
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew triazole-based hybrids as neurotropic agents†
Samvel N. Sirakanyan a,
Domenico Spinellib,
Athina Geronikaki*c,
Elmira K. Hakobyan*a,
Anti Petrouc,
Victor G. Kartsevd,
Hasmik A. Yegoryana,
Ervand G. Paronikyana,
Luca Zuppirolie,
Hasmik V. Jughetsyana,
Ruzanna G. Paronikyana,
Tatevik A. Arakelyana and
Anush A. Hovakimyana
a,
Domenico Spinellib,
Athina Geronikaki*c,
Elmira K. Hakobyan*a,
Anti Petrouc,
Victor G. Kartsevd,
Hasmik A. Yegoryana,
Ervand G. Paronikyana,
Luca Zuppirolie,
Hasmik V. Jughetsyana,
Ruzanna G. Paronikyana,
Tatevik A. Arakelyana and
Anush A. Hovakimyana
aScientific Technological Center of Organic and Pharmaceutical Chemistry of National Academy of Science of Republic of Armenia, Institute of Fine Organic Chemistry of A.L. Mnjoyan, 0014, Armenia. E-mail: shnnr@mail.ru
bDipartimento di Chimica G. Ciamician, Alma Mater Studiorum-Università di Bologna, Via F. Selmi 2, Bologna 40126, Italy. E-mail: domenico.spinelli@unibo.it
cAristotle University of Thessaloniki, School of Pharmacy, Thessaloniki-54124, Greece. E-mail: geronik@pharm.auth.gr
dInterBioScreen, Moscow 119019, Russia. E-mail: vkartsev@ibscreen.chg.ru
eDepartment of Industrial Chemistry ‘Toso Montanari’, Alma Mater Studiorum-Università di Bologna, Viale del Risorgimento 4, Bologna, 40136, Italy. E-mail: luca.zuppiroli@unibo.it
First published on 18th October 2024
Abstract
Herein, we describe the synthesis of new hybrids linked to 1,2,3- and 1,2,4-triazole units. Hybrids connected to a 1,2,3-triazole ring were synthesized using the well-known click reaction. The synthesis of the 1,2,4-triazole-based hybrids was carried out using 2-[(4-cyano-1-methyl(2-furyl)-5,6,7,8-tetrahydroisoquinolin-3-yl)oxy]acetohydrazides as starting compounds. The compounds were evaluated for their anticonvulsive activity via antagonism towards pentylenetetrazole (PTZ) – and thiosemicarbazide (TSC)-induced convulsion and maximal electroshock-induced seizure (MES). Furthermore, the most active compounds were studied for their locomotory and anxiolytic activity via the “open field” and elevated plus maze (EPM) assays. Finally, their antidepressant activity was studied via the “forced swim” method. All the hybrids displayed pentylenetetrazole antagonism, ranging from 40% to 80%, while in the TSC model, the most active compounds increased latency of thiosemicarbazide seizures to 1.9–4.65 times compared to that of the control. Some of the tested compounds exhibited a pronounced anxiolytic and antidepressant effect. Docking study demonstrated complete agreement with experimental pharmacological data. It was revealed that the most active compounds have a pyrano[3,4-c]pyridine ring in their structure.
1. Introduction
The wide prevalence of mental disorders, including epilepsy, one of the most severe diseases of the central nervous system, along with the lack of efficacy and side effects of existing anticonvulsants, makes the search and study of the mechanisms of action of new anticonvulsants imperative.1,2 Epilepsy is the most common neurological disease, with a prevalence ranging from 0.5% to 1% in developed countries.3 Convulsions arise as a result of an imbalance between the excitatory and inhibitory systems of the central nervous system.The main component in the complex treatment of epilepsy is pharmacotherapy—the use of anticonvulsants or antiepileptic drugs. Today, about 30 antiepileptic drugs are used in medicine. Such a number of medicines for the treatment of one disease is necessary because different medicines have different mechanisms of action. Each drug is most effective for only a certain form of epilepsy, which differs in the individual mechanism for the development of an attack in the brain. In recent years, in the treatment of epilepsy using antiepileptic drugs, mainly second-generation drugs, the tendency has been to optimize the treatment, aimed at the use of anticonvulsants with extended combined neurotropic properties.4
Heterocyclic compounds have attracted interest from the scientific community as a pool for development of new biologically active compounds. Thus, numerous publications refer to the neurotropic activity of pyridine derivatives.5–9 Another interesting scaffold is triazole derivatives, which besides their known antimicrobial activity,10–12 have been reported to possess antiepileptic/antidepressant activity.13–15
Recently, we synthesized some hybrid compounds16,17 via the click reaction18–20 and studied their neurotropic activity.17 The obtained results demonstrated that some compounds can be effective in various types of human epilepsy associated with mental disorders.17 It should be noted that starting compounds used for the synthesis of target hybrids are derivatives of biologically active compounds. In particular, compounds with a terminal triple bond were synthesized starting from 3(6)-hydroxypyridines 1, among which a compound with both strong antiplatelet and vasodilatory potential and with an IC50 two-times lower than that of clinically used acetylsalicylic acid was revealed.21 The preparation of compounds bearing azido group was performed using piperazinopyridines, which are known for their pronounced neurotropic22,23 and antimicrobial24 activities. Alternatively, the target hybrids were linked with 1,2,3- and 1,2,4-triazole rings, and their derivatives exhibited a wide range of bioactive properties.25–27
The structure of our initial compound bicyclic pyridine is very similar to that of the alkaloids gentianine I and gentianidine II,28 which are endowed with a wide range of bioactivities and used in medicine (Fig. 1). Alternatively, the piperazine core is included in various synthetic drugs such as befuraline III and ziprasidone IV, which are drugs approved by the FDA.29,30 Befuraline III (DIV-154) is a psychoactive drug, which has stimulant and antidepressant effects. Ziprasidone IV is an atypical antipsychotic drug used to treat schizophrenia, bipolar mania, and acute agitation in schizophrenic patients (Fig. 1).
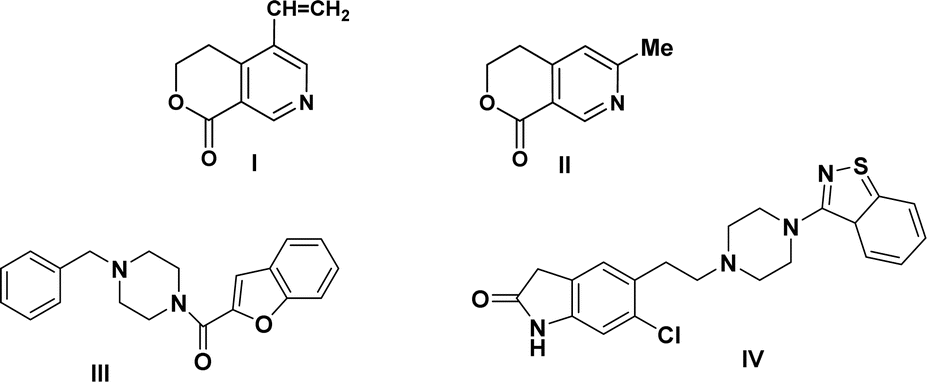 | ||
| Fig. 1 Alkaloids: gentianine I, gentianidine II and drugs bearing piperazine core III and IV. | ||
Considering all the above-mentioned factors and in continuation of our studies on the synthesis of potentially biologically active hybrid compounds, herein we report the synthesis of new representatives of these compounds (besides compounds 6a, j and m,16) and studied their neurotropic properties.
2. Results and discussion
2.1. Chemistry
The azido derivatives were synthesized starting from 3(6)-piperazinopyridines 1a–g.16,17 Compounds 1 were acylated with chloroacetyl chloride in benzene with the formation of 4-(chloroacetyl)piperazinopyridines 2a–g, respectively.16,17 Then, the obtained chlorides 2 were reacted with sodium azide in acetone and the corresponding 4-(azidoacetyl)piperazinopyridines 3a–g,16,17 were synthesized, respectively (Scheme 1).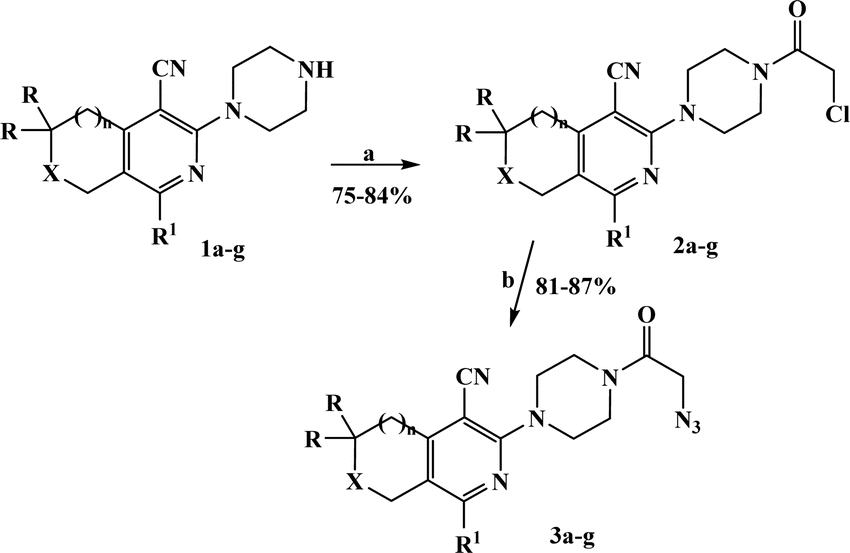 | ||
| Scheme 1 Synthesis of compounds 2a–g and 3a–g. (a) CICH2COCI, C6H6, Et3N, 35 °C, 6 h; (b) NaN3, MeCOMe, reflux, 15 h. 1–3. a, b: X = CH2, n = 0, R = H, a: R1 = n-Pr; b: R1 = i-Bu; c–e: X = CH2, n = 1, R = H, c: R1 = Me; d: R1 = i-Pr; e: R1 = 2-furyl; f, g: X = O, n = 1, R = Me, f: R1 = Me; g: R1 = Et. | ||
The terminal alkynes were synthesized starting from the 3(6)-hydroxy derivatives of cyclopenta[c]pyridine 4a,31 5,6,7,8-tetrahydroisoquinolines 4b–f,31 and pyrano[3,4-c]pyridines 4g–i.32 As shown in our previous paper,17 the alkylation of compounds 1 with propargyl bromide in basic medium proceeds almost regioselectively with the formation of O-alkylated derivatives in high yields and the traces of N-alkylated compounds were removed by recrystallization from ethanol (Scheme 2). Moreover, in our previous paper, in one instance we succeeded in separating the O- and N-alkylated compounds, also confirming their structure by X-ray data.17
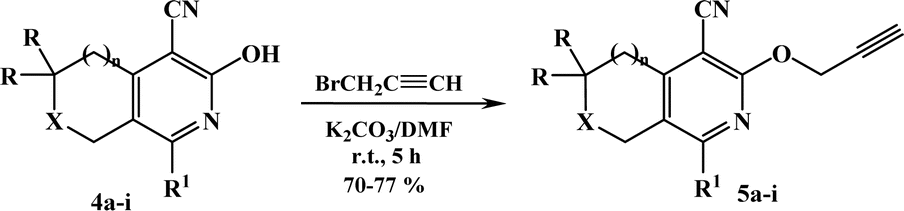 | ||
| Scheme 2 Synthesis of compounds 5a–i. 4, 5: a: X = CH2, n = 0, R = H, R1 = i-Pr; b: X = CH2, n = 1, R = H: R1 = Me; c: R1 = Et; d: R1 = i-Pr; e: R1 = Ph; f: R1 = 2-furyl; g: X = O, n = 1, R = Me: R1 = i-Pr; h: R1 = p-MeOC6H4; i: R1 = 2-furyl. | ||
Finally, the target hybrids 6a–q (resynthesis was carried out for compounds 6a, j and m,16) were synthesized in high yields via the click reaction16–20 between 4-(azidoacetyl)piperazinopyridines 3 and O-alkylated compounds 5 (Y = 71–87%; Scheme 3, Table 1). The structure of compounds 6 was defined spectroscopically and by elemental analysis. In the IR spectra of compounds 6a–q, the absorption bands of the azido group (2103–2111 cm−1) and triple bond (2121–2131 and 3268–3276 cm−1) disappeared, with the appearance of the singlet signal of the triazole ring at 7.92–8.02 ppm in the 1H NMR spectra. The MS spectra correspond to the expected structure of the compounds.
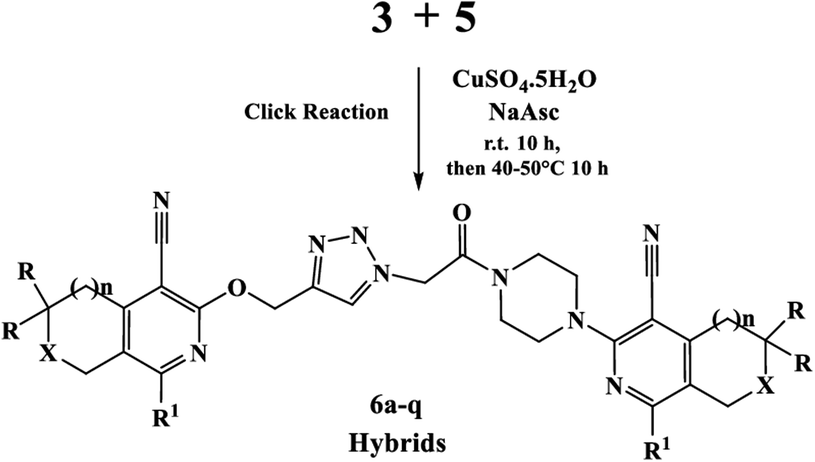 | ||
| Scheme 3 Synthesis of 1,4-disubstituted 1,2,3-triazoles 6a–q. | ||
| No. | Hybrids | No. | Hybrids |
|---|---|---|---|
| 6a | 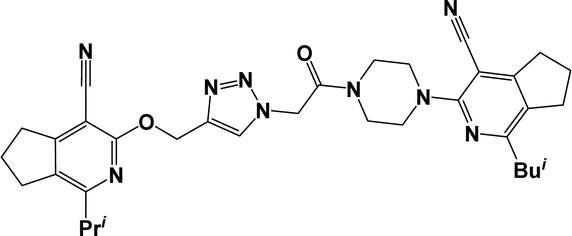 |
6j | 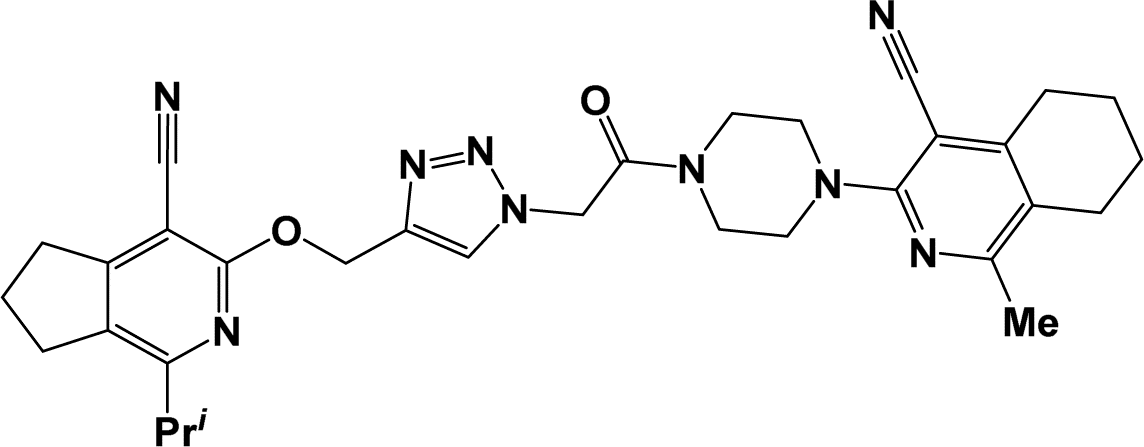 |
| 6b | 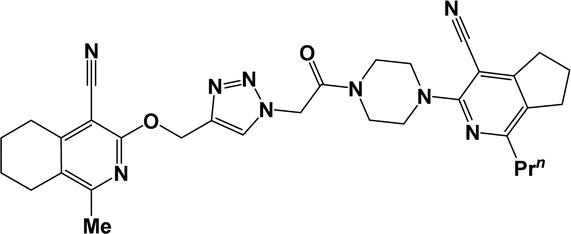 |
6k | 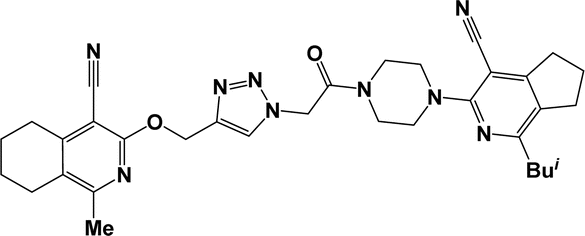 |
| 6c | 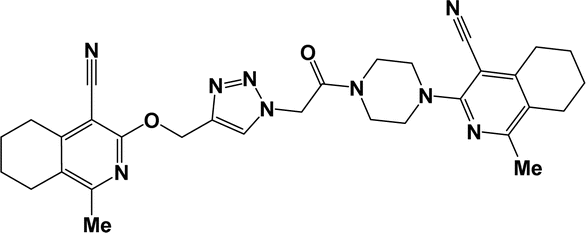 |
6l | 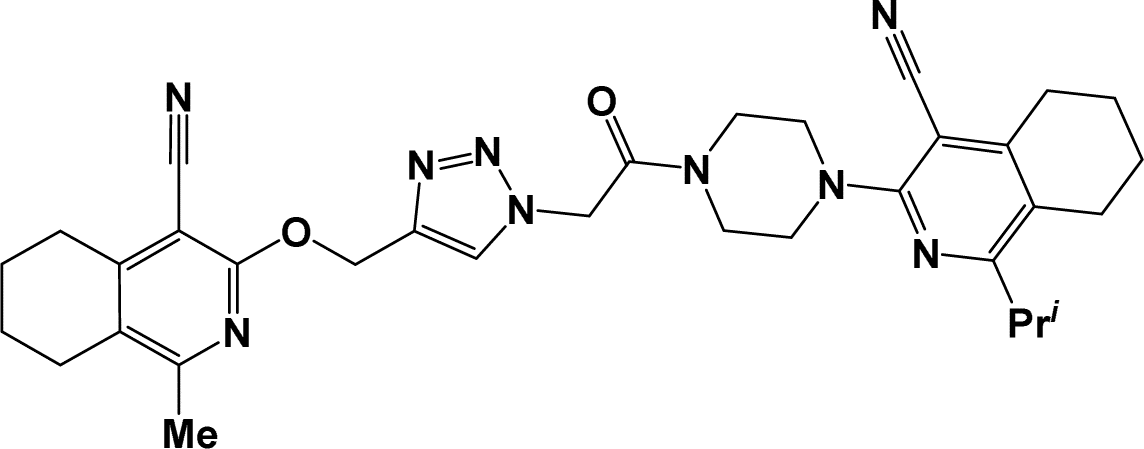 |
| 6d | 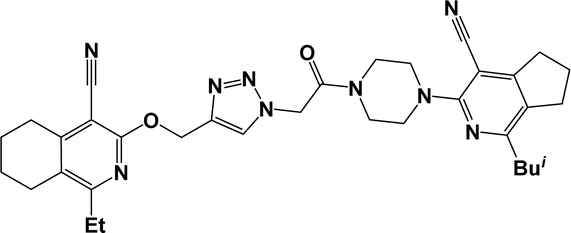 |
6m | 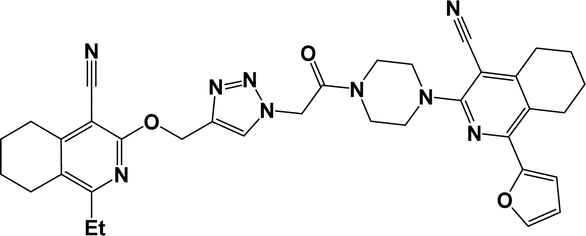 |
| 6e | 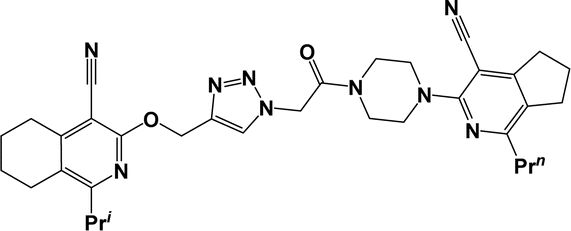 |
6n | 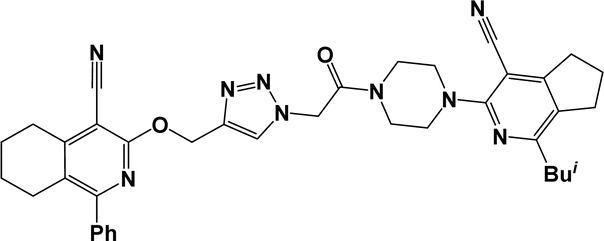 |
| 6f | 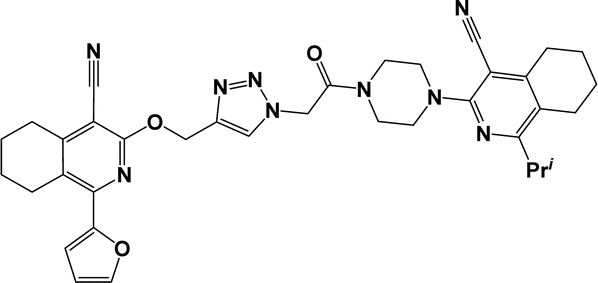 |
6o | 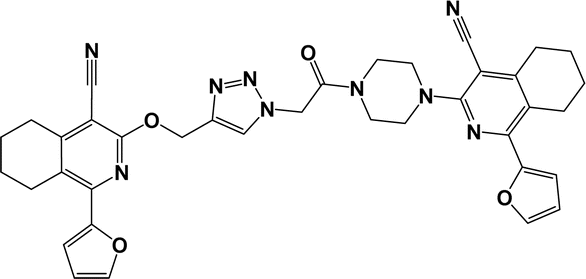 |
| 6g | 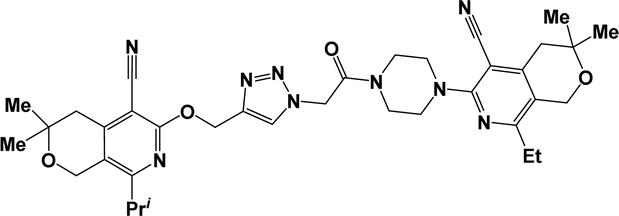 |
6p | 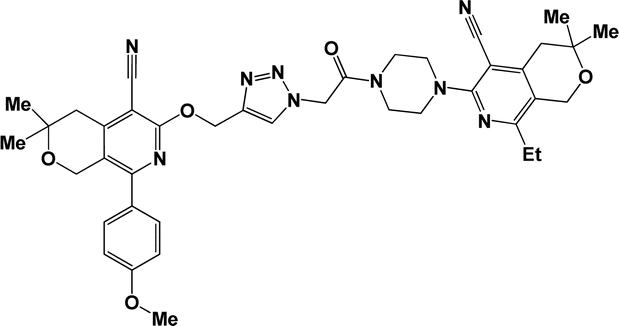 |
| 6h | 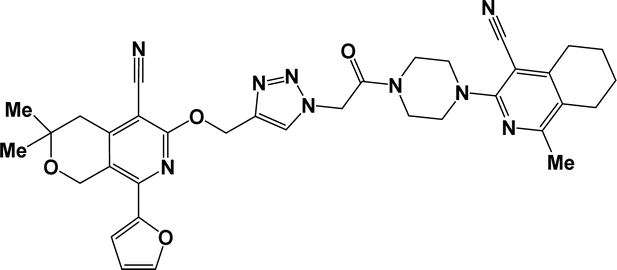 |
6q | 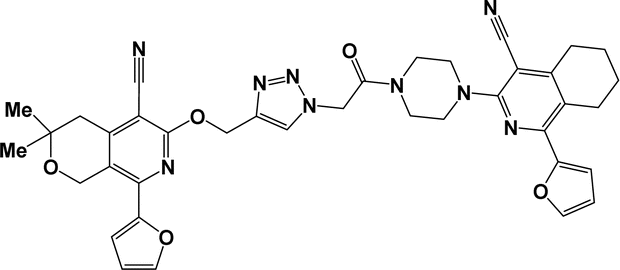 |
| 6i | 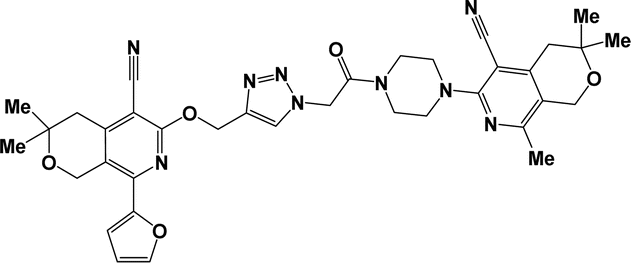 |
From both chemical and biological point of views, it was also interesting to synthesize hybrid compounds based on 1,2,4-triazole derivatives. For this purpose, the previously synthesized 2-[(4-cyano-1-methyl(2-furyl)-5,6,7,8-tetrahydroisoquinolin-3-yl)oxy]-acetohydrazides 7a and b,31 and 2-{[5-cyano-8-(2-furyl)-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridin-6-yl]oxy}acetohydrazide 7c,32 were used. Thus, compounds 7a–c upon reaction with phenylisothio-cyanate in absolute ethanol resulting the corresponding thioureido derivatives 8a–c, which were further cyclized into the corresponding 1,2,4-triazoles 9a–c in high yields, respectively (Scheme 4).
