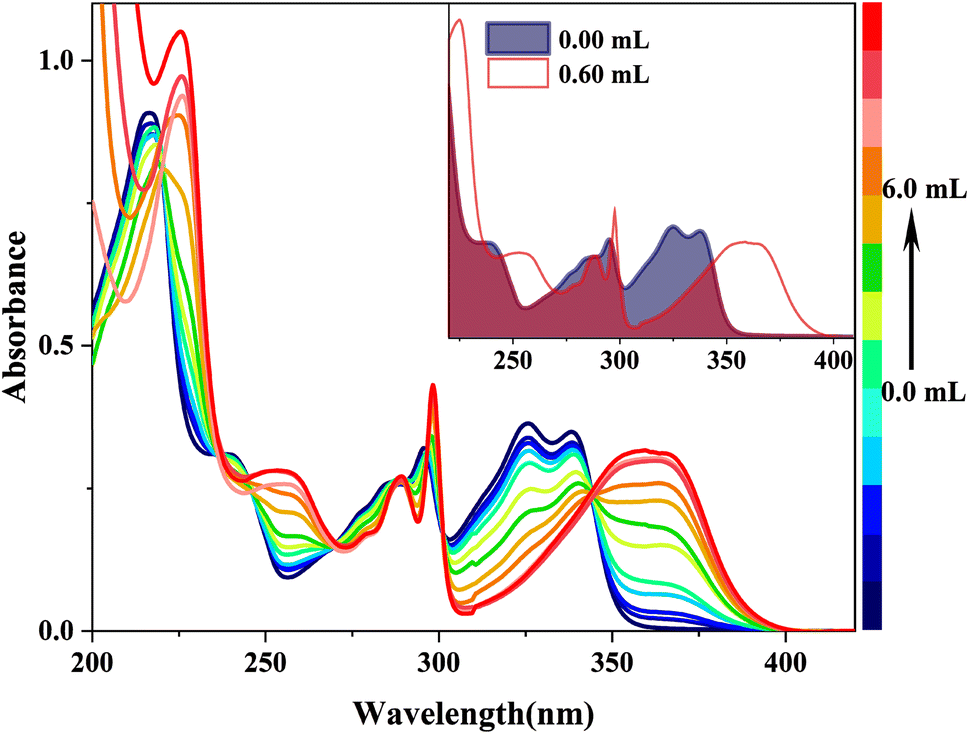 | ||
| Fig. 3 UV spectra of a 2.5 × 10−5 M HBI-pBr solution in CH3OH as a function of 2.0 × 10−4 M [(Bu)4N]OH solution in CH3OH. The absorbance evolution of HBI-pBr upon increasing concentration [(Bu)4N]OH from 0 to 6.00 mL. | ||
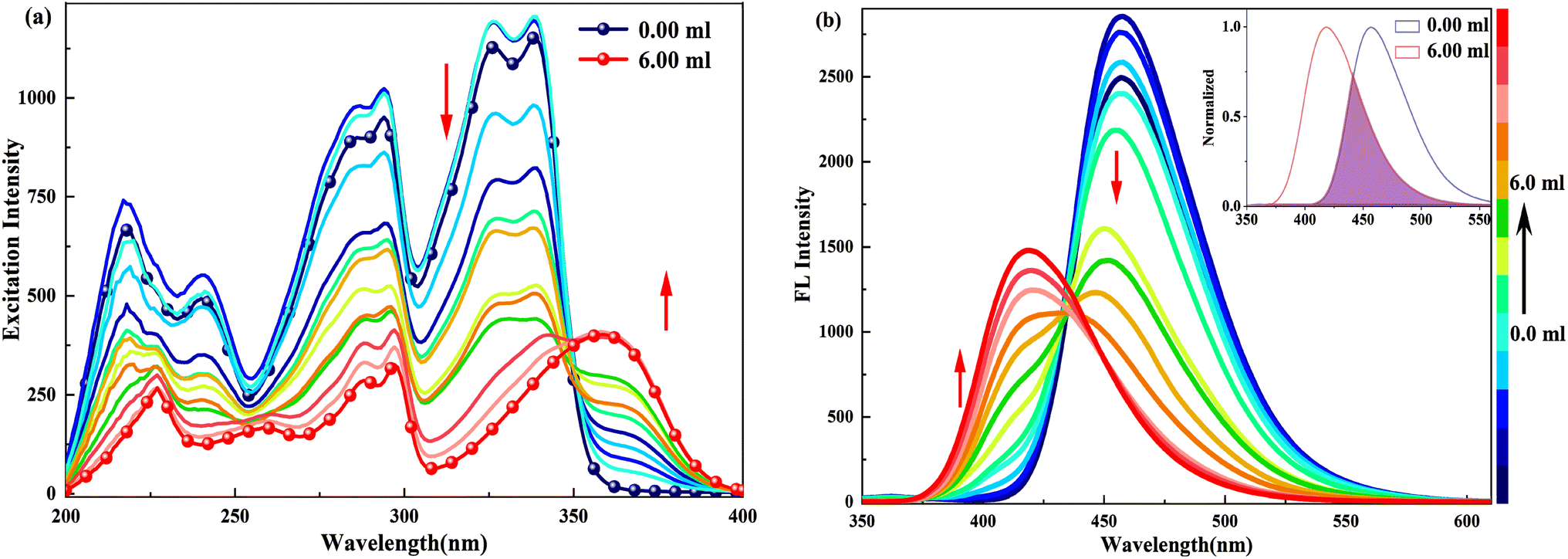 | ||
| Fig. 4 (a) Excitation (λem = 419.0 nm) and (b) emission spectra (λex = 340.0 nm) of 2.5 × 10−5 M HBI-pBr in CH3OH as a function of 2.0 × 10−4 M [(Bu)4N]OH solution in CH3OH from 0 to 6.00 mL. | ||
As shown in Fig. S5,† the ground state DA2 can spontaneously convert to ground DA1 through a very low barrier of only 0.7 kcal mol−1. Comparatively, in the first singlet excited state, the tautomeric reaction can occur from DA2 to DA1 when the barrier exceeds 8.9 kcal mol−1. To sum up, it is impossible to undergo from DA1 to DA2 in either a ground or excited state. DA1 should be easily detected in the base solution.
