
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNewly synthesized sulfonamide derivatives explored for DNA binding, enzyme inhibitory, and cytotoxicity activities: a mixed computational and experimental analyses†
Nasima Arshad *a,
Yasir Mehmooda,
Hammad Ismailb,
Fouzia Perveenc,
Aneela Javedd,
Pervaiz Ali Channare,
Aamer Saeedf,
Sadia Naseemf and
Fatima Naseerc
*a,
Yasir Mehmooda,
Hammad Ismailb,
Fouzia Perveenc,
Aneela Javedd,
Pervaiz Ali Channare,
Aamer Saeedf,
Sadia Naseemf and
Fatima Naseerc
aDepartment of Chemistry, Allama Iqbal Open University, 44000, Islamabad, Pakistan. E-mail: nasimaa2006@yahoo.com; nasima.arshad@aiou.edu.pk
bDepartment of Biochemistry & Biotechnology, University of Gujrat, 50700, Gujrat, Pakistan
cSchool of Interdisciplinary Engineering and Sciences (SINES), National University of Sciences & Technology-NUST, 44000, Islamabad, Pakistan
dHealthcare Biotechnology, Atta-ur-Rehman School of Applied Biosciences, National University of Sciences & Technology-NUST, 44000, Islamabad, Pakistan
eDepartment of Basic Sciences and Humanities, Faculty of Information Science and Humanities, Dawood University of Engineering and Technology, Karachi 74800, Pakistan
fDepartment of Chemistry, Quaid-i-Azam University, 45320, Islamabad, Pakistan
First published on 4th November 2024
Abstract
The current research work reports the synthesis of three 4-((3-arylthiazolo[3,4-d]isoxazol-5-yl)amino)benzene sulfonamide derivatives with a thaizaole(3,4-d)isoxazole-based fused ring heterocyclic system. The synthesized and characterized derivatives, namely, 4-(3-(2-hydroxy-3-methoxyphenyl)thiazolo[3,4-d]soxazole-5-ylamino)benzenesulfonamide (YM-1), 4-(3-(4-chlorophenyl)isoxazolo[3,4-d]thiazol-5-ylamino)benzenesulfonamide (YM-2), and 4-(3-(3-hydroxyphenyl)isoxazolo[3,4-d]thiazol-5-ylamino)benzenesulfonamide (YM-3) were further explored for their binding interactions with DNA and enzymes (urease and carbonic anhydrase). Cytotoxicity of these derivatives for both healthy (HEK-293) and cancerous (MG-U87) cells was determined by MTT analysis. Both experimental (UV-visible, fluorescence, cyclic voltammetry, and viscometry) and theoretical (molecular docking) profiles suggested that these derivatives are good DNA binders. All the derivatives interacted with DNA via mixed intercalative and groove binding interactions. However, the evaluated DNA binding parameters (Kb, ΔG, and n) were comparatively greater for YM-1. Docking data (Kb and ΔG) for binding of these derivatives with enzymes also supported that YM-1 was a comparatively better inhibitor for carbonic anhydrase. However, experimentally evaluated IC50 (1.90 ± 0.02 μM) and % inhibition (57.93%) were found to be greater for YM-2 against urease enzyme. All the derivatives show dose-dependent cytotoxicity (70–90%) against MG-U87 cancer cells. Conversely, only one concentration of YM-1 (120 μM) showed less toxicity (50.28% with IC50 of 1.154 ± 0.317 μM) than that of the positive control (52.22%) for healthy cells. Overall findings suggested sulfonamide derivative YM-1 is a better candidate for DNA binding, enzyme inhibition as well as anticancer activity.
1 Introduction
Sulfonamide is an important class of organic compounds known for its tremendous pharmacological activities. It was the first effective chemotherapeutic agent to be widely used for the treatment of bacterial infection in humans and animals.1 Sulfonamide was first tested in 1932 by Gerhard Johannes Paul Domagk (1895–1964) for treating streptococcal infection and was found to be effective. Sulfonamide is the drug of choice for the treatment of infections caused due to hemolytic Streptococcus, Meningococcus, Ducrey bacillus and Welch bacillus. The therapeutic value of sulfonamide is well established in certain infections, including urinary tract infection and infections in trachoma and lymphogranuloma inguinal.2Sulfonamide is also found to be effective in the treatment of oral disorders. It was reported by Sinclair in 1937 that this drug could be used in the treatment of a necrotic (dry) socket. In June of that year, Raper and Manser confirmed its effectiveness in a postoperative cellulitis case.3 Sulfonamide is the core skeleton for various classes of drugs and exhibits many outstanding biological applications, such as urease inhibitor,4 anticancer agent (1), anti-HIV agent (2), antibacterial agent (3), carbonic anhydrase inhibitor (4), and hypoglycemic agent (5),5 as shown in Fig. 1. Sulfonamide derivatives are also reported for their use in the treatment of certain ailments, including allergies, hypertension, schizophrenia, tumors, inflammations, fungal infections, Covid-19, and tuberculosis.6–9
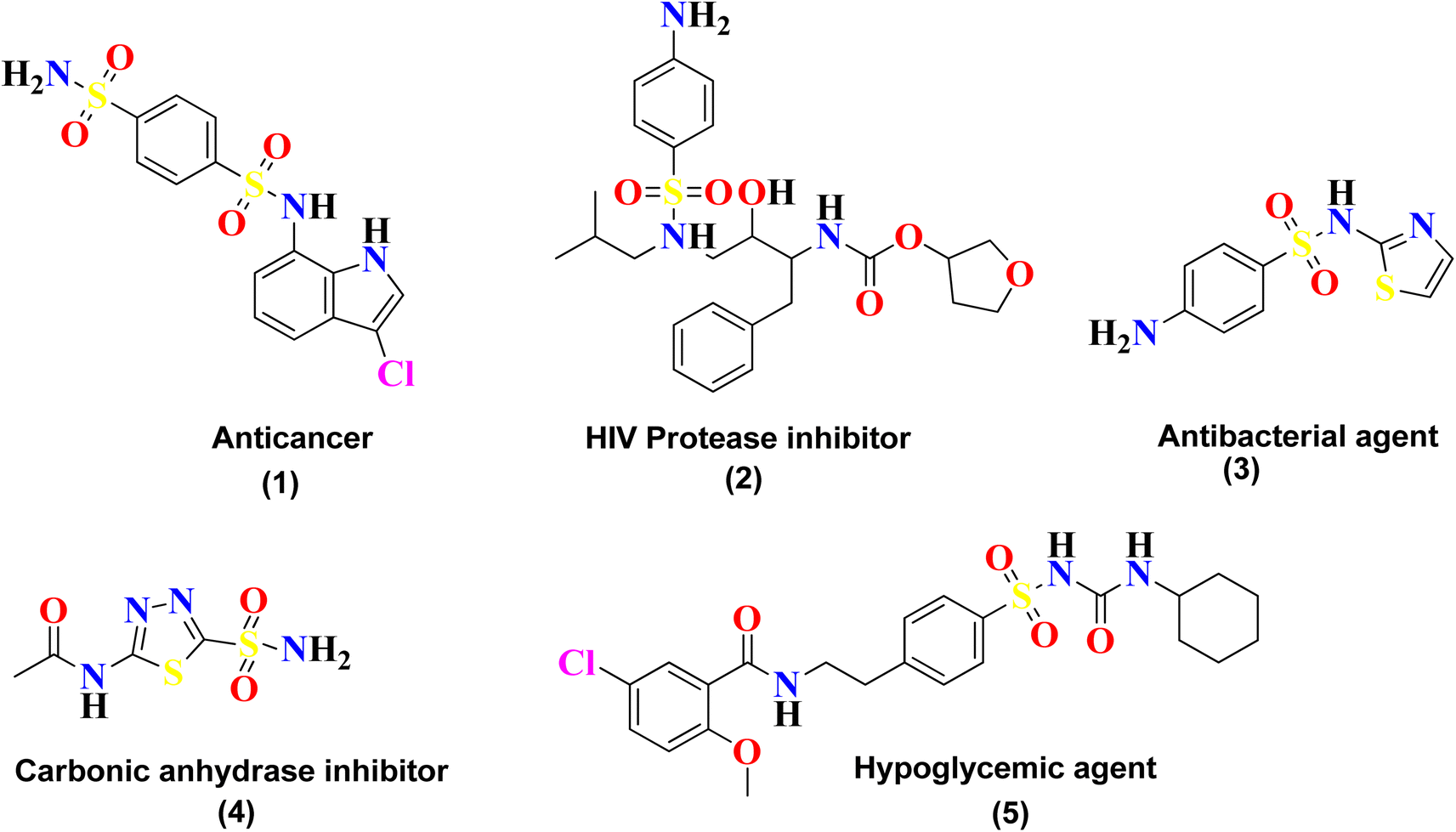 | ||
| Fig. 1 Pharmacological agents developed from sulfonamide. | ||
Heterocyclic compounds play an important role in regulating the biochemical processes in living systems, particularly five-membered heterocyclic compounds with N- and O-atoms such as isoxazoles.10 Isoxazole-based derivatives are largely employed in different pharmaceuticals due to their antibacterial, insecticidal, antibiotic, antitumor, antifungal, antituberculosis, ulcerogenic and anticancer properties. Its derivatives are used as COX-2 inhibitor, antagonist, antiplatelet, anti-HIV, antibacterial, antifungal, antioxidant, anticancer, antiarthritic, anti-inflammatory, anti-HRV-2, anti-coxsackievirus, analgesic, and antimicrobial drugs.10–21 The antibacterial activity of sulfamethoxazole and the antimicrobial activity of heavy metals in metal-sulfamethoxazole complexes make them an important field of research. Alkyldisulfanido complexes with transition metals have been synthesized, and their reactivity has been investigated against different compounds.21
Keeping in view the outstanding biological applications, it was decided to synthesize a new class of compounds with a core skeleton comprising sulfonamide and thaizaole (3,4-d)isoxazole. Since good DNA binding proficiency of a compound is often linked to its candidacy as a better anticancer agent, the synthesized sulfonamide derivatives namely YM-1, YM-2, and YM-3 were further investigated for their interaction with DNA, enzyme inhibition and anti-cancer activities.
2 Results and discussion
2.1 Chemistry
Sulfonamide derivatives possess important biological and non-biological applications. Three sulfonamide derivatives {YM-1 (5a), YM-2 (5b), YM-3 (5c)} were synthesized using synthetic approaches, which involved short reaction time and mild conditions and resulted in fine yield. Sulfonamide was converted into the corresponding acetylated product by treating it with chloroacetyl chloride. The freshly prepared acetylated product was reacted with potassium thiocyanate in dry acetone to obtain a key thiazol-4(5H)-one-based intermediate, which plays a pivotal role in the synthesis of the desired product. The key intermediate was condensed with suitably substituted aromatic aldehyde. In the last step, the condensed product was treated with hydroxylamine hydrochloride (anhydrous) to obtain the thiazole(4,5 c) isoxazole-based fused ring heterocyclic system. The crude product obtained was recrystallized using ethanol as a solvent to obtain the purified product in excellent yield. The schematic representation of the four-step synthesis of sulfonamide derivatives and the structures of newly synthesized derivatives are provided in Scheme 1.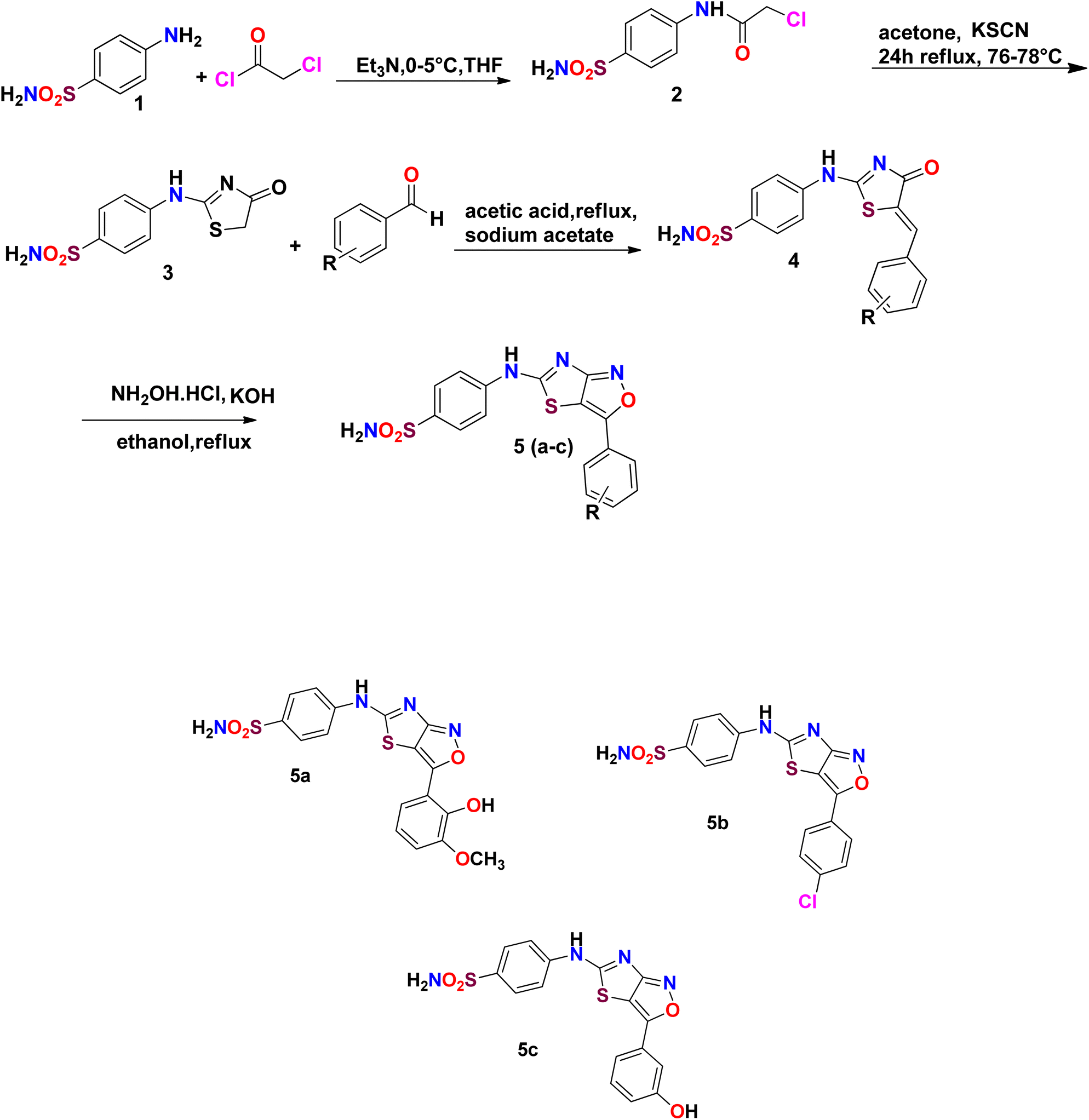 | ||
| Scheme 1 Schematic of the four-step synthesis of sulfonamide derivatives and their structures {YM-1 (5a), YM-2 (5b), YM-3 (5c)}. | ||
The synthesized aryl-substituted fused ring heterocyclic systems were characterized based on the spectroscopic data. The data was obtained from FT-IR, 1H-NMR, and 13C-NMR spectra of each derivative (provided in ESI as Fig. S1–S3†). In 1H-NMR spectra, singlet signals integrating 1H appeared in the range of δ 9–12 ppm, the most deshielded signal, corresponding to the N–H protons of the sulfonamide ring as this NH group is under the influence of two rings one sulfonamide and the second is fused ring heterocyclic moiety. Withdrawing groups always shift signals to higher ppm values, which justifies the bond between sulfonamide and thiazole[4,5 c]isoxazole heterocyclic system. The protons of the aromatic rings are observed in the typical region around δ 6.5–8.0 ppm. The 13C-NMR analysis further confirmed the structure of the title sulfonamide derivatives by displaying the characteristic signals for the (C–S) of thiazole ring, (C–O), (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and (CC) of the isoxazole ring appeared at 170.17, 165.32, 158.43 and 116.47 ppm, respectively. The most de-shielded signal appeared for (C–S) as this carbon was directly attached to three hetero atoms. (CC) appeared as the most shielded signal as both the atoms directly attached to it are carbons. These homo atoms shield the (CC) signal as compared to the others. The FTIR spectral data show the characteristic bands such as at 3430, 3223, 3145 cm−1 (NH2, NH), 1679 cm−1 (CN), 1430 cm−1 (CC), and 1279 cm−1 (C–O).
