
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanomolar detection of lurasidone hydrochloride in pharmaceutical formulations (Serodopamoun®) and spiked urine using a PVC/imprinted polymer/MWCNTs layer deposited onto polyaniline-coated screen-printed electrodes
Menna M. El-Beshlawya,
Ahmed Barhoum b and
Fatehy M. Abdel-Haleem*cd
b and
Fatehy M. Abdel-Haleem*cd
aDepartment of Chemistry, Faculty of Women, Ain Shams University, Cairo, Egypt. E-mail: Menna.El-Beshlawy@women.asu.edu.eg
bNanostructures Research Group, Department of Chemistry, Faculty of Science, Helwan University, Cairo, 11795, Egypt. E-mail: ahmed.barhoum@science.helwan.edu.eg
cDepartment of Chemistry, Faculty of Science, Cairo University, 12613 Giza, Egypt. E-mail: Fatehy@sci.cu.edu.eg
dDepartment of Chemistry, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh 11623, Saudi Arabia. E-mail: FMMohamed@imamu.edu.sa
First published on 18th December 2024
Abstract
This study developed potentiometric sensors for detecting lurasidone HCl (LSH), a vital drug for treating schizophrenia and bipolar I disorder, in pharmaceutical formulations and biological samples. The sensors are based on screen-printed electrodes (SPE) modified with a molecularly imprinted polymer (MIP) synthesized using lurasidone as a template, 1-vinyl-2-pyrrolidine (VP) as a functional monomer, ethylene glycol dimethacrylate (EGDMA) as a crosslinker, and benzoyl peroxide as an initiator. The SPE was further modified with a conductive polyaniline (PANI) film and a polyvinyl chloride (PVC) layer containing MIP as an ionophore and multiwalled carbon nanotubes (MWCNT) as a transducing material along with 2-nitrophenyl octyl ether (2-NPOE) as plasticizer. This configuration resulted in a sensor with a sensitive response and high selectivity for LSH. The electrochemical evaluation showed a Nernstian response slope of 57.3 ± 0.5 mV decade−1 in a concentration range of 10−4 to 10−8 M, with a detection limit of 10 nM and a response time of 2–3 minutes in Tris buffer (pH = 7.0). The optimized sensor possessed significantly enhanced accuracy, providing a cost-effective alternative to traditional methods. The accuracy, selectivity, precision, stability, and sensitivity of these potentiometric sensors make them valuable for detecting LSH in urine samples spiked with the pharmaceutical formulation Serodopamoun®.
1. Introduction
Lurasidone hydrochloride (LSH), a potent atypical antipsychotic, falls within the benzisothiazol derivative chemical class and is identified chemically as (3aR,4S,7R,7aS)-2-{(1R,2R)-2-[4-(1,2-benzisothiazol-yl)piperazin1ylmethyl]cyclohexylmethyl}hexahydro-4,7-methano-2H-isoindole-1,3-dione hydrochloride.1 Recognized by the brand name Latuda™, LSH has become a preferred treatment option for its minimal impact on body weight, low sedative potential, and limited effects on metabolic parameters.2 LSH is FDA-approved for treating schizophrenia and bipolar I disorder, including bipolar depression, benefiting approximately 10.4 million American adults.2 Swift and accurate detection of LSH in pharmaceuticals and biofluids is crucial in clinical and pharmaceutical settings. Excessive doses can lead to severe adverse effects such as cardiac arrhythmias, seizures, or respiratory depression, necessitating rapid intervention. Moreover, high doses or prolonged use of lurasidone can induce a serious, potentially irreversible movement disorder, particularly affecting women and older adults with extended use.2 Monitoring LSH levels plays a circular role in ensuring patient safety, evaluating treatment efficacy, and maintaining stringent quality control standards in healthcare settings.Electrochemical sensors integrating screen-printed electrodes (SPEs), multi-walled carbon nanotubes (MWCNTs), and molecularly imprinted polymers (MIPs) have been extensively employed for drug detection in pharmaceutical formulations and biological fluids. The employment of SPEs presents numerous advantages, including cost-effectiveness, scalable mass production, and portability, rendering it ideal for swift on-site analyses. SPEs also boast a substantial surface area, enhancing efficient interaction with analytes.3,4 Integration with MIPs allows for the creation of a customized recognition layer with high specificity, particularly for target drugs like LSH. MIPs, synthetic receptors emulating natural antibodies, provide outstanding selectivity and affinity.5 Furthermore, the incorporation of MWCNTs into SPEs enhances their conductivity and provides additional surface area, facilitating more efficient electron transfer and enhancing the sensitivity and selectivity of the sensor. However, challenges in this approach encompass the optimization of MIPs and MWCNTs for ideal binding properties and their secure integration onto SPEs, ensuring stability. Real-world applications of SPE-MIP systems may encounter issues such as sample matrix effects and interferences, demanding meticulous validation and calibration processes. Despite these challenges, the synergy between SPEs and MIPs stands out as a promising avenue for advancing drug detection methodologies, offering a potent combination of efficiency, specificity, and adaptability.4–6
This study explores the application of electrochemical sensors, specifically screen-printed electrodes (SPEs), for the precise detection of LSH in pharmaceutical formulations and biofluids (spiked urine). The sensor is based on SPEs coated first with a conductive polyaniline (PANI) film and secondly with a polyvinyl chloride (PVC) layer containing molecularly imprinted polymer (MIP) nanoparticles as ionophores, alongside multiwalled carbon nanotubes (MWCNTs) as transducing materials along with 2-nitrophenyl octyl ether (2-NPOE) as plasticizer. The synthesis of MIP nanoparticles involves LSH as a template molecule, 1-vinyl-2-pyrrolidone (VP) as functional monomers, ethylene glycol dimethacrylate (EGDMA) as a crosslinker, and benzoyl peroxide as an initiator. PANI serves as a conductive bridge, facilitating efficient electrical signal transfer between the recognition layer (PVC/MIP/MWCNTs) and the SPE. The integration of MWCNTs enhances sensor sensitivity, enabling the translation of chemical signals from LSH-MIP binding into measurable electrical responses. This innovative sensor design improves the accuracy, speed, and reliability of nanomolar LSH detection, demonstrating significant potential for advancing pharmaceutical and clinical applications.