In the excitation spectrum, as shown in Fig. 4(a), increasing [(Bu)4N]OH in CH3OH leads to the appearance and strengthening of new bands at 358 and 343 nm, which correspond to the broad absorption band observed at ∼360/363 nm, as shown in Fig. 3. Conversely, other bands weaken and even disappear. This indicates that the structure of the excited state derived from the ground-state (DA1)77 has changed. As depicted in Fig. 4(b), upon excitation at 340 nm, the emission peak at 458 nm gradually disappears; subsequently, a new emission peak arises at 419 nm, which originates from the absorption peak at 360 nm. Furthermore, the 419 nm emission peak aligns with the calculated emission peak at 433 nm in the S1 state of DA1, as shown in Fig. S6(a).† Thus, cis-phenol HBI-pBr undergoes deprotonation in solution to form a monovalent anion, DA1, with maximum absorption and emission peaks at 360 and 419 nm in an alkaline environment, respectively. Perhaps, the emission spectral shift may indicate a stronger interaction between the excited HBI-pBr and the charged OH− ions from [(Bu)4N]OH, which we ascribe to coulomb interactions.78
In the excited state, the energy barriers between DA1 and DA2 are 17.0 and 8.9 kcal mol−1, respectively, as shown in Fig. S5.† Compared with the ground state, both the forward and reverse barriers are enlarged in the first singlet excited state. The potential energy surface profiles indicate that large energy barriers exist for the change from DA1 to DA2 in both the ground and excited states, confirming that DA1 is the dominant species in a basic environment. To further interpret the emission, we compared the transition energy and oscillator strength (f) of DA1, DA2, DA3 and DA, as listed in Table S3.† It is found that the calculated transition energy of DA1 closely matches the experimentally observed maximum absorption. For example, the maximum absorption for DA1, calculated at 374 nm, matches well with that experimentally observed at 360 nm. Additionally, Table S4† indicates that the S0 → S1 transition of DA1 is a typical π → π* character with an intramolecular charge transfer (ICT) from the bromophenyl group to the benzimidazolyl unit. It is also evident that the additional absorption peaks at 298 and 289 nm are associated with the S4 and S5 states, respectively.
Similarly, when doped with trifluoroacetic acid (TFA) in HBI-pBr/CH3OH, a series of new absorption bands are produced at 346, 338, 300, 289, 240 and 217 nm, while the original bands at 339, 326, 296, 286, 236 and 217 nm disappear gradually, as shown in Fig. 5. Considering only one electronegative nitrogen (N3) position on the imidazole group, we tentatively identified the new species as a monovalent cation, PA. As depicted in Fig. 6(a), the excitation spectrum of HBI-pBr shows a broad band in the 310–370 nm region, accompanied by a pronounced tail that corresponds to the absorption band in the same region. This suggests that the excited state