
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A simple and cost-efficient route to prepare sulfonated dihalo-monomers for synthesizing sulfonated aromatic PEMs†
Nodar Dumbadze a,
Marco Vivianib,
Klaus-Dieter Kreuerbc and
Giorgi Titvinidze*ab
a,
Marco Vivianib,
Klaus-Dieter Kreuerbc and
Giorgi Titvinidze*ab
aAgricultural University of Georgia, 240 David Aghmashenebeli Alley, Tbilisi 0159, Georgia. E-mail: g.titvinidze@agruni.edu.ge
bHahn-Schickard Gesellschaft für Angewandte Forschung e.V, Georges-Koehler-Allee, 103, 79110, Freiburg Im Breisgau, Germany
cMax-Planck-Institute for Solid State Research, Heisenbergstraße 1, 70569 Stuttgart, Germany
First published on 21st November 2024
Abstract
We present a simple and cost-efficient route for the preparation of sulfonated dihalo-monomers for the synthesis of hydrocarbon ionomers. After conventional monomer sulfonation, excess sulfuric acid is quantitatively removed by neutralization with BaCO3. This leads to the precipitation of excess H2SO4 as insoluble BaSO4, which is easily separated from the sulfonated monomers in their soluble Ba-forms by filtration. Compared to conventional methods, the proposed approach leads to higher yields, drastically reduces the number of purification steps, and can easily be expanded to the preparation of other sulfonated monomers. The specific monomers presented here are suitable for the preparation of sulfonated polyarylenes and sulfonated polyphenylenes.
Introduction
For more than three decades, sulfonated aromatic polymers have been considered alternatives for perfluorosulfonic acid (PFSA) ionomers as membrane materials for fuel cells and electrolyzers. The interest in aromatic membranes was initially driven by cost considerations, but more recent concerns have been related to the use and production of poly- and perfluoro-alkyl substances (PFAS),1,2 further increasing the incentive to develop fluorine-free alternatives to benchmark PFSAs.3Initially, progress in this area of research was slow, and this had a lot to do with the unique chemical structure of PFSAs. This combines the extreme hydrophobicity of their PTFE backbones with the hydrophilicity of super-acidic –SO3H groups terminating the side chains. These side chains have some flexibility in contrast to the polymer backbone, with their high persistence length stemming from the characteristic helical conformation of PTFE. Over the years, it has become clear that one-to-one aromatic substitutes for PFSAs are surely out of reach: sulfonated aromatic polymers are a separate class of material with distinct properties, offering specific pros and cons compared to PFSAs.4 For example, sulfonated hydrocarbons generally have better gas-separating properties and lower electroosmotic water drag, but they can exhibit high swelling in water and lower conductivity at low humidification.
Interestingly, the diverse property profiles of the vast number of aromatic polymer electrolytes developed over the years, with different compositions and architectures, clearly show that specific solutions may meet the requirements of specific well-defined applications.5–9 This prospect is closely related to the enormous design-flexibility of sulfonated aromatic polymers, which is in stark contrast to the few degrees of freedom for the design of PFSAs (essentially their ion-exchange capacity and length of the side chains).7–9
Two main synthetic strategies enable the synthesis of sulfonated polymers: post-sulfonation of aromatic polymers and polymerization of sulfonated monomers. Historically, sulfonated polymer electrolytes were initially obtained by post-sulfonation. However, the use of sulfonating agents, such as fuming sulfuric acid, concentrated sulfuric acid, or chlorosulfonic acid, was found to cause degradation of the main backbone10 and did not allow for the controlled positioning and degree of sulfonation.8 To overcome post-sulfonation shortcomings, the synthesis of sulfonated aromatic polymers from presulfonated monomers has become the preferred approach.11
Polymerization using sulfonated monomers enables precise control of the sulfonation degree as well as the topological placement along the polymer backbone or side chain.7 The sulfonation pattern, i.e. the way in which the fixed-ionic groups (–SO3H) are distributed within the polymeric structure, has strong effects on the swelling, viscoelastic properties, chemical stability and proton transport. For example, the concentration of sulfonic groups in a domain with high connectivity allows for the combination of high conductivity, low swelling, and reasonable mechanical strength.12,13 At the same time, polymer electrolytes obtained from sulfonated monomers have superior stability compared to their postsulfonated analogues, owing to the nature of their sulfonation mechanism.6,14 The synthesis of highly sulfonated monomers allows increasing the local density of ionic groups. This may significantly impact the transport properties, especially when used for preparing new multiblock copolymers.7,13,15 Accordingly, the conceptualization and development of new sulfonated monomers can play a key role in enabling further progress while paving the way to commercialization.
