
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving the catalytic performance of Co/BaCeO3 catalyst for ammonia synthesis by Y-modification of the perovskite-type support†
Magdalena Zyberta,
Hubert Ronduda *a,
Wojciech Patkowskia,
Andrzej Ostrowskia,
Kamil Sobczakb and
Wioletta Raróg-Pileckaa
*a,
Wojciech Patkowskia,
Andrzej Ostrowskia,
Kamil Sobczakb and
Wioletta Raróg-Pileckaa
aWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, 00-664, Warsaw, Poland. E-mail: hubert.ronduda@pw.edu.pl; Tel: +48 22 234 76 02
bUniversity of Warsaw Biological and Chemical Research Centre, Żwirki i Wigury 101, 02-089 Warsaw, Poland
First published on 12th November 2024
Abstract
Y-modified perovskite-type oxides BaCe1−xYxO3−δ (x = 0–0.30) were synthesised and used as supports for cobalt catalysts. The influence of yttrium content on the properties of the support and catalyst performance in the ammonia synthesis reaction was examined using PXRD, STEM-EDX, and sorption techniques (N2 physisorption, H2-TPD, CO2-TPD). The studies revealed that the incorporation of a small amount of yttrium into barium cerate (up to 10 mol%) increased specific surface area and basicity. The catalyst testing under conditions close to the industrial ones (T = 400–470 °C, p = 6.3 MPa, H2/N2 = 3) showed that the most active catalyst was deposited on a support containing 10 mol% Y. The NH3 synthesis reaction rate was 15–20% higher than that of the undoped Co/BaCeO3 catalyst. The activity of the catalysts decreased with further increasing Y content in the support (up to 30 mol%). However, all the studied Co/BaCe1−xYxO3−δ catalysts exhibited excellent thermal stability, over 240 h of operation. The particularly beneficial properties of the catalyst containing 10 mol% of Y were associated with the highest basicity of the support surface, favourable adsorption properties (suitable proportion of weakly and strongly hydrogen-binding sites), and preferred size of cobalt particles (60 nm). The Co/BaCe0.90Y0.10O3−δ catalyst showed better ammonia synthesis performance compared to the commercial iron catalyst (ZA-5), giving prospects for process reorganisation towards energy-efficient ammonia production.
1. Introduction
Ammonia is one of the world's most important mass-produced chemical products, mainly due to its use in producing fertilisers responsible for supporting almost half of global food production for the world's ever-growing population. Interest in ammonia has been growing recently also due to the significant potential of ammonia as a hydrogen vector1–4 related to its high energy density (12.8 GJ m−3), bulk density of hydrogen (0.107 kg H2 per L), mass hydrogen content (17.8 wt%) and no CO2 emissions into the atmosphere during hydrogen recovery from ammonia. Moreover, hydrogen storage in the form of ammonia does not require the use of high-pressure or cryogenic tanks, and its transport methods are well-developed. Ammonia is also seen as a promising carbon-free fuel for fuel cell technology.5–7 Ammonia can power fuel cells directly or indirectly by supplying hydrogen from ammonia thermal decomposition.8,9 Ammonia owes its significant potential to be an excellent fuel to its suitable storage and transport properties, among others, which lead it to be used for combustion in car engines and turbines. For example, it is estimated that carbon emissions from shipping can be reduced by 50% by 2050 using ammonia as a fuel.10 It means a lot considering that the maritime industry is responsible for approx. 3% of total anthropogenic CO2 emissions.11 However, the lack of understanding of the NH3 combustion characteristics, methods of enhancement, and optimisation of NOx formation in combustion are still great limits for utilising NH3 as a fuel on a large scale.12–14Ammonia production on an industrial scale is carried out using the catalytic Haber–Bosch process, the main challenge of which is activating a very stable N–N triple bond (the rate-determining step of ammonia synthesis). The generally accepted mechanism for catalytic ammonia synthesis relies on a dissociative route, in which adsorbed N2 dissociates directly, and the N atom is gradually hydrogenated until ammonia is produced and released.15 Therefore, a high temperature (>400 °C) and pressure (>15 MPa) are required for the reaction to proceed effectively. However, the optimal range of conditions under which ammonia synthesis can be carried out efficiently is determined by the catalyst used. The most commonly used iron catalyst operates at temperatures of 400–500 °C and at pressures of 15–30 MPa. Using ruthenium as the active phase of the catalyst allowed the process pressure to be effectively reduced to 9 MPa.16 Despite its many meaningful applications, the industrial Haber–Bosch process is highly polluting and energy-intensive. Demanding reaction conditions cause the process to consume over 1.5% of annual energy production (30–60 GJ per ton of NH3) and emit 1.9 tons of CO2 per ton of ammonia produced,17 which, taking into account the annual production of about 176 million tons, gives about 1.8% of global CO2 emissions. Facing the deepening climate crisis and environmental requirements to reduce dependence on fossil fuels and greenhouse gas emissions, the obvious need to develop the ammonia synthesis process with reduced CO2 emissions, carried out under milder temperature and pressure conditions and powered by energy from renewable sources (green ammonia) appeared. There are several approaches to decarbonise ammonia production.18–21 In the first scenario, producers could still use natural gas as a feedstock to steam methane reforming process (SMR). However, carbon capture and storage (CCS) technology is needed to be developed. In the second scenario, steam methane reforming units can be replaced by electrolysers utilising water as a source of hydrogen and powered by renewable energy. Nevertheless, regardless of how the process is reorganised, it is still crucial to develop new catalytic systems enabling more sustainable ammonia production under milder pressure and temperature conditions to correspond well with an economy based on hydrogen and renewable energy such as solar, hydro, or wind power.
The theoretical model linking the surface activity of transition metals with the nitrogen adsorption energy indicates a significant potential of transition metals other than iron and ruthenium, particularly cobalt, to catalyse the ammonia synthesis reaction.22 Cobalt-based catalysts are, therefore, a constantly developing issue. Hagen et al.23 showed that at a temperature of 440 °C the activity of the barium-promoted cobalt catalyst supported on carbon was almost twice as high compared to the iron catalyst, and a small activity decrease resulting from increased product concentration in the gas was observed. Despite favourable catalytic properties, these systems were not stable under reaction conditions due to the carbon support methanation. Even though there is a possibility of limiting this unfavourable phenomenon by adding a barium promoter,24 cobalt catalysts supported on carbon materials have not found practical applications. In recent years, many attempts have been increasingly made to improve the properties and activity of cobalt catalysts by using new supports.25–32 Since the rate-determining step of ammonia synthesis is the chemisorptive dissociation of the nitrogen molecule on the catalyst surface,33 the support's electron-donating (basic) properties are of paramount importance. The ability to donate electrons from the support to the active metal and then to the adsorbed N2 molecules facilitates their dissociation, thus accelerating the reaction. In this context, supports such as rare earth oxides25,26,28,34,35 and mixed magnesium oxide–rare earth oxide31,32,36,37 become the object of greater interest of researchers focused on the development of new ammonia synthesis catalysts.
Perovskite-type oxides are a particularly promising group of new support materials for ammonia synthesis catalysts.38–44 They are characterised by high surface basicity and thermal stability in a wide temperature range and different conditions.45,46 Ruthenium catalysts using supports of perovskite structure, i.e., BaCeO3,38,39,42,44 BaZrO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 40,42 and BaTiO3,41 have been described in the literature. These supports allowed for good ruthenium dispersion and optimal particle size (2–3 nm). The strong basicity of the support surface resulted in an increase in the electron transfer rate to Ru and acceleration of the N2 dissociation. One of the ways to enhance the properties of perovskite-type supports is their modification with rare earth metal ions (e.g., La, Pr, Y), which in the case of ruthenium catalysts resulted in a significant improvement in their catalytic performance.42–44 The main factors responsible for the enhanced performance of modified ruthenium catalysts were better metal dispersion, increased strength of metal–support interactions and enhanced electronic conductivity of the support.
40,42 and BaTiO3,41 have been described in the literature. These supports allowed for good ruthenium dispersion and optimal particle size (2–3 nm). The strong basicity of the support surface resulted in an increase in the electron transfer rate to Ru and acceleration of the N2 dissociation. One of the ways to enhance the properties of perovskite-type supports is their modification with rare earth metal ions (e.g., La, Pr, Y), which in the case of ruthenium catalysts resulted in a significant improvement in their catalytic performance.42–44 The main factors responsible for the enhanced performance of modified ruthenium catalysts were better metal dispersion, increased strength of metal–support interactions and enhanced electronic conductivity of the support.
The high potential of BaCeO3 to be an effective catalyst support, as well as the possibility for boosting properties of this support and catalysts by modification with rare earth metal ions, have become a reason for conducting the study on the influence of yttrium modification on the properties of the BaCe1−xYxO3−δ (x = 0–0.30) supports and, consequently, on the catalytic performance of the cobalt catalyst in ammonia synthesis reaction. The supports and catalysts were examined using PXRD, STEM-EDX, and various sorption techniques (N2 physisorption, H2-TPD, CO2-TPD). The catalyst activity was investigated under conditions close to the industrial ones (T = 400–470 °C, p = 6.3 MPa, H2/N2 = 3). In addition, it is worth noting that there have been no previous literature reports on using BaCeO3 and Y-modified BaCeO3 as the support for cobalt catalysts for the ammonia synthesis process.
2. Experimental section
2.1. Support and catalyst preparation
Yttrium-modified BaCe1−xYxO3−δ (where x = 0–0.30) supports were synthesised by a co-precipitation method. The Y content range of 0–0.30 was selected based on the literature reports. According to Takeuchi et al.47 and Wu et al.,48 Y-doped BaCeO3 is prepared as almost single-phase perovskite compounds with a Y content up to 0.3 mole. In the case of BaCe1−xYxO3−δ (x = 0), aqueous ammonium carbonate solution was added dropwise to a solution of barium and cerium nitrates under vigorous stirring at 90 °C. The precipitation was conducted at a double molar excess of ammonium carbonate relative to barium and cerium nitrates. Then, the slurry was aged at 90 °C for 1 h, filtered, washed with distilled water, and dried at 90 °C for 24 h. Finally, the sample was calcined at 1100 °C for 10 h in air. The calcination conditions were established based on the thermal analysis results of the obtained precipitate (Fig. S1†). The yttrium-modified BaCe1−xYxO3−δ (x = 0.05–0.30) supports were prepared according to the same procedure as described above, with the difference that during the co-precipitation step, the ammonium carbonate was added to the mixture of barium, cerium, and yttrium nitrates (in a fixed proportion). The series of BaCe1−xYxO3−δ (x = 0–0.30) supports containing 0–30 mol% of yttrium was obtained and then used to prepare a series of Co/BaCe1−xYxO3−δ (x = 0–0.30) catalysts using a deposition–precipitation method. A 40 wt% Co loading was chosen based on our previous works.32 The support was dispersed in an aqueous solution of cobalt nitrate. Then, aqueous potassium carbonate solution was added dropwise under vigorous stirring at 90 °C. The precipitation was carried out until pH of about 9 was reached. The obtained slurry was aged at 90 °C for 1 h, filtered, washed with distilled water, and dried at 120 °C for 18 h. Then, the sample was calcined at 500 °C for 5 h in air. The precursor samples were activated at 600 °C under hydrogen or hydrogen/nitrogen flow before the characterisation studies and catalytic testing. The reduction temperature was chosen based on the H2-TPR studies (see Section 3.2). The benchmark catalysts supported on BaCe1−xYxO3−δ (where x = 0.10) using various transition metals as the active phase (Co, Fe, Mo, Ni) were also prepared using the conventional wet-impregnation method. The active metal content was fixed at 10 wt%. Preliminary research on the activity of these catalysts in NH3 synthesis reaction (Table S1†) allowed us to select the most active metal (cobalt), which was then used to prepare the series of catalysts described above, which are the subject of this work.2.2. Support and catalyst characterisation
N2 physisorption was performed on a Micromeritics ASAP2020 instrument at −196 °C. Before the measurement, a sample (0.5 g) was degassed under vacuum at 90 °C for 1 h and then at 300 °C for 4 h. The specific surface area (SSA) was calculated using the BET method, and the total pore volume (PV) was calculated using the BJH method. In order to determine the specific surface area of the reduced catalysts, the catalyst precursor sample (0.5 g) was firstly reduced in situ at 600 °C for 10 h under H2 flow (40 mL min−1) and purged at 620 °C for 2 h under He flow (40 mL min−1). Then, the sample was degassed under vacuum at 200 °C for 2 h and N2 physisorption measurements were performed.Powder X-ray diffraction profiles were recorded on a Bruker D8 Advance diffractometer equipped with a LYNXEYE position-sensitive detector, using CuKα radiation (λ = 0.15418 nm). The data were collected in the Bragg–Brentano (θ/θ) horizontal geometry between 20° and 70° (2θ) in a continuous scan using 0.03° steps 10 s per step. The diffractometer incident beam path was equipped with a 2.5° Soller slit and a 1.14° fixed divergence slit, while the diffracted beam path was equipped with a programmable anti-scatter slit (fixed at 2.20°), a Ni β-filter, and a 2.5° Soller slit. Data were collected under standard laboratory conditions.
