
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDomino dehydration/intermolecular (enantioselective) ketone-ene reactions catalysed by a simple solid in batch and in flow†
Miguel Espinosa and
Antonio Leyva-Pérez *
*
Instituto de Tecnología Química (UPV-CSIC), Universidad Politècnica de València-Agencia Estatal Consejo Superior de Investigaciones Científicas, Avda. de los Naranjos s/n, 46022 Valencia, Spain. E-mail: anleyva@itq.upv.es
First published on 18th October 2024
Abstract
The intermolecular carbonyl-ene reaction of ketones is still considered a challenge in organic chemistry, particularly with reusable solid catalysts, and implemented in a domino reaction. Herein, we show that the extremely cheap and non-toxic solid salt MgCl2 catalyzes the reaction of trifluoromethyl pyruvates not only during the conventional carbonyl-ene reaction with various aromatic and alkyl alkenes (in very high yields, up to >99%) but also in a domino reaction with the corresponding alcohols (precursors to the alkenes) in similar good yields. The solid can be reused in both cases without any erosion of the catalytic activity and can be employed in an in-flow process to maximize the reaction throughput. Besides, the reaction can be performed under solventless reaction conditions. Addition of a catalytic amount of chiral binaphthyl hydrogen phosphate allows carrying out the reaction with a reasonable enantiomeric excess (up to >70%) and in flow, in a rare example of enantioselective solid-catalyzed domino carbonyl-ene reaction using a cheap, simple, readily available and physically mixed catalytic solid. The MgCl2-catalytic system is also active in the industrially relevant citronellal-to-isopulegol carbonyl-ene reaction. These results pave the way to design sustainable domino carbonyl-ene reactions with extremely cheap solid catalysts.
1 Introduction
Use of reusable solid catalysts in relatively complex organic transformations is a key step towards the development of a sustainable organic synthesis.1 However, these solid catalysts should ideally be readily available, non-toxic and as cheap as possible;2 otherwise, researchers tend to employ much easier-to-prepare soluble catalysts, although the latter cannot be recovered at the end of the reaction. A representative example of the lack of simple solid catalysts in organic transformations is the carbonyl-ene reaction, which typically employs soluble catalysts, such as metal salts, complexes or organocatalysts (Fig. 1).3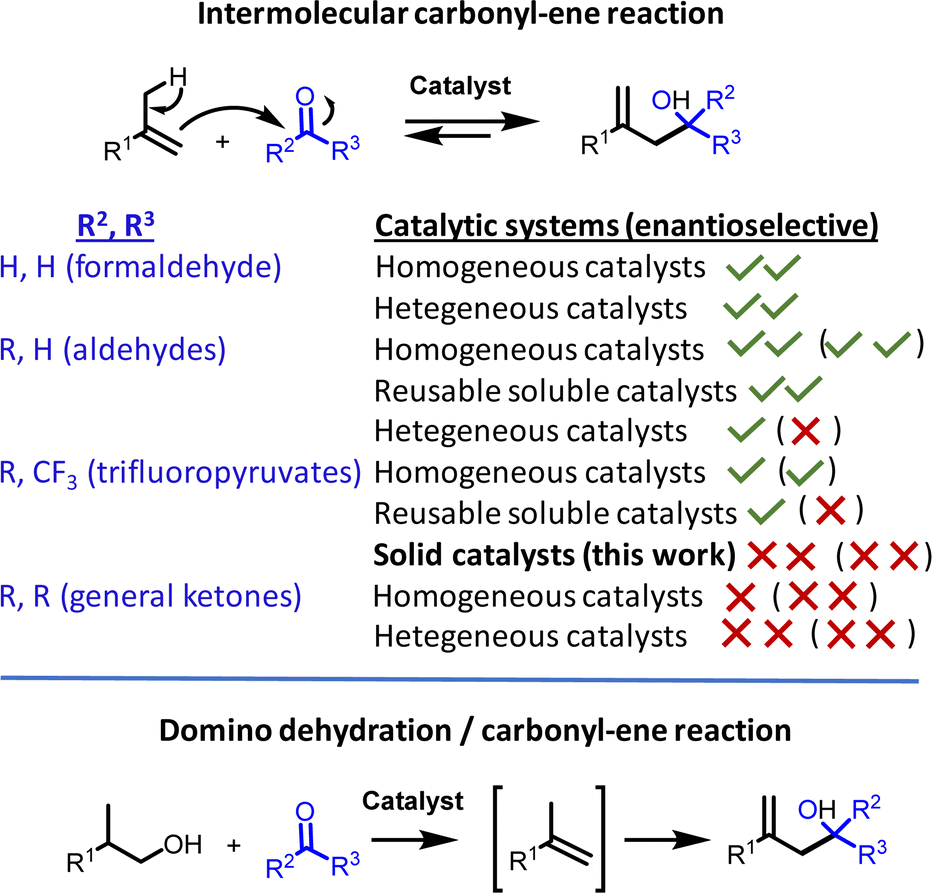 | ||
| Fig. 1 Overall view of the catalytic maturity during the intermolecular carbonyl-ene reaction of the different reactive carbonyl groups and the envisioned domino reaction with ketones. ✓✓ well-resolved, ✓ several examples, ✗ few examples, ✗✗ rare examples or none. | ||
The carbonyl-ene reaction is a fundamental reaction in organic chemistry, with industrial implementation in the multi-ton manufacturing of isopulegol to menthol,4a,b the synthesis of drugs4c or the production of pheromones,4d to name a few.4e This [3,3]-sigmatropic transformation proceeds with complete atom economy, but it is highly dependent on the nature of the carbonyl group since aldehydes react much faster than ketones. While these reactive differences are not so relevant for intramolecular reactions, where the entropic gain by cyclic formation overcomes not only the reactivity resistance but also the potential reversible reaction and undesired alkene isomerization reactions, the latest undesired issues become dominant in the intermolecular carbonyl-ene reaction.5 Therefore, it is not surprising that aldehydes are, by far, the most studied starting materials for the intermolecular reaction and that various soluble catalysts have been reported in both racemic and enantioselective versions,3,6 including strategies such as ionic liquid solvents and selective precipitation to recover the catalyst (Fig. 1).7
Solid catalysts have been employed successfully for the intermolecular aldehyde-ene reaction, in particular for the coupling with formaldehyde, where simple solid catalysts such as zeolites are able to catalyze the transformation.8 However, for less reactive aldehydes, more complex solids are typically employed,4b,9 which often involve elaborated hybrid materials where a soluble catalyst has been immobilized by covalent anchoring on the solid surface or physically entrapped into a micro-structured structure,10 and in well-designed catalytic sites inserted into metal–organic frameworks.11 These solid catalysts of course merit acknowledgement and pave new ways to perform sustainable aldehyde-ene reactions; however, their limited availability and high price hamper wide-ranging catalytic applications (Fig. 1).
The intermolecular ketone-ene reaction is much less developed than the aldehyde reaction, in all types of versions to be considered, i.e. racemic,12 enantioselective,13 with reusable soluble catalysts7,14a,b or solid-catalyzed,14c with most of the examples reported for trifluoromethyl pyruvates. Racemic intermolecular trifluoromethyl pyruvate-ene reactions are catalysed by either Cu,12a Ni12a or Fe12b salts, and enantioselective reactions have been reported for Cu,13a Pt,13b Pd,13b,c Rh,13d Ti13e and Mg (solventless) chiral complexes,13f and also with organocatalysts.13g–i Regarding reusable soluble metal catalysts, the ionic liquid approach has been studied for Pd7 and In14a complexes, and the selective precipitation approach, for Cu complexes.14b However, trifluoromethyl pyruvate-ene reactions with solid catalysts are particularly devoid of examples in the literature: we just found a 20 years-old study on entrapped Cu complexes in zeolites.14c Thus, any solid catalyst able to catalyze the intermolecular ketone-ene reaction, even for activated ketones such as trifluoromethyl pyruvates, would constitute a step forward in the field. Furthermore, if this solid is readily available, cheap and non-toxic, in other words really sustainable, the advance will be more relevant, since it is plausible that any other easier carbonyl-ene reaction, i.e. with aldehydes or intramolecular, will also be feasible with this solid catalyst. The enantioselective version with this type of cheap and available solid will only make sense, in our opinion, with a relatively simple and cheap chiral substance, after a physical mixture with the solid, since an otherwise elaborated complex material synthesis will avoid any wide access to researchers.
Domino reactions are desirable in organic synthesis to minimize the isolation of intermediates and maximize the throughput of the reaction, i.e. to intensify the chemical process.15 The domino reaction makes further sense if different reactions are catalysed by the same compound. The alkenes employed in the carbonyl-ene reaction come from the corresponding cheaper alcohols, after dehydration, thus a domino reaction from the alcohol to the final carbonyl-ene product would be of interest. However, this domino reaction is not straightforward with ketones since the equivalent of H2O generated during the dehydration reaction can degrade most of the metal catalysts employed so far for the carbonyl-ene reaction; indeed, we could not find in the literature any example of domino dehydration/intermolecular carbonyl-ene reaction with ketones, although other domino carbonyl-ene reactions have been reported.16 A simple solid catalyst for the intermolecular carbonyl-ene reaction of trifluoropyruvates, compatible with water, would open the door for this envisioned domino reaction.
MgCl2 is one of the cheapest solids available, massively used in industrial catalysis as a support/modifier for the Ziegler–Natta catalyst.17 However, the catalytic behaviour of MgCl2 in organic synthesis is yet to be exploited.18 Herein, we show that MgCl2 catalyzes not only the intermolecular ketone-ene reaction of trifluoromethyl pyruvates in high yields, but also the domino dehydration/intermolecular (enantioselective) carbonyl-ene reaction with tertiary alcohols, and that the solid is fully reusable (up to ten uses for the ketone-ene reaction). MgCl2 has apparently not been reported as a catalyst but as an additive in carbonyl-ene reactions.19 Besides, this process can be run in flow. The use of an amorphous solid catalyst such as MgCl2 enables the suppression of the solvent, in contrast to some elaborated or micro-structured solid catalysts, where the diffusion of reactants and products plays a key role during the catalytic process.20 Thus, our protocol here can be carried out solventless and with a 1/1 molar ratio of both reactants (the ketone with either the alcohol or the alkene), which maximizes the sustainable value of the process and also makes a substantial difference with many of the catalysts reported for the intermolecular carbonyl-ene reaction. In addition, the enantioselective version is achieved by mixing MgCl2 with binaphthyl hydrogen phosphate,21 one of the cheapest chiral proton substances available, to be run in flow with decent enantioselective excess.
