
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLevoglucosenone as a starting material for cascade continuous-flow synthesis of (R)-γ-carboxy-γ-butyrolactone†
Caio M. Pacheco a,
Wesley Limaa,
Fernanda A. Limaa,
Mauro R. B. P. Gomeza,
Isabela G. da Silvaa,
Leandro S. M. Mirandaa,
Pierre M. Estevesb,
Ivaldo Itabaiana Jrc,
Robert Wojcieszakde,
Raquel A. C. Leãoa and
Rodrigo O. M. A. de Souza*a
a,
Wesley Limaa,
Fernanda A. Limaa,
Mauro R. B. P. Gomeza,
Isabela G. da Silvaa,
Leandro S. M. Mirandaa,
Pierre M. Estevesb,
Ivaldo Itabaiana Jrc,
Robert Wojcieszakde,
Raquel A. C. Leãoa and
Rodrigo O. M. A. de Souza*a
aBiocatalysis and Organic Synthesis Group, Chemistry Institute, Federal University of Rio de Janeiro, Rio de Janeiro, 21941910, Brazil
bInstituto de Química, Universidade Federal do Rio de Janeiro, Av. Athos da Silveira Ramos, 149, CT, A-622, Cid. Univ., Rio de Janeiro, RJ 21941-909, Brazil
cDepartment of Biochemical Engineering, School of Chemistry, Federal University of Rio de Janeiro, Rio de Janeiro, 21941909, Brazil
dUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, Lille, France
eUniversité de Lorraine, CNRS, L2CM UMR 7053, Nancy, F-54000, France. E-mail: souzarod21@gmail.com
First published on 29th October 2024
Abstract
The global imperative to shift towards renewable and sustainable resources has spurred significant interest in exploring and utilizing platform chemicals derived from renewable feedstocks. Among these, levoglucosenone (LGO) and Cyrene™ have emerged as promising candidates. LGO, derived from the pyrolysis of cellulose and hemicellulose, exhibits structural versatility, making it an attractive starting material for various valuable products. Its chemical transformations can yield a diverse array of derivatives, including levulinic acid, furan derivatives, and intermediates for pharmaceutical and agrochemical synthesis, as well as bio-based materials such as bioplastics and resins. Cyrene™, produced through the hydrogenation of LGO, serves as a renewable, biodegradable, and non-toxic dipolar aprotic solvent, offering sustainability advantages for green chemistry applications. Herein we report our results on the continuous-flow cascade transformation of LGO into Cyrene™ and then (R)-γ-carboxy-γ-butyrolactone in good yields with an additional mechanistic investigation for the Baeyer–Villiger oxidation of Cyrene™.
Introduction
The shift toward renewable and sustainable resources is a global imperative. With this in mind, the exploration and utilization of platform chemicals derived from renewable feedstocks have garnered significant interest.1–6 Platform chemicals serve as building blocks for various valuable products, ranging from pharmaceuticals to advanced materials. Among the emerging stars we can find levoglucosenone (LGO, 1) and Cyrene™, two bio-based compounds with great potential. LGO (1) is derived from the pyrolysis of cellulose and hemicellulose, two abundant polysaccharides found in biomass.7–10 One of the key characteristics that makes LGO (1) an attractive starting material is its structural versatility, being able to be readily modified to yield a diverse array of derivatives.11–13 This versatility opens up a wide range of synthetic pathways, allowing for the production of various value-added products. The chemical transformations of LGO (1) can lead to the synthesis of valuable chemicals such as levulinic acid and its esters, furan derivatives, and a variety of intermediates for pharmaceutical and agrochemical synthesis.4,8,11,14,15 Additionally, LGO (1) has been explored as a platform for the production of bio-based materials, including bioplastics and resins.9 Its ability to undergo selective chemical transformations while maintaining its structural integrity makes LGO (1) an excellent candidate for sustainable chemical processes. Cyrene™ can be produced by hydrogenation of LGO (1), and it has been used recently as a bio-based dipolar aprotic solvent derived from renewable feedstock.15 Furthermore, Cyrene™ is biodegradable and non-toxic, making it a sustainable option for various chemical processes. The unique properties of Cyrene™ (2) make it suitable for a wide range of applications, positioning it as a promising solvent for green chemistry applications.15–19Among the possibilities, the synthesis of (R)-γ-carboxy-γ-butyrolactone (4), which is an intermediate for the synthesis of substituted lactones with potential biological activity is of growing interest.19–22 Herein we report our efforts on (R)-γ-carboxy-γ-butyrolactone (4) synthesis starting from LGO (1). Our group has envisioned a strategic approach that begins with continuous-flow hydrogenation of levoglucosenone (LGO, 1), resulting in the production of Cyrene™ (2). Subsequently, a cascade continuous-flow Baeyer–Villiger/oxidation protocol is employed, culminating in the production of (R)-γ-carboxy-γ-butyrolactone (4) (Scheme 1). The Baeyer–Villiger reaction of Cyrene™ (2) has been previously documented by Bonneau and colleagues.18 However, the unusual product from this transformation prompted us to undertake a theoretical investigation of the reaction mechanism.18 This study was conducted to gain a deeper understanding of this reaction for further developments.
 | ||
| Scheme 1 (R)-γ-Carboxy-γ-butyrolactone (4) synthesis starting from levoglucosenone (LGO, 1). | ||
Results and discussion
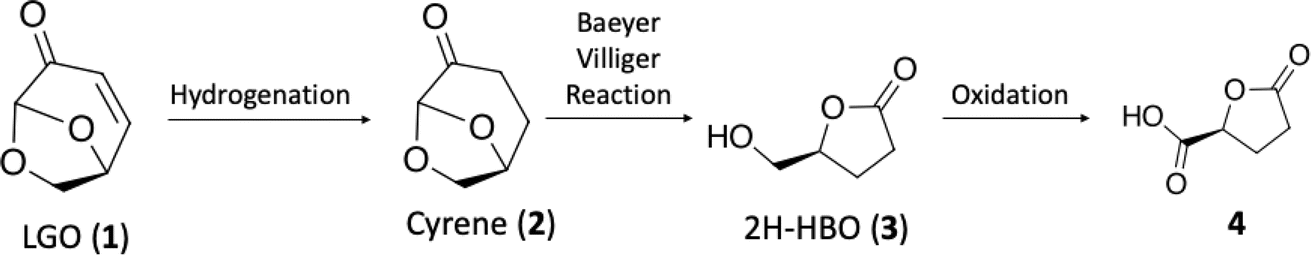
The research started with the optimization of LGO (1) hydrogenation under continuous-flow conditions. To accomplish this, we employed the H-Cube Mini reactor (see Fig. S1 at ESI†) was employed, which generates in situ hydrogen via water electrolysis, ensuring a safe operational environment for process optimization. For the initial screening, the concentration of LGO (1) in the reaction solution was kept at 0.1 M, and the impact of reaction variables like temperature, residence time, and reaction selectivity was evaluated to identify the most favorable reaction conditions (Table 1).
|
|||||
|---|---|---|---|---|---|
| Entry | T (°C) | Rt (sec) | Conv. (%)b | Selectivity (%)b | |
| Cyrene™ (2) | 5 | ||||
| a Reaction conditions: A solution of levoglucosenone (LGO, 1) in water (0.1 M), 250 mg of Pd/C 10% catalyst cartridge (Thales Nano), 200 μL internal volume, 15–30 bar H2.b Conversion (%) and selectivity (%) values based on GC/FID absolute area analysis (chromatographic profile: Fig. S4 to S9, see ESI). | |||||
| 1 | 40 | 100 | >99 | >99 | — |
| 2 | 50 | >99 | >99 | — | |
| 3 | 60 | 100 | >99 | 95 | 5 |
| 4 | 50 | >99 | 87 | 13 | |
| 5 | 80 | 100 | >99 | 40 | 60 |
| 6 | 50 | >99 | 50 | 50 | |
The condition with maximized Cyrene™ (2) production were observed at 40 °C and a flow rate of 1.0 mL min−1 (entry 2, Table 1). Under these conditions, Cyrene™ (2) was produced with a selectivity of 99% after only 50 seconds (entry 2, Table 1). When the temperature was raised to 60 and 80 °C, the formation of a single side-product was observed, and its appearance was greatly affected by increasing temperatures (entries 3–6, Table 1). The single side product observed was characterized as the 1,4-addition of water product 5, previously reported by other groups,23 and NMR spectroscopy (Fig. S37–S40 in the ESI†) has confirmed the chemical structure.
Further reduction of the residence time at 40 °C did not permit to reach full conversion limiting the reaction to a 50 seconds reaction. The limiting concentration that could be used for this reaction has also been evaluated and reproducible results (>99% conversion and selectivity) were achieved until 1.5 M at 40 °C and 100 seconds residence time.
