
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of pyrazole-based analogs as CDK2 inhibitors with apoptotic-inducing activity: design, synthesis and molecular dynamics study†
Ghada M. E. Alia,
Menna A. Ewidab,
Amira M. Elmetwalia,
Heba A. Ewidacd,
Riham F. George e,
Walaa R. Mahmoude,
Nasser S. M. Ismail*fb,
Mahmoud S. Ahmed*d and
Hanan H. Georgeyeg
e,
Walaa R. Mahmoude,
Nasser S. M. Ismail*fb,
Mahmoud S. Ahmed*d and
Hanan H. Georgeyeg
aCentral Administration of Drug Control, EDA, P.O. Box: 29, Cairo, Egypt
bDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Future University in Egypt, Cairo 11835, Egypt. E-mail: Nasser.saad@fue.edu.eg
cDepartment of Pharmacology and Biochemistry, Faculty of Pharmacy, Future University in Egypt, Cairo 11835, Egypt
dPharmaceutical Sciences Department, School of Pharmacy, Texas Tech University Health Science Center, Amarillo, Texas, USA. E-mail: mahmoudsalama.ahmed@ttuhsc.edu
ePharmaceutical Chemistry Department, Faculty of Pharmacy, Cairo University, Cairo 11562, Egypt
fPharmaceutical Chemistry Department, Faculty of Pharmacy, Ain-Shams University, Cairo 11566, Egypt
gPharmaceutical Chemistry Department, Faculty of Pharmacy and Drug Technology, Egyptian Chinese University, 11786, Cairo, Egypt
First published on 29th October 2024
Abstract
The discovery of novel CDK2 inhibitors is crucial for developing targeted anticancer therapies. Thus, in this study, we aimed to design, synthesize, and evaluate a series of novel pyrazole derivatives (2a–g, 7a–d, 8a and b, 9, and 10) for their potential as CDK2/cyclin A2 enzyme inhibitors. The newly synthesized compounds were screened in vitro at 50 μM for CDK2 inhibition, followed by IC50 profiling of the most promising candidates. Compounds 4, 7a, 7d, and 9 exhibited the strongest inhibition, with IC50 values of 3.82, 2.0, 1.47, and 0.96 μM, respectively. To assess their anti-proliferative effects, all target compounds were further screened against a panel of 60 National Cancer Institute (NCI) cell lines representing various carcinoma types. Among them, compound 4 demonstrated exceptional anti-proliferative activity with a mean growth inhibition (GI) of 96.47% across the panel, while compound 9 showed a mean GI of 65.90%. Additionally, compounds 2b and 7c exhibited notable inhibition against MCF7 breast cancer cells, with GI rates of 86.1% and 79.41%, respectively. Compound 4 was selected for further five-dose concentration evaluations, displaying a full-panel GI50 value of 3.81 μM, with a subpanel range of 2.36–9.17 μM. Western blot analysis of compounds 4 and 9 in HCT-116 cell lines confirmed their inhibitory effects on CDK2. Furthermore, compound 4 induced significant cell cycle arrest at the G1 phase and promoted apoptosis. In silico molecular docking studies revealed that compounds 4, 7a, 7d, and 9 adopt a similar binding mode as AT7519 (I) within the CDK2 binding site. Molecular dynamics simulations further validated the stability of these compounds within the catalytic domain of CDK2. ADME/TOPKAT analyses indicated their favorable pharmacokinetic profiles, which were confirmed by their low toxicity in normal cell lines. Based on these findings, it was concluded that the synthesized pyrazole derivatives, particularly compound 4, show potent CDK2 inhibition and significant anticancer activity, with promising drug-like properties and minimal toxicity. This positions them as strong candidates for further development as CDK2-targeting anticancer agents.