In this work, we present a new simple, versatile, and adaptable method for the preparation of new and previously reported sulfonated monomers with a high sulfonation degree. Four of these are suitable for the polymerization of sulfonated polyarylenes, such as poly(ether sulfone)s, poly(ether ether ketone)s, poly(phenylene sulfone)s, while three more types of monomer may be used for making sulfonated polyphenylenes.
The presented synthetic route reduces the number of purification steps, and eliminates the need for costly recrystallization and extraction with organic solvents, while minimizing waste production to just a solid barium sulfate cake and a neutral water solution.
Results and discussion
Monomers for sulfonated polyarylenes
Heteroatom-containing sulfonated polyarylenes are generally prepared via step-growth polymerization using sulfonated dihalo-monomers. The most commonly used dihalo-monomers are 3,3′-disulfonate-4,4′-dichlorodiphenylsulfone (sDCDPS) and 3,3′-disulfonate-4,4′-difluorodiphenylsulfone (sDFDPS).16 Both monomers are prepared via electrophilic sulfonation of the corresponding precursors using fuming sulfuric acid (Scheme 1).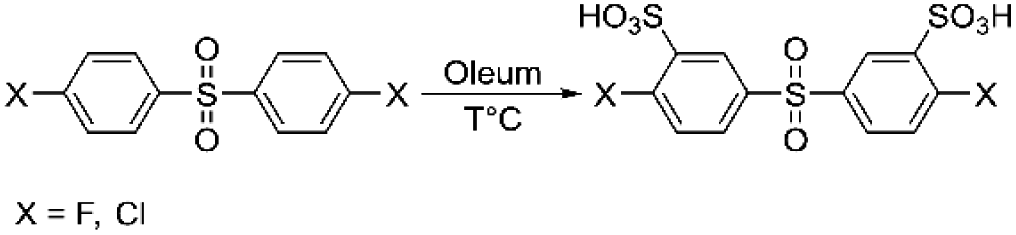 | ||
| Scheme 1 General synthesis scheme for sDCDPS or sDFDPS dihalo-monomers. | ||
Sulfonated dichlorodiphenylsulfone has practical importance because of its relatively low price, while difluoro analogues are more reactive, leading to higher molecular weight polymers with enhanced mechanical properties.17 The stronger stabilization of the intermediate Meisenheimer complex is responsible for the higher reactivity of difluoromonomers compared to dichloromonomers.
The synthesis and purification of sDFDPS are well established.18–20 Despite many years of optimization, the procedure involves several steps and at least three recrystallizations,18 leading to a minimum of 13 steps (Scheme 2a) and often a questionable purity of the monomer. To reach an acceptable purity for polycondensation reactions, the total yield is usually reduced to around 55 mol%. Here we report an innovative preparation route for sDFDPS and the use of the same method for the synthesis of another six highly sulfonated dihalo-monomers. A new purification process was developed exploiting the solubility of sulfonated monomers in barium form, which we recently reported as a method for the determination of sodium sulfate contamination in sDFDPS.21
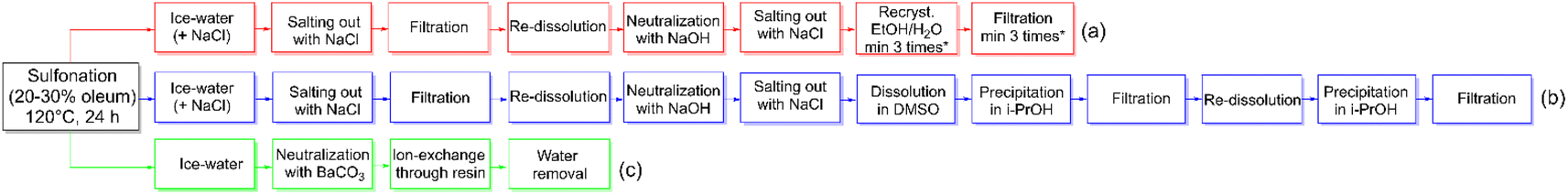 | ||
| Scheme 2 Synthesis of sDFDPS by (a) "conventional" (b) DMSO and (c) BaCO3 methods. | ||
An alternative to the multiple recrystallizations, the monomer can be separated from the salts using an organic solvent, such as dimethyl sulfoxide (DMSO).22 In this case, the insolubility of the salts in DMSO allowed their removal by simple filtration after dissolution of the impure sDFDPS. The monomer obtained by precipitation of the organic solution suffered from DMSO contamination, even after vacuum drying, and needed additional re-precipitation. DMSO contamination is a common problem in this procedure and it can easily be overlooked in 1H NMR analysis using DMSO-d6. However, the contamination was clearly visible in the 1H NMR spectrum recorded in D2O (ESI Fig. S1†). Although this method effectively removes inorganic salts without the need for recrystallization, it still consists of minimum 12 steps requiring organic solvents (Scheme 2b). The overall yield was increased up to 65%, roughly 10% more than the conventional method.6
The new approach developed and proposed here drastically reduces the purification procedure without compromising the purity and yield of the sulfonated monomer. The sulfonation of DFDPS under conventional conditions was followed by direct neutralization with BaCO3. The use of carbonate led to a complete removal of excessive H2SO4, which precipitated as BaSO4 and liberated CO2, while the filtrate consisted of water-soluble Ba-sDFDPS. After filtration, the monomer was transformed to the desired ion-form by passing the solution through an ion-exchange resin before water removal. The purity of the monomer obtained by this new route (Scheme 2c) was confirmed by 1H NMR and it was up to the standard required for polycondensation reactions (ESI Fig. S2–S4†). Possible incomplete exchange from Ba to the Na-form or Li-form was checked by adding Na2SO4, which did not lead to any precipitate. Overall, this process reduced the work-up times while the total yield was more than 84%. The economic gain of this process is both represented by the saved time and the increased yield.