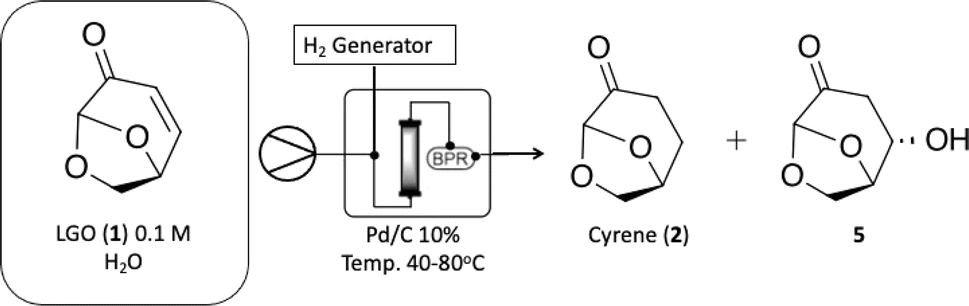
With Cyrene™ (2) in hands we proceeded to the Baeyer–Villiger protocol developed by Bonneau and co-workers.18 While the Baeyer–Villiger protocol has been efficiently optimized under batch conditions, its translation to a continuous-flow system necessitates the adaptation of specific reaction parameters, especially reaction temperature, to fully leverage the potential benefits of process intensification. We initially focused on increasing reaction temperature over different residence times to observe the effect on reaction conversion. Then we tested the limiting concentration of the starting material (Table 2). In batch conditions, the Baeyer–Villiger protocol employs a three-temperature setup, beginning with the addition of hydrogen peroxide at 0 °C, followed by a temperature increase to 50 °C for lactone formation followed by another temperature increase to 90 °C for the decomposition of any remaining H2O2 and deformylation of the final intermediate towards 2H-HBO (3) (see batch conditions Section 1.1 in the ESI†). Due to the rapid heat transfer characteristic of continuous-flow reactors, we opted to forego the three-temperature approach, as depicted on Table 2.
|
|||
|---|---|---|---|
| Entry | Rt (min) | T (°C) | Conv. (%)b |
| a Reaction conditions: solution A: Cyrene™ (4.61 mL, 45 mmol) in 2.84 mL of H2O (6.04 M), solution B: H2O2 30% (v/v) (5.57 mL, 54 mmol, 1.2 eq) in 0.715 mL of H2O (8 M). The solutions were pumped with a Syrris® syringe pump, mixed in a T connector, and passed through a 12 mL PTFE coil reactor with 1/16′ inner diameter, the line was pressurized with a back pressure regulator set at 4.0 bar.b Analysis by CG/FID absolute area (chromatographic profile: Fig. S11–S18, at ESI). | |||
| 1 | 30 | 100 | 99 |
| 2 | 15 | 99 | |
| 3 | 7.5 | 94 | |
| 4 | 3.75 | 72 | |
| 5 | 15 | 150 | 99 |
| 6 | 7.5 | 99 | |
| 7 | 3.75 | 99 | |
| 8 | 5 | 99 | |
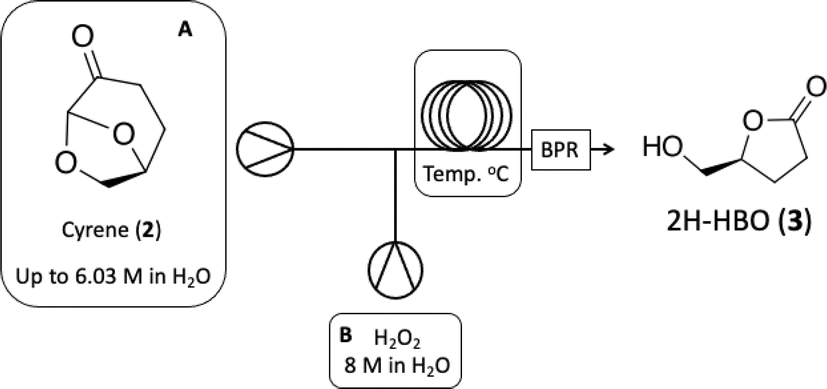
Table 2 demonstrates that at 100 °C, 7.5 and 3.75 minutes of residence times can result in moderate to good conversions (entries 3 and 4). A further increase in reaction temperature can enhance the reaction conversion at 3.75 minutes of residence time and increase reaction concentration to 6.03 M without sacrificing efficiency (entries 7 and 8). Product 3 can be easily isolated without compromising reaction yield. At 100 °C, where we found 72% conversion, we could identify the final formylated intermediate (FBO) as a single by-product. While the FBO intermediate is known in the literature,18 no mechanistic investigation has been conducted to shed light on this unusual Baeyer–Villiger reaction.
The next step involves the oxidation of 2H-HBO (3) to form a carboxylic acid derivative 4 (see batch conditions Section 1.2 in the ESI†). To optimize this step, we chose to use the crude reaction product from the previous step to mimic the reaction contents as closely as possible, facilitating the implementation of a cascade reaction. First, we decided to explore the residence time based on a standard protocol that uses TEMPO (0.075 eq), NaBr (0.8 eq), and NaOCl (1.5 eq) in order to optimize the production of 4. Results are presented in Table 3.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
||
|---|---|---|
| Entry | Rt (min) | Conv. 4 (%)b |
| a Reaction conditions: solution A: 2H-HBO (3) (610 μL, 6.46 mmol) in 2 mL of H2O (3.2 M); solution B: TEMPO (0.075 g, 0.075 eq), NaBr (0.531 g, 0.8 eq), NaClO (650 μL, 1.5 eq), 40 μL of NaOH solution 1 M in 2 mL of H2O (pH: 10). The solutions were pumped with a Syrris® syringe pump, mixed in a T connector and passed through a 12 mL coil reactor with 1/16′ inner diameter a temperature of 25 °C, the line was pressurized with back pressure regulator set at 4.0 bar.b Analysis by CG/FID absolute area (chromatographic profile: Fig. S20–S24, at ESI). | ||
| 1 | 60 | >99 |
| 2 | 30 | >99 |
| 3 | 20 | 84 |
| 4 | 15 | 68 |
| 5 | 5 | 31 |
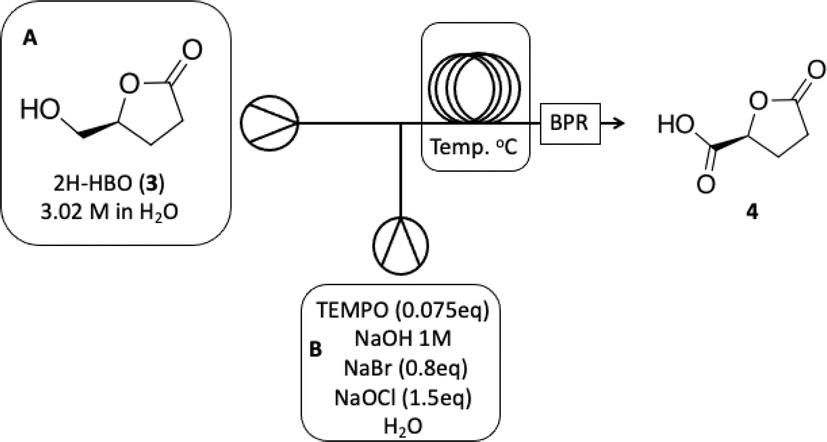
Based on this initial evaluation, we could observe that short residence times could be achieved by standard TEMPO oxidation protocol, where after only 20 minutes, a good conversion could still be obtained (entries 1–3, Table 3). Further decrease in the residence time has a dramatic effect on reaction conversion (entries 4 and 5, Table 3). Although 30 min of residence time provided excellent conversion, we decided to optimize the reaction conditions, keeping the residence time at 20 minutes and improving the reaction efficiency through this operational window. The oxidation step was optimized based on the number of equivalents of TEMPO, NaBr, and NaOCl. Results are presented in Table 4.
| Entry | TEMPO (eq) | NaBr (eq) | NaOCl (eq) | Conv. 4 (%)b |
|---|---|---|---|---|
| a Reaction conditions: solution A: 2H-HBO (3) (610 μL, 6.46 mmol) in 2 mL of H2O (3.2 M); solution B: TEMPO (0.075 g, 0.075 eq), NaBr (0.531 g, 0.8 eq), NaClO (866 μL, 2.0 eq), 40 μL of NaOH solution 1 M in 2 mL of H2O (pH: 10). The solutions were pumped with a Syrris® syringe pump, mixed in a T connector and passed through a 12 mL PTFE coil reactor with 1/16′ inner diameter a temperature of 25 °C with a residence time of 20 minutes, the line was pressurized with a back pressure regulator set at 4.0 bar.b Analysis by CG/FID absolute area (chromatographic profile: Fig. S25–S31, at ESI). | ||||
| 1 | 0.075 | 0.8 | 1.5 | 84 |
| 2 | 0.075 | 0.8 | 2.0 | >99 |
| 3 | 0.11 | 0.8 | 1.5 | 93 |
| 4 | 0.04 | 0.8 | 1.5 | 84 |
| 5 | 0.075 | 1.2 | 1.5 | 66 |
| 6 | 0.075 | 0.8 | 0.7 | 68 |
| 7 | 0.05 | 1 | 2 | 78 |
As observed in Table 4, decreasing the amount of TEMPO or/and increasing the number of equivalents of NaBr has a deleterious effect on reaction conversion (entries 4, 5 and 7, Table 4), as well as the reduction in NaOCl equivalents (entry 6, Table 4). In our experiments, we were able to note that a slightly increase in the amount of NaOCl, from 1.5 to 2.0 equivalents, led us from 84% of conversion to >99% (entries 1 and 2, Table 4). It is important to note that the increased amount of TEMPO did not have the same effect where since conversion has raised to 93% (entry 3, Table 4). Isolation of the product to establish the isolated yield was performed as mentioned in the experimental section (see Experimental section). The optimized conditions obtained for the oxidation step were then used to couple both steps on a cascade protocol (Scheme 2).