2. Experimental
2.1. Chemicals and reagents
LSH, sourced from Amoun Pharmaceutical Co., S.A.E., Egypt, possessed a purity level of 99.0% (Fig. 1). Key chemicals, including glycine (C2H5NO2, 98.0%), citric acid (C6H8O7, 97.0%), barbituric acid (C4H4N2O3, 96.0%), and D-glutamic acid (C5H9NO4, 96.0%), were supplied from Dr Ehrenstorfer GmbH, Berlin, Germany. Polyaniline (emeraldine salt, Mwt >15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 3–100 μm particle size), ethylene glycol dimethacrylate (EGDMA C10H14O4, 96.0%), 1-vinyl-2-pyrrolidine (VP, C6H9NO, 96.0%), multi-walled carbon nanotubes (MWCNT, 98% carbon basis), and benzoyl peroxide (BPO, C14H10O4, 95.0%) were obtained from Sigma-Aldrich Inc., St. Louis, MO, USA. Additional materials such as poly(vinyl chloride) (PVC, (CH2CHCl)n, high Mwt, 98%), tetrahydrofuran (THF, C4H8O, 95.0%), methanol (99%), ethanol (99.9%) ferrous ammonium sulfate ((NH4)2Fe(SO4)2·6H2O, 96%), potassium nitrate (KNO3, 97%), sodium chloride (NaCl, 98%), sodium hydroxide (99.9%) and 2-nitrophenyl octyl ether (2-NPOE, C14H21NO3, 96.0%) were sourced from Fluka AG, Buchs, Switzerland. For the preparation of a 10−2 M LSH solution, the pure drug was dissolved in 100 mL of Tris base buffer solution (TBB, pH = 7, Sigma-Aldrich). Various concentrations of LSH, ranging from 10−3 to 10−8 M, were prepared through dilution in TBB at pH 7.
000, 3–100 μm particle size), ethylene glycol dimethacrylate (EGDMA C10H14O4, 96.0%), 1-vinyl-2-pyrrolidine (VP, C6H9NO, 96.0%), multi-walled carbon nanotubes (MWCNT, 98% carbon basis), and benzoyl peroxide (BPO, C14H10O4, 95.0%) were obtained from Sigma-Aldrich Inc., St. Louis, MO, USA. Additional materials such as poly(vinyl chloride) (PVC, (CH2CHCl)n, high Mwt, 98%), tetrahydrofuran (THF, C4H8O, 95.0%), methanol (99%), ethanol (99.9%) ferrous ammonium sulfate ((NH4)2Fe(SO4)2·6H2O, 96%), potassium nitrate (KNO3, 97%), sodium chloride (NaCl, 98%), sodium hydroxide (99.9%) and 2-nitrophenyl octyl ether (2-NPOE, C14H21NO3, 96.0%) were sourced from Fluka AG, Buchs, Switzerland. For the preparation of a 10−2 M LSH solution, the pure drug was dissolved in 100 mL of Tris base buffer solution (TBB, pH = 7, Sigma-Aldrich). Various concentrations of LSH, ranging from 10−3 to 10−8 M, were prepared through dilution in TBB at pH 7.
 | ||
| Fig. 1 Chemical structures of the reagents used in the synthesis of MIP nanoparticles: (a) template (lurasidone hydrochloride), (b) monomer (1-vinyl-2-pyrrolidone), (c) crosslinker (ethylene glycol dimethacrylate), and (d) initiator (benzoyl peroxide). | ||
2.2. Synthesis and characterization of MIP and NIP nanoparticles
The MIP nanoparticles were synthesized through the bulk polymerization technique. In detail, 1.0 mmol of LSH (template, Fig. 1a) was mixed with 3.0 mmol of VP (monomer, Fig. 1b) and 3.0 mmol of EGDMA (cross-linker, Fig. 1c).These components were dissolved in 20 mL of acetonitrile (solvent). To this mixture, 60.0 mg of BPO (initiator, Fig. 1d) was added, and the entire solution was placed in a 30 mL glass-capped bottle. After purging the solution with a stream of N2 gas for 10 minutes to eliminate dissolved oxygen, solution was sonicated using a Q500 Ultrasonic Sonicator for 15 minutes to ensure homogenization. Polymerization was initiated when the glass-capped container was immersed in an oil bath heated to 80 °C and left for 18 hours. Subsequently, the template (LSH) was extracted from the polymer using a methanol solution of pH of 13 adjusted by addition of 0.1 M NaOH, employing a Soxhlet extraction method for duration of 6 hours. After extraction, the resulting polymer was thoroughly washed with methanol and left to dry overnight at room temperature. Simultaneously, a non-imprinted polymer (NIP) was prepared under analogous conditions, excluding LSH (template) from the synthesis process.
The bonding structure of both MIP and NIP nanoparticles was investigated through comprehensive characterization techniques. Fourier transform infrared spectrophotometry (FTIR) using the Nicolet 6700 instrument from Thermo Fisher Scientific, Waltham, MA, USA, was employed to analyze the functional groups and chemical bonds present in the synthesized materials. Solid KBr was employed for sample preparation, and spectra were recorded at a high resolution of 4 cm−1 in the range of 400–4000 cm−1. Morphological features of the prepared MIP and NIP nanoparticles were examined using scanning electron microscopy (SEM), JEOL JSM-5510LV instrument in Akishima, Tokyo, with 100000× magnification. Prior to imaging, a conductive Au/Pd coating was applied to the particles to enhance conductivity and facilitate clear visualization of their surface characteristics and morphology. These characterization methods provided detailed insights into the structural and morphological properties of both MIP and NIP nanoparticles, essential for evaluating their suitability in molecular recognition and selective binding for LSH.