Another monomer of interest for the synthesis of sulfonated polyarylene is sulfonated bis(4-fluorophenyl)phenyl phosphine oxide (sBFPPO). Due to its high sulfonation degree and higher water solubility, its conventional purification is more complex than for sDFDPS requiring column chromatography. We applied the aforementioned method for synthesizing the sBFPPO monomer (Scheme 3a). The benefit of the proposed method is its versatility for different sulfonated monomers prepared by direct sulfonation and the high yield obtained compared to conventional methods. In the case of sBFPPO, the phosphine oxide instead of sulfone group and the third ring attached to it resulted in a less pure product using the same method. As proven by the 1H, 13C, and 19F NMR spectra (ESI Fig. S5–S7†), an almost pure product was obtained following the same purification procedure. However, small impurities (approx. 2% according to the 19F NMR spectrum) were present and this could be assigned to partial sulfonation or sulfonation in different places of the monomer (ESI Fig. S6†). Despite the impurities still being acceptable for condensation polymerization, their presence underlines the impact of the starting monomer on the quality of the resulting product and the need for optimization of the sulfonation step. sBFPPO was obtained in a higher yield of 61.3% compared to previous reports of 50% obtained after column chromatography.23
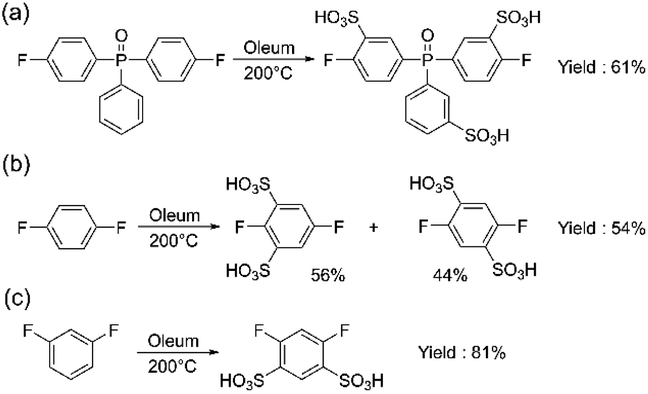 | ||
| Scheme 3 Syntheses of (a) sBFPPO (2), (b) ds-1,4-DFB (3) and (c) ds-1,3-DFB (4). | ||
Aiming to expand the monomer portfolio available for the synthesis of sulfonated polyarylenes, we applied the newly developed method to obtain disulfonated difluorophenylenes as potential new building blocks for polymers and block copolymers. A high local sulfonation degree is beneficial for achieving high proton conductivity and nanophase separation, although a trade-off with water solubility is needed for the final polymers. Since “hypersulfonated” units are not beneficial due to counter-ion condensation,24 in this work, we aimed for disulfonated monomers without sulfonic groups in the ortho position (Schemes 3b and c).
To achieve double sulfonation on the same ring, higher temperatures and longer sulfonation times were required compared to DFDPS or DCDPS (the commonly used temperature range of 110–140 °C for monomers containing a single benzene ring led to a mixture of mono- and disulfonated products, which will not be discussed in detail). The same synthetic steps reported in Scheme 3a were followed. The obtained products and their relative compositions depended on the starting non-sulfonated monomer. In the case of p-difluorobenzene, a mixture with a molar ratio of 0.56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.44 of 2,5-difluorobenzene-1,3-disulfonate and 2,5-difluorobenzene-1,4-disulfonate was obtained (Scheme 3b) (see ESI Fig. S8†). In order to separate the isomers, we carried out recrystallization from an i-PrOH/H2O mixture, which led to the pure 2,5-difluorobenzene-1,3-disulfonate (54%) (3), while we were not able to separate 2,5-difluorobenzene-1,4-disulfonate in a pure form. On the other hand, m-difluorobenzene yielded only 4,6-difluorobenzene-1,3-disulfonate (4) (Scheme 3c) with a high yield of 81%.