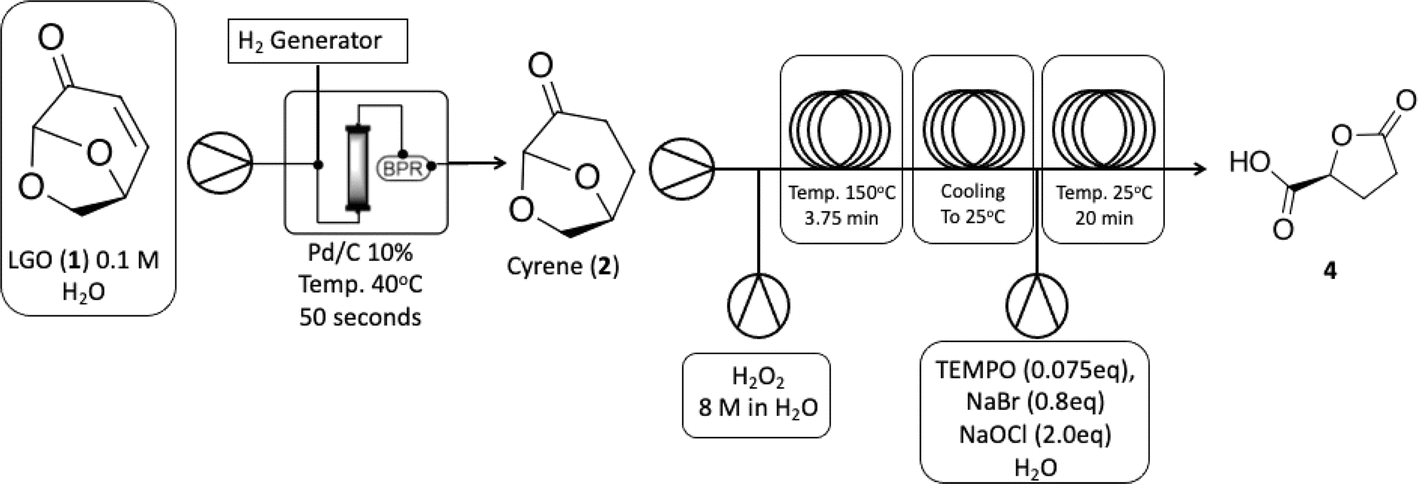 | ||
| Scheme 2 Continuous-flow synthesis of carboxylic acid 4 starting from LGO (1). | ||
We initiated the process by producing Cyrene™ (2) via hydrogenation of LGO (1) under mild conditions (using water as a solvent at 40 °C) and a very short residence time. Under these conditions, we achieved complete selectivity for the desired product. However, the first step is limited by the use of 0.1 M of LGO (1), whereas steps 2 (Baeyer–Villiger) and 3 (oxidation) can be conducted at significantly higher concentrations, up to 6.03 M. This disparity directly impacts the space-time-yield of the process. Subsequently, a 6.03 M solution of the synthesized Cyrene™ (2) was pumped through the cascade reactor. Initially, it reacted with H2O2 at 150 °C via the Baeyer–Villiger protocol to produce 2H-HBO (3), which then was cooled down in a heat exchange coil to react with the oxidation reagents and deliver carboxylic acid 4 in quantitative isolated yields, the whole cascade process led to a productivity of 5.11 g h−1.
Aiming to better understand this unusual Baeyer–Villiger reaction, we investigated the reaction mechanism both by DFT calculations (IEFPCM(H2O)/M06-2X/6-311++G(d,p)) and some control experiments (see Computational details for further information). In addition to the unusual Baeyer–Villiger rearrangement, the Cyrene™ (2) was also reported to participate in an abnormal Beckmann rearrangement, yielding a nitrile as the major product, instead of the usual amide.24 In the case of this abnormal Beckmann rearrangement, such behavior is reported when a carbocation stabilizing group is present anti to the departing leaving group. We hypothesized that such a stabilizing effect may also be the reason for the observed lactone formation in the BV reaction of Cyrene™ (2). This mechanistic rationale is depicted in Scheme 3.
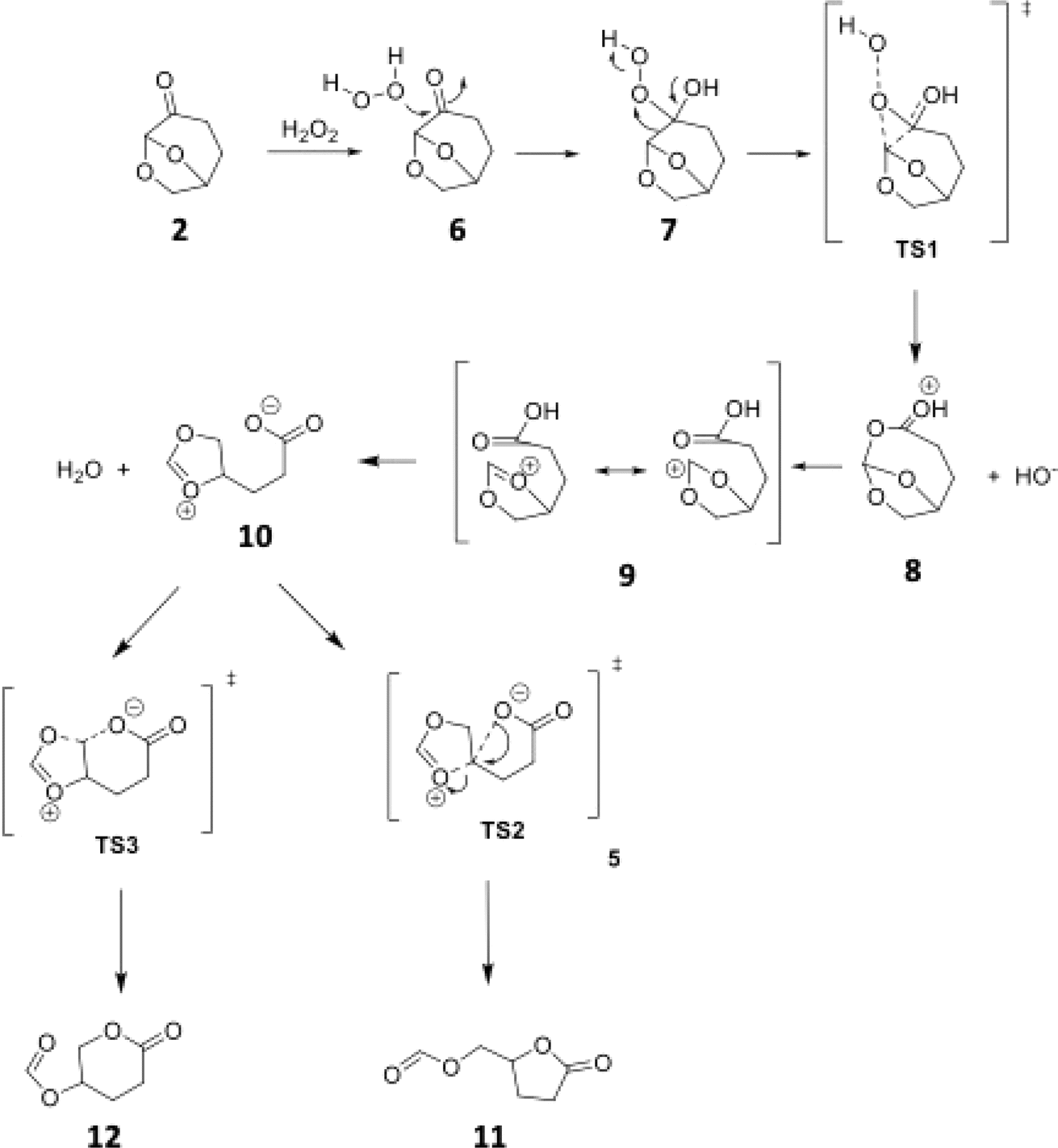 | ||
| Scheme 3 Pictorial representation of a mechanistic proposal for the Baeyer–Villiger rearrangement of Cyrene™ (2). | ||
In this mechanistic hypothesis, after the classical Baeyer–Villiger step, the presence of an acetal adjacent to the ester would stabilize the carbocation, which leads to the carboxylate intermediate 10, resulting then in the observed product after a 5-exo-tet cyclization of 10.