2 Experimental section
2.1 Materials
All reagents were obtained from commercial sources and used as received. Anhydrous MgCl2 was obtained from Merck-Sigma. The list of synthesized compounds and their abbreviations (3–29) is given in the ESI†.2.2 Instrumentals
See ESI† for details.2.3 Catalytic tests
3 Results and discussion
3.1 Intermolecular carbonyl-ene reaction with trifluoromethyl pyruvates
|
|||||
|---|---|---|---|---|---|
| Entry | Metal catalyst (5 mol%) | 2 (eq.) | Solvent | Drying agenta | 3b (%) |
| a MS: molecular sieves.b GC yields, double-checked by 1H-NMR, and referred to the starting material 1.c Polymerization of 1. | |||||
| 1c | CuCl2·2H2O | 1.5 | DCM | — | <1 |
| 2 | CuCl2 | 4 Å MS | >99 | ||
| 3 | Cu(OTf)2 | >99 | |||
| 4 | Cu(OAc)2 | 65 | |||
| 5 | CuO | 66 | |||
| 6 | CuCl | 87 | |||
| 7 | FeCl2 | 91 | |||
| 8 | CuCl2 | — | >99 | ||
| 9 | Cu(OTf)2 | >99 | |||
| 10 | — | 4 Å MS | 80 | ||
| 11 | CH(OEt)3 | 47 | |||
| 12 | — | 55 | |||
| 13 | MgSO4 | 80 | |||
| 14 | MgCl2 | — | >99 | ||
| 15 | MgO | 67 | |||
| 16 | MgCO3 | 30 | |||
| 17 | CaCO3 | 38 | |||
| 18 | MgCl2 | 1.0 | >99 | ||
| 19 | Toluene | 86 | |||
| 20 | Acetone | 3 | |||
| 21 | EtOAc | 19 | |||
| 22 | EtOH | <1 | |||
| 23 | Et2O | 56 | |||
| 24 | ACN | 61 | |||
| 25 | THF | <1 | |||
| 26 | No solvent | >99 | |||
Other Cu salts such as Cu(OAc)2, CuO and CuCl gave intermediate yields of product 3 (65–87%, entries 4–6) after 18 h of reaction time, and the reactions did not progress further beyond an additional 5–10% after monitoring the reaction for 96 h (4 days). These results suggest that an intermediate acidity of the metal salt is positive to catalyze the reaction. The iron salt FeCl2 gave 91% yield (entry 7) and the more active Cu salts also gave >99% yield of 3 without the 4 Å MS (entries 8 and 9). A blank experiment with 4 Å MS gave 80% yield of 3 (entry 10),22a higher than the blank experiment with an organic drying agent (ethyl acetal, 47%, entry 11) or without any additive or catalyst (55%, entry 12). The use of another inorganic drying agent such as MgSO4 gave 80% yield of 3 (entry 13), which not only confirms that the removal of water is beneficial for the reaction, but also suggests that water can be present during the reaction and that inorganic drying agents might also catalyse the reaction.22a To check this, MgCl2 was used as a catalyst without any other drying agent, and product 3 was obtained >99% yield (entry 14). Other Mg2+ salts such as MgO and MgCO3 were much less active for the reaction (30–67%, entries 15 and 16) as well as CaCO3 (38%, entry 17). The fact that basic metal salts decrease the yield below the blank experiment (compare entries 16, 17 and 12) indicates that neutral to acid conditions are necessary for the carbonyl-ene reaction to proceed.
The reaction was repeated without any excess of pyruvate 2, i.e. with equimolecular amounts of 1 and 2, and >99% yield of 3 was still obtained (entry 18). The kinetic experiment shown in Fig. 2 clearly showcases the catalytic action of MgCl2. At this point, the screening of different solvents was performed (entries 19–26). Toluene was the only solvent to give a high yield of 3 (86%, entry 19) with equimolecular amounts of 1 and 2, while acetone, ethyl acetate (EtOAc), ethanol (EtOH), diethyl ether (Et2O), acetonitrile (ACN) and tetrahydrofuran (THF) gave poorer results (>1–63%, entries 20–25). It seems that somewhat polar aprotic solvents are beneficial for the reaction.22b Gratifyingly, the solventless reaction gave >99% yield of product 3. Notice that neither the reactants nor the solvents were thoroughly dried, indicating that the reaction conditions were compatible with water in the reaction medium, of interest for a possible domino dehydration/carbonyl-ene reaction. A kinetic experiment confirms the catalytic action of MgCl2 under solventless reaction conditions (Fig. S1†). This result is satisfactory from both an economic and sustainable point of view, since the carbonyl-ene reaction proceeds quantitatively without any type of waste (nor solvent) and with an extremely cheap and easy-to-remove solid catalyst (see ahead), to give the final product 3 after simple filtration. It is worthy to comment here that these catalytic results with Mg(II) are drastically different from those previously reported with some soluble Mg(II) triflate salts,13f since our study is focused on a simple, non-toxic, inexpensive and reusable solid catalyst. MgCl2 acts here as a general acid catalyst for the carbonyl-ene reaction, the mechanism of which is well known (see Fig. 1).
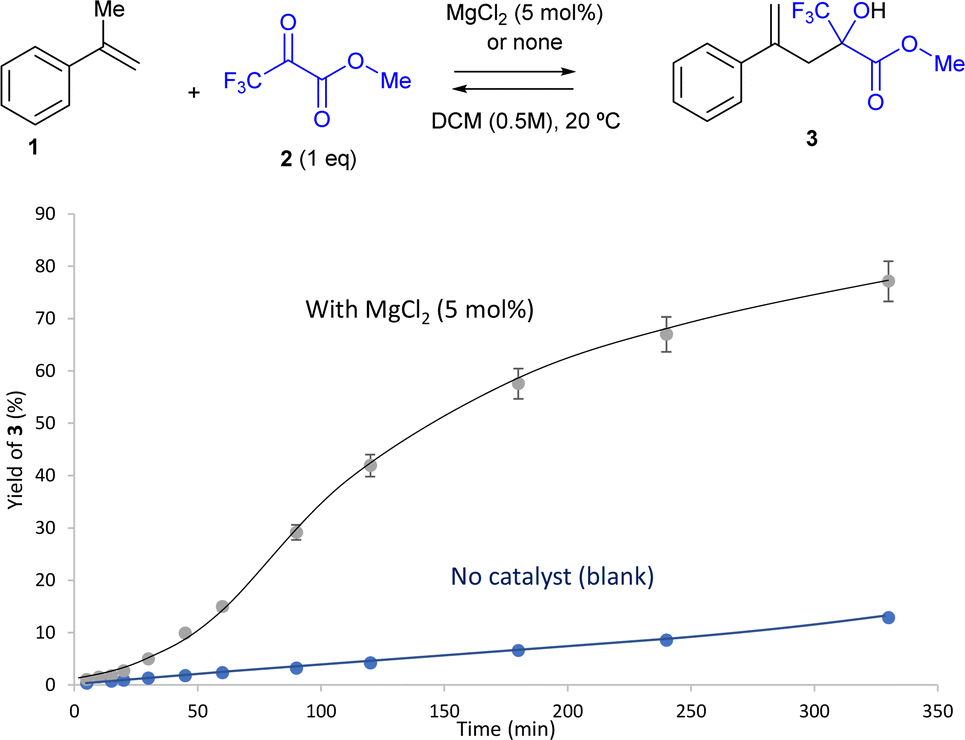 | ||
| Fig. 2 Kinetic plot of the intermolecular ketone-ene reaction between α-methyl styrene 1 and methyl pyruvate 2 (1 equivalent) catalyzed by MgCl2 (5 mol%, black line) or none (blue line) in a DCM (0.5 M) solvent at room temperature. Lines are a guide to the eye. GC yields. Error bars account for 5% uncertainty. | ||
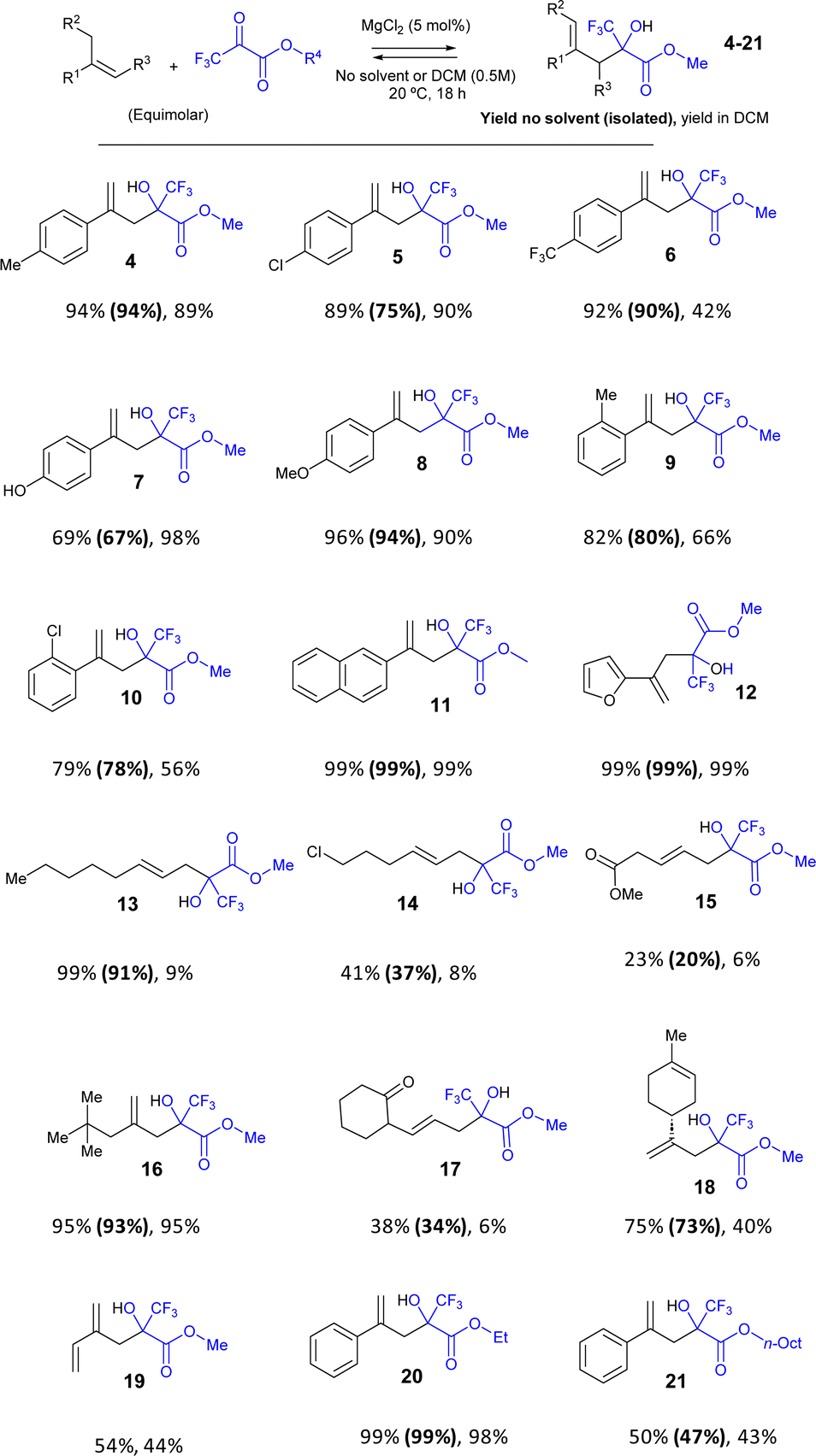 | ||
| Fig. 3 Scope of the carbonyl-ene reaction of different alkenes and pyruvates catalyzed by MgCl2 (5 mol%), at room temperature for 18 h, without any solvent or DCM (0.5 M). The results from left to right indicate: GC yield for the solventless reaction, isolated yield (between parentheses) and GC yield in DCM. Reactions were performed twice, showing very good reproducibility. | ||
A variety of para-substituted α-methyl styrenes engage well with pyruvate 2 to give the carbonyl-ene methyl-, chloro-, trifluoro-, hydroxy- and methoxy-substituted products 4–8 in very good isolated yields (67–94%) under solventless reaction conditions. The ortho-substituted products 9–10 were also obtained in good isolated yields (78–80%). It is noteworthy that some of these products are obtained in significantly lower yields when DCM is used as a solvent (products 6, 9 and 10). On the contrary, only in one case (product 7), the yield was higher in DCM, probably due to the lower solubility of the phenol group. Other aromatic rings such as naphthalene and furan are tolerated, to give products 11–12 in quantitative (99%) isolated yields.