1. Introduction
Cell proliferation is regulated by an integrated network of proteins, which dictate the cell cycle events from order and timing perspectives. Cell cycle phases are controlled by a family of related serine/threonine proteins called cyclin-dependent kinases (CDKs), which become active upon association with their respective cyclin regulatory partners.1 Cell cycle progression is controlled by the formation of a CDK/cyclin complex, which is responsible for the phosphorylation of target genes, such as the tumor suppressor protein retinoblastoma (Rb). A CDK/cyclin complex is activated by mitogenic signals and inhibited by the activation of cell-cycle checkpoints in response to DNA damage.2 Cyclin-dependent kinase inhibitors (CKIs) negatively regulate CDKs/cyclin as the inhibitor of CDK4 (INK4) proteins (p16INK4a, p15INK4b, p18INK4c, and p19INK4d) and CDK2 (p21 and p27).3CDK2-cyclin E is mainly responsible for the complete phosphorylation of retinoblastoma (Rb) in late G1, which allows the initiation of the S phase of the cell cycle, while CDK2-cyclin A eases S/G2 transition. Moreover, CDK2 plays an additional role in apoptosis, cell differentiation, immune response and the repair of normal DNA.4–7 Overexpression of CDK2 is predominant in many cancer types such as melanoma, glioblastoma, lymphoid tumor tissues and metastasis of prostate cancer. The irregular expression of CDK2 is often accompanied with the augmentation of its partner cyclins A and E in many human cancers as breast, endometrial, lung and thyroid carcinomas, melanoma, and osteosarcoma.8,9
In recent years, computational methods have become invaluable tools in cancer diagnosis and treatment, enabling the identification of potential therapeutic targets and facilitating the design of inhibitors through molecular modeling and dynamic simulation studies. These techniques provide significant insights into the binding interactions and stability of drug candidates, accelerating the drug discovery process.10–13