STEM-EDX investigations were performed on a Talos F200X (FEI) microscope operated at 200 kV. All STEM images were collected using a high-angle annular dark-field (HAADF) detector and energy-dispersive X-ray spectroscopy on a Bruker BD4 spectrometer. Before the analysis, the catalyst precursor sample (0.5 g) was reduced ex situ at 600 °C for 18 h under H2 flow (40 mL min−1) and then crushed and powdered. The powdered catalyst was dispersed in ethanol, and a few drops of the dispersion were dropped on a carbon-coated copper-mesh TEM grid and dried overnight at room temperature.
CO2 temperature-programmed desorption (CO2-TPD) was performed on a Micromeritics AutoChem II 2920 chemisorption analyser. Before the measurement, a support sample (0.2 g) was purged at 300 °C for 3 h under He flow (40 mL min−1). After cooling to 40 °C, CO2 was introduced (40 mL min−1) for 2 h. Next, the sample was purged with He flow (40 mL min−1) at 40 °C until the baseline was stable. CO2-TPD was carried out in a flow of He (40 mL min−1) with a ramp of 10 °C min−1 up to 900 °C, and the TCD signal was recorded continuously.
H2 temperature-programmed reduction (H2-TPR) studies were performed using a Micromeritics AutoChem II 2920 chemisorption analyser. A catalyst precursor sample (0.2 g) was loaded into a U-shape quartz reactor and heated to 800 °C at a ramping rate of 5 °C min−1 in a 10% H2/Ar mixture (40 mL min−1) flow. The consumption of hydrogen was determined by a thermal conductivity detector (TCD).
H2 temperature-programmed desorption (H2-TPD) was performed on a Micromeritics AutoChem II 2920 chemisorption analyser. Prior to the measurement, a catalyst precursor sample (0.5 g) was reduced at 600 °C for 18 h under H2 flow (40 mL min−1) and purged at 620 °C for 2 h under Ar flow (40 mL min−1). After cooling to 150 °C, H2 gas was introduced (40 mL min−1) for 15 min, then cooled to 0 °C and kept for 15 min. Next, the sample was purged with Ar at 0 °C until the baseline was stable. H2-TPD was carried out in a flow of Ar (40 mL min−1) with a ramp of 5 °C min−1 up to 700 °C, and the TCD signal was recorded continuously.
2.3. Catalytic performance testing
The activity of the catalysts in the ammonia synthesis reaction was evaluated in a tubular flow reactor. Prior to the catalytic testing, the catalyst precursor sample (0.5 g) was reduced at the following temperature program: 470 °C (72 h) → 520 °C (24 h) → 550 °C (48 h) → 600 °C (24 h) in a mixture of H2/N2 (75/25 mol%, 30 L h−1) at atmospheric pressure. After the catalyst activation, the system was pressurised to 6.3 MPa and heated to different target temperatures (400, 430, and 470 °C). The NH3 content in the outlet gas was analysed interferometrically. The ammonia synthesis rate (rNH3) was calculated from the mass balance for a plug-flow differential reactor. A thermal stability testing was performed by overheating the catalysts at 600 °C for 240 hours in total in a mixture of H2/N2 (75/25 mol%, 30 L h−1) at atmospheric pressure and repeating the catalytic testing at 470 °C and 6.3 MPa after 24, 96, 192 and 240 hours of catalysts overheating.3. Results and discussion
3.1. Effect of yttrium content on the properties of the support
The textural properties of the BaCe1−xYxO3−δ (x = 0–0.30) supports were measured by nitrogen physisorption (Table 1). The unmodified BaCe1−xYxO3−δ (x = 0) support was characterised by low specific surface area (2.5 m2 g−1). Modification of the support by introducing yttrium in the range of 5–30 mol% resulted in an increase in the specific surface area by approx. 35–70%. The support containing 10 mol% of yttrium had the highest surface area (4.2 m2 g−1), but it is still a relatively underdeveloped surface area. Fig. S2† presents the exemplary N2 adsorption–desorption isotherms for the BaCeO3 and BaCe0.90Y0.10O3−δ supports. According to the IUPAC classification,49 the adsorption isotherms of the supports are typical of type II isotherms without a hysteresis loop ascribed to non-porous materials. The low pore volume of the obtained supports (about 0.01 cm3 g−1 for all the supports) is related to the presence of only a small number of macropores (Fig. S3†).| Support | SSAa (m2 g−1) | Number of basic sitesb (μmol g−1) | Density of basic sites (μmol m−2) | |||
|---|---|---|---|---|---|---|
| LT | MT | HT | Total | |||
| a Specific surface area (SSA) determined using the BET isotherm model.b Calculated based on the total amount of CO2 desorbed from the support surface. | ||||||
| BaCeO3 | 2.5 | 5 | 52 | 97 | 154 | 61 |
| BaCe0.95Y0.05O3−δ | 3.4 | 17 | 88 | 7 | 112 | 33 |
| BaCe0.90Y0.10O3−δ | 4.2 | 16 | 134 | 249 | 399 | 95 |
| BaCe0.85Y0.15O3−δ | 3.9 | 8 | 90 | 60 | 158 | 40 |
| BaCe0.70Y0.30O3−δ | 3.4 | 9 | 155 | 63 | 227 | 67 |
The effect of the Y dopant on the crystal structure of BaCe1−xYxO3−δ (x = 0–0.30) support has been studied using powder X-ray diffraction. For the unmodified BaCe1−xYxO3−δ (x = 0) support, the orthorhombic structure (space group Pmcn) of barium cerate was detected, which is more stable at room temperature than other possible barium cerate structures. On the diffraction profile (Fig. 1), the characteristic, sharp peaks of barium cerium oxide were visible, which indicated the high crystallinity of the support material obtained under the synthesis conditions. In the case of Y-modified BaCe1−xYxO3−δ (x = 0.05–0.30) supports, some changes in the crystal structure were observed depending on the yttrium content. In the range of x = 0.05–0.15, stable single-phase supports of orthorhombic structure (Pmcn) was achieved. For the support with the highest yttrium content (x = 0.30), a monoclinic (I2/m) phase was identified, which is in accordance with the literature,47 indicating a phase transition occurring for Y-doped BaCeO3. The diffractograms of the studied supports showed mostly signals which can be ascribed to the barium cerium oxide phase. No additional reflections related to the presence of separate phases containing yttrium were observed. Only reflections of a very low intensity indicated the presence of a small amount of impurities, most likely barium carbonate. However, as the dopant content increased in the range x = 0.05–0.30, changes in the diffraction profiles of the supports became more visible. In the marked areas (Fig. 1), for the supports with lower Y content (x = 0.05–0.10), the overlap of (022)/(400) and (233)/(611) reflections in the 2 theta range of 40–42° and 66–68° was observed, respectively. A clear shift of these reflections towards lower angles can be seen with a higher Y content (x = 0.15–0.30). This is related to the increase in the c parameter of the cell unit (Table S2†) as the Y content increased, especially visible for the support with the highest Y content (x = 0.30).
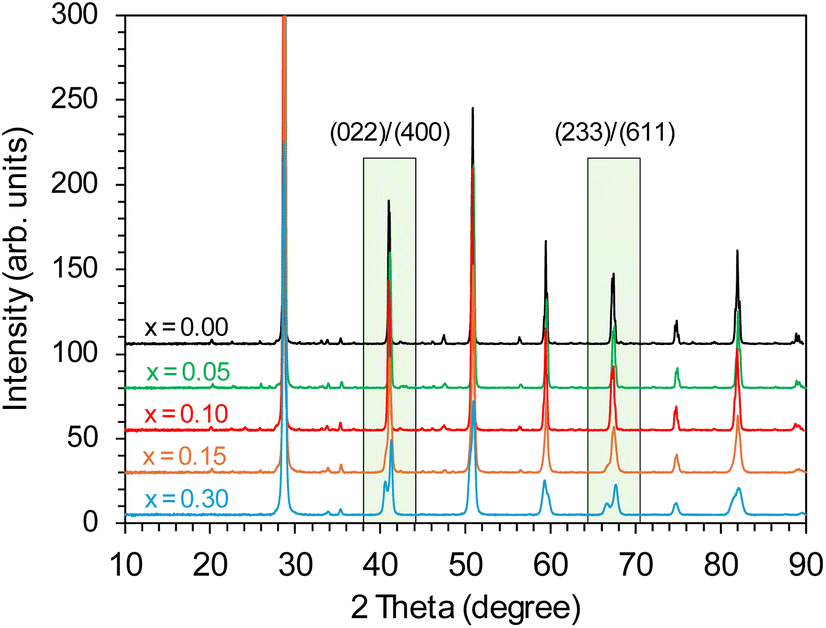 | ||
| Fig. 1 PXRD patterns of the BaCe1−xYxO3−δ (x = 0–0.30) supports. | ||
To study the surface basicity of the prepared supports, CO2 temperature desorption was employed. The CO2 desorption profiles, presented in Fig. 2, reflect the nature and strength of basic sites on the support surface and can be divided into three types: the low-temperature (LT) in the temperature range of 40–200 °C, mid-temperature (MT) in the temperature range of 200–650 °C, and high-temperature (HT) in the temperature >650 °C. The desorption peaks observed in these temperature regions were ascribed to weak, medium, and strong basic sites.39,40,43 For the unmodified BaCe1−xYxO3−δ (x = 0) support, a broad signal in the medium-temperature range and a clear peak in the high-temperature range with a maximum at 720 °C were observed. This proves the presence of Lewis basic sites of medium and strong strength, recognised as electron donors to the transition metal active sites, thus facilitating the dissociation of N2 molecules. These sites are of paramount importance from the point of view of the mechanism of ammonia synthesis reaction.50 The profiles were changed for Y-modified BaCe1−xYxO3−δ (x = 0.05–0.30) supports, with dominating broad signals in the mid-temperature (MT) range. However, the intensity of these signals increased compared to the unmodified BaCe1−xYxO3−δ (x = 0) support. The most visible differences in the CO2-TPD profile occurred for the support modified with 10 mol% of yttrium (x = 0.10), for which an increase in the intensity of the mid-temperature (MT) signal and a clear, sharp, intensive high-temperature (HT) peak with a maximum at 780 °C were observed. The shift of the maximum of the HT peak towards higher temperatures may also indicate that this support has stronger basic sites than the unmodified BaCe1−xYxO3−δ (x = 0) support. Quantitative analysis of desorption profiles was also conducted (Table 1). In general, modification of the BaCeO3 support with yttrium caused an increase in the overall basicity of the support surface (except for the support with the addition of 5 mol% of yttrium). In the case of the modification with 10 mol% of yttrium, the total number of basic sites increased by 2.5 times compared to unmodified BaCe1−xYxO3−δ (x = 0) support, with the dominance of strong basic sites. Whereas for supports with higher Y content (15 and 30 mol%), medium-strength sites dominated. The highest density of basic sites (95 μmol m−2) was observed for the support modified with 10 mol% of yttrium, and it was 1.5 times higher than that for unmodified BaCe1−xYxO3−δ (x = 0) support (61 μmol m−2).