N) and (CC) of the isoxazole ring appeared at 170.17, 165.32, 158.43 and 116.47 ppm, respectively. The most de-shielded signal appeared for (C–S) as this carbon was directly attached to three hetero atoms. (CC) appeared as the most shielded signal as both the atoms directly attached to it are carbons. These homo atoms shield the (CC) signal as compared to the others. The FTIR spectral data show the characteristic bands such as at 3430, 3223, 3145 cm−1 (NH2, NH), 1679 cm−1 (CN), 1430 cm−1 (CC), and 1279 cm−1 (C–O).
2.2 Structural analysis through DFT
The optimized geometries of YM-1, YM-2 and YM-3 by GGA:PBE/DZ are illustrated in Fig. 2(a–c), whereas the distribution of symmetrical charge can be seen on each individual atom of studied sulfonamide derivatives. Further, the molecular orbital frontier analysis (FMOs) was performed to anticipate chemical transitions,22,23 as shown in Fig. 2(d–f). To identify the electron donating and accepting characteristics, the isodensity distribution of the HOMO and LUMO surfaces was calculated, which depicted most of the isodensity distribution on heteroatoms.24,25 The EHOMO and ELUMO values were found to be −6.22 eV and −3.58 eV for YM-1, −6.83 eV and −3.71 eV for YM-2, and −6.50 eV and −3.72 eV for YM-3. The above values indicated that the electron transfer is comparatively more feasible in YM-1 (ΔE = 2.64 eV) than in YM-2 (ΔE = 3.12 eV) and YM-3 (ΔE = 2.78 eV) due to the little HOMO–LUMO gap. Hence, YM-1 could be predicted to possess comparatively greater reactivity than the other two sulfonamide derivatives. The binding energy Ebinding (eV) was evaluated to be −251.56, −222.49, and −230.48 for YM-1, YM-2, and YM-3, respectively.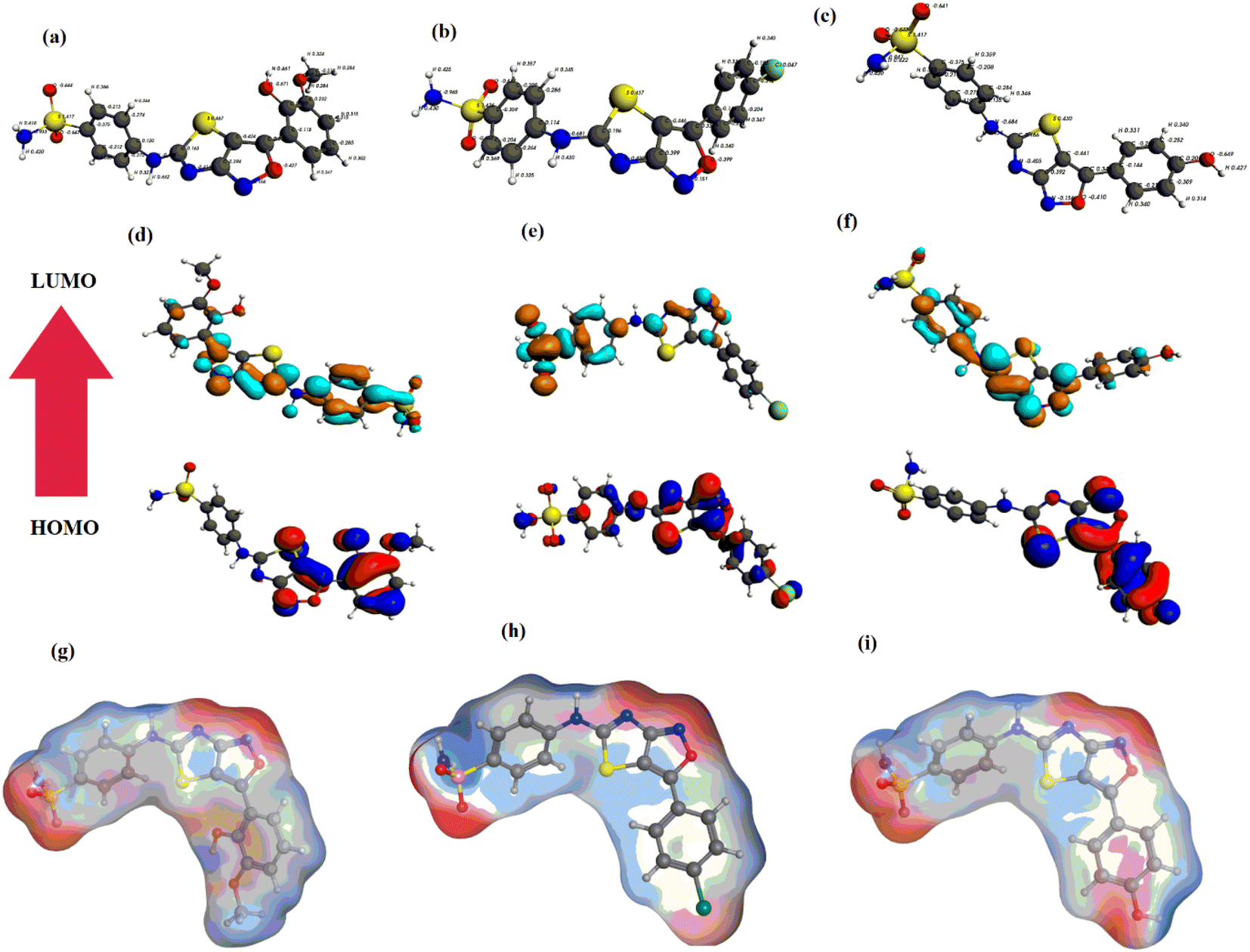 | ||
| Fig. 2 (a–c) Optimized geometries, (d–f) HOMO, LUMO, and (g–i) MESP of sulfonamide derivatives YM-1, YM-2, and YM-3, respectively. | ||
Furthermore, the optimized geometries were utilized to map the molecular electrostatic potential surfaces (MESP) of the sulfonamide derivatives, as shown in Fig. 2(g–i). It is obvious from the figure that negative potential is limited to nitrogen, oxygen, and chlorine atoms which indicates electron transfer from N, O, and Cl, and. the blue color specifies the contribution of –C, –S, and –H atoms as electrophilic regions, whereas red color shows the contribution of –O, –Cl, and –N centers as nucleophilic regions.
2.3 Molecular docking studies
Pose view analysis and conformations of sulfonamide derivatives (YM-1, -2, and -3) with DNA having the lowest free energies along with ligplots are shown in Fig. 3. These sulfonamide derivatives showed mixed mode type partial intercalation and groove binding interactions with DNA. Ligplot demonstrated 2D interactions of all sulfonamide derivatives with DNA. In the case of YM-1, the DNA base-pair (DT B19) developed hydrogen bond interactions with the sulfur of YM-1. For YM-2, DNA base pairs (DA A5) and (DA A6) showed H–bonding with –O atom and –NH2 of YM-2, respectively. The –S atom and –O atom of YM-3 also depicted hydrogen bonding with DNA base pairs (DT A7) on one hand and with (DA A5) on the other hand. Among all sulfonamide derivatives, YM-1 exhibited greater binding constant “Kb” and more negative binding free energy “ΔG”, which revealed its comparatively stronger and more spontaneous binding with the DNA, Table 1. The data obtained from docking and the binding constant suggest that all three compounds YM-1, YM-2, and YM-3 interact with DNA through a mixed-mode binding mechanism incorporating partial intercalation and groove binding. In general, the results for YM-1 suggest that this compound interacts more strongly with DNA as indicated by more positive Kb and more negative ΔG values, in general this may be linked to a more stable and spontaneous interaction of the compound that would have more significant effects on the stability and function of DNA.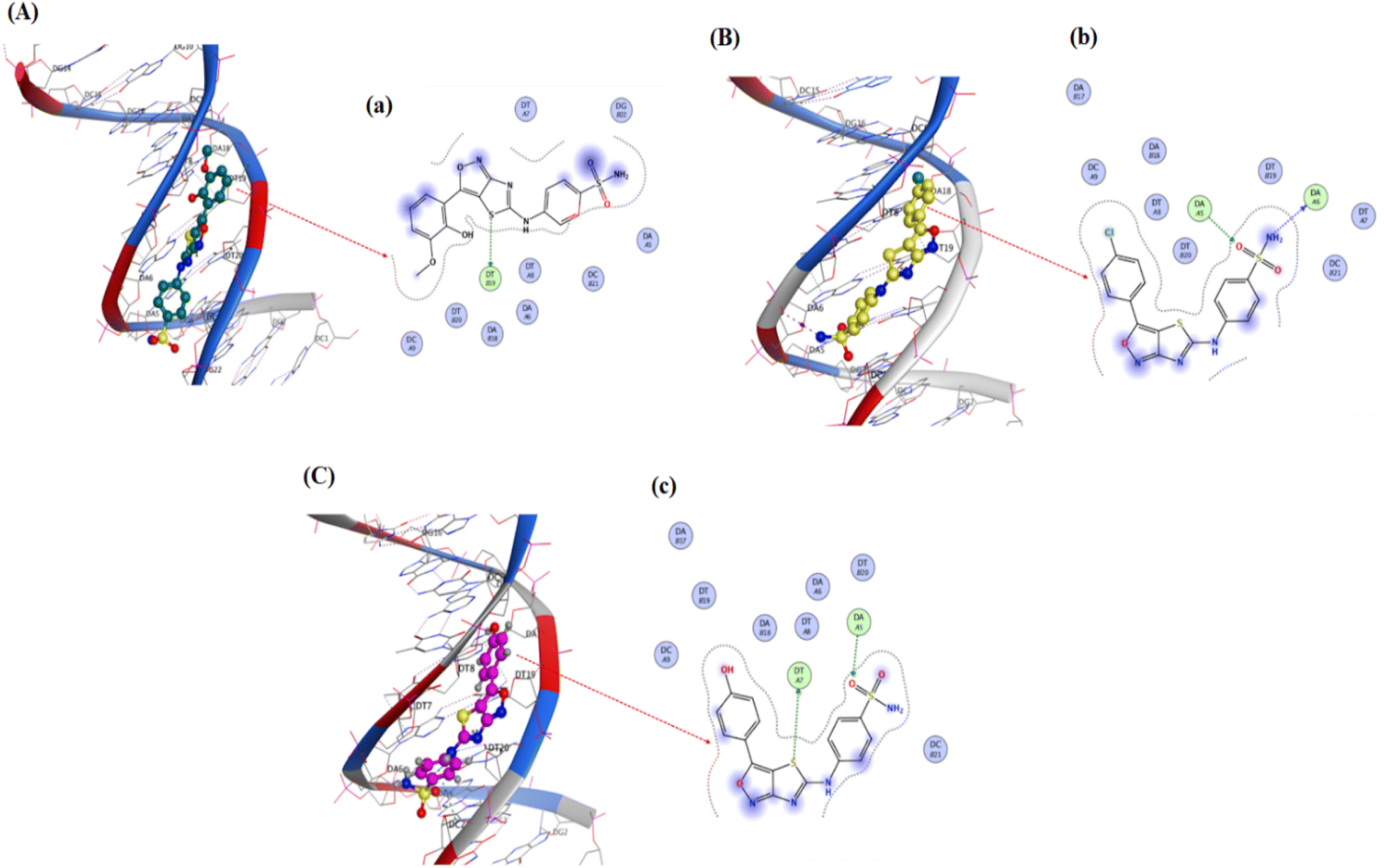 | ||
| Fig. 3 YM-1, YM-2 and YM-3: (A–C) molecular docked complexes with DNA; (a–c) ligplots with DNA base-pairs calculated at PM3 semi-empirical level of theory. | ||
| Compounds | DNA binding | Carbonic anhydrase binding | Urease binding | |||
|---|---|---|---|---|---|---|
| Kb/M−1 | ΔG/kJ mol−1 | Kb/M−1 | ΔG/kJ mol−1 | Kb/M−1 | ΔG/kJ mol−1 | |
| YM-1 | 6.2 × 105 | −33.29 | 1.3 × 104 | −23.66 | 4.4 × 105 | −32.43 |
| YM-2 | 4.4 × 105 | −32.43 | 2.8 × 103 | −19.83 | 6.2 × 105 | −33.29 |
| YM-3 | 2.2 × 105 | −30.71 | 2.6 × 103 | −19.69 | 2.2 × 105 | −30.71 |
The molecular docked complexes of all sulfonamide derivatives (YM-1, -2, and -3) with carbonic anhydrase and urease enzymes and their ligplots are provided in Fig. 4. For YM-1, interactions were explored with carbonic anhydrase, which indicated hydrogen bond interactions between His-160 residue and –NH2 of YM-1 and between –O atom of YM-1 and Thr-254 and Thr-255 residue. Whereas arene–H interactions between the aromatic ring of YM-1 and Gln-138 residue were also observed. In the case of YM-2, arene–arene interactions showed between His-360 residue and the aromatic ring of YM-2. While exhibiting H-bonding between His-17 His-360 residue with –O atom of YM-2. YM-3 showed hydrogen bonding between the Met-169 residue and –O atom of YM-3 with the Asp-275 residue and –OH group of YM-3. Carbonic anhydrase is a carboxylase that catalyzes the reversible reaction of carbon dioxide and water to bicarbonate with the generation of protons. One of its major roles is playing a central part in regulating pH as well as in the transport of CO2 in tissues. In general, inhibition by carbonic anhydrase is brought about by blocking critical residues within the active site for critical sulfonamide derivatives, which breaks its catalytic function.
