
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Physicochemical properties and mechanism of action of a new copper(II) pyrazine-based complex with high anticancer activity and selectivity towards cancer cells†
B. Rogalewicz *a,
T. Sierańskia,
M. Szczesioa,
A. Olczaka,
K. Gobisb,
C. Orlewskab,
I. Korona-Głowniakc,
A. Korga-Plewkod,
M. Iwane,
M. Michalczukd,
J. Kubikd,
G. Adamczukd,
M. Korgad,
N. Rutkowskaf,
T. Borutag,
K. Gash,
M. Sawickih,
E. Poleszaki,
W. Maniukiewicza,
M. Świątkowskia and
A. Czylkowska*a
*a,
T. Sierańskia,
M. Szczesioa,
A. Olczaka,
K. Gobisb,
C. Orlewskab,
I. Korona-Głowniakc,
A. Korga-Plewkod,
M. Iwane,
M. Michalczukd,
J. Kubikd,
G. Adamczukd,
M. Korgad,
N. Rutkowskaf,
T. Borutag,
K. Gash,
M. Sawickih,
E. Poleszaki,
W. Maniukiewicza,
M. Świątkowskia and
A. Czylkowska*a
aInstitute of General and Ecological Chemistry, Faculty of Chemistry, Lodz University of Technology, Żeromskiego 116, Lodz, 90-924, Poland. E-mail: bartlomiej.rogalewicz@dokt.p.lodz.pl; agnieszka.czylkowska@p.lodz.pl
bDepartment of Organic Chemistry, Faculty of Pharmacy, Medical University of Gdansk, Gen. Hallera 107, Gdańsk, 80-416, Poland
cDepartment of Pharmaceutical Microbiology, Medical University of Lublin, Lublin, 20-093, Poland
dIndependent Medical Biology Unit, Medical University of Lublin, Jaczewskiego 8b, Lublin, 20-093, Poland
eDepartment of Toxicology, Medical University of Lublin, Chodźki 8b, Lublin, 20-093, Poland
fInstitute of Molecular and Industrial Biotechnology, Lodz University of Technology, Stefanowskiego 2/22, Lodz, 90-537, Poland
gFaculty of Process and Environmental Engineering, Department of Bioprocess Engineering, Lodz University of Technology, ul. Wolczanska 213, Lodz, 93-005, Poland
hInstitute of Physics, Polish Academy of Sciences, Aleja Lotnikow 32/46, Warsaw, PL-02668, Poland
iLaboratory of Preclinical Testing, Chair and Department of Applied and Social Pharmacy, Medical University of Lublin, Chodzki 1, Lublin, 20-093, Poland
First published on 12th November 2024
Abstract
Two compounds, benzyl-2-(amino(pyrazin-2-yl)methylene)-1-methylhydrazine-1-carbodithioate (L) and its copper(II) complex Cu(L) were synthesized and studied in terms of their physicochemical properties, including single crystal, spectroscopic and magnetic properties; in silico simulations, including DFT calculations and pharmacokinetic profile analysis; and in vitro biological activity. The Cu(L) compound was found to exhibit good anticancer activity against A375, PANC-1, MKN-74, T-47D, HeLa, and NCI-H1563 cells, with the IC50 value against the HeLa cell line reaching 17.50 μM, significantly surpassing the activity of the organic ligand. Moreover, at the same time, the Cu(L) complex did not exhibit significant toxicity towards healthy cells. Mechanism of action studies revealed that its activity is connected with the oxidative stress and redox imbalance caused by the upregulation of genes encoding superoxide dismutase (SOD2) and catalase (CAT) antioxidant enzymes. The reported results further underscore the anticancer potential of pyrazine-based copper(II) complexes.
1. Introduction
With 24.6 million estimated new cases in 2030, cancer remains one of the deadliest diseases in the modern world.1 The most common therapeutic approaches include chemotherapy, radiation therapy, surgery, and hormonal therapy. Despite several disadvantages, chemotherapy remains at the forefront of the treatment, especially when alternative approaches such as radiation or surgery cannot be applied.2,3 With the growing resistance to some of the currently used drugs and collateral damage to healthy tissues, which result in severe adverse side effects, the search for new, more efficient, and safer therapeutics seems to be of superior importance. One of the most promising approaches is the utilization of coordination compounds containing metals lighter than platinum or ruthenium, which are mostly regarded as less toxic than heavier ones, which decreases the possibility of the occurrence of severe side effects. Current leading metals in this effort include copper and iron.4,5 One of the advantages of coordination chemistry, in terms of medicinal chemistry, is the possible synergistic effect of metal cations and organic ligands. This in turn allows for tailoring the properties of compounds and maximizing the possibility of obtaining compounds with good activity and low toxicity towards healthy cells. Currently, nitrogen- and sulfur-based compounds have attracted scientists' attention owing to their significant anticancer potential and remarkable coordinating properties.6–8Copper complexes are known for their redox-active properties under physiological conditions, which have led to the synthesis and biological characterization of a number of copper-containing anticancer drugs and radiopharmaceuticals. Intracellular generation of reactive oxygen species (ROS) via thiol-mediated reduction of copper(II) to copper(I) has been assumed as the major mechanism underlying the anticancer activity of copper(II) complexes. Cancer cells have higher basal levels of ROS in comparison to normal cells. However, their redox homeostasis is maintained by the increased capacity of the antioxidant defense system, which involves several compounds and enzymes such as glutathione (GSH), peroxiredoxin (Prx), thioredoxin (Trx), superoxide dismutase (SOD) and catalase. Thus, an increase in ROS levels and/or inhibition of antioxidant processes causes redox imbalance and disrupts cancer cells homeostasis, which in turn leads to oxidative stress as well as lipid, protein and DNA damage, and ultimately cell death.9–11
NNS-coordinating ligands containing pyrazine or dithiocarbazate moieties have attracted ever-growing attention due to their excellent coordinating and biological properties. Their Cu(II) complexes are known to exhibit significant anticancer activity.12,13 At the same time, only three structures containing both pyrazine and dithiocarbazate moieties were reported, although their biological potential was not investigated.14 NNS-coordinating Cu(II) dithiocarbazate complexes bearing the pyridine moiety have been better characterized in the literature, with some of those complexes exhibiting strong activity against pancreatic, breast, and brain cancer cells.15–17
Considering the above rationale, we designed, synthesized, and studied the physicochemical properties and biological activity of a new copper(II) pyrazine-based complex bearing both pyrazine and dithiocarbazate moieties, Cu(L). Analytical techniques used to study the complex included single crystal and powder X-ray diffraction analyses (SC-XRD and PXRD); elemental analysis (EA); inductively coupled plasma (ICP), infrared (FTIR), UV-visible (UV-vis) and fluorescence spectroscopies; and magnetic studies. The physicochemical analysis was supported with in silico methods, like Hirshfeld surface and fingerprint plots analyses, electrostatic potential maps and DFT calculations. In silico ADME analysis gave insight into the biological potential and pharmacokinetic profile of ligand L. In the last step, the in vitro activity against cancer and normal cells was studied, as well as on a panel of different bacterial strains and yeasts. For anticancer activity, mechanism of action was studied in detail, including apoptosis/necrosis detection, cell cycle analysis, thiol levels, ROS-related gene expression and DNA damage analysis.