2.3. Modification of the screen-printed electrodes
Screen-printed electrodes, Metrohm (SPE), featuring a 4 mm diameter, were sonicated using a Q500 Ultrasonic Sonicator in deionized water, for cleaning, which was followed by a subsequent 10 minutes immersion in absolute ethanol. The electrodes were then air-dried for an hour. To coat a conductive PANI layer on the SPE, a drop-casting method was employed. Specifically, a 10 μL solution of 2 mg mL−1 PANI in ethanol was meticulously applied. After the drying phase, this procedure resulted in a conductive PANI polymer layer with an approximate thickness of 0.25 mL, Fig. 2.4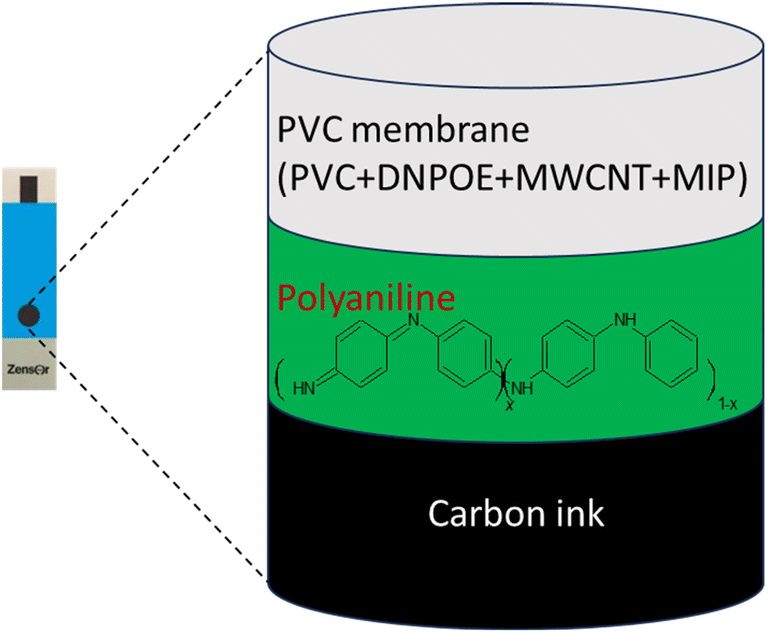 | ||
| Fig. 2 Schematic representation of the SPEs illustrating the sequential coating process: first with a conductive PANI layer, followed by a PVC layer containing MIP nanoparticles as ionophores, and incorporating MWCNTs as transducing materials, along with 2-nitrophenyl octyl ether (2-NPOE) as plasticizer. | ||
The four SPEs (sensors 1–4) detailed in Table 1 exhibit distinct variations in their polymer compositions and the presence of additional layers. The formulation of the recognition layer involved dissolving 100 mg of specific constituents in 2.0 mL of THF. Sensor 1 uses PVC (33.0 wt%) and DNPOE (62.0 wt%) as a plasticizer with MIP (5.0 wt%) as the ionophore, without incorporating MWCNT or a PANI layer. Sensor 2 shares a similar composition but integrates 2.0 wt% MWCNT alongside MIP (5.0 wt%), while excluding PANI. Sensor 3 includes the same components as sensor 2 but includes a PANI layer (√), potentially enhancing stability and selectivity. Sensor 4 mirrors sensor 3 composition but employs a different MIP formulation (5.0 wt%) and incorporates a PANI layer (√), affecting its sensitivity and detection limits. These variations exemplify customized strategies aimed at optimizing sensitivity, selectivity, and operational performance for detecting lurasidone hydrochloride in Tris buffer at pH 7.0. The membrane cocktail, totaling 100 μL, was dispensed onto the dried PANI layer and allowed to dry for two hours. Similar protocols were used to fabricate the SPE, with the exclusion of PANI incorporation, as depicted in Fig. 2. Subsequently, the coated electrodes were immersed in 10−3 M (LSH) solution for preservation.
| Sensor | PVC, wt% | DNPOE, wt% | MWCNT, wt% | Ionophore, wt% | PANI layer | Slope, mV decade−1 | Concentration range, M | Detection limit, M |
|---|---|---|---|---|---|---|---|---|
| 1 | 33.0 | 62.0 | — | 5.0 M | X | 61.3 | 10−5–10−4 | 1.0 × 10−5 |
| 2 | 33.0 | 60.0 | 2.0 | 5.0 M | X | 52.3 | 10−8–10−4 | 1.0 × 10−8 |
| 3 | 33.0 | 60.0 | 2.0 | 5.0 M | √ | 57.1 | 10−8–10−4 | 1.0 × 10−8 |
| 4 | 33.0 | 60.0 | 2.0 | 5.0 N* | √ | 19.0 | 10−7–10−5 | 1.0 × 17 |
2.4. Electromotive force (EMF) measurements
The electromotive force (EMF) of the constructed cell was determined at a constant temperature of 25 ± 1 °C using a pH/mV meter (Orion 720/SA, Cambridge, MA, USA). Potentiometric measurements were conducted after immersing the modified SPEs (sensors 1–4) along with a reference electrode (Ag/AgCl reference electrode with Double-Junction System, Orion 720/SA) into a stirred solution containing LSH (10−4 to 10−8 M). The performance characteristics of the fabricated SPEs were assessed in accordance with the standards set by the International Union of Pure and Applied Chemistry (IUPAC).7 Each experiment was carried out a minimum of three times for reliability and consistency.2.5. Water layer test and pH sensitivity
The water layer test, also called the Morf test, was performed to study the potential drift; it indicates a water layer formation between the conductive layer (PANI) and the recognition layer (PVC/MIP/MWCNTs/2-NPOE).8 Following a 30 minutes recording of electrode potential in a 10−4 M LSH in Tris buffer, the electrode was exposed to a 10−4 M KCl in Tris buffer, as an interfering ion, for another 30 minutes before returning to the LSH solution. This technique tests the probability of a water layer formation and evaluates the sensor stability.The pH sensitivity of the sensor, ranging from pH 3 to 9, was assessed at 25 °C using a 10−4 M LSH in Tris buffer, showing potential stability at different pH conditions, ensuring the reliability and robustness of the electrochemical sensing platform.