To theoretically evaluate this hypothesis, the geometries of reactant, intermediates, transition states, and products were optimized. Neutral and protonated reactants were considered in the calculations, as well as the explicit solvation of water molecules.
According to the theoretical model used to evaluate the reaction coordinate, under neutral conditions, the product of the addition of hydrogen peroxide to Cyrene™ is a slightly exergonic process when compared to the pre-association complex 6 between hydrogen peroxide and Cyrene™. Interestingly, intermediate 7 leads to intermediate carboxylate 10 in a highly exergonic process that does not pass through intermediate 8 but through 9. Such a sequence of events represents a Grob-like fragmentation24,25 of the addition intermediate 7 instead of the classical Bayer–Villiger mechanism which would lead to 8. From the kinetic point of view, the most significant barrier is the one related to the ring-opening step with a reaction barrier (ΔGTS1≠) of 34.2 kcal mol−1. In this transition state (TS2) the interatomic distance between the carbocation carbon and the carbonyl carbon is 2.009 Å (Fig. 3). The observed lactone product 11 is then derived from intermediate 10 through a 5-exo-tet cyclization (evidence by NMR 1H spectra in Fig. S44†), kinetically favored over the concurrent 6-exo-tet cyclized product 12 (Fig. 1).
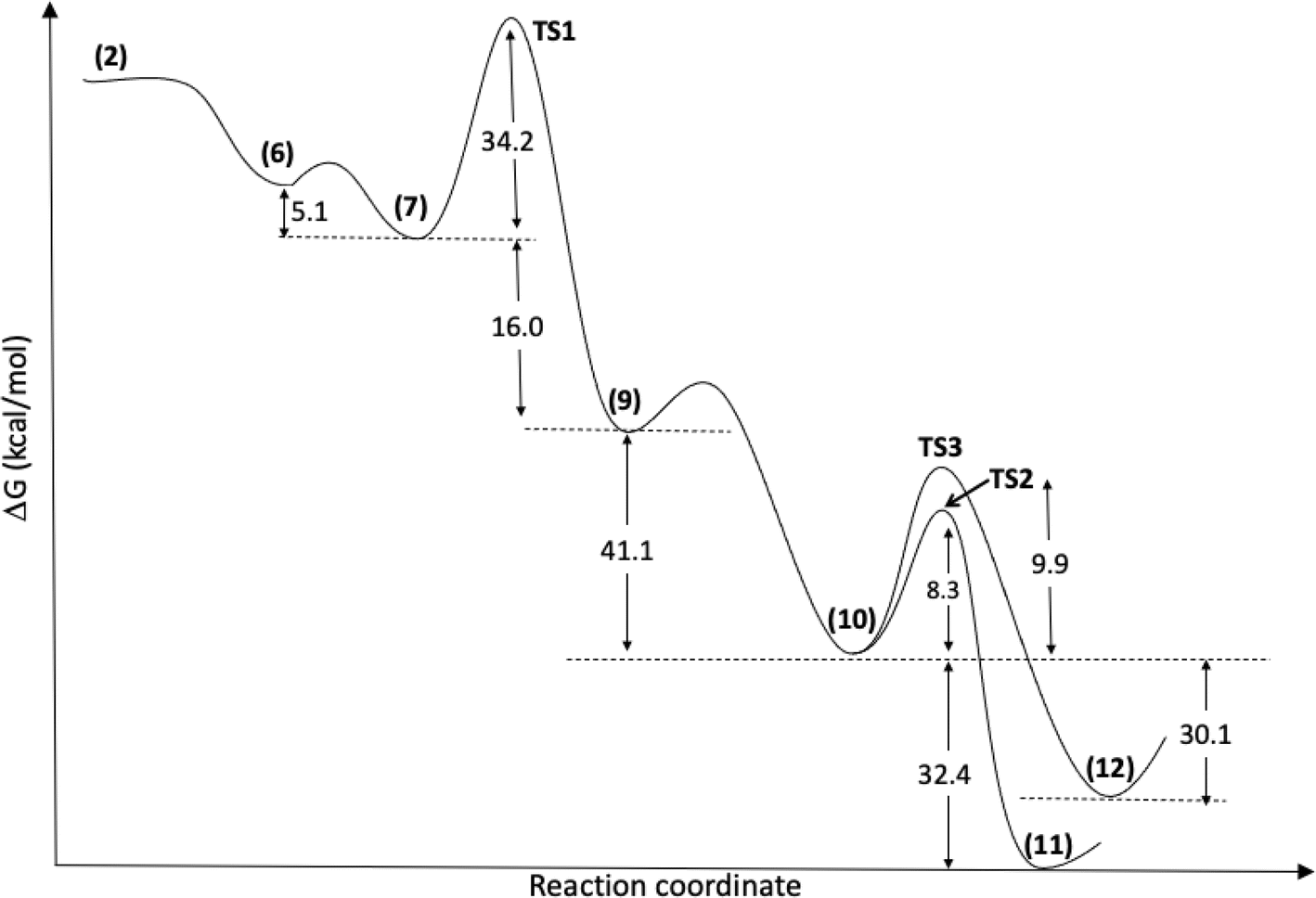 | ||
| Fig. 1 Reaction mechanism profile, with optimized geometries at IEFPCM(H2O)/M06-2X/6-311++G(d,p) level, for the species shown in Scheme 3. | ||
The influence of acid catalysis and explicit solvent assistance effects on the fragmentation step was then investigated. The presence of an acid leads to the elimination of water instead of the hydroxide anion, dramatically decreasing the reaction barrier, with a reaction barrier (ΔG≠TS1) of only 1.1 kcal mol−1, with a much earlier transition state, Fig. 2b. The presence of explicit solvation water molecules also impacts this step, however, in different directions in the acid-catalyzed and non-catalyzed reactions. The presence of explicit solvent molecules in the non-catalyzed process decreases the reaction barrier of TS1 to ΔGTS1≠ = 30.9 kcal mol−1), probably through solvation (and stabilization) of the hydroxide leaving group, which increases its nucleofugacity, Fig. 2a. The interaction of the explicit solvent molecules also impacts the acid-catalyzed reaction, where the reaction barrier is now increased from ΔGTS1≠ = 25.9 kcal mol−1 to ΔGTS1≠ of 12.7 kcal mol−1, a result of the increased stabilization of the intermediate 3 due to solvation, Fig. 2c. These reaction barriers in the presence of explicit solvent molecules represent a more realistic scenario once a reaction barrier of only 1.1 kcal mol−1 is highly unlikely.