2. Experimental
2.1 Chemistry and synthesis
All reagents used for the syntheses were purchased from Sigma-Aldrich and Avantor Performance Materials Poland companies, and used without further purification. For the reaction control, thin-layer chromatography was performed on Merck silica gel 60F254 plates and visualized with UV light. 1H and 13C NMR spectra were recorded on an INOVA-500 spectrometer (Fig. S1 and S2†). The melting point was determined on a Boetius apparatus (HMK, Dresden) and is uncorrected. The contents of C, H, N, and S were determined by a Vario MICRO company Elementar Analysensysteme GmbH. The content of Cu was determined by ICP spectroscopy. A standard solution from Merck (1000 mg L−1) was used for the preparation of the diluted solutions used for calibration. For analysis, distilled water (electrical conductivity = 0.05 μS cm−1) was used (obtained with the Polwater system). Solid samples were decomposed using the Anton Paar Multiwave 3000 closed-system instrument. Mineralization was carried out for 45 min at 240 °C under a pressure of 60 bar. Accurate mass measurements were obtained with the use of a high-resolution Q-TOF mass spectrometer SYNAPT G2 (Waters Corporation, Milford, MA, USA) under ESI+ mode (Fig. S3†). The following analytical conditions were applied: capillary 3.00 kV, sampling cone 25 V, extraction cone 4.0 V, source 120 °C, desolvation gas 500 L h−1. UV-vis spectra were acquired using a Jasco V-660 spectrophotometer, with solid samples compressed between two quartz plates. An integrating sphere was employed for this measurement. Solution measurements were performed for both compounds at a concentration of 5 μM in a DMSO/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture in a time-dependent manner to investigate their stability after 24 and 48 h. These solutions were analyzed using quartz cuvettes with a 1 cm optical path length. Three-dimensional fluorescence spectra were collected on a Jasco FP-6300 spectrofluorometer (Jasco, Easton, MD, USA), positioning the solid samples at a 30° angle relative to the incident beam. Both monochromators were set with a data pitch and bandwidth of 1 nm. The FTIR spectra were recorded using an IR Tracer-100 Shimadzu Spectrometer (Shimadzu Corporation, Kyoto, Japan) (4000–600 cm−1) with an accuracy of recording of 1 cm−1, using KBr pellets. Spectra were processed and analyzed using Spectragryph software.18
:1) mixture in a time-dependent manner to investigate their stability after 24 and 48 h. These solutions were analyzed using quartz cuvettes with a 1 cm optical path length. Three-dimensional fluorescence spectra were collected on a Jasco FP-6300 spectrofluorometer (Jasco, Easton, MD, USA), positioning the solid samples at a 30° angle relative to the incident beam. Both monochromators were set with a data pitch and bandwidth of 1 nm. The FTIR spectra were recorded using an IR Tracer-100 Shimadzu Spectrometer (Shimadzu Corporation, Kyoto, Japan) (4000–600 cm−1) with an accuracy of recording of 1 cm−1, using KBr pellets. Spectra were processed and analyzed using Spectragryph software.18
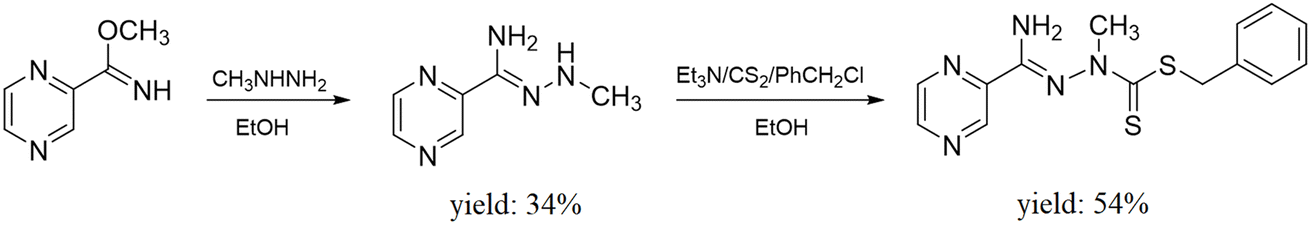 | ||
| Fig. 1 The synthesis route of the ligand L. | ||
Ligand L (C14H15N5S2) (benzyl-2-(amino(pyrazin-2-yl)methylene)-1-methylhydrazine-1-carbodithioate) (317.43 g mol−1) yield (54%) m.p. 140–141 °C. 1H NMR (500 MHz, DMSO-d6): δ 3.60 (s, 3H, NCH3), 4.36 (s, 2H, CH2), 7.20 (t, 1H, Ph, J = 7.33 Hz), 7.26 (t, 2H, Ph, J = 7.32 Hz), 7.34 (d, 2H, Ph, J = 7.32 Hz), 7.71 (br s, 2H, NH2), 8.73 (br s, 1H, pyrazine), 8.82 (d, 1H, pyrazine, J = 2.44 Hz), 9.25 (d, 1H, pyrazine, J = 0.97 Hz) ppm; 13C NMR (125 MHz, DMSO-d6): δ 42.29, 127.44, 128.80 (2C), 129.58 (2C), 137.81, 143.66, 144.01, 145.07, 147.39, 157.06, 191.37 ppm, signal of CH3 carbon overlapped with the solvent. Anal. calculated (%): C, 52.97; H, 4.76; N, 22.06. Found (%): C, 52.78; H, 4.64; N, 21.39. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C14H16N5S2: 318.0847; found: 318.0854 FTIR spectra (KBr, cm−1): ν(NH): 3410, 3309; ν(CH): 3024, 3000, 2970, 2931; ν(CN)aliph: 1647; ν(CC), δ(CH), δ(NH): 1580, 1561, 1452, 1416, 1367; ν(CN)arom: 1339; ν(NN): 1018; ν(CSS): 985.
 | ||
| Fig. 2 The synthesis route of the copper(II) complex Cu(L). | ||
Complex Cu(L) (C14H15Cl2CuN5S2) (451.88 g mol−1) yield (73%) Anal. calculated (%): C, 37.21; H, 3.35; N, 15.50; S, 14.19; Cu, 14.06. Found (%): C, 37.11; H, 3.39; N, 15.28; S, 14.83; Cu, 14.51. HRMS (ESI/Q-TOF) m/z: [M − Cl]+ calculated for C14H15N5S2CuCl: 414.9753; found: 414.9760. FTIR spectra (KBr, cm−1): ν(NH): 3316; ν(CH): 2974, 2874; ν(CN)aliph: 1651; ν(CC), δ(CH), δ(NH): 1602, 1574, 1483, 1460, 1428, 1391; ν(CN)arom: 1364; ν(NN): 1038; ν(CSS): 1005.