A simple alkyl alkene also works satisfactorily in the reaction (product 13, 91% isolated yield after 72 h reaction time), although the presence of a chloro-substitution significantly decreases the final yield (product 14, 37% isolated yield) as well as with an ester substituted alkene (product 15, 20% isolated yield) after 6 days. However, a tert-butyl-substituted alkyl alkene is obtained in very high yields (product 16, 93% isolated yield). An unsaturated ketone is also reactive (product 17, 34% isolated yield) and particularly remarkable is the reaction with the natural compound limonene, to give product 18 in 73% isolated yield. It is noticeable that a good tolerance to different functional groups is found during the reaction, and also that most of the alkyl alkenes showed lower yields in the DCM solution, in some cases <10% GC yield.
A diene such as isoprene also reacts to give product 19 in 54% yield; however, the Diels–Alder reaction competes under the present reaction conditions. Since both reactions are in equilibrium, the products of both reactions may interconvert during the course of the reaction. In order to assess this hypothesis, kinetic experiments were performed. The results (Fig. S2†) show that, indeed, the Diels–Alder product of isoprene with pyruvate 2 rapidly appears at the beginning of the reaction, to then slow and finally decrease, and give the carbonyl-ene product 19. The formation of 19 is only possible by the action of MgCl2, since the reaction without catalyst gives the Diels–Alder product in >98% yield, at the same initial reaction rate than with MgCl2 (Fig. S3†). In other words, MgCl2 takes advantage of the equilibrium of the Diels–Alder reaction to drive the reactants to the carbonyl-ene product 19 under the present reaction conditions.
The pyruvate methyl ester can be changed, and ethyl and n-octyl pyruvates were also reactive with alkene 1, to give products 20 and 21 in 99% and 47% isolated yields, respectively. These results, together, demonstrate the reasonable generality of the MgCl2-catalyzed pyruvate-ene reaction under solventless reaction conditions and employing equimolecular amounts of reactants, which results in the higher yields, to directly isolate the product after simple filtration.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Use | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Yield (%) | 92 | 86 | 94 | 84 | 84 | 86 | 80 | 82 | 94 | 84 |
The used MgCl2 catalyst was analyzed by powder X-ray diffraction (PXRD), Fourier transformed infrared (FT-IR) spectroscopy and X-ray photoelectron spectroscopy (XPS). The results show that the same diffractogram was obtained for the fresh and recovered MgCl2 catalyst (Fig. S4†), and also a very similar FT-IR spectrum (Fig. S5†). Besides, the Mg 1s (Fig. S6†) and Cl 2p (Fig. S7†) XPS spectra of the fresh and recovered MgCl2 coincide, indicating that any change in the oxidation state or bonding in the solid does not occur during the reaction. A filtration test shows that the reaction does not evolve further after removing the solid MgCl2 catalyst, beyond the blank experiment (see Fig. 2). These results confirm the robustness of MgCl2 during the intermolecular ketone-ene reaction between 1 and 2.
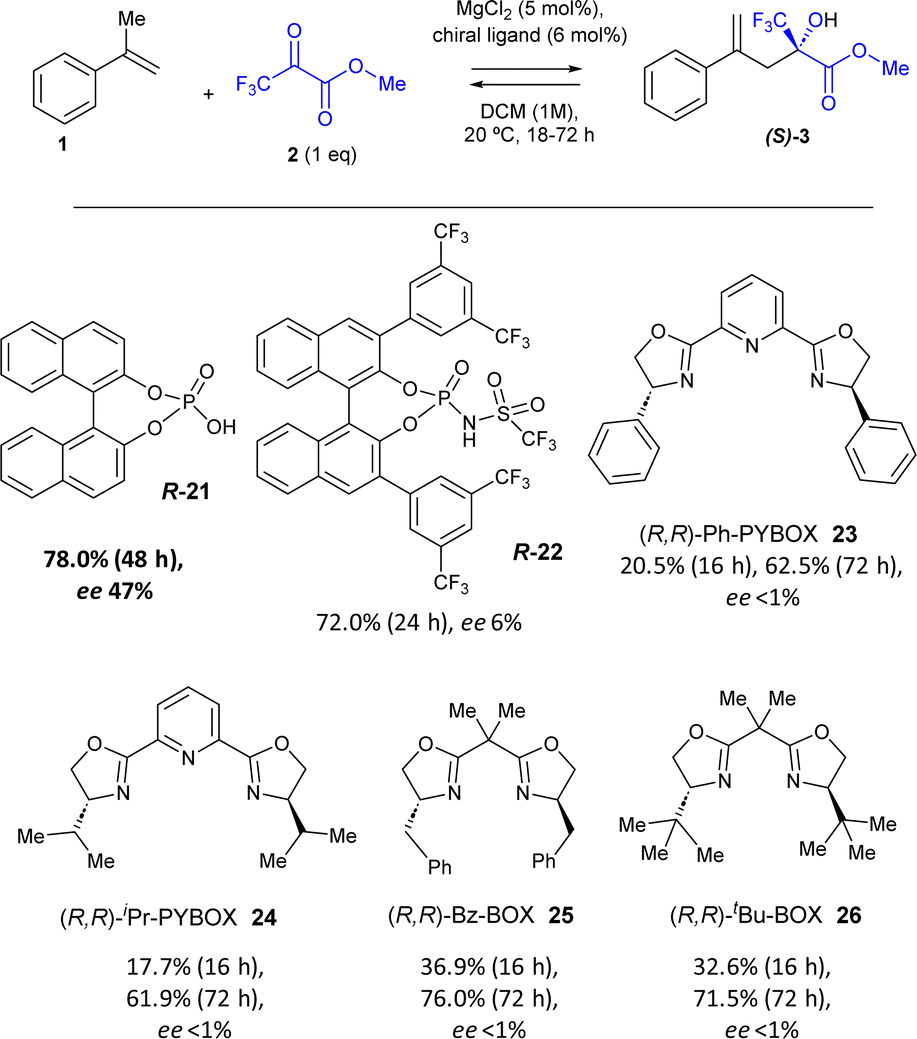 | ||
| Fig. 4 Enantioselective catalytic results for the intermolecular ketone-ene reaction between α-methyl styrene 1 and methyl pyruvate 2 (1 equivalent) catalyzed by MgCl2 (5 mol%) and the chiral (R) ligands 21–26 (6 mol%), in DCM (0.5 M) at room temperature. Yields calculated by GC and enantiomeric excess (ee) calculated by chiral HPLC resolution. | ||
The addition of the simple (R)-(−)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate chiral phosphoric acid 21 (6 mol%) to the MgCl2-catalysed reaction yielded the product (S)-3 in 78% yield and induced an enantiomeric excess (ee) of 47%, according to high-pressure liquid chromatography (HPLC) in chiral columns after subtracting the blank experiment. Kinetic experiments show that the catalysis is caused by MgCl2 and that the ligand only slows down the reaction (Fig. S8†). The much more hindered and acidic triflurophosphoramide naphthol (R) ligand 22 (ref. 24) gave product 3 in a similar yield (72%) but with just 6% ee. Different PyBox (R,R) ligands 23–26 also gave product 3 in 62–76% yields but without any ee. These results strongly support the coordination of the chiral ligand 21 with the MgCl2 solid catalyst to induce enantioselectivity, and the poorer interaction of MgCl2 with the rest of ligands 22–26.