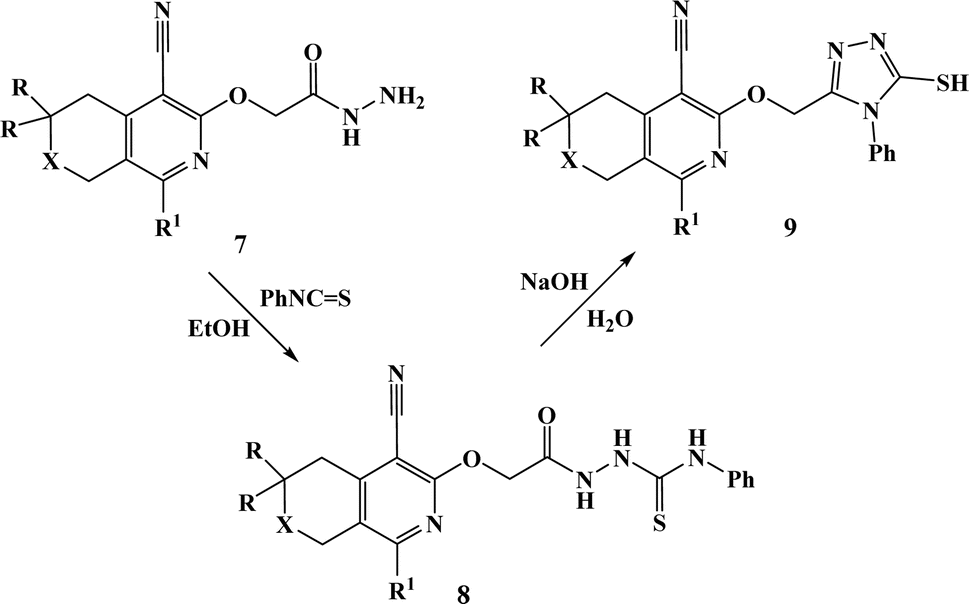 | ||
| Scheme 4 Synthesis of compounds 9a–c. 7–9. a: X = CH2, R = H, R1 = Me; b: X = CH2, R = H, R1 = 2-furyl; c: X = O, R = Me, R1 = 2-furyl. | ||
Finally, alkylation of the obtained 1,2,4-triazoles 9a–c using compounds 2 as alkylating agents led to the designed 3,5-disubstituted 1,2,4-triazoles (Scheme 5 and Table 2).
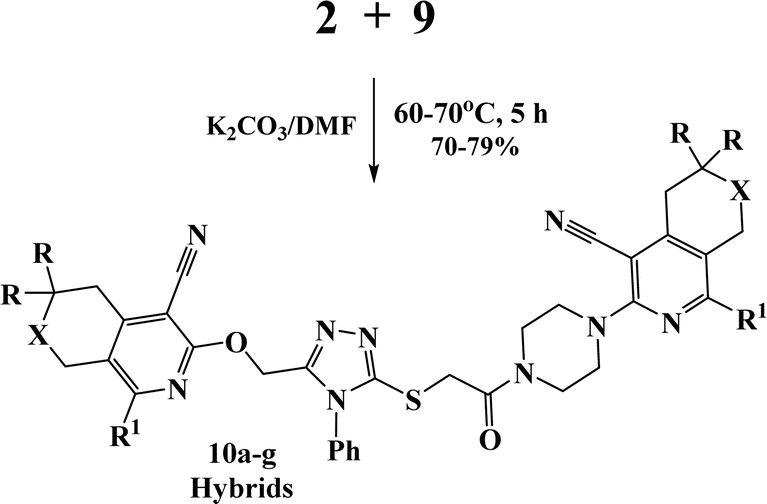 | ||
| Scheme 5 Synthesis of 3,5-disubstituted 1,2,4-triazoles 10a–g. | ||
| No. | Hybrids | No. | Hybrids |
|---|---|---|---|
| 10a | 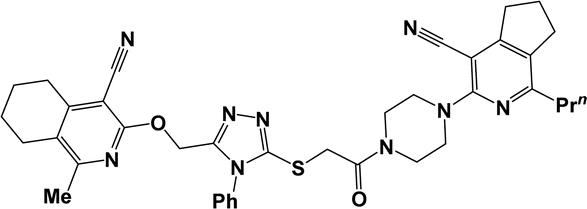 |
10e | 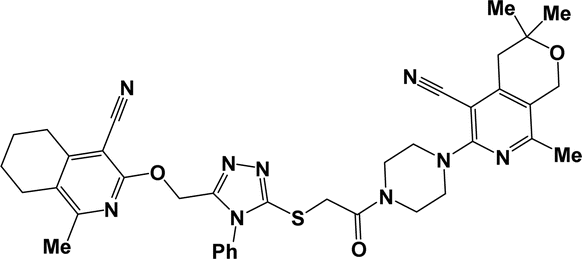 |
| 10b | 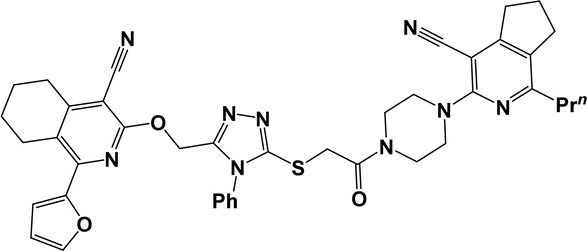 |
10f | 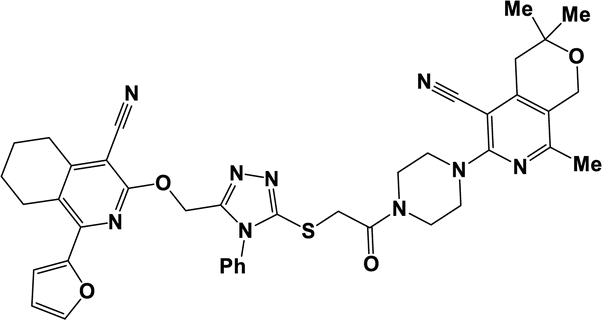 |
| 10c | 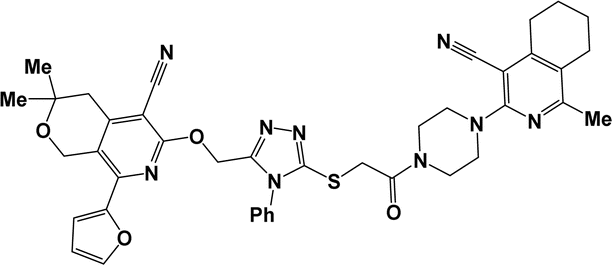 |
10g | 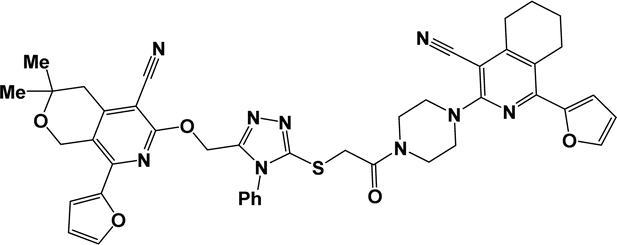 |
| 10d | 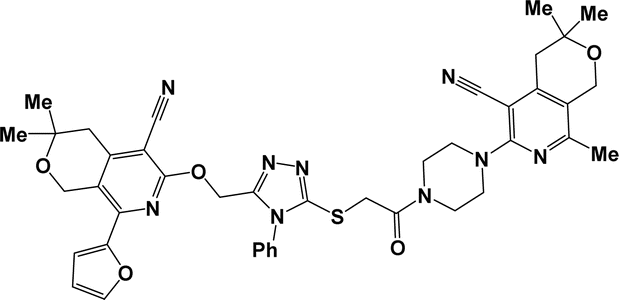 |
In the 1H NMR spectra of the obtained 3,5-disubstituted 1,2,4-triazoles 10a–g, the singlet signals of the OCH2 and SCH2 groups appeared at 5.36–5.60 ppm and 4.30–4.33 ppm, respectively.
2.2. Biological evaluation
Twenty-four newly synthesized heterocyclic compounds, 6a–q and 10a–g, were evaluated for their neurotropic activities and side effects.The preclinical development of new chemical agents for the treatment of epilepsy is routinely based on the use of animal models that imitate the given type of human seizures.
For the evaluation of the anticonvulsant activity of the hybrids, they were assessed based on their antagonism to pentylenetetrazole (PTZ), thiosemicarbazide (TSC) convulsion and maximal electroshock-induced seizure (MES).33–36 The MES and PTZ models have been proven useful for detecting the anti-seizure effects of drugs in healthy rodents.37,38 The MES test is one of the most useful pharmacological tools for the discovery of new anticonvulsants, given that it enables the identification of compounds that can prevent the spread of seizures. From a clinical point of view, the MES test is an animal model of human tonic–clonic epilepsy.39 The PTZ test is another very common rodent model of generalized absence epilepsy in humans. This test detects the ability of a test compound to increase the chemoconvulsant-induced seizure threshold of an animal, thus protecting it from exhibiting clonic, forebrain seizures.40 PTZ is a common convulsant used in animal models to investigate the mechanisms of seizures. The model of TSC seizures (affecting the exchange of GABA) causes clonic seizures similar to pentylenetetrazole seizures. The side effects of the compounds, neurotoxicity (movement coordination disorder, myorelaxation and ataxia), were also studied using the “rotating rod”33 test on mice. The well-known antiepileptic drug ethosuximide and the tranquilizer diazepam were used as reference drugs.41
The investigation revealed that all the hybrids displayed pentylenetetrazole antagonism, which ranged from 40% to 80%. Compounds 6d, k, and m and 10a–g at the studied doses of 25 and 50 mg kg−1 showed weak activity of up to 40%, respectively, and therefore they were not studied more deeply in other tests and are not presented in the tables. Compounds 6a–c, e–j, l, and n–q showed superior anticonvulsant activity compared to ethosuximide in the PTZ antagonism test, but were inferior to diazepam (Table 3 and Chart 1). Furthermore, at higher doses (50, 200, and 500 mg kg−1), they did not induce muscle relaxation, being low-toxic with daily toxicity between 630–870 mg kg−1 and therapeutic index (TI) much better that of ethosuximide (Table 3).
| Compound | ED50* mg kg−1 (by PTZ antagonism) | TD50* mg kg−1 | LD50* mg kg−1 | TI | Latency of convulsions, induced by TSC, min | |
|---|---|---|---|---|---|---|
| M ± M | I** | |||||
| a *indicates P ≤ 0.05 at a probability level; ** index of latent period of TSC seizures. | ||||||
| Control | — | — | — | 13.0 ± 2.59 | 1.0 | |
| 6a | 34 (28.3 ÷ 40.8) | >500 | 800 (666.7 ÷ 960) | 23.5 | 39.0 ± 4.2 | 3.0 |
| 6b | 35 (29.7 ÷ 41.3) | >500 | 750 (630 ÷ 892.5) | 21.4 | 25.0 ± 4.1 | 1.92 |
| 6c | 28 (24.3 ÷ 32.2) | >500 | 820 (683 ÷ 984) | 29.3 | 41.2 ± 1.61 | 3.17 |
| 6e | 22 (18.9 ÷ 25.5) | >500 | 815 (690.6 ÷ 962) | 37.0 | 52.6 ± 1.11 | 4.05 |
| 6f | 32 (26.7 ÷ 38.4) | >500 | 650 (541.6 ÷ 780) | 20.3 | 28.4 ± 1.67 | 2.18 |
| 6g | 30 (25.2 ÷ 35.7) | >500 | 720 (600 ÷ 864) | 24.0 | 25.2 ± 1.2 | 1.9 |
| 6h | 34 (28.1 ÷ 41.1) | >500 | 630 (538.5 ÷ 737) | 18.5 | 41.0 ± 2.75 | 4.1 |
| 6i | 41 (34.2 ÷ 49.2) | >500 | 790 (675 ÷ 924) | 19.3 | 41.0 ± 3.95 | 3.15 |
| 6j | 27 (22.9 ÷ 31.9) | >500 | 830 (686 ÷ 1004) | 30.7 | 36.4 ± 1.11 | 2.8 |
| 6l | 28 (22.8 ÷ 34.4) | >500 | 850 (692 ÷ 1020) | 30.4 | 41.2 ± 1.61 | 3.17 |
| 6n | 20 (16.7 ÷ 24.0) | >500 | 870 (757 ÷1001) | 43.5 | 30.4 ± 1.1 | 2.34 |
| 6o | 30 (25.0 ÷ 36.0) | >500 | 800 (656 ÷ 891) | 26.7 | 60.4 ± 5.2 | 4.65 |
| 6p | 22 (18.6 ÷ 26.0) | >500 | 730 (598 ÷ 845) | 33.2 | 39.4 ± 1.11 | 3.03 |
| 6q | 29 (24.4 ÷ 34.5) | >500 | 750 (615 ÷ 891) | 25.9 | 35.8 ± 4.34 | 2.75 |
| Ethosuximide | 155 (117.5 ÷ 204.5) | 520 (413 ÷ 655) | 1325 (1200 ÷ 1462) | 8.5 | 28.0 ± 1.4 | 2.15 |
| Diazepam | 0.5 (0.4 ÷ 0.7) | 2.7 (1.4 ÷ 5.5) | 180 (128.5 ÷ 252) | 360 | 9.0 ± 2.5 | 0.7 |
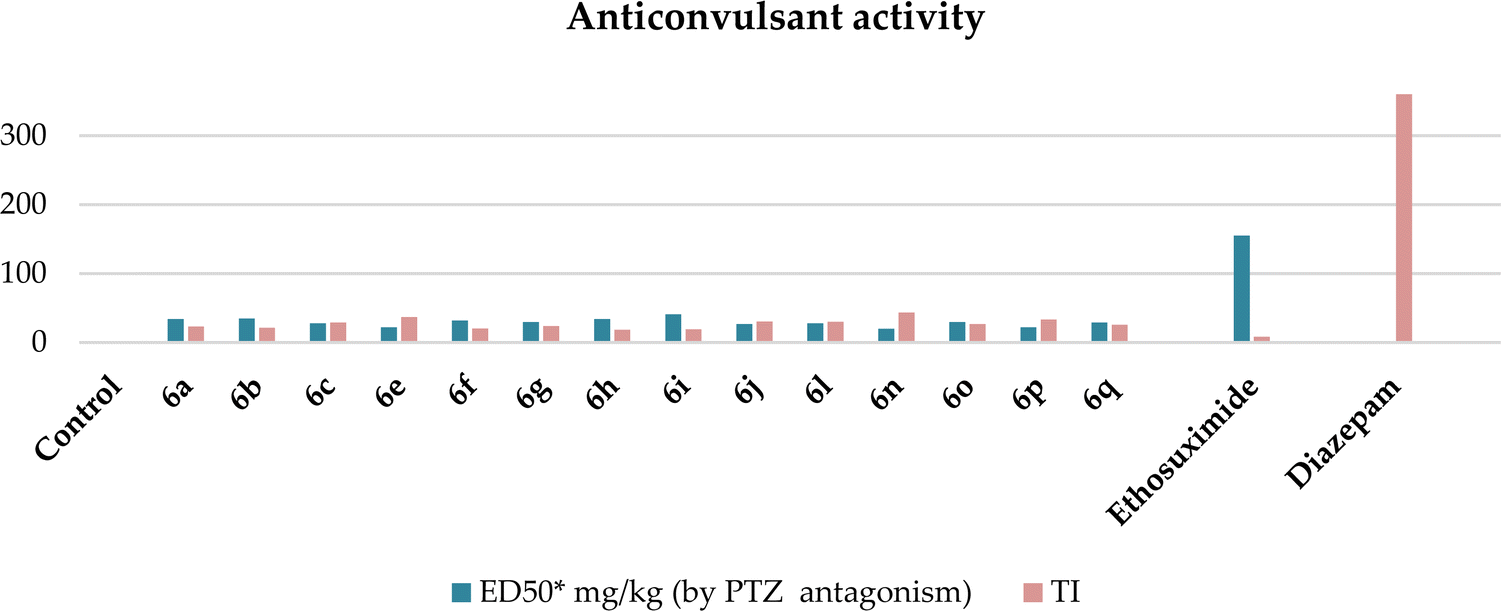 | ||
| Chart 1 Graphical representation of the anticonvulsant activity and TI of hybrids 6a–c, e–j, l, and n–q. The ordinate axis shows the obtained data on PTZ activity and the therapeutic index values, and the abscissa axis shows the number of compounds. | ||
The structure–activity relationship for anticonvulsant activity by antagonism to pentylenetetrazole revealed that the presence of pyrano[3,4-c]pyridine as a substituent on both piperazine and the triazole ring (compounds 6n) is beneficial for the anticonvulsant activity. The introduction of 8-(furan-2-yl)-6-methoxy-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile in the triazole ring and 1-(furan-2-yl)-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile on the triazole ring (compound 6p) slightly decreased the activity. The same activity was observed for compound 6e. Compound 6e has a 5,6,7,8-tetrahydroisoquinoline substituent on the piperazine ring. Replacement of the methyl group with furanyl in compound 6e from 3-methoxy-1-methyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile and Me group of 1-methyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile by propyl or furanyl led to the formation of compounds 6c and 6l with decreased activity, respectively. The introduction of a bulkier substituent in position 1 of the pyridine ring as well as cyclohexane ring by cyclopentane in general led to less active compounds (Fig. 2).