originates from the ground state structure, correlating with PA. As shown in Fig. 6(b), the emission peak at 458 nm gradually diminishes, while a new emission peak emerges at 391 nm in the insert of Fig. 6(b). To accurately assign the new species, DFT and TD-DFT calculations were performed to determine the stable structures and elucidate the spectral behaviour. Fig. S7† presents the optimization structures of the PA in both the ground and first excited electronic states. Table S5† lists the maximal transition energies calculated at 339, 300, 295, 236 and 218 nm, which align well with the experimentally observed peaks at 338, 300, 289, 240 and 217 nm, respectively. Table S6† demonstrates that the above-mentioned light transition is associated with the typical π → π*. As shown in Fig. S6(b),† the calculated emission peak of PA at 395 nm is consistent with that observed at 391 nm. Therefore, we can deduce that in acidic environments, HBI-pBr can attract one H+ to a cationic HBO-pBr, PA.
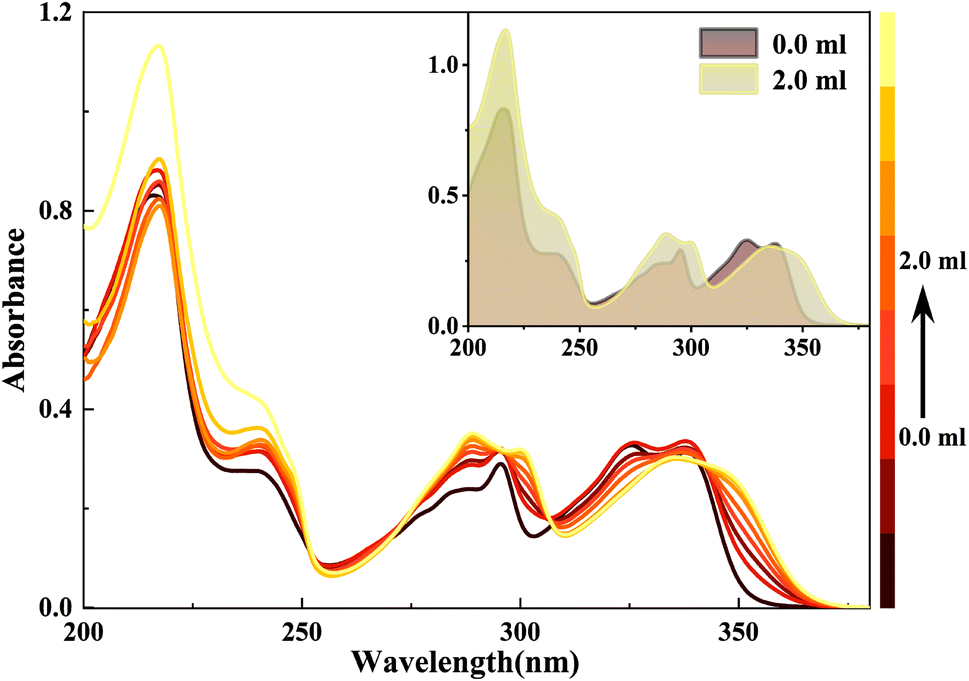 | ||
| Fig. 5 UV-Vis spectra of a 2.5 × 10−5 M HBI-pBr solution in CH3OH as a function of CF3COOH (TFA, 2.0 × 10−4 M) in CH3OH from 0 to 2.00 mL. | ||
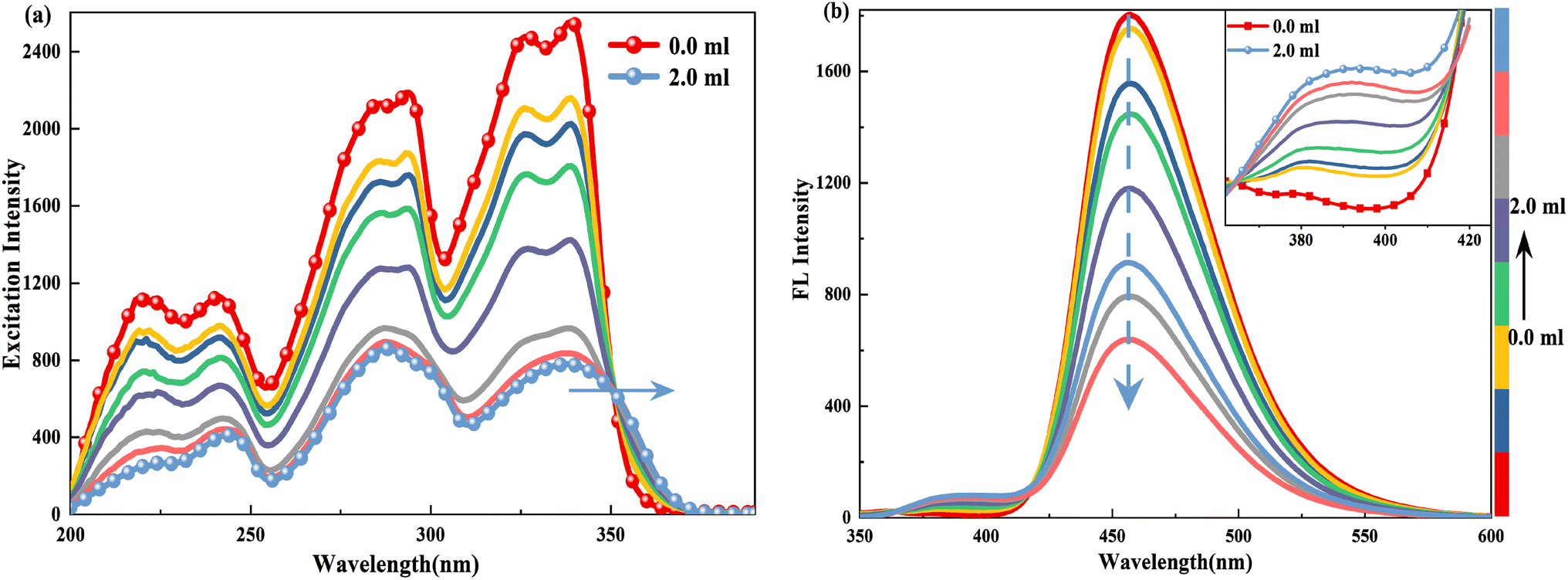 | ||
| Fig. 6 (a) Excitation (λem = 391.0 nm) and (b) emission spectrum (λex = 340.0 nm) of 2.5 × 10−5 M HBI-pBr in CH3OH doped with different volumes of as a function of 2.0 × 10−4 TFA solution in CH3OH from 0 to 2.00 mL. Note: Inset shows normalized fluorescence emission spectra from VTFA = 0 to 2.00 mL. | ||
ESIPT mechanism
To gain deeper insight into the aforementioned spectral results, NICS(1)_ZZ values are predicted, as shown in Table 3. For the ground state cis-phenol isomer, the value of NICS(1)_ZZ is −71.80 ppm, while the total NICS(1)_ZZ value of excited cis-phenol* increases to 38.03 ppm, demonstrating lower stability due to the change from aromaticity to antiaromaticity. After the ESIPT reaction, the total NICS(1)_ZZ value of the excited keto* decreases to −15.99 ppm from the cis-phenol*, suggesting that the ESIPT reaction in HBI-pBr is favorable, as it enhances stability by restoring aromaticity from antiaromaticity, consistent with Baird's rule.61–63 This is why a large Stokes' shift is observed in HBI-pBr. Moreover, both the anion and cation forms of HBI-pBr exhibit aromaticity in the ground state, with the total NICS(1)_ZZ values of −69.44 and −69.66 ppm, respectively. However, the reversal of aromaticity occurred using NICS(1)_ZZ values of 3.77 and 10.64 ppm in the excited DA1 and PA, respectively, which shows that the S1 state would return to the ground state by fluorescence emission. For the ground state, the total NICS(1)_ZZ values of cis-phenol HBI-pBr are similar to DA1 and PA in S0; thus, the structures experience minimal loss when adding or losing protons. This explains why only small Stokes' shifts are observed for DA1 and PA because no significant change is observed in the equilibrium structure and NICS(1)_ZZ values from the ground to the electronic singlet excited cation and anion, as shown in Fig. 7. Thus, in terms of (anti)-aromaticity rationalized, the large Stokes' shifts observed can be attributed to significant changes in the structures and NICS(1)_ZZ values following the ESIPT reaction.79,80| System | S0 | S1 | Diffa | Diffb | |