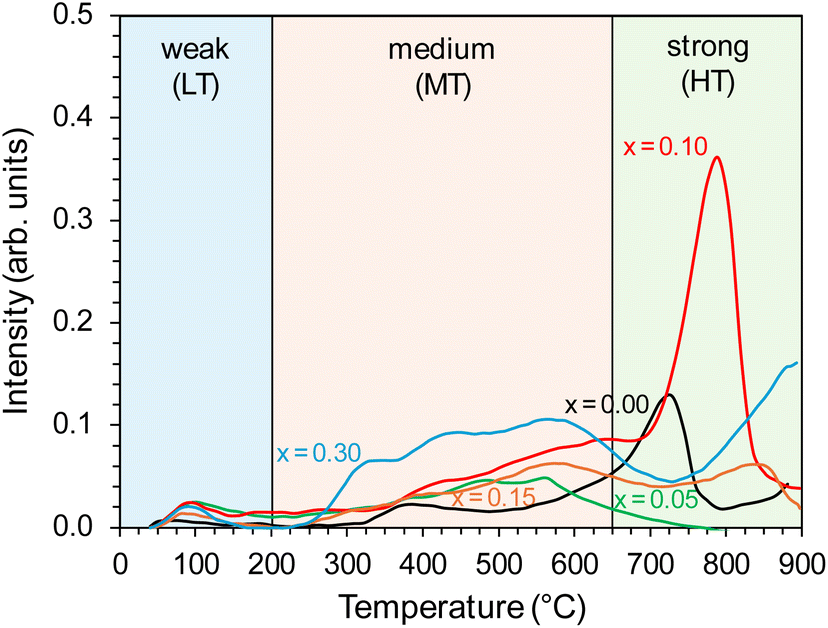 | ||
| Fig. 2 CO2-TPD profiles of the BaCe1−xYxO3−δ (x = 0–0.30) supports. | ||
3.2. Effect of the support on the cobalt catalyst properties
The textural characterisation of the obtained cobalt catalysts was conducted for the catalyst precursors and the catalysts in the reduced form (i.e., after reduction in hydrogen at 600 °C). Deposition of the active phase (about 40 wt% of Co), according to the procedure described in Section 2.1, on low-surface area supports resulted in a significant increase in the specific surface area of the catalyst precursors (Table 2). As a result of the deposition and calcination process, cobalt oxide Co3O4 was formed on the surface of the supports with a more developed surface and porosity (Table 2 and Fig. S3†) than the supports themselves, which resulted in an increase in the SSA parameter value for the catalyst precursors from 5 times for samples with a lower Y content to even 10 times for the sample with the highest Y content. As a result of the reduction of catalyst precursors (reduction of Co3O4 to metallic Co), there was a significant decrease in the specific surface area of the catalysts, which ranged from 1.2 to 6.6 m2 g−1 with the increasing content of yttrium in the support.| Catalyst | Co contenta (wt%) | SSAb (m2 g−1) | PVc (cm3 g−1) | H2 desorbedd (μmol g−1) | FEd (%) | dCod (nm) |
|---|---|---|---|---|---|---|
| a Determined by XRF analysis for the catalysts precursors.b Specific surface area (SSA) determined using the BET isotherm model for the catalysts precursors and catalysts in the reduced form (values in brackets).c Total pore volume (PV) determined using BJH isotherm model for the catalysts precursors.d Amount of H2 desorbed from the catalysts and FE (fraction exposed) and dCo (average Co particle size) estimated based on H2-TPD experiments. | ||||||
| Co/BaCeO3 | 39.1 | 19.3 (1.2) | 0.09 | 84 | 2.5 | 50 |
| Co/BaCe0.95Y0.05O3−δ | 38.4 | 17.2 (1.3) | 0.10 | 71 | 2.2 | 58 |
| Co/BaCe0.90Y0.10O3−δ | 39.2 | 20.8 (1.6) | 0.10 | 70 | 2.1 | 60 |
| Co/BaCe0.85Y0.15O3−δ | 37.8 | 29.0 (3.8) | 0.10 | 96 | 3.0 | 42 |
| Co/BaCe0.70Y0.30O3−δ | 38.6 | 33.2 (6.6) | 0.13 | 127 | 3.9 | 33 |
The phase composition of the Co/BaCe1−xYxO3−δ (x = 0–0.30) catalysts in the reduced form (after activation in hydrogen at 600 °C) is presented in Fig. 3. For all the catalysts, reflections ascribed mainly to BaCeO3 (PDF 22-0074) were detected. However, the degree of crystallinity of this phase decreased significantly compared to the reflections of the BaCeO3 phase identified in the diffractograms of the supports before the deposition of the active phase (Fig. 1), which in the reduced catalysts occurred mainly in the form of face-centred cubic (fcc) cobalt (PDF 15-0806) (Fig. 3). The intensity of Co cubic signals was the highest for Co/BaCe1−xYxO3−δ (x = 0–0.10) catalysts and gradually decreased as the yttrium content increased in the range of x = 0.15–0.30. It is also likely to occur in a hexagonal close-packed (hcp) structure for metallic cobalt. However, in this case, the reflections of Co hexagonal were very weak and partially overlayed with the reflections of other phases, which did not allow us to determine the presence of this phase unequivocally.
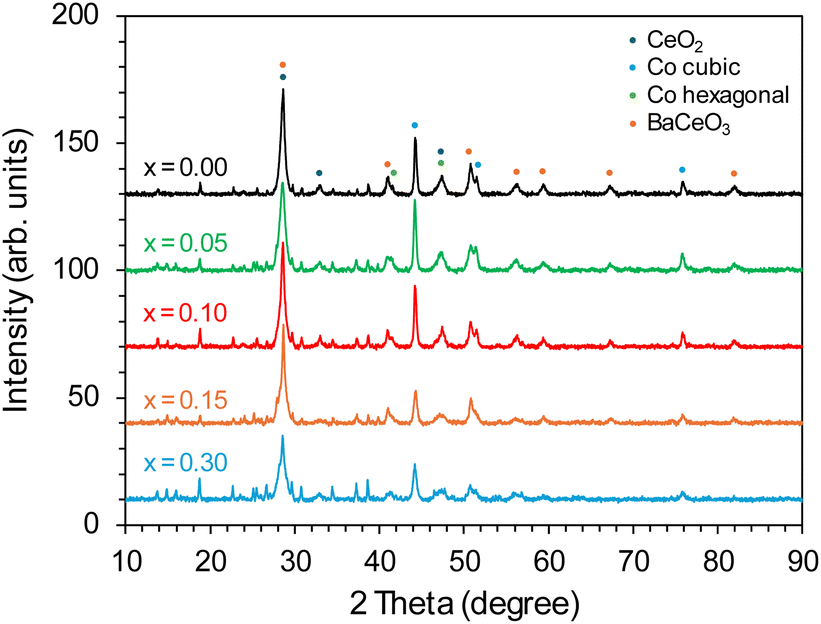 | ||
| Fig. 3 PXRD patterns of the Co/BaCe1−xYxO3−δ (x = 0–0.30) catalysts after reduction in hydrogen at 600 °C. | ||
The morphology and chemical composition of the catalyst surfaces were determined using STEM-EDX studies. The images revealed that the morphology of the catalyst particles changed significantly as the yttrium content in the support increased. The Co/BaCe1−xYxO3−δ catalysts with a lower yttrium content (x = 0–0.10) (Fig. 4a–c) consisted of relatively large grains with irregular shapes tending to aggregate into larger particles. In the case of the Co/BaCe1−xYxO3−δ catalysts with a higher yttrium content (x = 0.15–0.30) (Fig. 4d and e), the particles were smaller, better dispersed, and the shape of the particles seems to be much more regular. This is consistent with the results of physisorption measurements (Table 2), indicating that the specific surface area of reduced catalysts with high yttrium content (x = 0.15–0.30) was several times higher compared to systems with low yttrium content (x = 0–0.10).
 | ||
| Fig. 4 STEM images of the Co/BaCe1−xYxO3−δ catalysts after reduction in hydrogen at 600 °C, where (a) x = 0, (b) x = 0.05, (c) x = 0.10, (d) x = 0.15, (e) x = 0.30. | ||
The HAADF-STEM images and EDX mapping of the main catalyst components are presented in Fig. 5–9. For the Co/BaCe1−xYxO3−δ (x = 0) catalyst, the areas of occurrence of main support components, i.e., Ba and Ce, perfectly overlapped (Fig. 5a, d and e). No separate particles containing distinct Ba or Ce phases were observed. An even distribution of cobalt on the catalyst surface was observed (Fig. 5c). For the Co/BaCe1−xYxO3−δ (x = 0.05–0.30) catalysts, the distribution of the support components (Ba, Ce, Y) was even. There were no areas with separate particles containing Ba, Ce or Y phases (Fig. 6–9d–f, and S4†). It can be stated that the dopant (yttrium) was, therefore, incorporated into the structure of the supports, which is in accordance with the results of phase analysis by PXRD (Fig. 1). Fig. 6–9f clearly illustrate the increasing content of yttrium in the BaCe1−xYxO3−δ (x = 0.05–0.30) supports. Despite the relatively high active phase loading (about 40 wt%), cobalt was evenly distributed on the surface of the support particles. However, the decreasing size of cobalt particles with the increasing yttrium content in the support was visible (Fig. 6–9c).