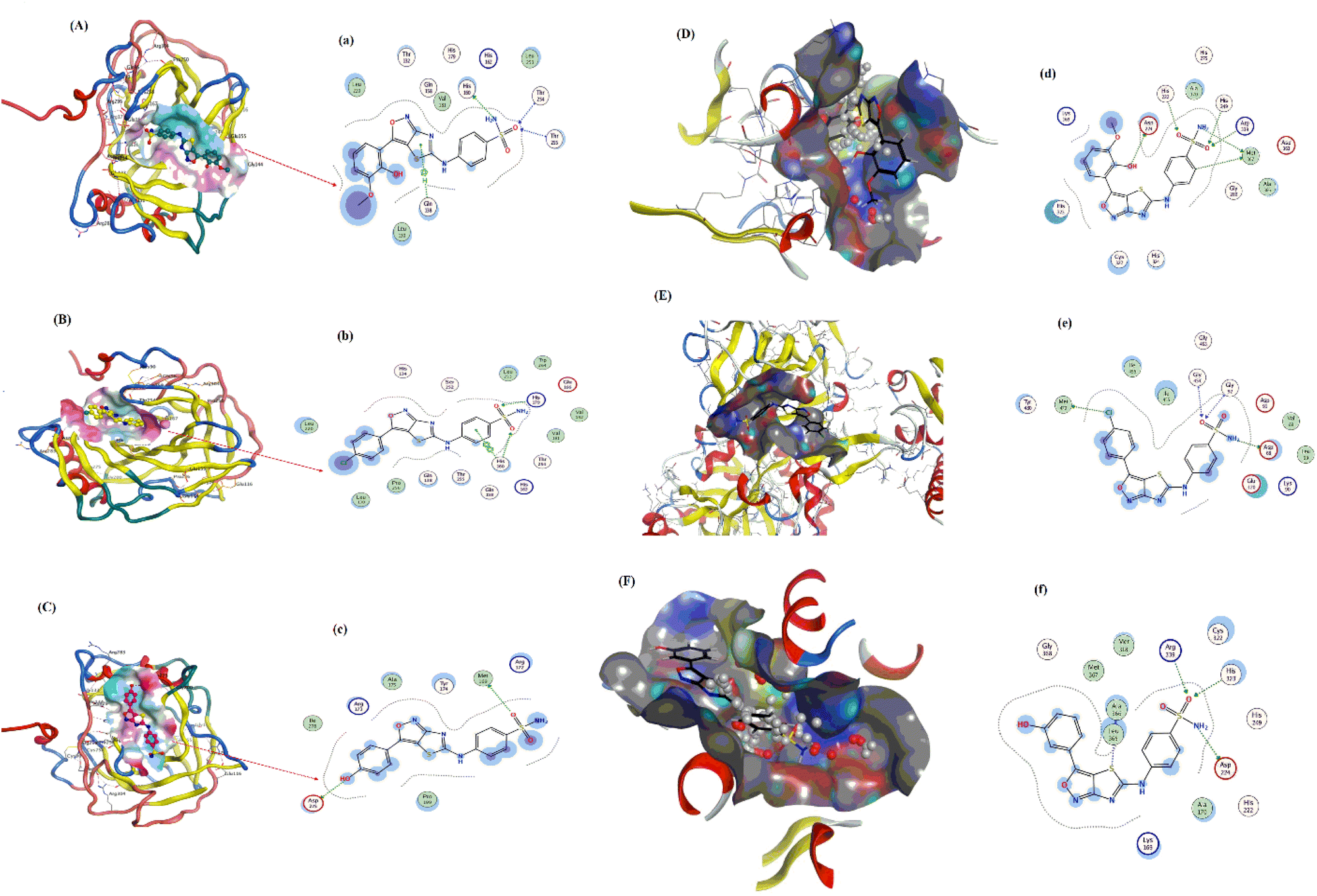 | ||
| Fig. 4 YM-1, YM-2 and YM-3: (A–C) molecular docked complexes with carbonic anhydrase and (D–F) urease; ligplots with residues of (a–c) carbonic anhydrase and (d–f) urease enzyme calculated at PM3 semi-empirical level of theory. | ||
YM-1–urease interaction indicated bond interactions between Met-367 residue and –NH2 at one end and arene–H at another end of YM-1. At the same time, the –O atom of YM-1 indicated interactions with Asp-224, His-249 and His-222 residues. In the case of YM-2, –NH2 and arene interactions showed between His-249 residue and the aromatic ring of YM-2. While exhibiting H-bonding between the Met-367 residue with the –O atom of YM-2. YM-3 showed hydrogen bonding between His-323 and Arg-339 residue of urease with the –O atom of YM-3 alongside the Asp-224 residue and the –NH2 group of YM-3. At the same time, the –S atom of YM-1 indicated interactions with Ala-366 of urease. An enzyme that catalyzes the hydrolysis of urea to ammonia and carbon dioxide, it plays a significant role in nitrogen metabolism, contributes to soil alkalinity and grows pathogenic bacteria. To put it in a nutshell, the sulfonamide derivatives inhibit the urease by occupying its active site and forming strong interactions with key catalytic residues, thereby blocking the enzymatic breakdown of urea.
The binding data demonstrated that YM-1 binds to carbonic anhydrase, while YM-2 binds to urease more strongly and has comparatively higher Kb and more negative ΔG values as shown in Table 1.
2.4 DNA binding experimental studies
The absorption spectrum of each sulfonamide derivative was recorded, individually, at different concentrations, which showed broader absorption peaks within the range of 300–450 nm, and the molar extinction coefficients (ε) were determined using varying concentrations of each derivative, separately, and plotting them against their absorbance, as shown in Fig. S4(a–f) in the ESI.† The absorbance maxima were found to be 371.94 nm, 353.19 nm, and 352.04 nm for YM1, YM-2, and YM-3, respectively. The ε (M−1 cm−1) values were evaluated to be 2.34 × 104, 1.5 × 104, and 2.36 × 104 for YM-1, YM-2, and YM-3, respectively, indicating π–π* transitions in all the derivatives.26,27The spectral responses (UV-visible) of sulfonamide derivatives after DNA titration are shown in Fig. 5(a–c), which indicate a drop in the absorption peak intensity with DNA additions. The drop in peak intensities of YM-1, YM-2, and YM-3 spectra upon maximum addition of DNA were found to be 40.78%, 28.81%, and 23.40%, respectively. Wavelength shifts (red) of 5.97, 5.12, and 4.54 nm were also observed for each sulfonamide derivative-DNA adduct. These changes in a compound's spectrum after DNA additions have been reported for intercalation of a compound's planner part within the DNA base pairs.28–32 The DNA binding constant (Kb) was obtained by using absorbance values, before (Ao) and after (A) DNA titrations, in the Hildebrand equation 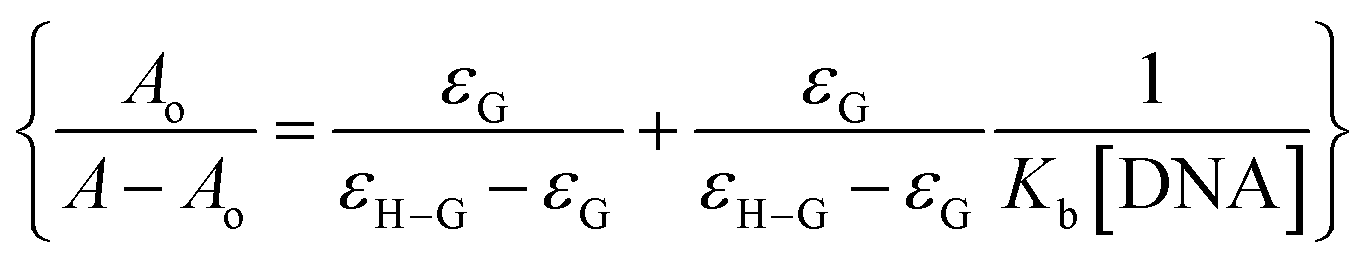 .33 In this straight-line equation, a graph between
.33 In this straight-line equation, a graph between 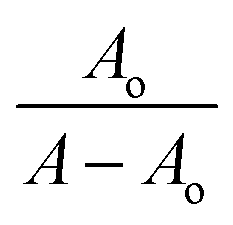 and
and 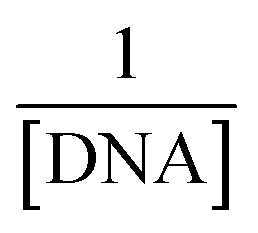 provide the Kb value as the intercept/slope ratio, Fig. 5(a–c) (inset).27 To assure reaction spontaneity, thermodynamic analysis was carried out by evaluating the free energy change (ΔG). For that, the Kb value was used in the van't Hoff equation {ΔG = −RT
provide the Kb value as the intercept/slope ratio, Fig. 5(a–c) (inset).27 To assure reaction spontaneity, thermodynamic analysis was carried out by evaluating the free energy change (ΔG). For that, the Kb value was used in the van't Hoff equation {ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKb}. Both Kb and ΔG values for the three DNA-bound complexes are provided in Table 2. The binding constants were found in 104 orders of magnitude for all DNA-bound complexes, while ΔG values were found to be negative. However, greater, and more spontaneous binding with DNA was found for YM-1. The appearance of an isosbestic point showed the development of a dynamic equilibrium among each sulfonamide derivative and DNA and further assured the formation of the DNA-bound complex via spontaneous intercalation into the DNA base pairs.34
lnKb}. Both Kb and ΔG values for the three DNA-bound complexes are provided in Table 2. The binding constants were found in 104 orders of magnitude for all DNA-bound complexes, while ΔG values were found to be negative. However, greater, and more spontaneous binding with DNA was found for YM-1. The appearance of an isosbestic point showed the development of a dynamic equilibrium among each sulfonamide derivative and DNA and further assured the formation of the DNA-bound complex via spontaneous intercalation into the DNA base pairs.34
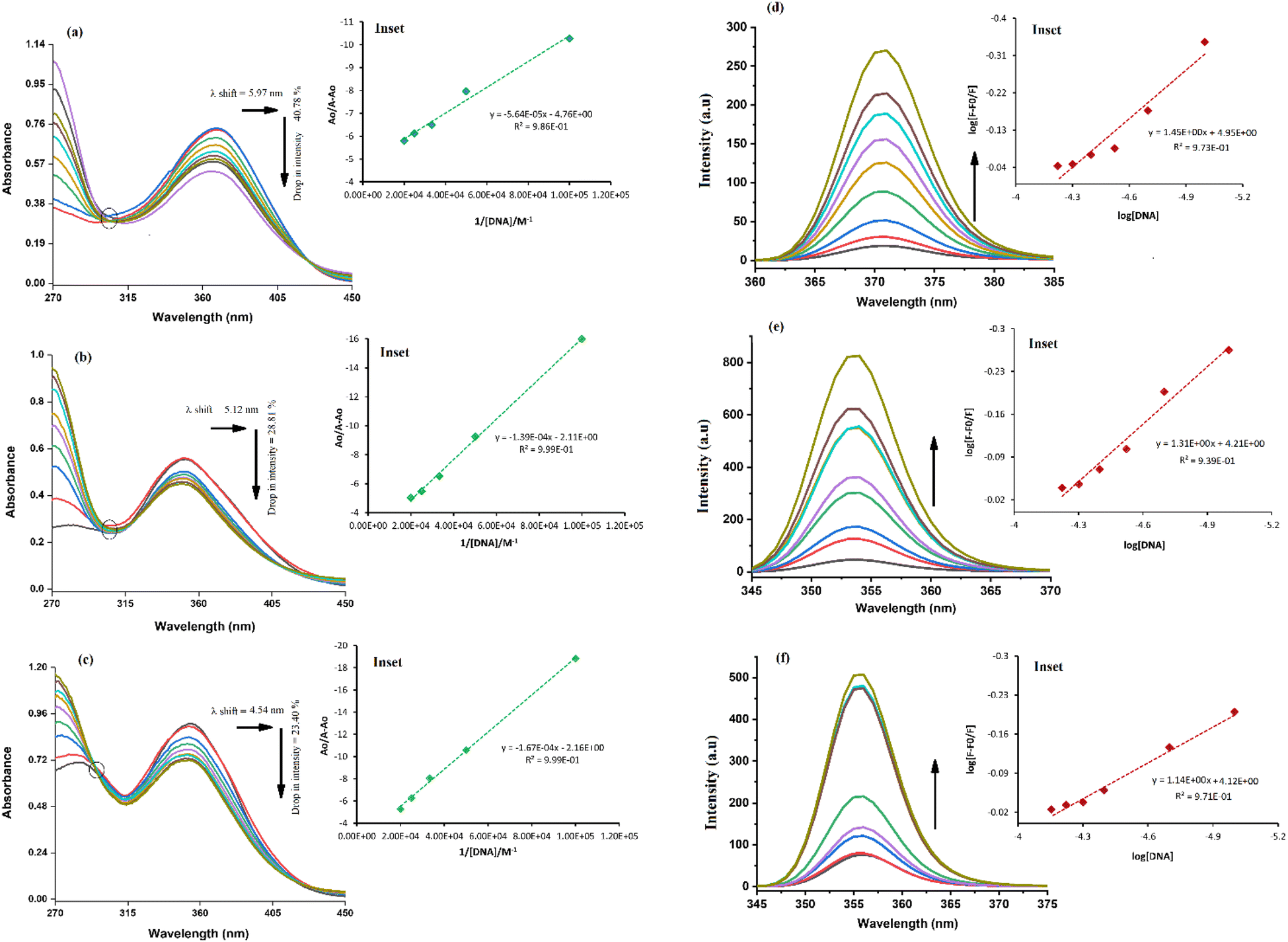 | ||