|---|---|---|---|---|---|
| S0 | S1 | ||||
| a The difference of total NICS(1)_ZZ values from S1 minus S0 in the corresponding keto and phenol isomers.b The difference of total NICS(1)_ZZ values with the phenol isomer in the corresponding minima of S0,min and S1,min, respectively. The values in parentheses corresponds with the keto. | |||||
| Phenol | −71.80 | 38.03 | 109.83 | +10.57 | −54.02 |
| Keto | −61.23 | −15.99 | 45.24 | ||
| Anion | −69.44 | 3.77 | 73.21 | +2.36(−8.21) | −34.26(−8.21) |
| Cation | −69.66 | 10.64 | 80.30 | +2.14(−8.43) | −27.39(+26.63) |
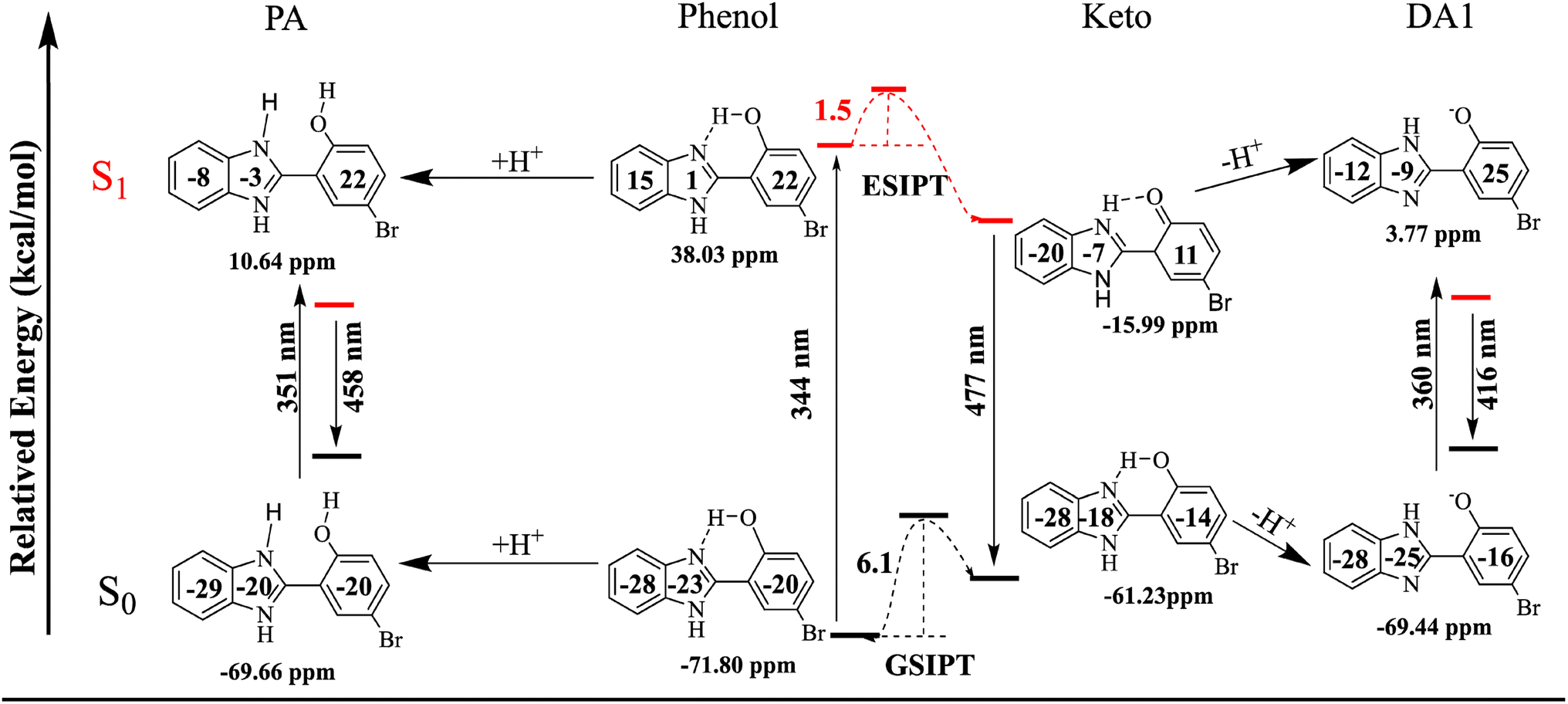 | ||
| Fig. 7 Total and separately NICS(1)_ZZ values (in ppm) of HBI-pBr and ions in the S0 and S1 states and the maximal absorption and emission bands observed experimentally. | ||
Conclusion
In summary, the ground-state and excited-state proton transfer in 2-(2′-hydroxy-5′-bromo)phenylbenzimidazole (HBI-pBr) are significantly influenced by solvent and pH conditions. A combination of spectroscopic techniques and a time-dependent density functional theory (TDDFT) approach offers comprehensive mechanistic insights into this intricate process. The mechanism of the ground-state and excited-state proton transfer of HBI-pBr was proposed experimentally and theoretically. The maximum absorptions at ∼340.0 and 330.0 nm were assigned to the ground cis-phenol isomer. The strong FL emission band observed at ∼470.0 nm was initiated from the keto of HBI-pBr. The monovalent anion and cation were characterized in CH3OH doped with acid and base, which displayed 416 and 458 nm emission, respectively. Para-Bromine substitution localizes the electron distribution in the HBI framework, thereby inhibiting charge transfer.Finally, both the potential energy surface curves and NICS(1)_ZZ values interpreted the mechanism of ESIPT reaction and formation of cation and anion. This study opens a chapter on the mechanistic understanding of ESIPT through NICS(1)_ZZ values, laying a solid foundation for further designing and developing applications of ESIPT-based fluorescent probes in related fields.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 22273086, 21873020 and 21473162). Y. Z. is grateful for support from the Project Grants 521 Talents Cultivation of Zhejiang Sci-Tech University. This work is also supported by the Zhejiang Provincial Top Key Academic Discipline of Chemical Engineering and Technology.References
- L. He, B. Dong, Y. Liu and W. Lin, Fluorescent Chemosensors Manipulated by Dual/Triple Interplaying Sensing Mechanisms, Chem. Soc. Rev., 2016, 45(23), 6449–6461 RSC.