2.6. Selectivity assessment against interfering ions
The selectivity was conducted according to the separated solutions method (SSM).9 This method involved the potential measurement of the cell with two solutions of both LSH and interfering ion (10−4 to 10−8 M), maintaining the same activity aA = aB, and the selectivity coefficient against interfering species, was calculated by eqn (1):
was calculated by eqn (1):
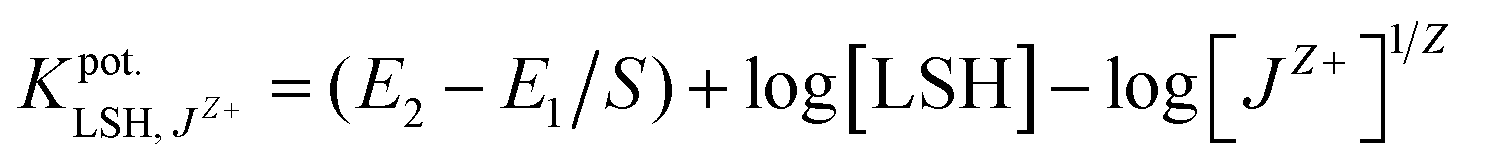 | (1) |
2.7. Response time
The time needed to achieve about 95% of the final signal is the intended response time, which is related to the equilibrium chemistry of the sensor. Response time is recorded for the different LSH concentrations (10−4 to 10−8 M LSH in Tris buffer), to confirm its suitability for routine work.2.8. Applications to pharmaceutical and body fluids samples
In the analysis of real samples (urine spiked with LSH), the procedure included decanting the supernatant liquid from the particulate matter, which was then transferred to a 10 mL measuring flask. To complete the measurement, a Tris base buffer solution was added to the flask. After achieving electrode potential stabilization, EMF readings were recorded with the cell electrodes submerged in the buffer solution. The quantity of LSH was then calculated using the calibration plot established between log[LSH+] and potential values. The efficiency of the developed sensor in detecting LSH in real samples was assessed. These samples, obtained from Amoun Co., Egypt, were in the form of Tablets (Serodopamoun®, 40 mg). To prepare the samples for analysis, five tablets of (Serodopamoun®) were ground, an amount equivalent to one tablet was weighed, and dissolved in (100 mL) of Tris buffer, followed by a two-hour sonication period. After filtration, the resulting filtrate was adjusted with buffer to the desired concentration and subjected directly to analysis using the proposed potentiometric cell or used for spiking of urine sample which was determined also using direct potentiometry.The obtained results were compared with those obtained using the liquid chromatography with tandem mass spectrometry (LC-MS-MS) technique.10 The analysis of Serodopamoun® tablets, containing 40 mg of lysergic acid hydroxyethylamide (LSH), was performed using LC-MS-MS for precise quantification. The preparation involved grinding five tablets to a uniform powder, dissolving an equivalent of one tablet in 100 mL of Tris buffer, and enhancing dissolution with two hours of sonication. After filtering out particulates, the solution was ready for LC-MS-MS analysis, which used a C18 column and a mobile phase of water and acetonitrile with 0.1% formic acid to separate LSH. Electrospray ionization (ESI) in positive mode was used, and the triple quadrupole mass spectrometer in the Multiple Reaction Monitoring (MRM) mode provided high sensitivity. Quantification was done by comparing sample peak areas against a standard calibration curve. This method underscores LC-MS-MS's efficiency and accuracy in detecting LSH in pharmaceutical samples. This comparative analysis aimed to validate the effectiveness and accuracy of the developed ISEs in real-world sample analysis.
3. Results and discussion
3.1. Chemical boding and morphology of MIP and NIP nanoparticles
The synthesis of MIPs and NIPs involves meticulous control over polymerization conditions to achieve specific molecular recognition capabilities. In the case of MIPs for LSH, the process begins with the mixture of VP monomers and EGDMA crosslinker in acetonitrile solvent. BPO acts as the initiator, generating free radicals under heat to initiate polymerization. The template molecule, LSH, interacts with VP monomers through non-covalent bonds, including hydrogen bonding and van der Waals forces. This interaction guides the formation of selective binding sites within the polymer matrix that mimic the size, shape, and functional groups of LSH. Concurrently, EGDMA crosslinks the polymer chains, forming a 3D network that stabilizes the imprinted cavities. Post-polymerization, the removal of LSH from the polymer matrix is crucial to unveil the imprinted sites. Soxhlet extraction with a NaOH in methanol solution dissolves and removes the template molecule, leaving behind specific binding sites that retain molecular recognition characteristics. This step ensures that MIPs retain high selectivity and affinity towards LSH even after template removal. In contrast, NIPs are synthesized under identical conditions but without LSH, resulting in polymers with randomly distributed binding sites and with no selectivity. The absence of the template molecule in NIPs leads to a lack of specific molecular recognition capabilities, highlighting the critical role of molecular imprinting in enhancing polymer selectivity and sensitivity for targeted analytes like LSH.Bonding structure of the MIP and NIP was studied using FTIR, Fig. 3; the spectra of MIP before and after washing confirm the interaction between the polymer and LSH via hydrogen bonding. MIP before washing contains LSH molecules in cavities, where LSH is removed from MIP to form cavities in washed MIP. It is shown in Fig. 3 that the broad band assigned to both N–H and O–H at 3620 cm−1 in the absence of LSH was shifted to 3400 cm−1 in the presence of LSH in the unwashed MIP, which confirms the hydrogen bonding;11–13 also, band at 1734, 2961, 1263, and 751 cm−1 assigned to ketone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, aromatic C–H, C–N of the amine, and C–Cl were shifted to 1688, 2956, 1272, 721 cm−1 after interaction with LSH in the unwashed MIP, respectively;11–13 these shifts to lower or higher values confirms the hydrogen bonding between LSH and the MIP.11–13 The key bands of LSH assigned to N–H or O–H, aromatic C–H and C–Cl were completely absent or very weak in the case of NIP spectra, where the peak of aryl CO existed at a different place in the case of NIP from that of washed and unwashed MIP, which can be assigned to keto group of monomers. Also, the C–Cl band is very weak in the case of washed MIP and absent in the case of NIP, so the washing process for LSH removal to form MIP was good.
O, aromatic C–H, C–N of the amine, and C–Cl were shifted to 1688, 2956, 1272, 721 cm−1 after interaction with LSH in the unwashed MIP, respectively;11–13 these shifts to lower or higher values confirms the hydrogen bonding between LSH and the MIP.11–13 The key bands of LSH assigned to N–H or O–H, aromatic C–H and C–Cl were completely absent or very weak in the case of NIP spectra, where the peak of aryl CO existed at a different place in the case of NIP from that of washed and unwashed MIP, which can be assigned to keto group of monomers. Also, the C–Cl band is very weak in the case of washed MIP and absent in the case of NIP, so the washing process for LSH removal to form MIP was good.
 | ||
| Fig. 3 FTIR spectrum of unwashed MIP (pink), washed MIP (blue), and NIP (red). | ||
The SEM images of MIPs and NIPs highlight clear differences in their morphology and structure, underscoring the impact of the imprinting process. As depicted in Fig. 4, MIP particles appear spherical and uniform, with diameters of 425 nm, ensuring an even distribution across the sensor membrane, optimizing accessible binding sites, and reducing membrane resistance. In contrast, Fig. 4 shows NIP particles with varied shapes and smaller sizes (250 nm), illustrating the lack of uniformity typical in NIPs compared to the consistent morphology of MIPs. The larger particle size of MIPs results from the organized and structured polymerization process driven by the LSH template, which interacts with monomers through non-covalent bonds, guiding the formation of selective binding sites and promoting a cohesive polymer network.14 This organized growth, facilitated by efficient crosslinking around the template, results in larger, uniform particles. Conversely, NIPs, synthesized without the template, undergo random polymerization, leading to a less structured network and smaller, irregular particles.15 The elemental mapping of the MIP in Fig. 4 shows the presence of carbon, oxygen, nitrogen, and notably sulfur, a characteristic element indicating the presence of LSH, confirming the successful imprinting of LSH within the MIP and validating its high selectivity and specificity for LSH.