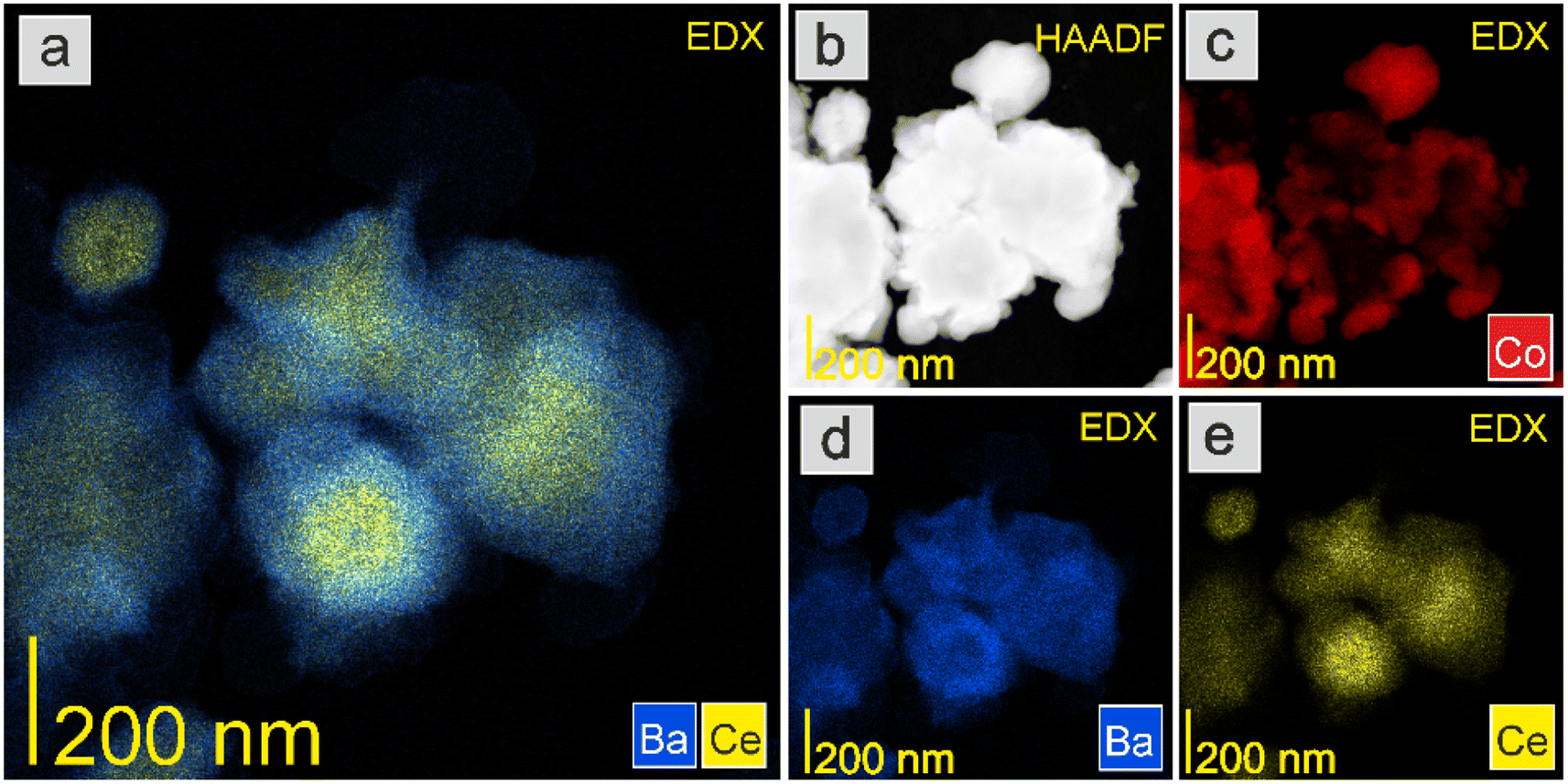 | ||
| Fig. 5 HAADF-STEM image and EDX maps of the Co/BaCe1−xYxO3−δ (x = 0) catalyst after reduction in hydrogen at 600 °C. (a) Overlay EDX map of Ba and Ce. (b) HAADF-STEM image. EDX maps of (c) Co, (d) Ba, and (e) Ce. | ||
 | ||
| Fig. 6 HAADF-STEM image and EDX maps of the Co/BaCe1−xYxO3−δ (x = 0.05) catalyst after reduction in hydrogen at 600 °C. (a) Overlay EDX map of Y, Ba, and Ce. (b) HAADF-STEM image. EDX maps of (c) Co, (d) Ba, (e) Ce, and (f) Y. | ||
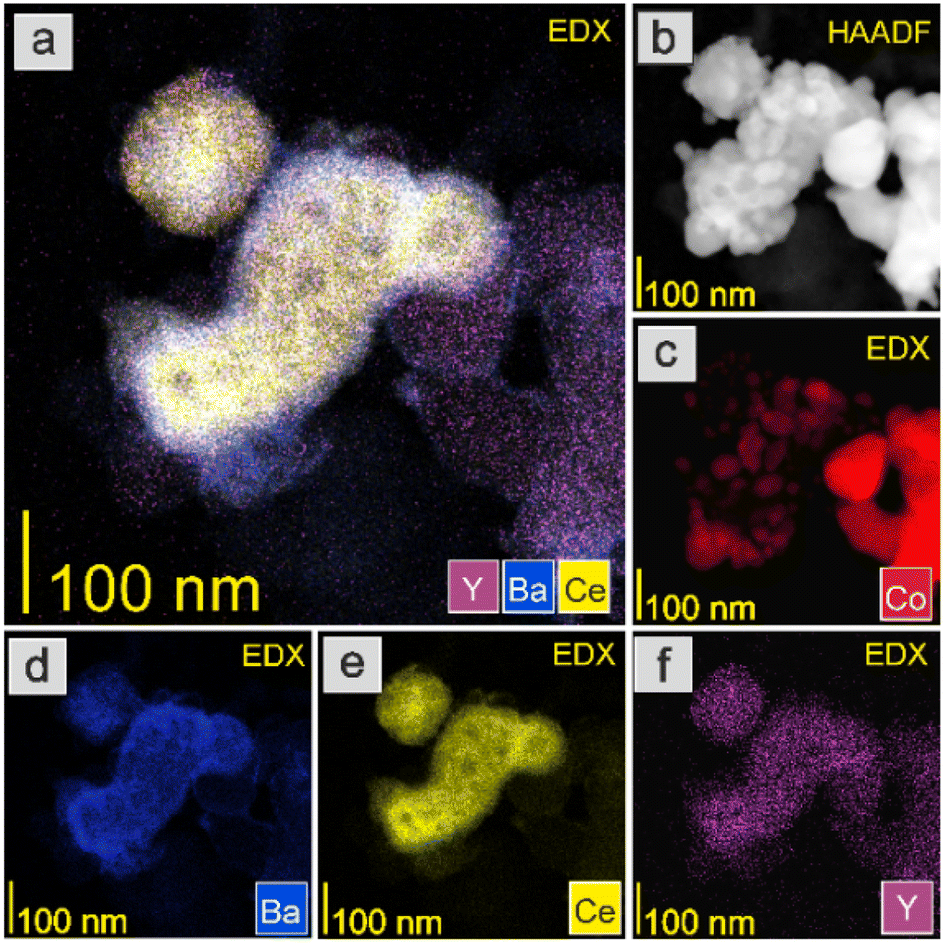 | ||
| Fig. 7 HAADF-STEM image and EDX maps of the Co/BaCe1−xYxO3−δ (x = 0.10) catalyst after reduction in hydrogen at 600 °C. (a) Overlay EDX map of Y, Ba, and Ce. (b) HAADF-STEM image. EDX maps of (c) Co, (d) Ba, (e) Ce, and (f) Y. | ||
 | ||
| Fig. 8 HAADF-STEM image and EDX maps of the Co/BaCe1−xYxO3−δ (x = 0.15) catalyst after reduction in hydrogen at 600 °C. (a) Overlay EDX map of Y, Ba, and Ce. (b) HAADF-STEM image. EDX maps of (c) Co, (d) Ba, (e) Ce, and (f) Y. | ||
 | ||
| Fig. 9 HAADF-STEM image and EDX maps of the Co/BaCe1−xYxO3−δ (x = 0.30) catalyst after reduction in hydrogen at 600 °C. (a) Overlay EDX map of Y, Ba, and Ce. (b) HAADF-STEM image. EDX maps of (c) Co, (d) Ba, (e) Ce, and (f) Y. | ||
The catalyst reduction process was examined using H2 temperature-programmed reduction (H2-TPR). The support reducibility was also evaluated, and the results are presented in Fig. S5.† The peaks at 450 and 600 °C were attributable to the reduction of surface oxygen, whereas the peak at around 800 °C was associated with the reduction of lattice oxygen.43 However, for the catalyst precursors, the major reaction occurring during the H2-TPR was a two-step reduction process, visible as two distinct peaks on the H2-TPR profiles (Fig. 10). According to literature reports,51,52 cobalt(II,III) oxide reduction usually proceeds according to the following scheme: Co3O4 → CoO → Co. For the tested Co/BaCe1−xYxO3−δ catalysts (x = 0–0.30), a similar course of the first step of the reduction process was observed. The peak maximum for all the catalysts was recorded at around 320 °C. In the second step, a change in the course of the process was observed depending on the Y content in the support. The maximum reduction peak for the Co/BaCe1−xYxO3−δ (x = 0) catalyst occurred at a temperature of 436 °C. For catalysts with a low Y content (x = 0.05–0.10), a slight shift of the maximum temperature of this peak towards lower temperatures was observed (433 °C and 400 °C when x = 0.05 and 0.10, respectively). However, for catalysts with a higher Y content, there was a clear shift of the maximum reduction peak towards higher temperatures, i.e., 454 °C and 507 °C when x = 0.15 and 0.30, respectively. Moreover, the peak broadening was also observed for these catalysts. This can be related to the smaller size of cobalt oxide particles in these catalysts, which is one of the important factors determining the course of the reduction process of catalysts containing cobalt as the active phase. There are reports53 indicating that the reduction of small cobalt particles deposited on support is possible only at elevated temperatures, which the authors explained by the increase in metal–support interactions with decreasing metal particle size. The above observations correspond well with the increased specific surface area (SSA) of these catalysts (Table 2) and the significantly better particle dispersion visible in the microscopic images of these catalysts (Fig. 8 and 9).