- G.-J. Zhao and K.-L. Han, Hydrogen Bonding in the Electronic Excited State, Acc. Chem. Res., 2012, 45(3), 404–413 CrossRef CAS.
- C.-H. Wang, Z.-Y. Liu, C.-H. Huang, C.-T. Chen, F.-Y. Meng, Y.-C. Liao, Y.-H. Liu, C.-C. Chang, E. Y. Li and P.-T. Chou, Chapter Open for the Excited-State Intramolecular Thiol Proton Transfer in the Room-Temperature Solution, J. Am. Chem. Soc., 2021, 143(32), 12715–12724 CrossRef CAS.
- D. Schilter, pHotoacids Jump Further, Nat. Rev. Chem, 2020, 4(10), 505 CrossRef CAS.
- P. Thordarson and J. E. Beves, Confining Photoacidity, Chem, 2019, 5(6), 1366–1368 CAS.
- T. Sun, L. Kang, H. Zhao, Y. Zhao and Y. Gu, Photoacid Generators for Biomedical Applications, Adv. Sci., 2024, 11(5), e2302875 CrossRef.
- H. Gu, W. Wang, W. Wu, M. Wang, Y. Liu, Y. Jiao, F. Wang, F. Wang and X. Chen, Excited-State Intramolecular Proton Transfer (ESIPT)-Based Fluorescent Probes for Biomarker Detection: Design, Mechanism, and Application, Chem. Commun., 2023, 59(15), 2056–2071 RSC.
- C. Bai, J. Zhang, Y. Qin, H. Huang, Z. Xia, Q. Zheng, H. Dai, P. Lu, H. Miao and C. Qu, et al., Precise Probe Design Based ESIPT Coupled AIE Mechanism Toward Endogenous Cyanide in Food Detection and Bioimaging, Chem. Eng. J., 2022, 443, 136445 CrossRef CAS.
- X. Wang and C. Ma, A Near-Infrared Fluorescent Chemosensor with a Remarkably Large Stokes Shift for the Ultrasensitive Detection of Tyrosinase Activity and Bioimaging in Living Cells and Mouse Xenograft Model, Sens. Actuators, B, 2022, 354, 131211 CrossRef CAS.
- C.-C. Yan, X.-D. Wang and L.-S. Liao, Organic Lasers Harnessing Excited State Intramolecular Proton Transfer Process, ACS Photonics, 2020, 7(6), 1355–1366 CrossRef CAS.
- K. Wu, T. Zhang, Z. Wang, L. Wang, L. Zhan, S. Gong, C. Zhong, Z. Lu, S. Zhang and C. Yang, De Novo Design of Excited-State Intramolecular Proton Transfer Emitters via a Thermally Activated Delayed Fluorescence Channel, J. Am. Chem. Soc., 2018, 140(28), 8877–8886 CrossRef CAS PubMed.
- Y. Wu, R. Wang, R. Lin, X. Xu, X. Zhang, O. Alsalman, Y. Qiu, A. Uddin and X. Ouyang, Excited-State Intramolecular Proton Transfer Emitter for Efficient Violet-Blue Organic Light-Emitting Diodes with Hybridized Local/Charge Transfer Channel, Chem. Eng. J., 2023, 465, 142929 CrossRef CAS.
- G. Xiao, Y. J. Ma, X. Fang and D. Yan, Quadruple Anticounterfeiting Encryption: Anion-Modulated Forward and Reverse Excitation-Dependent Multicolor Afterglow in Two-Component Ionic Crystals, ACS Appl. Mater. Interfaces, 2022, 14(26), 30246–30255 CrossRef CAS.
- Q. Zhang, L. Yang, Y. Han, Z. Wang, H. Li, S. Sun and Y. Xu, A pH-Sensitive ESIPT Molecule with Aggregation-Induced Emission and Tunable Solid-State Fluorescence Multicolor for Anti-Counterfeiting and Food Freshness Detection, Chem. Eng. J., 2022, 428, 130986 CrossRef CAS.
- L. Wu, C. Huang, B. P. Emery, A. C. Sedgwick, S. D. Bull, X.-P. He, H. Tian, J. Yoon, J. L. Sessler and T. D. James, Förster Resonance Energy Transfer (Fret)-Based Small-Molecule Sensors and Imaging Agents, Chem. Soc. Rev., 2020, 49(15), 5110–5139 RSC.
- R. Salaeh and R. Daengngern, Theoretical Insights into Ultrafast Excited State Proton Transfer Coupled with Twisted Intramolecular Charge Transfer Mechanism in 7-(2′-Pyridyl)indole: Effect of Hydrogen Bonding, J. Mol. Liq., 2023, 388, 122707 CrossRef CAS.
- S. Ríos Vázquez, J. L. Pérez Lustres, F. Rodríguez-Prieto, M. Mosquera and M. C. Ríos Rodríguez, Excited-State Proton and Charge Transfer in Protonated Amino and Methylated Derivatives of 2-(2'-Hydroxyphenyl)benzimidazole, J. Phys. Chem. B, 2015, 119(6), 2475–2489 CrossRef.
- S.-Z. Yi, B.-N. Li, P.-Y. Fu, M. Pan and C.-Y. Su, Interplay of Dual-Proton Transfer Relay to Achieve Full-Color Panel Luminescence in Excited-State Intramolecular Proton Transfer (ESIPT) Fluorophores, ACS Appl. Mater. Interfaces, 2023, 15(2), 3172–3181 CrossRef CAS.
- Y. Guo, X. Li, J. Ma and D. L. Phillips, Reaction Mechanisms of Photoinduced Quinone Methide Intermediates Formed via Excited-State Intramolecular Proton Transfer or Water-Assisted Excited-State Proton Transfer of 4-(2-Hydroxyphenyl)pyridine, J. Phys. Chem. Lett., 2021, 12(48), 11666–11672 CrossRef CAS.