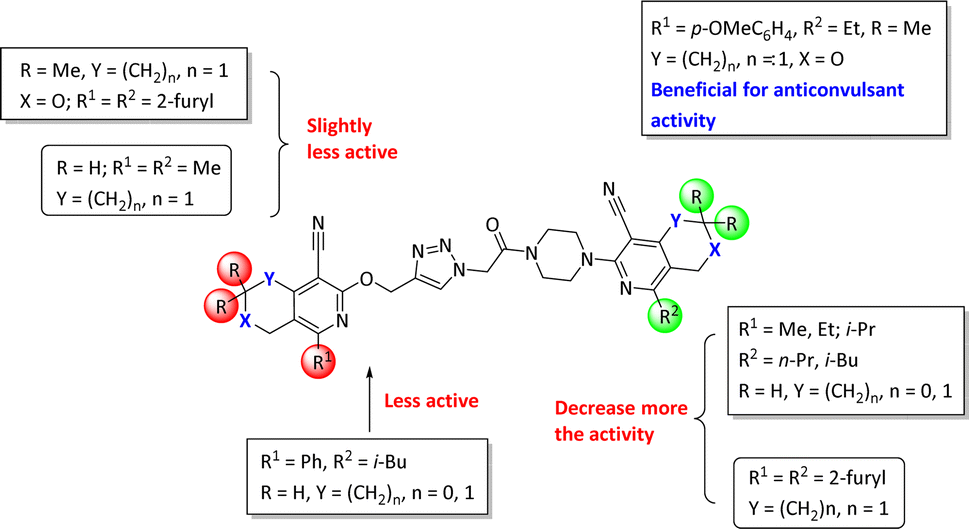 | ||
| Fig. 2 Structure–activity relationship for the anticonvulsant activity via antagonism towards pentylenetetrazole for compounds 6. | ||
The study of 14 the most active compounds (6a–c, e–j, l, and n–q) on the model of thiosemicarbazide (TSC) seizures demonstrated that at the dose of 100 mg kg−1 they increased the latency of thiosemicarbazide seizures to 1.9–4.65 times compared to the control, similar to ethosuximide (Table 3). Diazepam was not effective in this model. The most active compounds in this test appeared to be 6o and 6e. Both compounds have a 1-methyl-5,6,7,8-tetrahydroisoquinoline substituent on the piperazine ring. Alternatively, these compounds did not cause any protection tonic and clonic seizures in the MES test.
To evaluate the influence of the most active compounds on the locomotory activity and anxiety of animals, the “open field test”42,43 and “elevated plus maze” (EPM)44,45 were used. The results for the activity of the compounds in the “open field” model are shown in Table 4. An increase in the number of horizontal (especially compounds 6b, c, and f) and vertical movements (especially compounds 6h, n, and o) was observed, indicating the activating effect of the compounds. The same behavior was observed for diazepam, whereas not ethosuximide. In contrast to the control and ethosuximide, similar to diazepam, all the studied compounds showed an increase the number of cells examined. This indicates the anxiolytic (anti-anxiety) activity of the compounds, which was especially pronounced in compounds 6f, h, j, l, n and p. Diazepam at a dose of 2 mg kg−1 has the same properties, in contrast to ethosuximide, but is inferior to all these compounds. The most active compound, as in the previous PTZ test, showing the greatest effect was compound 6n, with two pyrano[3,4-c]pyridine rings. The second active compound was 6p, which has a 2-furyl substituent on both pyridine rings (Chart 2).
| Compound, dose 50 mg kg−1 | Amount (absolute data during 5 min)* | ||
|---|---|---|---|
| Horizontal displacement | Vertical displacement | Cells examination | |
| a *P ≤ 0.05 at a probability level. **The differences are statistically significant compared with the control. | |||
| Control | 10.6 ± 2.25 | 2.0 ± 0.5 | 1.6 ± 0.6 |
| 6a | 50.8 ± 8.1** | 5.2 ± 2.3** | 6.8 ± 2.4** |
| 6b | 192.0 ± 10.8** | 5.4 ± 1.03** | 6.2 ± 1.6** |
| 6c | 196.6 ± 15.8** | 4.8 ± 2.1** | 5.4 ± 1.2** |
| 6e | 49.8 ± 4.9** | 5.4 ± 1.7** | 6.0 ± 1.7** |
| 6f | 216.0 ± 17.2** | 8.2 ± 2.3** | 8.6 ± 3.5** |
| 6g | 105.0 ± 12.1** | 5.6 ± 0.8** | 6.2 ± 2.2** |
| 6h | 58.4 ± 7.5** | 9.0 ± 4.0** | 9.4 ± 1.9** |
| 6i | 37.0 ± 6.3** | 5.0 ± 1.3** | 3.8 ± 1.2** |
| 6j | 49.0 ± 9.58** | 7.8 ± 1.9** | 11.4 ± 3.5** |
| 6l | 71.2 ± 12.2** | 8.4 ± 2.5** | 11.8 ± 2.8** |
| 6n | 39.4 ± 5.4** | 13.2 ± 1.36** | 19.2 ± 3.8** |
| 6o | 30.2 ± 4.61** | 8.8 ± 1.61** | 7.4 ± 2.42** |
| 6p | 58.8 ± 7.4** | 3.8 ± 0.4** | 15.8 ± 1.1** |
| 6q | 47.8 ± 7.37** | 5.0 ± 0.8** | 6.6 ± 1.8** |
| Ethosuximide (200 mg kg−1) | 16.8 ± 5.51 | 2.6 ± 1.8 | 0.8 ± 0.5 |
| Diazepam (2 mg kg−1) | 33.6 ± 6.1** | 6.4 ± 1.4** | 5.0 ± 1.2** |
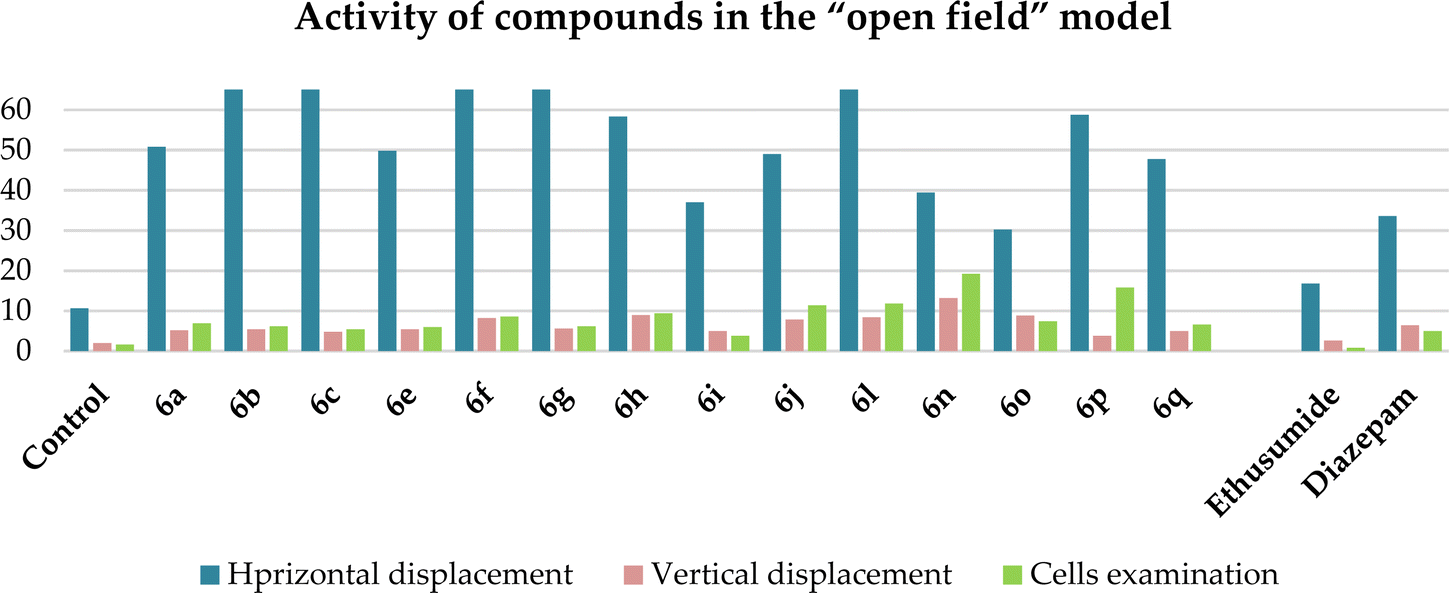 | ||
| Chart 2 Graphical representation of the activity of compounds 6a–c, e–j, l, and n–q, ethosuximide and diazepam in the “open field” model. The ordinate axis shows the obtained data in the ‘’open field’’ model and the abscissa axis shows the number of compounds. | ||
Regarding the elevated plus-maze (EPM) model, the control animals were mainly situated in the closed arms (Table 5), while the compounds (except compounds 6a, f and l) as well as both reference drugs increased the time spent by the experimental animals in the center. Simultaneously, they reduced the stay and number of entries into the closed arms (Chart 3).
| Compound, 50 mg kg−1 | Time spent in closed arms/s* | Number of entries into the closed arms* | Time spent in the center/s* | Time spent in the open arms/s* |
|---|---|---|---|---|
| a *P ≤ 0.05 at a probability level. ** The differences are statistically significant compared with the control. | ||||
| Control | 275.0 ± 9.4 | 4.8 ± 1.6 | 25.0 ± 3.1 | 0 |
| 6a | 245.6 ± 14.6** | 3.4 ± 1.1 | 31.4 ± 4.0 | 23.0 ± 2.1** |
| 6b | 173.0 ± 15.67** | 2.8 ± 1.4 | 64.0 ± 4.7** | 63.0 ± 6.1** |
| 6c | 169.8 ± 8.12** | 3.6 ± 0.8 | 105.2 ± 23.62** | 25.0 ± 3.2** |
| 6e | 217.4 ± 16.3** | 3.0 ± 0.8 | 65.0 ± 7.1** | 17.6 ± 2.0** |
| 6f | 236.0 ± 13.5** | 2.8 ± 1.1** | 27.0 ± 5.6 | 37.0 ± 5.3** |
| 6g | 250.0 ± 12.4** | 1.2 ± 0.6** | 35.0 ± 2.4** | 15.0 ± 2.4** |
| 6h | 175.0 ± 10.2** | 2.6 ± 0.6 | 57.0 ± 5.6** | 68.0 ± 8.43** |
| 6i | 200.0 ± 24.2** | 2.8 ± 2.1** | 81.0 ± 5.8** | 19.0 ± 3.1** |
| 6j | 207.8 ± 16.2** | 2.0 ± 2.1 | 53.4 ± 5.8** | 38.8 ± 4.7** |
| 6l | 231.0 ± 13.1** | 3.0 ± 1.2 | 30.4 ± 5.2 | 39.0 ± 5.4** |
| 6n | 206.0 ± 11.2** | 3.2 ± 1.03 | 62.2 ± 13.8** | 31.8 ± 3.78** |
| 6o | 237.0 ± 13.7** | 2.4 ± 1.2 | 38.8 ± 3.4** | 24.2 ± 3.5** |
| 6p | 226.4 ± 13.4** | 3.0 ± 1.59 | 44.2 ± 5.1** | 29.4 ± 4.59** |
| 6q | 227.0 ± 15.1** | 3.0 ± 0.67 | 50.0 ± 7.2** | 23.0 ± 5.51** |
| Ethosuximide (200 mg kg−1) | 247.2 ± 15.0 | 8.1 ± 2.5 | 52.8 ± 4.7** | 0 |
| Diazepam (2 mg kg−1) | 257.5 ± 15.2 | 5.5 ± 1.1** | 42.5 ± 3.9** | 57.0 ± 4.2** |
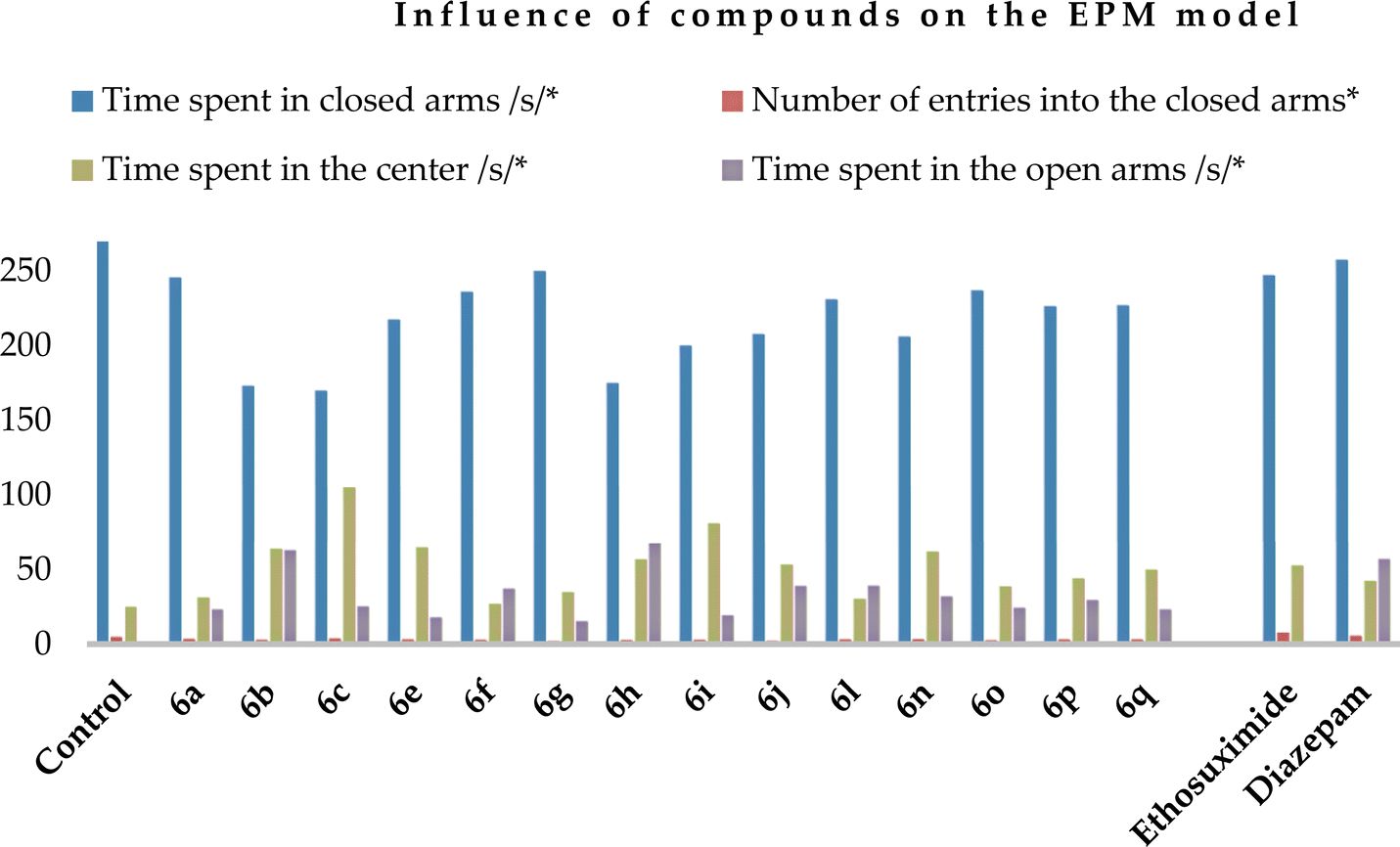 | ||
| Chart 3 Graphical representation of the influence of compounds 6a–c, e–j, l, and n–q and compared drugs on the EPM model (5 min of research). The ordinate axis shows the obtained data on the EPM model and the abscissa axis shows the number of compounds. | ||
Administration of all the compounds was beneficial given that the experimental animals entered the open arms and stayed there for 15.0 (6g) to 68.0 (6h) s, in contrast to the control animals and those who received ethosuximide at a dose of 200 mg kg−1. This is an indication of an anxiolytic effect. The most active compound was found to be 6h, which contains two 5,6,7,8-tetrahydroisoquinoline rings in its structure.
The antidepressant activity of selected compounds was studied using the “forced swim” method46 at a dose of 50 mg kg−1. It was found that compounds 6a, f, g, i, o and p increased the latent period of the first immobilization (Table 6) compared to the control mice (52 s after the first immobilization), which indicates a certain antidepressant effect. Diazepam at a dose of 2 mg kg−1 acted similarly to these compounds, increasing the latent time of first immobilization, while in the case of ethosuximide at a dose of 200 mg kg−1, it behaved similarly to the control. Alternatively, compounds 6a, e, and h and ethosuximide increased, while diazepam reduced the total immobilization time, increasing the total swimming time. However, compounds 6b, c, f, g, j, n and q exhibited a pronounced antidepressant effect, leading to an increase in the total swimming time, among which, after the introduction of compounds 6b, c, j, n and q, there was complete swimming for 6 min. Also, compounds 6n and 6q were the most active in this test (Chart 4).
| Compound, dose 50 mg kg−1 | Latent period first immobilization (s) | Total time of active swimming (s)* | Total time of immobilization (s)* |
|---|---|---|---|
| a *P ≤ 0.05 at a probability level. **The differences are statistically significant compared with the control. | |||
| Control | 52.0 ± 5.4 | 318.0 ± 8.36 | 42.0 ± 7.24 |
| 6a | 79.0 ± 7.8** | 276.4 ± 6.2** | 84.0 ± 3.2** |
| 6b | 0** | 360.0 ± 0.2** | 0** |
| 6c | 0** | 360.0 ± 0.2** | 0** |
| 6e | 52.0 ± 6.3 | 296.0 ± 16.85 | 66.0 ± 4.48** |
| 6f | 72.0 ± 6.3** | 357.0 ± 13.5** | 3.0 ± 0.6** |
| 6g | 73.0 ± 5.8** | 358.0 ± 8.2** | 2.0 ± 0.8** |
| 6h | 58.0 ± 7.1 | 288.0 ± 8.35 | 84.0 ± 5.04** |
| 6i | 66.9 ± 5.3** | 346.0 ± 11.35** | 14.0 ± 3.37** |
| 6j | 0** | 360.0 ± 0.2** | 0** |
| 6l | 53.0 ± 7.4 | 324.0 ± 12.4 | 42.0 ± 12.04 |
| 6n | 0** | 360.0 ± 0.2** | 0** |
| 6o | 68.5 ± 7.2** | 336.0 ± 25.3** | 24.0 ± 2.9** |
| 6p | 70.0 ± 7.3** | 345.0 ± 18.55 | 15.0 ± 2.55** |
| 6q | 0** | 360.0 ± 1.64** | 0** |
| Ethosuximide (200 mg kg−1) | 55.0 ± 10.1 | 262 ± 25.0 | 98.0 ± 9.2 |
| Diazepam (2 mg kg−1) | 74.0 ± 8.7** | 336.0 ± 8.1** | 24.0 ± 8.2** |
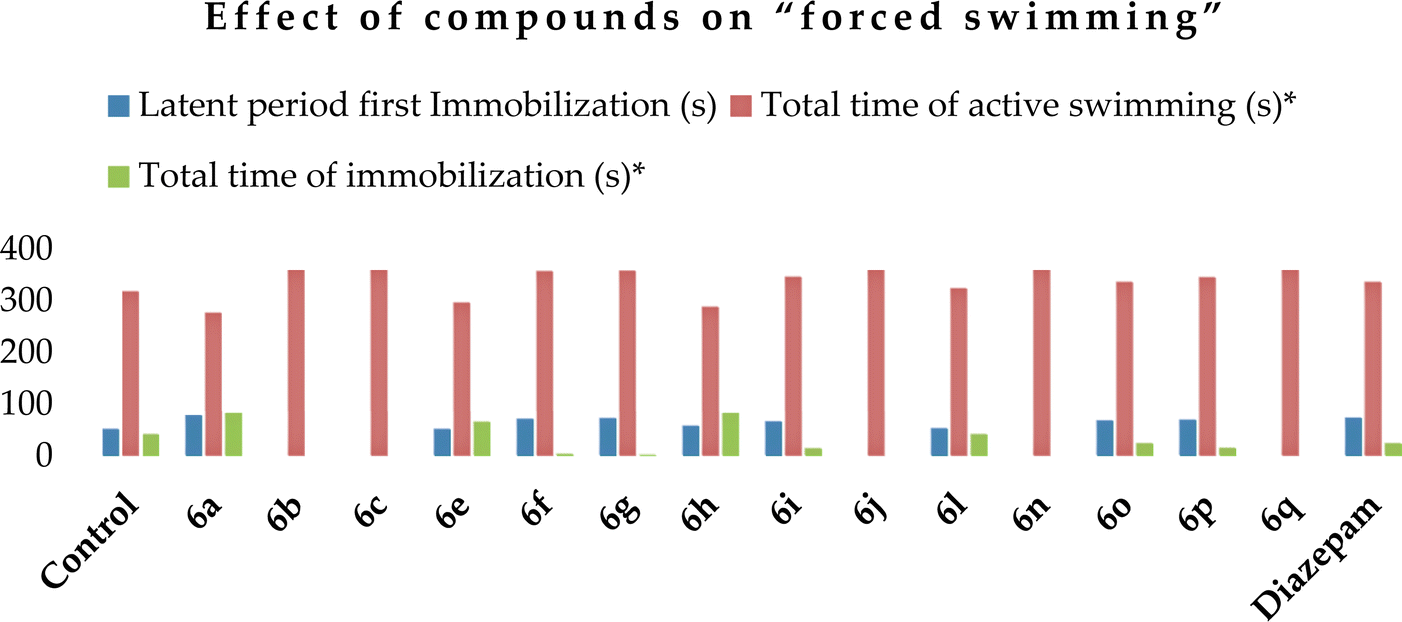 | ||
| Chart 4 Graphical representation of the effect of compounds 6a–c, e–j, l, and n–q and reference preparations on “forced swim” (study for 6 min). The ordinate axis shows the obtained data on “forced swim” and the abscissa axis shows the number of compounds. | ||
For the evaluation of learning and memory in rodent models of CNS disorders, the model of electroshock retrograde amnesia, conditioned response of passive avoidance (CRPA) (Table 7), was used.47 During 6 min of the experiment, the control rats on the 1st and 2nd day remained in the light compartment for almost the entire period (280.0 s), while with the administration of the compounds at a dose of 50 mg kg−1, they somehow increased the time of reproduction of the reflex in animals similar to the nootropic drug piracetam in a day (Table 7). This indicates the anti-amnestic effect of the chosen hybrid derivatives. The results obtained are very close, although they are statistically significant, and therefore are not presented in graphical form.
| Compound, dose 50 mg kg−1 | The time spent in the light chamber during the training of CRPA (s), first day | The time spent in the light chamber when playing CRPA + MESH after 24 hours (s), second day |
|---|---|---|
| a CRPA + MESh – conditioned reaction of passive avoidance + maximal electric shock; * significant differences in values from the intact control without MES; ** significant differences in values from the control with MES at P ≤ 0.05. | ||
| Control | 280.0 ± 7.5 | 281.0 ± 6.1 |
| 6a | 283.0 ± 9.3 | 297.4 ± 6.1** |
| 6b | 281.0 ± 8.6 | 297.0 ± 6.5** |
| 6c | 281.0 ± 7.4 | 299.0 ± 8.1** |
| 6e | 285.8 ± 5.2 | 301.0 ± 7.1** |
| 6f | 287.0 ± 7.2 | 297.2 ± 5.8** |
| 6g | 282.0 ± 7.7 | 299.0 ± 8.4** |
| 6h | 283.9 ± 8.9 | 300.0 ± 7.7** |
| 6i | 280.0 ± 8.0 | 298.4 ± 5.3** |
| 6j | 286.0 ± 7.3 | 297.0 ± 5.3** |
| 6l | 285.0 ± 6.5 | 296.4 ± 5.3** |
| 6n | 285.0 ± 8.1 | 295.4 ± 5.3** |
| 6o | 281.0 ± 7.4 | 297.0 ± 5.3** |
| 6p | 283.0 ± 5.8 | 298.0 ± 5.3** |
| 6q | 284.0 ± 6.3 | 296.2 ± 3.3** |
| Diazepam (2 mg kg−1) | 187.0 ± 6.7* | 126.0 ± 6.5** |
| Piracetam (1000 mg kg−1) | 158.0 ± 5.9* | 243.7 ± 8.4** |
2.3. Molecular docking
In the docking studies, the crystal structure of the GABAA receptor was acquired from the Protein Data Bank (PDB) using PDB ID: 4COF.50 The resolution, R, and R free values for the X-ray diffraction structure of the GABAA receptor were 2.97, 0.206, and 0.226, respectively. The initial co-crystal ligand benzamidine was removed and re-docked at the catalytic site of the protein using the same parameters and preparation procedure of as that used for the tested compounds as the first step in the docking studies and for their validation. The root-mean-square deviation (RMSD) of the co-crystal and re-docked pose was found to be 0.34 (Fig. 3).