2.2 SC-XRD analysis
Single crystals of the studied compound that were suitable for X-ray diffraction were obtained by slow solvent evaporation at room temperature from an ethanol solution. Diffraction measurements were performed using a XtaLAB Synergy, Dualflex diffractometer, (Rigaku Oxford Diffraction, Poland-Japan-UK), with a Pilatus 300 K detector at low temperature (100.0(2) K) using Mo Kα radiation (0.71073 Å). Diffraction data were processed using CrysAlisPRO software (Rigaku Oxford Diffraction. CrysAlisPRO; Rigaku Oxford Diffraction Ltd: Yarnton, Oxfordshire, England). Solving and refinement of the crystal structure were performed with SHELX22,23 using full-matrix least-squares minimization on F2. All H atoms were geometrically optimized and allowed as riding atoms, with C–H = 0.95 Å for aromatic CH groups, and 0.99 Å for secondary CH2 groups with Uiso(H) = 1.2 Ueq(C). In all studied structures, the methyl H atoms were refined with C–H = 0.98 Å and with Uiso(H) = 1.5 Ueq(C). ShelXle software24 was used to visualize the molecular structure. Graphical representation of the crystal structures was performed using the Mercury program.25 As a result of refinement of the crystal structure of the tested compound, a Hooft parameter equal to 0.5 was obtained. It turned out that this crystal is a merohedral twin, in which the two equivalent components are rotated 180° relative to each other around the OX or OY axis. Unfortunately, it is generally impossible to determine which rotation we are dealing with based on the diffraction data. The studied structure was refined with the matrix −1 0 0/0 −1 0/0 0 −1 because it is formally an inversion twin, but the same results can be obtained using the matrix 1 0 0/0 −1 0/0 0 −1 (rotation around the OX axis) or matrix −1 0 0/0 1 0/0 0 −1 (rotation around the OY axis). CCDC 2292685† contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/structures. Hirshfeld surfaces and fingerprint plots were calculated and visualized using Crystal Explorer 17.5 (University of Western Australia, Nedlands 6009, Australia).262.3 PXRD analysis
Room temperature powder X-ray diffraction patterns of Cu(L) were collected using a PANalytical X'Pert Pro MPD diffractometer in the Bragg–Brentano reflection geometry (Cu Kα radiation, λ = 1.54059 Å, 40 kV/30 mA). The pattern was acquired in the 2θ range of 4–90° in a continuous scan mode with 0.0167° step size and an exposure per step of 30 s. The sample was rotated during the measurement to reduce the effect of preferred orientation in the crystalline phase.2.4 Quantum-mechanical calculations
The excited states for the organic ligand L and its coordination compound Cu(L) were calculated using time-dependent density functional theory (TD-DFT) based on coordinates determined via X-ray measurements. Input structural models were prepared using Mercury 2022.3.0 software (Cambridge Crystallography Data Centre, Cambridge, UK).25 Hydrogen atom positions were normalized by adjusting them along the covalent bond vector (X → H) to a distance equivalent to the average neutron diffraction value. For the coordination compound, only the coordination unit was initially used as input. Calculations were carried out using Gaussian09 rev. E.01,27 employing the B3LYP density functional and 6-31g(2d,2p) basis set.28–30 To ensure the inclusion of all experimentally determined maxima, the number of calculated transitions was set to 500. Assignment of the calculated excited states to the observed experimental maxima was performed by comparing excitation energies and oscillator strengths/intensities of the corresponding maxima.Furthermore, the Cu(L) complex was optimized and calculated in dimethyl sulfoxide (DMSO) as a solvent, utilizing the polarizable continuum model (PCM) to account for solvent effects.31 To assess the energetic stability of the compound in an aqueous environment, optimization of Cu(L) was also performed in water using the same solvent model. The computational methods, including the basis set, remained consistent with those used for the systems with X-ray derived coordinates.
The nature of the specific orbital excitations was analysed based on orbital contour plots. All orbital transitions were further examined using Chemissian v4.67 software.32 Molecular electrostatic potential maps (MESP) of the studied compounds were also generated, based on electron density calculations performed using the B3LYP/6-31g(2d, 2p) method.
2.5 Magnetic properties
5.6(2) and 2.1(2) mg sized samples of the ligand L and its coordination compound Cu(L), respectively, were magnetically investigated in a superconducting quantum interference device (SQUID) magnetometer MPMS XL of Quantum Design (San Diego, CA, USA) in magnetic field H up to several tenths of kOe and in temperature T down to 2 K. For the measurements, the powders were transferred into polycarbonate capsules (obtained also from Quantum Design). To facilitate accurate magnetic measurements of such miniscule powder specimens in much larger and heavier capsules (with a typical mass of about 33 mg), a recently described method of in situ compensation of the magnetic signal of the sample-carrying capsules was applied.33 By a careful selection of the abutting capsules, a compensation factor of above 40 has been obtained. During these measurements, the SQUID magnetometer was registering only 1/40 of the capsule carrying the powder specimen, thus reducing in the same proportion all uncertainties connected with determination of the searched signal of the powder, including very sizable errors.33 All the results analysed here are corrected for the (small) signals exerted by the empty sample holder, which are established separately. The measurements have been carried out following the code described for high-sensitivity studies of minute magnetic signals.342.6 In silico biological properties predictions
For in silico biological properties predictions, freely available online tools were used. The pharmacokinetic profile and druglikeness of the ligand L were analysed using the SwissADME service (Swiss Institute of Bioinformatics 2021).35,36 The overall toxicity was evaluated using ProTOX II service,37 and the cytotoxicity predictions towards cancer cells were calculated using CLC-Pred 2.0 (Cell Line Cytotoxicity Predictor).382.7 Anticancer activity
342 for 15 min at 37 °C. Next, the cells were washed with annexin V binding buffer and the cell pellets were resuspended in 100 μL of annexin V binding buffer supplemented with 10 μg mL−1 of propidium iodide (PI), and the annexin V Assay was performed immediately. Cellular fluorescence was quantified using a NucleoCounter® NC-3000™ image cytometer (ChemoMetec USA Inc., Lillerød, Denmark).×g for 5 min at room temperature. Then, 1 mL of TRIzol™ Reagent (Invitrogen, Carlsbad, CA, USA) was added to lyse the cells. Lysates were centrifuged for 5 min at 12000×g at 4 °C, and clear supernatants were processed according to the manufacturer's protocol. For cDNA synthesis, all obtained samples (OD260/280 ratio of app. 2.0) were reverse-transcribed using NG dART RT-PCR reagents (EURx, Gdansk, Poland) and a Mastercycler gradient thermocycler (Eppendorf, Hamburg, Germany) according to the manufacturer's instructions, maintaining a reaction thermal profile of 10 min at 25 °C, 50 min at 50 °C and 5 min at 85 °C. The qPCR reaction was performed using Fast SG/ROX qPCR Master Mix reagents (2×) (EURx, Gdansk, Poland) in a 7500 fast real-time PCR system (Applied Biosystems, USA) in triplicate, according to the manufacturer's instructions. The thermal profile of the reactions was as follows: 20 s at 95 °C, followed by 40 cycles of 3 s at 95 °C and 30 s at 60 °C. The sequences of primers used in the qPCR are presented in Table S2.† RNA18SN5 and ACTB were used as reference genes, and the relative expression of the tested genes was determined by the ΔΔCt method. Analysis was performed using RQ values (relative quantification, RQ = 2−ΔΔCt). The data have been obtained from two independent experiments.2.8 Antimicrobial and antifungal activity