In this study, pyrazole was chosen as a fundamental framework for drug exploration. This decision was driven by its molecular diversity, which accounts for the wide range of biological activities observed in different pyrazole derivatives, including antibacterial, anti-inflammatory, and anticancer activities.14–17 Fig. 1 illustrates a set of differently substituted pyrazole-containing compounds with outstanding CDK inhibitory activity.
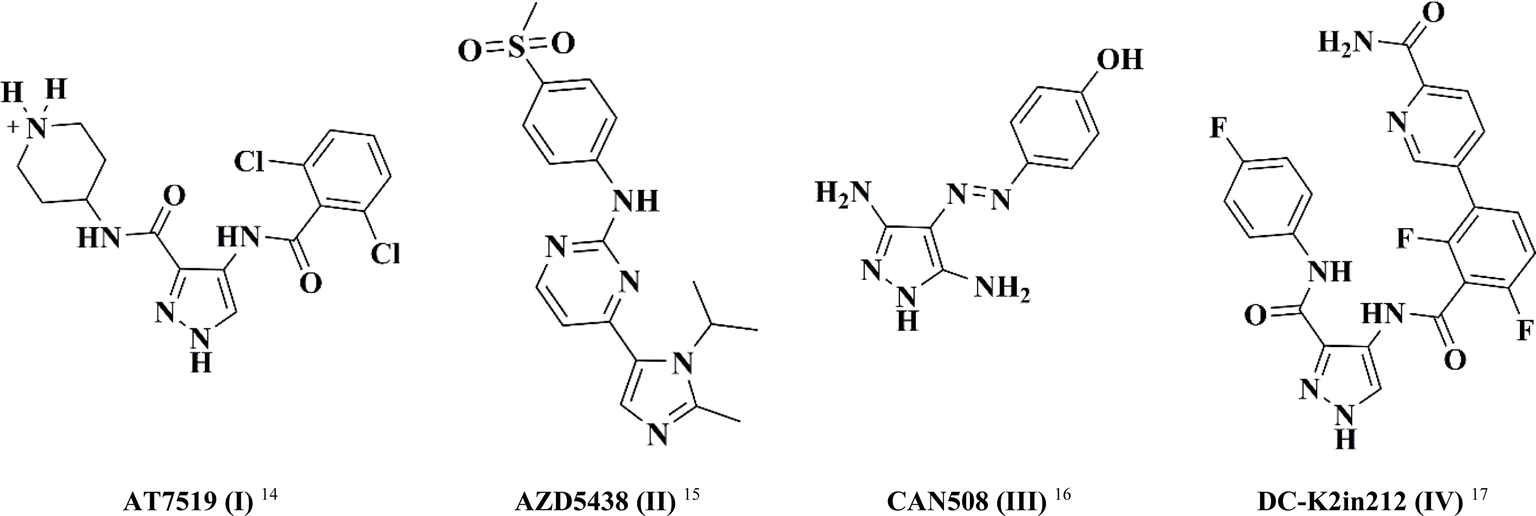 | ||
| Fig. 1 Structure of clinically approved CDK2 inhibitors (I–IV). | ||
AT7519 (I) is a multi CDK inhibitor for CDK 1, 2, 4, 6 and 9 isoforms with IC50 values in the range of 10–210 nM.18 Interestingly, the imidazo-pyrmidine derivative AZD5438 (II) displayed potent CDK2 inhibition with an IC50 value of 6 nM.19 Furthermore, the di-amino pyrazole derivative CAN508 (III) exhibited selective inhibition activity against CDK2 with an IC50 value of 0.35 μM.20 Besides, 4-benzoylamino-1H-pyrazole-3-carboxamide derivative DC-K2in212 (IV) displayed 17-fold selectivity against CDK2 over CDK1 with an IC50 value of 0.295 μM.21
The binding mode of AT7519 (I) to the CDK2 ATP binding site disclosed the importance of the pharmacophoric pyrazole nucleus to occupy the adenine region of the ATP binding pocket. The H-bonding interactions mediated by the pyrazole core NH with Glu82 anchored AT7519 (I) tightly into the CDK2 hinge region. Both the carboxamide side chain N atom and pyrazole N displayed two H-bonding with Leu83. The 2,6-dichlorobenzamide moiety exhibited hydrophobic interactions with the Asp145 residue, as shown in Fig. 2.
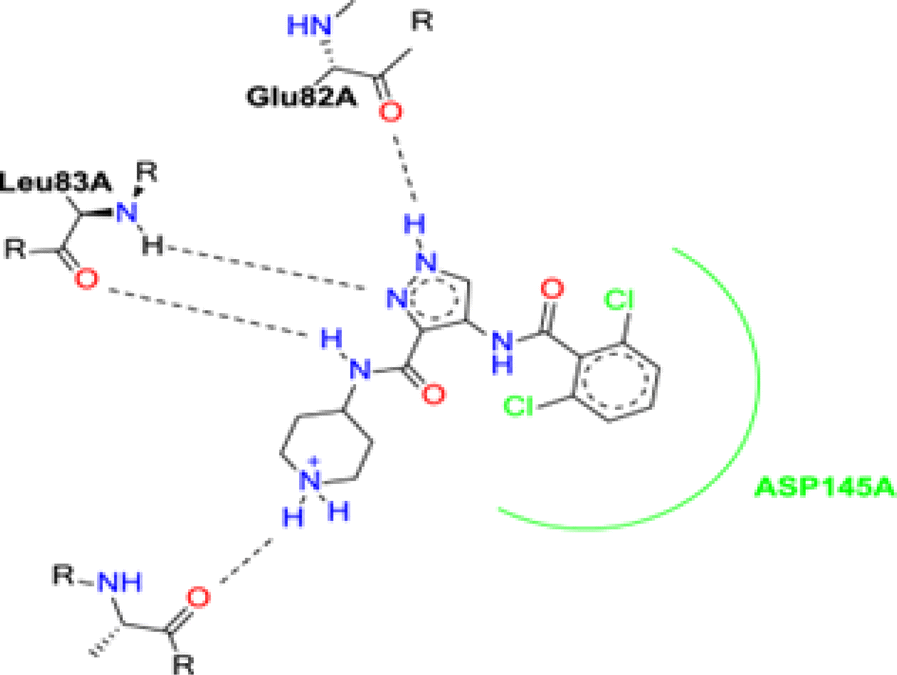 | ||
| Fig. 2 Schematic of the binding mode of the lead compound AT7519 (I) to CDK2 ATP binding site. | ||
Herein, the design of the target compounds with a pyrazole core, i.e., 2a–g, 4, 7a–d, 8a–b, 9 and 10, was derived from the structure optimization of the reference compound AT7519 (I) based on its reported structure–activity relationship (SAR), as follows.18
The pharmacophoric pyrazole core in the lead compound, which fills the adenine region of the ATP binding pocket is preserved. The H-bonding and hydrophobic interaction of our lead compound AT7519 (I) was furnished by the functional groups illustrated in Fig. 3.