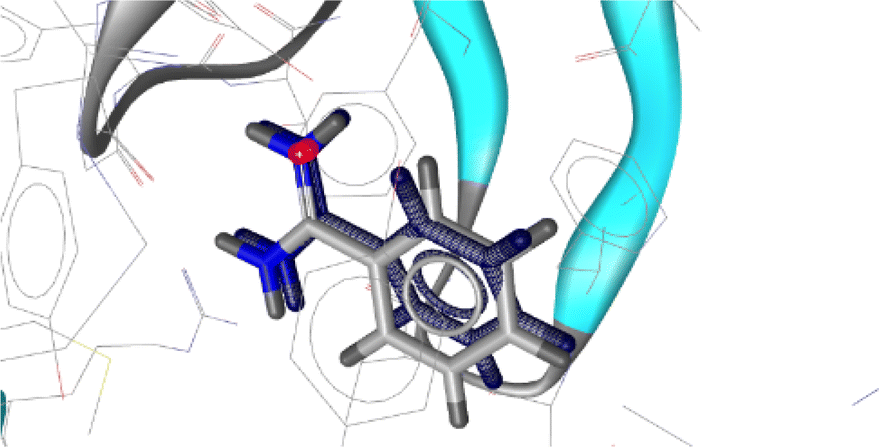 | ||
| Fig. 3 Alignment of the docked conformation of the re-docked initial inhibitor benzamidine (blue) and the co-crystalized ligand to the 4COF structure (RMSD: 0.34 Å). | ||
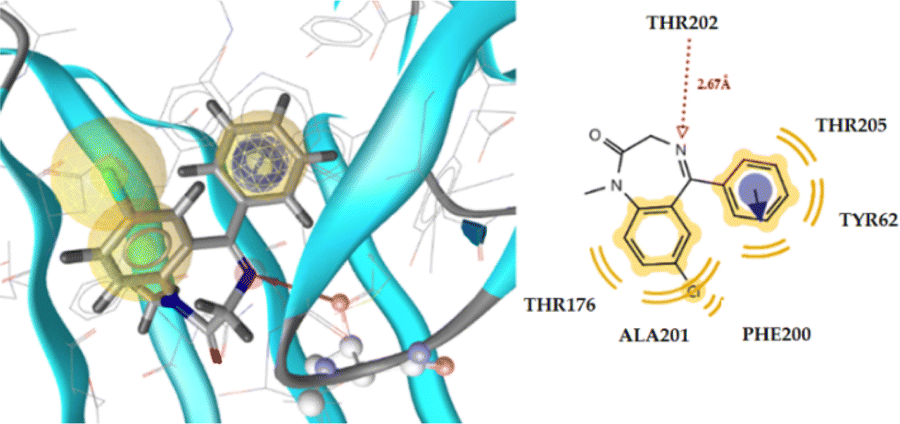
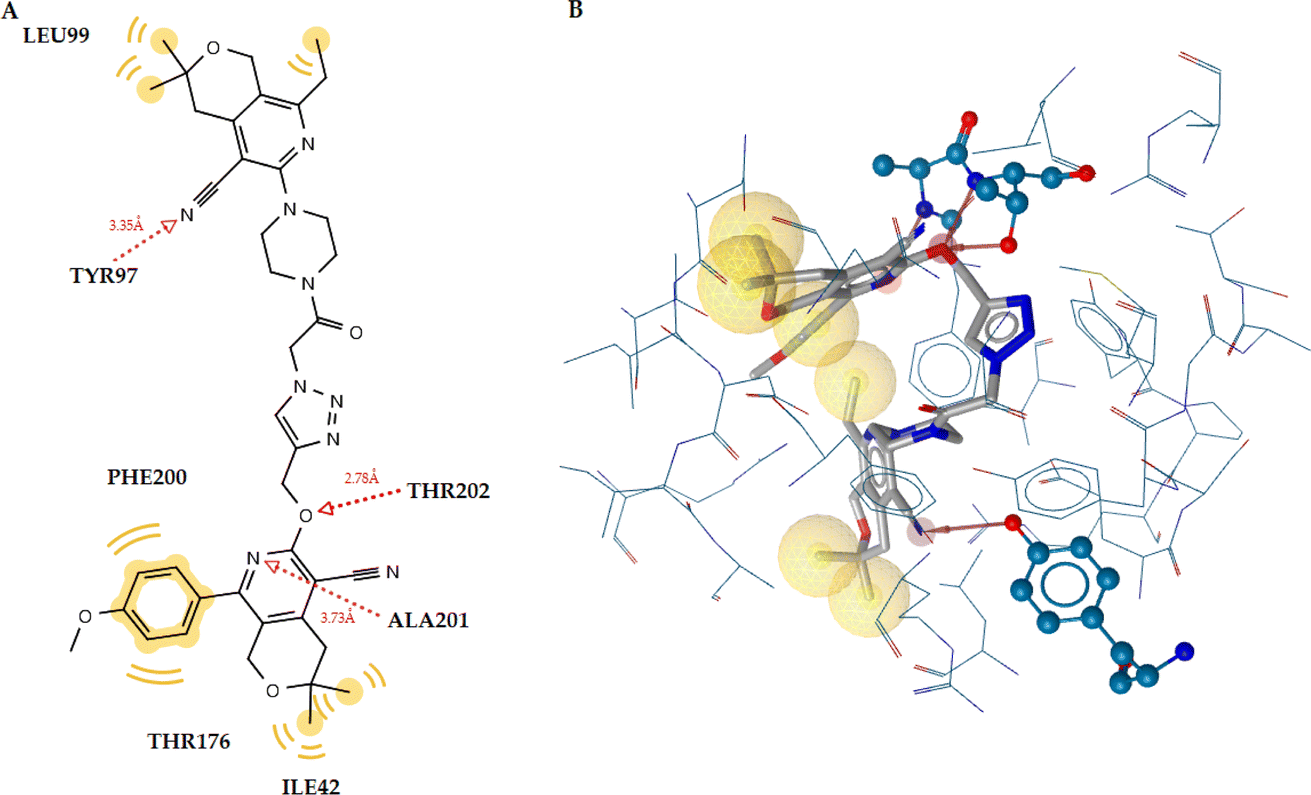
Table 8 displays the docking analysis results. According to the obtained results, compound 6n forms three hydrogen bonds with the Tyr97 (N⋯H, 3.35), Thr202 (O⋯H, 2.78), and Ala201 (N⋯H, 3.73) residues, while diazepam only forms one hydrogen bond with Thr202 (N⋯H, 2.67), as shown in Fig. 4 for the latter and Fig. 5 for the former. The benzene moiety of the molecule interacts hydrophobically with the Thr176 and Phe200 residues, further stabilizing the complex ligand-enzyme.
| No. | Est. binding energy (kcal mol−1) | I–H | Residues involved in hydrogen bond formation | Hydrophobic interactions | Aromatic interactions |
|---|---|---|---|---|---|
| 6a | −6.89 | 1 | Thr202 (O⋯H, 3.98 Å), Tyr205 (N⋯H, 2.77 Å) | Ala25, Leu99, Phe200 | — |
| 6b | −6.35 | — | — | Thr176, Tyr205 | — |
| 6c | −8.30 | 1 | Thr202 (O⋯H, 3.29 Å) | Thr176, Phe200, Ala201 | Phe200 |
| 6d | −6.45 | — | — | Thr176, Phe200 | — |
| 6e | −10.72 | 3 | Tyr97 (N⋯H, 3.25 Å), Thr202 (O⋯H, 3.56 Å), Tyr205 (N⋯H, 2.94 Å) | Thr176, Ala201, Phe200 | — |
| 6f | −7.02 | 1 | Tyr97 (O⋯H, 3.42 Å) | Phe200 | — |
| 6g | −7.10 | 1 | Thr202 (N⋯H, 3.20 Å) | Tyr62, Phe200 | — |
| 6h | −6.78 | — | — | Thr176, Ala201, Tyr205 | — |
| 6i | −6.20 | — | — | Ty157, Tyr205 | — |
| 6j | −9.81 | 2 | Tyr97 (N⋯H, 3.78 Å), Thr202 (O⋯H, 3.26 Å) | Ala201, Tyr205 | — |
| 6k | |||||
| 6l | −9.48 | 2 | Thr202 (N⋯H, 2.79 Å), Tyr205 (N⋯H, 2.88 Å) | Ile42, Tyr62, Leu99, Phe200, Ala201 | Phe200 |
| 6m | |||||
| 6n | −11.54 | 3 | Tyr97 (N⋯H, 3.35 Å), Thr202 (O⋯H, 2.78 Å), Ala201 (N⋯H, 3.73 Å) | Ile42, Tyr97, Leu99Thr176, Phe200, Ala201 | — |
| 6o | −8.11 | 1 | Thr202 (O⋯H, 3.76 Å) | Thr176, Phe200 | — |
| 6p | −9.97 | 2 | Tyr97 (N⋯H, 3.54 Å), Thr202 (O⋯H, 3.11 Å) | Thr176, Ala201, Phe200, Tyr205 | — |
| 6q | −8.25 | 1 | Thr202 (O⋯H, 3.49 Å) | Phe200, Ala201 | — |
| Diazepam | −8.90 | 1 | Thr202 (N⋯H, 2.67 Å) | Tyr62, Thr176, Phe200, Ala201, Tyr205 | Phe200 |
 | ||
| Fig. 4 Docked conformation of diazepam and the GABAA receptor complex. | ||
 | ||
| Fig. 5 Docked pose of compound 6n and the GABAA receptor complex. Red lines show hydrogen bonds and yellow spheres represent hydrophobic interactions. | ||
After accounting for all the previously mentioned variables, we proceeded with the docking studies of SERT and 5-HT1A receptor to ascertain whether the investigated compounds function as dual inhibitors of the serotonin transporter (SERT) and pre-synaptic auto inhibitory 5-HT1A receptors.
We used the X-ray crystal structure of LeuT coupled to L-tryptophan (PDB code: 3F3A), a prokaryotic homologue of SERT, given that no crystal structure of SERT is available in the Protein Data Bank (PDB).54 Table 9 displays the results of the docking studies in SERT.
| No. | Est. binding energy (kcal mol−1) | I–H | Residues involved in hydrogen bond formation | Residues involved in hydrophobic interactions | Residues involved in aromatic interactions |
|---|---|---|---|---|---|
| 6a | −4.92 | — | — | — | — |
| 6b | −9.11 | — | Arg11 (N⋯H, 2.52 Å) | His7, Arg431 | — |
| 6c | −9.48 | — | Arg11 (N⋯H, 3.57 Å), Lys443 (N⋯H, 3.54 Å) | His7, Arg431 | — |
| 6d | −5.24 | — | — | His7, Arg431 | — |
| 6e | −6.95 | 1 | His7 (N⋯H, 3.51 Å) | Asp267, Gly433 | — |
| 6f | −6.45 | 1 | His7 (N⋯H, 3.85 Å) | Asp267 | — |
| 6g | −7.08 | 1 | His7 (N⋯H, 3.23 Å) | Asp267, Gly433 | Lys264 |
| 6h | −5.52 | — | — | Asp267, Arg431 | — |
| 6i | −5.20 | — | — | Gly432 | — |
| 6j | −8.26 | 1 | Gln266 (N⋯H, 2.96 Å) | Asp267, Gly432, Gly433 | — |
| 6k | −6.58 | 1 | His7 (N⋯H, 3.23 Å) | Asp267, Gly433 | — |
| 6l | −6.79 | 1 | Gln266 (N⋯H, 3.54 Å) | His7, Asp267 | — |
| 6m | −5.02 | — | — | Arg431 | — |
| 6n | −9.65 | — | Arg11 (N⋯H, 3.02 Å), His7 (O⋯H, 3.16 Å) | Asp267, Ile441 | — |
| 6o | −4.26 | — | — | His7, Gly433 | — |
| 6p | −4.15 | — | — | — | — |
| 6q | −9.87 | — | Arg11 (N⋯H, 2.43 Å), Asp272 (O⋯H, 3.12 Å) | Ala9, Ile441 | — |
Initially, as a step in the docking experiments and for the validation of the docking parameters, the co-crystal ligand L-tryptophan was removed and re-docked at the protein binding site using the same parameters as that utilized for the tested drugs. The RMSD of 0.86 was found between the co-crystal and re-docked poses, as shown in Fig. 6.
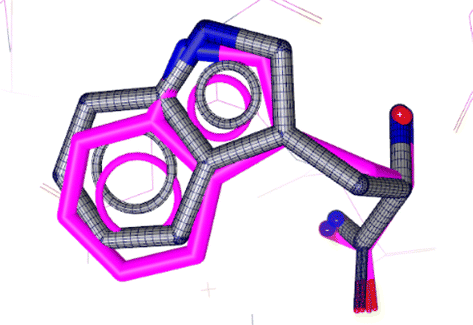 | ||
| Fig. 6 Alignment of the docked conformation of the re-docked initial inhibitor L-tryptophan (blue) and the co-crystallized 3F3A structure (magenta, RMSD: 0.86 Å). | ||
Compound 6q, which forms two hydrogen bonds, had the best docking score. The first hydrogen bond is 2.43 Å between the nitrogen atom of the side chain of Arg11 and the hydrogen atom, and the other is 3.12 Å between the oxygen atom of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and the hydrogen of the side chain of Asp272 (Fig. 7). Thus, the experimental data and the docking results are in agreement.
O group and the hydrogen of the side chain of Asp272 (Fig. 7). Thus, the experimental data and the docking results are in agreement.
 | ||
| Fig. 7 (A) Docked pose of compound 6q and the SERT complex. (B) 2D ligand interaction diagram for the docked ligand. | ||
Docking to the 5-HT1A receptor was done using the crystal structure of the human 2-adrenergic receptor in conjunction with the antagonist alprenolol (PDB code: 3NYA).55,56 To validate the docking parameters, the original co-crystal ligand alprenolol was re-docked at the catalytic region of the protein. The RMSD (root-mean-square deviation) between the co-crystal and re-docked poses was 0.98 (Fig. 8).
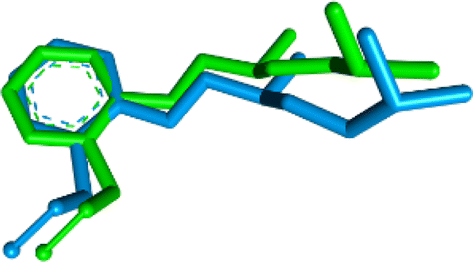 | ||
| Fig. 8 Alignment of the docked conformation of the re-docked initial inhibitor alprenolol (blue) and the co-crystalized ligand (green) to the 3NYA structure (RMSD: 0.98 Å). | ||
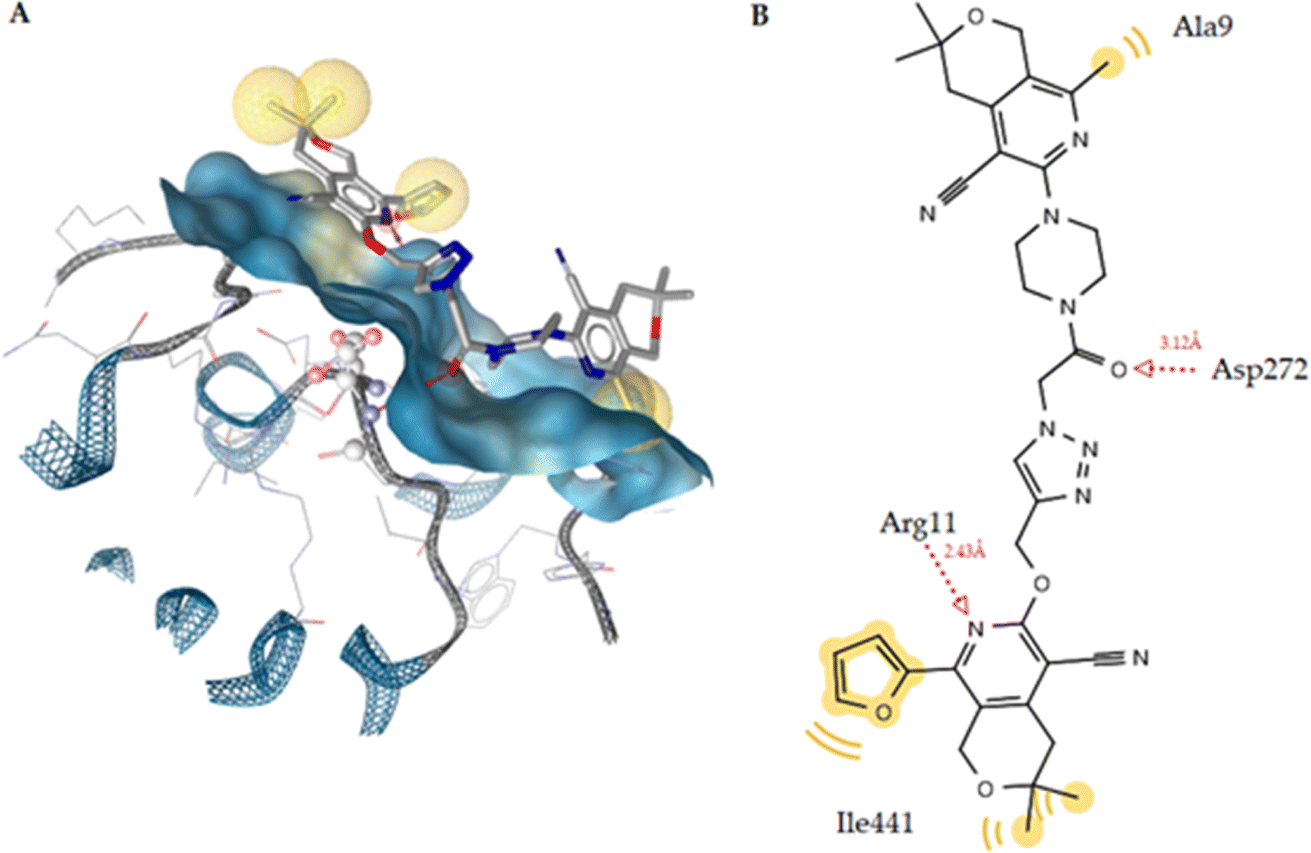
The orthosteric binding site of the 5-HT1A receptor was docked with all the substances studied (Table 10), and compound 6q (−12.12 kcal mol−1) was formed the best docking score because it forms four hydrogen bonds with the Phe193, Thr195, and Tyr316 residues. Alprenolol also forms a hydrogen bond with Tyr316. This compound also shows hydrophobic interactions with the Trp109, Phe193, Tyr199, Thr110, Ala200, Phe289, and Tyr316 residues (Fig. 9A). According to Fig. 9B, it is obvious that compound is situated in the same cavity of the enzyme, similar to alprenolol, which can explain the high activity of compound 6q. Finally, according to the docking results, this compound can be a dual target given that it is a good inhibitor of SERT as well as 5-HT1A receptor binder.