| Fig. 5 (a–c) UV-visible and (d–f) fluorescence-spectrophotometric responses of sulfonamide derivatives YM-1, YM-2, and YM-3, respectively, before and after DNA additions along with the graphs as their insets for the evaluation of binding parameters. | ||
| Binding parameters | YM-1-DNA | YM-2-DNA | YM-3-DNA | ||||||
|---|---|---|---|---|---|---|---|---|---|
| UV- | Fluo- | CV | UV- | Fluo- | CV | UV- | Fluo- | CV | |
| Kb/M−1 | 8.43 × 104 | 8.91 × 104 | 3.23 × 104 | 1.52 × 104 | 1.62 × 104 | 1.66 × 104 | 1.29 × 104 | 1.32 × 104 | 1.22 × 104 |
| ΔG/kJ mol−1 | −29.23 | −29.38 | −26.76 | −24.82 | −24.98 | −25.05 | −24.39 | −24.45 | 24.26 |
| n | — | 1.45 | 2.16 | — | 1.31 | 1.88 | — | 1.14 | 1.34 |
The fluorescence spectra of YM-1, -2, and -3 are shown in Fig. S4(g) in the ESI.† All the sulfonamide derivatives were found to be luminescent as emission peaks were observed having intensities at 458, 432, and 425 nm, respectively for YM-1, -2, and -3. Therefore, their binding with DNA was studied directly by adding varying DNA concentrations to the compound's fixed concentration. The emission responses (Fluo-) of sulfonamide derivatives after DNA titration are shown in Fig. 5(d–f), which indicated a heightening in the emission peak intensity with DNA additions. Upon maximum DNA addition, the emission peak intensities of YM-1, -2, and -3 were evaluated to be 16.4, 13.5, and 6.66 times larger, respectively. The peak enlargement could be associated with the compound's binding with DNA via intercalation.35–37 The Kb was evaluated by using the values of emission peak intensities of each derivative before (Fo) and after (F) DNA addition in the equation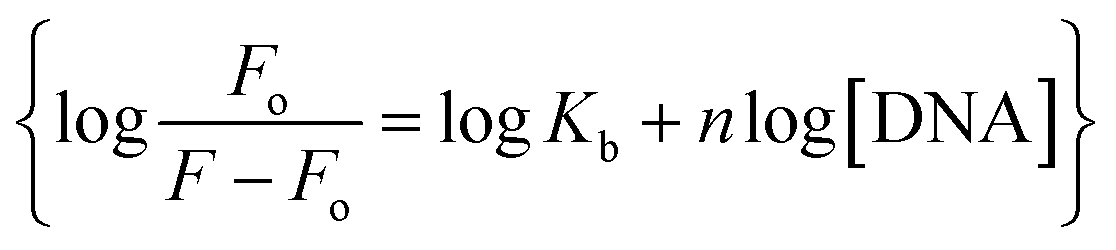 .38 A graph between
.38 A graph between 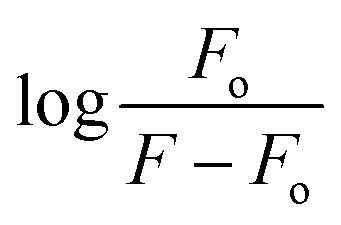 and log[DNA] provided the Kb value as the antilog of the intercept, while the slope value represented the binding site size (n), Fig. 5(d–f) (inset).37 The evaluated fluorescence data of Kb and n along with ΔG values are provided in Table 2. The Kb and ΔG values evaluated from UV- and Flou- spectroscopies have shown similar trends, while the binding site size (n) was found to be greater than one that indicated more site availability for interaction along with intercalation.
and log[DNA] provided the Kb value as the antilog of the intercept, while the slope value represented the binding site size (n), Fig. 5(d–f) (inset).37 The evaluated fluorescence data of Kb and n along with ΔG values are provided in Table 2. The Kb and ΔG values evaluated from UV- and Flou- spectroscopies have shown similar trends, while the binding site size (n) was found to be greater than one that indicated more site availability for interaction along with intercalation.
To complement the spectroscopic findings for DNA binding, the work was further extended to electrochemical studies using cyclic voltammetry. Cyclic voltammetry (CV) experiments were run for each sulfonamide derivative with and without DNA at various scan rates (30–130 mV s−1), as shown from the data in Fig. S5 in the ESI.† A broad irreversible reduction peak was observed within the potential scan range of −600–−800 mV for each sulfonamide derivative (YM-1, -2, and -3). Cyclic voltammetric responses of sulfonamide derivatives without and in the presence of DNA are provided in Fig. 6. A drop in current with a slight shift, towards less negative potential, was observed in the CV response of each sulfonamide derivative with DNA additions. The drop in the peak current was evaluated to be 31.71%, 27.57%, and 20.78%, respectively, for the adducts of YM-1, YM-2, and YM-3 with DNA. These variations in the compound's spectrum are indicative of the interaction between a compound and DNA via intercalation.39–44
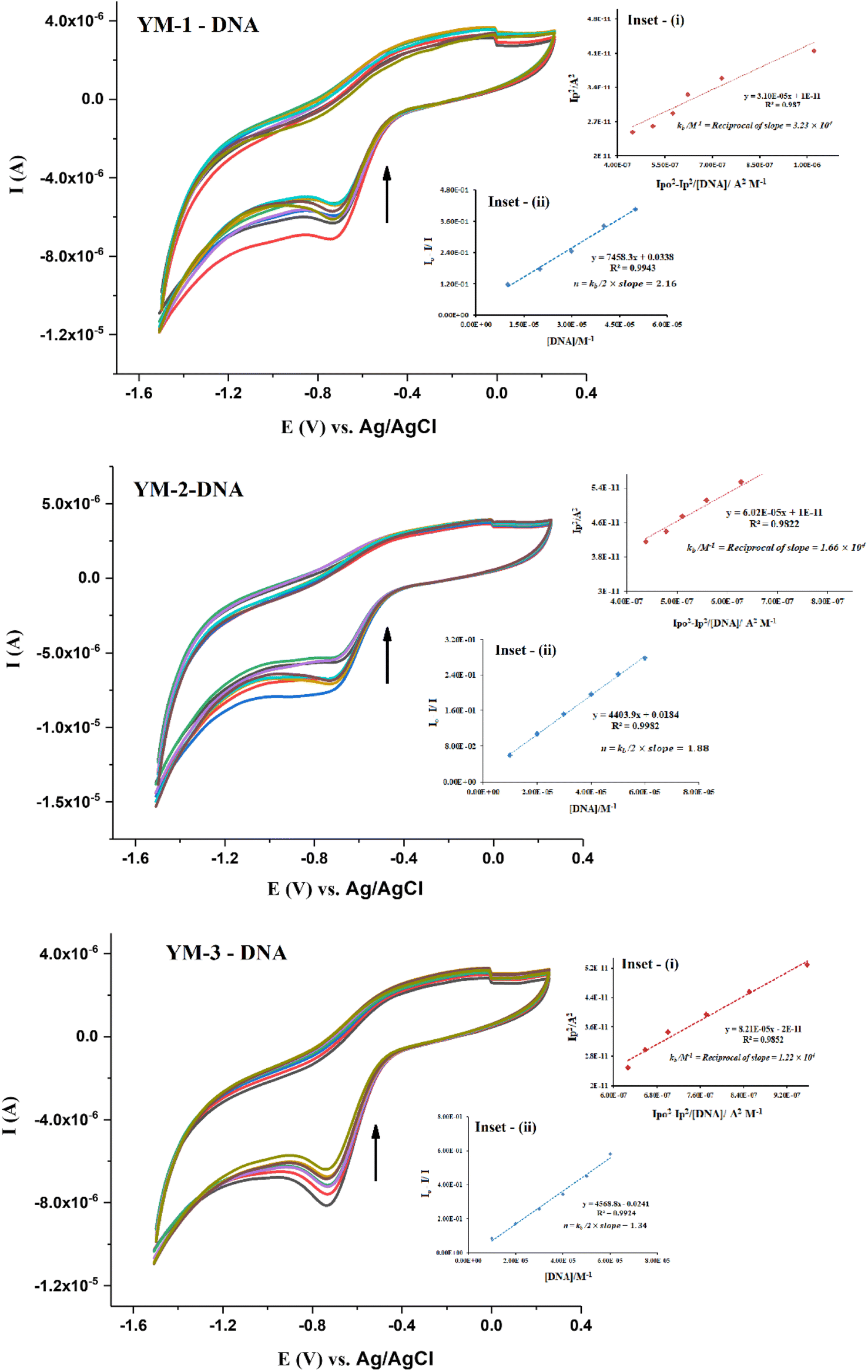 | ||
| Fig. 6 CV responses of sulfonamide derivatives (YM-1–3) before and after DNA additions, along with graphs as inset (i) and inset (ii) for the calculation of binding constant and binding site size, respectively. | ||
The binding parameters Kb and n were evaluated from the current variations by using the equations 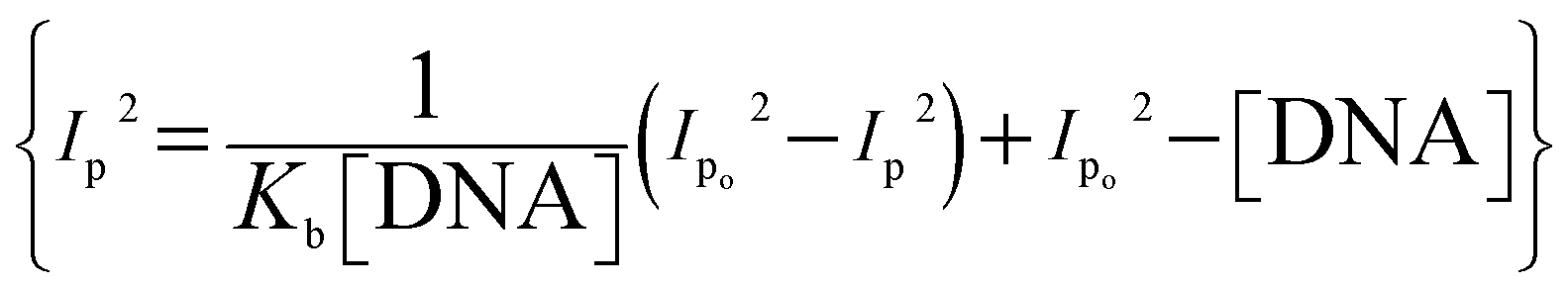 , and {I − IDNA/IDNA = Kb[DNA]/2n}.39,41 The plots are shown as inset (i) and (ii) in Fig. 6 and the evaluated values are provided in Table 2 along with the evaluated ΔG values. The electrochemical findings have shown similar preferences in the values as evaluated from spectroscopic measurements. The binding order from all experimental work was found to be YM-1–DNA > YM-2–DNA > YM-3–DNA.
, and {I − IDNA/IDNA = Kb[DNA]/2n}.39,41 The plots are shown as inset (i) and (ii) in Fig. 6 and the evaluated values are provided in Table 2 along with the evaluated ΔG values. The electrochemical findings have shown similar preferences in the values as evaluated from spectroscopic measurements. The binding order from all experimental work was found to be YM-1–DNA > YM-2–DNA > YM-3–DNA.