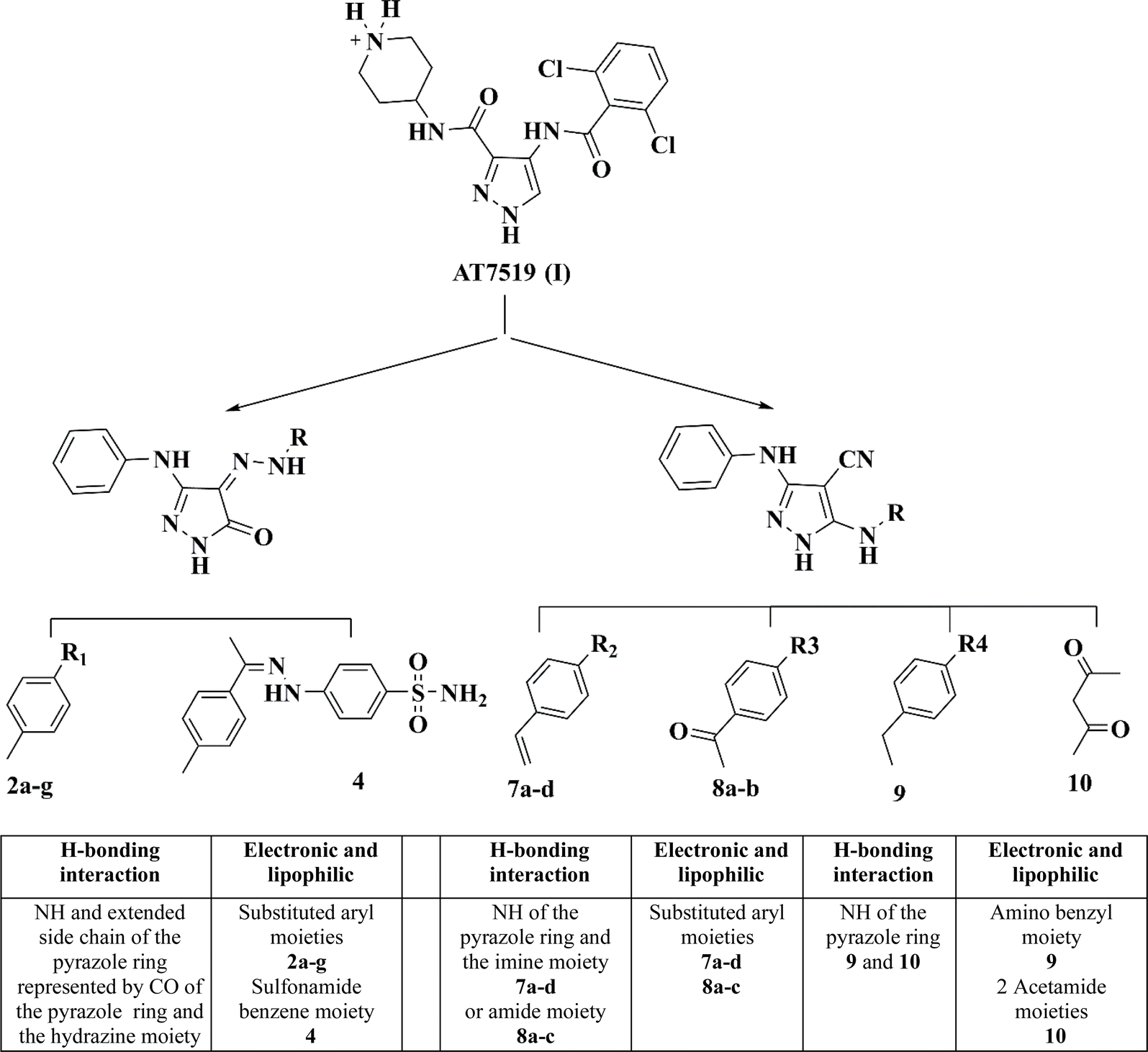 | ||
| Fig. 3 Similarities of the pharmacophoric features between AT7519 (I) and the target compounds. | ||
Consequently, a nitrile group was introduced at position 4 in compounds 7a–d, 8a and b, 9, and 10 to afford hydrogen bond acceptor functionality and form non-specific dipole interactions with the amino acid residues, promoting their binding with the sterically occluded protein kinases, and consequently enhancing their activity. The phenyl amino group at position 3 of the pyrazole ring acts as a hydrogen bond donor and provides occupation of a small hydrophobic pocket.