:0.44 of 2,5-difluorobenzene-1,3-disulfonate and 2,5-difluorobenzene-1,4-disulfonate was obtained (Scheme 3b) (see ESI Fig. S8†). In order to separate the isomers, we carried out recrystallization from an i-PrOH/H2O mixture, which led to the pure 2,5-difluorobenzene-1,3-disulfonate (54%) (3), while we were not able to separate 2,5-difluorobenzene-1,4-disulfonate in a pure form. On the other hand, m-difluorobenzene yielded only 4,6-difluorobenzene-1,3-disulfonate (4) (Scheme 3c) with a high yield of 81%.
The purity of the monomers was confirmed by their 1H, 13C, and 19F NMR spectra (ESI Fig. S9–S14†). The single signal in 19F NMR spectrum of 4,6-difluorobenzene-1,3-disulfonate (4) indicated a symmetric substitution (ESI Fig. S13†), while the 19F NMR spectrum of 2,5-difluorobenzene-1,3-disulfonate (3) displayed two signals (ESI Fig. S10†). Similar to sDFDPS, full exchange from Ba to the Na- or Li-form was checked by adding Na2SO4, leading to no precipitation.
Monomers for sulfonated polyphenylenes
The synthesis of sulfonated polyphenylenes (sPP) often involves monomers, such as sulfonated dibromo or dichloro monomers (Scheme 4).25–33 These polymers are generally prepared via metal-catalyzed reactions, including Ullmann coupling or Ni(0)-mediated coupling.31,34,35 Among them, the sulfonated polyphenylenes prepared using 2,5-dichlorobenzene sulfonic acid by Miyatake's group at the University of Yamanashi are the most promising.29,36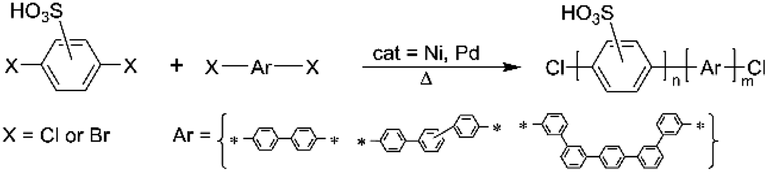 | ||
| Scheme 4 General scheme for the synthesis of sulfonated poly(phenylenes). | ||
However, there are a few reports using disulfonated dichloro- or dibromo-benzenes.32,33,37–42 In contrast to 2,5-dichlorobenzene sulfonic acid, the disulfonated analogue is not commercially available, which could be due to the complex purification procedure. The high-water solubility of the monomer hinders the efficient salting-out purification step after neutralization, and, for this reason, sometimes this step is simply skipped. However, to remove inorganic salts, extraction with DMF is common practice,38 even if sodium sulfate and sodium carbonate are somewhat soluble in DMF,43 thereby requiring subsequent Soxhlet extraction with ethanol.38
In this work, we implemented the new synthetic approach also to single-ring dihalobenzene compounds to provide a solution to the aforementioned shortcomings.
After the sulfonation of p-dibromobenzene, the excess sulfonic acid was removed by neutralization with BaCO3. The water solution containing the sulfonated monomer was passed through an ion-exchange resin to obtain the desired ion-form, in this case the Na-form. The crude product consisted of a mixture of 2,5-dibromobenzene-1,3-disulfonate, 2,5-dibromobenzene-1,4-disulfonate, and a minor amount of degradation products. Due to the overlap of the main signals in the 1H NMR spectrum (ESI Fig. S15†), it was impossible to identify the isomers, while the 13C NMR spectrum clearly showed the presence of two isomers (ESI Fig. S16†). To separate the isomers, we carried out recrystallization from ethanol/water. This yielded pure 2,5-dibromobenzene-1,4-disulfonate (Scheme 5a) (5) (38%), but separation of pure 2,5-dibromobenzene-1,3-disulfonate was not possible. The purity of the monomer was confirmed by its 1H and 13C NMR spectra (ESI Fig. S17 and S18†).