An irreversible redox activity was observed for each sulfonamide derivative and its DNA-bound complex at various scan rates (30–130 mV s−1) and provided as Fig. S5 in the ESI.† The shift in the peak potential with the increase in the scan rate further assured the irreversible nature of redox reactions with and without DNA. The rise in current with the scan rate before and after DNA additions was further used to verify the formation of the sulfonamide derivative–DNA complex. Using the Randles–Sevick equation {ip = 2.99 × 105n(αna)1/2A0C0 × D01/2ν1/2},45 ip was plotted against the square root of the scan rate and diffusion coefficient (Do) of each sulfonamide derivative was evaluated from the slope before and after the formation of the DNA complex, Fig. 7(a–c). The slopes for YM-1, YM-2, and YM-3 were found to be 2.11 × 10−5, 2.13 × 10−5, and 2.14 × 10−5, respectively, and for YM-1–DNA, YM-2–DNA, and YM-3–DNA the values were 1.82 × 10−5, 1.57 × 10−5, and 1.81 × 10−5, respectively. Using the slope values, Do (cm2 s−1) of YM-1, YM-2, and YM-3 were evaluated to be 3.25 × 10−3, 3.32 × 10−3, and 3.35 × 10−3, while, for their DNA complexes YM-1–DNA, YM-2–DNA, and YM-3–DNA, the values were evaluated to be 2.42 × 10−3, 1.80 × 10−3, and 2.39 × 10−3, respectively. The transfer coefficient (α) was taken at 0.5, while the surface area of the working electrode (glassy carbon) was 0.070 cm2. A linear dependency of current with the square root of scan rate was observed from the graphs for all sulfonamide derivatives without and in the presence of DNA. However, the diffusion coefficient value for each sulfonamide derivative was evaluated as comparatively low after DNA addition, which indicated slow diffusion due to the formation of a bulky sulfonamide derivative–DNA complex.36,37,46
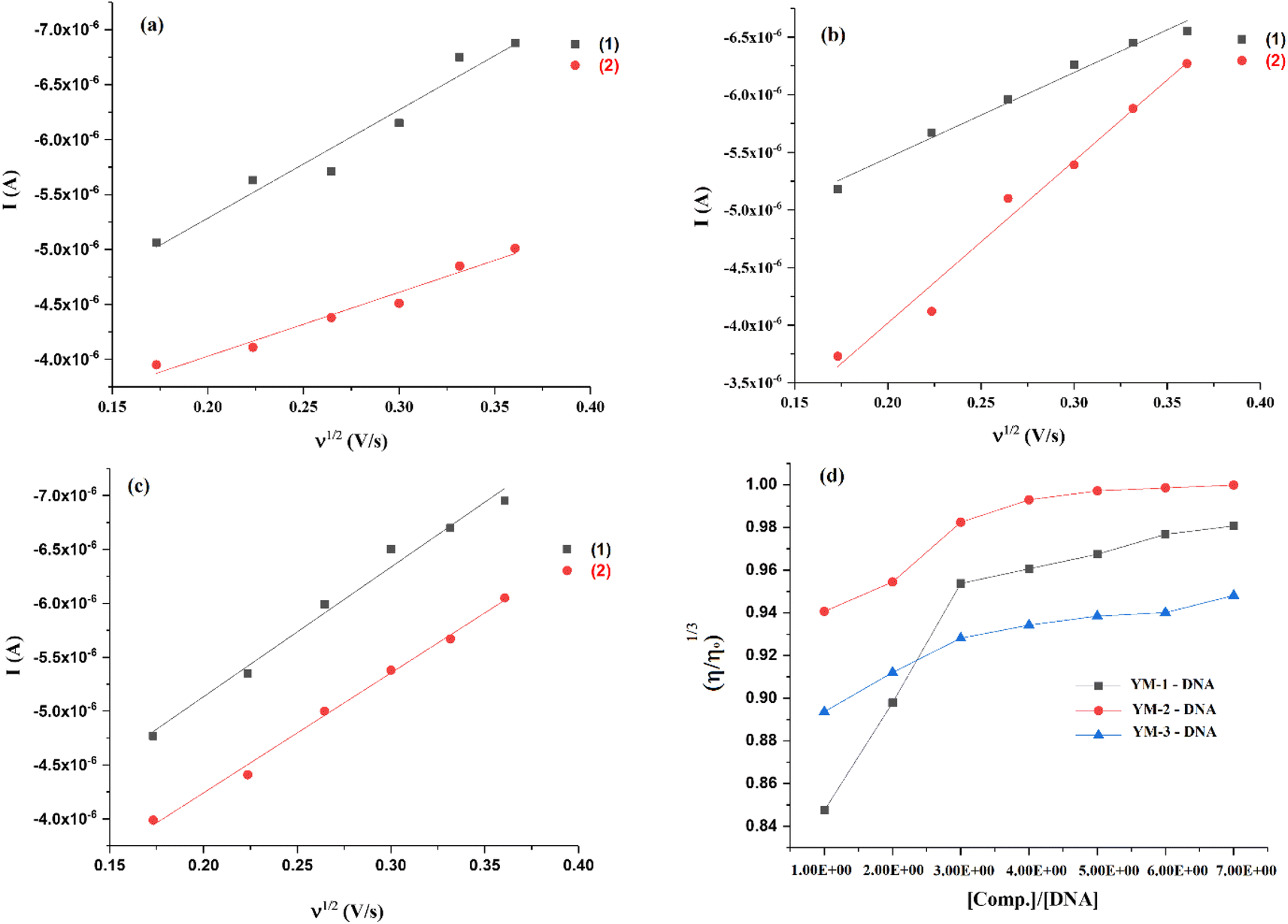 | ||
| Fig. 7 (a–c) Plots for the measurements of diffusion coefficient (Do) for (1) YM-1, YM-2, YM-3, and (2) YM-1-DNA, YM-2-DNA, YM-3-DNA complexes, and (d) viscosity plots for binding mode verification. | ||
DNA viscosity measurements after the compound's additions is another method that verifies the formation of the compound–DNA complex via various modes of interaction.47 The DNA viscosity was monitored without and in the presence of varying concentrations of each sulfonamide derivative and then graphs between cube root of relative viscosities and concentration ratios were plotted as shown in Fig. 7(d). Viscosity graphs indicated an increase in the DNA viscosity with sulfonamide's addition. However, DNA viscosity stopped rising after 30–40 μM sulfonamide's addition, and no significant change was further observed. This rise and then consistency in the DNA viscosity in the presence of each sulfonamide derivative verified the formation of a complex via a mixed mode that could be attributed to partial intercalation and groove binding.48 The same binding modes were anticipated in molecular docking analysis. DNA denaturing was not observed as it results in a decrease in the relative viscosity of DNA due to the separation of bases in denatured DNA, which is attributed to the lowering in DNA viscosity.48 In the current study, two reasons could be emphasized to ignore the chance of denaturing of DNA; (i) low DMSO concentration as stock solutions of sulfonamide derivatives were prepared in the aqueous DMSO (DMSO:water; 1:9 ratio) solution mixture, and (ii) rise in DNA viscosity was observed at initially added concentrations of each sulfonamide derivative.49,50
Overall, theoretical and experimental observations and data analyses revealed that all the sulfonamide derivatives have an affinity towards DNA via mixed binding interactions, while, based on binding parameters, YM-1 was found to be comparatively better DNA binder than YM-2, and YM-3.
2.5 Enzyme inhibition activity studies
Anti-urease assay is based on the principle that free ammonia is generated and then used by the compound.51 The ability of recently synthesized sulfonamide derivatives to inhibit the urease enzyme was examined at different concentrations; the IC50 ± SD values were evaluated, and the % inhibition findings are shown in Fig. 8. Significant enzyme inhibition activity was observed by thiourea (positive control), with an IC50 equal to 0.92 ± 0.03 μM. YM-2's IC50 results showed 1.90 ± 0.02 μM, which was comparatively less than YM-1's (1.98 ± 0.02 μM) and YM-3's (2.02 ± 0.01 μM) IC50 values. The IC50 graphs are provided in Fig. 8(b) and the data is provided in a tabulated form as Fig. S6 in the ESI.† Furthermore, all sulfonamide derivatives exhibited anti-urease activity that was dependent on concentration and showed significant results with p-value <0.05. The highest percentage inhibition (57.93%) activity was exhibited by YM-2's while YM-1's and YM-3's exhibited 52.61% and 49.17% urease inhibition, respectively, at 100 μM concentration.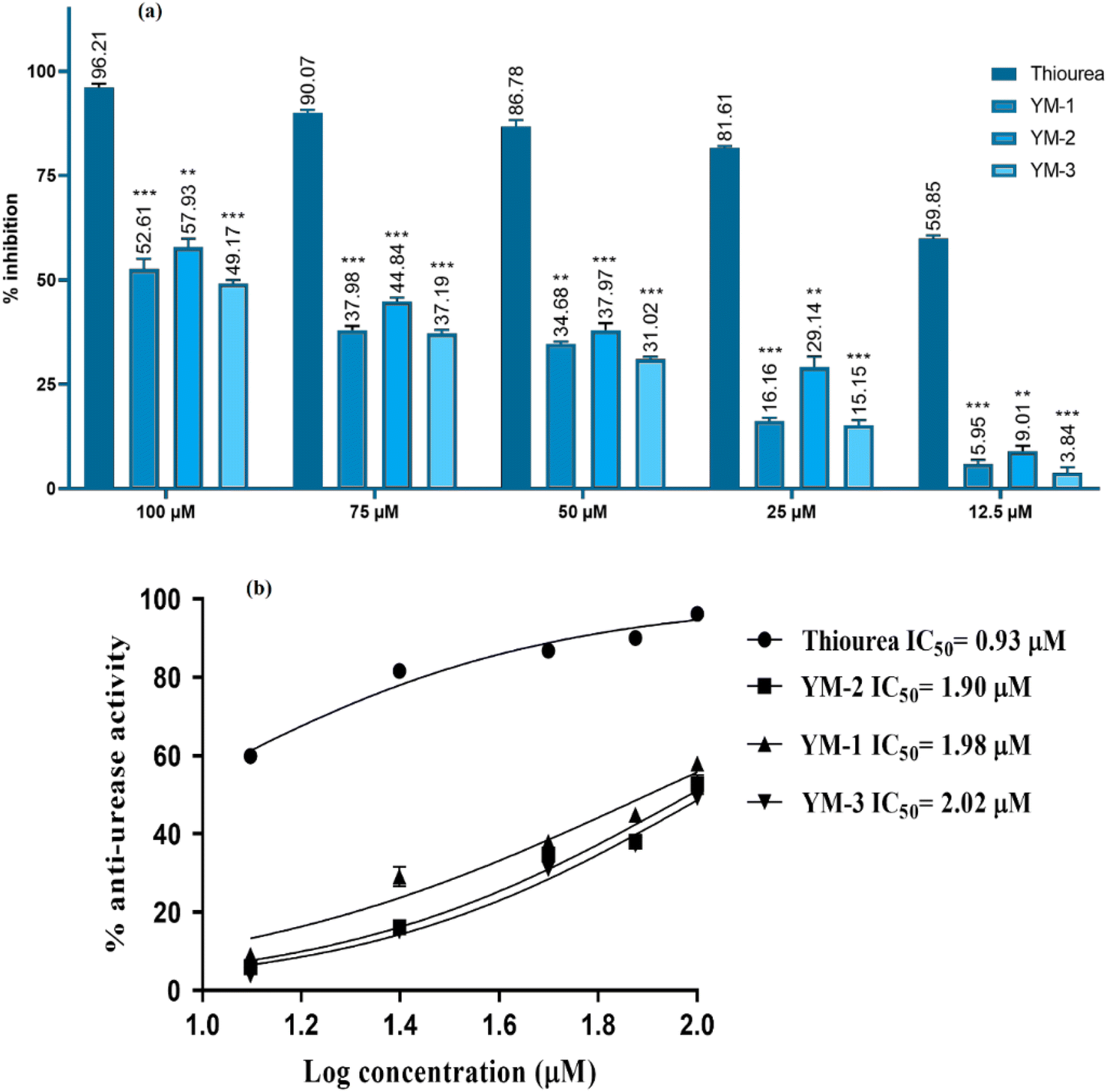 | ||
| Fig. 8 (a) Results of % anti-urease activity at different concentrations of sulfonamide derivatives (YM-1, -2, and -3). Values are stated as mean (n = 3) ± standard deviation (SD) and thiourea was used as a positive control. Values were statistically significant having ***<0.001 and **<0.01 with respect to thiourea. (b) Graphs for IC50 values calculations. | ||
2.6 Cytotoxicity activity by cell line studies
In vitro cell studies were performed by MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay and cytotoxicity activity of each sulfonamide derivative was approximated for healthy and cancer (HEK-293, MG-U87) cell lines. The dose-dependent percentage cytotoxicity results are given in Fig. 9. As obvious from the % cytotoxicity results, HEK-293 showed no resistance against any concentration (40–400 μM) of YM-2, and YM-3, which indicated that these derivatives not only killed the cancer cells but were toxic to the normal cells as well. Among both derivatives (YM-2, and -3), YM-2 showed comparatively greater toxicity against healthy cells and the cytotoxicity results are almost parallel to cancer cells' cytotoxicity at all concentrations. However, YM-1 showed some concentration-dependent tolerance against healthy cells, and the % cytotoxicity values were found to be comparatively less than those for MG-U87 cells. Among all the doses of YM-1, 120 μM concentration showed less % cytotoxicity (50.28%) for healthy cells, which was even less than that evaluated for the positive control (doxorubicin HCl) for healthy cells (52.22%), while the % cytotoxicity of YM-1 was found to be 20% greater than that of the positive control towards cancer cells. Moreover, the % cytotoxicity results for the cancer cell lines were found to be greater for all the sulfonamide derivatives (YM-1, -2, and -3) at all concentrations than those of the positive control. The IC50 values further assured better performance of YM-1 against the studied cell lines with comparatively low IC50 value for cancer cells and high IC50 value for healthy cells. The order of IC50 values for cancer and healthy cells was found to be {YM-1IC50 (1.154 ± 0.317 μM) < YM-2IC50 (7.024 ± 0.374 μM) < YM-3IC50 (9.512 ± 0.668 μM)} and {YM-1IC50 (15.29 ± 0.926 μM) > YM-3IC50 (9.304 ± 0.386 μM) > YM-2IC50 (7.938 ± 0.345 μM)}, respectively. The data for IC50 values are provided in a tabulated form as Fig. S7 in the ESI.†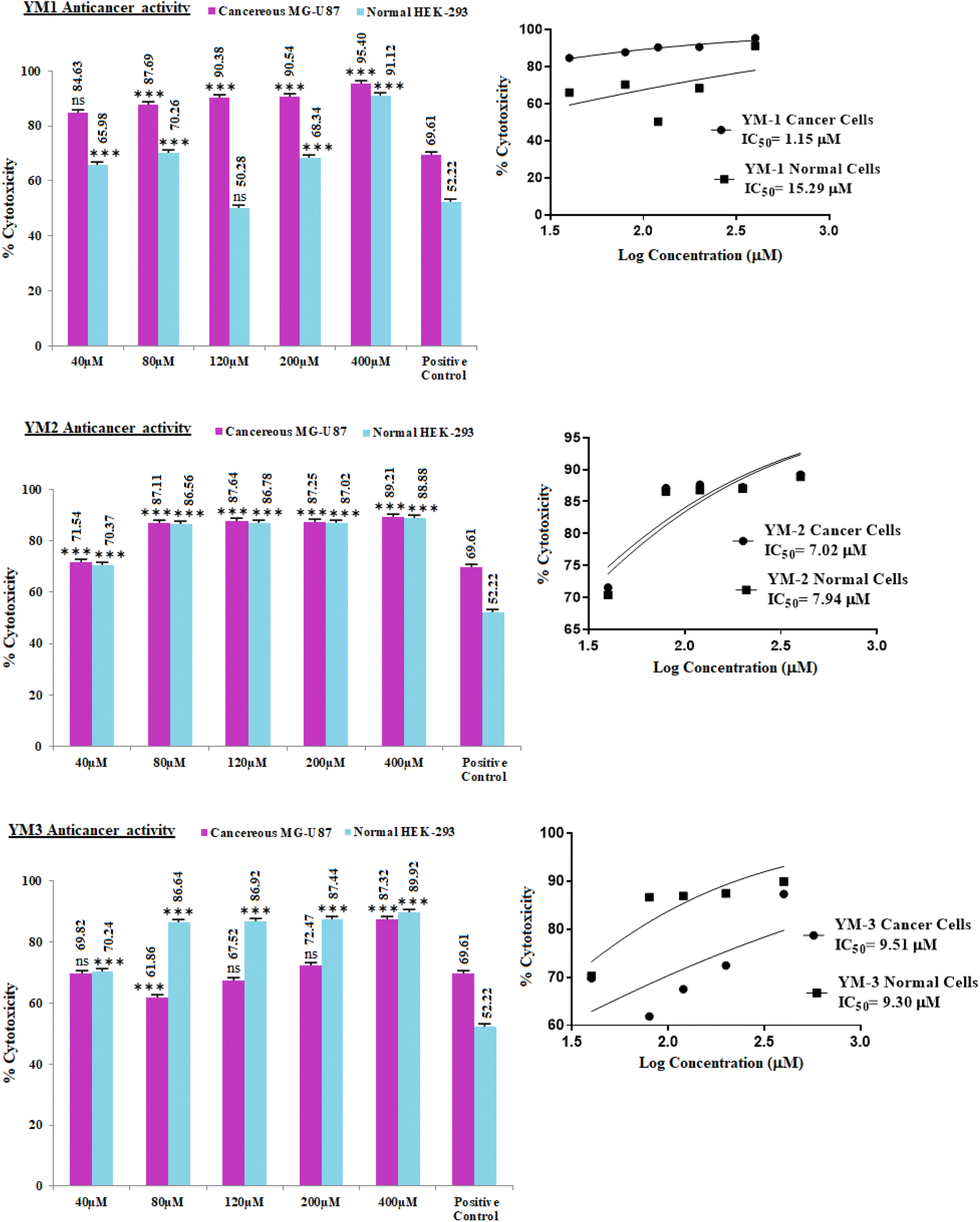 | ||
| Fig. 9 Dose-dependent % cytotoxicity of sulfonamide derivatives (YM-1, -2, and -3) on HEK-293 and MG-U87 cell lines along with the graphs for IC50 calculations. Positive reference control (doxorubicin HCl), and solvent control (PBS with 10% DMSO). ns presents non-significance and values were statistically significant having ***<0.001 with respect to a positive control (doxorubicin HCl). | ||