- Z. Li, Z. Tang, W. Li, H. Zhan, X. Liu, Y. Wang, J. Tian and X. Fei, Substituents Effect on the Methanol-Assisted Excited-State Intermolecular Proton Transfer of 7-Aminoquinoline: A theoretical study, J. Mol. Liq., 2021, 341, 116920 CrossRef CAS.
- Y. Chen, Y. Fang, H. Gu, J. Qiang, H. Li, J. Fan, J. Cao, F. Wang, S. Lu and X. Chen, Color-Tunable and ESIPT-Inspired Solid Fluorophores Based on Benzothiazole Derivatives: Aggregation-Induced Emission, Strong Solvatochromic Effect, and White Light Emission, ACS Appl. Mater. Interfaces, 2020, 12(49), 55094–55106 CrossRef CAS.
- G. K. Weragoda, N. M. Abdelaziz, D. Govorov, R. Merugu, L. J. Patton, J. E. Grabo, R. A. A. U. Ranaweera, A. C. Ratliff, W. D. Mendis and N. Ahmed, et al., Excited-State Intramolecular Proton Transfer in Salicylidene-α-Hydroxy Carboxylate Derivatives: Direct Detection of the Triplet Excited State of the cis-Keto Tautomer, J. Phys. Chem. A, 2023, 127(12), 2765–2778 CrossRef CAS PubMed.
- B. C. Dickinson and C. J. Chang, A Targetable Fluorescent Probe for Imaging Hydrogen Peroxide in the Mitochondria of Living Cells, J. Am. Chem. Soc., 2008, 130(30), 9638–9639 CrossRef CAS.
- S. Zhong, S. Huang, B. Feng, T. Luo, F. Chu, F. Zheng, Y. Zhu, F. Chen and W. Zeng, An ESIPT-Based AIE Fluorescent Probe to Visualize Mitochondrial Hydrogen Peroxide and Its Application in Living Cells and Rheumatoid Arthritis, Org. Biomol. Chem., 2023, 21(24), 5063–5071 RSC.
- Z. Li, Y. Wu, Y. Shen and B. Gu, Simple NIR-Emitting ESIPT Fluorescent Probe for Thiophenol with a Remarkable Stokes Shift and Its Application, ACS Omega, 2020, 5(19), 10808–10814 CrossRef CAS.
- G. Zeng, Z. Liang, X. Jiang, T. Quan and T. Chen, An ESIPT-Dependent AIE Fluorophore Based on HBT Derivative: Substituent Positional Impact on Aggregated Luminescence and Its Application for Hydrogen Peroxide Detection, Chem.–Eur. J., 2022, 28(5), e202103241 CrossRef CAS PubMed.
- G. Mazzeo, M. Fusè, A. Evidente, S. Abbate and G. Longhi, Circularly Polarized Luminescence of Natural Products Lycorine and Narciclasine: Role of Excited-State Intramolecular Proton-Transfer and Test of pH Sensitivity, Phys. Chem. Chem. Phys., 2023, 25(34), 22700–22710 RSC.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Ultrafast Elementary Photochemical Processes of Organic Molecules in Liquid Solution, Chem. Rev., 2017, 117(16), 10826–10939 CrossRef CAS.
- V. Khorwal and R. J. S. C. Kathuria, Protonation–Deprotonation Dynamics of 2-(4′-Pyridyl)benzimidazole Derivative with Cucurbit[6]uril at Two Different pH, Supramol. Chem., 2021, 33, 452–459 CrossRef CAS.
- M. Das, M. Brahma and G. Krishnamoorthy, Controlling the Photoswitching of 2-(4′-Diethylamino-2′-hydroxyphenyl)-1H-imidazo-[4,5-b]pyridine by pH, J. Photochem. Photobiol., A, 2021, 421, 113504 CrossRef CAS.
- S. K. Dogra, Spectral Characteristics of 2-(2′-Hydroxy-3′-pyridyl)benzimidazole: Effects of Solvents and Acid or Base Concentrations, J. Mol. Struct., 2005, 734(1), 51–60 CrossRef CAS.
- C. Prommin, N. Kanlayakan, W. Chansen, R. Salaeh, K. Kerdpol, R. Daengngern and N. Kungwan, Theoretical Insights on Solvent Control of Intramolecular and Intermolecular Proton Transfer of 2-(2′-Hydroxyphenyl)benzimidazole, J. Phys. Chem. A, 2017, 121(31), 5773–5784 CrossRef CAS PubMed.
- X. Liu, M. Cheng, L. Yang, M. Wang, Y. Yang and J. Han, Theoretical Study on the Effect of Naphthalene Modifications on the ESIPT Mechanism and Photophysical Properties of Benzimidazoles, J. Lumin., 2024, 269, 120495 CrossRef CAS.
- A. Malakar, F. A. S. Chipem and G. Krishnamoorthy, Perturbation of Proton Transfer of 2-(2′-Hydroxyphenyl)benzimidazole and Its Nitrogenous Analogues by Nanoparticles, J. Mol. Struct., 2020, 1217, 128352 CrossRef CAS.
- W.-Z. Bi, Y. Geng, W.-J. Zhang, C.-Y. Li, C.-S. Ni, Q.-J. Ma, S.-X. Feng, X.-L. Chen and L.-B. Qu, Highly Sensitive and Selective Detection of Triphosgene with a 2-(2′-Hydroxyphenyl)benzimidazole Derived Fluorescent Probe, RSC Adv., 2023, 13(44), 30771–30776 RSC.
- Z.-J. Li, W.-J. Zhang, W.-Z. Bi, Q.-J. Ma, S.-X. Feng, X.-L. Chen and L.-B. Qu, An Amino-Substituted 2-(2′-Hydroxyphenyl)benzimidazole for the Fluorescent Detection of Phosgene Based on an ESIPT Mechanism, RSC Adv., 2021, 11(18), 10836–10841 RSC.