| No. | Est. binding energy (kcal mol−1) | I–H | Residues involved in hydrogen bond formation | Residues involved in hydrophobic interactions | Residues involved in aromatic interactions |
|---|---|---|---|---|---|
| 6a | −4.40 | — | — | — | — |
| 6b | −10.12 | 1 | Asn312 (N⋯H, 2.89 Å) | Trp109, Phe193, Ala200, Phe289, Tyr316 | — |
| 6c | −9.94 | 2 | Thr195 (O⋯H, 3.15 Å), Tyr316 (O⋯H, 2.88 Å) | Thr118, Ala200, Asn312 | — |
| 6d | −5.08 | — | — | Trp286, Asn312 | — |
| 6e | −5.37 | — | — | — | — |
| 6f | −6.10 | — | — | Trp286 | — |
| 6g | −7.02 | 1 | Tyr316 (O⋯H, 3.16 Å) | Thr118, Ala200, Asn312 | — |
| 6h | −5.03 | — | — | Ala200, Tyr308 | — |
| 6i | −4.19 | — | — | Trp286 | — |
| 6j | −9.25 | 1 | Tyr308 (N⋯H, 2.53 Å) | Trp286, Asn312 | — |
| 6k | −4.10 | — | — | — | — |
| 6l | −2.79 | — | — | — | — |
| 6m | −5.02 | — | — | Arg431 | — |
| 6n | −11.65 | 3 | Tyr118 (O⋯H, 2.72 Å), Ser204 (N⋯H, 3.53 Å), Tyr316 (N⋯H, 3.71 Å) | Trp109, Ile309, Tyr308, Phe193, Val114, Tyr110, Ile201, Ala200 | — |
| 6o | −4.20 | — | — | Thr118, Ala200 | — |
| 6p | −4.82 | — | — | Thr118, Ala200, Asn312 | — |
| 6q | −12.12 | 4 | Phe193 (O⋯H, 2.75 Å), Phe193 (N⋯H, 2.62 Å), Thr195 (O⋯H, 3.02 Å), Tyr316 (O⋯H, 2.53 Å) | Trp109, Phe193, Tyr199, Thr110, Ala200, Phe289, Tyr316 | Phe193 |
| Alprenolol | −13.19 | 4 | Asp113, Asn312, Tyr316 | Tyr118, Ala200, Tyr308 | — |
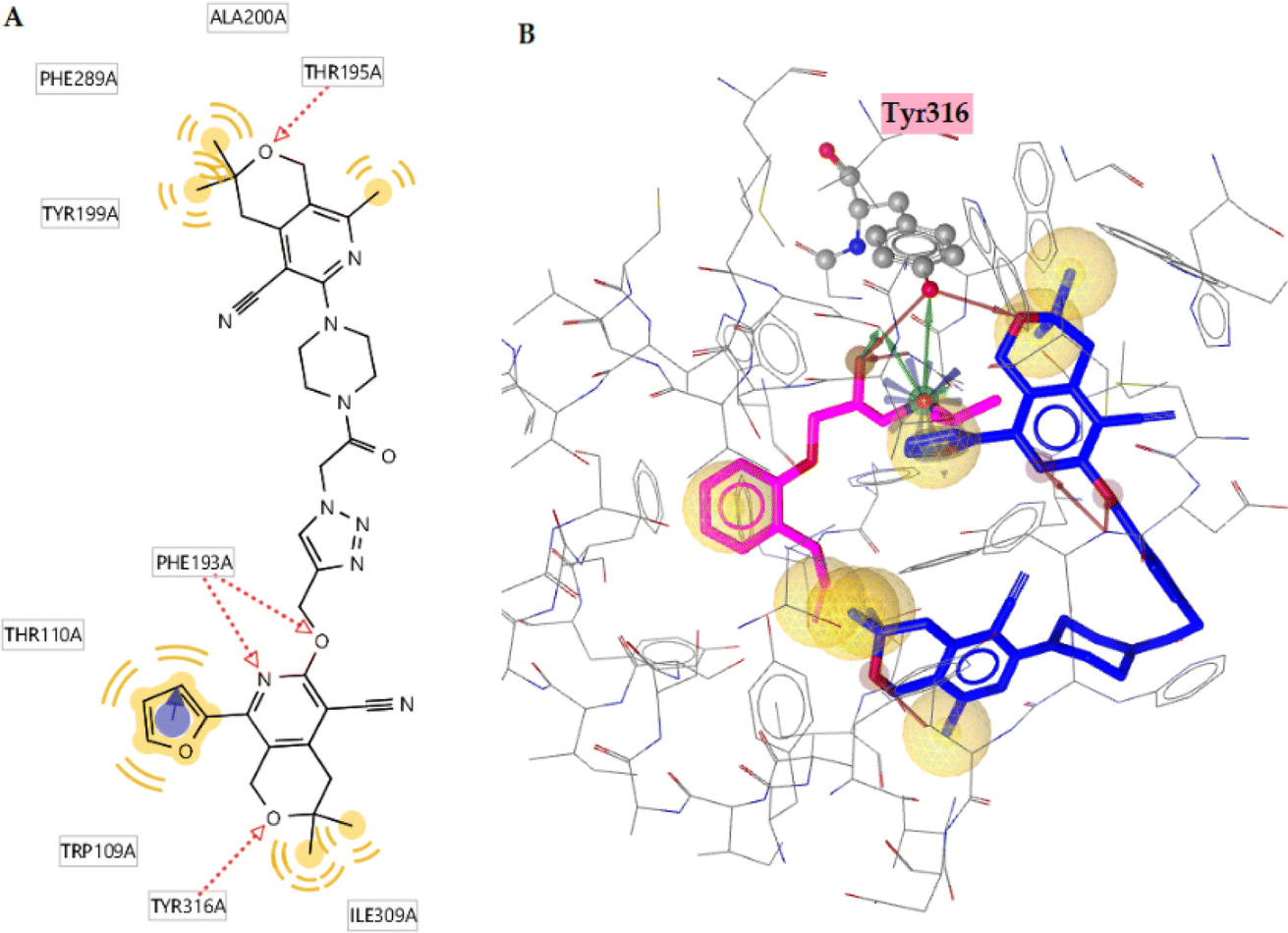 | ||
| Fig. 9 (A) 2D ligand interaction diagram for docked compound 6q. (B) Docked pose of compound 6q (blue) and alprenolol (magenta) into the 5-HT1A receptor. | ||
3. Materials and methods
3.1. Chemistry
3.1.1.1. General procedure for the synthesis of compounds 2a and c–e. To a stirred solution of compound 1 (5 mmol) and triethylamine (0.84 mL, 6 mmol) in benzene (50 mL), chloroacetyl chloride (4.8 mL, 6 mmol) was added dropwise. The reaction mixture was maintained at 35 °C for 6 h. Then the reaction mixture was cooled at room temperature, the solvent was removed under vacuum and water (50 mL) was added. The resulting crystals were filtered, washed with water, dried and recrystallized from ethanol.
3.1.1.1.1. 3-[4-(Chloroacetyl)piperazin-1-yl]-1-propyl-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbonitrile (2a). The reaction between compound 1a and chloroacetyl chloride produced a white solid in 77% yield; mp 114–116 °C; IR ν/cm−1: 1654 (C
O), 2208 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 0.97 (t, J = 7.4 Hz, 3H, CH2CH3), 1.64–1.77 (m, 2H, CH2CH3), 2.09–2.20 (m, 2H, 6-CH2), 2.55–2.61 (m, 2H, CH2C2H5), 2.84 (t, J = 7.5 Hz, 2H, 7-CH2), 3.01 (t, J = 7.6 Hz, 2H, 5-CH2), 3.54–3.70 (m, 8H, C4H8N2), 4.22 (s, 2H, CH2Cl). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 13.50, 20.41, 23.52, 29.14, 32.43, 37.05, 40.79, 41.10, 45.09, 47.69, 48.32, 90.31, 115.97, 129.08, 158.52, 159.08, 160.13, 164.18. Anal. calcd for C18H23ClN4O: C 62.33%; H 6.68%; N 16.15%. Found: C 62.73%; H 6.90%; N 16.44%.
N). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 0.97 (t, J = 7.4 Hz, 3H, CH2CH3), 1.64–1.77 (m, 2H, CH2CH3), 2.09–2.20 (m, 2H, 6-CH2), 2.55–2.61 (m, 2H, CH2C2H5), 2.84 (t, J = 7.5 Hz, 2H, 7-CH2), 3.01 (t, J = 7.6 Hz, 2H, 5-CH2), 3.54–3.70 (m, 8H, C4H8N2), 4.22 (s, 2H, CH2Cl). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 13.50, 20.41, 23.52, 29.14, 32.43, 37.05, 40.79, 41.10, 45.09, 47.69, 48.32, 90.31, 115.97, 129.08, 158.52, 159.08, 160.13, 164.18. Anal. calcd for C18H23ClN4O: C 62.33%; H 6.68%; N 16.15%. Found: C 62.73%; H 6.90%; N 16.44%.
3.1.1.1.2. 3-[4-(Chloroacetyl)piperazin-1-yl]-1-methyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (2c). The reaction between compound 1c and chloroacetyl chloride produced a white solid in 77% yield; mp 128–130 °C; IR ν/cm−1: 1655 (C
O), 2215 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.76–1.88 (m, 4H, 6,7-CH2), 2.35 (s, 3H, CH3), 2.53–2.59 (m, 2H, 8-CH2), 2.80–2.86 (m, 2H, 5-CH2), 3.49–3.70 (m, 8H, C4H8N2), 4.20 (s, 2H, CH2Cl). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 21.10, 21.90, 22.12, 24.64, 27.92, 40.76, 41.18, 45.17, 47.86, 48.37, 94.03, 115.74, 122.64, 150.83, 158.62, 158.81, 164.16. Anal. calcd for C17H21ClN4O: C 61.35%; H 6.36%; N 16.83%. Found: C 61.67%; H 6.53%; N 17.08%.
3.1.1.1.3. 3-[4-(Chloroacetyl)piperazin-1-yl]-1-isopropyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (2d). The reaction between compound 1d and chloroacetyl chloride produced a white solid in 75% yield; mp 119–121 °C; IR ν/cm−1: 1661 (C
O), 2209 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.17 (t, J = 6.7 Hz, 6H, CH(CH3)2), 1.77–1.85 (m, 4H, 6,7-CH2), 2.61–2.68 (m, 2H, 8-CH2), 2.81–2.88 (m, 2H, 5-CH2), 3.16 (sp, J = 6.7 Hz, 1H, CH(CH3)2), 3.52–3.70 (m, 8H, C4H8N2), 4.21 (s, 2H, CH2Cl). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.90, 21.07, 22.02, 23.82, 28.23, 30.30, 40.76, 41.12, 45.16, 47.65, 48.36, 93.86, 115.79, 121.11, 151.42, 158.87, 164.15, 165.98. Anal. calcd for C19H25ClN4O: C 63.24%; H 6.98%; N 15.53%. Found: C 63.62%; H 7.19%; N 15.81%.
3.1.1.1.4. 3-[4-(Chloroacetyl)piperazin-1-yl]-1-(2-furyl)-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (2e). The reaction between compound 1e and chloroacetyl chloride produced a brown solid in 84% yield; mp 186–188 °C; IR ν/cm−1: 1676 (C
O), 2210 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.77–1.90 (m, 4H, 6,7-CH2), 2.88–3.01 (m, 4H, 8,5-CH2), 3.54–3.73 (m, 8H, C4H8N2), 4.23 (s, 2H, CH2Cl), 6.56 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.08 (dd, J = 3.5, 0.8 Hz, 1H, 3-CHfur), 7.68 (dd, J = 1.7, 0.8 Hz, 1H, 5-CHfur). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.90, 22.00, 25.51, 28.59, 40.83, 41.12, 45.12, 47.77, 48.36, 94.94, 111.30, 113.52, 115.70, 121.17, 143.80, 147.19, 153.15, 153.20, 158.44, 164.23. Anal. calcd for C20H21ClN4O2: C 62.42%; H 5.50%; N 14.56%. Found: C 62.79%; H 5.96%; N 14.83%.
3.1.1.2. General procedure for the synthesis of compounds 3a and c–e. A mixture of compound 2 (5 mmol) and sodium azide (0.65 g, 10 mmol) in acetone (50 mL) was heated at refluxing for 15 h. After filtration, the solvent was evaporated and water (50 mL) was added. The resulting crystals were filtered, washed with water, dried, and recrystallized from ethanol.
3.1.1.2.1. 3-[4-(Azidoacetyl)piperazin-1-yl]-1-propyl-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbonitrile (3a). The reaction between compound 2a and sodium azide produced a white solid in 84%; mp 122–124 °C; IR ν/cm−1: 1651 (C
O), 2109 (N3), 2210 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 0.96 (t, J = 7.4 Hz, 3H, CH2CH3), 1.64–1.77 (m, 2H, CH2CH3), 2.09–2.20 (m, 2H, 6-CH2), 2.55–2.61 (m, 2H, CH2C2H5), 2.84 (t, J = 7.5 Hz, 2H, 7-CH2), 3.00 (t, J = 7.6 Hz, 2H, 5-CH2), 3.49–3.71 (m, 8H, C4H8N2), 4.05 (s, 2H, CH2N3). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 13.50, 20.42, 23.52, 29.14, 32.44, 37.06, 40.97, 43.81, 47.73, 48.19, 49.56, 90.22, 116.00, 129.06, 158.53, 159.07, 160.14, 165.27. Anal. calcd for C18H23N7O: C 61.17%; H 6.56%; N 27.74%. Found: C 61.51%; H 6.73%; N 27.99%.
3.1.1.2.2. 3-[4-(Azidoacetyl)piperazin-1-yl]-1-methyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (3c). The reaction between compound 2c and sodium azide produced a white solid in 81% yield; mp 100–102 °C; IR ν/cm−1: 1655 (C
O), 2106 (N3), 2210 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.76–1.87 (m, 4H, 6,7-CH2), 2.35 (s, 3H, CH3), 2.52–2.59 (m, 2H, 8-CH2), 2.79–2.85 (m, 2H, 5-CH2), 3.49–3.56 (m, 6H, C4H8N2), 3.64–3.71 (m, 2H, C4H8N2), 4.05 (s, 2H, CH2N3). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 21.11, 21.90, 22.14, 24.64, 27.93, 41.04, 43.88, 47.86, 48.27, 49.55, 93.95, 115.78, 122.61, 150.84, 158.65, 158.81, 165.26. Anal. calcd for C17H21N7O: C 60.16%; H 6.24%; N 28.89%. Found: C 60.55%; H 6.46%; N 29.17%.
3.1.1.2.3. 3-[4-(Azidoacetyl)piperazin-1-yl]-1-isopropyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (3d). The reaction between compound 2d and sodium azide produced a white solid in 87% yield; mp 116–118 °C; IR ν/cm−1: 1645 (C
O), 2111 (N3), 2202 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.17 (t, J = 6.7 Hz, 6H, CH(CH3)2), 1.77–1.85 (m, 4H, 6,7-CH2), 2.61–2.68 (m, 2H, 8-CH2), 2.81–2.88 (m, 2H, 5-CH2), 3.16 (sp, J = 6.7 Hz, 1H, CH(CH3)2), 3.51–3.60 (m, 6H, C4H8N2), 3.66–3.72 (m, 2H, C4H8N2), 4.04 (s, 2H, CH2N3). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.90, 21.06, 22.01, 23.81, 28.22, 30.29, 40.97, 43.84, 47.70, 48.23, 49.55, 93.80, 115.79, 121.07, 151.41, 158.87, 165.19, 165.96. Anal. calcd for C19H25N7O: C 62.10%; H 6.86%; N 26.68%. Found: C 62.42%; H 7.04%; N 26.92%.
3.1.1.2.4. 3-[4-(Azidoacetyl)piperazin-1-yl]-1-(2-furyl)-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (3e). The reaction between compound 2e and sodium azide produced a yellow solid in 85% yield; mp 174–176 °C; IR ν/cm−1: 1655 (C
O), 2103 (N3), 2213 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.77–1.90 (m, 4H, 6,7-CH2), 2.88–2.94 (m, 2H, 8-CH2), 2.95–3.00 (m, 2H, 5-CH2), 3.52–3.74 (m, 8H, C4H8N2), 4.06 (s, 2H, CH2N3), 6.57 (dd, J = 3.5,1.7 Hz, 1H, 4-CHfur), 7.08 (dd, J = 3.5, 0.8 Hz, 1H, 3-CHfur), 7.68 (dd, J = 1.7, 0.8 Hz, 1H, 5-CHfur). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.89, 22.00, 25.51, 28.57, 40.97, 43.82, 47.80, 48.22, 49.56, 94.86, 111.30, 113.51, 115.71, 121.13, 143.80, 147.18, 153.15, 153.18, 158.44, 165.30. Anal. calcd for C20H21N7O2: C 61.37%; H 5.41%; N 25.05%. Found: C 61.73%; H 5.61%; N 25.32%.
3.1.1.3. General procedure for the synthesis of compounds 5b, c, h and i. To a stirred suspension of compound 4 (5 mmol) and potassium carbonate (1.38 g, 10 mmol) in absolute DMF (25 mL), propargyl bromide (0.42 mL, 5.5 mmol) was added dropwise. The reaction mixture was maintained at room temperature for 5 h. After water was added (50 mL), the resulting crystals were filtered, washed with water, dried and recrystallized from ethanol.
3.1.1.3.1. 1-Methyl-3-(prop-2-yn-1-yloxy)-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (5b). The reaction between compound 4b and propargyl bromide produced a white solid in 71% yield; mp 94–96 °C; IR ν/cm−1: 2128 (C
CH), 2221 (CN), 3246, 3269 (CH). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3): δ 1.76–1.89 (m, 4H, 6,7-CH2), 2.41 (s, 3H, CH3), 2.57–2.62 (m, 2H, 8-CH2), 2.83–2.88 (m, 2H, 5-CH2), 2.91 (t, J = 2.4 Hz, 1H, CH), 5.02 (d, J = 2.4 Hz, 2H, OCH2). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.84, 21.80, 21.87, 24.62, 27.83, 53.10, 75.85, 78.02, 92.89, 113.29, 124.21, 152.01, 158.32, 159.30. Anal. calcd for C14H14N2O: C 74.31%; H 6.24%; N 12.38%. Found: C 74.69%; H 6.46%; N 12.67%.
3.1.1.3.2. 1-Ethyl-3-(prop-2-yn-1-yloxy)-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (5c). The reaction between compound 4c and propargyl bromide produced a white solid in 75% yield; mp 84–86 °C; IR ν/cm−1: 2131 (C
CH), 2220 (CN), 3276 (CH). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.27 (t, J = 7.4 Hz, 3H, CH2CH3), 1.76–1.88 (m, 4H, 6,7-CH2), 2.59–2.65 (m, 2H, 8-CH2), 2.70 (q, J = 7.4 Hz, 2H, CH2CH3), 2.85–2.88 (m, 2H, 5-CH2), 2.87 (t, J = 2.4 Hz, 1H, CH), 5.04 (d, J = 2.4 Hz, 2H, OCH2). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 11.04, 20.86, 21.85, 24.02, 27.04, 27.95, 53.06, 75.62, 78.12, 92.73, 113.33, 123.54, 152.12, 159.51, 162.26. Anal. calcd for C15H16N2O: C 74.97%; H 6.71%; N 11.66%. Found: C 75.30%; H 6.88%; N 11.89%.
3.1.1.3.3. 8-(4-Methoxyphenyl)-3,3-dimethyl-6-(prop-2-yn-1-yloxy)-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (5h). The reaction between compound 4h and propargyl bromide produced a white solid in 70% yield; mp 131–133 °C; IR ν/cm−1: 2121 (C
CH), 2225 (CN), 3268 (CH). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.33 (s, 6H, C(CH3)2), 2.86 (s, 2H, CH2), 3.00 (t, J = 2.4 Hz, 1H, CH), 3.87 (s, 3H, OCH3), 4.69 (s, 2H, OCH2), 5.10 (d, J = 2.4 Hz, 2H, OCH2), 6.95–7.00 (m, 2H, C6H4), 7.50–7.57 (m, 2H, C6H4). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 26.18, 38.16, 53.50, 54.66, 59.84, 69.15, 76.16, 78.00, 93.62, 112.99, 113.24, 121.54, 129.44, 129.97, 150.66, 155.06, 159.84, 160.04. Anal. calcd for C21H20N2O3: C 72.40; H 5.79; N 8.04%. Found: C 72.75; H 5.98; N 8.30%.
3.1.1.3.4. 8-(2-Furyl)-3,3-dimethyl-6-(prop-2-yn-1-yloxy)-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (5i). The reaction between compound 4i and propargyl bromide produced a light-yellow solid in 72% yield; mp 183–185 °C; IR ν/cm−1: 2127 (C
CH), 2219 (CN), 3276 (CH). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.31 (s, 6H, C(CH3)2), 2.83 (s, 2H, CH2), 2.98 (t, J = 2.4 Hz, 1H, CH), 5.00 (s, 2H, OCH2), 5.10 (d, J = 2.4 Hz, 2H, OCH2), 6.63 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.28 (dd, J = 3.5, 0.7 Hz, 1H, 3-CHfur), 7.76 (dd, J = 1.7, 0.7 Hz, 1H, 5-CHfur). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 25.71, 38.34, 53.61, 59.77, 68.43, 76.13, 77.74, 93.86, 111.85, 112.89, 114.30, 119.42, 143.31, 144.94, 151.07, 152.49, 159.49. Anal. calcd for C18H16N2O3: C 70.12%; H 5.23%; N 9.09%. Found: C 70.43%; H 5.39%; N 9.31%.