 | ||
| Scheme 5 Sulfonation of (a) 1,4-dibromobenzene (5), (b) 1,4-dichlorobenzene (6) and (c) 1,3-dichlorobenzene (7). | ||
Similar to difluoro and dibromo analogues, high temperature and a relatively long sulfonation time were applied for the sulfonation of p- and m-dichlorobenzenes (Scheme 5b and c). The purification of both monomers was carried out using the aforementioned method. Here, p-dichlorobenzene yielded a mixture of 1,4-sulfonated and 1,3 sulfonated products (6) with a ratio of 0.91:0.09 and a yield of 73.1%. Further separation of the mixture was not carried out. For m-dichlorobenzene, however, the yield of the pure 1,3-sulfonated product (7) was higher (80.6%), similar to the difluoro analogue. For both products, no further purification was needed. The purity of the monomers was confirmed by their 1H and 13C NMR spectra (ESI Fig. S19–S22†).
In the IR spectra (ESI Fig S23–S29†) of all the synthesized monomers, a new characteristic band at 1025–1041 cm−1, assigned to the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO stretching, appeared, indicating successful sulfonation, along with the previously discussed NMR spectral data.
SO stretching, appeared, indicating successful sulfonation, along with the previously discussed NMR spectral data.
Experimental section
Materials
4,4′-Difluorodiphenylsulfone (DFDPS) was supplied by Fumatech BWT GmbH (Germany). 1,3-Difluorobenzene (99%, Alfa Aesar), 1,4-difluorobenzene (99+%, Thermo Scientific), 1,4-dibromobenzene (99%, Thermo Scientific), 1,3-dichlorobenzene (98%, Thermo Fischer), 1,4-dichlorobenzene (99+%, Thermo Scientific), and sulfuric acid fuming (20–30%, Thermo Scientific) were used without further purification. Bis(4-fluorophenyl)phenyl phosphine oxide (BFPPO) was synthesized according to the reported procedure.44NMR spectroscopy
1H, 13C, and 19F NMR spectra were recorded using Bruker 250 MHz NMR and Bruker 300 MHz NMR spectrometers at room temperature with D2O as a solvent and internal standard.FT-IR analysis
Infrared spectra were recorded on a Bruker Alpha II FT-IR spectrophotometer with ZnSe-ATR.Disodium 3,3′-disulfonate-4,4′-difluorodiphenylsulfone (sDFDPS) (1)
A 50 ml round-bottom flask equipped with a magnetic stirring bar and condenser was charged with DFDPS (5.04 g, 19.8 mmol) and fuming sulfuric acid (11 ml, 20–30% SO3). The mixture was heated for 24 h at 140 °C. To avoid a loss of SO3, the system was connected to a gas-bubbler filled with silicon oil. After 24 h, the reaction mixture was cooled down and slowly poured into 100 ml of ice water. To the solution, BaCO3 was added to precipitate excess sulfuric acid. The solution containing barium 3,3′-disulfonate-4,4′-difluorodiphenylsulfone was concentrated using a rotary evaporator and then passed through an ion-exchange resin (Na-form) leading to disodium 3,3′-disulfonate-4,4′-difluorodiphenylsulfone. Water was distilled off, and the white-coloured monomer was dried in a vacuum oven at 145 °C. The yield was 7.64 g (84%).1H NMR (300 MHz, D2O) δ 8.41 (dd, J = 6.3, 2.4 Hz, 1H), 8.19 (ddd, J = 8.8, 4.4, 2.5 Hz, 1H), 7.53 (dd, J = 9.6, 8.8 Hz, 1H). 13C NMR (75 MHz, D2O) δ 162.01 (d, J = 261.2 Hz), 135.53 (d, J = 3.6 Hz), 133.68 (d, J = 10.8 Hz), 131.67 (d, J = 17.1 Hz), 128.67 (d, J = 3.2 Hz), 118.99 (d, J = 23.9 Hz). 19F NMR (282 MHz, D2O) δ −102.55 (ddd, J = 9.6, 6.4, 4.5 Hz).
Sulfonated bis(4-fluorophenyl)phenyl phosphine oxide (sBFPPO) (2)
sBFPPO was prepared following the same procedure described above. The yield was 61.3%.1H NMR (300 MHz, D2O) δ 8.23–8.04 (m, 2H), 7.98–7.72 (m, 2H), 7.55 (ddd, J = 10.5, 8.5, 2.2 Hz, 1H). 13C NMR (75 MHz, D2O) δ 161.88 (dd, J = 259.9, 3.3 Hz), 143.65 (d, J = 12.9 Hz), 138.01 (dd, J = 12.2, 10.2 Hz), 134.77 (d, J = 11.1 Hz), 131.27 (dd, J = 16.0, 13.6 Hz), 130.52, 130.25 (d, J = 12.6 Hz), 129.82 (d, J = 110.2 Hz), 128.65 (d, J = 11.6 Hz), 125.33 (dd, J = 109.9, 3.9 Hz), 118.45 (dd, J = 22.7, 13.6 Hz). 19F NMR (282 MHz, D2O) δ −104.13 (dt, J = 10.6, 5.5 Hz).