Table 3 shows the optimization of reaction conditions with the catalytic combination of MgCl2/ligand 21. The results show that the addition of 20 mol% of ligand 21 is beneficial for the ee of the reaction, to give 85% yield and 61% ee after 72 h reaction time and subtraction of the blank (entry 1). If the reaction is run solventless, the yield increases up to 94% but the ee decreases to 46%, since the solubility of ligand 21 and its interaction with the solid metal salt diminishes (entry 2). Indeed, when a higher amount of MgCl2 is added (50 mol%), the yields of 3 are nearly quantitative and the ee increases to 57% in DCM but not without any solvent (entries 3 and 4). A decrease in the reaction temperature to 0 °C slightly improves the ee after 24 h reaction time (53% in entry 5 vs. 48% in entry 3), but not that much to justify a low temperature set-up. These results demonstrate the possibility of achieving a significant enantioselectivity with the solid MgCl2 in combination with the simple and commercially available ligand 21. It is worthy to comment herein that better enantioselective results (up to 99%) can be found in the literature with optimized homogeneous catalysts and ligands;13f however, they are often much expensive, toxic and unrecoverable (see ahead).
|
|||||
|---|---|---|---|---|---|
| Entry | MgCl2 + 21 (mol%) | Solvent | T (°C) | 3 (%) [ee] (%) | |
| 24 h | 72 h | ||||
| a GC yield and chiral HPLC ee. N.D.: not determined. | |||||
| 1 | 5 + 20 | DCM | 20 | 65 [44] | 85 [61] |
| 2 | — | 89 [41] | 94 [46] | ||
| 3 | 50 + 20 | DCM | 20 | 92 [48] | >99 [57] |
| 4 | — | 96 [29] | >99 [27] | ||
| 5 | DCM | 0 | 77 [53] | N.D. | |
| 6 | Reuse from entry 1 | N.D. | 85 [61] | ||
| 7 | Reuse from entry 6 | N.D. | 87 [72] | ||
| 8 | Reuse from entry 7 | N.D. | 90 [70] | ||
Table 3 also shows the reusability studies for the catalytic combination of MgCl2 (5 mol%) and ligand 21 (20 mol%). The catalytic mixture was recovered after fully precipitating the ligand with n-hexane, filtering and drying. The recovered mixture of MgCl2 and 21 was placed for a second run, to achieve 85% yield of 3 with 61% ee after 72 h at room temperature (entry 6). Two more uses of the catalytic solid mixture were carried out, with even higher yields and ee (up to 90% and 70%, respectively, entries 7 and 8). These results showcase the ability of a simple solid salt such as MgCl2 to bring enantioselectivity throughout several reuses with the solid ligand 21.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 wt% ratio) embedded between 2 g of silicon carbide. Then, an equimolar mixture of 1 and 2 was passed through the solid catalyst using a syringe pump, contragravity wise to avoid excessive solid aggregation, and at a rate of 2 mL h−1. The product mixture at the reactor exit was periodically analysed by GC-MS and chiral HPLC, and the results are shown in Fig. 5.
:2 wt% ratio) embedded between 2 g of silicon carbide. Then, an equimolar mixture of 1 and 2 was passed through the solid catalyst using a syringe pump, contragravity wise to avoid excessive solid aggregation, and at a rate of 2 mL h−1. The product mixture at the reactor exit was periodically analysed by GC-MS and chiral HPLC, and the results are shown in Fig. 5.
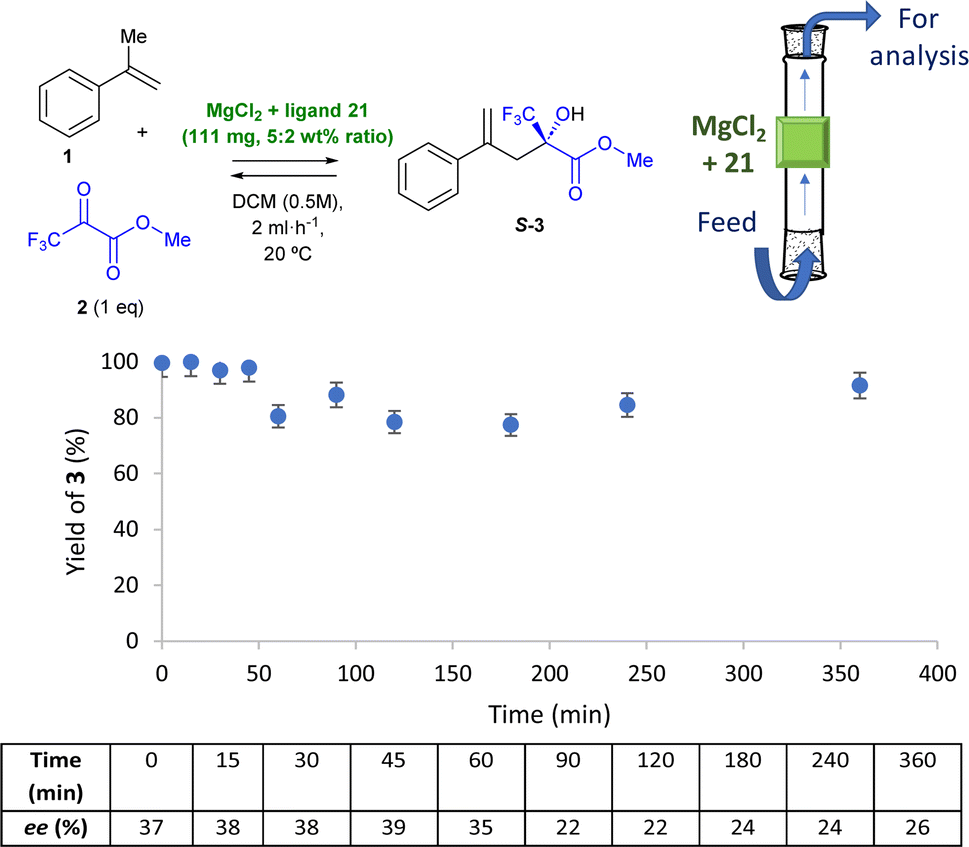 | ||
| Fig. 5 In-flow reaction with MgCl2 + chiral ligand 21 as a catalyst (111 mg in total, 5:2 wt% ratio) for the intermolecular ketone-ene reaction between α-methyl styrene 1 and methyl pyruvate 2 (1 equivalent) in DCM (0.5 M) at room temperature. The feed flow rate is 2 mL h−1. GC yields and HPLC ees. | ||
The in-flow reaction operates well for >6 h, with >90% yield of 3 on average during that time. Although the ees obtained are not high (≈30%), they add value to the in-flow experiment, since they are also consistent with the corresponding in-batch reaction (see entry 4 in Table 3). These results pave the way to perform intermolecular carbonyl-ene reactions in continuous flows, catalysed by MgCl2.
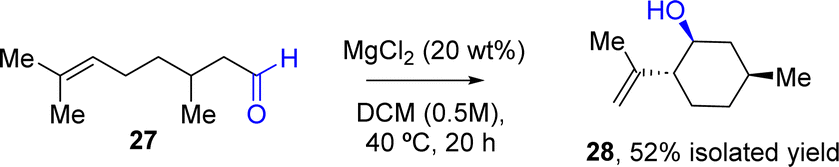 | ||
| Fig. 6 Results of the intramolecular carbonyl-ene reaction of citronellal 27 (0.25 mmol) with MgCl2 as a catalyst (20 wt%) in DCM (0.5 M) at 40 °C. Isolated yield. | ||
MgCl2 catalyzes well the carbonyl-ene reaction of citronellal 27 into isopulegol 28 (neo-isoisopulegol is the only isolated isomer, and the one with the higher conversion) with almost full diastereoselectivity,31 starting from a racemic mixture of 27. It is true that higher amounts of MgCl2 (20 wt%) and a slightly higher reaction temperature (40 °C) were required, but the desired compound could be isolated in a reasonable isolated yield of 52% as the major diastereoisomer. Two different conformers were detected in GC in a 25% ratio for each of them. These results showcase the feasibility of the MgCl2-catalysed protocol in industrially interesting processes.
3.2 Domino dehydration/intermolecular carbonyl-ene reaction
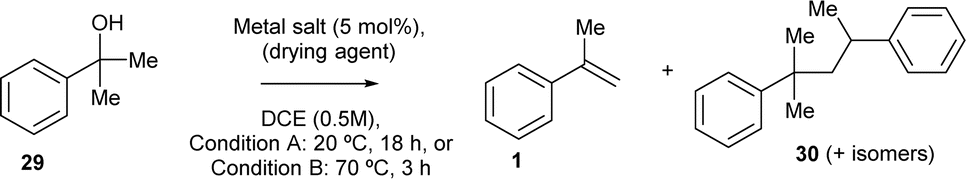
The dehydration reaction was, first, independently studied with some of the most active catalysts for the ketone-ene reaction (Table 1). The results are shown in Table 4, for two different reaction temperatures and reaction times (20 °C and 18 h, and 75 °C and 3 h, named conditions A and B, respectively). It can be seen that all the catalysts were inactive at room temperature (20 °C) except FeCl2 (entry 11), which however generates significant amounts of by-products 30. This mixture of by-products (the structure for the head-to-tail product is shown, but the other potential dimers are also formed) comes from either the dimerization of alkene 1 or the coupling of the carbocation intermediate of alcohol 29, which may be expected with strong Lewis acids, as those typically required for the carbonyl-ene reaction of reluctant carbonyl group such as ketones.12 Indeed, when heating at 70 °C, the complete conversion to by-products 30 was realized with FeCl2 (entry 12). In contrast, Cu salts with enough Lewis acidity were able to catalyse the desired dehydration reaction to form product 1 after heating (entries 4 and 6), although unavoidably generating some by-products 30. Gratifyingly, 4 Å MS (entry 14) and, particularly, MgCl2 (entry 18), only gave product 1 without any by-products 30. The high selectivity of MgCl2 towards 1 is explained by an intermediate Lewis acidity, circumventing the formation of 30 while reasonably giving the desired product 1. The relatively low yield of the former (24%) should not be a problem since, in the domino reaction, 1 (as an intermediate) will rapidly react with pyruvate 2 to the desired carbonyl-ene reaction product 3, while still at low concentrations. Besides, the non-reversible nature of the dehydration reaction will shift the equilibrium towards the domino reaction; indeed, an independent experiment shows the lack of reactivity of 1 with H2O under the optimized reaction conditions (Fig. S9†).