3.1.1.4. General procedure for the synthesis of compounds 6b–i, k, l, n and o–q. Compounds 5 (2 mmol) and the corresponding azides 3 (2.2 mmol) were suspended in a 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of water and tert-butyl alcohol (30 mL). Sodium ascorbate (0.04 g, 0.2 mmol, of freshly prepared solution in water) was added, followed by copper(II) sulfate pentahydrate (0.05 g, 0.2 mmol, in water). The mixture was stirred for 10 h at room temperature, and then 10 h at 40–50 °C. After cooling, water was added (50 mL), the precipitate was filtered, washed with water and recrystallized from a mixture of ethanol-chloroform (1:3).
:1 mixture of water and tert-butyl alcohol (30 mL). Sodium ascorbate (0.04 g, 0.2 mmol, of freshly prepared solution in water) was added, followed by copper(II) sulfate pentahydrate (0.05 g, 0.2 mmol, in water). The mixture was stirred for 10 h at room temperature, and then 10 h at 40–50 °C. After cooling, water was added (50 mL), the precipitate was filtered, washed with water and recrystallized from a mixture of ethanol-chloroform (1:3).
3.1.1.4.1. 3-{4-[(4-{[(4-Cyano-1-isopropyl-6,7-dihydro-5H-cyclopenta[c]pyridin-3-yl)oxy]methyl}-1H-1,2,3-triazol-1-yl)acetyl]piperazin-1-yl}-1-methyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (6b). The reaction between compound 3c and 5a produced a light-yellow solid in 72% yield; mp 207–209 °C; IR ν/cm−1: 1660 (C
O), 2193, 2231 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.27 (d, J = 6.8 Hz, 6H, CH(CH3)2), 1.77–1.87 (m, 4H, 6′,7′-CH2), 2.13–2.24 (m, 2H, 6-CH2), 2.36 (s, 3H, CH3), 2.53–2.60 (m, 2H, 8′-CH2), 2.80–2.93 (m, 4H, 7,5′-CH2), 3.03 (t, J = 7.6 Hz, 2H, 5-CH2), 3.05 (sp, J = 6.8 Hz, 1H, CH(CH3)2), 3.51–3.74 (m, 8H, C4H8N2), 5.47 (s, 2H, OCH2), 5.55 (s, 2H, NCH2), 7.93 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.60, 21.11, 21.91, 22.17, 23.89, 24.64, 27.93, 28.68, 32.00, 32.70, 41.24, 44.00, 47.79, 48.25, 50.33, 59.64, 89.76, 93.89, 113.80, 115.83, 122.58, 125.19, 129.58, 141.78, 150.83, 158.66, 158.82, 161.00, 161.22, 162.60, 163.71. Anal. calcd for C32H37N9O2: C 66.30%; H 6.43%; N 21.75%. Found: C 66.68%; H 6.65%; N 22.03%.
3.1.1.4.2. 3-[(1-{2-[4-(4-Cyano-1-propyl-6,7-dihydro-5H-cyclopenta[c]pyridin-3-yl)piperazin-1-yl]-2-oxoethyl}-1H-1,2,3-triazol-4-yl)methoxy]-1-methyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (6c). The reaction between compound 3a and 5b produced a white solid in 86% yield; mp 136–138 °C; IR ν/cm−1: 1673 (C
O), 2217, 2218 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 0.97 (t, J = 7.4 Hz, 3H, CH2CH3), 1.65–1.77 (m, 2H, CH2CH3), 1.77–1.88 (m, 4H, 6,7-CH2), 2.10–2.21 (m, 2H, 6′-CH2), 2.45 (s, 3H, CH3), 2.55–2.63 (m, 4H, 8-CH2, CH2C2H5), 2.82–2.89 (m, 4H, 5,7′-CH2), 3.02 (t, J = 7.5 Hz, 2H, 5′-CH2), 3.58–3.75 (m, 8H, C4H8N2), 5.47 (s, 2H, OCH2), 5.52 (s, 2H, NCH2), 7.95 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 13.50, 20.41, 20.87, 21.84, 21.94, 23.51, 24.61, 27.81, 29.14, 32.44, 37.06, 41.17, 43.93, 47.62, 48.15, 50.32, 59.30, 90.15, 92.71, 113.65, 116.02, 123.74, 125.51, 128.97, 141.77, 151.93, 158.45, 158.52, 159.05, 160.13, 160.17, 163.73. Anal. calcd for C32H37N9O2: C 66.30%; H 6.43%; N 21.75%. Found: C 66.61%; H 6.59%; N 21.98%. ESI + MS; [C32H37N9O2+Na+] found: 602.2968.
3.1.1.4.3. 3-[(1-{2-[4-(4-Cyano-1-isobutyl-6,7-dihydro-5H-cyclopenta[c]pyridin-3-yl)piperazin-1-yl]-2-oxoethyl}-1H-1,2,3-triazol-4-yl)methoxy]-1-methyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (6d). The reaction between compound 3b and 5b produced a white solid in 79% yield; mp 166–168 °C; IR ν/cm−1: 1673 (C
O), 2206, 2217 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 0.94 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.77–1.88 (m, 4H, 6,7-CH2), 2.07–2.22 (m, 3H, 6′-CH2, CH(CH3)2), 2.45 (s, 3H, CH3), 2.48 (d, J = 7.1 Hz, 2H, CHCH2), 2.57–2.63 (m, 2H, 8-CH2), 2.81–2.88 (m, 4H, 5,7′-CH2), 3.02 (t, J = 7.6 Hz, 2H, 5′-CH2), 3.56–3.74 (m, 8H, C4H8N2), 5.47 (s, 2H, OCH2), 5.52 (s, 2H, NCH2), 7.95 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.90, 21.86, 21.97, 22.17, 23.55, 24.63, 27.26, 27.84, 29.37, 32.51, 41.19, 43.95, 44.13, 47.66, 48.22, 50.35, 59.31, 90.24, 92.72, 113.69, 116.03, 123.78, 125.55, 129.45, 141.77, 151.97, 158.08, 158.49, 158.95, 160.15, 160.18, 163.75. Anal. calcd for C33H39N9O2: C 66.76%; H 6.62%; N 21.23%. Found: C 67.11%; H 6.81%; N 21.48%. ESI + MS; [C33H39N9O2+Na+] found: 616.3124.
3.1.1.4.4. 3-{4-[(4-{[(4-Cyano-1-methyl-5,6,7,8-tetrahydroisoquinolin-3-yl)oxy]methyl}-1H-1,2,3-triazol-1-yl)acetyl]piperazin-1-yl}-1-methyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (6e). The reaction between compound 3c and 5b produced a white solid in 76% yield; mp 198–200 °C; IR ν/cm−1: 1658 (C
O), 2202, 2219 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.77–1.88 (m, 8H, 6,6′,7,7′-CH2), 2.36 (s, 3H, CH3), 2.45 (s, 3H, CH3), 2.53–2.63 (m, 4H, 8,8′-CH2), 2.80–2.89 (m, 4H, 5,5′-CH2), 3.51–3.74 (m, 8H, C4H8N2), 5.47 (s, 2H, OCH2), 5.52 (s, 2H, NCH2), 7.96 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.85, 21.10, 21.82, 21.85, 21.89, 21.94, 22.15, 24.59, 24.62, 27.80, 27.92, 41.21, 43.99, 47.77, 48.22, 50.32, 59.28, 92.67, 93.86, 113.65, 115.83, 122.56, 123.74, 125.58, 150.83, 151.93, 158.45, 158.66, 158.79, 160.14, 163.73. Anal. calcd for C31H35N9O2: C 65.82%; H 6.24%; N 22.29%. Found: C 66.19%; H 6.45%; N 22.56%. ESI + MS; [C31H35N9O2+Na+] found: 588.2811.
3.1.1.4.5. 3-[(1-{2-[4-(4-Cyano-1-isopropyl-5,6,7,8-tetrahydroisoquinolin-3-yl)piperazin-1-yl]-2-oxoethyl}-1H-1,2,3-triazol-4-yl)methoxy]-1-methyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (6f). The reaction between compound 3d and 5b produced a white solid in 82% yield; mp 119–121 °C; IR ν/cm−1: 1668 (C
O), 2210 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.19 (d, J = 6.6 Hz, 6H, CH(CH3)2), 1.77–1.87 (m, 8H, 6,6′,7,7′-CH2), 2.45 (s, 3H, CH3), 2.58–2.69 (m, 4H, 8,8′-CH2), 2.82–2.89 (m, 4H, 5,5′-CH2), 3.17 (sp, J = 6.6 Hz, 1H, CH(CH3)2), 3.55–3.76 (m, 8H, C4H8N2), 5.47 (s, 2H, OCH2), 5.52 (s, 2H, NCH2), 7.94 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.87, 20.90, 21.07, 21.83, 21.94, 22.01, 23.81, 24.60, 27.80, 28.22, 30.29, 41.15, 41.19, 43.98, 47.59, 48.20, 50.31, 59.29, 92.70, 93.70, 113.64, 115.84, 121.03, 123.74, 125.54, 141.7, 151.42, 151.93, 158.44, 158.86, 160.16, 163.69, 165.98. Anal. calcd for C33H39N9O2: C 66.76%; H 6.62%; N 21.23%. Found: C 67.09%; H 6.80%; N 21.47%. ESI + MS; [C33H39N9O2+Na+] found: 616.3124.
3.1.1.4.6. 3-[(1-{2-[4-(4-Cyano-1-isobutyl-6,7-dihydro-5H-cyclopenta[c]pyridin-3-yl)piperazin-1-yl]-2-oxoethyl}-1H-1,2,3-triazol-4-yl)methoxy]-1-ethyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (6g). The reaction between compound 3b and 5c produced a white solid in 77% yield; mp 140–142 °C; IR ν/cm−1: 1661 (C
O), 2205, 2220 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 0.94 (d, J = 6.6 Hz, 6H, CH(CH3)2), 1.30 (t, J = 7.5 Hz, 3H, CH2CH3), 1.77–1.88 (m, 4H, 6,7-CH2), 2.08–2.22 (m, 3H, 6′-CH2, CH(CH3)2), 2.48 (d, J = 7.1 Hz, 2H, CHCH2), 2.61–2.67 (m, 2H, 8-CH2), 2.74 (q, J = 7.5 Hz, 2H, CH2CH3), 2.82–2.89 (m, 4H, 5,7′-CH2), 3.02 (t, J = 7.6 Hz, 2H, 5′-CH2), 3.56–3.75 (m, 8H, C4H8N2), 5.47 (s, 2H, OCH2), 5.55 (s, 2H, NCH2), 7.93 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 11.25, 20.89, 21.87, 22.16, 23.52, 24.00, 27.16, 27.24, 27.92, 29.35, 32.49, 41.16, 43.94, 44.10, 47.64, 48.20, 50.31, 59.23, 90.22, 92.58, 113.70, 115.99, 123.13, 125.30, 129.43, 141.83, 152.05, 158.05, 158.92, 160.13, 160.37, 162.45, 163.70. Anal. calcd for C34H41N9O2: C 67.19%; H 6.80%; N 20.74%. Found: C 67.58%; H 7.02%; N 21.01%. ESI + MS; [C34H41N9O2+Na+] found: 630.3281.
3.1.1.4.7. 3-{4-[(4-{[(4-Cyano-1-ethyl-5,6,7,8-tetrahydroisoquinolin-3-yl)oxy]methyl}-1H-1,2,3-triazol-1-yl)acetyl]piperazin-1-yl}-1-(2-furyl)-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (6h). The reaction between compound 3e and 5c produced a brown solid in 74% yield; solid; mp 216–218 °C; IR ν/cm−1: 1678 (C
O), 2204, 2215 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.30 (t, J = 7.4 Hz, 3H, CH2CH3), 1.77–1.88 (m, 8H, 6,6′,7,7′-CH2), 2.60–2.66 (m, 2H, 8-CH2), 2.74 (q, J = 7.4 Hz, 2H, CH2CH3), 2.81–2.87 (m, 2H, 8′-CH2), 2.88–2.94 (m, 2H, 5-CH2), 2.94–3.00 (m, 2H, 5′-CH2), 3.56–3.77 (m, 8H, C4H8N2), 5.49 (s, 2H, OCH2), 5.54 (s, 2H, NCH2), 6.59 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.10 (dd, J = 3.5, 0.8 Hz, 1H, 3-CHfur), 7.72 (dd, J = 1.7, 0.8 Hz, 1H, 5-CHfur), 7.96 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO/CCl4, 1/3) δ 11.27, 20.83, 21.80, 21.94, 23.93, 25.52, 27.16, 27.93, 28.58, 41.15, 43.86, 47.77, 48.17, 50.40, 59.05, 92.34, 94.68, 111.47, 113.76, 113.96, 115.94, 121.20, 123.41, 125.73, 144.23, 147.22, 152.24, 152.91, 153.30, 158.45, 160.31, 162.64, 163.94. Anal. calcd for C35H37N9O3: C 66.54%; H 5.90%; N 19.95%. Found: C 66.85%; H 6.06%; N 20.18%. ESI + MS; [C35H37N9O3+Na+] found: 654.2917.
3.1.1.4.8. 3-[(1-{2-[4-(4-Cyano-1-propyl-6,7-dihydro-5H-cyclopenta[c]pyridin-3-yl)piperazin-1-yl]-2-oxoethyl}-1H-1,2,3-triazol-4-yl)methoxy]-1-isopropyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (6i). The reaction between compound 3a and 5d produced a white solid in 80% yield; mp 211–213 °C; IR ν/cm−1: 1674 (C
O), 2101, 2218 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 0.98 (d, J = 7.4 Hz, 3H, CH2CH3), 1.25 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.65–1.78 (m, 2H, CH2CH3), 1.78–1.87 (m, 4H, 6,7-CH2), 2.10–2.21 (m, 2H, 6′-CH2), 2.56–2.62 (m, 2H, CH2C2H5), 2.66–2.72 (m, 2H, 8-CH2), 2.82–2.90 (m, 4H, 5,7′-CH2), 3.02 (t, J = 7.6 Hz, 2H, 5′-CH2), 3.22 (sp, J = 6.7 Hz, 1H, CH(CH3)2), 3.57–3.75 (m, 8H, C4H8N2), 5.47 (s, 2H, OCH2), 5.56 (s, 2H, NCH2), 7.92 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 13.50, 20.41, 20.84, 20.89, 20.92, 21.98, 23.52, 23.82, 28.11, 29.14, 30.35, 32.44, 37.06, 41.16, 43.94, 47.63, 48.17, 50.32, 59.20, 90.16, 92.72, 113.68, 116.01, 122.21, 125.09, 128.96, 141.86, 152.51, 158.51, 159.05, 160.12, 160.48, 163.69, 166.02. Anal. calcd for C34H41N9O2: C 67.19; H 6.80; N 20.74%. Found: C 67.59; H 7.02; N 21.03%. ESI + MS; [C34H41N9O2+Na+] found: 630.3281.
3.1.1.4.9. 3-(4-{[4-({[4-Cyano-1-(2-furyl)-5,6,7,8-tetrahydroisoquinolin-3-yl]oxy}methyl)-1H-1,2,3-triazol-1-yl]acetyl}piperazin-1-yl)-1-isopropyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (6k). The reaction between compound 3d and 5f produced a white solid in 87% yield; mp 192–194 °C; IR ν/cm−1: 1684 (C
O), 2199, 2219 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.18 (d, J = 6.6 Hz, 6H, CH(CH3)2), 1.77–1.87 (m, 8H, 6,6′,7,7′-CH2), 2.61–2.68 (m, 2H, 8-CH2), 2.82–3.04 (m, 6H, 5,5′,8′-CH2), 3.17 (sp, J = 6.6 Hz, 1H, CH(CH3)2), 3.54–3.74 (m, 8H, C4H8N2), 5.45 (s, 2H, OCH2), 5.59 (s, 2H, NCH2), 6.62 (dd, J = 3.5,1.7 Hz, 1H, 4-CHfur), 7.25 (dd, J = 3.5, 0.8 Hz, 1H, 3-CHfur), 7.73 (dd, J = 1.7, 0.8 Hz, 1H, 5-CHfur), 8.01 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.95, 21.81, 21.90, 23.69, 25.36, 28.11, 28.33, 30.18, 41.05, 43.86, 47.48, 48.07, 50.28, 59.35, 93.48, 93.58, 111.47, 113.49, 114.25, 120.91, 121.96, 125.74, 144.02, 146.39, 151.30, 152.49, 154.01, 158.73, 159.58, 163.54, 165.85. Anal. calcd for C36H39N9O3: C 66.96%; H 6.09%; N 19.52%. Found: C 67.28%; H 6.26%; N 19.77%. ESI + MS; [C36H39N9O3+Na+] found: 668.3074.
3.1.1.4.10. 3-(4-{[4-({[4-Cyano-1-(2-furyl)-5,6,7,8-tetrahydroisoquinolin-3-yl]oxy}methyl)-1H-1,2,3-triazol-1-yl]acetyl}piperazin-1-yl)-1-(2-furyl)-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (6l). The reaction between compound 3e and 5f produced a light-yellow solid in 78% yield; mp 170–172 °C; IR ν/cm−1: 1671 (C
O), 2204, 2224 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.79–1.90 (m, 8H, 6,6′,7,7′-CH2), 2.85–3.04 (m, 8H, 8,8′,5,5′-CH2), 3.56–3.76 (m, 8H, C4H8N2), 5.47 (s, 2H, OCH2), 5.60 (s, 2H, NCH2), 6.57 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 6.62 (dd, J = 3.5, 1.7 Hz, 1H, 4′-CHfur), 7.09 (dd, J = 3.5, 0.8 Hz, 1H, 3-CHfur), 7.25 (dd, J = 3.5, 0.7 Hz, 1H, 3′-CHfur), 7.68 (dd, J = 1.7, 0.8 Hz, 1H, 5-CHfur), 7.68 (dd, J = 1.7, 0.7 Hz, 1H, 5′-CHfur), 8.02 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.62, 20.90, 21.93, 22.00, 25.50, 28.45, 28.57, 41.13, 41.17, 43.96, 47.73, 48.18, 50.39, 59.45, 93.61, 94.80, 111.32, 111.59, 113.54, 113.62, 114.38, 115.74, 121.07, 122.10, 125.63, 143.79, 144.16, 146.53, 147.19, 152.60, 153.13, 153.16, 154.15, 158.43, 159.72, 163.75. Anal. calcd for C37H35N9O4: C 66.35%; H 5.27%; N 18.82%. Found: C 66.65%; H 5.48%; N 19.11%. ESI + MS; [C37H35N9O4+Na+] found: 692.2710.
3.1.1.4.11. 6-[(1-{2-[4-(5-Cyano-8-ethyl-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridin-6-yl)piperazin-1-yl]-2-oxoethyl}-1H-1,2,3-triazol-4-yl)methoxy]-8-(4-methoxyphenyl)-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (6n). The reaction between compound 3g and 5h produced a white solid in 83% yield; mp 128–130 °C; IR ν/cm−1: 1650 (C
O), 2218 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.24 (t, J = 7.4 Hz, 3H, CH2CH3), 1.28 (s, 6H, C(CH3)2), 1.33 (s, 6H, C(CH3)2), 2.57 (q, J = 7.4 Hz, 2H, CH2CH3), 2.72 (s, 2H, 4-CH2), 2.85 (s, 2H, 4′-CH2), 3.61–3.75 (m, 8H, C4H8N2), 3.87 (s, 3H, OCH3), 4.60 (s, 2H, 1-CH2), 4.68 (s, 2H, 1′-CH2), 5.49 (s, 2H, OCH2), 5.61 (s, 2H, NCH2), 6.97–7.02 (m, 2H, C6H4), 7.52–7.58 (m, 2H, C6H4), 7.96 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 10.90, 25.76, 26.20, 26.46, 37.87, 38.16, 41.21, 43.97, 47.41, 47.93, 50.38, 54.70, 58.84, 59.59, 59.88, 69.01, 69.19, 93.04, 93.57, 113.32, 113.38, 115.63, 118.79, 121.18, 125.49, 129.62, 130.01, 141.64, 147.87, 150.56, 155.32, 159.15, 159.42, 160.01, 160.75, 163.832. Anal. calcd for C40H45N9O5: C 65.65%; H 6.20%; N 17.23%. Found: C 66.01%; H 6.39%; N 17.50%. ESI + MS; [C40H45N9O5+Na+] found: 754.3441.