Enzyme inhibition versus CDK2 was performed for all the compounds. Furthermore, the targeted compounds were screened for their anticancer activity versus 60 human cancer cell lines from the NCI-USA. The most promising compounds were subjected to further assessment at five doses against a full NCI 60 cell panel assay. A molecular modeling study was also performed to explore their possible binding modes within the CDK2/ATP binding site. The active analogs were further investigated for their dynamic stability through a dynamic simulation process. Eventually, the ADMET computational parameters were assessed to predict the drug-likeness characteristics of the target compounds.
2. Results and discussion
2.1. Chemistry
Ethyl acetoacetate was reacted with phenyl isothiocyanate in a solution of sodium methylate, followed by cyclization using hydrazine hydrate, resulting in 5-phenylamino-2,4-dihydro-pyrazol-3-one 1.22 Subsequently, the target 4-(2-(4-un/substitutedphenyl)hydrazono)-5-(phenylamino)-2,4-dihydro-3H-pyrazol-3-ones 2a–g were obtained via the coupling of 1 with the respective diazonium salts of different aromatic amines, as shown in Scheme 1.23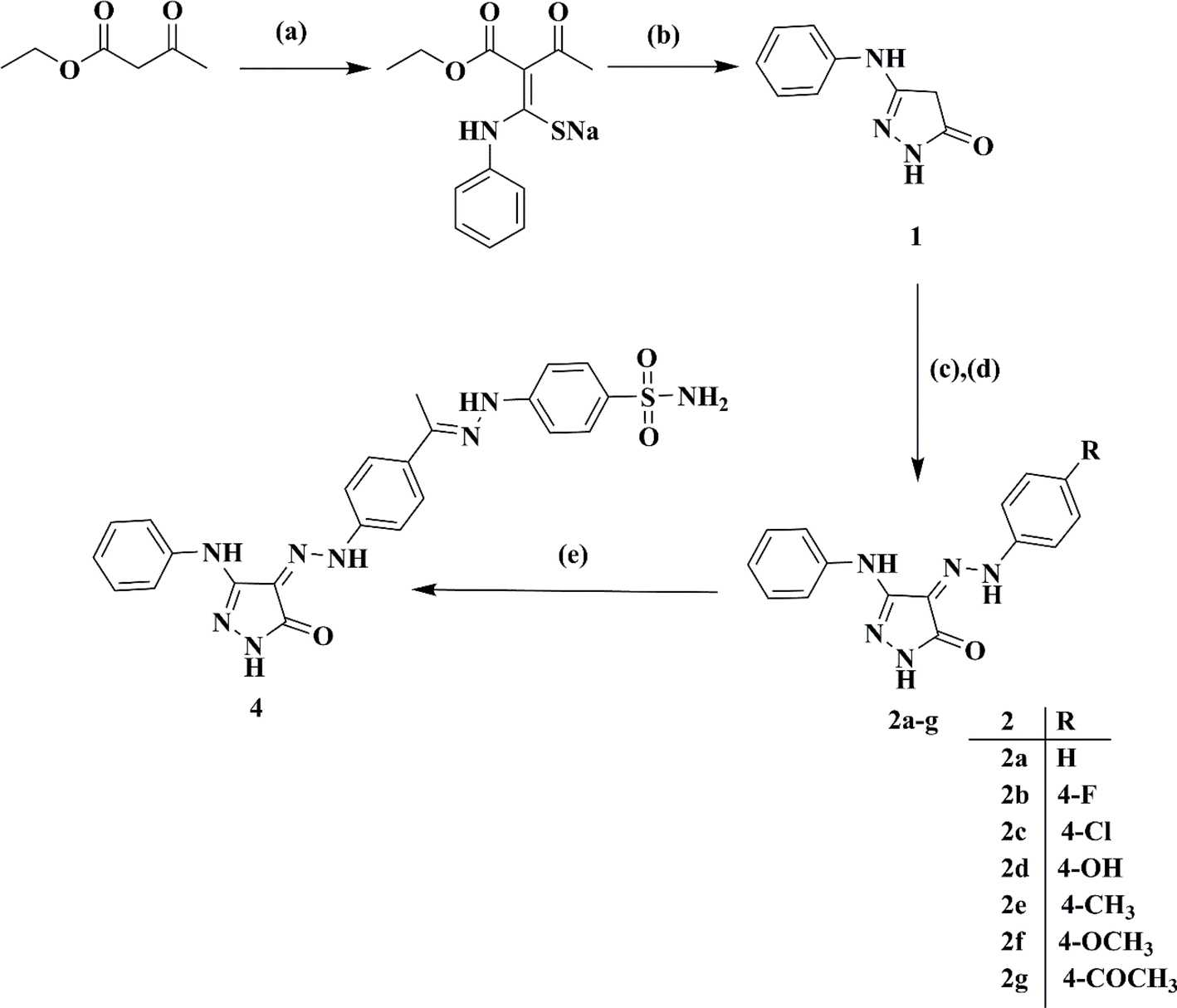 | ||
| Scheme 1 Synthetic route for target compounds 2a–g and 4. Reagents and conditions: (a) Na, CH3OH, phenyl isothiocyanate, reflux, 30 min, cool to 50 °C. (b) Hydrazine hydrate, reflux, 1 h. (c) Ar–NH2, HCl, sodium nitrite solution, stirring, 0–5 °C, 15 min. (d) Diazonium salt, 2 N HCl, sodium acetate, stirring, RT, 24 h. (e) 2g, 4-hydrazineylbenzenesulfonamide 3, C2H5OH, acetic acid, stirring, RT, 24 h. | ||