|
|||||
|---|---|---|---|---|---|
| Entry | Metal catalyst (5 mol%) | Condition | 1b (%) | 30b (%) | Ratio 1/30 |
| a MS: molecular sieves.b GC yields, mass balances are complete with unreacted starting material 29. | |||||
| 1 | — | A | <1 | <1 | — |
| 2 | B | 5 | <1 | 5.0 | |
| 3 | CuCl2 | A | <1 | <1 | — |
| 4 | B | 40 | 4 | 10.0 | |
| 5 | Cu(OTf)2 | A | 8 | 2 | 4.0 |
| 6 | B | 52 | 20 | 2.6 | |
| 7 | Cu(OAc)2 | A | <1 | <1 | — |
| 8 | B | <1 | <1 | — | |
| 9 | CuCl | A | <1 | <1 | — |
| 10 | B | <1 | <1 | — | |
| 11 | FeCl2 | A | 19 | 3 | 6.3 |
| 12 | B | <1 | >99 | 0.01 | |
| 13 | 4 Å MS | A | 2 | <1 | 2 |
| 14 | B | 15 | <1 | 15 | |
| 15 | MgO | A | <1 | <1 | — |
| 16 | B | <1 | <1 | — | |
| 17 | MgCl2 | A | <1 | <1 | — |
| 18 | B | 24 | <1 | 24 | |
|
||||||
|---|---|---|---|---|---|---|
| Entry | Metal catalyst | Solvent | 1a (%) | 30a (%) | 3a (%) | Select. to 3 (%) |
| a GC yields, mass balances are complete with unreacted starting material 29.b GC, weight and 1H NMR yield for an experiment on a 9 g scale (30 mmol of 29 and 45 mmol of 2). | ||||||
| 1 | — | DCE | 3 | 4 | 7 | 50 |
| 2 | MgCl2 | DCE | 10 | 14 | 60 | 71 |
| 3 | CuCl2 | DCE | <1 | 84 | 15 | 15 |
| 4b | MgCl2 | None | 3 (<)1 | 12 (9) | 85 (91) | 85 (91) |
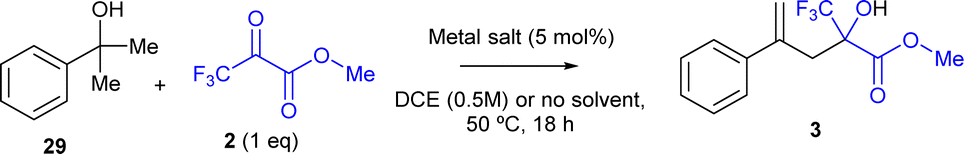
The above-mentioned results show that the domino reaction indeed proceeds well with MgCl2 as a catalyst, to give 60% yield of the carbonyl-ene reaction product 3 when DCE is used as a solvent (entry 2), and 85% yield when any solvent is not used (entry 4). A hot filtration test shows that there is no significant amount of catalytically active species in the solution for the domino reaction since, after hot filtration of the MgCl2 solid catalyst, the conversion is only ≈10% (Fig. S10†), similar to the blank experiment without any catalyst (entry 1 in Table 5), in contrast to the 50% conversion found in the presence of the catalyst after 3.5 h. Thus, any leaching can be discarded. A 9 g scale experiment with catalytic MgCl2 and without any solvent yields 91% of product 3 (using 1.5 equiv. of 2 in this case), according not only to GC but also to the recovered and weighted crude, as well as to the corresponding 1H, 13C, 19F and DEPT NMR characterization (Fig. S11,† top). The photograph of the mixture at the end of the reaction shows the good separation of the MgCl2 solid catalyst from the liquid phase, and also the separation of the water generated during the domino reaction (Fig. S11,† bottom). In contrast, CuCl2 only gave 15% of the desired product 3, and the rest was by-products 30 (entry 3 in Table 5), in accordance with the low activity towards the carbonyl-ene reaction of the pre-hydrated CuCl2 complex catalyst (entry 1 in Table 1).
|
|||
|---|---|---|---|
| Entry | Product | Solvent | Yielda (%) |
| a GC yields, mass balances are complete with unreacted starting materials, alkene and ether by-products.b Reaction carried out at 100 °C. | |||
| 1 | 3 | DCE | 60 |
| 2 | None | 85 | |
| 3 | 4 | DCE | 15 |
| 4 | None | 40 | |
| 5 | 5 | DCE | 20 |
| 6 | None | 76 | |
| 7 | 6 | DCE | 2 |
| 8 | None | 35 | |
| 9 | 8 | DCE | 92 |
| 10 | None | 99 | |
| 11b | 16 | DCE | >99 |
| 12b | None | >99 | |
| 13b | 18 | DCE | 13 |
| 14b | None | 75 | |
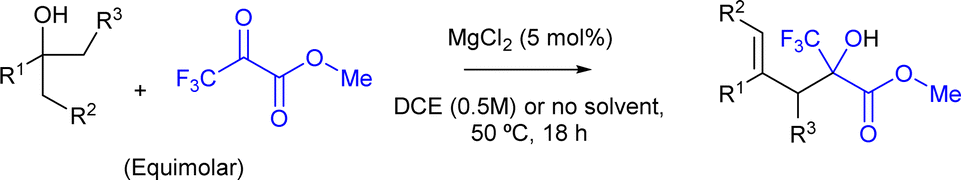
The results show that the domino dehydration/intermolecular carbonyl-ene reaction performs successfully for various aromatic and aliphatic alcohols, using the same MgCl2 catalyst under both dichloroethane (for comparative purposes) and solventless conditions, obtaining the best results under the latter. The reaction temperature had to be slightly increased to 50 °C in almost all cases (entries 1 to 10), in order to enhance the dehydration reaction rate. Different para-substituted alcohols were tested under the optimized reaction condition giving the expected ene-product with excellent to good yields for the chloro (entry 6, 76% yield), trifluoromethyl (entry 8, 35% yield) and the methoxy-substituted products (entry 10, 99% yield). In contrast, α,α-trimethylbenzyl alcohol yielded the desired ene-product in moderate yields (entry 4, 40% yield). Regarding aliphatic alcohols such as 2,4,4-trimethyl-2-pentanol (entries 11 and 12) and α-terpineol (entries 13 and 14), the reaction temperature had to be increased to 100 °C, giving, nevertheless, the corresponding products in high yields (99% of 16 and 75% of 18, respectively). The catalyst amount could be maintained to 5 mol%.
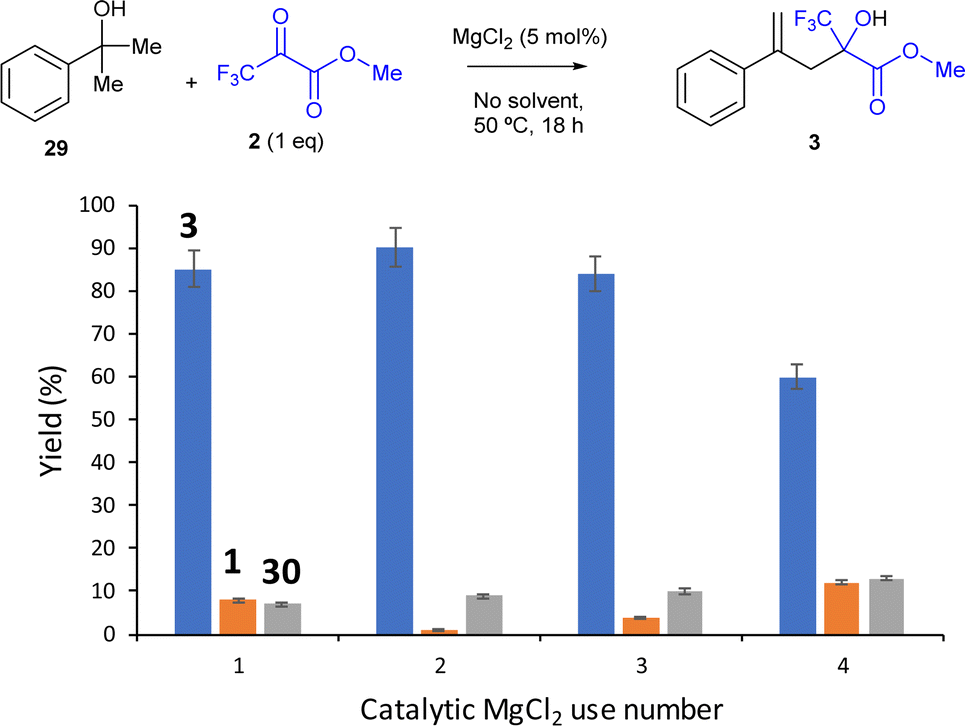 | ||
| Fig. 7 Results of the domino dehydration/intermolecular carbonyl-ene reaction of alcohol 29 with trifluoromethyl pyruvate 2 (1 equiv.) catalysed by MgCl2 (5 mol%) without any solvent, at 50 °C for 18 h, after recovering the MgCl2 catalyst by decantation and reuse. GC yields, mass balances are complete with unreacted starting material 29. | ||
Furthermore, analogously to the carbonyl-ene reaction, the one-pot reaction was performed enantioselectively, using the same chiral phosphoric acid, (R)-(−)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate chiral phosphoric acid 21 (6 mol%).
Different ratios of alcohol 29 and pyruvate 2 were tested along with MgCl2, as shown in Table 7. Equimolar amounts of both reagents 29 and 2 led mostly to the formation of product 30 either in the absence or presence of DCE as a solvent (entries 1 and 4). However, increasing the amount of pyruvate to 3 equivalents led to a severe decrease in the undesired by-product formation (entries 2 and 5), even though this amount is not negligible in the case of the solventless reaction. As a result, and due to any small difference in performance between the reaction using 3 or 5 equivalents of 2 (entries 3 and 6), the use of 3 equivalents of 2 was selected for the application of the reaction conditions for the in-flow reaction.
The same conditions as in the ene-reaction were tested for the in-flow domino dehydration/intermolecular carbonyl-ene reaction, as shown in Fig. 8. Although conversions towards the desired ene-product 3 did not exceed 20% at a flow rate of 2 mL h−1 (Fig. S12†), a slight decrease in the flow rate to 1.5 mL h−1 led to good conversions with moderate enantiomeric excesses during the first hour of the flow reaction (Fig. S13†). The conversion decreased drastically after that time. Consequently, flow was further decreased to 0.7 mL h−1, leading to the best results, as shown in Fig. 8.