3.1.1.4.12. 6-[(1-{2-[4-(4-Cyano-1-methyl-5,6,7,8-tetrahydroisoquinolin-3-yl)piperazin-1-yl]-2-oxoethyl}-1H-1,2,3-triazol-4-yl)methoxy]-8-(2-furyl)-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (6o). The reaction between compound 3c and 5i produced a brown solid in 75% yield; mp 215–217 °C; IR ν/cm−1: 1667 (C
O), 2205, 2223 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.31 (s, 6H, C(CH3)2), 1.77–1.87 (m, 4H, 6′,7′-CH2), 2.36 (s, 3H, CH3), 2.53–2.59 (m, 2H, 8′-CH2), 2.80–2.86 (m, 2H, 5′-CH2), 2.83 (s, 2H, 4-CH2), 3.50–3.73 (m, 8H, C4H8N2), 5.00 (s, 2H, 8-CH2), 5.47 (s, 2H, OCH2), 5.63 (s, 2H, NCH2), 6.66 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.36 (dd, J = 3.5, 0.7 Hz, 1H, 3-CHfur), 7.79 (dd, J = 1.7, 0.7 Hz, 1H, 5-CHfur), 8.01 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 21.14, 21.91, 22.17, 24.65, 25.72, 27.94, 38.33, 41.25, 43.99, 47.76, 47.79, 48.23, 50.39, 59.69, 59.82, 68.46, 93.80, 93.86, 111.97, 113.26, 114.44, 115.85, 119.04, 122.58, 125.59, 141.56, 143.46, 144.91, 150.85, 150.97, 152.42, 158.67, 158.81, 160.32, 163.73. Anal. calcd for C35H37N9O4: C 64.90%; H 5.76%; N 19.46%. Found: C 65.24%; H 5.93%; N 19.71%. ESI + MS; [C35H37N9O4+Na+] found: 670.2866.
3.1.1.4.13. 6-{[1-(2-{4-[4-Cyano-1-(2-furyl)-5,6,7,8-tetrahydroisoquinolin-3-yl]piperazin-1-yl}-2-oxoethyl)-1H-1,2,3-triazol-4-yl]methoxy}-8-(2-furyl)-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (6p). The reaction between compound 3e and 5i produced a white solid in 71% yield; mp 220–222 °C; IR ν/cm−1: 1678 (C
O), 2214 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.31 (s, 6H, C(CH3)2), 1.78–1.90 (m, 4H, 6,7-CH2), 2.82 (s, 2H, 5-CH2), 2.86–3.01 (m, 4H, 8′,5′-CH2), 3.56–3.77 (m, 8H, C4H8N2), 5.00 (s, 2H, 8-CH2), 5.48 (s, 2H, OCH2), 5.62 (s, 2H, NCH2), 6.57 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 6.66 (dd, J = 3.5, 1.7 Hz, 1H, 4′-CHfur), 7.08–7.11 (m, 1H, 3-CHfur), 7.34–7.38 (m, 1H, 3′-CHfur), 7.68–7.70 (m, 1H, 5-CHfur), 7.78–7.80 (m, 1H, 5′-CHfur), 8.02 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.81, 21.91, 25.43, 25.64, 28.50, 38.23, 41.11, 43.85, 43.88, 47.67, 48.07, 50.38, 59.61, 59.66, 59.74, 68.38, 93.67, 94.65, 111.28, 111.93, 113.24, 113.52, 114.40, 115.73, 118.94, 120.98, 125.75, 143.35, 143.79, 144.90, 147.08, 150.88, 152.32, 153.07, 158.32, 160.22, 163.70. Anal. calcd for C38H37N9O5: C 65.22%; H 5.33%; N 18.01%. Found: C 65.61%; H 5.56%; N 18.29%. ESI + MS; [C38H37N9O5+Na+] found: 722.2816.
3.1.1.4.14. 6-(4-{[4-({[5-Cyano-8-(2-furyl)-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridin-6-yl]oxy}methyl)-1H-1,2,3-triazol-1-yl]acetyl}piperazin-1-yl)-3,3,8-trimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (6q). The reaction between compound 3f and 5i produced a white solid in 80% yield; mp 243–245 °C; IR ν/cm−1: 1672 (C
O), 2209, 2225 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.28 (s, 6H, C(CH3)2), 1.31 (s, 6H, C(CH3)2), 2.31 (s, 3H, CH3), 2.71 (s, 2H, 5-CH2), 2.83 (s, 2H, 5′-CH2), 3.57–3.75 (m, 8H, C4H8N2), 4.57 (s, 2H, 8-CH2), 5.00 (s, 2H, 8′-CH2), 5.48 (s, 2H, OCH2), 5.63 (s, 2H, NCH2), 6.66 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.36 (dd, J = 3.5, 0.7 Hz, 1H, 3-CHfur), 7.80 (dd, J = 1.7, 0.8 Hz, 1H, 5-CHfur), 8.01 (s, 1H, CHtriazole). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 21.01, 25.74, 37.74, 41.25, 43.97, 47.48, 47.97, 50.40, 59.20, 59.69, 59.82, 68.47, 69.06, 93.23, 93.80, 111.99, 113.29, 114.46, 115.57, 119.07, 119.43, 125.60, 141.56, 143.48, 144.94, 147.76, 151.00, 152.42, 155.42, 159.09, 160.34, 163.82. Anal. calcd for C36H39N9O5: C 63.80%; H 5.80%; N 18.60%. Found: C 64.12%; H 5.98%; N 18.84%. ESI + MS; [C36H39N9O5+Na+] found: 700.2972.
3.1.1.5. General procedure for the synthesis of compounds 8a–c. To compound 7 (5 mmol) in absolute ethanol, phenylisothiocyanate (5 mmol) was added and the mixture was stirred for 5 h at room temperature, and then 5 h at 35–40 °C. After the mixture was cooled, the precipitate was filtered, washed with ethanol, dried and recrystallized from ethanol.
3.1.1.5.1. 2-{[(4-Cyano-1-methyl-5,6,7,8-tetrahydroisoquinolin-3-yl)oxy]acetyl}-N-phenylhydrazinecarbothioamide (8a). The reaction between compound 7a and phenylisothiocyanate produced a colorless solid in 83% yield; mp 167–169 °C. 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.78–1.88 (m, 4H, 6,7-CH2), 2.33 (s, 3H, CH3), 2.53–2.60 (m, 2H, 8-CH2), 2.84–2.90 (m, 2H, 5-CH2), 4.96 (s, 2H, OCH2), 7.08–7.15 (m, 1H, Ph), 7.25–7.32 (m, 2H, Ph), 7.45–7.51 (m, 2H, Ph), 9.24–9.87 (m, 2H, 2NH), 10.09 (br, 1H, NH). 13C NMR (75 MHz, DMSO/CCl4, 1/3) δ 20.90, 21.84, 21.88, 21.92, 24.62, 27.88, 63.15, 92.89, 113.79, 124.21, 124.33, 124.37, 127.46, 138.78, 151.91, 158.45, 159.90, 180.42. Anal. calcd for C20H21N5O2S: C 60.74%; H 5.35%; N 17.71%. Found: C 61.03%; H 5.53%; N 17.96%.
3.1.1.5.2. 2-({[4-Cyano-1-(2-furyl)-5,6,7,8-tetrahydroisoquinolin-3-yl]oxy}acetyl)-N-phenylhydrazinecarbothioamide (8b). The reaction between compound 7b and phenylisothiocyanate produced a colorless solid in 87% yield; mp 192–194 °C. 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.80–1.90 (m, 4H, 6,7-CH2), 2.92–3.04 (m, 4H, 8,5-CH2), 5.02 (s, 2H, OCH2), 6.55 (m, 1H, 4-CHfur), 7.06–7.68 (m, 7H, Ph, 3,5-CHfur), 9.15–9.92 (m, 2H, 2NH), 10.18 (br, 1H, NH). Anal. calcd for C23H21N5O3S: C 61.73%; H 4.73%; N 15.65%. Found: C 62.05%; H 4.92%; N 15.94%.
3.1.1.5.3. 2-({[5-Cyano-8-(2-furyl)-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridin-6-yl]oxy}acetyl)-N-phenylhydrazinecarbothioamide (8c). The reaction between compound 7c and phenylisothiocyanate produced a colorless solid in 81% yield; mp 176–178 °C. 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.32 (s, 6H, C(CH3)2), 2.86 (s, 2H, 5-CH2), 4.98 (s, 2H, 8-CH2), 5.04 (s, 2H, OCH2), 6.55–6.60 (m, 1H, 4-CHfur), 7.06–7.72 (m, 7H, Ph, 3,5-CHfur), 9.15–9.96 (m, 2H, 2NH), 10.20 (br, 1H, NH). Anal. calcd for C24H23N5O4S: C 60.36%; H 4.85%; N 14.67%. Found: C 60.67%; H 5.05%; N 14.91%.
3.1.1.6. General procedure for the synthesis of compounds 9a–c. To a stirred solution of NaOH (10 mmol) in water (30 mL), the corresponding thiosemicarbazide 8 (5 mmol) was added and the mixture was stirred for 5 h at room temperature, and then 5 h at 40–50 °C. After cooling, the mixture was diluted with water and acidified with hydrochloric acid to pH 5–6. The precipitated crystals were filtered, washed with water, dried and recrystallized from ethanol.
3.1.1.6.1. 3-[(5-Mercapto-4-phenyl-4H-1,2,4-triazol-3-yl)methoxy]-1-methyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (9a). The reaction between compound 8a and NaOH solution produced a colorless solid in 86% yield; mp 209–211 °C; IR ν/cm−1: 1700 (C
S), 2226 (CN), 3101 (NH). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.73–1.86 (m, 4H, 6,7-CH2), 2.32 (s, 3H, CH3), 2.52–2.57 (m, 2H, 8-CH2), 2.79–2.84 (m, 2H, 5-CH2), 5.23 (s, 2H, OCH2), 7.41–7.53 (m, 5H, Ph), 13.88 (s, 1H, NH). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.77, 21.72, 21.80, 24.57, 27.81, 57.14, 92.67, 113.17, 124.57, 127.97, 128.64, 128.86, 132.92, 146.88, 152.11, 158.30, 158.91, 168.69. Anal. calcd for C20H19N5OS: C 63.64%; H 5.07%; N 18.55%. Found: C 63.94%; H 5.25%; N 18.78%.
3.1.1.6.2. 1-(2-Furyl)-3-[(5-mercapto-4-phenyl-4H-1,2,4-triazol-3-yl)methoxy]-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (9b). The reaction between compound 8b and NaOH solution produced a light-yellow solid in 91% yield; mp 235–237 °C; IR ν/cm−1: 1700 (C
S), 2229 (CN), 3219 (NH). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.77–1.88 (m, 4H, 6,7-CH2), 2.86–3.00 (m, 4H, 8,5-CH2), 5.33 (s, 2H, OCH2), 6.60 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.13 (dd, J = 3.5, 0.7 Hz, 1H, 3-CHfur), 7.39–7.52 (m, 5H, Ph), 7.71 (dd, J = 1.7, 0.7 Hz, 1H, 5-CHfur), 13.88 (s, 1H, NH). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.53, 21.83, 25.43, 28.45, 57.33, 93.42, 111.58, 113.12, 114.56, 122.77, 127.93, 128.65, 128.84, 132.92, 144.29, 146.30, 146.90, 152.32, 154.32, 158.51, 168.73. Anal. calcd for C23H19N5O2S: C 64.32%; H 4.46%; N 16.31%. Found: C 64.65%; H 4.67%; N 16.54%.
3.1.1.6.3. 8-(2-Furyl)-6-[(5-mercapto-4-phenyl-4H-1,2,4-triazol-3-yl)methoxy]-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (9c). The reaction between compound 8c and solution of NaOH produced a colorless solid in 83% yield; mp 222–224 °C; IR ν/cm−1: 1700 (C
S), 2220 (CN), 3167 (NH). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.29 (s, 6H, C(CH3)2), 2.79 (s, 2H, 5-CH2), 4.95 (s, 2H, 8-CH2), 5.39 (s, 2H, OCH2), 6.63 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.23 (dd, J = 3.5, 0.7 Hz, 1H, 3-CHfur), 7.38–7.50 (m, 5H, Ph), 7.76 (dd, J = 1.7, 0.7 Hz, 1H, 5-CHfur), 13.90 (s, 1H, NH). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 25.69, 38.31, 57.42, 59.73, 68.43, 93.57, 111.93, 112.72, 114.61, 119.67, 127.93, 128.64, 128.81, 132.93, 143.20, 145.05, 146.79, 151.10, 152.13, 159.08, 168.78. Anal. calcd for C24H21N5O3S: C 62.73%; H 4.61%; N 15.24%. Found: C 63.09%; H 4.83%; N 15.47%.
3.1.1.7. General procedure for the synthesis of compounds 10a–g. To a stirred suspension of compound 9 (5 mmol) and potassium carbonate (1.38 g, 10 mmol) in absolute DMF (30 mL), the corresponding chloride 2 (5.5 mmol) was added and the reaction mixture was maintained at 65–70 °C for 5 h. After cooling, water was added and the resulting crystals were filtered, washed with water, dried and recrystallized from ethanol.
3.1.1.7.1. 3-{[5-({2-[4-(4-Cyano-1-propyl-6,7-dihydro-5H-cyclopenta[c]pyridin-3-yl)piperazin-1-yl]-2-oxoethyl}thio)-4-phenyl-4H-1,2,4-triazol-3-yl]methoxy}-1-methyl-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (10a). The reaction between compound 2a and 9a produced a cream solid in 74% yield; mp 164–166 °C; IR ν/cm−1: 1658 (C
O), 2207, 2212 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 0.97 (t, J = 7.4 Hz, 3H, CH2CH3), 1.64–1.86 (m, 6H, CH2CH3, 6,7-CH2), 2.09–2.20 (m, 2H, 6′-CH2), 2.34 (s, 3H, CH3), 2.52–2.61 (m, 4H, 8-CH2, CH2C2H5), 2.79–2.90 (m, 4H, 5,7′-CH2), 3.01 (t, J = 7.5 Hz, 2H, 5′-CH2), 3.53–3.76 (m, 8H, C4H8N2), 4.31 (s, 2H, SCH2), 5.36 (s, 2H, OCH2), 7.47–7.57 (m, 5H, Ph). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 13.51, 20.39, 20.79, 21.75, 21.81, 23.52, 24.58, 27.80, 29.14, 32.44, 35.93, 37.05, 41.13, 45.06, 47.64, 48.25, 57.04, 90.14, 92.59, 113.33, 116.01, 124.35, 126.98, 128.94, 129.16, 129.45, 132.24, 150.66, 151.47, 152.00, 158.36, 158.49, 159.03, 159.18, 160.10, 164.64. Anal. calcd for C38H41N9O2S: C 66.35%; H 6.01%; N 18.33%. Found: C 66.70%; H 6.21%; N 18.63%.
3.1.1.7.2. 6-(4-{[(5-{[(4-Cyano-1-methyl-5,6,7,8-tetrahydroisoquinolin-3-yl)oxy]methyl}-4-phenyl-4H-1,2,4-triazol-3-yl)thio]acetyl}piperazin-1-yl)-3,3,8-trimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (10b). The reaction between compound 2f and 9a produced a cream solid in 70% yield; mp 119–121 °C; IR ν/cm−1: 1647 (C
O), 2218 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.27 (s, 6H, C(CH3)2), 1.74–1.86 (m, 4H, 6,7-CH2), 2.30 (s, 3H, CH3), 2.34 (s, 3H, CH3), 2.52–2.58 (m, 2H, 8-CH2), 2.71 (s, 2H, 5′-CH2), 2.78–2.86 (m, 2H, 5-CH2), 3.54–3.76 (m, 8H, C4H8N2), 4.31 (s, 2H, SCH2), 4.56 (s, 2H, 8′-CH2), 5.36 (s, 2H, OCH2), 7.47–7.57 (m, 5H, Ph). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.78, 20.99, 21.74, 21.82, 24.57, 25.74, 27.80, 35.91, 37.72, 41.18, 45.07, 47.50, 48.01, 57.04, 59.20, 69.05, 92.58, 93.22, 113.35, 115.53, 119.36, 124.37, 126.98, 129.17, 129.45, 132.24, 147.73, 150.69, 151.47, 152.01, 155.37, 158.38, 159.07, 159.18, 164.71. Anal. calcd for C38H41N9O3S: C 64.84%; H 5.87%; N 17.91%. Found: C 65.21%; H 6.06%; N 18.19%. ESI + MS; [C38H41N9O3S+H+] found: 704.3135.
3.1.1.7.3. 3-{[5-({2-[4-(4-Cyano-1-propyl-6,7-dihydro-5H-cyclopenta[c]pyridin-3-yl)piperazin-1-yl]-2-oxoethyl}thio)-4-phenyl-4H-1,2,4-triazol-3-yl]methoxy}-1-(2-furyl)-5,6,7,8-tetrahydroisoquinoline-4-carbonitrile (10c). The reaction between compound 2a and 9b produced a cream solid in 76% yield; mp 160–162 °C; IR ν/cm−1: 1648 (C
O), 2207, 2220 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 0.97 (t, J = 7.4 Hz, 3H, CH2CH3), 1.64–1.79 (m, 2H, CH2CH3), 1.78–1.88 (m, 4H, 6,7-CH2), 2.09–2.20 (m, 2H, 6′-CH2), 2.55–2.61 (m, 4H, 8-CH2, CH2C2H5), 2.81–2.89 (m, 2H, 7′-CH2), 2.96–3.04 (m, 4H, 5,5′-CH2), 3.53–3.76 (m, 8H, C4H8N2), 4.31 (s, 2H, SCH2), 5.47 (s, 2H, OCH2), 6.60 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.21 (dd, J = 3.5, 0.7 Hz, 1H, 3-CHfur), 7.45–7.56 (m, 5H, Ph), 7.70 (dd, J = 1.7, 0.7 Hz, 1H, 5-CHfur). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 13.51, 20.39, 20.55, 21.86, 23.52, 25.38, 28.43, 29.13, 32.44, 35.94, 37.05, 41.12, 45.03, 47.63, 48.25, 57.28, 90.13, 93.34, 111.56, 113.24, 114.62, 116.01, 122.55, 126.93, 128.92, 129.16, 129.42, 132.24, 144.18, 146.38, 150.69, 151.50, 152.50, 154.20, 158.49, 158.74, 159.02, 160.10, 164.62. Anal. calcd for C41H41N9O3S: C 66.56%; H 5.59%; N 17.04%. Found: C 66.87%; H 5.79%; N 17.32%.
3.1.1.7.4. 6-[4-({[5-({[4-Cyano-1-(2-furyl)-5,6,7,8-tetrahydroisoquinolin-3-yl]oxy}methyl)-4-phenyl-4H-1,2,4-triazol-3-yl]thio}acetyl)piperazin-1-yl]-3,3,8-trimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (10d). The reaction between compound 2f and 9b produced a cream solid in 73% yield; mp 193–195 °C; IR ν/cm−1: 1647 (C
O), 2211, 2219 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.27 (s, 6H, C(CH3)2), 1.78–1.87 (m, 4H, 6,7-CH2), 2.30 (s, 3H, CH3), 2.70 (s, 2H, 5′-CH2), 2.86–2.93 (m, 2H, 8-CH2), 2.96–3.02 (m, 2H, 5-CH2), 3.53–3.76 (m, 8H, C4H8N2), 4.30 (s, 2H, SCH2), 4.56 (s, 2H, 8′-CH2), 5.46 (s, 2H, OCH2), 6.60 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.20 (dd, J = 3.5, 0.7 Hz, 1H, 3-CHfur), 7.45–7.56 (m, 5H, Ph), 7.70 (dd, J = 1.7, 0.7 Hz, 1H, 5-CHfur). 13C NMR (75 MHz, DMSO/CCl4, 1/3) δ 20.56, 21.00, 21.87, 25.40, 25.75, 28.45, 35.88, 37.73, 41.20, 45.07, 47.50, 48.01, 57.29, 59.20, 69.06, 93.23, 93.35, 111.59, 113.27, 114.64, 115.53, 119.37, 122.57, 126.95, 129.18, 129.45, 132.25, 144.21, 146.40, 147.74, 150.72, 151.49, 152.50, 154.21, 155.36, 158.74, 159.08, 164.71. Anal. calcd for C41H41N9O4S: C 65.15%; H 5.47%; N 16.68%. Found: C 65.48%; H 5.65%; N 16.97%. [C41H41N9O4S+H+] found: 756.3082.
3.1.1.7.5. 6-{[5-({2-[4-(4-Cyano-1-methyl-5,6,7,8-tetrahydroisoquinolin-3-yl)piperazin-1-yl]-2-oxoethyl}thio)-4-phenyl-4H-1,2,4-triazol-3-yl]methoxy}-8-(2-furyl)-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (10e). The reaction between compound 2b and 9c produced a cream solid in 71% yield; mp 154–156 °C; IR ν/cm−1: 1626 (C
O), 2209, 2228 (CN). 1H NMR (300 MHz, DMSO-d6) δ 1.24 (s, 6H, C(CH3)2), 1.68–1.78 (m, 4H, 6′,7′-CH2), 2.34 (s, 3H, CH3), 2.50–2.55 (m, 2H, 8′-CH2), 2.74–2.80 (m, 2H, 5′-CH2), 2.79 (s, 2H, 5-CH2), 3.40–3.65 (m, 8H, C4H8N2), 4.33 (s, 2H, SCH2), 4.91 (s, 2H, 8-CH2), 5.60 (s, 2H, OCH2), 6.75 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.29 (dd, J = 3.5, 0.7 Hz, 1H, 3-CHfur), 7.45–7.48 (m, 5H, Ph), 7.99 (dd, J = 1.7, 0.7 Hz, 1H, 5-CHfur). 13C NMR (75 MHz, DMSO-d6) δ 21.10, 21.83, 22.55, 24.54, 25.75, 28.06, 36.28, 38.25, 41.36, 45.06, 48.03, 48.34, 57.70, 59.82, 68.93, 93.23, 93.56, 112.51, 113.71, 115.37, 116.65, 119.71, 123.23, 127.04, 129.58, 129.92, 130.60, 132.41, 143.39, 146.36, 151.41, 151.45, 151.55, 151.62, 151.80, 159.04, 159.41, 159.47, 165.30. Anal. calcd for C41H41N9O4S: C 65.15%; H 5.47%; N 16.68%. Found: C 65.47%; H 5.33%; N 16.89%.