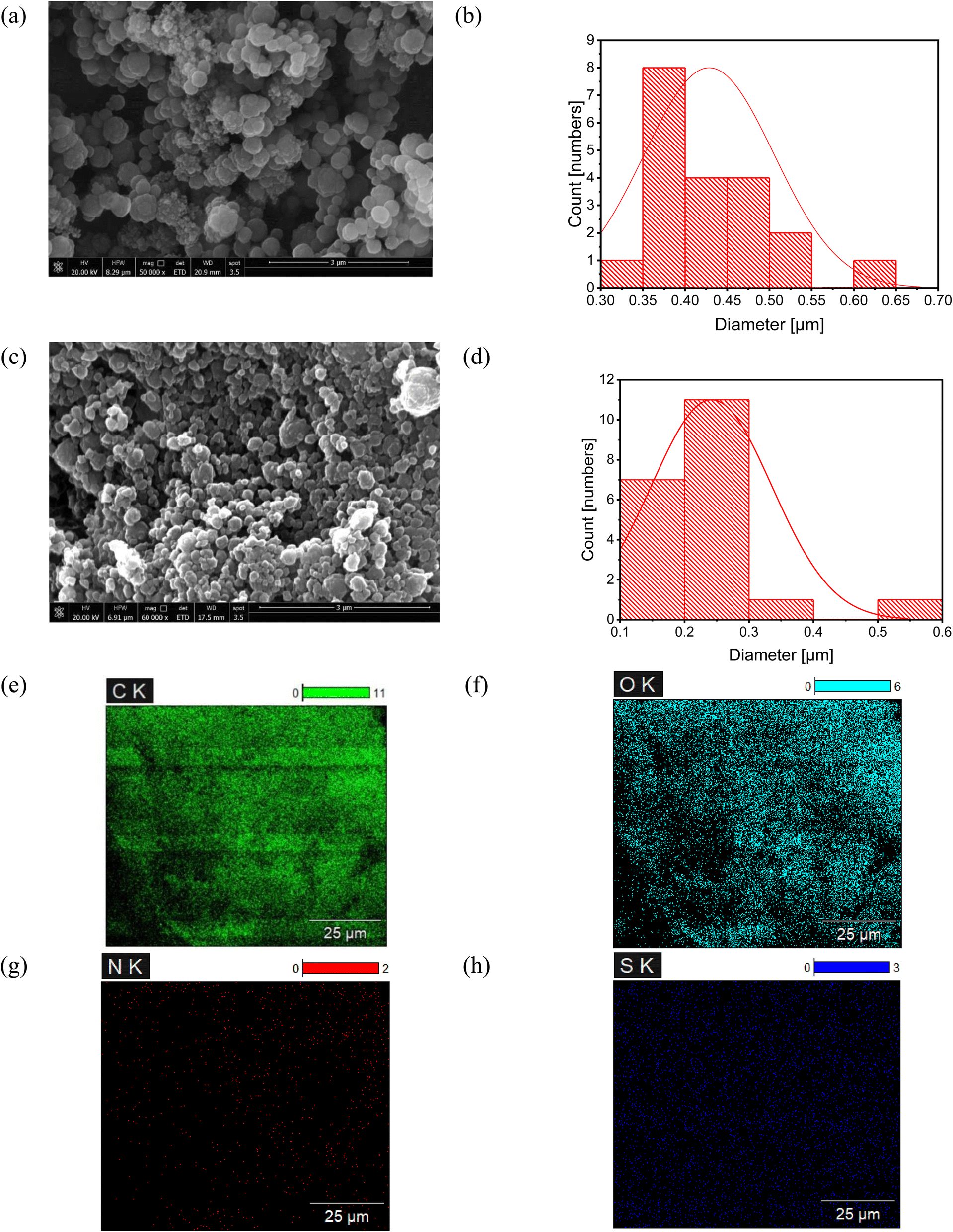 | ||
| Fig. 4 SEM analysis and elemental mapping: SEM images reveal the morphologies and particle size distributions of MIP and NIP, with (a) showing the uniform spherical morphology of MIP and (c) displaying the irregular spherical morphology of NIP. Their corresponding histograms indicate average particle sizes of 425 nm for MIP (b) and 250 nm for NIP (d). Additionally, SEM-EDS elemental mapping of unwashed MIP (loaded with LSH) illustrates the presence and distribution of elements, specifically (e) carbon, (f) oxygen, (g) nitrogen, and (h) sulfur. | ||
3.2. Sensor design and performance evaluation
The modification of SPEs (working electrodes) incorporates innovative elements that collectively enhance their performance for detecting LSH and other analytes. Beginning with a conductive PANI layer, chosen for its excellent electrical conductivity and stability, the initial coating provides a stable foundation for subsequent layers, contributing to overall sensor sensitivity and response time. The subsequent PVC layer plays a pivotal role by embedding MIP nanoparticles designed to selectively bind LSH molecules. PVC not only supports mechanical stability but also enhances electrical properties crucial for signal transmission during electrochemical detection. 2-2-NPOE, acting as a plasticizer within PVC, improves flexibility, workability, and long-term integrity, crucial for uniform coating and preventing brittleness.16 In addition, MWCNTs integrated as transducing materials further amplify sensor performance by enhancing electrical conductivity on the electrode surface.17,18 This augmentation accelerates electron transfer kinetics, thereby boosting sensitivity and response time. This composite approach not only addresses selectivity and stability challenges but also aligns with advancements in sensor technology utilizing nanomaterial enhancements for precise analytical detection in complex matrices. These innovations collectively underscore the potential of SPEs in pharmaceutical analysis, environmental monitoring, and biomedical diagnostics, where accurate and reliable detection is paramount.The electrochemical capabilities of the various sensors were evaluated through analysis of their potentiometric response to LSH across a concentration range from 10−9 to 10−4 M. In sensor 1, the carbon ink layer of the SPE is coated with a PVC membrane layer containing 33 wt% PVC and 62 wt% DNPOE, 5 wt% MIP ionophore; it showed nearly Nernstian response in the narrow concentration range of 10−5–10−4 M, with a high detection limit of 10−5 M and slope of 61.3 mV decade−1, Table 1. Sensor 2 was modified by addition of 2 wt% MWCNTs for the layer by reducing the DNPOE to 60 wt%; the concentration range was increased to 4 orders of magnitude (concentration range of 10−8–10−4) with achieving a very low detection limit of 10 nM, but with a slight decline in the sub-Nernstian response of 52.3 mV decade−1, Fig. 5. The addition of 2 wt% MWCNT improved the electron transfer within the PVC/MIP/DNPOE layer, that resulted in the improved detection limit and wider concentration range.19,20 In sensor 3, the slope was further improved by the addition of PANI coating between the base SPE and PVC/MIP/DNPOE/MWCNTs layer to 57.1 mV decade−1 in the concentration range of 10−8–10−4 M and a detection limit of 10 nM, Table 1 and Fig. 5, due to ease of electron transfer by PANI layer.4,21,22
 | ||
| Fig. 5 Calibration curves with error bars for the various SPEs, the modified SPE was used as the working electrode, Ag/AgCl as the reference electrode, with 10−4 to 10−9 M LSH in Tris buffer as the electrolyte solution. | ||
For comparison performance of MIP and NIP, sensor 4 was prepared with the same composition as sensor 3 using NIP as an ionophore instead of MIP. The NIP in sensor 4 demonstrates some sensitivity to LSH but lacks selectivity compared to MIP-based sensors. Sensor 4, using NIP as the ionophore instead of MIP, showed a significantly reduced slope (19 mV decade−1) and limited detection capability (10−7 M) in a narrow concentration range (10−7–10−5 M), as observed in Fig. 5. This indicates that while NIP can interact with LSH to some extent, its response lacks the specificity and efficiency achieved by MIPs. Based on these promising results, sensor 3 underwent further examination to assess its performance regarding interference from ions, stability over time, and its ability to detect LSH in spiked urine samples spiked with the pharmaceutical formulation.