The minimal inhibitory concentration (MIC) and the minimum bactericidal/fungicidal concentration (MBC/MFC) of the tested complexes were evaluated for antibacterial and antifungal activity using the microdilution broth method in Mueller–Hinton broth or RPMI with MOPS for the growth of bacteria and fungi, respectively. The panel of reference microorganisms, including Gram-negative bacteria (Escherichia coli ATCC 25922, Salmonella typhimurium ATCC 14028, Klebsiella pneumoniae ATCC 13883, Pseudomonas aeruginosa ATCC 9027, Proteus mirabilis ATCC 12453), Gram-positive bacteria (Staphylococcus aureus ATCC 25923, Staphylococcus aureus ATCC BAA-1707, Staphylococcus epidermidis ATCC 12228, Micrococcus luteus ATCC 10240, Enterococcus faecalis ATCC 29212, Bacillus cereus ATCC 10876), and fungi (Candida albicans ATCC 10231, Candida parapsilosis ATCC 22019, Candida glabrata ATCC 90030) was investigated. The sterile 96-well polystyrene microtitration plates (Nunc, Denmark) were prepared by dispensing 100 μL of an appropriate dilution of the tested compounds in broth medium or per well by serial two-fold dilutions to obtain final concentrations of the tested compounds, ranging from 500 to 3.9 mg L−1. The inocula were prepared with fresh microbial cultures in sterile 0.85% NaCl to match the turbidity of 0.5. The McFarland standard was added to wells to obtain a final density of 5 × 105 CFU mL−1 for bacteria and 5 × 104 CFU mL−1 for yeasts (CFU, colony forming units). After incubation (35 °C for 24 h), the MICs were assessed visually and spectrophotometrically at 600 nm as the lowest concentration of the compounds that shows complete growth inhibition of the reference microbial strains. An appropriate DMSO control (at a final concentration of 10%), a strain growth control (inoculum without the tested compounds), and negative control (the tested compounds without inoculum) were included on each microplate. MBC or MFC was obtained by culture of 5 μL from each well that showed through growth inhibition, from the last positive one, and from the growth control onto recommended agar plates. The plates were incubated at 35 °C for 24 h for all microorganisms. The MBC/MFC was defined as the minimum concentration of compounds that kills 99.9% of the test microorganisms in the original inoculum. Vancomycin (Van), ciprofloxacin (Cip), and nystatin (Nys) were used as the standard reference antibiotics.3. Results
3.1 SC-XRD analysis
The compound crystallized in the space group Pna21 with two molecules of the neutral complex in the asymmetric unit (Fig. 3). In both molecules, the copper atom is coordinated by two nitrogen atoms of the organic ligand, a thione sulfur atom, and two chlorine ions (Table 1). The neighborhood of the Cu2+ coordination atom may be described as distorted trigonal bipyramidal. Chlorine atoms and a hydrazine nitrogen atom occupy the equatorial positions. The structure is stabilized by hydrogen bonds of the N–H⋯Cl type (Fig. 4 and Table 2), which form the R2,4(8) ring according to the hydrogen-bond graph-set theory.41 | ||
| Fig. 3 Molecular structure of the compound showing atom-labeling schemes. Displacement ellipsoids are drawn at the 50% probability level. | ||
| Distance (Å) | Distance (Å) | ||
|---|---|---|---|
| Cu1A—Cl1A | 2.2420 (15) | Cu1–Cl1 | 2.2587 (14) |
| Cu1A—Cl2A | 2.4832 (14) | Cu1–Cl2 | 2.4789 (14) |
| Cu1A—S1A | 2.3030 (15) | Cu1–S1 | 2.3060 (15) |
| Cu1A—N3A | 1.976 (5) | Cu1–N3 | 1.991 (5) |
| Cu1A—N42A | 2.062 (5) | Cu1–N42 | 2.050 (5) |
|
|||
| Angle (°) | Angle (°) | ||
| Cl1A—Cu1A—Cl2A | 100.18 (5) | Cl1–Cu1–Cl2 | 99.83 (5) |
| Cl1A—Cu1A—S1A | 95.33 (6) | Cl1–Cu1–S1 | 95.06 (6) |
| S1A—Cu1A—Cl2A | 102.28 (5) | S1–Cu1–Cl2 | 102.29 (5) |
| N3A—Cu1A—Cl1A | 157.38 (14) | N3–Cu1–Cl1 | 146.63 (14) |
| N3A—Cu1A—Cl2A | 102.18 (14) | N3–Cu1–Cl2 | 113.14 (14) |
| N3A—Cu1A—S1A | 83.22 (16) | N3–Cu1–S1 | 83.21 (16) |
| N3A—Cu1A—N42A | 78.2 (2) | N3–Cu1–N42 | 77.2 (2) |
| N42A—Cu1A—Cl1A | 93.96 (14) | N42–Cu1–Cl1 | 93.80 (15) |
| N42A—Cu1A—Cl2A | 102.01 (14) | N42–Cu1–Cl2 | 98.08 (14) |
| N42A—Cu1A—S1A | 151.97 (14) | N42–Cu1–S1 | 156.01 (14) |
 | ||
| Fig. 4 Intermolecular hydrogen bonds in the Cu(L) compound with atom labeling (left) and coordination polyhedra (right). | ||
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| a Symmetry codes: (i) x + 1/2, −y + 5/2, z; (ii) x, y − 1, z; (iii) x − 1/2, −y + 3/2, z. | ||||
| N4A—H4AA⋯Cl2 | 0.88 | 2.50 | 3.300 (5) | 152 |
| N4A—H4AB⋯Cl2Ai | 0.88 | 2.37 | 3.199 (5) | 158 |
| N4–H4A⋯Cl2Aii | 0.88 | 2.48 | 3.289 (5) | 154 |
| N4–H4B⋯Cl2iii | 0.88 | 2.45 | 3.275 (5) | 156 |
3.2 Hirshfeld surface analysis and electrostatic potential maps
Hirshfeld surface analysis is a useful tool for evaluating the intermolecular interactions with the neighboring molecules within crystals. For both compounds, L and Cu(L), Hirshfeld surfaces and the fingerprint plots were generated and studied (Fig. S5–S7†). For all described contacts, enrichment ratio (ER) values were calculated to determine whether or not a contact is favored in a crystalline environment.42 For the L compound, four main contacts were detected: H⋯H (44.6%), H⋯C/C⋯H (17.5%), H⋯S/S⋯H (17.2%), and H⋯N/N⋯H (12.2%). With ER = 1.42, the H⋯S/S⋯H contacts seem to play a key role in the crystal structure of L, as these contacts are the most favored ones. The ER values for the remaining contacts are close to 1. The key difference in the Cu(L) contacts is the crucial role of the H⋯Cl/Cl⋯H contacts in the structural assembly, both in terms of the percentage share (26.6%) and high ER value (1.53). The distribution of the remaining contacts is similar, with H⋯H (29.2%) and H⋯C/C⋯H (13.2%), and the highly favored H⋯S/S⋯H (10.9%) and H⋯N/N⋯H (9.6%) contacts.For both compounds, electrostatic potential maps (MEPS) were generated and analyzed (Fig. 5). MEPS offer a three-dimensional illustration of a molecule's charge distribution. For the organic ligand alone, a negative charge is predominantly localized on the sulfur atoms. However, in the coordination compound containing chlorine atoms, there is a noticeable shift, with the negative charge becoming more pronounced and concentrated on the chlorine atoms. The coordination induces a change in the molecule's geometry and, consequently, its charge distribution. In both scenarios, the NH2 group emerges as the most positively charged region. This pronounced positive charge is due to its electron deficiency, which is far more evident in the coordination compound.