2,5-Difluorobenzene-1,3-disulfonate (ds-1,4-DFB) (3)
A 50 ml round-bottom flask equipped with a magnetic stirring bar and condenser was charged with 1,4-difluorobenzene (1,4-DFB) (5.00 g, 43.9 mmol) and fuming sulfuric acid (20 ml, 20–30% SO3). The mixture was slowly heated to 80 °C and kept for 1 h at this temperature to prevent the evaporation of 1,4-DFB, and then it was heated for 48 h at 200 °C. The further work-up was done as described for sDFDPS. After ion-exchange, the mixture consisted of two isomers: 2,5-difluorobenzene-1,3-disulfonate and 2,5-difluorobenzene-1,4-disulfonate with a ratio of 0.56:0.44. To separate the isomers, we carried out recrystallization from an i-PrOH/H2O mixture (8:1), which yielded pure 2,5-difluorobenzene-1,3-disulfonate with a 54% yield (white powder).
1H NMR (300 MHz, D2O) δ 7.80 (dd, J = 7.6, 5.0 Hz, 1H). 13C NMR (75 MHz, deuterium oxide) δ 156.67 (dd, J = 247.8, 3.1 Hz), 151.33 (dd, J = 252.9, 3.0 Hz), 132.85 (dd, J = 18.7, 6.5 Hz), 118.70 (dd, J = 26.7, 2.0 Hz). 19F NMR (282 MHz, deuterium oxide) δ −115.17 (dt, J = 18.8, 7.6 Hz), −118.11 (dt, J = 18.9, 5.1 Hz).
4,6-Difluorobenzene-1,3-disulfonate (ds-1,3-DFB) (4)
ds-1,3-DFB was prepared following the procedure described above, but with a shorter reaction time (15 h). In contrast to above, a 50 ml round-bottom flask equipped with a magnetic stirring bar and condenser was charged with 1,3-difluorobenzene (1,3-DFB) (5.01 g, 43.9 mmol) and fuming sulfuric acid (20 ml, 20–30% SO3). The mixture was slowly heated to 80 °C and kept for 1 h at this temperature to prevent the evaporation of 1,3-DFB, and then it was heated for 15 h at 200 °C. The further work-up was done as described for sDFDPS. The yield was 11.31 g (81%) white powder.1H NMR (300 MHz, D2O) δ 8.29 (t, J = 7.8 Hz, 1H), 7.37 (t, J = 9.8 Hz, 1H). 13C NMR (75 MHz, D2O) δ 160.90 (dd, J = 259.5, 13.6 Hz), 128.91, 126.54 (dd, J = 11.8, 8.4 Hz), 107.08 (t, J = 26.8 Hz). 19F NMR (282 MHz, D2O) δ −102.40 (dd, J = 9.8, 7.9 Hz).
2,5-Dibromobenzene-1,4-disulfonate (ds-1,4-DBB) (5)
ds-1,4-DBB was prepared following the procedure described above, but with a reaction time of 24 h. After ion-exchange, the mixture consisted of two isomers: 2,5-dibromobenzene-1,3-disulfonate and 2,5-dibromobenzene-1,4-disulfonate. To separate the isomers, we carried out recrystallization from an EtOH/H2O mixture (7:1), which yielded pure 2,5-dibromobenzene-1,4-disulfonate with a 38% yield.
1H NMR (300 MHz, D2O) δ 8.39 (s). 13C NMR (75 MHz, D2O) δ 144.77, 135.06, 118.14.
2,5-Dichlorobenzene-1,4-disulfonate (1,4-ds-2,5-DCB)/2,5-dichlorobenzene-1,3-disulfonate (1,3-ds-2,5-DCB) (6)
The sulfonation of 1,4-dichlorobenzene was carried out following the procedure described above. After ion-exchange, the obtained mixture consisted of 1,4-sulfonated and 1,3-sulfonated products with a ratio of 0.91:0.09. No further purification was done. The yield was 73.1%.
1H NMR (300 MHz, D2O) δ 8.24 (s) (1,4-ds-2,5-DCB), δ 8.20 (s) (1,3-ds-2,5-DCB). 13C NMR (75 MHz, D2O) δ 143.28, 132.64, 131.61 (1,4-ds-2,5-DCB) and 143.00, 131.71, 129.62, 127.03 (1,3-ds-2,5-DCB).