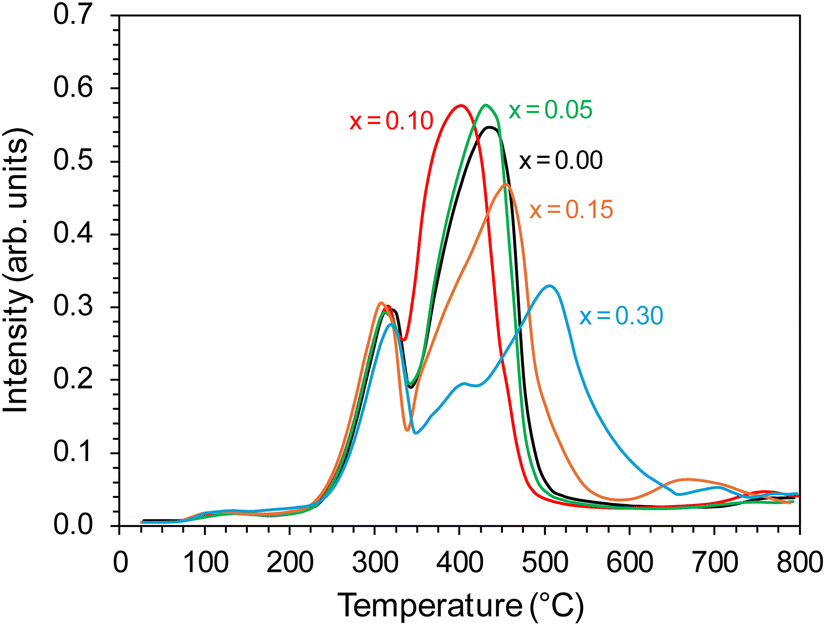 | ||
| Fig. 10 H2-TPR profiles of the Co/BaCe1−xYxO3−δ (x = 0–0.30) catalysts. | ||
The properties of the active phase (metallic cobalt) surface were characterised using the temperature-programmed hydrogen desorption (H2-TPD) method. For all the tested catalysts, two distinct peaks were visible in the hydrogen desorption profiles (Fig. 11), i.e., low-temperature (LT) in the range of 0–300 °C, corresponding to the desorption of hydrogen weakly bound to the surface of the active phase, and high-temperature (HT) in the range of 300–700 °C, related to desorption of hydrogen strongly bound to the cobalt surface. The desorption curves did not change significantly with the change in the yttrium content in the catalyst. However, a slight shift of the desorption peaks towards lower temperatures was observed.
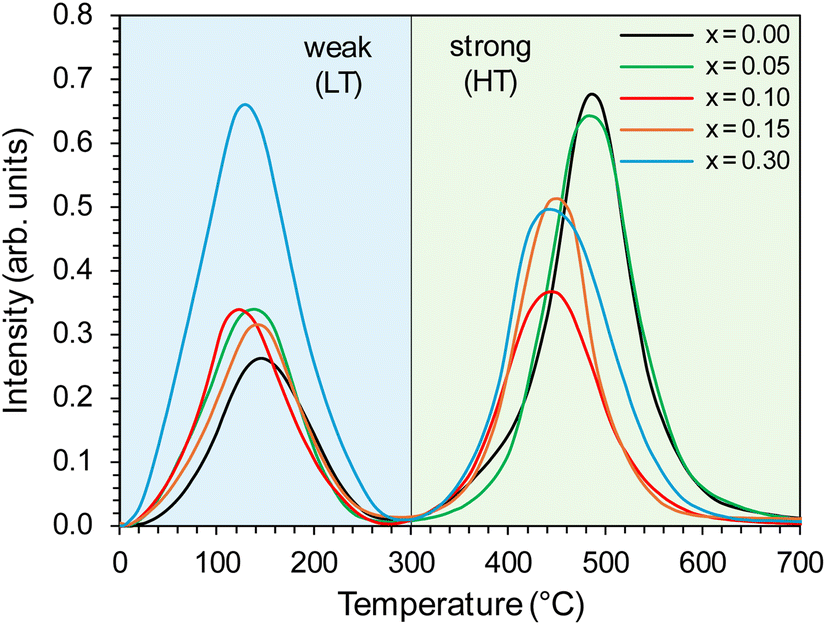 | ||
| Fig. 11 H2-TPD profiles of the Co/BaCe1−xYxO3−δ (x = 0–0.30) catalysts. | ||
There were also clear differences in the peak intensity for individual catalysts. The LT/HT peak ratio is presented in Fig. 12. As yttrium content in the catalyst increased, the predominance of weakly binding sites (LT) over strongly binding sites (HT) increased until their proportions became almost equal for the catalyst containing 10 mol% Y. For the system containing 15 mol% of Y, an increase in the share of strongly binding sites was observed, while for the system with the highest yttrium content (30 mol%), the low-temperature sites became dominant. The total amount of hydrogen desorbed from the surface of the catalysts was the total number of hydrogen adsorption sites, which for systems with low yttrium content (x = 0.05 and 0.10) was lower than for the unmodified system. For higher yttrium contents (x = 0.15 and 0.30), the number of adsorption sites increased and reached the highest value for the system containing 30 mol% of Y. This was also reflected in the dispersion of the active phase. Systems with low yttrium content (x = 0.05 and 0.10) were characterised by slightly lower cobalt dispersion (FE) and similar Co particle size (dCo) compared to the unmodified system. This means they had a slightly reduced surface area of the active phase available to the reactants. However, for systems with a higher content of yttrium (x = 0.15 and 0.30), a significant increase in dispersion and a decrease in the average size of Co particles were observed. For systems with low yttrium content, the average Co particle size (dCo) was in the range of 50–60 nm, while for the systems with higher yttrium content, the particle size was in the range of 30–40 nm (Table 2). This is consistent with the results of STEM-EDX analyses indicating a gradual decrease in the size of cobalt particles with the increase in the yttrium content in the catalyst (Fig. 5–9c).
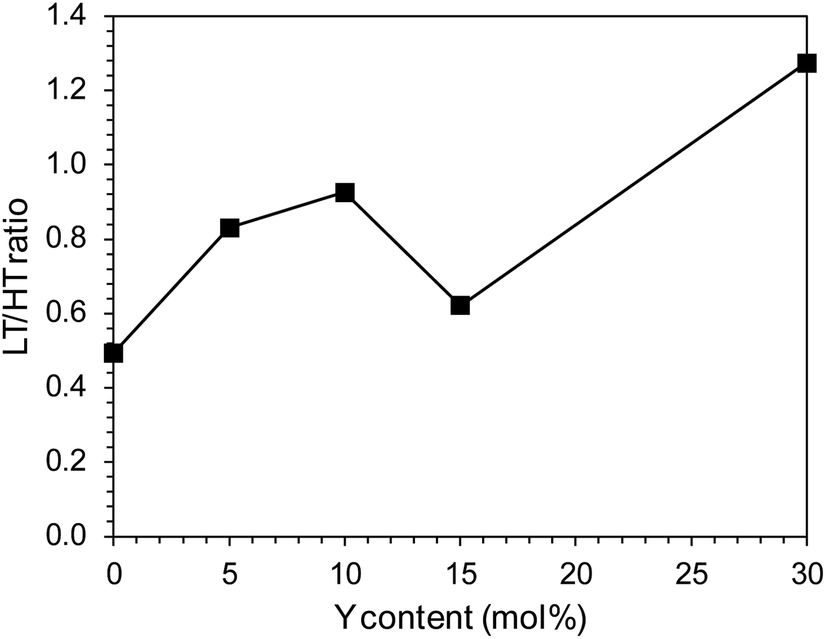 | ||
| Fig. 12 Low temperature (LT) to high temperature (HT) hydrogen desorption peak ratio of the Co/BaCe1−xYxO3−δ (x = 0–0.30) catalysts. | ||
The catalytic performance of the catalysts was evaluated in ammonia synthesis reaction under conditions close to the industrial ones, i.e., in the temperature range of 400–470 °C, pressure of 6.3 MPa, stoichiometric H2/N2 ratio. Fig. 13a and b show the dependence of the ammonia synthesis rate (rNH3) on the yttrium content in the support. For the catalysts with a low yttrium content (x = 0.05 and 0.10), the reaction rate was approx. 15–20% higher than that for the unmodified Co/BaCe1−xYxO3−δ (x = 0) catalyst. With the increasing yttrium content, a decrease in the activity of the catalysts was observed. The reaction rate for the catalysts containing 30 mol% of Y was approx. 13% lower than for the unmodified Co/BaCe1−xYxO3−δ (x = 0) catalyst. The same relationship was observed when the activity of catalysts was expressed in relation to a single active site of the catalyst surface (TOF, i.e., turnover frequency). The activity of a single active site increased with increasing yttrium content, reaching the maximum value (0.212 s−1) for the Co/BaCe1−xYxO3−δ (x = 0.10) catalyst containing 10 mol% yttrium. When the yttrium content was further increased, the value of the TOF parameter decreased. This is in accordance with the results of chemisorption studies. The increasing catalyst activity with increasing yttrium content corresponded well with the increasing LT/HT ratio (Fig. 12). It reached the highest value for the system containing 10 mol% of Y, for which the ratio of the weakly and strongly hydrogen-binding sites was almost equal. The activation energy of the catalysts was determined based on the Arrhenius plots (Fig. 13c). It was revealed (Table 3) that the catalysts containing a small amount of yttrium (x = 0.05 and 0.10) were characterised by lower activation energy (the value of Ea was approximately 55 kJ mol−1) compared to the unmodified Co/BaCe1−xYxO3−δ (x = 0) catalyst. While for the catalysts with higher yttrium content (x = 0.15 and 0.30), an increase in the activation energy to over 60 kJ mol−1 was observed. Long-term overheating of the catalysts at a temperature of 600 °C, i.e., a temperature higher than the standard temperature of their operation in the ammonia synthesis reaction, revealed high thermal stability of the tested catalysts. Catalytic tests performed after 24, 96, 192 and 240 hours of catalysts overheating did not show any sign of catalysts deactivation (Fig. 13d). It is consistent with literature reports indicating good stability and chemical resistance of BaCeO3 supports of a perovskite structure.39,43,46
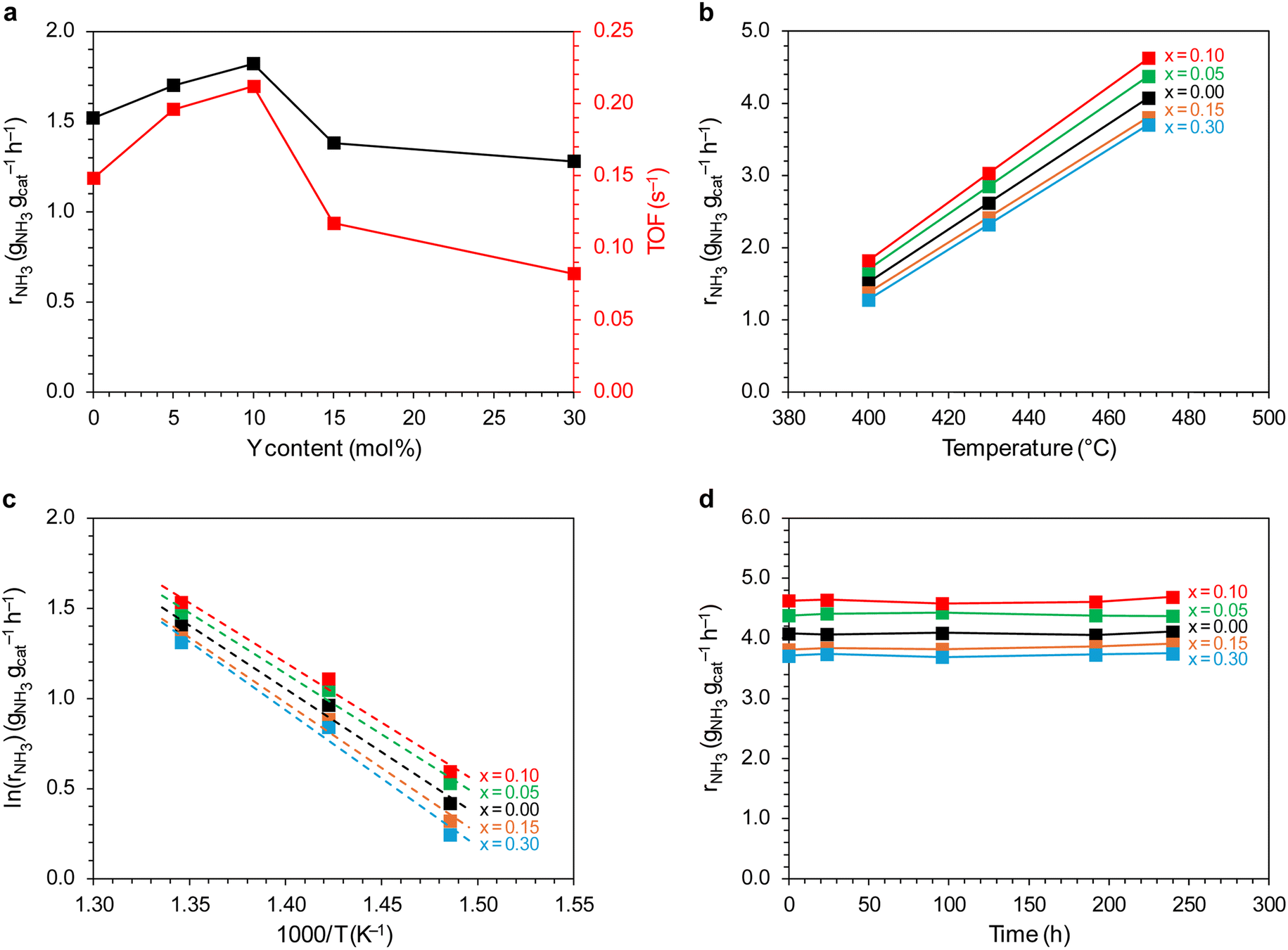 | ||
| Fig. 13 Evaluation of the catalytic performance of Co/BaCe1−xYxO3−δ (x = 0–0.30) catalysts in the ammonia synthesis reaction. (a) Dependence of the ammonia synthesis rate (400 °C, 6.3 MPa, H2/N2 = 3) on the yttrium content in the support. (b) Temperature dependence of the ammonia synthesis rate at 6.3 MPa. (c) Arrhenius plots of ammonia synthesis over studied catalysts (6.3 MPa, H2/N2 = 3). (d) Time-on-stream performance of the studied catalysts (470 °C, 6.3 MPa, H2/N2 = 3). The catalysts were kept at 600 °C between measurements (240 h in total). | ||
| Catalyst | Ea (kJ mol−1) |
|---|---|
| Co/BaCeO3 | 58.3 |
| Co/BaCe0.95Y0.05O3−δ | 55.9 |
| Co/BaCe0.90Y0.10O3−δ | 55.2 |
| Co/BaCe0.85Y0.15O3−δ | 60.0 |
| Co/BaCe0.70Y0.30O3−δ | 62.8 |
Perovskite-type oxides are currently attracting much attention as promising supports for ammonia synthesis catalysts. In the paper,43 Li et al. showed that better catalytic properties in ammonia synthesis are achieved by using BaCeO3 modified with lanthanum or yttrium ions as a support for ruthenium. The particularly high activity of the 2.5% Ru/BaCe0.9La0.1O3−δ system was explained by the increased number of oxygen vacancies caused by the introduction of the lanthanum dopant. Moreover, the BaCe0.9La0.1O3−δ support was characterised by a 1.4 times higher number of basic sites on the surface than the undoped BaCeO3. The introduction of lanthanum ions into the BaCeO3 structure also increased the strength of ruthenium–support interactions, which resulted in a better distribution of the metal on the support surface, i.e., the reduction of the average size of the active phase (Ru particles were two times smaller compared to the 2.5% Ru/BaCeO3 catalyst). Yang et al.44 tested ruthenium catalysts on a BaCeO3 support doped with yttrium, the activity of which was 1.6 times higher than that of the catalyst supported on BaCeO3 without the addition of yttrium. Similarly to Li et al.43 results, the increased catalytic activity was explained by favourable electronic metal–support interactions and enhanced electronic conductivity by doping with Y3+. In the study by Shimoda et al.,42 a positive effect of adding yttrium was observed only in the case of the BaZrO3 support. For the 2% Ru/BaZr1−xYxO3−δ (where x = 0–0.30) systems, only the catalysts with a substitution of 10 mol% Y in the support exhibited NH3 synthesis performance higher than that of the catalysts without the Y doping. Further replacement of Zr with Y by 20 mol% or more caused a significant decrease in the catalytic performance. Interestingly, in the case of the BaCeO3 support, the addition of Y caused a significant decrease in the activity of the ruthenium catalysts deposited on this support, regardless of the amount of yttrium introduced.42