The sulfonamide diazonium salt was reduced using a mild reducing agent, stannous chloride, to the corresponding 4-hydrazineylbenzenesulfonamide 3. The latter was condensed with acetyl derivative 2g (generated from Scheme 1) in glacial acetic acid to attain pyrazole benzene sulfonamide derivative 4.24
The synthesis of 5-((4-un/substitutedbenzylidene)amino)-3-(phenylamino)-1H-pyrazole-4-carbonitriles 7a–d was initiated via the preparation of the key 2-((methylthio)(phenylamino)methylene)malononitrile intermediate 5 by the nucleophilic addition reaction of C2 of malononitrile to phenyl isothiocyanate, followed by the methylation reaction using dimethyl sulfate.25,26 Subsequently, the latter was cyclized directly upon the fusion with hydrazine hydrate to afford the key intermediate 5-amino-3-phenylamino-1H-pyrazole-4-carbonitrile 6,27 which was refluxed with different aromatic aldehydes in glacial acetic acid to afford the final target compounds 7a–d, as illustrated in Scheme 2.28
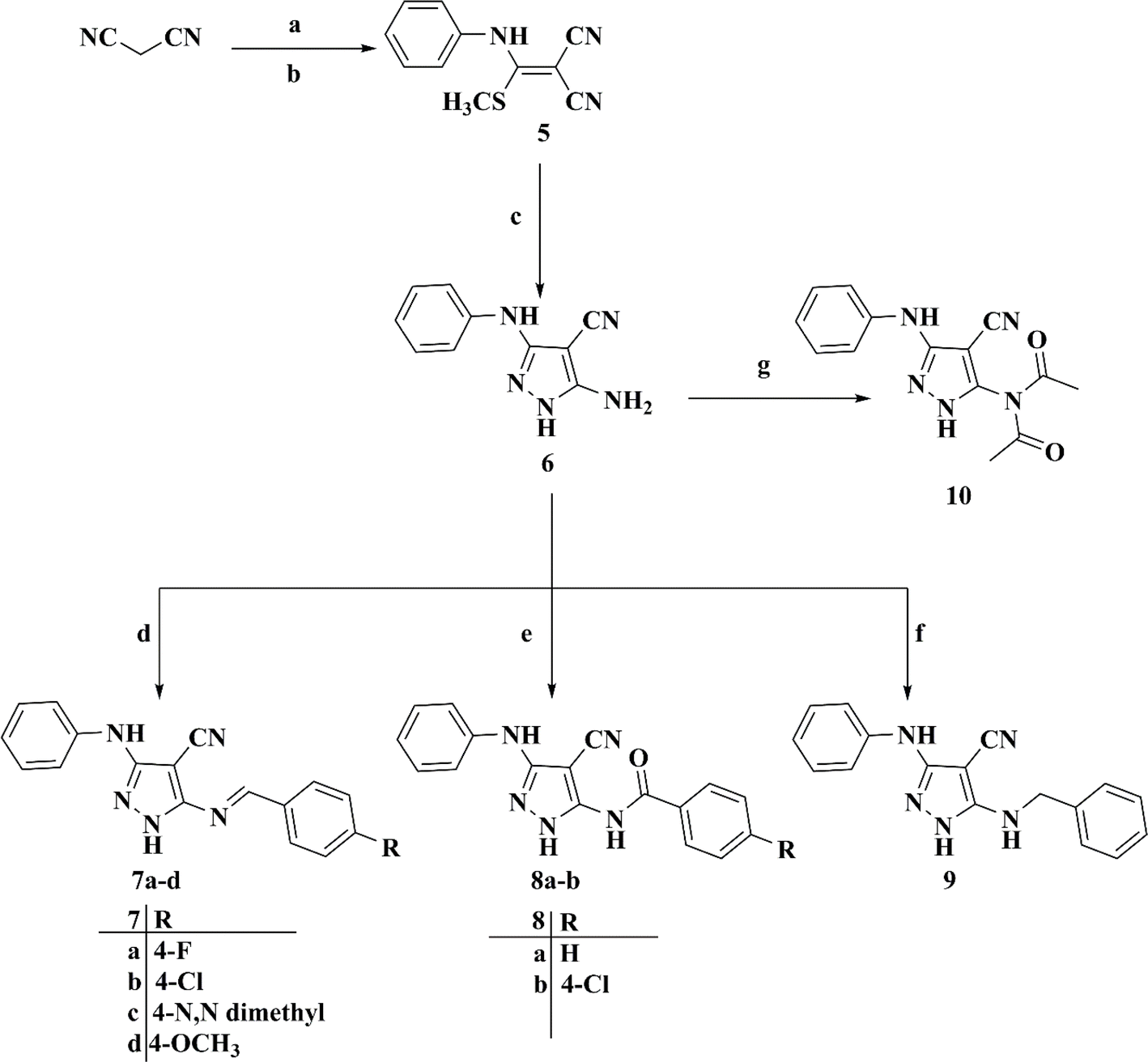 | ||
| Scheme 2 Synthetic route for target compounds 7a–d, 8a and b, 9, and 10. Reagents and reaction conditions: (a) KOH, DMF, phenyl isothiocyanate, stirring, RT, 24 h. (b) Dimethyl sulphate, stirring, RT, 8 h. (c) Hydrazine hydrate, W. B., 3–4 h. (d) Ar–CHO, acetic acid, reflux, 5 h. (e) Ar–CO–Cl, N(CH2CH3)3, dry benzene, stirring, RT, 6 h. (f) Ar–CH2–Cl, anhydrous K2CO3, dry benzene, reflux, 6 h. (g) (CH3CO)2O, acetic acid, stirring, RT, 24 h. | ||