 | ||
| Fig. 5 Molecular electrostatic potential maps (MESP) of the studied compounds imposed on electron density at an isovalue of 0.005. | ||
3.3 Quantum-mechanical calculations, UV-vis, fluorescence and FTIR measurements
In addition to solid-state measurements, the UV-vis spectrum for Cu(L) was also recorded in DMSO. Five distinct maxima were observed (Fig. S11 and Table S3†), closely corresponding to the calculated values. More pronounced differences are visible for transitions at high wavenumbers (Fig. S11 and Table S3†), which are typical for TD-DFT calculations in this range. This is related to the fact that the observed maximum in the spectrum is actually associated with several different energy transitions. The nature of the transitions is similar to those observed in the solid state and mainly concerns transitions in which the acceptor orbital is an anti-bonding π orbital (Fig. S11†). However, in aqueous solution, the influence of the d orbitals of the central atom (copper) becomes more apparent. It can be observed that in most cases, the electron density of the donor orbital is also localized around the copper atom. All described transitions can therefore be classified as n → π* and d → π* transitions, except for the transition associated with the first maximum, where the acceptor orbital is the anti-bonding σ orbitals of the organic ligand. In general, the transitions have the character of a metal-to-ligand charge transfer (MLCT). Additionally, optimization of the Cu(L) complex was performed in a water environment. A decrease in energy was observed during the optimization process, and no dissociation of the complex occurred (Fig. S12†), suggesting that the obtained coordination compound remains intact in aqueous solutions. To assess the stability of both ligand L and its complex, Cu(L) in mixed DMSO/H2O solutions, their absorption spectra in DMSO/H2O (1:1) mixture were recorded in a time-dependent manner (Fig. S13†). No significant changes were observed after 24 and 48 hours, confirming the high stability of both compounds.
The organic ligand L also exhibits weak fluorescence with a single pronounced maximum, observed at an excitation wavelength of 470 nm and an emission wavelength of 481 nm (Table S3 and Fig. S14†). In contrast, the coordination compound Cu(L) essentially shows no fluorescence. This quenching effect is attributed to the copper cation, which has an open-shell electronic structure, and aligns with the general findings in the literature.43 Fluorescence quenching may be useful in applications such as biosensors, where specific quenching could serve as an indicator of the analyte presence.
3.4 Magnetic properties
In the first step, the molar magnetic susceptibility χm of the ligand L was established. The slope of its linear response in H at T = 300 K yields χmL = −1.63 × 10−4 emu mol−1. Under the same experimental conditions, the coordination compound Cu(L) exhibits a net paramagnetic response, which grows steadily on lowering T. This behavior can be assigned to the presence of spin S = ½ Cu ions with configuration 3d9 in the investigated compound. To evaluate the molar spin susceptibility of these ions, χmS = ½, we corrected the experimental data for the magnitudes of χmL (noting that the L ligand constitutes only 70% of the mass of Cu(L) compound) and diamagnetic corrections for (i) the Cu2+ cation, χmCu = −1.1 × 10−5 emu mol−1 and two Cl− anions, χmCl− = −2.34 × 10−5 emu mol−1.46 The resulting temperature dependence of the inverse of χmS = ½ is presented in Fig. S17.† Indeed, the experimental points follow the paramagnetic T−1 dependency very closely, as indicated by the solid straight line. The slope of this line yields the magnitude of the inverse of the Curie constant Cmol−1 = 3 kB/μeff2/NA, from which the magnitude of the effective spin moment μeff2 ≅ 8/Cmol of the Cu ions is readily obtained. Here, μeff is given in units of Bohr magnetons μB, kB is the Boltzmann constant, and NA is the Avogadro number. We obtained μeffCu = 1.6(1) μB, which correlates remarkably well with the spin-only magnitude of 3d9 configuration, for which μeff = 1.73 μB is expected. This finding confirms the presence of a single unpaired electron on the Cu site and its 3d9 configuration. The uncertainty of μeffCu is determined predominantly by the uncertainty of the masses of the powder specimens after transferring into the capsules.3.5 In silico biological properties predictions
In silico tests covered the ADME bioavailability analysis and predictions of overall toxicity, as well as cytotoxicity towards cancer cells of the ligand L. Its pharmacokinetic profile was evaluated based on six basic properties (lipophilicity, size, polarity, solubility, flexibility, and saturation; Fig. 6a), and allowed to classify the ligand L as a potential drug candidate. It meets the rules of Lipinski,47 Ghose,48 Egan,49 Veber,50 and Muegge51 for druglikeness. The BOILED-egg diagram analysis suggests moderate solubility in water with high gastrointestinal absorption and no blood–brain-barrier permeation. Thus, compound L is predicted not to be the P-glycoprotein substrate (Fig. 6b). Therefore, it may be a suitable drug candidate with oral administration and a poor drug candidate against brain tumors. According to the overall toxicity predictions, compound L belongs to the 4th toxicity class. The predicted LD50 value was equal to 400 mg kg−1. The evaluation of the cytotoxicity towards cancer cell lines revealed good results for the tested compound, with the highest probability of anticancer activity against skin, cervix, and lung cancer cells (Fig. S18†). Moderate activity was predicted against pancreas, breast, stomach, prostate, and urinary bladder cancers, suggesting the above-mentioned diseases as potential targets. | ||
| Fig. 6 (a) Bioavailability graph generated using the SwissADME service. The red-colored zone is considered physicochemically suitable for oral bioavailability. LIPO, lipophilicity (−0.7 < XlogP3 < +5.0); SIZE, molecular weight (150 g mol−1 < MW < 500 g mol−1); POLAR, polarity (20 Å2 < TPSA < 130 Å2); INSOLU, insolubility (0 < logS < 6); INSATU – insaturation (0.25 < fraction Csp3 < 1); FLEX – flexibility (0 < num. of rotatable bonds < 9). (b) The BOILED-egg diagram for the compound L. The white area represents high gastrointestinal absorption; the yellow area represents blood–brain barrier permeation. | ||
3.6 Anticancer activity and mechanism of action
The in vitro cytotoxicity of both ligand L and complex Cu(L) was investigated on cell lines of different origin, such as A-375 (malignant melanoma), PANC-1 (pancreatic cancer), MKN-74 (gastric cancer), T-47D (breast cancer), HeLa (cervical cancer) and NCI-H1563 (non-small cell lung cancer), with BJ (normal human fibroblast) as the non-cancer control cell line. Results were compared with those of the widely used drugs (dacarbazine, gemcitabine, doxorubicin and cisplatin), as well as copper(II) chloride alone, under the same conditions.Cells were exposed to 0–100 μM of L or Cu(L) for 48 h, and cell viability was assessed using the MTT assay. Results were determined according to the dose values of the potential drug needed to reduce the cell lines growth to 50% (IC50). In the range of the concentrations used, the ligand L was slightly cytotoxic against the tested cell lines. The IC50 value was calculated only in the case of the MKN-74 gastric cancer line (Table 3 and Fig. S19†). Complexation of L with copper(II) ions substantially increased the cytotoxic activity against cancer cells, without toxicity towards normal fibroblasts. Significant differences were observed starting from the concentration of 25 μM in the case of the HeLa cell line, and 50 μM for the other cell lines (Fig. S19†). Similarly, for the HeLa line, the lowest IC50 value was obtained. For A375 and HeLa cell lines, the cytotoxicity of Cu(L) was comparable to the cytotoxicity of the reference drugs, dacarbazine and cisplatin, respectively. At the same time, copper(II) chloride alone did not exhibit significant toxicity in the tested concentrations.