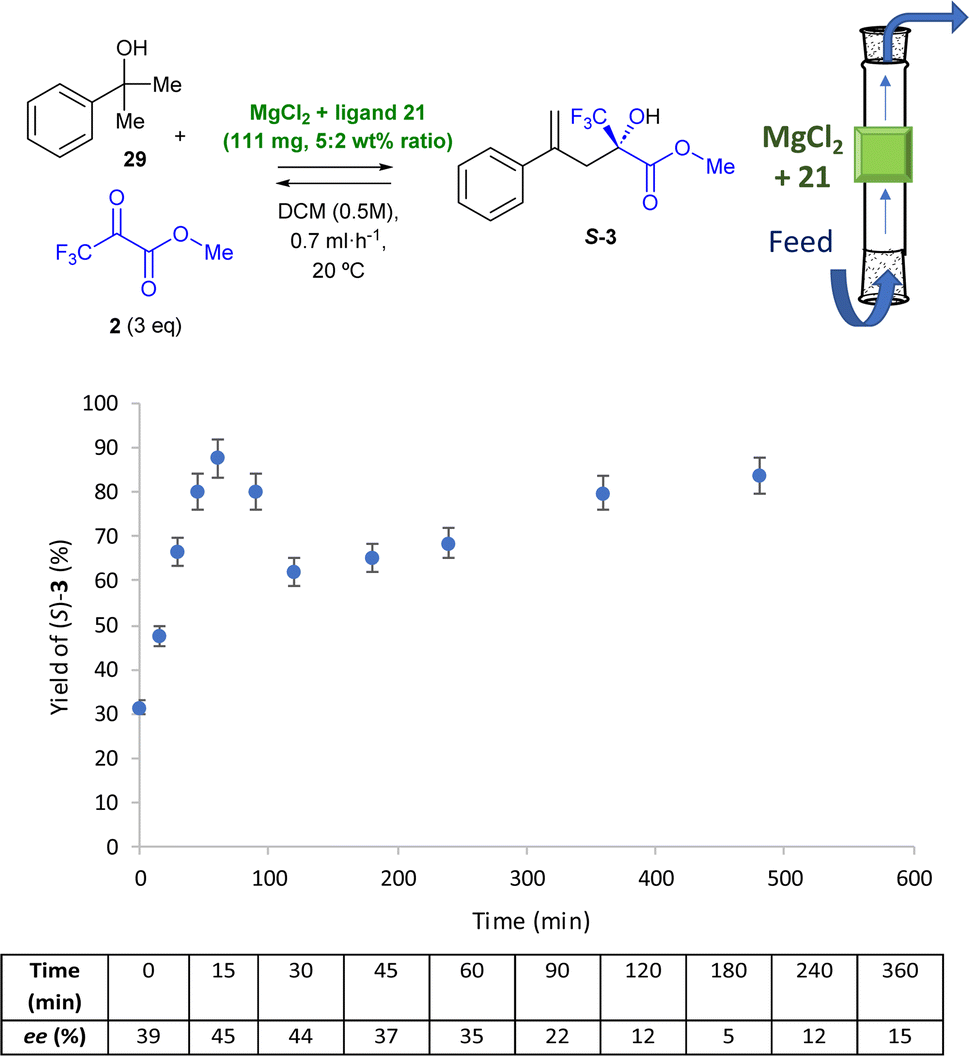 | ||
| Fig. 8 In-flow reaction with MgCl2 + chiral ligand 21 as a catalyst (111 mg in total, 5:2 wt% ratio) for the domino reaction, in DCE (0.5 M) at room temperature. The feed flow rate is 0.7 mL h−1. GC yields and chiral GC ees. Error bars account for a 5% uncertainty. | ||
The in-flow reaction operates well for >6 h; after stabilization of the reactive system during the first 30 min, the yields range from 70 to 90%. Furthermore, a 44% ee is achieved after 30 min reaction, which is consistent with the enantioselectivity obtained in the batch experiment and demonstrates that the in-flow chiral version of the domino dehydration/intermolecular carbonyl-ene reaction can be achieved with moderate ees during the first hour, similarly to the intermolecular carbonyl-ene.
4 Conclusions
The intermolecular ketone-ene reaction of trifluoromethyl pyruvates with alkenes and the corresponding domino dehydration/ketone-ene reaction with tertiary alcohols are catalysed by the simple salt MgCl2 in high yields (up to >99%) under solventless, mild reaction conditions. The solid salt can be reused in batch or implemented in flow, to give very good yields (>80%) in both cases of the ketone-ene product after extensive use (up to 10 uses in batch and 6 h reaction time in flow for the ketone-ene reaction). The use of the simple and commercially available chiral phosphoric acid ligand 21 allows performing the enantioselective version of the reaction, in very high yields (>85%) and with significant enantiomeric excess (up to 70%) when the solid mixture is reused in batch (the in-flow enantiomeric excess is ≈30%). The industrial synthesis of isopulegol from citronellal is also viable with MgCl2 as a solid catalyst. This study brings the extremely inexpensive and non-toxic solid MgCl2 as an efficient reusable catalyst for carbonyl-ene reactions, preferable in tandem with the previous dehydration reaction of the corresponding tertiary alcohols.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. E. performed the experimental work and analysis, including all product characterization, and interpreted the results. A. L.-P. conceived the idea, interpreted the results and supervised the project. Both authors wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the projects PID2020-115100GB-I00 (funded by Spanish MCIINN, MCIN/AEI/10.13039/501100011033MICIIN) and Severo Ochoa centre of excellence program (CEX2021-001230-S, also from MCIINN) is acknowledged. The work has also been funded by Generalitat Valenciana (Prometeo Grupos de Investigación de Excelencia PROMETEU/2021/054) and M. E. thanks to the Universitat de València (PROMETEU/2021/054) for the concession of a contract.References
- (a) Y. Peng and M. Beller, Geminal–atom catalysts: Advanced single–atom catalysis for cross–coupling reactions, Chem Catal., 2023, 3(11), 100814 CrossRef CAS; (b) R. J. Lewis and G. J. Hutchings, Selective oxidation using in situ–generated hydrogen peroxide, Acc. Chem. Res., 2024, 57(1), 106–119 CrossRef CAS PubMed.
- (a) J. R. Cabrero-Antonino, A. Leyva-Pérez and A. Corma, Beyond Acid Strength in Zeolites: Soft Framework Counteranions for Stabilization of Carbocations on Zeolites and Its Implication in Organic Synthesis, Angew. Chem., Int. Ed., 2015, 54(19), 5658–5661 CrossRef CAS PubMed; (b) X. Frogneux, F. Borondics, S. Lefrançois, F. D'Accriscio, C. Sanchez and S. Carenco, Surprisingly high sensitivity of copper nanoparticles toward coordinating ligands: consequences for the hydride reduction of benzaldehyde, Catal. Sci. Technol., 2018, 8(19), 5073–5080 RSC; (c) L. R. Smith, P. J. Smith, K. S. Mugford, M. Douthwaite, N. F. Dummer, D. J. Willock, M. Howard, D. W. Knight, S. H. Taylor and G. J. Hutchings, New insights for the valorisation of glycerol over MgO catalysts in the gas–phase, Catal. Sci. Technol., 2019, 9(6), 1464–1475 RSC; (d) F. Garnes-Portolés, R. Greco, J. Oliver-Meseguer, J. Castellanos-Soriano, M. C. Jiménez, M. López-Haro, J. C. Hernández-Garrido, M. Boronat, R. Pérez-Ruiz and A. Leyva-Pérez, Regioirregular and catalytic Mizoroki–Heck reactions, Nat. Catal., 2021, 4, 293–303 CrossRef; (e) V. A Ghadge, R. Krishnan, D. R. Naikwadi, P. B. Shinde and A. V. Biradar, Natural eumelanin–based porous N–doped carbon as an active bio–catalyst for base– and initiator–free aerobic oxidation of olefins and alkyl aromatic hydrocarbons, Green Chem., 2023, 25(7), 2863–2871 RSC; (f) P. Mingueza-Verdejo, J. C. Hernández-Garrido, A. Vidal-Moya, J. Oliver-Meseguer and A. Leyva-Pérez, Zeolites catalyze the halogen exchange reaction of alkyl halides, Catal. Sci. Technol., 2023, 13(8), 2308–2316 RSC; (g) L. A. Gomez, C. Q. Bavlnka, T. E. Zhang, D. E. Resasco and S. P. Crossley, Revealing the mechanistic details for the selective deoxygenation of carboxylic acids over dynamic MoO3 catalysts, ACS Catal., 2023, 13, 8455–8466 CrossRef CAS; (h) A. S. Singh, H. G. Kachgunde, R. Krishnan, D. R. Naikwadi and A. V. Biradar, Bifunctional heterogeneous catalyst: A sustainable route for cyclic acetals synthesis through tandem hydroformylation–acetalization reaction, Mol. Catal., 2024, 555, 113859 CrossRef CAS.
- M. L. Clarke and M. B. France, The carbonyl ene reaction, Tetrahedron, 2008, 64(38), 9003–9031 CrossRef CAS.
- (a) E. D. Anderson, J. J. Ernat, M. P. Nguyen, A. C. Palma and R. S. Mohan, Environment friendly organic synthesis using bismuth compounds. An efficient method for carbonyl–ene reactions catalyzed by bismuth triflate, Tetrahedron Lett., 2005, 46(45), 7747–7750 CrossRef CAS; (b) S. Imachi, K. Owada and M. Onaka, Intramolecular carbonyl–ene reaction of citronellal to isopulegol over ZnBr2–loading mesoporous silica catalysts, J. Mol. Catal. A: Chem., 2007, 272(1–2), 174–181 CrossRef CAS; (c) S. Baeurle, M. Berger, S. Jaroch, K. Krolikiewicz, D. Nguyen, H. Rehwinkel, H. Schaecke, N. Schmees and W. Skuballa, Alkylidene tetrahydronaphthalene derivatives, method for their production and their use as anti–inflammatory agents, World Intellectual Property Organization, 2006 Search PubMed; (d) M. Terada and K. Mikami, Periselective and enantioselective carbonyl–ene reaction of isoprene with fluoroalkyl glyoxylate catalysed by modified binaphthol–titanium complex: asymmetric catalytic synthesis of enantiomerically pure ipsdienol, J. Chem. Soc., Chem. Commun., 1995, 23, 2391–2392 RSC; (e) L. E. Overman and D. Lesuisse, The synthesis of indolizidines by intramolecular ene cyclizations. Preparation of (E)–alkylidene analogs of pumiliotoxin A, Tetrahedron Lett., 1985, 26(35), 4167–4170 CrossRef CAS.
- (a) M. L. Clarke, C. E. S. Jones and M. B. France, The first organocatalytic carbonyl–ene reaction: isomerization–free C–C bond formations catalysed by H–bonding thio–ureas, Beilstein J. Org. Chem., 2007, 3, 24 Search PubMed; (b) P. Tremel, C. Iacobucci, L. Massi, S. Olivero, J.-F. Gal and E. Duñach, Catalytic intramolecular carbonyl–ene reaction with ketones: evidence for a retro–ene process, New J. Chem., 2015, 39(9), 7453–7458 RSC.
- (a) F. Alonso, M. Rodríguez-Fernández, D. Sánchez and M. Yus, Synthesis of perhydrofuro[2,3–b]furans from isopentenyl alcohol through carbonyl–ene and Wacker–type reactions, Eur. J. Org. Chem., 2011, 32, 6459–6469 CrossRef; (b) A. Crespo-Peña, D. Monge, E. Martín-Zamora, E. Alvarez, R. Fernández and J. M. Lassaletta, Asymmetric formal carbony–ene reactions of formaldehyde tert–butyl hydrazone with α–keto esters: dual activation by bis–urea catalysts, J. Am. Chem. Soc., 2012, 134(31), 12912–12915 CrossRef PubMed; (c) X. Xu, X. Wang, Y. Liu and M. P. Doyle, Enantioselective carbonyl–ene reactions catalyzed by chiral cationic dirhodium(II,III) carboxamidates, J. Org. Chem., 2014, 79(24), 12185–12190 CrossRef CAS PubMed; (d) L. Liu, M. Leutzsch, Y. Zheng, M. W. Alachraf, W. Thiel and B. List, Confined acid–catalyzed asymmetric carbonyl–ene cyclization, J. Am. Chem. Soc., 2015, 137(41), 13268–13271 CrossRef CAS PubMed; (e) H. A. Rodríguez, D. A. Cruz, J. I. Padrón and I. Fernández, Lewis Acid–catalyzed carbonyl–ene reaction: Interplay between aromaticity, synchronicity, and Pauli repulsion, J. Org. Chem., 2023, 88(15), 11102–11110 CrossRef PubMed.