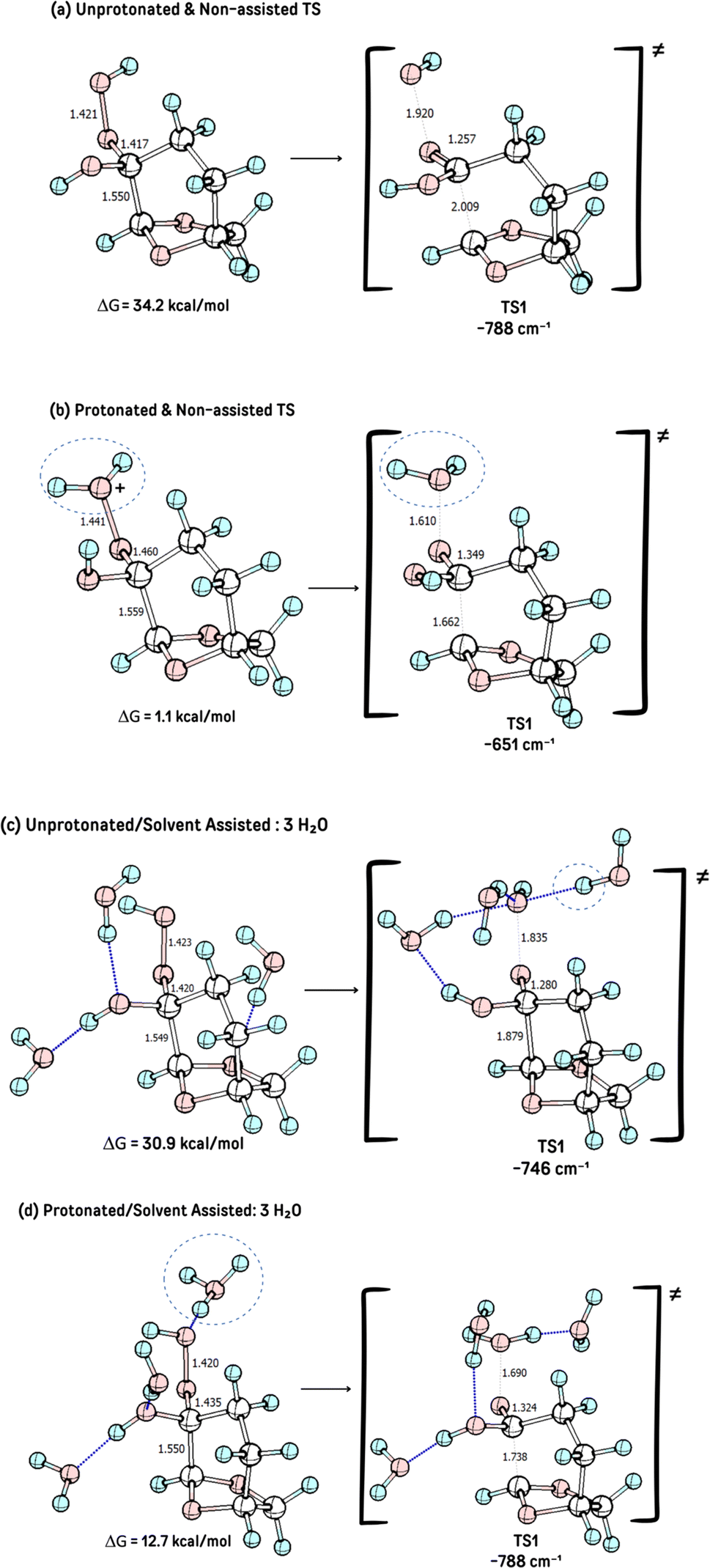 | ||
| Fig. 2 The TS1 for structures for the different transitions stated studied: (a) uprotonated/non-asssited, (b) acid-catalyzed/non-assisted, (c) unprotonated/non-assisted and (d) acid-catalyzed/solvent-assisted (3H2O). | ||
The IBO analysis26,27 of the rearrangement step shows that the C–C bond density flows in order to displace the leaving group, confirming the BV nature of the transformation. However, the reactive intermediate, an oxygen-stabilized carbocation (or oxonium ion), is formed and can undergo internal rotations that afford the different products, formed by an intramolecular substitution, Fig. 3.
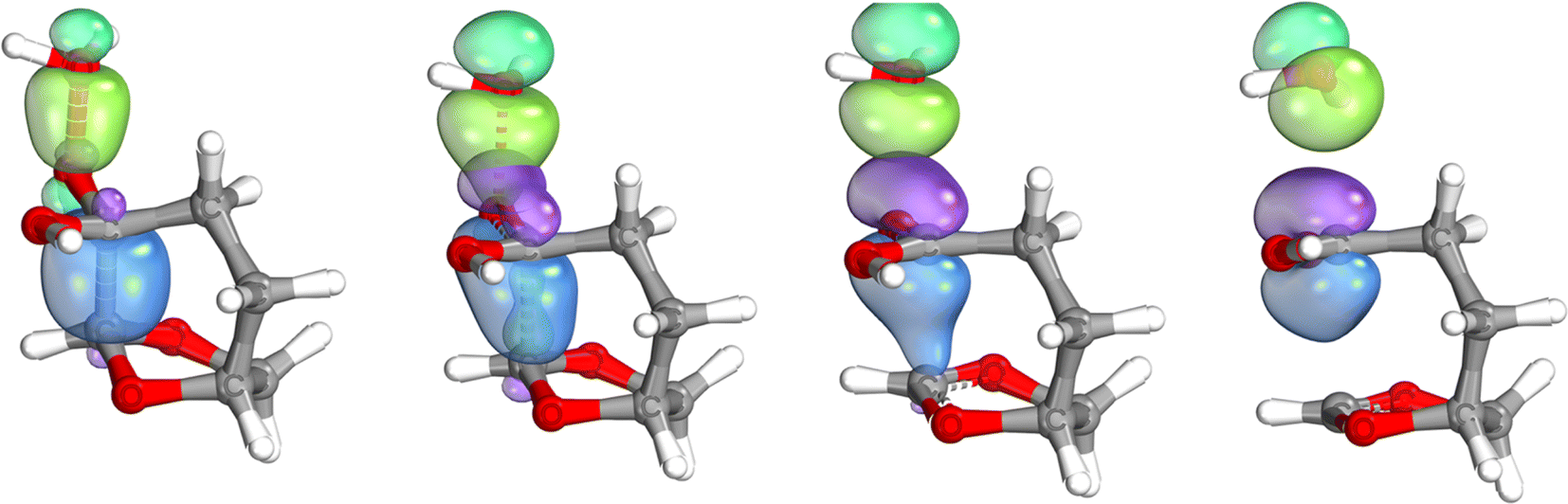 | ||
| Fig. 3 IBO analysis of the BV step, showing the progressive flow of electron density from the CC bond (blue) to the oxygen group from the leaving OH. | ||
In conclusion, we have optimized two crucial steps in the synthesis of (R)-γ-carboxy-γ-butyrolactone (4), beginning with the hydrogenation of levoglucosenone (LGO, 1) to produce Cyrene™ (2), followed by the Baeyer–Villiger (BV) protocol and subsequent oxidation steps. The hydrogenation step, employing continuous-flow conditions with the H-Cube Mini reactor achieved a remarkable 99% selectivity towards the desired product after only 50 seconds. Upscale results were attained to 1.5 M concentration at 40 °C and 50 seconds residence time, providing valuable insights for scalability. Regarding the Baeyer–Villiger protocol, increasing reaction temperature to expedite ester hydrolysis, led us to achieve moderate to good conversions at 100 °C with varying residence times and concentrations. In the final oxidation step, we optimized conditions based on the TEMPO oxidation protocol, adjusting the equivalents of reagents to maximize conversion. By balancing reagent ratios, we achieved over 99% conversion, highlighting the importance of fine-tuning reaction parameters for optimal outcomes.
Furthermore, our mechanistic investigations shed light on the unusual Baeyer–Villiger rearrangement observed during 2H-HBO (3) synthesis, providing valuable insights into the mechanism for product formation. Through computational modeling, we proposed a mechanism for the formation of lactone product 3, underscoring the significance of understanding reaction mechanisms for rational design and optimization.
Material and methods
Materials
All reagents and solvents, were purchased from commercial sources and used without further purification. Cyrene was purchased from Sigma Aldrich, and hydrogen peroxide 30% was purchased from ISOFAR Indústria e Processos Química.Flow synthesis was performed using a 3-module setup: a syringe pump utilizing a set of 1 and 0.5 mL syringes; a PTFE coil reactor with 12 mL volume and 1/8′′ OD; and a back pressure regulator.
NMR spectra were recorded on a Bruker 400 MHz spectrometer in a deuterated solvent. The values of chemical shifts (δ) are expressed in ppm with reference to tetramethylsilane (TMS) for 1H NMR and chloroform signal for 13C NMR. Coupling constants (J) are expressed in hertz (Hz). Gas chromatography analyses were carried out in GC-MS using Shimadzu GC 2010 (gas chromatography/flame-ionization detection/helium was used as carrier gas) and GC/FID using Shimadzu GC 2010 – DB5 HT column, 30 m, with automatic injector.
Gas chromatography GC/FID analysis
Injector at 310 °C, 20.0 split ratio, pressure: 72.3 kPa, column flow: 1.76 mL min−1. The oven starts at 60 °C and then increases to 230 °C at a 15 °C min−1 rate. Flame temperature: 380 °C. Analysis preparation: To 30 μL of analyte was added 500 μL of CHCl3. Then, in a 1.5 mL flask, an aliquot of 20 μL of the initial solution and 980 μL of CHCl3 was added.Gas chromatography-mass spectrometry (GC-MS) analysis
Gas chromatography analyses were obtained on a Shimadzu GC-2010 MS with a DB-5 column (30 m × 0.25 mm × 0.25 μm) (Agilent Technologies). Injection temperature 280 °C, injection split ratio 1:50, helium (99.9992% purity) was used as carrier gas at 2 mL min−1 in constant flow mode, pressure 92.0 kPa, column flow 1.09 mL min−1. The oven temperature setting was: 50 °C and heated at 10 °C min−1 to 250 °C. Solvent cut time 2 minutes. Mass spectra were obtained in scan mode (45−800 Da).