| A375 | PANC-1 | MKN-74 | T-47D | HeLa | NCI-H1563 | BJ | |
|---|---|---|---|---|---|---|---|
| IC50 (μM) | |||||||
| Ligand L | >100 | >100 | 89.2 ± 4.47 | >100 | >100 | >100 | >100 |
| Complex Cu(L) | 40.81 ± 1.18 | 46.90 ± 3.64 | 38.1 ± 0.78 | 44.71 ± 4.75 | 17.50 ± 2.44 | 39.11 ± 3.99 | >100 |
| Reference | 47.12 ± 2.78 (dacarbazine) | 12.05 ± 1.12 (gemcitabine) | 15.02 ± 0.98 (cisplatin) | 5.28 ± 0.32 (doxorubicin) | 13.68 ± 1.21 (cisplatin) | 2.25 ± 0.30 (cisplatin) | — |
| CuCl2 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
In addition to the MTT assay, the cytotoxic activities of ligand L and its complex, Cu(L), were verified by apoptosis detection using annexin V and image cytometry. Annexin V binds specifically to phosphatidylserine, which is externalized on the outer surface of the cell membrane to mark the cell to undergo apoptosis process.52 Analysis of the ligand L activity confirmed its low cytotoxicity against the majority of the cell lines – only in the case of MKN-74, ligand L induced apoptosis at a concentration equal to 100 μM (Fig. 7 and S20†). The Cu(L) complex induced apoptosis, with a predominance of cells in the early apoptosis stage in the A375 and PANC-1 lines and late apoptosis predominance in the MKN-74, T46D, HeLa, and NCI-H1563 cell cultures (Fig. 7 and S20†).
 | ||
| Fig. 7 Cancer cell populations based on apoptosis/necrosis detection via annexin V and image cytometry. | ||
As the lowest IC50 value was obtained for the complex Cu(L) against the HeLa cell line, it was subjected to cell cycle analysis. The concentration of Cu(L) corresponded to the IC50 value obtained in the MTT test, with the ligand L equal to 100 μM or DMSO used as a vehicle in the control cultures, and the culture was analyzed after 48 hours of incubation. Cell cycle analysis showed that treatment of HeLa cells with ligand L and complex Cu(L) resulted in an increase in the population of cells in the G2/M phase at the expense of the population in the S phase (Fig. 8 and S21†). This pattern was much more pronounced for Cu(L). Cell cycle arrest in the G2 phase indicates that the cell has halted its progression through the cell cycle due to various reasons, such as DNA damage, allowing the cell to undergo the DNA repair pathway.
 | ||
| Fig. 8 Cell cycle distribution, showing the populations of HeLa cells in different phases (subG1, G1, S, and G2/M) after 48 h of incubation with ligand L (100 μM), complex Cu(L) (17.50 μM), and a positive control. | ||
Copper(II) complexes are known to induce oxidative stress in cancer cells via redox cycling, which leads to the production of superoxide radicals (˙O2−) and hydrogen peroxide (H2O2).12,53 Cellular thiols, particularly glutathione (GSH), play a crucial role in protecting cells from oxidative stress.11 The cycle of oxidation and regeneration of glutathione (GSH) involves GSH donating electrons to neutralize reactive oxygen species (ROS), becoming oxidized (GSSG), and then being reduced back to its active form, GSH. A decrease in the level of reduced thiols shifts the cellular redox potential towards a more oxidative state, which can trigger oxidative damage to lipids, proteins, and DNA.11 Therefore, the level of cellular thiols was examined in HeLa cells, which showed the greatest sensitivity to the tested compounds.
Analysis of the thiol level distribution by image cytometry revealed the presence of a subpopulation of cells with decreased thiol levels after treatment with ligand L and its complex Cu(L), equal to 32% and 53%, respectively, vs. 4% in the control culture (Fig. 9 and S22†).
 | ||
| Fig. 9 Thiol level distribution in HeLa cells after 48 h of incubation with the ligand L (100 μM), complex Cu(L) (17.50 μM), and positive control. | ||
Reduced thiol levels found in the studied cells indicate a redox imbalance. Thus, it is possible that the cycle arrest is related to oxidative stress and oxidative DNA damage. The expression of enzymes related to the first line of cell defense against reactive oxygen species (ROS) and DNA damage was evaluated. It was observed that Cu(L) complex treatment significantly upregulated expression of the NF2L2 gene, which is a transcription factor for genes encoding antioxidant enzymes (such as superoxide dismutase (SOD2) and catalase (CAT)) neutralizing superoxide radicals and hydrogen peroxide, respectively. These two genes exhibited an approximately a 3-fold increase in the expression level. The expression of enzymes involved in the GSH oxidation and regeneration cycle (GPX and GSR), as well as the genes associated with oxidative DNA damage (ATM, ATR, OGG1, PARP), increased to a lesser extent. For comparison, the changes in the tested gene expression in HeLa cells after incubation with the ligand L were minor, with only SOD expression being increased by almost 1.5-fold (Fig. 10).
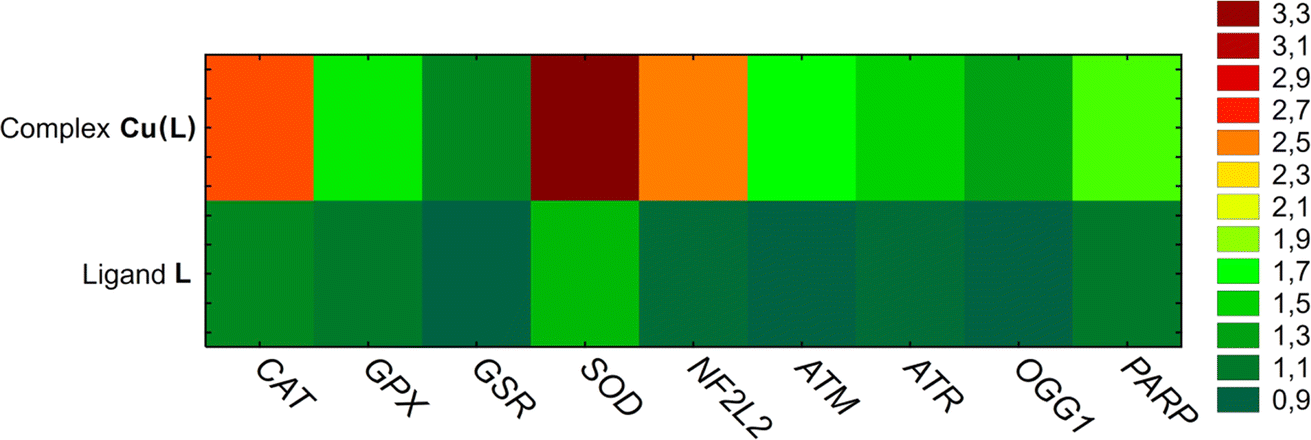 | ||
| Fig. 10 The results of mRNA gene expression analysis in HeLa cells treated with ligand L (100 μM) and complex Cu(L) (at concentrations corresponding to an IC50 value of 17.50 μM). Mean RQ levels are marked with a color scale. | ||
To determine the genotoxic effect of ligand L and its copper complex Cu(L), the comet assay was used, presented as the tail moment parameter (tail length × DNA amount in the tail). The tail moment values of the negative control (the untreated HeLa cells) indicated a low level of spontaneous DNA damage (TM: 7.22 ± 3.25) (Fig. 11). A higher degree of DNA degradation than that observed in the control was noted in cells treated with ligand L (14.01 ± 4.18) and the studied complex Cu(L) (29.1 ± 8.08), indicating both their genotoxic and cytotoxic properties. In addition, complex Cu(L) induces twice as much DNA damage as compared to ligand L.