3.3. Stability against the pH change
The investigation into the pH sensitivity of sensor 3 provided insightful findings, as depicted in Fig. 6. Across the tested pH range of 3 to 9, the sensor's potential remained stable, except for a noticeable decrease observed between pH 5 to 6.5, as illustrated in Fig. 6. This pH dependency correlates with the pKa of LSH, which is 7.6. The decrease in potential within this pH range is influenced by LSH's solubility, where the drug is less soluble and less available for detection in its non-ionized form. The observed potential decrease as pH increases from 5 to 6.5 can be attributed to the rising concentration of hydroxyl ions, confirming the sensor's anionic response, which aligns with the MIP's ability to interact with hydroxide ions through hydrogen bonding.4,14,23 Previous reports indicated that LSH exhibits maximum solubility at pH 3.8 using acetate buffer; however, this pH may introduce interference due to LSH existing in different forms.23 Given LSH's pKa aligns closely with the pH maintained by Tris buffer (around pH 7.0), Tris buffer proves suitable for this study. The pH stability provided by Tris-buffer ensures LSH predominantly exists in its ionized form, optimizing the accuracy and stability of detection using electrochemical sensors. Moreover, the Tris-buffer minimizes interference from ions or substances in complex biological samples like urine, thereby enhancing the sensor's selectivity and sensitivity towards LSH. These observations underscore sensor 3's effectiveness in detecting LSH within the typical pH range of Tris buffer. | ||
| Fig. 6 Effect of pH variation from 3 to 9, with error bars, on sensor 3. The modified SPE was used as the working electrode, Ag/AgCl as the reference electrode, with 10−4 M LSH as the electrolyte solution. | ||
3.4. Stability against water layer formation
The formation of a water layer between the PVC, PANI layer, and the carbon surface of the SPEs can potentially hinder or adversely affect the conductivity and potential response of the sensor.8 To assess this possibility for sensor 2 and sensor 3, the Morf test was conducted. The results, as shown in Fig. 7, revealed a slight potential drift for both sensors when alternating between the 10−4 M LSH in Tris buffer. Specifically, sensor 3 exhibited a smaller potential shift (approximately 7 mV) compared to sensor 2, which showed a drift of about 15 mV. This difference suggests that the PANI layer in sensor 3 plays a crucial role due to its lipophilic nature, which resists the formation of a water layer. The PANI layer's compatibility with both the SPE and PVC layers facilitates electronic conduction and enhances the stability of potential readings.8 These findings underscore the PANI layer's effectiveness in mitigating potential drift caused by water layer formation, thereby improving the sensor's reliability and performance in electrochemical detection applications.4 | ||
| Fig. 7 Water layer test (Morf test) for sensor 2 (red) and sensor 3 (black), the E measurements alternating between 10−4 M LSH in Tris buffer and Tris buffer (pH 7) as an interfering ion. | ||
3.5. Selectivity against interfering species
The sensor selectivity is so important property that defines the applicability of the sensor in real samples, as it shows the sensor preference for the analyte over different interfering ions existing in sample. The response for LSH was compared to the response of the selected interfering ions; citric acid, barbituric acid, glycine, acetic acid, glutamic acid, urea, glucose, Fe2+, K+, Na+, and phosphate; these interfering ions were selected as it may exist with LSH in its pharmaceutical formulation or biological samples of urine or serum. The selectivity of sensor 3, for LSH over the selected interfering ions (10−4 to 10−8 M), was tested using SSM method, Fig. 8. The results obtained showed minimum response of sensor 3 for all interfering ions, and very low response of glutamic acid at high concentration, shown in Fig. 8; this is attributed to the existence of MIP ionophore that is designed for the target LSH as key-lock principle; interaction of LSH, that act as key, through the formation of hydrogen bonding on surface and within cavities of MIP nanoparticles which act as lock.4 The other interfering ions can form hydrogen bonding only on MIP surface, where the MIP cavities are not suitable for this interaction, i.e. key-lock principle is not obeyed; so, all interfering ions showed minimum response. Glutamic acid exhibited low response at the highest measured concentration, which may be due to its small size and ability to form hydrogen bonding within MIP particles cavities and on the MIP surface.24 | ||
| Fig. 8 Selectivity profile of sensor 3 showing its response to (10−4 to 10−8 M in Tris buffer, pH = 7) and various interfering ions (10−4 to 10−8 M in Tris buffer, pH = 7) using SSM methods. | ||
3.6. Response time and lifetime
The time interval between electrode immersion in solution and 90% of the stable reading is the true response time for the sensor.25 Sensor 3 exhibited a response time of 2–3 minutes, which is lower than that of sensor 2, which extends for 10 minutes in high concentration region, Fig. 9. This improvement can be attributed to the inclusion of the PANI layer in sensor 3. The PANI layer reduces the formation of a water layer between the PVC, PANI layer, and the carbon surface of the SPEs, which is known to cause potential instability and longer response times. Additionally, the PANI layer enhances the electronic conductivity and stability of the sensor due to its lipophilic nature and compatibility with both the SPE and PVC layers, facilitating faster and more stable potential readings.4 Sensor 3 was found to operate properly for 15 days, and after that its response was deteriorated; so shelf life is 15 days. | ||