Experimental procedure
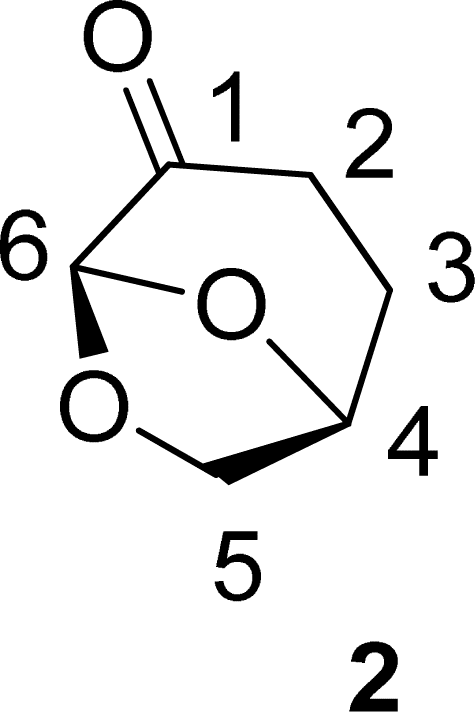 . Continuous-flow experiments were performed in a high-pressure continuous-flow hydrogenator reactor H-Cube® Mini Plus (ThalesNano; see Fig. S1 in the ESI†). Levoglucosenone (LGO, 1) solutions in water (0.1 and 1.5 M) were pumped through the reactor module containing a 70 mm catalyst cartridge filled with 250 mg of 10% Pd/C catalyst. The total flow rates were 0.5, 1.0, and 2.3 mL min−1, corresponding to residence times of 100, 50, and 20 s, respectively. Reaction temperatures of 40, 60, and 80 °C were investigated. The system pressure varied depending on the chosen temperature and flow rate, ranging from 15 to 30 bar. Conversion (%) and selectivity (%) were monitored using GC-MS (see ESI, Fig. S4 and S9†), considering the absolute peak areas. IR νmax (KBr) cm−1: 2969, 1742, 1419, 1286, 1245, 1110, 986, 882, 800. 1H NMR (500 MHz, CDCl3) δ 5.11 (s, H6), 4.71 (d, J = 3.4 Hz, H5b), 4.05 (dd, J = 7.4, 0.6 Hz, H5a), 3.99–3.95 (m, H4), 2.66 (ddd, J = 16.4, 11.6, 8.6 Hz, H2a), 2.40 (ddd, J = 16.0, 4.9, 3.9 Hz, H3a), 2.36–2.27 (m, H2b), 2.06–2.00 (m, H3b). 13C NMR (126 MHz, CDCl3) δ 200.38 (s, C1), 101.54 (s, C6), 73.18 (s, C4), 67.59 (s, C5), 31.15 (s, C4), 29.92 (s, C3). The IR, 1H and 13C NMR can be seen at ESI, Fig. S34, S35 and S36,† respectively.
. Continuous-flow experiments were performed in a high-pressure continuous-flow hydrogenator reactor H-Cube® Mini Plus (ThalesNano; see Fig. S1 in the ESI†). Levoglucosenone (LGO, 1) solutions in water (0.1 and 1.5 M) were pumped through the reactor module containing a 70 mm catalyst cartridge filled with 250 mg of 10% Pd/C catalyst. The total flow rates were 0.5, 1.0, and 2.3 mL min−1, corresponding to residence times of 100, 50, and 20 s, respectively. Reaction temperatures of 40, 60, and 80 °C were investigated. The system pressure varied depending on the chosen temperature and flow rate, ranging from 15 to 30 bar. Conversion (%) and selectivity (%) were monitored using GC-MS (see ESI, Fig. S4 and S9†), considering the absolute peak areas. IR νmax (KBr) cm−1: 2969, 1742, 1419, 1286, 1245, 1110, 986, 882, 800. 1H NMR (500 MHz, CDCl3) δ 5.11 (s, H6), 4.71 (d, J = 3.4 Hz, H5b), 4.05 (dd, J = 7.4, 0.6 Hz, H5a), 3.99–3.95 (m, H4), 2.66 (ddd, J = 16.4, 11.6, 8.6 Hz, H2a), 2.40 (ddd, J = 16.0, 4.9, 3.9 Hz, H3a), 2.36–2.27 (m, H2b), 2.06–2.00 (m, H3b). 13C NMR (126 MHz, CDCl3) δ 200.38 (s, C1), 101.54 (s, C6), 73.18 (s, C4), 67.59 (s, C5), 31.15 (s, C4), 29.92 (s, C3). The IR, 1H and 13C NMR can be seen at ESI, Fig. S34, S35 and S36,† respectively.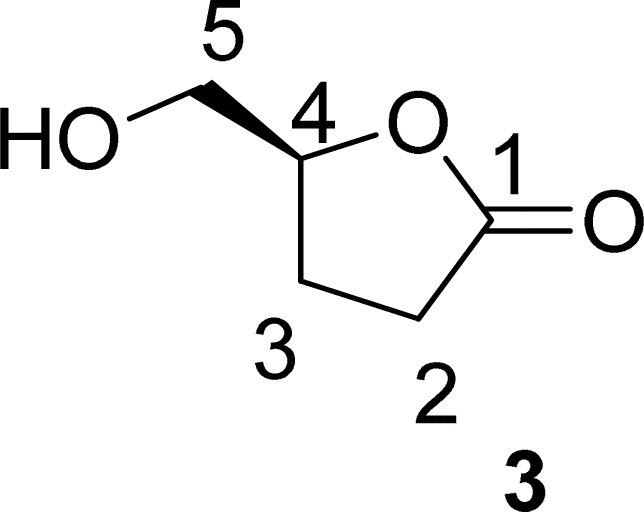 . Solution A: Cyrene™ (4.61 mL, 45 mmol) in 2.84 mL of H2O (6.04 M), solution B: H2O2 30% (v/v) (5.57 mL, 54 mmol, 1.2 eq) in 0.715 mL of H2O (8 M). The solutions were pumped with a Syrris® syringe pump, mixed in a T connector, and passed through a 12 mL coil reactor with 1/16′ inner diameter, the line was pressurized with a back pressure regulator set at 4.0 bar. At the end of the reaction, the reaction medium was distilled under reduced pressure, obtaining a colorless oil with a yield of >99% and purity >99%, analyzed by GC/FID (see ESI, Fig. S11–S18†), considering the absolute peak areas. IR νmax (KBr) cm−1: 3382, 1754, 1640, 1364, 1198, 1062. 1H NMR (400 MHz, CDCl3) δ 4.59 (tdd, J = 7.4, 4.6, 2.8 Hz, H4), 3.86 (dd, J = 12.5, 2.8 Hz, H5a), 3.64–3.55 (m, H5a), 2.89 (s, H6), 2.64–2.45 (m, H2), 2.27–2.04 (m, H3). 13C NMR (126 MHz, CDCl3) δ 177.84 (C
. Solution A: Cyrene™ (4.61 mL, 45 mmol) in 2.84 mL of H2O (6.04 M), solution B: H2O2 30% (v/v) (5.57 mL, 54 mmol, 1.2 eq) in 0.715 mL of H2O (8 M). The solutions were pumped with a Syrris® syringe pump, mixed in a T connector, and passed through a 12 mL coil reactor with 1/16′ inner diameter, the line was pressurized with a back pressure regulator set at 4.0 bar. At the end of the reaction, the reaction medium was distilled under reduced pressure, obtaining a colorless oil with a yield of >99% and purity >99%, analyzed by GC/FID (see ESI, Fig. S11–S18†), considering the absolute peak areas. IR νmax (KBr) cm−1: 3382, 1754, 1640, 1364, 1198, 1062. 1H NMR (400 MHz, CDCl3) δ 4.59 (tdd, J = 7.4, 4.6, 2.8 Hz, H4), 3.86 (dd, J = 12.5, 2.8 Hz, H5a), 3.64–3.55 (m, H5a), 2.89 (s, H6), 2.64–2.45 (m, H2), 2.27–2.04 (m, H3). 13C NMR (126 MHz, CDCl3) δ 177.84 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 80.87 (C4), 64.10 (C5), 28.70 (C2), 23.15 (C3). The 1H and 13C NMR can be seen at ESI, Fig. S41, S42 and S43,† respectively.