- J. Wang, Y. Li and Y. Pang, A Step Toward an NIR-Emitting ESIPT Probe for Smart Zn2+ Sensing in Different Environments, Chem. Biomed. Imaging, 2023, 1(6), 537–540 CrossRef CAS.
- G. Sun and H. Fang, Computational Insights into Sensing Mechanism for Al3+ in a New Acylhydrazone Fluorescent Probe Based on Excited-State Intramolecular Proton Transfer (ESIPT) and Twisted Intramolecular Charge Transfer (TICT), J. Phys. Chem. A, 2023, 127(8), 1857–1865 CrossRef CAS.
- N. Nehra and R. Kaushik, ESIPT-Based Probes for Cations, Anions and Neutral Species: Recent Progress, Multidisciplinary Applications and Future Perspectives, Anal. Methods, 2023, 15(40), 5268–5285 RSC.
- M. Coehlo, G. Clavier and G. Pieters, Excited State Intramolecular Proton Transfer Based Fluorophores with Circularly Polarized Luminescence Emission, Adv. Opt. Mater., 2022, 10(4), 2101774 CrossRef CAS.
- X. Lv, J. Song, S. Guo, J. Gu, L. Meng and C.-Z. Lu, Enhancing Reverse Intersystem Crossing in Triptycene-TADF Emitters: Theoretical Insights into Reorganization Energy and Heavy Atom Effects, J. Phys. Chem. A, 2024, 128(9), 1611–1619 CrossRef CAS PubMed.
- R. Okumura, H. Tanaka, K. Shizu, S. Fukushima, Y. Yasuda and H. Kaji, Development of an Organic Emitter Exhibiting Reverse Intersystem Crossing Faster than Intersystem Crossing, Angew. Chem., Int. Ed., 2024, e202409670 CAS.
- M. G. Holler, L. F. Campo, A. Brandelli and V. Stefani, J. Photochem. Photobiol., A, 2002, 149, 217–225 CrossRef CAS.
- A. D. Becke, Density-Functional Thermochemistry. III. The Role of Exact Exchange, J. Phys. Chem., 1993, 98(7), 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- A. D. Becke, Density-Functional Thermochemistry. IV. A New Dynamical Correlation Functional and Implications for Exact-Exchange Mixing, J. Phys. Chem., 1996, 104(3), 1040–1046 CrossRef CAS.
- E. Runge and E. K. U. Gross, Density-Functional Theory for Time-Dependent Systems, Phys. Rev. Lett., 1984, 52(12), 997–1000 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Treatment of Electronic Excitations Within the Adiabatic Approximation of Time Dependent Density Functional Theory, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, Molecular Excitation Energies to High-Lying Bound States from Time-Dependent Density-Functional Response Theory: Characterization and Correction of the Time-Dependent Local Density Approximation Ionization Threshold, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, An Efficient Implementation of Time-Dependent Density-Functional Theory for the Calculation of Excitation Energies of Large Molecules, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- F. Furche and R. Ahlrichs, Adiabatic Time-Dependent Density Functional Methods for Excited State Properties, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef CAS.
- M. A. L. Marques and E. K. U. Gross, Time-Dependent Density Functional Theory, Annu. Rev. Phys. Chem., 2004, 55, 427–455 CrossRef CAS PubMed.
- M. Chiba, T. Tsuneda and K. Hirao, Excited State Geometry Optimizations by Analytical Energy Gradient of Long-Range Corrected Time-Dependent Density Functional Theory, J. Chem. Phys., 2006, 124, 144106 CrossRef PubMed.
- M. E. Casida, Time-Dependent Density-Functional Theory for Molecules and Molecular Solids, J. Mol. Struct.:THEOCHEM, 2009, 914, 3–18 CrossRef CAS.
- M. A. Marques and E. K. Gross, Time-Dependent Density Functional Theory, Annu. Rev. Phys. Chem., 2004, 55, 427–455 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, Self-Consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets, J. Chem. Phys., 1984, 80(7), 3265–3269 CrossRef CAS.
- C. Amovilli, V. Barone, R. Cammi, E. Cancès, M. Cossi, B. Mennucci, C. S. Pomelli and J. Tomasi, Recent Advances in the Description of Solvent Effects with the Polarizable Continuum Model, Adv. Quantum Chem., 1998, 32, 227–261 CrossRef.
- (a) K. Fukui, Formulation of the Reaction Coordinate, J. Phys. Chem., 1970, 74(23), 4161–4163 CrossRef CAS; (b) K. Fukui, The Path of Chemical Reactions-the IRC Approach, Acc. Chem. Res., 1981, 14(12), 363–368 CrossRef CAS.
- R. West, J. J. Buffy and M. Haaf, Chemical Shift Tensors and NICS Calculations for Stable Silylenes, J. Am. Chem. Soc., 1998, 120(7), 1639–1640 CrossRef CAS.
- J. Polkaehn, R. Molenda and M. A. Cordero, Synthesis and Properties of 5,7-Diazaullazines, J. Org. Chem., 2024, 89(4), 2169–2181 CrossRef CAS PubMed.
- N. C. Baird, Quantum Organic Photochemistry. II. Resonance and Aromaticity in the Lowest 3ππ* State of Cyclic Hydrocarbons, J. Am. Chem. Soc., 1972, 94(14), 4941–4948 CrossRef CAS.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118(26), 6317–6318 CrossRef CAS PubMed.
- D. Dunlop, L. Ludvíková, A. Banerjee, H. Ottosson and T. Slanina, Excited-State (Anti)Aromaticity Explains Why Azulene Disobeys Kasha's Rule, J. Am. Chem. Soc., 2023, 145(39), 21569–21575 CrossRef CAS PubMed.