| Fig. 9 Response time comparison of sensor 2 (blue) and sensor 3 (red) for 10−4 M LSH in Tris buffer solution. | ||
3.7. Analysis of tablets and spiked urine
The optimized SPE (sensor 3) was applied to determine LSH in its pharmaceutical formulation (Serodopamoun® 40 mg) and a urine sample spiked with LSH. Using a reference HPLC method,26 the sample concentrations and recovery rates were calculated, as shown in Table 2. The experimental concentrations obtained were 51.5 mg L−1 for Serodopamoun® and 52.0 mg L−1 for the spiked urine sample in Tri's buffer (1 M, pH = 7), with theoretical concentrations of 52.9 mg L−1 (10−4 M) for both. The recovery percentages were 97.3 ± 0.1% for the pharmaceutical formulation and 98.2 ± 0.3% for the spiked urine sample. These results, compared to the reference HPLC method, show high accuracy, with recovery values exceeding 97.0%. The reproducibility of the measurements was confirmed by the low standard deviation values, indicating that the optimized sensor can be successfully applied for the determination of LSH in real samples.| Sample | Theoretical conc. | HPLC method % recovery | Sensor 3 | |
|---|---|---|---|---|
| Concentration | % Recovery | |||
| Serodopamoun® | 52.9 mg L−1 (10−4 M) | 101.6 ± 0.2 | 51.5 mg L−1 | 97.3 ± 0.1 |
| Spiked urine | 52.9 mg L−1 (10−4 M) | — | 52.0 mg L−1 | 98.2 ± 0.3 |
4. Conclusion
The potentiometric biosensors developed for the determination of Lurasidone Hydrochloride (LSH) in pharmaceutical formulations demonstrate significant improvements in performance due to the incorporation of specific materials. The sensor is based on screen-printed electrodes (SPEs) coated first with a conductive polyaniline (PANI) film and secondly with a polyvinyl chloride (PVC) layer containing molecularly imprinted polymer (MIP) nanoparticles as ionophores, alongside multiwalled carbon nanotubes (MWCNTs) as transducing materials and 2-nitrophenyl octyl ether (2-NPOE) as a plasticizer. The optimized SPE composition, including 33% PVC, 60% DNPOE, 2% MWCNTs, and 5% MIP, achieved a slope of 57.1 mV decade−1 within a concentration range of 10−8–10−4 M and the lowest reported detection limit of 7.94 × 10−9 M. MWCNTs enhance electron transfer, improving sensitivity and detection limits, while the lipophilic PANI layer resists water layer formation, maintaining potential stability and reducing response time. The MIP, tailored for LSH, ensures high selectivity by providing numerous binding sites that match the target molecule's shape and functional groups. Adjusting the pH to 7 using Tris buffer optimizes sensor response, ensuring LSH exists in its unionized form for accurate detection. This setup minimizes interference from other ions in complex biological samples, such as urine. The sensor's ability to accurately determine LSH in Serodopamoun® tablets with high reproducibility highlights its potential for pharmaceutical and biological fluid analysis, exhibiting high stability, selectivity, short response time, and effective performance against interfering species like citric acid, barbituric acid, glycine, acetic acid, glutamic acid, urea, glucose, Fe2+, K+, Na+, and phosphate ions.Data availability
All data supporting the results are available as part of the article, and no additional data is available.Conflicts of interest
There are no conflicts to declare.References
- S. Shah, B. Parmar, M. Soniwala and J. Chavda, Design, Optimization, and Evaluation of Lurasidone Hydrochloride Nanocrystals, AAPS PharmSciTech, 2016, 17, 1150–1158, DOI:10.1208/S12249-015-0449-Z/TABLES/4.
- F. Cder, LATUDA (Lurasidone Hydrochloride) Tablets.
- S. Singh, J. Wang and S. Cinti, Review—An Overview on Recent Progress in Screen-Printed Electroanalytical (Bio)Sensors, ECS Sens. Plus, 2022, 2(1), 023401, DOI:10.1149/2754-2726/ac70e2.
- M. M. El-Beshlawy, F. M. Abdel-Haleem, A. H. Kamel and A. Barhoum, Screen-Printed Sensors Coated with Polyaniline/Molecularly Imprinted Polymer Membranes for the Potentiometric Determination of 2,4-Dichlorophenoxyacetic Acid Herbicide in Wastewater and Agricultural Soil, Chemosens, 2023, 11, 3, DOI:10.3390/CHEMOSENSORS11010003.
- D. Y. Choi, J. C. Yang, S. W. Hong and J. Park, Molecularly Imprinted Polymer-Based Electrochemical Impedimetric Sensors on Screen-Printed Carbon Electrodes for the Detection of Trace Cytokine IL-1β, Biosens. Bioelectron., 2022, 204, 114073, DOI:10.1016/j.bios.2022.114073.
- S. S. Soliman, A. M. Mahmoud, M. R. Elghobashy, H. E. Zaazaa and G. A. Sedik, Point-of-Care Electrochemical Sensor for Selective Determination of Date Rape Drug “Ketamine” Based on Core-Shell Molecularly Imprinted Polymer, Talanta, 2023, 254, 124151, DOI:10.1016/j.talanta.2022.124151.
- W. E. Morf, K. Seiler, B. Lehmann, C. Behringer, K. Hartman and W. Simon, Carriers for Chemical Sensors: Design Features of Optical Sensors (Optodes) Based on Selective Chromoionophores, Pure Appl. Chem., 1989, 61, 1613–1618, DOI:10.1351/PAC198961091613/MACHINEREADABLECITATION/RIS.
- M. Fibbioli, W. E. Morf, M. Badertscher, N. F. de Rooij and E. Pretsch, Potential Drifts of Solid-Contacted Ion-Selective Electrodes Due to Zero-Current Ion Fluxes Through the Sensor Membrane, Electroanalysis, 2000, 12, 1286–1292, DOI:10.1002/1521-4109(200011)12:16<1286::AID-ELAN1286>3.0.CO;2-Q.