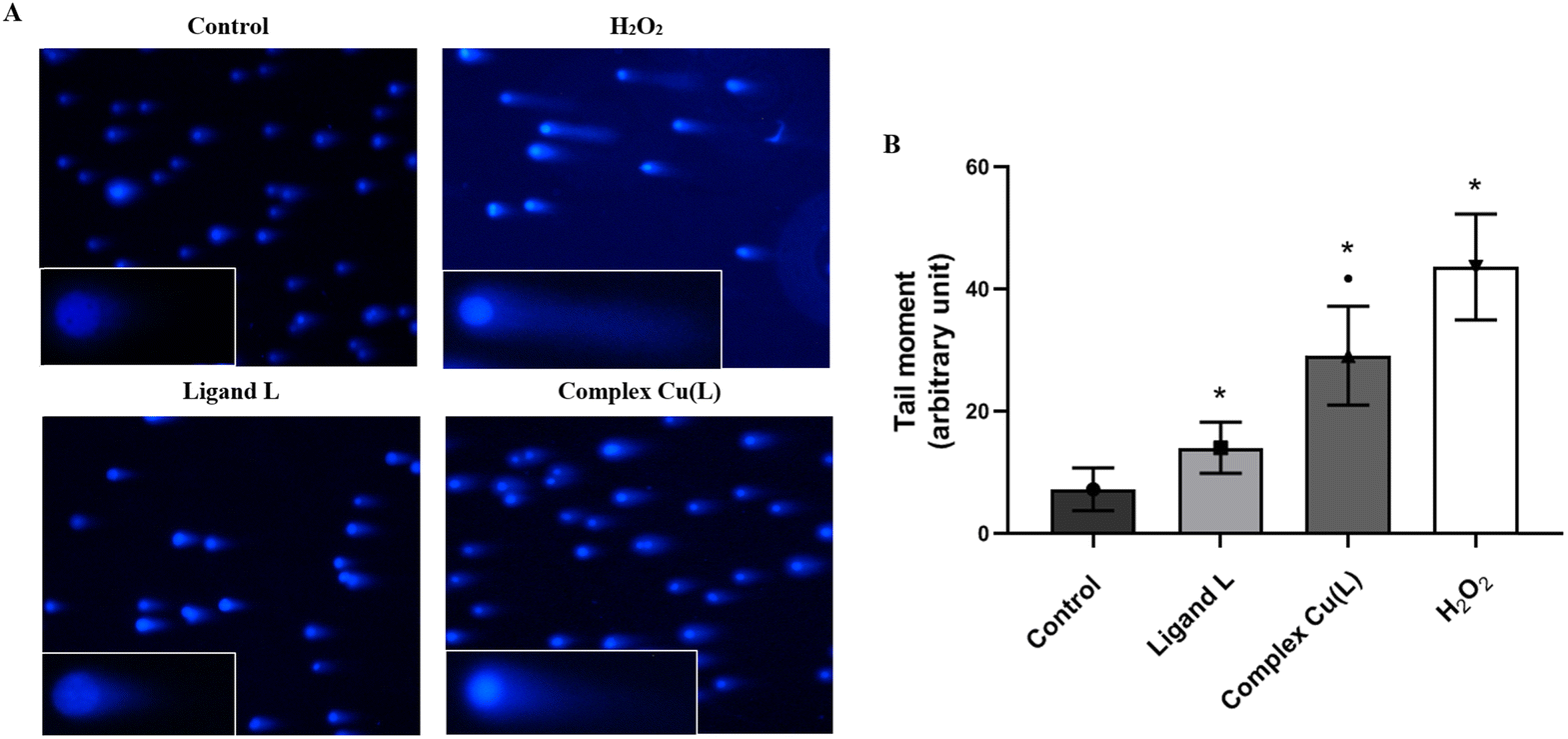 | ||
| Fig. 11 DNA damage (tail moment) induced by ligand L (100 μM) and complex Cu(L) (at concentration corresponding to an IC50 value of 17.50 μM) detected by the neutral comet assay in HeLa cells. (A) Representative images from the neutral comet assay (100× and 150× magnification). (B) Representative graph obtained by plotting the means of the tail moment (whose magnitude reflects the frequency of DNA strand breaks per nucleus) of the HeLa cells comets. The values are presented as the mean of three independent experiments ± SD. *p < 0.05 vs. control. ■ p < 0.05 vs. ligand L. | ||
3.7 Antimicrobial and antifungal activity
Antimicrobial activities of the free ligand L and its copper(II) complex Cu(L) were tested against a wide panel of Gram-negative bacteria, Gram-positive bacteria, and yeasts (Table S4†). Whereas the ligand L showed a lack of activity in the tested range of concentrations, Cu(L) revealed negligible activity against the tested Gram-positive bacteria (MIC 250 mg L−1). Interestingly, comparable activity was observed against methicillin-resistant Staphylococcus aureus (MRSA). It was found that the Cu(L) complex exhibited bactericidal activity since the MBC/MIC index for most of the tested Gram-positives was <4. Cu(L) was also slightly active against two out of the three tested yeasts. Activity against Gram-negative bacteria was not observed for ligand L or for the Cu(L) complex. Vancomycin (Van), ciprofloxacin (Cip), and nystatin (Nys) were used as the standard reference antibiotics.4. Conclusions
In the presented study, a new copper(II) pyrazine-based complex bearing a dithiocarbazate moiety Cu(L) was synthesized and studied in terms of its physicochemical properties, in silico simulations, and biological activity. The NNS-coordinated Cu(L) complex is stabilized by hydrogen bonds formed between NH2 hydrogen atoms and chlorine atoms, as highlighted through SC-XRD, Hirshfeld surface and fingerprint plot analysis. Both the Cu(L) complex and its corresponding ligand L were tested for anticancer activity against A375, PANC-1, MKN-74, T-47D, HeLa, and NCI-H1563 cancer cells, and normal human fibroblasts BJ. Along with the increasing concentration of Cu(L), a significant increase in activity against all cancer cells was observed in comparison with the ligand L alone. The lowest IC50 value (17.50 μM) was reached against HeLa lines. For this cell line, the studies of the mechanism of action revealed that the high activity of this compound is related to the redox imbalance and oxidative stress induced in the treated cells, caused by the upregulation of genes encoding superoxide dismutase (SOD2) and catalase (CAT) antioxidant enzymes. Structures of a similar Cu(II) NNS-coordinated dithiocarbazate complex bearing pyrazine moiety were reported, although their biological activity has not been studied.14 In contrast, complexes containing pyridine moieties have been more thoroughly investigated, often reaching sub-micromolar or near-sub-micromolar IC50 values.15,16 While ligand L was found to be mostly inactive, Cu(L) exhibited good anticancer activity. This trend can also be observed in the abovementioned pyridine-based complexes, highlighting the potential of the novel metalorganic drugs concept, especially in terms of the application of metals lighter than platinum or ruthenium (like copper), which are generally considered significantly less toxic. While the synthesized Cu(L) complex exhibited good anticancer activity, its antibacterial activity was found to be negligible.Data availability
Crystallographic data for Cu(L) has been deposited at the CCDC as CCDC 2292685.†Conflicts of interest
The Authors declare no conflict of interest.Acknowledgements
This article was completed while the two authors were doctoral candidates in the Interdisciplinary Doctoral School at the Lodz University of Technology, Poland (B.R., N.R.). The calculations mentioned in this paper were performed using TUL Computing & Information.References
- Cancer Tomorrow, https://gco.iarc.fr/tomorrow/en/dataviz/isotype?years=2040%26single_unit=500000%26sexes=0, accessed 24 August 2023.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra, D. K. Pandey, V. De Falco, A. Upadhyay, R. Kandimalla, A. Chaudhary, J. K. Dhanjal, S. Dewanjee, J. Vallamkondu and J. M. Pérez de la Lastra, Genes Dis., 2023, 10, 1367–1401 CrossRef CAS.