4,6-Dichlorobenzene-1,3-disulfonate (ds-1,3-DCB) (7)
ds-1,3-DCB was prepared following the procedure described above. After ion-exchange, no further purification was needed. The yield was 80.6%.1H NMR (250 MHz, D2O) δ 8.61 (s, 1H), 7.95 (s, 1H). 13C NMR (75 MHz, D2O) δ 138.64, 134.50, 133.89, 129.15.
Conclusions
The newly developed method for the synthesis of mono-, di-, and tri-sulfonated monomers offers a simpler, more economical, and sustainable purification route after a common sulfonation procedure. The new method offers three main advantages compared to conventional purification systems: (i) simplicity, since no recrystallization is needed, no extractions and no organic solvents are employed; (ii) use of cheap and easily available chemicals with a reduction of waste, which is simply limited to a solid cake and a neutral water solution; and (iii) drastic reduction of the number of purification steps from 13 to 6, which comes with a significant reduction of the time and costs. Overall, the proposed monomer synthesis appears to be versatile and adaptable to different sulfonated monomers independent of the degree of sulfonation and the presence of heteroatoms. This synthetic strategy will enable a faster and simpler way to expand the portfolio of sulfonated monomers for the preparation of sulfonated aromatic polymers and block copolymers. As substitutes for PFSAs, they will play a key role in the future of the emerging hydrogen economy.Data availability
The data supporting this article have been included as part of the ESI.† This includes 1H, 13C and 19F NMR and IR spectral data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research [PHDF-22-299] has been supported by Shota Rustaveli National Science Foundation of Georgia (SRNSFG) and German Federal Ministry of Education and Research project “Fluorfreie-MEA” (Grant No. O3HY106A).Notes and references
- B. Barhoumi, S. G. Sander and I. Tolosa, Environ. Res., 2022, 206, 112595 CrossRef CAS PubMed.
- R. Lohmann, I. T. Cousins, J. C. DeWitt, J. Glüge, G. Goldenman, D. Herzke, A. B. Lindstrom, M. F. Miller, C. A. Ng, S. Patton, M. Scheringer, X. Trier and Z. Wang, Environ. Sci. Technol., 2020, 54, 12820–12828 CrossRef CAS PubMed.
- M. Adamski, N. Peressin and S. Holdcroft, Mater. Adv., 2021, 2, 4966–5005 RSC.
- K. D. Kreuer, J. Membr. Sci., 2001, 185, 29–39 CrossRef CAS.
- N. Li and M. D. Guiver, Macromolecules, 2014, 47, 2175–2198 CrossRef CAS.
- M. Schuster, K. D. Kreuer, H. T. Andersen and J. Maier, Macromolecules, 2007, 40, 598–607 CrossRef CAS.
- D. W. Shin, M. D. Guiver and Y. M. Lee, Chem. Rev., 2017, 117, 4759–4805 CrossRef CAS PubMed.
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4612 CrossRef CAS PubMed.
- E. J. Park, P. Jannasch, K. Miyatake, C. Bae, K. Noonan, C. Fujimoto, S. Holdcroft, J. R. Varcoe, D. Henkensmeier, M. D. Guiver and Y. S. Kim, Chem. Soc. Rev., 2024, 53, 5704–5780 RSC.
- M. Rikukawa and K. Sanui, Prog. Polym. Sci., 2000, 25, 1463–1502 CrossRef CAS.
- F. Wang, M. Hickner, Y. S. Kim, T. A. Zawodzinski and J. E. McGrath, J. Membr. Sci., 2002, 197, 231–242 CrossRef CAS.
- K. D. Kreuer, Chem. Mater., 2014, 26, 361–380 CrossRef CAS.
- G. Titvinidze, K. D. Kreuer, M. Schuster, C. C. de Araujo, J. P. Melchior and W. H. Meyer, Adv. Funct. Mater., 2012, 22, 4456–4470 CrossRef CAS.
- Y. Hu, L. Yan and B. Yue, ACS Omega, 2020, 5, 13219–13223 CrossRef CAS PubMed.
- M. Viviani, S. P. Fluitman, K. Loos and G. Portale, Polym. Chem., 2021, 12, 2563–2571 RSC.
- M. Ueda, H. Toyota, T. Ouchi, J. I. Sugiyama, K. Yonetake, T. Masuko and T. Teramoto, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 853–858 CrossRef CAS.
- F. Wang, J. Mecham, W. Harrison and J. E. McGrath, in American Chemical Society, Polymer Preprints, Division of Polymer Chemistry, 2000, vol. 41, pp. 1401–1402 Search PubMed.
- Y. Li, R. A. VanHouten, A. E. Brink and J. E. McGrath, Polymer, 2008, 49, 3014–3019 CrossRef CAS.