Remarkably, 4-substituted-N-(4-cyano-3-(phenylamino)-1H-pyrazol-5-yl)benzamides 8a and b were attained by the treatment of 5-amino-3-(phenylamino)-1H-pyrazole-4-carbonitrile 6 with the appropriate benzoyl chloride derivative with the aid of triethylamine as a catalyzing agent in dry benzene.29,30 Curiously, the reaction of compound 6 with benzyl chloride produced 5-(benzylamino)-3-(phenylamino)-1H-pyrazole-4-carbonitrile 9, whereas the reaction of our intermediate of interest 6 with a catalytic amount of acetic anhydride in acetic acid produced N-acetyl-N-(4-cyano-3-(phenylamino)-1H-pyrazol-5-yl)acetamide 10, as illustrated in Scheme 2.31
The spectroscopic characteristics of compounds 2a–g, 7a–d, 8a and b, 9, and 10 confirmed their structures. In the case of compounds 2a–g, the IR spectrum of compound 2d revealed a broad OH band at the v max of 3479 cm−1, while the 1H-NMR spectra of compounds 2e, 2f and 2g revealed singlet bands corresponding to the Ar–CH3, Ar–OCH3 and Ar–COCH3 protons at δ of 2.31, 3.78, 2.58 ppm, respectively. The 13C-NMR spectra of compounds 2f and 2g showed bands located at δ of 55.86 and 27.06 for Ar–OCH3 and Ar–CH3, respectively. The IR spectrum of compound 4 revealed bands for the expected new SO2 group at 1338 and 1177 cm−1, respectively. The 1H-NMR spectrum of compound 4 confirmed the appearance of a singlet band for the aliphatic protons of CH3 at δ of 4.39 ppm, protons of Ar–CH in the δ range of 6.93–7.9 ppm and identical singlet peak at δ of 6.94 ppm for the NH2 protons. The 1H NMR spectra of compounds 