O), 80.87 (C4), 64.10 (C5), 28.70 (C2), 23.15 (C3). The 1H and 13C NMR can be seen at ESI, Fig. S41, S42 and S43,† respectively.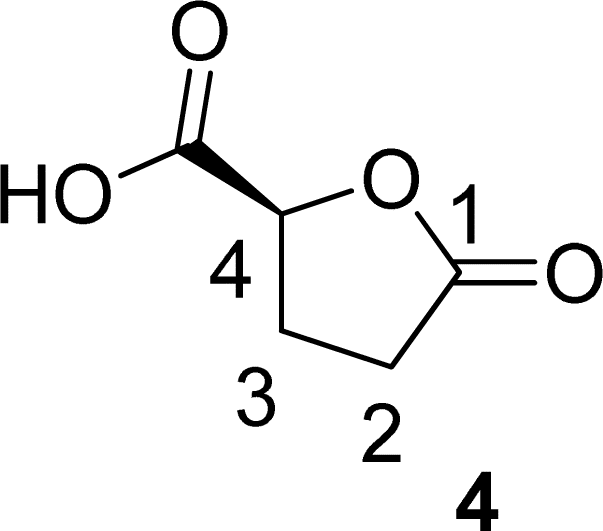 . Solution A: 2H-HBO (610 μL, 6.46 mmol) in 2 mL of H2O (3.2 M); solution B: TEMPO (0.075 g, 0.075 eq), NaBr (0.531 g, 0.8 eq), NaClO (866 μL, 2.0 eq), 40 μL of NaOH solution 1 M in 2 mL of H2O (pH: 10). The solutions were pumped with a Syrris® syringe pump, mixed in a T connector and passed through a 12 mL coil reactor with 1/16′ inner diameter a temperature of 25 °C with a residence time of 20 minutes, the line was pressurized with a back pressure regulator set at 4.0 bar. At the end, methanol (1 mL) was added, followed by a 6 M solution of HCl (60 μL, pH adjustment = 1). Then, 3 mL of methanol was added, and the mixture was distilled under reduced pressure. To the obtained solid, 3 mL of methanol heated at 60 °C during 15 minutes, followed by vacuum filtration to remove inorganic salts. Subsequently, methanol was distilled under reduced pressure, yielding a white solid with a 90% yield. For analysis in GC/FID, 0.01 g of the obtained solid was weighed into a 4 mL vial. 90 μL of MSTFA was added and the mixture was heated at 60 °C for 30 minutes. At the end, 1 mL of DCM was added. An aliquot of 50 μL from this solution was withdrawn and added to a 1.5 mL via L with 0.95 mL of DCM, resulting in purity >99% in absolute area. IR νmax (KBr) cm−1: 3550–3200, 1775, 1723, 1175. 1H NMR (400 MHz, D2O) δ (ppm) δ 2.4–2.6 (4H, m, –CH2–CH2), 4.85–5.15 (H, m, ring-H), 5.1 (H, s, –COOH).
. Solution A: 2H-HBO (610 μL, 6.46 mmol) in 2 mL of H2O (3.2 M); solution B: TEMPO (0.075 g, 0.075 eq), NaBr (0.531 g, 0.8 eq), NaClO (866 μL, 2.0 eq), 40 μL of NaOH solution 1 M in 2 mL of H2O (pH: 10). The solutions were pumped with a Syrris® syringe pump, mixed in a T connector and passed through a 12 mL coil reactor with 1/16′ inner diameter a temperature of 25 °C with a residence time of 20 minutes, the line was pressurized with a back pressure regulator set at 4.0 bar. At the end, methanol (1 mL) was added, followed by a 6 M solution of HCl (60 μL, pH adjustment = 1). Then, 3 mL of methanol was added, and the mixture was distilled under reduced pressure. To the obtained solid, 3 mL of methanol heated at 60 °C during 15 minutes, followed by vacuum filtration to remove inorganic salts. Subsequently, methanol was distilled under reduced pressure, yielding a white solid with a 90% yield. For analysis in GC/FID, 0.01 g of the obtained solid was weighed into a 4 mL vial. 90 μL of MSTFA was added and the mixture was heated at 60 °C for 30 minutes. At the end, 1 mL of DCM was added. An aliquot of 50 μL from this solution was withdrawn and added to a 1.5 mL via L with 0.95 mL of DCM, resulting in purity >99% in absolute area. IR νmax (KBr) cm−1: 3550–3200, 1775, 1723, 1175. 1H NMR (400 MHz, D2O) δ (ppm) δ 2.4–2.6 (4H, m, –CH2–CH2), 4.85–5.15 (H, m, ring-H), 5.1 (H, s, –COOH).Computational details
Quantum chemical calculations were carried out using the Gaussian16 package28 at M06-2X/6-311++G(d,p) level29 considering solvation through the IEFPCM modification30 of the PCM method for simulating solvation in H2O, which is the solvent used experimentally. The optimized geometries of the reagents, the corresponding transition states, intermediates, and products were computed and further characterized by vibrational analysis and are shown in Scheme 4, with selected geometric parameters shown. Reactants, reaction intermediates, and products have no imaginary frequency, while transition states are characterized as having a single imaginary frequency. In this last case, the imaginary will be shown below the figure as vimag. Inspection of the animation the imaginary frequency of each transition state, indicates the correct reaction outcome. Intrinsic reaction coordinate (IRC)31 calculations were employed for the analysis of the transition states related to TS1, to show that the product of the rearrangement is the carbocation.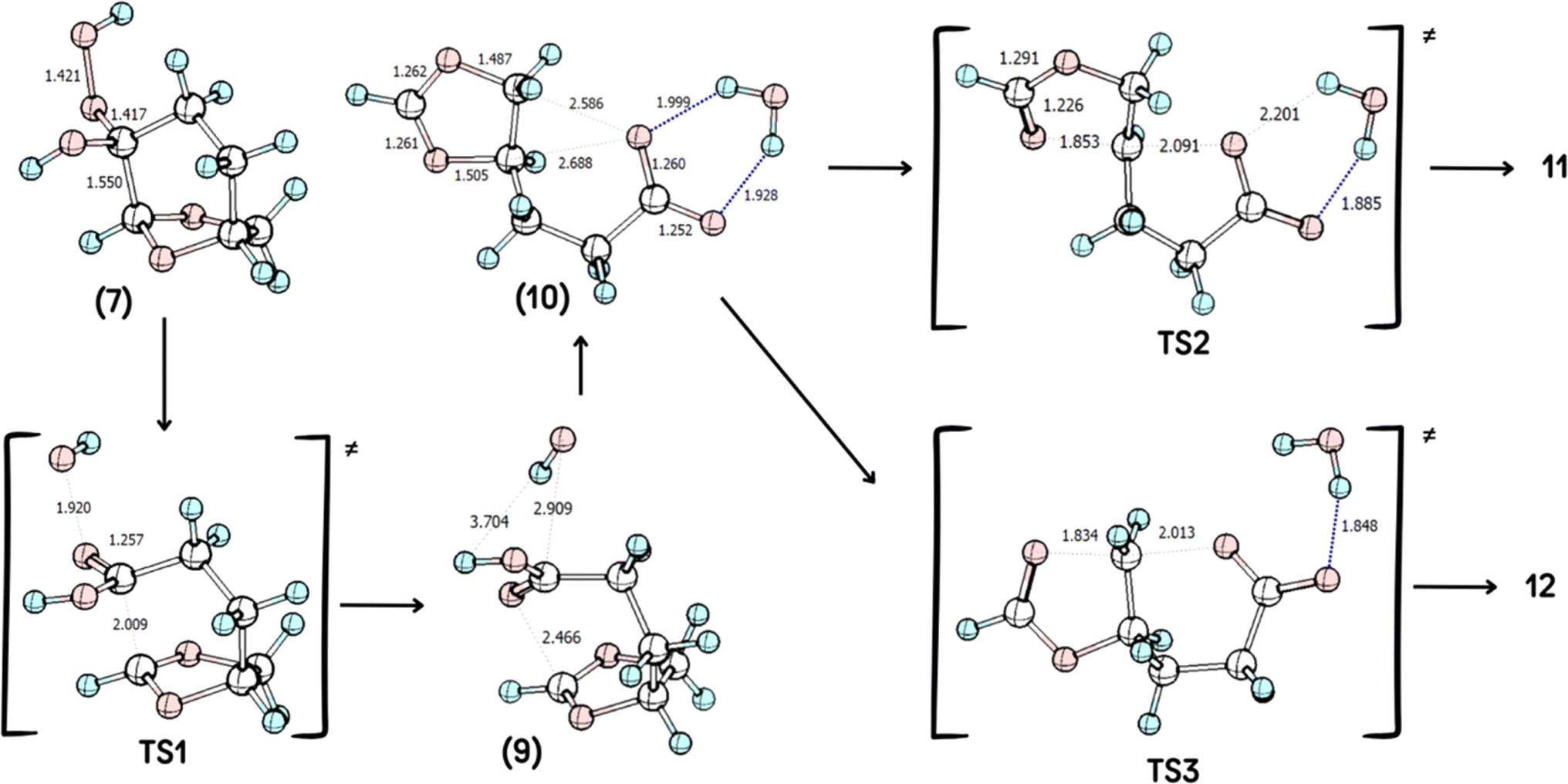 | ||
| Scheme 4 Geometries of the species involved in the reaction, computed at IEFPCM(H2O)/M06-2X/6-311++G(d,p) level. Selected geometric parameters are shown, when appropriate. | ||
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- S. I. Mussatto, I. L. Motta, R. M. Filho, L. Van der Wielen, R. Capaz, J. Seabra, P. Osseweijer, J. Posada, M. F. Gonçalves, P. R. Scorza and G. Dragone, 5.16-Sustainable aviation fuels: production, use and impact on decarbonization, Compr. Renew. Energy, 2022, 348–371 CrossRef.