- R. G. Gore, T.-K.-T. Truong, M. Pour, L. Myles, S. J. Connon and N. Gathergood, Tandem ionic liquid antimicrobial toxicity and asymmetric catalysis study: carbonyl–ene reactions with trifluoropyruvate, Green Chem., 2013, 15(10), 2727–2739 RSC.
- (a) J. Tateiwa, A. Kimura, M. Takasuka and S. Uemura, Metal cation–exchanged montmorillonite(Mn+–Mont)–catalysed carbonyl–ene reactions, J. Chem. Soc., Perkin Trans. 1, 1997, 2169–2174 RSC; (b) T. Okachi and M. Onaka, Formaldehyde encapsulated in zeolite: A long–lived, highly activated one–carbon electrophile to carbonyl–ene reactions, J. Am. Chem. Soc., 2004, 126(8), 2306–2307 CrossRef CAS PubMed; (c) A. Corma and M. Renz, Sn–Beta zeolite as diastereoselective water–resistant heterogeneous Lewis–acid catalyst for carbon–carbon bond formation in the intramolecular carbonyl–ene reaction, Chem. Commun., 2004, 5, 550–551 RSC; (d) A. Corma and M. Renz, Water–resistant Lewis–acid sites: carbonyl–ene reactions catalyzed by tin–containing hydrophobic molecular sieves, ARKIVOC, 2007, 8, 40–48 Search PubMed; (e) S. Wannakao, P. Khongpracha and J. Limtrakul, Density functional theory study of the carbonyl–ene reaction of encapsulated formaldehyde in Cu(I), Ag(I), and Au(I) exchanged FAU zeolites, J. Phys. Chem. A, 2011, 115(45), 12486–12492 CrossRef CAS PubMed; (f) H. Fu, S. Xie, A. Fu and T. Ye, Theoretical study of the carbonyl–ene reaction between formaldehyde and propylene on the MgY zeolite, Comput. Theor. Chem., 2012, 982(15), 51–57 CrossRef CAS; (g) S. Yoshimura, D. Kiriyama and T. Kurata, Carbonyl–ene reaction of styrene derivatives catalyzed by H–Beta zeolite in water, J. Jpn. Pet. Inst., 2014, 57(2), 84–87 CrossRef CAS; (h) T. Maihom, M. Probst and J. Limtrakul, Computational study of the carbonyl–ene reaction between formaldehyde and propylene encapsulated in coordinatively unsaturated metal–organic frameworks M3(btc)2 (M = Fe, Co, Ni, Cu and Zn), Phys. Chem. Chem. Phys., 2019, 21(5), 2783–2789 RSC.
- (a) R. Annunziata, M. Benaglia, M. Cinquini, F. Cozzi and M. Pitillo, Poly(ethylene glycol)–supported bisoxazolines as ligands for catalytic enantioselective synthesis, J. Org. Chem., 2001, 66(9), 3160–3166 CrossRef CAS PubMed; (b) S. Kobayashi, R. Akiyama and H. Kitagawa, Polymer–supported glyoxylate and alpha–imino acetates. Versatile reagents for the synthesis of alpha–hydroxycarboxylic acid and alpha–amino acid libraries, J. Comb. Chem., 2001, 3(2), 196–204 CrossRef CAS PubMed; (c) J. H. Koh, A. O. Larsen, P. S. White and M. R. Gagné, Disparate roles of chiral ligands and molecularly imprinted cavities in asymmetric catalysis and chiral poisoning, Organometallics, 2002, 21(1), 7–9 CrossRef CAS; (d) Y. M. A. Yamada, M. Ichinohe, H. Takahashi and S. Ikegami, Assembled catalysts of titanium and non–cross–linked chiral copolymers for and enantioselective carbonyl–ene reaction, Tetrahedron Lett., 2002, 43(18), 3431–3434 CrossRef CAS; (e) S. Takizawa, H. Somei, D. Jayaprakash and H. Sasai, Metal–bridged polymers as insoluble multicomponent asymmetric catalysts with high enantiocontrol: An approach for the immobilization of catalysts without using any support, Angew. Chem., Int. Ed., 2003, 42(46), 5711–5714 CrossRef CAS PubMed; (f) H. Guo, X. Wang and K. Ding, Assembled enantioselective catalysts for carbonyl–ene reactions, Tetrahedron Lett., 2004, 45(9), 2009–2012 CrossRef CAS; (g) Y. Huang, Synthesis of new polymer–supported C2–symmetric chiral bis(oxazoline)s and their applications in asymmetric reactions, Chem. J. Internet, 2004, 6(11), 79 Search PubMed; (h) A. Mandoli, S. Orlandi, D. Pini and P. Salvadori, Insoluble polystyrene–bound bis(oxazoline): Batch and continuous–flow heterogeneous enantioselective glyoxylate–ene reaction, Tetrahedron: Asymmetry, 2004, 15(20), 3233–3244 CrossRef CAS; (i) F. Ono, S. Kanemasa and J. Tanaka, Reusable nano–sized chiral bisoxazoline catalysts, Tetrahedron Lett., 2005, 46(44), 7623–7626 CrossRef CAS; (j) K. Ding, Z. Wang, X. Wang, Y. Liang and X. Wang, Self–supported chiral catalysts for heterogeneous enantioselective reactions, Chem.–Eur. J., 2006, 12(10), 5188–5197 CrossRef CAS PubMed.
- X. Wang, X. Wang, H. Guo, Z. Wang and K. Ding, Self–supported heterogeneous titanium catalysts for enantioselective carbonyl–ene and sulfoxination reactions, Chem.–Eur. J., 2005, 11(14), 4078–4088 CrossRef CAS PubMed.
- (a) L. Mitchell, B. Gonzalez-Santiago, J. P. S. Mowat, M. E. Gunn, P. Williamson, N. Acerbi, M. L. Clarke and P. A. Wright, Remarkable Lewis acid catalytic performance of the scandium trimesate metal organic framework MIL–100(Sc) for C–C and C=N bond–forming reactions, Catal. Sci. Technol., 2013, 3(3), 606–617 RSC; (b) L. Mitchell, P. Williamson, B. Ehrlichová, A. E. Anderson, V. R. Seymour, S. E. Ashbrook, N. Acerbi, L. M. Daniels, R. L. Walton, M. L. Clarke and P. A. Wright, Mixed–metal MIL(Sc, m) (M=Al, Cr, Fe) for Lewis acid catalysis and tandem C–C bond formation and alcohol oxidation, Chem.–Eur. J., 2014, 20(51), 17185–17197 CrossRef CAS PubMed; (c) J. Han, M. S. Lee, P. K. Thallapally, M. Kim and N. Jeong, Identification of reaction sites on metal–organic framework–based asymmetric catalysts for carbonyl–ene reactions, ACS Catal., 2019, 9(5), 3969–3977 CrossRef CAS; (d) R. R. R. Prasad, S. E. Seidner, D. B. Cordes, M. M. Lozinska, D. M. Dawson, M. J. Thompson, T. Düren, K. K. Chakarova, M. Y. Mihaylov, K. I. Hadjiivanov, F. Hoffmann, A. m. Z. Slawin, S. E. Ashbrook, M. L. Clarke and P. A. Wright, STA–27, a porous Lewis acidic scandium MOF with an unexpected topology type prepared with 2,3,5,6–tetrakis(4–carboxyphenyl)pyrazine, J. Mater. Chem. A, 2019, 7(10), 5685–5701 RSC.
- (a) B. Xiang, T.-F. Xu, L. Wu, R.-R. Liu, J.-R. Gao and Y.-X. Jia, Lewis Acid Catalyzed Friedel–Crafts alkylation of alkenes with trifluoropyruvates, J. Org. Chem., 2016, 81(9), 3929–3935 CrossRef CAS PubMed; (b) D. Meng and T. Ollevier, Fe(BF4)2–catalyzed inter– and intramolecular carbonyl–ene reaction of trifluoropyruvate, Synlett, 2018, 29(5), 640–644 CrossRef CAS.
- (a) D. A. Evans, S. W. Tregay, C. S. Burgey, N. A. Paras and T. Vojkovsky, C2–symmetric Copper(II) complexes as chiral Lewis acids. Catalytic enantioselective carbonyl–ene reactions with glyoxylate and pyruvate esters, J. Am. Chem. Soc., 2000, 122(33), 7936–7943 CrossRef CAS; (b) S. Doherty, J. G. Knight and H. Mehdi-Zodeh, Asymmetric carbonyl–ene and Friedel–Crafts reactions catalysed by Lewis acid platinum group metal complexes of the enantiopure atropisomeric biaryl–like diphosphine (S)–Me2–CATPHOS: A comparison with BINAP, Tetrahedron: Asymmetry, 2012, 23(3–4), 209–216 CrossRef CAS; (c) L. Jiang, B. Hu, X. Xie and Z. Zhang, Asymmetric carbonyl–ene reaction of trifluoropyruvate catalyzed by Pd(II)–SunPhos complex, Tetrahedron, 2017, 73(49), 6901–6905 CrossRef CAS; (d) T. Wang, X.-Q. Hao, J.-J. Huang, J.-L. Niu, J.-F. Gong and M.-P. Song, Chiral bis(imidazolinyl)phenyl NCN pincer rhodium(III) catalysts for enantioselective allylation of aldehydes and carbonyl–ene reaction of trifluoropyruvates, J. Org. Chem., 2013, 78(17), 8712–8721 CrossRef CAS PubMed; (e) Y. Yuan, X. Zhang and K. Ding, Quasi solvent–free enantioselective carbonyl–ene reaction with extremely low catalyst loading, Angew. Chem., Int. Ed., 2003, 42(44), 5478–5480 CrossRef CAS PubMed; (f) K. Zheng, Y. Yang, J. Zhao, C. Yin, L. Lin, X. Liu and X. Feng, The magnesium(II)–catalyzed asymmetric ketone–ene reaction under solvent–free conditions: Stereocontrolled access to enantioenriched trifluoromethyl–substituted compounds, Chem.–Eur. J., 2010, 16, 9969–9972 CrossRef CAS PubMed; (g) J. Lv, Q. Zhang, X. Zhing and S. Luo, Asymmetric latent carbocation catalysis with chiral trityl phosphate, J. Am. Chem. Soc., 2015, 137(49), 15576–15583 CrossRef CAS PubMed; (h) Y.-B. Shen, S.-S. Li, Y.-M. Sun, L. Yu, Z.-H. Hao, Q. Liu and J. Xiao, Organocatalytic C(sp3)–H functionalization of 5–methyl–2,3–dihydrofuran derivatives with trifluoropyruvates via a sequential exo–tautomerization/carbonyl–ene process, J. Org. Chem., 2019, 84(5), 2779–2785 CrossRef CAS PubMed; (i) H. Ishihara, J. Huang, T. Mochizuki, M. Hatano and K. Ishihara, Enantio– and diastereoselective carbonyl–ene cyclization–acetalization tandem reaction catalyzed by tris(pentafluorophenyl)borane–assisted chiral phosphoric acids, ACS Catal., 2021, 11(10), 6121–6127 CrossRef CAS.