- C. M. Tilsed, S. A. Fisher, A. K. Nowak, R. A. Lake and W. J. Lesterhuis, Front. Oncol., 2022,(12), 960317 CrossRef CAS PubMed.
- W. A. Wani, U. Baig, S. Shreaz, R. A. Shiekh, P. F. Iqbal, E. Jameel, A. Ahmad, S. H. Mohd-Setapar, M. Mushtaque and L. T. Hun, New J. Chem., 2016, 40, 1063–1090 RSC.
- U. Ndagi, N. Mhlongo and M. Soliman, Drug Des. Dev. Ther., 2017, 11, 599–616 CrossRef CAS.
- R. Paprocka, M. Wiese-Szadkowska, T. Kosmalski, D. Frisch, M. Ratajczak, B. Modzelewska-Banachiewicz and R. Studzińska, Pharmacy, 2022, 15, 1219 CAS.
- B. Shakya and P. N. Yadav, Mini Rev. Med. Chem., 2020, 20, 638–661 CrossRef CAS.
- D. K. Lang, R. Kaur, R. Arora, B. Saini and S. Arora, Anticancer Agents Med. Chem., 2020, 20, 2150–2168 CrossRef CAS PubMed.
- S. J. Kim, H. S. Kim and Y. R. Seo, Oxid. Med. Cell. Longev., 2019, 2019, e5381692 Search PubMed.
- G.-K. Marioli-Sapsakou and M. Kourti, Anticancer Res., 2021, 41, 5881–5902 CrossRef CAS.
- S. P. Baba and A. Bhatnagar, Curr. Opin. Toxicol., 2018, 7, 133–139 CrossRef.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS.
- N. K. Singh, A. A. Kumbhar, Y. R. Pokharel and P. N. Yadav, J. Inorg. Biochem., 2020, 210, 111134 CrossRef CAS PubMed.
- M. H. S. A. Hamid, M. Akbar Ali, A. H. Mirza, P. V. Bernhardt, B. Moubaraki and K. S. Murray, Inorg. Chim. Acta, 2009, 362, 3648–3656 CrossRef CAS.
- Y. Gou, M. Chen, S. Li, J. Deng, J. Li, G. Fang, F. Yang and G. Huang, J. Med. Chem., 2021, 64, 5485–5499 CrossRef CAS PubMed.
- C. D. Q. O. Cavalcante, E. Garcia, T. H. A. da Mota, D. M. de Oliveira and C. C. Gatto, J. Inorg. Biochem., 2022, 237, 112015 CrossRef CAS.
- A. Santra, P. Brandao, G. Mondal, P. Bera, A. Jana, I. Bhattacharyya, C. Pramanik and P. Bera, Polyhedron, 2020, 176, 114277 CrossRef CAS.
- F. Menges, ‘Spectragryph – Optical Spectroscopy Software’, Version 1.2.16.1, 2022, http://www.effemm2.de/spectragryph/ Search PubMed.
- A. Czylkowska, B. Rogalewicz, M. Szczesio, A. Raducka, K. Gobis, P. Szymański, K. Czarnecka, B. C. Camargo, J. Szczytko, A. Babich, S. Dubkov and P. Lazarenko, Int. J. Mol. Sci., 2022, 23, 2980 CrossRef CAS.
- H. Foks, L. Balewski, K. Gobis, M. Dabrowska-Szponar and K. Wisniewska, Heteroat. Chem., 2012, 1, 49–58 CrossRef.
- A. Olczak, M. Szczesio, J. Gołka, C. Orlewska, K. Gobis, H. Foks and M. L. Główka, Acta Crystallogr., C, 2011, 67, o37–o42 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., A, 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., A, 2008, 64, 112–122 CrossRef CAS PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09 (Revision E.01), 2013 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. S. Gordon, J. S. Binkley, J. A. Pople, W. J. Pietro and W. J. Hehre, J. Am. Chem. Soc., 1982, 104, 2797–2803 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- S. Lenoid, Chemissian v4.67, http://www.chemissian.com/ Search PubMed.
- K. Gas and M. Sawicki, Materials, 2022, 15, 495 CrossRef CAS.
- M. Sawicki, W. Stefanowicz and A. Ney, Semicond. Sci. Technol., 2011, 26, 064006 CrossRef.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- A. Daina and V. Zoete, ChemMedChem, 2016, 11, 1117–1121 CrossRef CAS PubMed.
- P. Banerjee, A. O. Eckert, A. K. Schrey and R. Preissner, Nucleic Acids Res., 2018, 46, W257–W263 CrossRef CAS.
- A. A. Lagunin, V. I. Dubovskaja, A. V. Rudik, P. V. Pogodin, D. S. Druzhilovskiy, T. A. Gloriozova, D. A. Filimonov, N. G. Sastry and V. V. Poroikov, PLOS ONE, 2018, 13, e0191838 CrossRef.
- Quest Graph™ IC50 Calculator, AAT Bioquest, Inc., 2024, https://www.aatbio.com/tools/ic50-calculator Search PubMed.
- P. L. Olive and J. P. Banáth, Nat. Protoc., 2006, 1, 23–29 CrossRef CAS PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed., 1995, 34, 1555–1573 CrossRef CAS.
- C. Jelsch, K. Ejsmont and L. Huder, IUCrJ, 2014, 1, 119–128 CrossRef CAS.
- S. S. Tan, S. J. Kim and E. T. Kool, J. Am. Chem. Soc., 2011, 133, 2664–2671 CrossRef CAS.
- M. A. Ali, A. H. Mirza, C. C. Mei, P. V. Bernhardt and M. R. Karim, Polyhedron, 2013, 49, 277–283 CrossRef CAS.
- M. Akbar Ali and S. E. Livingstone, Coord. Chem. Rev., 1974, 13, 101–132 CrossRef.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Deliv. Rev., 2001, 46, 3–26 CrossRef CAS.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- W. J. Egan, K. M. Merz and J. J. Baldwin, J. Med. Chem., 2000, 43, 3867–3877 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS.
- I. Muegge, S. L. Heald and D. Brittelli, J. Med. Chem., 2001, 44, 1841–1846 CrossRef CAS PubMed.
- M. van Engeland, L. J. W. Nieland, F. C. S. Ramaekers, B. Schutte and C. P. M. Reutelingsperger, Cytometry, 1998, 31, 1–9 CrossRef CAS.
- Q. Peña, G. Sciortino, J.-D. Maréchal, S. Bertaina, A. J. Simaan, J. Lorenzo, M. Capdevila, P. Bayón, O. Iranzo and Ò. Palacios, Inorg. Chem., 2021, 60, 2939–2952 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2292685. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra06874b |
| This journal is © The Royal Society of Chemistry 2024 |