- J. J. Blackner, O. M. Schneider and W. O. Wong, Removing Neighboring Ring Influence in Monocyclic B–OH Diazaborines: Properties and Reactivity as Phenolic Bioisosteres with Dynamic Hydroxy Exchange, J. Am. Chem. Soc., 2024, 146(28), 19499–19508 CrossRef CAS PubMed.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; B. Mennucci; G. A. Petersson; H. Nakatsuji; M. Caricato; X. Li; H. P. Hratchian; A. F. Izmaylov; J. Bloino; G. Zheng; J. L. Sonnenberg; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; J. A. Jr. Montgomery; J. E. Peralta; F. Ogliaro; M. Bearpark; J. J. Heyd; E. Brothers; K. N. Kudin; V. N. Staroverov; R. Kobayashi; J. Normand; K. Raghavachari; A. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; N. Rega; J. M. Millam; M. Klene; J. E. Knox; J. B. Cross; V. Bakken; C. Adamo; J. Jaramillo; R. Gomperts; R. E. Stratmann; O. Yazyev; A. J. Austin; R. Cammi; C. Pomelli; J. W. Ochterski; R. L. Martin; K. Morokuma; V. G. Zakrzewski; G. A. Voth; P. Salvador; J. J. Dannenberg; S. Dapprich; A. D. Daniels; Ö. Farkas; J. B. Foresman; J. V. Ortiz; J. Cioslowski; D. J. Fox Gaussian 09, Revision D.01; Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y. Guo, Z. Cai, F. Yan, D. Lei, Y. Guo, S. Zhang and X. Dou, Precise Electron-Withdrawing Strength Modulation of ESIPT Probes for Ultrasensitive and Specific Fluorescence Sensing, Anal. Chem., 2023, 95(23), 9014–9024 CrossRef CAS.
- H. Ren, F. Huo, X. Wu, X. Liu and C. Yin, An ESIPT-Induced NIR Fluorescent Probe to Visualize Mitochondrial Sulfur Dioxide During Oxidative Stress in Vivo, Chem. Commun., 2021, 57(5), 655–658 RSC.
- R. Omidyan and M. Iravani, Excited State Proton Transfer and Deactivation Mechanism of 2-(4′-Amino-2′-hydroxyphenyl)-1H-imidazo-[4,5-c]pyridine and Its Analogues: A Theoretical Study, J. Phys. Chem. A, 2016, 120(7), 1012–1019 CrossRef CAS PubMed.
- M. Das, M. Brahma and G. Krishnamoorthy, Light-Driven Switching between Intramolecular Proton-Transfer and Charge-Transfer States, J. Phys. Chem. B, 2021, 125(9), 2339–2350 CrossRef CAS.
- M. Vidal, M. C. Rezende, C. Pastene, C. Aliaga and M. Domínguez, Solvatochromism of Conjugated 4-N,N-Dimethylaminophenyl-pyridinium Donor-Acceptor Pairs, New J. Chem., 2018, 42(6), 4223–4231 RSC.
- Y. Yang, Y. Ding and W. Shi, The effects of amino group meta-and para-substitution on ESIPT mechanisms of amino 2-(2'-hydroxyphenyl)benzazole derivatives, J. Lumin., 2020, 218, 116836–116846 CrossRef CAS.
- C. Li, Y. Yang and C. Ma, Effect of amino group on the excited-state intramolecular proton transfer (ESIPT) mechanisms of 2-(2'-hydroxyphenyl)benzoxazole and its amino derivatives, RSC Adv., 2016, 6, 5134–5140 RSC.
- Y. Li, Y. Wang, X. Feng and Y. Zhao, Spectroscopic and Mechanistic Insights into Solvent Mediated Excited-State Proton Transfer and Aggregation-Induced Emission: Introduction of Methyl Group onto 2-(o-Hydroxyphenyl)benzoxazole, Phys. Chem. Chem. Phys., 2022, 24(42), 26297–26306 RSC.
- P.-Y. Fu, B.-N. Li, Q.-S. Zhang, J.-T. Mo, S.-C. Wang, M. Pan and C.-Y. Su, Thermally Activated Fluorescence vs Long Persistent Luminescence in ESIPT-Attributed Coordination Polymer, J. Am. Chem. Soc., 2022, 144(6), 2726–2734 CrossRef CAS PubMed.
- J. Hao and Y. Yang, Theoretical Investigation of the Excited-State Dynamics Mechanism of the Asymmetric Two-Way Proton Transfer Molecule BTHMB, J. Phys. Chem. A, 2021, 125(48), 10280–10290 CrossRef CAS PubMed.
- N. A. Shekhovtsov and M. B. Bushuev, Enol or keto? Interplay Between Solvents and Substituents as a Factor Controlling ESIPT, J. Mol. Liq., 2022, 361, 119611 CrossRef CAS.
- M.-A. Codescu, M. Weiß, M. Brehm, O. Kornilov, D. Sebastiani and E. T. J. Nibbering, Switching Between Proton Vacancy and Excess Proton Transfer Pathways in the Reaction between 7-Hydroxyquinoline and Formate, J. Phys. Chem. A, 2021, 125(9), 1845–1859 CrossRef CAS PubMed.
- M. K. Saroj, R. Payal, S. K. Jain and R. C. Rastogi, Study of Prototropic Reactions of Indole Chalcone Derivatives in Ground and Excited States Using Absorption and Fluorescence Spectroscopy, J. Mol. Liq., 2020, 302, 112164 CrossRef.
- C.-H. Wu, L. J. Karas, H. Ottosson and J. I. C. Wu, Excited-State Proton Transfer Relieves Antiaromaticity in Molecules, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(41), 20303–20308 CrossRef CAS.
- B. Oruganti, J. Wang and B. Durbeej, Modulating the Photocyclization Reactivity of Diarylethenes through Changes in the Excited-State Aromaticity of the π-Linker, J. Org. Chem., 2022, 87(17), 11565–11571 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06147k |
| ‡ Contributed equally by Mingxia Hu and Yumeng Wang. |
| This journal is © The Royal Society of Chemistry 2024 |