The literature reports on using perovskite-type oxides modified with rare earth metal ions as supports for ruthenium catalysts for ammonia synthesis indicate several key factors causing their performance to be superior to many other supports in this reaction. The most important include strong basicity (the strength of basic sites and their density), a large number of oxygen vacancies and favourable dispersion of metal particles.38–44 The analysis of the characterisation studies results presented in this paper confirms these relationships also in the case when cobalt (as the active metal) was deposited on the BaCeO3 perovskite-type support modified with yttrium. It is worth noting that there have been no previous reports of this type of catalytic cobalt system for the ammonia synthesis process. Similarly to the papers concerning Ru-based catalysts,42–44 in the case of the studied Co-based catalysts, the amount of yttrium introduced as a modifier into the BaCeO3 support structure is of paramount importance for its properties. The studies revealed that introducing small amounts of yttrium (up to 10 mol%) improved the physicochemical properties and performance of the cobalt catalyst under the conditions of the ammonia synthesis reaction. The most active catalyst was deposited on a support containing 10 mol% Y. The reasons for the particularly favourable catalytic properties of this system are as follows:
(1) Highest basicity of the support surface, manifested by the highest total number of basic sites, the highest density of basic sites and the dominance of strong basic sites (Table 1 and Fig. 2), particularly desirable from the point of view of the ammonia synthesis reaction mechanism, i.e., electron donation from the support to the active metal. BaCeO3 doped with Y3+ ions was characterised by strong electron-donating ability. Next to the presence of surface oxygen species of the electron-donating ability (Lewis basic sites), the Y incorporation into BaCeO3 resulted in the enriched electron density on the support due to the formation of oxygen vacancies (OVs). Replacing the Ce4+ cation with a trivalent rare earth ion (Y3+) leads to the formation of a highly reactive radical (Ȯ−) according to the reaction: [2Ce4+,2O2−] → [Ce4+,Y3+,O2−,Ȯ−]. Formation of oxygen vacancies (OVs) accompanied by the reduction of Ce4+ to Ce3+ and the associated delocalisation of electrons according to the reaction: [Ce4+,Y3+,O2−,Ȯ−] → [Ce3+,Y3+,O2−,VO++] + ½O2 + 2e−, caused a locally increased electron density. Furthermore, as revealed during H2-TPR studies (Fig. S5†), the oxygen vacancies were also formed due to the partial reduction of Ce4+ to Ce3+ by hydrogen. These all led to the increased electron density and charge transfer from the support to the active sites, enhancing the rate of N2 molecule dissociation, which is the rate-determining step in the ammonia synthesis reaction.54,55
(2) Favourable adsorption properties, manifested by an almost equal ratio of weakly and strongly hydrogen-binding adsorption sites (LT/HT ratio, Fig. 12). As stated by Tarka et al.,56 both types of adsorption sites are usually present for ammonia synthesis cobalt catalysts. However, weak adsorption sites may be undesirable because they bind hydrogen too weakly, and hydrogen desorption may occur at temperatures much lower than the temperature of the reaction on the catalyst. As a result, a small amount of H atoms remain adsorbed on the surface in the conditions of N2 dissociation, thus the hydrogenation is less likely to occur. On the other hand, the presence of a significant number of strongly hydrogen-binding adsorption sites may also have a detrimental effect on the cobalt catalyst performance. These sites can bind hydrogen so strongly that its desorption is possible only at high temperatures. As a result the active sites may be permanently blocked and not participate in the reaction (so-called hydrogen poisoning). The study by Tarka et al.56 showed that for cobalt catalysts promoted with barium and cerium, BaCeO3 (formed in situ under the conditions of catalyst activation) is the third promoter with the ability to differentiate the character of hydrogen adsorption sites. It was stated that the co-existence of weakly and strongly binding sites on the active phase surface is needed to conduct the NH3 synthesis process effectively. However, the proper ratio of weakly and strongly hydrogen-binding sites is required.
(3) Preferred size of cobalt particles (Table 2). Ammonia synthesis reaction is a prime example of a structure-sensitive reaction highly dependent on the active phase structure when it is conducted in the presence of iron,57 ruthenium58 and cobalt also.59,60 The hexagonal close-packed (hcp) phase of cobalt is known as twice more active (expressed as TOF) than the face-centred cubic (fcc) phase.61 Moreover, Kitakami et al.62 revealed the following relationship between the cobalt particle size and the crystal phase: the fcc Co phase for particles of diameter ≤20 nm, a mixture of the fcc Co and hcp Co phases for particles with a diameter of ca. 30 nm, and the hcp phase (with only a small amount of the fcc phase) for particles with a diameter ≥40 nm. In this context, the favourable cobalt particle size of 60 nm for the tested Co/BaCe1−xYxO3−δ (x = 0.10) catalyst may significantly impact its catalytic performance. Although the PXRD analysis of the catalysts (Fig. 3) did not demonstrate the presence of the hcp Co phase unequivocally, the possibility of its occurrence in a catalyst with this size of cobalt particles cannot be completely ruled out.
Finally, the comparison presented in Table 4 revealed that the newly developed Co/BaCe0.90Y0.10O3−δ catalyst was characterised by higher activity compared to that of a commercially available iron catalyst (ZA-5) reported in the paper by Zhou et al.64 Moreover, the activity of the Co/BaCe0.90Y0.10O3−δ catalyst was comparable or higher to those based on Co and Ru.32,63,65–67
| Catalyst | Metal | T (°C) | p (MPa) | rNH3 (gNH3 gcat−1 h−1) | Ref. |
|---|---|---|---|---|---|
| Co/BaCe0.90Y0.10O3−δ | Co (39 wt%) | 400 | 6.3 | 1.80 | This work |
| Co(40)/Mg–La | Co (41 wt%) | 400 | 6.3 | 0.68 | 32 |
| Co/Ba | Co (73 wt%) | 400 | 6.3 | 0.87 | 63 |
| Fe1−xO (ZA-5) | Fe | 400 | 10 | 0.32 | 64 |
| Fe3O4 | Fe (70 wt%) | 430 | 3 | 0.15 | 65 |
| 10% Cs-FePc | Fe (19 wt%) | 400 | 7 | 0.44 | 66 |
| Ru/CeO2-r | Ru (10 wt%) | 400 | 10 | 1.96 | 67 |
The development of this catalyst conceives a perspective for a reorganisation of the ammonia production process by significantly reducing the reaction pressure, enabling a more sustainable and energy-efficient production of this important chemical. The presented study corresponds well with the current trend of developing a hydrogen-based economy. Hydrogen production and storage methods are currently one of the priority research areas, and the importance of ammonia and its synthesis process in this context is significant. Finding a catalyst with favourable properties would open the prospect of further optimisation of these systems in terms of their potential use as an alternative or supplement to currently used catalysts. Obtaining an active and stable catalyst that can work effectively under milder conditions is an important step towards reducing the energy consumption of the process.