- Y. Umezawa, P. Bühlmann, K. Umezawa, K. Tohda and S. Amemiya, Potentiometric Selectivity Coefficients of Ion-Selective Electrodes Part I. Inorganic Cations (Technical Report), Pure Appl. Chem., 2000, 72(10), 1851–2082, DOI:10.1351/pac200072101851.
- M. Y. Katteboina, N. R. Pilli, R. Mullangi, R. R. Seelam and S. R. Satla, LC-MS/MS Assay for the Determination of Lurasidone and Its Active Metabolite, ID-14283 in Human Plasma and Its Application to a Clinical Pharmacokinetic Study, Biomed. Chromatogr., 2016, 30, 1065–1074, DOI:10.1002/BMC.3651.
- J. R. Madan, K. T. Pawar and K. Dua, Solubility Enhancement Studies on Lurasidone Hydrochloride Using Mixed Hydrotropy, Int. J. Pharm. Invest., 2015, 5, 114, DOI:10.4103/2230-973X.153390.
- (14) (PDF) Development and in vitro assessment of melt in mouth tablets of lurasidone hydrochloride, https://www.researchgate.net/publication/319164407_DEVELOPMENT_AND_IN_VITRO_ASSESSMENT_OF_MELT_IN_MOUTH_TABLETS_OF_LURASIDONE_HYDROCHLORIDE/figures?lo=1, accessed 9 June 2024.
- K. J. Devi, C. Shilpaja, K. Umasankar and K. Sushma, J. Global Trends Pharm. Sci., 2017, 8(2), 3917–3926 CAS.
- M. M. El-Beshlawy, F. M. Abdel-Haleem and A. Barhoum, Molecularly Imprinted Potentiometric Sensor for Nanomolar Determination of Pioglitazone Hydrochloride in Pharmaceutical Formulations, Electroanalysis, 2021, elan.202060141, DOI:10.1002/elan.202060141.
- K. Y. Goud, S. K. Kalisa, V. Kumar, Y. F. Tsang, S. E. Lee, K. V. Gobi and K. H. Kim, Progress on Nanostructured Electrochemical Sensors and Their Recognition Elements for Detection of Mycotoxins: A Review, Biosens. Bioelectron., 2018, 121, 205–222, DOI:10.1016/J.BIOS.2018.08.029.
- I. Md Isa, N. Mohd Sohaimi, N. Hashim, A. Kamari, A. Mohamed, M. Ahmad and S. A. Ghani, Determination of Salicylate Ion by Potentiometric Membrane Electrode Based on Zinc Aluminium Layered Double Hydroxides-4(2,4-Dichlorophenoxy) Butyrate Nanocomposites, Int. J. Electrochem. Sci., 2013, 8(2), 2112–2121, DOI:10.1016/S1452-3981(23)14295-7.
- F. M. Abdel-Haleem, E. Gamal, M. S. Rizk, A. Madbouly, R. M. El Nashar, B. Anis, H. M. Elnabawy, A. S. G. Khalil and A. Barhoum, Molecularly Imprinted Electrochemical Sensor-Based Fe2O3@MWCNTs for Ivabradine Drug Determination in Pharmaceutical Formulation, Serum, and Urine Samples, Front. Bioeng. Biotechnol., 2021, 9, 1–16, DOI:10.3389/fbioe.2021.648704.
- F. M. Abdel-Haleem, E. Gamal, M. S. Rizk, R. M. El Nashar, B. Anis, H. M. Elnabawy, A. S. G. Khalil and A. Barhoum, T-Butyl Calixarene/Fe2O3@MWCNTs Composite-Based Potentiometric Sensor for Determination of Ivabradine Hydrochloride in Pharmaceutical Formulations, Mater. Sci. Eng. C, 2020, 116, 111110, DOI:10.1016/j.msec.2020.111110.
- F. M. Abdel-Haleem, M. S. Rizk and I. H. A. Badr, Potentiometric Determination of Ciprofloxacin in Physiological Fluids Using Carbon Paste and Nano-Composite Carbon Paste Electrodes, Electroanalysis, 2017, 29(4), 1172–1179, DOI:10.1002/elan.201600735.
- D. Yuan, A. H. C. Anthis, M. Ghahraman Afshar, N. Pankratova, M. Cuartero, G. A. Crespo and E. Bakker, All-Solid-State Potentiometric Sensors with a Multiwalled Carbon Nanotube Inner Transducing Layer for Anion Detection in Environmental Samples, Anal. Chem., 2015, 87(17), 8640–8645, DOI:10.1021/acs.analchem.5b01941.
- K. L. Bhowmik, K. Deb, A. Bera, R. K. Nath and B. Saha, Charge Transport through Polyaniline Incorporated Electrically Conducting Functional Paper, J. Phys. Chem. C, 2016, 120, 5855–5860, DOI:10.1021/acs.jpcc.5b08650.
- C. Dhand, M. Das, M. Datta and B. D. Malhotra, Recent Advances in Polyaniline Based Biosensors, Biosens. Bioelectron., 2011, 26, 2811–2821, DOI:10.1016/j.bios.2010.10.017.
- Y. Hu, Y. Guo, B. Li, R. Xu, X. Fang, Y. Cao, Z. Liu, C. Jiang and S. Lu, Influence of the PKa Value of Cinnamic Acid and P-Hydroxycinnamic Acid on the Solubility of a Lurasidone Hydrochloride-Based Coamorphous System, ACS Omega, 2021, 6(4), 3106–3119, DOI:10.1021/acsomega.0c05510.
- P. Screenivasulu Reddy, T. Kobayashi, M. Abe and N. Fujii, Molecular Imprinted Nylon-6 as a Recognition Material of Amino Acids, Eur. Polym. J., 2002, 38, 521–529, DOI:10.1016/S0014-3057(01)00212-9.
- E. Bakker, P. Bühlmann and E. Pretsch, Carrier-Based Ion-Selective Electrodes and Bulk Optodes. 1. General Characteristics, Chem. Rev., 1997, 97(8), 3083–3132, DOI:10.1021/cr940394a.
- M. D. Vaja, R. R. Patel, B. D. Patel and A. B. Chaudhary, Development and Validation of RP-HPLC Method for Estimation of Lurasidone and its impurities Lurasidone 1 and Lurasidone 8, Res. J. Pharm. Technol., 2022, 15(11), 4999–5004, DOI:10.52711/0974-360X.2022.00840.
| This journal is © The Royal Society of Chemistry 2024 |