- D. Carpenter, T. L. Westover, S. Czernik and W. Jablonski, Biomass feedstocks for renewable fuel production: a review of the impacts of feedstock and pretreatment on the yield and product distribution of fast pyrolysis bio-oils and vapors, Green Chem., 2014, 16, 384–406 RSC.
- S. Verma, S. Verma, A. Agrwal and S. Kumar, Use of renewable feedstocks for chemical synthesis, in Green Chemistry Approach Environmental Sustainability, 2024, ch. 11, pp. 219–237 Search PubMed.
- I. Itabaiana Junior, M. A. Do Nascimento, R. O. M. A. De Souza, A. Dufour and R. Wojcieszak, Levoglucosan: a promising platform molecule?, Green Chem., 2020, 22, 5859–5880 RSC.
- K. Y. Sen and S. Baidurah, Renewable biomass feedstocks for production of sustainable biodegradable polymer, Curr. Opin. Green Sustainable Chem., 2021, 27, 100412–100418 CrossRef CAS.
- T. Raj, K. Chandrasekhar, A. N. Kumar and S.-H. Kim, Lignocellulosic biomass as renewable feedstock for biodegradable and recyclable plastics production: a sustainable approach, Renew. Sustainable Energy Rev., 2022, 158, 112130–112152 CrossRef CAS.
- J. H. Clark, M. Bruyn and L. V. Budarin, Method for Producing Levoglucosenone, WO2016170329A1, 2016 Search PubMed.
- F. Cao, T. J. Schwartz, D. J. McClelland, S. H. Krishna, J. A. Dumesica and G. W. Huber, Dehydration of cellulose to levoglucosenone using polar aprotic solvents, Energy Environ. Sci., 2015, 8, 1808–1815 RSC.
- S. Adhikari, H. Nam and J. P. Chakraborty, Conversion of solid wastes to fuels and chemicals through pyrolysis, in Waste Biorefinery: Potential and Perspectives, 2018, ch. 8, pp. 239–263 Search PubMed.
- M. A. Do Nascimento, B. Haber, M. R. B. P. Gomez, R. A. C. Leão, M. Pietrowski, M. Zieliński, R. O. M. A. De Souza, R. Wojcieszak and I. Itabaiana Junior, Optimization of 5-hydroxymethylfurfural oxidation via photo-enzymatic cascade process, Green Chem., 2024, 26, 8211–8219 RSC.
- B. T. Sharipov, A. N. Davydova, L. K. Faizullina and F. A. Valeev, Preparation of the diastereomerically pure 2S-hydroxy derivative of dihydrolevoglucosenone (Cyrene™), Mendeleev Commun., 2019, 29, 200–202 CrossRef CAS.
- L. K. Faizullina, Y. S. Galimova, Y. A. Khalilova, S. M. Salikhov and F. A. Valeev, Aldol-type transformations of levoglucosenone-derived medium-sized keto lactones, Mendeleev Commun., 2018, 28, 482–484 CrossRef CAS.
- V. K. Brel, A. V. Samet, L. D. Konyushkin, A. I. Stash, V. K. Belskyd and V. V. Semenov, Levoglucosenone-derived precursors for the stereoselective synthesis of methylene-expanded analogues of C-nucleosides, Mendeleev Commun., 2015, 25, 44–46 CrossRef CAS.
- L. Qiang, H. Bin, Z. Zhen-Xi, W. Yu-Ting, C. Min-Shu, L. Ding-Jia, D. Chang-Qing and Y. Yong-Ping, Mechanism of cellulose fast pyrolysis: the role of characteristic chain ends and dehydrated units, Combust. Flame, 2018, 198, 267–277 CrossRef.
- F. Diot-Néant, L. Mouterde, S. Fadlallah, S. A. Miller and F. Allais, Sustainable synthesis and polycondensation of levoglucosenone-Cyrene™ based bicyclic diol monomer: access to renewable polyesters, ChemSusChem, 2020, 13, 2613–2620 CrossRef PubMed.
- J. Sherwood, M. De bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunta and J. H. Clark, Dihydrolevoglucosenone (Cyrene™) as a bio-based alternative for dipolar aprotic solvents, Chem. Commun., 2014, 50, 9650–9652 RSC.
- A. R. S. Teixeira, A. L. Flourat, A. A. M. Peru, F. Brunissen and F. Allais, Lipase-catalyzed baeyer-villiger oxidation of cellulose-derived levoglucosenone into (S)-γ-hydroxymethyl-α,β-butenolide: optimization by response surface methodology, Front. Chem., 2016, 4, 1–11 Search PubMed.
- G. Bonneau, A. A. M. Peru, A. L. Flourat and F. Allais, Organic solvent- and catalyst-free Baeyer–Villiger oxidation of levoglucosenone and dihydrolevoglucosenone (Cyrene™): a sustainable route to (S)-γ-hydroxymethyl-α,β-butenolide and (S)-γ-hydroxymethyl-γ-butyrolactone, Green Chem., 2018, 20, 2455–2458 RSC.
- A. Díaz-Rodríguez, Y. S. Sanghvi, S. Fernandéz, R. F. Schinazi, E. A. Theodorakis, M. Ferreroa and V. Gotor, Synthesis and anti-HIV activity of conformationally restricted bicyclic hexahydroisobenzofuran nucleoside analogs, Org. Biomol. Chem., 2009, 7, 1415–1423 RSC.
- T. Ishiguro, H. Mizuguchi, K. Tomioka and K. Koga, Stereoselective reactions. VIII. Stereochemical requirement for the benzylic oxidation of lignan lactone. A highly selective synthesis of the antitumor lignan lactone steganacin by the oxidation of stegane, Chem. Pharm. Bull., 1985, 33, 609–617 CrossRef CAS PubMed.
- D. Enders and M. M. Milovanovic, Asymmetric synthesis of (+)-hinokinin, (+)-dihydrocubebin and cubebin dimethyl ether, a new lignan from phyllanthus niruri, Z. Naturforsch, 2007, 62, 117–120 CrossRef CAS.
- M. S. Miftakhov, F. A. Valeev and I. N. Gaisina, Levoglucosenone: the properties, reactions, and use in fine organic synthesis, Russ. Chem. Rev., 1994, 63, 922–936 CAS.
- L. M. M. Mouterde, F. Allais and J. D. Stewart, Enzymatic reduction of levoglucosenone by an alkene reductase (OYE 2.6): a sustainable metal- and dihydrogen-free access to the bio-based solvent Cyrene®, Green Chem., 2018, 20, 5528–5532 RSC.
- K. Prantz and J. Mulzer, Synthetic applications of the carbonyl generating grob fragmentation, Chem. Rev., 2010, 110, 3741–3766 CrossRef CAS PubMed.
- A. Alhifthi, B. L. Harris, L. Goerigk, J. M. White and S. J. Williams, Structure-reactivity correlations of the abnormal beckmann reaction of dihydrolevoglucosenone oxime, Org. Biomol. Chem., 2017, 15, 10105–10115 RSC.
- G. Knizia and J. E. M. N. Klein, Electron flow in reaction mechanisms—revealed from first principles, Angew. Chem., Int. Ed., 2015, 54, 5518–5522 CrossRef CAS PubMed.
- G. Knizia, Intrinsic atomic orbitals: an unbiased bridge between quantumtheory and chemical concepts, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; G. A. Petersson; H. Nakatsuji; X. Li; M. Caricato; A. V. Marenich; J. Bloino; B. G. Janesko; R. Gomperts; B. Mennucci; H. P. Hratchian; J. V. Ortiz; A. F. Izmaylov; J. L. Sonnenberg; D. Williams-Young; F. Ding; F. Lipparini; F. Egidi; J. Goings; B. Peng; A. Petrone; T. Henderson; D. Ranasinghe; V. G. Zakrzewski; J. Gao; N. Rega; G. Zheng; W. Liang; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; K. Throssell; J. A. Montgomery Jr.; J. E. Peralta; F. Ogliaro; M. J. Bearpark; J. J. Heyd; E. N. Brothers; K. N. Kudin; V. N. Staroverov; T. A. Keith; R. Kobayashi; J. Normand; K. Raghavachari; A. P. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; J. M. Millam; M. Klene; C. Adamo; R. Cammi; J. W. Ochterski; R. L. Martin; K. Morokuma; O. Farkas; J. B. Foresman and D. J. Fox. Gaussiañ16 Revision C.01, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-Class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- E. Cancès, B. Mennucci and J. Tomasi, A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- K. Fukui, The path of chemical reactions – the IRC approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06403h |
| This journal is © The Royal Society of Chemistry 2024 |