2.7 Structure–activity relationship
Among all sulfonamide derivatives, the DNA binding, cytotoxicity, and enzyme inhibition (carbonic anhydrase) activities were found to be greater for YM-1, which could be attributed to its structure. The less bulky –OCH3 and –OH functionalities are present on the phenyl ring and the disubstitution of these electron donating groups (at ortho and or meta position) may result in the enhancement of its activity. The bulkier electron withdrawing group (–Cl) is present at the para position in YM-2, which may produce a deficiency of electrons on the aromatic ring at its C center and mono substitution of electron-donating substituents –OH at the meta position in YM-3 that does not produce a profound withdrawing effect; hence making these two sulfonamide derivatives to have comparatively less activity than that of YM-1.3 Conclusions
Our work aimed to synthesize three new derivatives (YM-1,-2, and -3) with a basic framework containing sulfonamide and thaizaole (3,4-d)isoxazole. These sulfonamide derivatives revealed interesting results when explored for their DNA binding, enzyme inhibition and cytotoxicity effects against healthy and cancer cells. Both experimental and theoretical studies were carried out to authenticate our claim for the better candidacy of these derivatives as DNA binder, enzyme inhibitory, and anticancer potentials. The binding interactions with DNA were found to be mixed type and the evaluated data revealed comparatively greater, more spontaneous binding of YM-1 with DNA. Low values of the diffusion coefficient, as evaluated from CV data for each derivative–DNA adduct, further assured the complex formation that made it bulky, and it diffused slowly than the sulfonamide derivative. Mixed-type binding (via intercalation + groove) was further proven by measuring variation in the DNA viscosity in the presence of each sulfonamide derivative and it also justified the size of the binding site (n) that was evaluated >1. YM-1 also showed greater binding affinity for carbonic anhydrase enzyme, while YM-2 was found to have comparatively greater anti-urease activity. Cell line studies were carried out in a dose-dependent way for both healthy and cancer cells. All the derivatives showed greater cytotoxicity against cancer MG-U87 cells, but the tolerance was found insignificant against healthy cells HEK-293 at all concentrations of YM-1, 2, and -3 except 120 μM concentration of YM-1. Hence YM-1 at this concentration, with greater cytotoxicity for MG-U87 (90.38%) and less cytotoxicity for HEK-293 (50.28%) than that of positive control, could be anticipated to have better capability towards anticancer drug candidacy. The same derivative showed a better DNA binding profile that further validated YM-1 candidacy against cancer cells. Current work was performed according to the expertise and facilities available for this research. In vivo investigations could be suggested further to endorse our in vitro work.4 Experimental
4.1 Materials
All the reagents and chemicals utilized in the synthesis and experimental work were of analytical grade and purchased from Sigma-Aldrich and used without further purification. The drying and distillation of solvents were made prior to their use by adopting standard procedures. The reaction mixture, during the synthesis, was examined by thin layer chromatography (TLC) in a 9:1 ratio chloroform and methanol using silica gel F254 (Merck) coated aluminum sheets, and detection was carried out under UV-light at 254 and 360 nm.
4.2 Instrumentation
Melting points of the synthesized sulfonamide derivatives were found on a Stuart SMP3 melting point apparatus. Their NMR spectra were recorded on a Bruker 300 (1H-NMR at 300 MHz and 13C-NMR at 75.5 MHz). The chemical shifts are stated in ppm in DMSO-d6 solvent. Moreover, tetramethyl silane (TMS) was used as the standard internal reference. Shimadzu (UV-1800), Hitachi (F-7000), Metrohm potentiostat, (Autolab PGSTAT-302), and a viscometer (Schott-Gerate AVS@310, automated) were the instrumentations used in DNA binding studies by electronic absorption (UV-visible; UV-) and emission (fluorescence; Fluo-) spectroscopies, cyclic voltammetry (CV-), and by viscosity methods. A double-walled electrochemical cell and quartz cells were used in CV and spectroscopic experiments. CV scanning was carried out on a smooth, clean, and shiny surface of a glassy carbon GC electrode having a surface area of 0.070 cm2. Ag/AgCl (3.5 M KCl filling) and platinum wire (0.5 mm diameter) were employed as reference and counter, respectively. Electrochemical experiments were run under the inert atmosphere by purging argon gas for a few minutes in the solution mixture to evacuate the oxygen. For all DNA binding experiments, temperature controller devices were used with the instruments to maintain the required temperature within the cell.4.3 Synthesis protocol
The desired products were synthesized via four steps. The first step was simply an acetylation reaction of labile proton of NH2 of sulfonamide 1 ring with chloroacetyl chloride in the presence of triethylamine base. The second step involved the cyclization of acetylated product 2-chloro-N-(4-sulfamoylphenyl)acetamide 2 with potassium thiocyanide in acetone solvent. In the third step cyclized product 4-(4-oxo-4,5-dihydrothiazol-2-ylamino)benzene sulfonamide 3 was condensed with different aromatic aldehydes. In the final step, the condensed product (Z)-4-(5-benzylidene-4-oxo-4,5-dihydrothiazol-2-ylamino)benzene sulfonamide 4 was converted into desired product 5 by treating with hydroxylamine hydrochloride.4.4 Characterization data
Yellow solid, yield; 70%, m. p.: >300 °C, Rf = 00.52 (chloroform
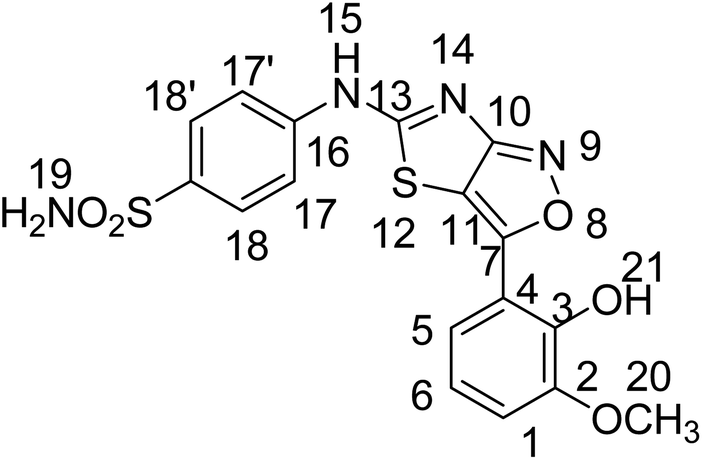 :methanol, 9:1), FTIR ν (cm−1) 3445, 3394, 3293, 3127 (OH, NH2, NH), 3027 (ArH), 2922, 2849 (CH3), 1642 (CN), 1471 (Ar–C), 1471 (CH3), 1221 (CS), 1182 (C–N), 1152 (C–S); 1HNMR (300 MHz, DMSO-d6); δ (ppm) 12.38 (s, 1H, 15), 9.47 (s, 1H, 21), 7.37 (s, 2H, 19), 7.85–6.93 (ArH), 3.79 (s, 3H, 20); 13CNMR (75 MHz, DMSO-d6); 0δ (ppm) 162.64 (13), 151.39 (7), 150.14 (10), 112.87 (11), 147.32, 140.46, 130.93, 127.74, 126.32, 123.84, 122.02, 120.73, 119.61, 115.67 (ArC), 56.08 (20). Anal. calcd for C17H14N4O5S2 C, 48.80; H, 3.37; N, 13.39; S, 15.33. Found: C, 48.82; H, 3.35; N, 13.32; S, 15.36. HRMS (ESI): m/z calcd for [C17H14N4O5S2 + H]+418.0406, found: 418.0408.
:methanol, 9:1), FTIR ν (cm−1) 3445, 3394, 3293, 3127 (OH, NH2, NH), 3027 (ArH), 2922, 2849 (CH3), 1642 (CN), 1471 (Ar–C), 1471 (CH3), 1221 (CS), 1182 (C–N), 1152 (C–S); 1HNMR (300 MHz, DMSO-d6); δ (ppm) 12.38 (s, 1H, 15), 9.47 (s, 1H, 21), 7.37 (s, 2H, 19), 7.85–6.93 (ArH), 3.79 (s, 3H, 20); 13CNMR (75 MHz, DMSO-d6); 0δ (ppm) 162.64 (13), 151.39 (7), 150.14 (10), 112.87 (11), 147.32, 140.46, 130.93, 127.74, 126.32, 123.84, 122.02, 120.73, 119.61, 115.67 (ArC), 56.08 (20). Anal. calcd for C17H14N4O5S2 C, 48.80; H, 3.37; N, 13.39; S, 15.33. Found: C, 48.82; H, 3.35; N, 13.32; S, 15.36. HRMS (ESI): m/z calcd for [C17H14N4O5S2 + H]+418.0406, found: 418.0408.Yellow solid, yield; 71%, m. p.: >300 °C, Rf = 00.57 (chloroform
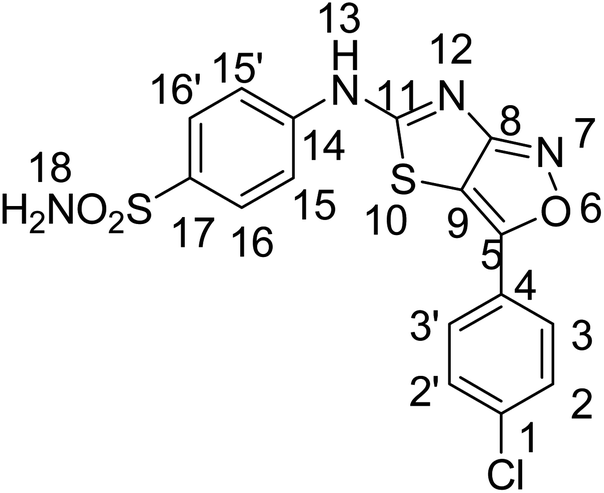 :methanol, 9:1), FTIR ν (cm−1) 3373, 3189, 3102 (NH2, NH), 1696 (CN), 1451 (Ar–C), 1231 (CS), 1123 (C–N), 1076 (C–S); 1HNMR (300 MHz, DMSO-d6); 0δ0 (ppm) 12.6 (s, 1H, 13), 7.54 (s, 2H, 18), 7.85–7.20 (ArH); 13CNMR (75 MHz, DMSO-d6); δ0 (ppm) 171.7 (C–S), 167.7(CN), 151.1 (C–N), 141.4, 135.0, 132.54, 130.4, 129.8, 129.1, 127.7, 123.8 (ArC), 120.9(CC), anal. calcd for C16H11ClN4O3S2 C, 47.23; H, 2.73; N, 13.77; S, 15.76. Found: C, 47.25; H, 2.76; N, 13.75; S, 15.78. HRMS (ESI): m/z calcd for [C16H11ClN4O3S2 + H]+ 405.9961, found: 405.9963.