- Y. Zhang, J. Kim and K. Miyatake, J. Appl. Polym. Sci., 2016, 133, 44218 CrossRef.
- B. Bae, K. Miyatake and M. Watanabe, Macromolecules, 2009, 42, 1873–1880 CrossRef CAS.
- Z. Katcharava, T. Saatkamp, A. Muenchinger, N. Dumbadze, K. D. Kreuer, M. Schuster and G. Titvinidze, Polym. Adv. Technol., 2022, 33, 2336–2343 CrossRef CAS.
- A. Haragirimana, P. B. Ingabire, Y. Zhu, Y. Lu, N. Li, Z. Hu and S. Chen, J. Membr. Sci., 2019, 583, 209–219 CrossRef CAS.
- H. Liao, G. Xiao and D. Yan, Chem. Commun., 2013, 49, 3979 RSC.
- S. Takamuku, A. Wohlfarth, A. Manhart, P. Räder and P. Jannasch, Polym. Chem., 2015, 6, 1267–1274 RSC.
- Z. Long and K. Miyatake, iScience, 2021, 24, 102962 CrossRef CAS.
- K. Shiino, T. Otomo, T. Yamada, H. Arima, K. Hiroi, S. Takata, J. Miyake and K. Miyatake, ACS Appl. Polym. Mater., 2020, 2, 5558–5565 CrossRef CAS.
- J. Ahn, R. Shimizu and K. Miyatake, J. Mater. Chem., 2018, 6, 24625–24632 RSC.
- K. Shiino, J. Miyake and K. Miyatake, Chem. Commun., 2019, 55, 7073–7076 RSC.
- F. Liu, I. S. Kim and K. Miyatake, Sci. Adv., 2023, 9, 1–13 Search PubMed.
- R. Rulkens, G. Wegner, V. Enkelmann and M. Schulze, Ber. Bunsenges. Phys. Chem., 1996, 100, 707–714 CrossRef CAS.
- M. H. Litt, S. Granados-Focil and J. Kang, ACS Symp. Ser., 2010, 1040, 49–63 CrossRef CAS.
- S. Granados-focil, A New Class of Polyelectrolytes, Poly (Phenylene Sulfonic Acids) and Its Copolymers As Proton Exchange Membranes for PEMFC’S, PhD thesis, Case Western Reserve University, 2006.
- J. Kang, A new class of polyelectrolyte, poly(p-phenylene disulfonic acids), PhD thesis, Case Western Reserve University, 2008.
- S. Granados-Focil and M. H. Litt, Prepr. Symp. – Am. Chem. Soc., Div. Fuel Chem., 2004, 49, 528–529 CAS.
- K. Goto, I. Rozhanskii, Y. Yamakawa, T. Otsuki and Y. Naito, Polym. J., 2009, 41, 95–104 CrossRef CAS.
- J. Miyake, R. Taki, T. Mochizuki, R. Shimizu, R. Akiyama, M. Uchida and K. Miyatake, Sci. Adv., 2017, 3, 1–8 Search PubMed.
- O. Beyer, T. Homburg, M. Albat, N. Stock and U. Lüning, New J. Chem., 2017, 41, 8870–8876 RSC.
- M. Litt, S. Granados-Focil, J. Kang, K. Si and R. Wycisk, ECS Trans., 2010, 33, 695–710 CrossRef CAS.
- K. Si, R. Wycisk, D. Dong, K. Cooper, M. Rodgers, P. Brooker, D. Slattery and M. Litt, Macromolecules, 2013, 46, 422–433 CrossRef CAS.
- K. Si, D. Dong, R. Wycisk and M. Litt, J. Mater. Chem., 2012, 22, 20907 RSC.
- A. Künzel-Tenner, C. Kirsch, O. Dolynchuk, L. Rößner, M. Wach, F. Kempe, T. von Unwerth, A. Lederer, D. Sebastiani, M. Armbrüster and M. Sommer, Macromolecules, 2024, 57, 1238–1247 CrossRef.
- M. F. Teasley, Polyarylene Ionomeric Membranes, WO2011/082197A1, 2011.
- C. L. Forryan, R. G. Compton, O. V. Klymenko, C. M. Brennan, C. L. Taylor and M. Lennon, Phys. Chem. Chem. Phys., 2006, 8, 633–641 RSC.
- Y. Tan, K. Zhang, H. Liao, G. Xiao, Y. Yao, G. Sun and D. Yan, J. Membr. Sci., 2016, 508, 32–39 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra. See DOI: https://doi.org/10.1039/d4ra06283c |
| This journal is © The Royal Society of Chemistry 2024 |