4. Conclusions
In summary, the influence of yttrium content on the perovskite-type BaCeO3 support and cobalt catalysts for ammonia synthesis has been investigated. The studies revealed that introducing small amounts of yttrium (up to 10 mol%) into the BaCeO3 structure improved its physicochemical properties, increasing the specific surface area and basicity. The total number of basic sites increased by 2.5 times compared to unmodified BaCe1−xYxO3−δ (x = 0) support, with the dominance of strong basic sites. The NH3 synthesis reaction rate was approx. 15–20% higher than that for the unmodified Co/BaCe1−xYxO3−δ (x = 0) catalyst. The performance of the catalysts became poorer with increasing Y content in the support (up to 30 mol%). However, all the studied Co/BaCe1−xYxO3−δ catalysts exhibited excellent thermal stability over 240 h of operation under close to industrial conditions (470 °C, 6.3 MPa, H2/N2 = 3). The beneficial effect of Y3+ ions incorporated into the structure of BaCeO3 was mostly due to the strengthening of the electron-donating ability of the support, i.e., better charge transfer from the support to the active metal, enhancing the rate of N2 molecule dissociation, which is the rate-determining step in the NH3 synthesis reaction. Particularly favourable properties of the catalyst containing 10 mol% of yttrium into the support had been associated with (i) the highest basicity of the support surface (the highest total number of basic sites, the density of basic sites and the dominance of strong basic sites), (ii) favourable adsorption properties (proper proportion of weakly and strongly hydrogen-binding sites), and (iii) preferred size of cobalt particles (60 nm). Importantly, the Co/BaCe0.90Y0.10O3−δ catalyst showed better NH3 synthesis performance compared to the commercial iron catalyst (ZA-5). This creates a perspective for a process reorganisation by a significant reduction of the reaction pressure, enabling a more sustainable and energy-efficient ammonia production. Our work adopts a novel strategy to use perovskite-type oxide and Y-modified perovskite-type oxides as a support for cobalt catalyst. It is the first literature report describing BaCeO3 and Y-modified BaCeO3 as a support for cobalt catalyst for the ammonia synthesis process.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Wioletta Raróg-Pilecka: investigation, writing – review & editing. Kamil Sobczak: investigation, visualization. Andrzej Ostrowski: investigation. Wojciech Patkowski: investigation. Hubert Ronduda: investigation, visualization, methodology, writing – review & editing. Magdalena Zybert: writing – review & editing, writing – original draft, supervision, methodology, investigation, conceptualization, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Warsaw University of Technology under I-Chem.4 project of the Scientific Council for Chemical Engineering.References
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, Ammonia for hydrogen storage: challenges and opportunities, J. Mater. Chem., 2008, 18, 2304–2310, 10.1039/B720020J.
- Y. Kojima and M. Yamaguchi, Ammonia as a hydrogen energy carrier, Int. J. Hydrogen Energy, 2022, 47, 22832–22839, DOI:10.1016/j.ijhydene.2022.05.096.
- Z. Wan, Y. Tao, J. Shao, Y. Zhang and H. You, Ammonia as an effective hydrogen carrier and a clean fuel for solid oxide fuel cells, Energy Convers. Manage., 2021, 228, 113729, DOI:10.1016/j.enconman.2020.113729.
- K. E. Lamb, M. D. Dolan and D. F. Kennedy, Ammonia for hydrogen storage; A review of catalytic ammonia decomposition and hydrogen separation and purification, Int. J. Hydrogen Energy, 2019, 44, 3580–3593, DOI:10.1016/j.ijhydene.2018.12.024.
- A. Valera-Medina, F. Amer-Hatem, A. K. Azad, I. C. Dedoussi, M. de Joannon, R. X. Fernandes, P. Glarborg, H. Hashemi, X. He, S. Mashruk, J. McGowan, C. Mounaim-Rouselle, A. Ortiz-Prado, A. Ortiz-Valera, I. Rossetti, B. Shu, M. Yehia, H. Xiao and M. Costa, Review on Ammonia as a Potential Fuel: From Synthesis to Economics, Energy Fuels, 2021, 35, 6964–7029, DOI:10.1021/acs.energyfuels.0c03685.
- D. Erdemir and I. Dincer, A perspective on the use of ammonia as a clean fuel: Challenges and solutions, Int. J. Energy Res., 2021, 45, 4827–4834, DOI:10.1002/er.6232.
- O. Herbinet, P. Bartocci and A. Grinberg Dana, On the use of ammonia as a fuel – A perspective, Fuel Commun., 2022, 11, 100064, DOI:10.1016/j.jfueco.2022.100064.
- O. Siddiqui and I. Dincer, A review and comparative assessment of direct ammonia fuel cells, Therm. Sci. Eng. Prog., 2018, 5, 568–578, DOI:10.1016/j.tsep.2018.02.011.
- G. Jeerh, M. Zhang and S. Tao, Recent progress in ammonia fuel cells and their potential applications, J. Mater. Chem. A, 2021, 9, 727–752, 10.1039/D0TA08810B.
- K. Smart, Review of Recent Progress in Green Ammonia Synthesis, Johnson Matthey Technol. Rev., 2022, 66, 230–244, DOI:10.1595/205651322X16334238659301.
- K. Machaj, J. Kupecki, Z. Malecha, A. W. Morawski, M. Skrzypkiewicz, M. Stanclik and M. Chorowski, Ammonia as a potential marine fuel: A review, Energy Strategy Rev., 2022, 44, 100926, DOI:10.1016/j.esr.2022.100926.
- C. Tornatore, L. Marchitto, P. Sabia and M. de Joannon, Ammonia as Green Fuel in Internal Combustion Engines: State-of-the-Art and Future Perspectives, Front. Mech. Eng., 2022, 8, 944201, DOI:10.3389/fmech.2022.944201.
- J. Li, S. Lai, D. Chen, R. Wu, N. Kobayashi, L. Deng and H. Huang, A Review on Combustion Characteristics of Ammonia as a Carbon-Free Fuel, Front. Energy Res., 2021, 9, 760356, DOI:10.3389/fenrg.2021.760356.
- A. M. Elbaz, S. Wang, T. F. Guiberti and W. L. Roberts, Review on the recent advances on ammonia combustion from the fundamentals to the applications, Fuel Commun., 2022, 10, 100053, DOI:10.1016/j.jfueco.2022.100053.
- M. A. Shipman and M. D. Symes, Recent progress towards the electrosynthesis of ammonia from sustainable resources, Catal. Today, 2017, 286, 57–68, DOI:10.1016/j.cattod.2016.05.008.
- H. Liu, Ammonia synthesis catalyst 100 years: Practice, enlightenment and challenge, Chin. J. Catal., 2014, 35, 1619–1640, DOI:10.1016/S1872-2067(14)60118-2.
- J. Humphreys and S. Tao, Advancements in green ammonia production and utilization technologies, Johnson Matthey Technol. Rev., 2024, 68, 280–292, DOI:10.1595/205651324X16946999404542.
- A. E. Yüzbaşıoğlu, A. H. Tatarhan and A. O. Gezerman, Decarbonization in ammonia production, new technological methods in industrial scale ammonia production and critical evaluations, Heliyon, 2021, 7, e08257, DOI:10.1016/j.heliyon.2021.e08257.
- A. Isella, R. Ostuni and D. Manca, Towards the decarbonization of ammonia synthesis – A techno-economic assessment of hybrid-green process alternatives, Chem. Eng. J., 2024, 486, 150132, DOI:10.1016/j.cej.2024.150132.
- S. Rostami, R. Tayebee and B. Mahdavi, Photofixation of N2 to ammonia utilizing Ni@TPP–HPA nanocomposite under visible-light illumination, RSC Adv., 2023, 13, 31303–31313, 10.1039/D3RA03921H.
- L. Collado, A. H. Pizarro, M. Barawi, M. García-Tecedor, M. Liras and V. A. de la Peña O'Shea, Light-driven nitrogen fixation routes for green ammonia production, Chem. Soc. Rev., 2024 10.1039/D3CS01075A.
- C. J. H. Jacobsen, S. Dahl, B. S. Clausen, S. Bahn, A. Logadottir and J. K. Nørskov, Catalyst Design by Interpolation in the Periodic Table: Bimetallic Ammonia Synthesis Catalysts, J. Am. Chem. Soc., 2001, 123, 8404–8405, DOI:10.1021/ja010963d.
- S. Hagen, R. Barfod, R. Fehrmann, C. J. H. Jacobsen, H. T. Teunissen, K. Ståhl and I. Chorkendorff, New efficient catalyst for ammonia synthesis: barium-promoted cobalt on carbon, Chem. Commun., 2002, 11, 1206–1207, 10.1039/B202781J.
- A. Tarka, M. Zybert, E. Truszkiewicz, B. Mierzwa, L. Kępiński, D. Moszyński and W. Raróg-Pilecka, Effect of a Barium Promoter on the Stability and Activity of Carbon-Supported Cobalt Catalysts for Ammonia Synthesis, ChemCatChem, 2015, 7, 2836–2839, DOI:10.1002/cctc.201500309.
- B. Lin, Y. Qi, K. Wei and J. Lin, Effect of pretreatment on ceria-supported cobalt catalyst for ammonia synthesis, RSC Adv., 2014, 4, 38093–38102, 10.1039/C4RA06175F.
- B. Lin, Y. Liu, L. Heng, J. Ni, J. Lin and L. Jiang, Effect of ceria morphology on the catalytic activity of Co/CeO2 catalyst for ammonia synthesis, Catal. Commun., 2017, 101, 15–19, DOI:10.1016/j.catcom.2017.07.015.
- K. Sato, S.-I. Miyahara, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura, K. Inazu, H. Mohri, T. Iwasa, T. Taketsugu and K. Nagaoka, Barium Oxide Encapsulating Cobalt Nanoparticles Supported on Magnesium Oxide: Active Non-Noble Metal Catalysts for Ammonia Synthesis under Mild Reaction Conditions, ACS Catal., 2021, 11, 13050–13061, DOI:10.1021/acscatal.1c02887.
- W. Patkowski, M. Zybert, H. Ronduda, A. Albrecht, D. Moszyński, A. Fidler, P. Dłużewski, B. Mierzwa and W. Raróg-Pilecka, Towards green ammonia synthesis – exploring the influence of lanthanide oxides as supports on the cobalt catalysts properties, J. CO2 Util., 2024, 80, 102699, DOI:10.1016/j.jcou.2024.102699.
- Y. Jiang, R. Takashima, T. Nakao, M. Miyazaki, Y. Lu, M. Sasase, Y. Niwa, H. Abe, M. Kitano and H. Hosono, Boosted Activity of Cobalt Catalysts for Ammonia Synthesis with BaAl2O4–xHy Electrides, J. Am. Chem. Soc., 2023, 145, 10669–10680, DOI:10.1021/jacs.3c01074.
- Y. Onoue, M. Kitano, M. Tokunari, T. Taniguchi, K. Ooya, H. Abe, Y. Niwa, M. Sasase, M. Hara and H. Hosono, Direct Activation of Cobalt Catalyst by 12CaO·7Al2O3 Electride for Ammonia Synthesis, ACS Catal., 2019, 9, 1670–1679, DOI:10.1021/acscatal.8b03650.