- (a) J. F. Zhao, B.-H. Tan, M.-K. Zhu, T.-B. W. Tjan and T.-P. Loh, Enantioselective carbonyl–ene reactions of trifluoropyruvate in ionic liquid via a recyclable indium(III)–pybox complex, Adv. Synth. Catal., 2010, 352(11–12), 2085–2088 CrossRef CAS; (b) D. Didier and E. Schulz, Recycling chiral copper bis(oxazoline) complexes in an original multireaction procedure, ChemCatChem, 2011, 3(12), 1880–1884 CrossRef CAS; (c) N. A. Caplan, F. E. Hancock, P. C. B. Page and G. J. Hutchings, Heterogeneous enantioselective catalyzed carbonyl– and imini–ene reactions using copper bis(oxazoline) zeolite Y, Angew. Chem., Int. Ed., 2004, 43(13), 1685–1688 CrossRef CAS PubMed.
- A. Leyva-Pérez, P. García-García and A. Corma, Multisite organic–inorganic hybrid catalysts for the direct sustainable synthesis of GABAergic drugs, Angew. Chem., Int. Ed., 2014, 53, 8687–8690 CrossRef PubMed.
- (a) K. Mikami and H. Ohmura, One–pot synthesis of tropinone by tandem (domino) ene–type reactions of acetone silyl enol ethers, Chem. Commun., 2002, 22, 2626–2627 RSC; (b) S. Kaden and M. Hiersemann, The first catalytic asymmetric domino Claisen rearrangement/intramolecular carbonyl ene reaction, Synlett, 2002, 12, 1999–2002 Search PubMed; (c) B. T. Parr and H. M. L. Davies, Stereoselective synthesis of highly substituted cyclohexanes by a rhodium–carbene initiated domino sequence, Org. Lett., 2015, 17(4), 794–797 CrossRef CAS PubMed.
- (a) N. Kashiwa, The discovery and progress of MgCl2–supported TiCl4 catalysts, J. Polym. Sci., Part A: Polym. Chem., 2004, 42(1), 1–8 CrossRef CAS; (b) T. B. Mikenas, V. A. Zakharov, V. E. Nikitin, L. G. Echevskaya and M. A. Matsko, New generation of supported Ziegler–type catalysts for polyethylene production, Russ. J. Appl. Chem., 2010, 83, 2210–2219 CrossRef CAS.
- D. Yang, L. Wang, D. Li and R. Wang, Magnesium catalysis in asymmetric synthesis, Chem, 2019, 5(5), 1108–1166 CAS.
- C. Nadeau, F. Gosselin, P. D. O'Shea, I. W. Davies and R. P. Volante, A concise synthesis of (S)–γ–fluoroleucine ethyl ester, Synlett, 2006, 2, 291–295 CrossRef.
- (a) R. Greco, E. Tiburcio, B. Palomar-De Lucas, J. Ferrando-Soria, D. Armentano, E. Pardo and A. Leyva-Pérez, Click amidations, esterifications and one–pot reactions catalyzed by Cu salts and multimetal–organic frameworks (M–MOFs), Mol. Catal., 2022, 522, 112228 CrossRef CAS; (b) E. Tiburcio, Y. Zheng, C. Bilanin, J. C. Hernández-Garrido, A. Vidal-Moya, J. Oliver-Meseguer, N. Martín, M. Mon, J. Ferrando-Soria, D. Armentano, A. Leyva-Pérez and E. Pardo, MOF–triggered synthesis of subnanometer Ag02 clusters and Fe3+ single atoms: heterogenization led to efficient and synergetic one–pot catalytic reactions, J. Am. Chem. Soc., 2023, 145(18), 10342–10354 CrossRef CAS PubMed.
- Z. Mao, F. Mo and X. Lin, Diastereo– and enantioselective assembly of spirooxindole tetrahydroquinoline skeletons through asymmetric binary acid catalyzed hydride transfer–cyclization, Synlett, 2015, 27(04), 546–550 CrossRef.
- (a) T. Okachi, K. Fujimoto and M. Onaka, Practical carbonyl–ene reactions of α–methylstyrenes with paraformaldehyde promoted by a combined system of boron trifluoride and molecular sieves 4A, Org. Lett., 2002, 4(10), 1667–1669 CrossRef CAS PubMed; (b) C. Reichardt and T. Welto, Solvents and Solvent Effects in Organic Chemistry, Wiley, 2010, 692 pages, online ISBN:9783527632220 CrossRef.
- J. F. Zhao, T.-B. W. Tjan, B.-H. Tan and T.-P. Loh, Highly enantioselective ketone–ene reactions of trifluoropyruvate: Significant counterion effect of the In(III)–pybox complex, Org. Lett., 2009, 11(24), 5714–5716 CrossRef CAS PubMed.
- M. Rueping, T. Theissmann, A. Kuenkel and R. M. Koenigs, Highly enantioselective organocatalytic carbonyl–ene reaction with strongly acidic, chiral Brønsted acids as efficient catalysts, Angew. Chem., Int. Ed., 2008, 47(36), 6798–6801 CrossRef CAS PubMed.
- (a) F. Garnes-Portolés, C. López-Cruz, J. Sánchez-Quesada, E. Espinós-Ferri and A. Leyva-Pérez, Solid–catalyzed synthesis of isomers–free terpinen–4–ol, Mol. Catal., 2022, 533, 112785 CrossRef; (b) A. Martínez-Castelló, M. Tejeda-Serrano, A. Nowacka, J. Oliver-Meseguer and A. Leyva-Pérez, Solid–catalyzed esterification reaction of long–chain acids and alcohols in fixed–bed reactors at pilot plant scale, Chem. Eng. Process., 2022, 178, 109038 CrossRef; (c) P. Mingueza-Verdejo, J. P. Cerón-Carrasco, J. Oliver-Meseguer and A. Leyva-Pérez, a formal exchange reaction between ketones and vinyl ethers with solid catalysts, Synthesis, 2024, 56(3), 455–461 CrossRef CAS.
- Y. Guo, P. Gaczyński, K.-D. Becker and E. Kemnitz, Sol–gel synthesis and characterisation of nanoscopic FeF3–MgF2 heterogeneous catalysts with bi–acidic properties, ChemCatChem, 2013, 5(8), 2223–2232 CrossRef CAS.
- S. M. Coman, P. Patil, S. Wuttke and E. Kemnitz, Cyclisation of citronellal over heterogeneous inorganic fluorides–highly chemo– and diastereoselective catalysts for (±)–isopulegol, Chem. Commun., 2009, 4, 460–462 RSC.
- A. Negoi, S. Wuttket, E. Kemnitz, D. Macovei, V. I. Parvulescu, C. M. Teodorescu and S. M. Coman, One–pot synthesis of menthol catalyzed by a highly diastereoselective Au/MgF2 catalyst, Angew. Chem., Int. Ed., 2010, 49, 8134–8138 CrossRef CAS PubMed.
- Z. Yongzhong, N. Yuntong, S. Jaenickem and G. K. Chuah, Cyclisation of citronellal over zirconium zeolite Beta– a highly diastereoselective catalyst to (±)–isopulegol, J. Catal., 2005, 229(2), 404–413 CrossRef.
- (a) A. Corma and M. Renz, Sn–Beta zeolite as diastereoselective water–resistant heterogeneous Lewis–acid catalyst for carbon–carbon bond formation in the intramolecular carbonyl–ene reaction, Chem. Commun., 2004, 5, 550–551 RSC; (b) A. Corma, F. X. Llabres i Xamena, C. Prestipino, M. Renz and S. Valencia, Water resistant, catalytically active Nb and Ta isolated Lewis acid sites, homogeneously distributed by direct synthesis in a beta zeolite, J. Phys. Chem. C, 2009, 113(26), 11306–11315 CrossRef CAS.
- H. Itoh, H. Maeda, S. Yamada, Y. Hori, T. Mino and M. Sakamoto, Highly selective aluminium–catalysed intramolecular Prins reaction for L–menthol synthesis, RSC Adv., 2014, 4(106), 61619–61623 RSC.
- A. Petronilho, A. Vivancos and M. Albrecht, Ether formation through reductive coupling of ketones or aldehydes catalyzed by a mesoionic carbene iridium complex, Catal. Sci. Technol., 2017, 7(23), 5766–5774 RSC.
- (a) T. Kojima, K. Nakayama, K. Ikemura, T. Ogura and S. Fukuzumi, Formation of a ruthenium(IV)–oxo complex by electron–transfer oxidation of a coordinatively saturated ruthenium(II) complex and detection of oxygen–rebound intermediates in C–H bond oxygenation, J. Am. Chem. Soc., 2011, 133(30), 11692–11700 CrossRef CAS PubMed; (b) B. Wang, Y.-M. Lee, M. Clémancey, M. S. Seo, R. Sarangi, J.-M. Latour and W. Nam, Mononuclear nonheme high–spin iron(III)–acylperoxo complexes in olefin epoxidation and alkane hydroxylation reactions, J. Am. Chem. Soc., 2016, 138(7), 2426–2436 CrossRef CAS PubMed; (c) A. Malekafzali, K. Malinovska and F. W. Patureau, The cumene/O2 system: a very simple tool for the radical chain oxidation of some functional groups, New J. Chem., 2017, 41(15), 6981–6985 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental details, Fig. S1–S13, product characterisation, and NMR and HPLC copies. See DOI: https://doi.org/10.1039/d4ra06449f |
| This journal is © The Royal Society of Chemistry 2024 |