3.1.1.7.6. 6-({5-[(2-{4-[4-Cyano-1-(2-furyl)-5,6,7,8-tetrahydroisoquinolin-3-yl]piperazin-1-yl}-2-oxoethyl)thio]-4-phenyl-4H-1,2,4-triazol-3-yl}methoxy)-8-(2-furyl)-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (10f). The reaction between compound 2e and 9c produced a brown solid in 79% yield; mp 250–252 °C; IR ν/cm−1: 1649 (C
O), 2216 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.29 (s, 6H, C(CH3)2), 1.77–1.90 (m, 4H, 6′,7′-CH2), 2.78 (s, 2H, 5-CH2), 2.93–3.01 (m, 4H, 5′,8′-CH2), 3.52–3.79 (m, 8H, C4H8N2), 4.31 (s, 2H, SCH2), 4.95 (s, 2H, 8-CH2), 5.52 (s, 2H, OCH2), 6.56 (dd, J = 3.5, 1.7 Hz, 1H, 4′-CHfur), 6.63 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.08 (dd, J = 3.5, 0.7 Hz, 1H, 3′-CHfur), 7.30 (dd, J = 3.5, 0.7 Hz, 1H, 3-CHfur), 7.46–7.55 (m, 5H, Ph), 7.67 (dd, J = 1.7, 0.7 Hz, 1H, 5′-CHfur), 7.75 (dd, J = 1.7, 0.7 Hz, 1H, 5-CHfur). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.90, 22.00, 25.49, 25.68, 28.57, 35.93, 38.29, 41.12, 41.13, 45.04, 47.73, 48.25, 57.43, 59.73, 68.41, 93.46, 94.75, 111.32, 111.93, 112.88, 113.53, 114.73, 115.73, 119.45, 121.01, 126.94, 129.18, 129.43, 132.25, 143.27, 143.76, 145.00, 147.14, 150.66, 150.99, 151.55, 152.24, 153.13, 153.21, 158.38, 159.30, 164.65. Anal. calcd for C44H41N9O5S: C 65.41%; H 5.12%; N 15.60%. Found: C 65.53%; H 5.31%; N 15.88%.
3.1.1.7.7. 6-[4-({[5-({[5-Cyano-8-(2-furyl)-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridin-6-yl]oxy}methyl)-4-phenyl-4H-1,2,4-triazol-3-yl]thio}acetyl)piperazin-1-yl]-3,3,8-trimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carbonitrile (10g). The reaction between compound 2f and 9c produced a yellow solid in 73% yield; mp 168–170 °C; IR ν/cm−1: 1646 (C
O), 2220 (CN). 1H NMR (300 MHz, DMSO-d6/CCl4, 1/3) δ 1.27 (s, 6H, C(CH3)2), 1.30 (s, 6H, C(CH3)2), 2.30 (s, 3H, CH3), 2.70 (s, 2H, 5′-CH2), 2.79 (s, 2H, 5-CH2), 3.54–3.77 (m, 8H, C4H8N2), 4.30 (s, 2H, SCH2), 4.56 (s, 2H, 8′-CH2), 4.96 (s, 2H, 8-CH2), 5.52 (s, 2H, OCH2), 6.63 (dd, J = 3.5, 1.7 Hz, 1H, 4-CHfur), 7.31 (dd, J = 3.5, 0.7 Hz, 1H, 3-CHfur), 7.45–7.56 (m, 5H, Ph), 7.75 (dd, J = 1.7, 0.7 Hz, 1H, 5-CHfur). 13C NMR (75 MHz, DMSO-d6/CCl4, 1/3) δ 20.99, 25.69, 25.74, 35.89, 37.72, 38.30, 41.19, 45.09, 47.48, 48.01, 57.45, 59.20, 59.74, 68.41, 69.05, 93.22, 93.49, 111.94, 112.86, 114.73, 115.53, 119.34, 119.47, 126.95, 129.18, 129.43, 132.25, 143.28, 144.96, 147.74, 150.66, 151.00, 151.55, 152.27, 155.35, 159.05, 159.31, 164.68. Anal. calcd for C42H43N9O5S: C 64.19%; H 5.51%; N 16.04%. Found: C 64.54%; H 5.69%; N 16.31%. [C42H43N9O5S+H+] found: 786.3183.
3.2. Biological evaluation
All new 24 compounds were studied for their possible neurotropic activities as well as side effects on 550 outbred mice weighing 18–24 g and 75 male rats of the Wistar line weighing 120–140 g. As reference compounds, the known antiepileptic drugs ethosuximide and tranquilizer diazepam and nootropic drug piracetam57 were used. The psychotropic properties of the substances were investigated at a dose of 50 mg kg−1, given that the ED50 of these compounds were within 50 mg kg−1 at the confidence intervals. The doses of the reference compounds were chosen based on the literature and our experimental data including ethosuximide – 200 mg kg−1, diazepam – 2 mg kg−1 and piracetam – 1000 mg kg−1.57,58To determine the 50% effective dose (ED50, causing the anticonvulsant effect of 50% of animals, which is calculated by the antagonism to PTZ test), 50% toxic doses (TD50, causing a myorelaxant effect in 50% of animals) and lethal effect in 50% of animals (LD50), the statistical method of probit analysis by Litchfield and Wilcoxon was used.59 The acute toxicity was determined by calculating the number of dead animals after 24 h of exposure to doses of 400–1200 mg kg−1. In all the experiments, the results were statistically tested at a P ≤ 0.05 probability level.
The biological experiments were carried out in full compliance with the European Convention for the Protection of Vertebrate. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of (ETS No 123, Strasbourg, 03/18/1986): Strasbourg (France).
The MES test was used as an animal model for the generalized tonic seizures of epilepsy with the following parameters: 50 mA, duration of 0.2 s, and oscillation frequency of 50 imp per s. The anticonvulsant properties of the compounds was assessed by the prevention of the tonic-extensor phase of convulsions.
Thiosemicarbazide was administered subcutaneously to the mice at a dose of 18 mg kg−1 as a 0.5% solution and caused clonic convulsions in the animals. Anti-thiosemicarbazide activity was evaluated based on the latency time for the onset of seizures.
The exploratory-motor behavior of rats weighing 120–140 g was studied using a modification of the “open field” model. Therefore, an installation featuring a bottom divided into squares with holes (cells) was utilized. The experiments were carried out in the daytime under natural light. During the 5 min experiment, we assessed indicators of activating and sedative behavior, including standing, the number of horizontal and vertical movements, and sniffing the cells. Each compound, control, and comparison analogues in this model had 6 animals. The most effective dose for administering the test compound to rats was determined to be 50 mg kg−1, which was given intraperitoneally.
Anti-anxiety, antidepressant, and sedative effects were studied using the “elevated plus maze” model in mice developed by S. Pellow co-authors (1986). Typically, normal animals display a preference for spending the majority of their time in the closed arms of the maze. The anxiolytic effect of a drug is evaluated by an increase in the number of entries into the open sleeves and the time spent in them, without an increase in general motor activity. The duration spent in the closed arms and the number of attempts to enter the center of the installation are also recorded. In the above-mentioned model, the test compounds and comparison drugs were administered intraperitoneally prior to the experiments. The control animals were administered an emulsifier. The results were processed statistically (P ≤ 0.05).
The “forced swim” model was used to assess “despair and depression”. The experimental animals were required to swim in a glass container (height of 22 cm and diameter of 14 cm) filled 1/3 of the way with water. Initially, the intact mice displayed vigorous swimming activity, but eventually, they were forced to immobilize. The latency period of immobilization and the total duration of active swimming immobilization were fixed for 6 min.
Test for learning and memory. The anti-amnesic properties of the compounds were studied on the model of electroshock amnesia according to the method reported by Y. Buresh et al. with modification.47 In rats, a conditioned passive avoidance reaction (CRPA) of a dark–light setting was developed, while the time for training of the animals with CRPA was recorded. An increase in the time spent by the rats in the light compartment compared to the control on the second day indicates the presence of anti-amnestic properties of the compounds.
3.3. Docking studies
Docking studies were performed using AutoDock 4 (ver. 4.2.6) in the 3D structures of the GABAA receptor (PDB code: 4COF), SERT (PDB code: 3F3A) and 5-HT1A receptor (PDB code: 3NYA), retrieved from the Protein Data Bank (PDB). For the final preparation of both ligands and protein preparation, the Wizard of AutoDock tools 1.5.6 were used, as previously reported.174. Conclusions
In summary, new 1,2,3- and 1,2,4-triazole-based hybrids (besides compounds 6a, j, and m) were synthesized and evaluated for their anticonvulsant activity together with some psychotropic properties. The evaluation revealed that 14 out of the 24 synthesized compounds appeared to be potent anticonvulsants, showing antagonism to pentylenetetrazole and being superior to ethosuximide, but inferior to diazepam. Moreover, the compounds showed low toxicity with acute daily toxicity and greater therapeutic indexes (TI) than that of ethosuximide. In this test, the most active compound was found to be 6n, which contains two pyrano[3,4-c]pyridine rings with an ethyl and p-MeOC6H4 groups in its structure.Alternatively, in the TSC model, all the compounds increased the latency of thiosemicarbazide seizures in the same manner as ethosuximide, suggesting a GABAergic mechanism of action.
The evaluation of psychotropic activity revealed activating behavior and high anxiolytic effects for compounds 6b, n and p. A higher active swimming time and latent period of first immobilization for compounds 6b, c, j, n and q was observed, indicating their high antidepressant effect.
In the model of electroshock retrograde amnesia, the conditioned response of passive avoidance (CRPA), some compounds increased the time of reproduction of the reflex in the animals. Docking to the GABAA receptor, which is known as a mechanism of anticonvulsant and anxiolytic activities, as well as to SERT and 5-HT1a receptor, known as a possible mechanism of antidepressant activity, was performed. The data from the pharmacological studies were completely consistent with the data from the docking analysis. The most active compound 6n in terms of anticonvulsant and anxiolytic activity shows the greatest affinity for the GABAA receptor. Compounds 6b, c, j, n and q, and especially 6q, which exhibited a pronounced antidepressant effect, inhibited SERT and the 5-HT1a receptor.
Thus, in general, it can be stated that the 1,2,3-triazole-linked hybrids synthesized based on the pyrano[3,4-c]pyridine cycle are the more active compounds. In contrast, the 1,2,4-triazole-linked hybrids showed low activity.
Ethical statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of the Yerevan State Medical University (YSMU, Yerevan, Armenia) (protocol code 5 and date of approval 24.03.2016), followed the “Principles of laboratory animal care” and carried out in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC).Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, S. N. S., E. K. H., H. A. Y., H. V. J. and A. A. H. performed experiments on the synthesis of all compounds and analyzed the results. R. G. P. and T. A. A. performed the neurotropic activity experiments. A. G. and A. P. performed docking studies. L. Z. recorded the MS spectra. S. N. S., D. S., V. K. and E. G. P. reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work was supported by the Science Committee of RA, in the frames of the research project No. 24WS-1D019.References
- A. Golyala and P. Kwan, Seizure, 2017, 44, 147–156 CrossRef PubMed.
- J. Gierbolini, M. Giarratano and S. R. Benbadis, Expert Opin. Pharmacother., 2016, 17, 885–888 CrossRef.
- R. S. Fisher, C. Acevedo, A. Arzimanoglou, A. Bogacz, J. H. Cross, C. E. Elger, J. Engel, L. Forsgren and J. A. French, Epilepsia, 2014, 55, 475–482 CrossRef PubMed.
- K. V. Voronkova, G. S. Golosnaya, I. D. Lemeshko and A. S. Petrukhin, Epilepsy Paroxysmal Conditions, 2017, 9, 79–85 CrossRef.
- O. I. Abdel Salam, M. A. Al-Omar, N. M. Khalifa, A.-G. Amr and M. M. Abdallah, Z. Naturforsch., C: J. Biosci., 2013, 68, 264–268 CrossRef CAS.
- S. Wang, H. Liu, X. Wang, K. Lei, G. Li, X. Li, L. Wei and Z. Quan, Arch. Pharm., 2019, 352, 1900106 CrossRef CAS PubMed.
- G. Prasanthi, K. V. Prasad and K. Bharathi, Eur. J. Med. Chem., 2013, 66, 516–525 CrossRef CAS PubMed.
- G. Nkomba, G. Terre'Blanche, H. D. Janse van Rensburg and L. J. Legoabe, Med. Chem. Res., 2022, 31, 1277–1297 CrossRef CAS.
- W. Cheng-Xi, B. Ming and G. Guo-Hua, Molecules, 2015, 20, 20741–20776 CrossRef PubMed.
- M. Strzelecka and P. Świątek, Pharmaceuticals, 2021, 14, 224 CrossRef CAS PubMed.
- E. Stingaci, M. Zveaghinteva, S. Pogrebnoi, L. Lupascu, V. Valica, L. Uncu, A. Smetanscaia, M. Drumea, A. Petrou, A. Ciric, J. Glamoclija, M. Sokovic, V. Kravtsev, A. Geronikaki and F. Macaev, Bioorg. Med. Chem. Lett., 2020, 30, 127368 CrossRef CAS.
- S. Dawbaa, D. Nuha, A. E. Evren, M. Y. Cankiliç, L. Yurtta and G. Turan, J. Mol. Struct., 2023, 1282, 135213 CrossRef CAS.
- D. Kumudha, T. Kalavathi and B. A. Viswanath, World J. Pharm. Res., 2018, 7, 1614–1619 Search PubMed.
- M. Song, W. Zhao, Y. Zhu, W. Liu, X. Deng and Y. Huang, Front. Chem., 2022, 10, 925281 CrossRef CAS PubMed.
- M. X. Song and X. Q. Deng, J. Enzyme Inhib. Med. Chem., 2018, 33, 453–478 CrossRef CAS PubMed.
- S. N. Sirakanyan, T. V. Ghochikyan, D. Spinelli, A. S. Galstyan, A. Geronikaki, M. A. Samvelyan, E. K. Hakobyan and A. A. Hovakimyan, Arkivoc, 2022, 7–21 Search PubMed.
- S. N. Sirakanyan, D. Spinelli, A. Petrou, A. Geronikaki, V. G. Kartsev, E. K. Hakobyan, H. A. Yegoryan, L. Zuppiroli, R. Zuppiroli, A. G. Ayvazyan, R. G. Paronikyan, T. A. Arakelyan and A. A. Hovakimyan, Molecules, 2023, 28, 921 Search PubMed.
- D. B. Ramachary, A. B. Shashank and S. Karthik, Angew. Chem., Int. Ed., 2014, 53, 10420–10424 CrossRef CAS PubMed.
- Zh-J. Quan, Q. Xu, Zh. Zhang, Y.-X. Da and X.-C. Wang, Tetrahedron, 2013, 69, 881–887 CrossRef CAS.
- N. Seus, M. T. Saraiva, E. E. Alberto, L. Savegnago and D. Alves, Tetrahedron, 2012, 68, 10419–10425 CrossRef CAS.
- S. N. Sirakanyan, M. Hrubša, D. Spinelli, P. Dias, V. Kartsev, A. Carazo, A. A. Hovakimyan, J. Pourová, E. K. Hakobyan, J. Karlíčková, Sh. Parvin, J. Fadraersada, K. Macáková, A. Geronikaki and P. Mladěnka, J. Pharm. Pharmacol., 2022, 74, 887–895 CrossRef.
- S. N. Sirakanyan, E. K. Akopyan, R. G. Paronikyan, I. M. Nazaryan, A. G. Akopyan and A. A. Ovakimyan, Pharm. Chem. J., 2019, 53, 495–499 CrossRef CAS.
- S. N. Sirakanyan, A. S. Noravyan, I. A. Dzhagatspanyan, I. M. Nazaryan, A. A. Ovakimyan, A. G. Akopyan and N. G. Avetisyan, Pharm. Chem. J., 2013, 46, 591–594 CrossRef CAS.
- S. N. Sirakanyan, V. G. Kartsev, D. Spinelli, A. Geronikaki, A. Petrou, M. Ivanov, J. Glamoclija, M. Sokovic, E. Hakobyan and A. A. Hovakimyan, Arch. Pharm., 2021, 354, 2000208 CrossRef CAS.
- L. Gonnet, M. Baron and M. Baltas, Molecules, 2021, 26, 5667 CrossRef CAS.
- D. Pereira, M. Pinto, M. Correia-da-Silva and H. Cidade, Molecules, 2022, 27, 230 CrossRef CAS.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- G. V. Lavrenova and V. D. Onipko, Encyclopedia of Traditional Medicine, Neva, 2003, pp. 88–90 Search PubMed.
- J. F. Lassen, N. R. Holm, G. Stankovic, T. Lefevre, A. Chieffo, D. Hildick-Smith, M. Pan, O. Darremont, R. Albiero and M. Ferenc, EuroIntervention, 2014, 10, 545–560 CrossRef PubMed.
- M. E. A. Mekky and M. H. Sanad, Synth. Commun., 2019, 49, 1385–1395 CrossRef.
- S. N. Sirakanyan, D. Spinelli, A. Geronikaki, A. A. Hovakimyan and A. S. Noravyan, Tetrahedron, 2015, 71, 3263–3272 CrossRef CAS.
- S. N. Sirakanyan, E. K. Hakobyan and A. A. Hovakimyan, Russ. J. Org. Chem., 2018, 54, 929–932 CrossRef CAS.
- B. Katzung, Drugs Used in Generalized Seizures, Basic and Clinical Pharmacology, Large Medical Books, McGraw-Hill, 9th edn, 2003 Search PubMed.
- H. G. Vogel and W. H. Vogel, Psychotropic and neurotropic activity, in Drug Discovery and Evaluation: Pharmacological Assays, ed. Vogel, H. E., Springer, Berlin & N.-Y., 2008, pp. 569–874 Search PubMed.
- W. Loscher and D. Schmidt, Epilepsy Res., 1988, 2, 145–181 CrossRef CAS PubMed.
- E. A. Swinyard, Experimental Models of Epilepsy, ed. Purpura D. P., Penry J. K., Tower D., Woodbury D. M. and Walter R., Raven Press, New-York, 1992, pp. 433–458 Search PubMed.
- E. Yuen and I. Troconiz, Seizure, 2015, 24, 21–27 CrossRef PubMed.
- M. Bialera and H. S. White, Nat. Rev. Drug Discovery, 2010, 9, 68–82 CrossRef.
- W. Löscher, C. Fassbender and B. Nolting, Epilepsy Res., 1991, 8, 79–94 CrossRef.
- W. Löscher, Seizure, 2011, 20, 359–368 CrossRef.
- M. D. Mashkovsky, Medicines, New wave, 16th edn, 2021, p. 1216s Search PubMed.
- S. E. File, Behav. Brain Res., 2001, 125, 151–157 CrossRef CAS PubMed.
- L. Prut and C. Belzung, Eur. J. Pharmacol., 2003, 463, 3–33 CrossRef CAS PubMed.
- S. Pellow, P. Chopin, S. E. File and M. Briley, J. Neurosci. Methods, 1985, 14, 149–167 CrossRef CAS PubMed.
- F. G. Graeff, C. F. Netto and Jr. H. Zangrossi, Neurosci. Biobehav. Rev., 1998, 23, 237–246 CrossRef CAS.
- R. D. Porsolt, G. Anton, N. Blavet and M. Jalfre, Eur. J. Pharmacol., 1978, 47, 379–391 CrossRef CAS PubMed.
- Y. Buresh, O. Bureshova and D. P. Houston, Methods and Basic Experiments for Investigation of the Brain and Behavior [in Russian], Moscow, 1991, pp. 175–189 Search PubMed.
- M. A. Rogawski and W. Löscher, Nat. Rev. Neurosci., 2004, 5, 553–564 CrossRef CAS PubMed.
- M. Kammerer, M. P. Rassner, T. M. Freiman and T. J. Feuerstein, Naunyn-Schmiedeberg's Arch. Pharmacol., 2011, 384, 47–57 CrossRef CAS PubMed.
- P. S. Miller and A. R. Aricescu, Nature, 2014, 512, 270–275 CrossRef CAS.
- M. H. Saier Jr, Microbiol. Mol. Biol. Rev., 2000, 64, 354–411 CrossRef CAS.
- R. Perrone, F. Berardi, N. A. Colabufo, E. Lacivita and C. Larizza, J. Pharm. Pharmacol., 2005, 57, 1319–1327 CrossRef CAS.
- P. Blier, J. Clin. Psychiatry, 2001, 62, 12–17 CrossRef CAS.
- S. K. Singh, C. L. Piscitelli and A. Yamashita, eScience, 2008, 322, 1655–1661 CAS.
- W. Kuipers, R. Link, P. J. Standaar, A. R. Stoit, I. Van Wijngaarden, R. Leurs and A. P. Ijzerman, Mol. Pharmacol., 1997, 51, 889–896 CrossRef CAS PubMed.
- D. Wacker, G. Fenalti, M. A. Brown, V. Katritch, R. Abagyan, V. Cherezov and R. C. Stevens, J. Am. Chem. Soc., 2010, 132, 11443–11445 CrossRef CAS PubMed.
- B. Winbla, Piracetam: A Review of Pharmacological Properties and Clinical Uses, CNS Drug Rev., 2005, 11, 169–182 CrossRef.
- M. D. Mashkovsky, Lekarstvennyye Sredstvas [Medicines], Novaya Volna, Moscow, Russia, 16th edn, 2021, p. 1216 Search PubMed.
- T. Litchfield and F. Wilcoxon, J. Pharmacol. Exp. Ther., 1949, 96, 99–113 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06121g |
| This journal is © The Royal Society of Chemistry 2024 |