:methanol, 9:1), FTIR ν (cm−1) 3373, 3189, 3102 (NH2, NH), 1696 (CN), 1451 (Ar–C), 1231 (CS), 1123 (C–N), 1076 (C–S); 1HNMR (300 MHz, DMSO-d6); 0δ0 (ppm) 12.6 (s, 1H, 13), 7.54 (s, 2H, 18), 7.85–7.20 (ArH); 13CNMR (75 MHz, DMSO-d6); δ0 (ppm) 171.7 (C–S), 167.7(CN), 151.1 (C–N), 141.4, 135.0, 132.54, 130.4, 129.8, 129.1, 127.7, 123.8 (ArC), 120.9(CC), anal. calcd for C16H11ClN4O3S2 C, 47.23; H, 2.73; N, 13.77; S, 15.76. Found: C, 47.25; H, 2.76; N, 13.75; S, 15.78. HRMS (ESI): m/z calcd for [C16H11ClN4O3S2 + H]+ 405.9961, found: 405.9963.Yellow solid, yield; 72%, m. p.: >300 °C, Rf = 0.64 (chloroform
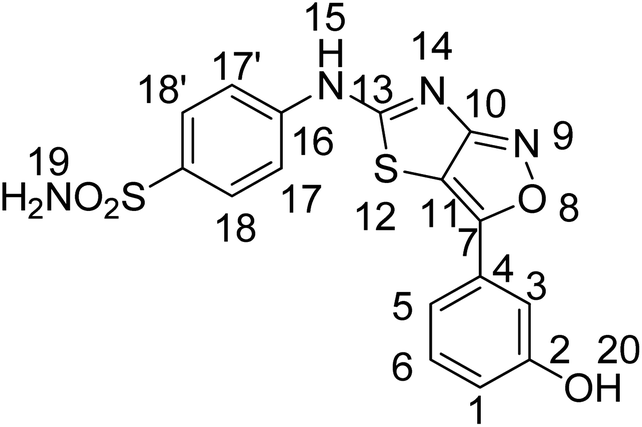 :methanol, 9:1), FTIR ν(cm−1) 3576, 3326, 3248, 3128 (OH, NH2, NH), 3046 (ArH), 1635 (CN), 1479 (Ar–C), 1244 (CS), 1183 (C–N), 1154 (C–S); 1HNMR (300 MHz, DMSO-d6); δ (ppm) 9.91 (s, 1H, 15), 7.92 (s, 1H, 20), 7.52 (s, 2H, 19), 8.00–6.920 (ArH); 13CNMR (75 MHz, DMSO-d6); δ (ppm) 170.17 (13), 165.32 (7), 158.43 (10), 116.47 (11), 167.32, 144.98, 136.24, 134.61, 133.98, 131.02, 129.22, 127.05, 121.95, 121.61 (ArC). Anal. calcd for C16H12N4O4S2 C, 49.47; H, 3.11; N, 14.42; S, 16.51. found: C, 49.44; H, 3.13; N, 14.40; S, 16.50, HRMS (ESI): m/z calcd for [C16H12N4O4S2 + H]+388.0300, found: 388.0302.
:methanol, 9:1), FTIR ν(cm−1) 3576, 3326, 3248, 3128 (OH, NH2, NH), 3046 (ArH), 1635 (CN), 1479 (Ar–C), 1244 (CS), 1183 (C–N), 1154 (C–S); 1HNMR (300 MHz, DMSO-d6); δ (ppm) 9.91 (s, 1H, 15), 7.92 (s, 1H, 20), 7.52 (s, 2H, 19), 8.00–6.920 (ArH); 13CNMR (75 MHz, DMSO-d6); δ (ppm) 170.17 (13), 165.32 (7), 158.43 (10), 116.47 (11), 167.32, 144.98, 136.24, 134.61, 133.98, 131.02, 129.22, 127.05, 121.95, 121.61 (ArC). Anal. calcd for C16H12N4O4S2 C, 49.47; H, 3.11; N, 14.42; S, 16.51. found: C, 49.44; H, 3.13; N, 14.40; S, 16.50, HRMS (ESI): m/z calcd for [C16H12N4O4S2 + H]+388.0300, found: 388.0302.4.5 Computational analysis
Structure building and graphics visualization were made possible with the Amsterdam density function (ADF) modeling suite.52 The optimization of sulfonamide derivatives' (YM-1, 2, and -3) was performed at GGA:PBE (generalized gradient approximation including Perdew–Burke–Ernzerhof) with DZ (double zeta) basis set, which is the most effective, reliable, accurate, and cost-effective technique and hence frequently employed for the structural characteristics of a compound.26 Further, FMOs, MEP and band gap were computed at this level of theory.To design and optimize the structures of sulfonamide derivatives (YM-1, -2, and -3) molecular operating environment (MOE 2015.10) in the MOPAC7 level of theory was employed. The structures were generated by following the geometry relaxation and saved into the MOE database. For the molecular docking simulations with (YM-1, -2, and -3), X-ray crystallographic structures of DNA possessing PDB ID:360D with a resolution of 1.85 A° and carbonic anhydrase and urease enzymes having PDB ID:4XIW and 4AC7, respectively, with a resolution of 2.60 A°, were picked up from the Protein Data Bank. Moreover, the molecules of water bound to base pairs of the DNA were eliminated. The carbonic anhydrase is a tetramer, only 4XIW·A was selected for docking after removing attached water molecules with the amino acid residues of 4XIW·A. While urease is a trimer, all three were used after removing its water and hetero molecules. All probable variations in DNA, carbonic anhydrase and urease were performed with the aid of an MOE sequence editor. Coordinates of the refined 360D 4XIW and 4AC7 were relaxed by using the approaches of PM3 (semi-empirical) and Amber forcefield to remove restrictions and interferences in the molecule due to the structure's protonation by protonate-3D menu in MOE. The relaxation of coordinates led to the least stability and energy for the optimal computation of the best-scoring function calculations. Site finder was used to initialize molecular docking, and the optimized structures were submitted to logical conformational search with RMS (0.01 kcal mol−1) gradient to locate active sites in 360D, 4XIW and 4AC7. All sulfonamide derivatives (YM-1, -2, and -3) were docked with 360D, 4XIW and 4AC7 using positioning as α tri-angle, improvement as induced fit with the score as London ΔG and affinity ΔG. Several docking simulations were run to acquire the most feasible and perfect dock position. The interactional energies of sulfonamide derivatives with 360D 4XIW and 4AC7 were assessed at all stages of the simulation. The rest of the parametric setting was maintained as a default.
4.6 Procedures for DNA binding
Double strands ds-DNA (calf thymus, Sigma-Aldrich) were used for sulfonamide derivative-DNA interactional/binding studies. The absorption spectrum of DNA was taken, which showed an absorbance peak with maxima at 260 nm. Its purity was further ascertained by taking a ratio (abs.@ 260 nm/abs.@ 280 nm), that was found 1.79, indicating protein-free DNA.27 Each sulfonamide derivative's stock solution was prepared in a DMSO–water (1:9) mixture. The concentration of each sulfonamide derivative was then optimized to 2.5 × 10−5 M, while titrating it with the DNA solution. For DNA binding investigations by UV-, Fluo-, and CV- titration experiments, the solution of a sulfonamide derivative was taken in its fixed concentration (2.5 × 10−5 M) and DNA was titrated in μL quantities to obtain their μM concentrations (10 up to 100 μM) within the solution mixture. The DNA viscosity at its fixed concentration was determined and then the viscosity of the sulfonamide derivative–DNA complex was obtained by monitoring the changes in DNA viscosity after adding varying concentrations of sulfonamide derivative. The pH was kept neutral, and all the experimental procedures were run at 37 °C.
4.7 Enzyme inhibition activity
Due to the unavailability of carbonic anhydrase enzyme, experimental work was only carried out with urease enzyme. To determine the anti-urease activity of sulfonamide derivative, experiments were performed in 200 μL 96-well plate by mixing 0.1 U enzyme, buffer of pH 8.2 (0.1 M urea, 0.01 M LiCl2, 0.0 mM EDTA, 0.01 M K2HPO4) and sample (sulfonamide derivative) at concentrations of 100, 75, 50, 25 and 12.5 μM followed by incubation at 37 °C for 15 minutes. After incubation 0.1% NaOCl, 0.5% NaOH and 0.005% Na2[Fe(CN)5NO] and 1% C6H5OH were added, and a second round of incubation was performed at 37 °C for 50 minutes. The experiment was run in triplicate, and a microplate reader was employed to recode the absorbance values at 625 nm. This data was further analyzed using Prism V-8 software and IC50 was calculated.4.8 MTT analysis
In vitro cytotoxicity activity via MTT assay was performed for both normal (human embryonic kidney; HEK-293) and cancer (malignant glioma; MG-U87) cell lines. 1% Penicillin Streptomycin, 10% Fetal Bovine Serum and DMEM (Dulbecco's modified eagle medium (all purchased from Gibco, Life Technologies)) were used in combination to maintain and replicate the cells exponentially in this medium. The growing cells were counted and plated (1 × 104 cells per well, in triplicate) on nunc 96 well culture plates (Fisher Scientific, Denmark). The per well cell volume was kept at 100 μL. A CO2 (5%) incubator was used to incubate the plates for 24 hours at physiological temperature (37 °C). 10% DMSO was used to prepare 1 mL of each sulfonamide derivative (YM-1, -2, and -3), separately. Then, 40, 80, 120, 200, and 400 μM concentrations of each sulfonamide derivative were prepared by the dilution method. 100 μL of each concentration was added, separately, into the prepared microplates so that the final volume per well reached 200 μL. Five concentrations of each derivative were examined, in three sets, on both MG-U87 and HEK-293 cells. The control (solvent control) wells and blank media were used without drugs and without cells, respectively. Finally, 15 μL of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; 5.0 mg mL−1 in PBS) was added per well, incubated for 3 hours at 37 °C, and then visualized under a microscope to ascertain formazan crystals formation. After draining the left solution, and after drying the plates at room temperature, DMSO (100 μL) was added for crystal dissolution, then the absorbance was measured at 550 nm. Doxorubicin hydrochloride was used as a positive reference control.Data availability
The data supporting this article has been included in the experimental part and as part of the ESI.†Author contributions
NA: conceptualization, project administration, supervision, methodology, visualization, validation, investigation, resources, writing—original draft, writing—review and editing. YM: investigation, data curation. HI: methodology, software, investigation, writing—review and editing. FP: software, writing—review and editing. AJ: methodology. PAC: formal analysis. AS: methodology, writing—review and editing. SN: investigation, data curation. FN: software.Conflicts of interest
All authors in the present work declare no conflict of interest.Acknowledgements
The major contribution and research facilities of the Physical Chemistry Laboratory at Allama Iqbal Open University are highly acknowledged. In addition, the contribution and facilitation of all authors' organizations in the present research work are also accredited and appreciated.References
- A. Ovung and J. Bhattacharyya, Sulfonamide drugs: structure, antibacterial property, toxicity, and biophysical interactions, Biophys. Rev., 2021, 13(2), 259–272, DOI:10.1007/%2Fs12551-021-00795-9.
- W. H. Barker, The Uses and Abuses of the Sulfonamide Drugs, Med. Clin. North Am., 1941, 25(2), 453–478, DOI:10.1016/S0025-7125(16)36603-2.
- H. R. Raper and J. G. Manser, Use of Sulfanilamide in Postoperative Acute Cellulitis, D. Digest, 43:434, September 1937, cross reference of; R. W. Edwards, Dental Uses of Sulfanilamide, J. Am. Dent. Assoc., 1940, 27(9), 1394–1397, DOI:10.14219/jada.archive.1940.0272.
- P. A. Channar, A. Saeed, F. Albericio, F. A. Larik, Q. Abbas, M. Hassan, H. Raza and S. Y. Seo, Sulfonamide-linked ciprofloxacin, sulfadiazine and amantadine derivatives as a novel class of inhibitors of jack bean urease; synthesis, kinetic mechanism and molecular docking, Molecules, 2017, 22(8), 1352, DOI:10.3390/molecules22081352.
- C. T. Supuran, A. Casini and A. Scozzafava, Protease inhibitors of the sulfonamide type: anticancer, anti-inflammatory, and antiviral agents, Med. Res. Rev., 2003, 23(5), 535–558, DOI:10.1002/med.10047.
- B. G. Alani, K. S Salim, A. S. Mahdi and A. A. Al-Temimi, Sulfonamide derivatives: Synthesis and applications, Int. J. Pharm. Res., 2024, 4(01), 001–015, DOI:10.53294/ijfcpr.2024.4.1.0021.
- S. Apaydın and M. Török, Sulfonamide Derivatives as Multi-target Agents for Complex Diseases, Bioorg. Med. Chem. Lett., 2019, 29(16), 2042–2050, DOI:10.1016/j.bmcl.2019.06.041.
- B. Blass, Sulfonamide Derivatives and Pharmaceutical Applications Thereof, ACS Med. Chem. Lett., 2016, 7(1), 12–14, DOI:10.1021/acsmedchemlett.5b00466.
- C. B. Scarim and F. R. Pavan, An overview of sulfonamide-based conjugates: Recent advances for tuberculosis treatment, Drug Dev. Res., 2022, 83, 567–577, DOI:10.1002/ddr.21913.
- M. Elagawany, L. Maram and B. Elgendy, Synthesis of 3-Aminoquinazolinones via a SnCl2-Mediated ANRORC-like Reductive Rearrangement of 1,3,4-Oxadiazoles, J. Org. Chem., 2023, 88(24), 17062–17068, DOI:10.1021/acs.joc.3c01973.
- W. Alam, H. Khan, M. S. Jan, U. Rashid, A. Abusharha and M. Daglia, Synthesis, in-vitro inhibition of cyclooxygenases and in silico studies of new isoxazole derivatives, Front. Chem., 2023, 11, 1222047, DOI:10.3389/fchem.2023.1222047.
- A. Sysak and B. O. Mrukowicz, Isoxazole ring as a useful scaffold in a search for new therapeutic agents, Eur. J. Med. Chem., 2017, 137, 292–309, DOI:10.1016/j.ejmech.2017.06.002.
- N. Daryabari, T. Akbarzadeh, M. Amini, R. M. H. Mirkhani and A. Shafieea, Synthesis and calcium channel antagonist activities of new derivatives of dialkyl 1, 4-dihydro-2, 6-dimethyl-4-(5-phenylisoxazol-3-yl) pyridine-3, 5-dicarboxylates, J. Iran. Chem. Soc., 2007, 4(1), 30–36, DOI:10.1007/BF03245800.