- H. Ronduda, M. Zybert, W. Patkowski, A. Ostrowski, P. Jodłowski, D. Szymański, L. Kępiński and W. Raróg-Pilecka, Development of cobalt catalyst supported on MgO–Ln2O3 (Ln = La, Nd, Eu) mixed oxide systems for ammonia synthesis, Int. J. Hydrogen Energy, 2022, 47, 6666–6678, DOI:10.1016/j.ijhydene.2021.12.022.
- H. Ronduda, M. Zybert, W. Patkowski, K. Sobczak, D. Moszyński, A. Albrecht, A. Sarnecki and W. Raróg-Pilecka, On the effect of metal loading on the performance of Co catalysts supported on mixed MgO–La2O3 oxides for ammonia synthesis, RSC Adv., 2022, 12, 33876–33888, 10.1039/D2RA06053A.
- A. Ozaki, H. S. Taylor and M. Boudart, Kinetics and mechanism of the ammonia synthesis, Proc. R. Soc. London, 1960, 258, 47–62, DOI:10.1098/rspa.1960.0174.
- P. Liu, R. Niu, W. Li, S. Wang and J. Li, Morphology Effect of Ceria on the Ammonia Synthesis Activity of Ru/CeO2 Catalysts, Catal. Lett., 2019, 149, 1007–1016, DOI:10.1007/s10562-019-02674-1.
- S. Miyahara, K. Sato, Y. Kawano, K. Imamura, Y. Ogura, K. Tsujimaru and K. Nagaoka, Ammonia synthesis over lanthanoid oxide–supported ruthenium catalysts, Catal. Today, 2021, 376, 36–40, DOI:10.1016/j.cattod.2020.08.031.
- R. Javaid, Y. Aoki and T. Nanba, Highly efficient Ru/MgO–Er2O3 catalysts for ammonia synthesis, J. Phys. Chem. Solids, 2020, 146, 109570, DOI:10.1016/j.jpcs.2020.109570.
- X. Wang, J. Ni, B. Lin, R. Wang, J. Lin and K. Wei, Highly efficient Ru/MgO–CeO2 catalyst for ammonia synthesis, Catal. Commun., 2010, 12, 251–254, DOI:10.1016/j.catcom.2010.09.024.
- X.-L. Yang, W.-Q. Zhang, C.-G. Xia, X.-M. Xiong, X.-Y. Mu and B. Hu, Low temperature ruthenium catalyst for ammonia synthesis supported on BaCeO3 nanocrystals, Catal. Commun., 2010, 11, 867–870, DOI:10.1016/j.catcom.2010.03.008.
- W. Li, S. Wang and J. Li, Highly Effective Ru/BaCeO3 Catalysts on Supports with Strong Basic Sites for Ammonia Synthesis, Chem.–Asian J., 2019, 14, 2815–2821, DOI:10.1002/asia.201900618.
- Z. Wang, B. Liu and J. Lin, Highly effective perovskite-type BaZrO3 supported Ru catalyst for ammonia synthesis, Appl. Catal., A, 2013, 458, 130–136, DOI:10.1016/j.apcata.2013.03.037.
- Z. Wang, J. Lin, R. Wang and K. Wei, Ammonia synthesis over ruthenium catalyst supported on perovskite type BaTiO3, Catal. Commun., 2013, 32, 11–14, DOI:10.1016/j.catcom.2012.11.024.
- N. Shimoda, Y. Kimura, Y. Kobayashi, J. Kubota and S. Satokawa, Ammonia synthesis over yttrium-doped barium zirconate and cerate-based perovskite-type oxide supported ruthenium catalysts, Int. J. Hydrogen Energy, 2017, 42, 29745–29755, DOI:10.1016/j.ijhydene.2017.10.108.
- W. Li, S. Wang and J. Li, Effect of rare earth elements (La, Y, Pr) in multi-element composite perovskite oxide supports for ammonia synthesis, J. Rare Earths, 2021, 39, 427–433, DOI:10.1016/j.jre.2020.06.006.
- X. Yang, C. Xia, X. Xiong, X. Mu and B. Hu, Preparation and catalytic properties of barium cerate and yttrium-doped barium cerate supported ruthenium for ammonia synthesis, Chin. J. Catal., 2010, 31, 377–379, DOI:10.1016/S1872-2067(09)60055-3.
- D. Medvedev, A. Murashkina, E. Pikalova, A. Demin, A. Podias and P. Tsiakaras, BaCeO3: Materials development, properties and application, Prog. Mater. Sci., 2014, 60, 72–129, DOI:10.1016/j.pmatsci.2013.08.001.
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, Perovskite Oxides: Preparation, Characterizations, and Applications in Heterogeneous Catalysis, ACS Catal., 2014, 4, 2917–2940, DOI:10.1021/cs500606g.
- K. Takeuchi, C.-K. Loong, J. W. Richardson, J. Guan, S. E. Dorris and U. Balachandran, The crystal structures and phase transitions in Y-doped BaCeO3: their dependence on Y concentration and hydrogen doping, Solid State Ionics, 2000, 138, 63–77, DOI:10.1016/S0167-2738(00)00771-2.
- J. Wu, R. A. Davies, M. S. Islam and S. M. Haile, Atomistic Study of Doped BaCeO3: Dopant Site-Selectivity and Cation Nonstoichiometry, Chem. Mater., 2005, 17, 846–851, DOI:10.1021/cm048763z.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 87, 1051–1069, DOI:10.1515/pac-2014-1117.
- H. Ronduda, M. Zybert, W. Patkowski, A. Ostrowski, P. Jodłowski, D. Szymański and W. Raróg-Pilecka, Co supported on Mg-La mixed oxides as an efficient catalyst for ammonia synthesis, Int. J. Hydrogen Energy, 2022, 47, 35689–35700, DOI:10.1016/j.ijhydene.2022.08.144.
- H. Wang, J. L. Ye, Y. Liu, Y. D. Li and Y. N. Qin, Steam reforming of ethanol over Co3O4/CeO2 catalysts prepared by different methods, Catal. Today, 2007, 129, 305–312, DOI:10.1016/j.cattod.2006.10.012.
- L. F. Liotta, G. Di Carlo, G. Pantaleo and G. Deganello, Co3O4/CeO2 and Co3O4/CeO2–ZrO2 composite catalysts for methane combustion: Correlation between morphology reduction properties and catalytic activity, Catal. Commun., 2005, 6, 329–336, DOI:10.1016/j.catcom.2005.02.006.
- T. Paryjczak, J. Rynkowski and S. Karski, Thermoprogrammed reduction of cobalt oxide catalysts, J. Chromatogr. A, 1980, 188, 254–256, DOI:10.1016/S0021-9673(00)88437-X.
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, J. Wu, M. Sasase, M. Kitano and H. Hosono, Dissociative and Associative Concerted Mechanism for Ammonia Synthesis over Co-Based Catalyst, J. Am. Chem. Soc., 2021, 143, 12857–12866, DOI:10.1021/jacs.1c06657.
- Y. Chen, J. He, H. Lei, Q. Tu, C. Huang, X. Cheng, X. Yang, H. Liu and C. Huo, Regulating oxygen vacancies by Zn atom doping to anchor and disperse promoter Ba on MgO support to improve Ru-based catalysts activity for ammonia synthesis, RSC Adv., 2024, 14, 13157–13167, 10.1039/D4RA01517G.
- A. Tarka, W. Patkowski, M. Zybert, H. Ronduda, P. Wieciński, P. Adamski, A. Sarnecki, D. Moszyński and W. Raróg-Pilecka, Synergistic Interaction of Cerium and Barium-New Insight into the Promotion Effect in Cobalt Systems for Ammonia Synthesis, Catalysts, 2020, 10, 658, DOI:10.3390/catal10060658.
- I. B. Parker, K. C. Waugh and M. Bowker, On the structure sensitivity of ammonia synthesis on promoted and unpromoted iron, J. Catal., 1988, 114, 457–459, DOI:10.1016/0021-9517(88)90049-8.
- C. J. H. Jacobsen, S. Dahl, P. L. Hansen, E. Törnqvist, L. Jensen, L. H. Topsøe, D. V. Prip, P. B. Møenshaug and I. Chorkendorff, Structure sensitivity of supported ruthenium catalysts for ammonia synthesis, J. Mol. Catal. A: Chem., 2000, 163, 19–26, DOI:10.1016/S1381-1169(00)00396-4.
- B. Y. Zhang, P.-P. Chen, J.-X. Liu, H.-Y. Su and W.-X. Li, Influence of Cobalt Crystal Structures on Activation of Nitrogen Molecule: A First-Principles Study, J. Phys. Chem. C, 2019, 123, 10956–10966, DOI:10.1021/acs.jpcc.9b00590.
- M. Zybert, A. Tarka, W. Patkowski, H. Ronduda, B. Mierzwa, L. Kępiński and W. Raróg-Pilecka, Structure Sensitivity of Ammonia Synthesis on Cobalt: Effect of the Cobalt Particle Size on the Activity of Promoted Cobalt Catalysts Supported on Carbon, Catalysts, 2022, 12, 1285, DOI:10.3390/catal12101285.
- G. Rambeau, A. Jorti and H. Amariglio, Catalytic activity of a cobalt powder in NH3 synthesis in relation with the allotropic transformation of the metal, J. Catal., 1985, 94, 155–165, DOI:10.1016/0021-9517(85)90091-0.
- O. Kitakami, H. Sato, Y. Shimada, F. Sato and F. M. Tanaka, Size effect on the crystal phase of cobalt fine particles, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 13849–13854, DOI:10.1103/PhysRevB.56.13849.
- M. Zybert, M. Karasińska, E. Truszkiewicz, B. Mierzwa and W. Raróg-Pilecka, Properties and activity of the cobalt catalysts for NH3 synthesis obtained by co-precipitation – the effect of lanthanum addition, Pol. J. Chem. Technol., 2015, 17, 138–143, DOI:10.1515/pjct-2015-0020.
- Y. Zhou, Y. Ma, G. Lan, H. Tang, W. Han, H. Liu and Y. Li, A highly stable and active mesoporous ruthenium catalyst for synthesis prepared by a RuCl3/SiO2-templated approach, Chin. J. Catal., 2019, 40, 114–123, DOI:10.1016/S1872-2067(18)63192-4.
- A. Jafari, A. Ebadi and S. Sahebdelfar, Effect of iron oxide precursor on the properties and ammonia synthesis activity of fused iron catalysts, React. Kinet., Mech. Catal., 2019, 126, 307–325, DOI:10.1007/s11144-018-1498-6.
- N. Morlanés, W. Almaksoud, R. K. Rai, S. Ould-Chikh, M. M. Ali, B. Vidjayacoumar, B. E. Al-Sabban, K. Albahily and J.-M. Basset, Development of catalysts for ammonia synthesis based on metal phthalocyanine materials, Catal. Sci. Technol., 2020, 10, 844–852, 10.1039/C9CY02326G.
- B. Lin, Y. Liu, L. Heng, X. Wang, J. Ni, J. Lin and L. Jiang, Morphology Effect of Ceria on the Catalytic Performances of Ru/CeO2 Catalysts for Ammonia Synthesis, Ind. Eng. Chem. Res., 2018, 57, 9127–9135, DOI:10.1021/acs.iecr.8b02126.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06251e |
| This journal is © The Royal Society of Chemistry 2024 |