- B. L. Deng, Y. Zhao, T. L. Hartman, K. Watson, R. W. Buckheit Jr, C. Pannecouque, E. De Clercq and M. Cushman, Synthesis of alkenyldiarylmethanes (ADAMs) containing benzo [d] isoxazole and oxazolidin-2-one rings, a new series of potent non-nucleoside HIV-1 reverse transcriptase inhibitors, Eur. J. Med. Chem., 2009, 44(3), 1210–1214, DOI:10.1016/j.ejmech.2008.09.013.
- V. E. Kuz'min, A. G. Artemenko, E. N. Muratov, I. L. Volineckaya, V. A. Makarov, O. B. Riabova, P. Wutzler and M. Schmidtke, Quantitative Structure−Activity Relationship Studies of [(Biphenyloxy)propyl]isoxazole Derivatives. Inhibitors of Human Rhinovirus 2 Replication, J. Med. Chem., 2007, 50(17), 4205–4213, DOI:10.1021/jm0704806M.
- G. C. Arya, K. Kaur and V. Jaitak, Isoxazole derivatives as anticancer agent: A review on synthetic strategies, mechanism of action and SAR studies, Eur. J. Med. Chem., 2021, 221, 113511, DOI:10.1016/j.ejmech.2021.113511.
- J. Wang, D.-B. Wang, L.-L. Sui and T. Luan, Natural products-isoxazole hybrids: A review of developments in medicinal chemistry, Arabian J. Chem., 2024, 17(6), 105794, DOI:10.1016/j.arabjc.2024.105794.
- E. Muratov, V. Kuz’min, A. Artemenko, E. Varlamova, V. Makarov, O. Riabova, P. Wutzler and M. Schmidtke, Hit QSAR Analysis of Anti-Coxsackievirus B3 Activity of [Biphenyloxy)Propyl]Isoxazole Derivatives, Antiviral Res., 2008, 78(2), A60–A61, DOI:10.1016/j.antiviral.2008.01.129.
- A. M. Eid, M. Hawash, J. Amer, A. Jarrar, S. Qadri, I. Alnimer, A. Sharaf, R. Zalmoot, O. Hammoudie, S. Hameedi and A. Mousa, Synthesis and Biological Evaluation of Novel Isoxazole-Amide Analogues as Anticancer and Antioxidant Agents, BioMed Res. Int., 2021, 2021, 6633297, DOI:10.1155/2021/6633297.
- R. Vaickelionienė, V. Petrikaitė, I. Vaškevičienė, A. Pavilonis and V. Mickevičius, Synthesis of novel sulphamethoxazole derivatives and exploration of their anticancer and antimicrobial properties, PLoS One, 2023, 18(3), e0283289, DOI:10.1371/journal.pone.0283289.
- M. Pervaiz, A. Riaz, A. Munir, Z. Saeed, S. Hussain, A. Rashid, U. Younas and A. Adnan, Synthesis and characterization of sulfonamide metal complexes as antimicrobial agents, J. Mol. Struct., 2020, 1202(10), 127284, DOI:10.1016/j.molstruc.2019.127284.
- F. Perveen, N. Arshad, R. Qureshi, J. Nowsherwan, A. Sultan, B. Nosheen and H. Rafique, Electrochemical, spectroscopic and theoretical monitoring of anthracyclines' interactions with DNA and ascorbic acid by adopting two routes: Cancer cell line studies, PLoS One, 2018, 13(10), e0205764, DOI:10.1371/journal.pone.0205764.
- F. Perveen, R. Qureshi, F. L. Ansari, S. Kalsoom and S. Ahmed, Investigations of drug–DNA interactions using molecular docking, cyclic voltammetry and UV–Vis spectroscopy, J. Mol. Struct., 2011, 1004(1–3), 67–73, DOI:10.1016/j.molstruc.2011.07.027.
- A. Arab and M. Habibzadeh, Comparative hydrogen adsorption on the pure Al and mixed Al–Si nano clusters: a first principle DFT study, Comput. Theor. Chem., 2015, 1068, 52–56, DOI:10.1016/j.comptc.2015.06.021.
- H. Zgou, S. Boussaidi, A. Eddiouane, H. Chaib, R. P. Tripathi, T. Ben Hadda, M. Bouachrine and M. Hamidi, New low band-gap conjugated organic materials based on fluorene, thiophene and phenylene for photovoltaic applications: Theoretical study, Mater. Today: Proc., 2016, 3(7), 2578–2586, DOI:10.1016/j.matpr.2016.04.005.
- N. Arshad, M. Shakeel, A. Javed, F. Perveen, A. Saeed, A. Ahmed, H. Ismail, P. A. Channar and F. Naseer, Exploration of newly synthesized amantadine-thiourea conjugates for their DNA binding, anti-elastase, and anti-glioma potentials, Int. J. Biol. Macromol., 2024, 263(1), 130231, DOI:10.1016/j.ijbiomac.2024.130231.
- N. Arshad, U. Parveen, P. A. Channar, A. Saeed, W. S. Saeed, F. Perveen, A. Javed, H. Ismail, M. I. Mir, A. Ahmed, B. Azad and I. Khan, Investigations on Newly Synthesized Bis-Acyl-Thiourea Derivatives of 4-Nitrobenzene-1,2-Diamine for Their DNA Binding, Urease Inhibition and Anti-Brain Tumor Activities, Molecules, 2023, 28(6), 2707, DOI:10.3390/molecules28062707.
- M. Sirajuddin, S. Ali and A. Badshah, Drug–DNA interactions and their study by UV–Visible, fluorescence spectroscopies and cyclic voltammetry, J. Photochem. Photobiol., B, 2013, 124, 1–19, DOI:10.1016/j.jphotobiol.2013.03.013.
- M. Ibrahim, H. U. Nabi, N. Muhammad, M. Ikram, M. Khan, M. Ibrahim, A. F. AlAsmari, M. Alharbi and A. Alshammari, Synthesis, Antioxidant, Molecular Docking and DNA Interaction Studies of Metal-Based Imine Derivatives, Molecules, 2023, 28, 5926, DOI:10.3390/molecules28155926.
- T. Göktürk, E. S. Çetin, T. Hökelek, H. Pekel, Ö. Şensoy, E. N. Aksu and R. Güp, Synthesis, Structural Investigations, DNA/BSA Interactions, Molecular Docking Studies, and Anticancer Activity of a New 1,4-Disubstituted 1,2,3-Triazole Derivative, ACS Omega, 2023, 8(35), 31839–31856, DOI:10.1021/acsomega.3c03355.
- S. Kumar and M. S. Nair, Deciphering the interaction of flavones with calf thymus DNA and octamer DNA sequence (CCAATTGG)2, RSC Adv., 2021, 11, 29354–29371, 10.1039/D1RA04101K.
- N. Arshad, A. Saeed, F. Perveen, R. Ujan, S. I. Farooqi, P. A. Channar, G. Shabir, H. R. El-Seedi, A. Javed, M. Yamin, M. Bolte and T. Hökelek, Synthesis, X-ray, Hirshfeld surface analysis, exploration of DNA binding, urease enzyme inhibition and anticancer activities of novel adamantane-naphthyl thiourea conjugate, Bioorg. Chem., 2021, 109(14), 104707, DOI:10.1016/j.bioorg.2021.104707.
- M. Shaldam, H. Tawfik, H. Elmansi, F. Belal, K. Yamaguchi, M. Sugiura and G. Magdy, Synthesis, crystallographic, DNA binding, and molecular docking/dynamic studies of a privileged chalcone-sulfonamide hybrid scaffold as a promising anticancer agent, J. Biomol. Struct. Dyn., 2023, 41(18), 8876–8890, DOI:10.1080/07391102.2022.2138551.
- K. Sakthikumar, B. K. Isamura and R. W. M. Krause, An Integrated Analysis of Mechanistic Insights into Biomolecular Interactions and Molecular Dynamics of Bio-Inspired Cu(II) and Zn(II) Complexes towards DNA/BSA/SARS-CoV-2 3CLpro by Molecular Docking-Based Virtual Screening and FRET Detection, Biomolecules, 2022, 12, 1883, DOI:10.3390/biom12121883.
- N. Arshad, N. Abbas, F. Perveen, B. Mirza, A. M. Almuhaini and S. Alkahtani, Molecular docking analysis and spectroscopic investigations of zinc(II), nickel(II) N-phthaloyl-balanine complexes for DNA binding: Evaluation of antibacterial and antitumor activities, J. Saudi Chem. Soc., 2021, 25, 101323, DOI:10.1016/j.jscs.2021.101323.
- S. I. Farooqi, N. Arshad, F. Perveen, P. A. Channar, A. Saeed and A. Javeed, Aroylthiourea derivatives of ciprofloxacin drug as DNA binder: Theoretical, spectroscopic and electrochemical studies along with cytotoxicity assessment, Arch. Biochem. Biophys., 2019, 666, 83–98, DOI:10.1016/j.abb.2019.03.021.
- S. I. Farooqi, N. Arshad, F. Perveen, P. A. Channar, A. Saeed, F. A. Larik and A. Javeed, Synthesis, theoretical, spectroscopic and electrochemical DNA binding investigations of 1, 3, 4-thiadiazole derivatives of ibuprofen and ciprofloxacin: Cancer cell line studies, J. Photochem. Photobiol., B, 2018, 189, 104–118, DOI:10.1016/j.jphotobiol.2018.10.006.
- A. Kosiha, C. Parthiban and K. P. Elango, Metal(II) complexes of bioactive aminonaphthoquinone-based ligand: synthesis, characterization and BSA binding, DNA binding/cleavage, and cytotoxicity studies, J. Coord. Chem., 2018, 7(10), 1560–1574, DOI:10.1080/00958972.2018.1461846.
- S. Ramotowska, A. Ciesielska and M. Makowski, What Can Electrochemical Methods Offer in Determining DNA-Drug Interactions?, Molecules, 2021, 26(11), 3478, DOI:10.3390/molecules26113478.
- A. Yahiaoui, B. Nabil, A. Messai, T. Lanez1 and E. Lanez, Voltametric and molecular docking investigations of ferrocenylmethylaniline and its N-acetylated derivative interacting with DNA, J. Electrochem. Sci. Eng., 2024, 14(2), 135–145, DOI:10.5599/jese.2061.
- N. Arshad, M. I. Mir, F. Perveen, A. Javed, M. Javaid, A. Saeed, P. A. Channar, S. I. Farooqi, S. Alkahtani and J. Anwar, Investigations on Anticancer Potentials by DNA Binding and Cytotoxicity Studies for Newly Synthesized and Characterized Imidazolidine and Thiazolidine-Based Isatin Derivatives, Molecules, 2022, 27(2), 354, DOI:10.3390/molecules27020354.
- S. I. Farooqi, N. Arshad, F. Perveen, P. A. Channar, A. Saeed, A. Javed, T. Hökelek and U. Flörke, Structure and surface analysis of ibuprofen-organotin conjugate: Potential anti-cancer drug candidacy of the compound is proven by in-vitro DNA binding and cytotoxicity studies, Polyhedron, 2020, 192, 114845, DOI:10.1016/poly.2020.114845.
- S. I. Farooqi and N. Arshad, Cyclic Voltammetric DNA Binding Investigations on Some Anticancer Potential Metal Complexes: a Review, Appl. Biochem. Biotechnol., 2018, 186, 1090–1110, DOI:10.1007/s12010-018-2818-z.
- H. H. AL-Gedany, A. A. Shoukry and S. R. Al-Mhayawi, Synthesis, Speciation, DNA Binding, Electrochemical and Antiproliferative Properties of Pd(II) Complexes Designed to Improve the Interaction with DNA, Egypt. J. Chem., 2021, 64(7), 3913–3926, DOI:10.21608/EJCHEM.2021.74711.3679.
- D. V. Thomaz and P. A. d. Santos, The Electrochemical Behavior of Methotrexate upon Binding to the DNA of Different Cell Lines, Med. Sci. Forum, 2021, 3(1), 16, DOI:10.3390/IECC2021-09215.
- N. Arshad, U. Yunus, S. Razzque, M. Khan, S. Saleem, B. Mirza and N. Rashid, Electrochemical and spectroscopic investigations of isoniazide and its analogs with ds.DNA at physiological pH: evaluation of biological activities, Eur. J. Med. Chem., 2012, 47, 452–461, DOI:10.1016/j.ejmech.2011.11.014.
- N. Kasyanenko, E. Belyi, I. Silanteva, V. Demidov and A. Komolkin, DNA Interaction with Coordination Compounds of Cd(II)containing 1,10-Phenanthroline, Int. J. Mol. Sci., 2024, 25, 1820, DOI:10.3390/ijms25031820.
- N. K. Janjua, A. Shaheen, A. Yaqub, F. Perveen, S. Sabahat, M. Mumtaz, C. Jacob, L. A. Ba and H. A. Mohammed, Flavonoid–DNA binding studies and thermodynamic parameters, Spectrochim. Acta, Part A, 2011, 79(5), 1600–1604, DOI:10.1016/j.saa.2011.05.018.
- N. Arshad, P. A. Channar, A. Saeed, S. I. Farooqi, A. Javeed, F. A. Larik, W. A. Abbasi and U. Flörke, Structure elucidation, DNA binding, DFT, molecular docking and cytotoxic activity studies on novel single crystal ( E )-1-(2-fluorobenzylidene)thiosemicarbazide, J. Saudi Chem. Soc., 2018, 22(8), 1003–1013, DOI:10.1016/j.jscs.2018.05.002.
- M. Xu, T. Dai, Y. Wang and G. Yang, The incipient denaturation mechanism of DNA, RSC Adv., 2022, 12(36), 23356–23365, 10.1039/d2ra02480b.
- R. Muhammad, H. Rafique, S. Roshan, S. Shamas, Z. Iqbal, Z. Ashraf, Q. Abbas, M. H. Z. R. Qureshi and M. H. H. B. Asad, Enzyme Inhibitory Kinetics and Molecular Docking Studies of Halo-Substituted Mixed Ester/Amide-Based Derivatives as Jack Bean Urease Inhibitors, BioMed Res. Int., 2020, 2020, 1–11, DOI:10.1155/2020/8867407.
- (a) M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 09, Rev. C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed; (b) R. Dennington, T. Keith and J. Millam, GaussView 05, Semichem Inc., 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06412g |
| This journal is © The Royal Society of Chemistry 2024 |