7a–d showed a characteristic singlet signal assigned to the (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) proton, which appeared in the δ range of 8.75–9.01 ppm. Compound 7c showed a singlet signal at δ of 3.88 for OCH3, where compound 7d revealed a singlet signal at δ of 3.07 ppm for the 6 protons of 2CH3. Alternatively, the 1H NMR spectra of compounds 8a and b revealed bands for NH and CO. Compound 8b showed a singlet signal at δ of 3.89 for the OCH3 protons. The 1H NMR spectrum of compound 9 showed a singlet signal assigned to the benzyl proton at δ of 4.51 ppm. Compound 10 showed 2 extra CO bands. In contrast, compound 10 exhibited 2 singlet signals for the acetyl protons at 2.19 and 2.62 pm. Finally, the mass spectra of all the compounds were harmonized with their calculated molecular weights.
CH) proton, which appeared in the δ range of 8.75–9.01 ppm. Compound 7c showed a singlet signal at δ of 3.88 for OCH3, where compound 7d revealed a singlet signal at δ of 3.07 ppm for the 6 protons of 2CH3. Alternatively, the 1H NMR spectra of compounds 8a and b revealed bands for NH and CO. Compound 8b showed a singlet signal at δ of 3.89 for the OCH3 protons. The 1H NMR spectrum of compound 9 showed a singlet signal assigned to the benzyl proton at δ of 4.51 ppm. Compound 10 showed 2 extra CO bands. In contrast, compound 10 exhibited 2 singlet signals for the acetyl protons at 2.19 and 2.62 pm. Finally, the mass spectra of all the compounds were harmonized with their calculated molecular weights.
2.2. Biological screening
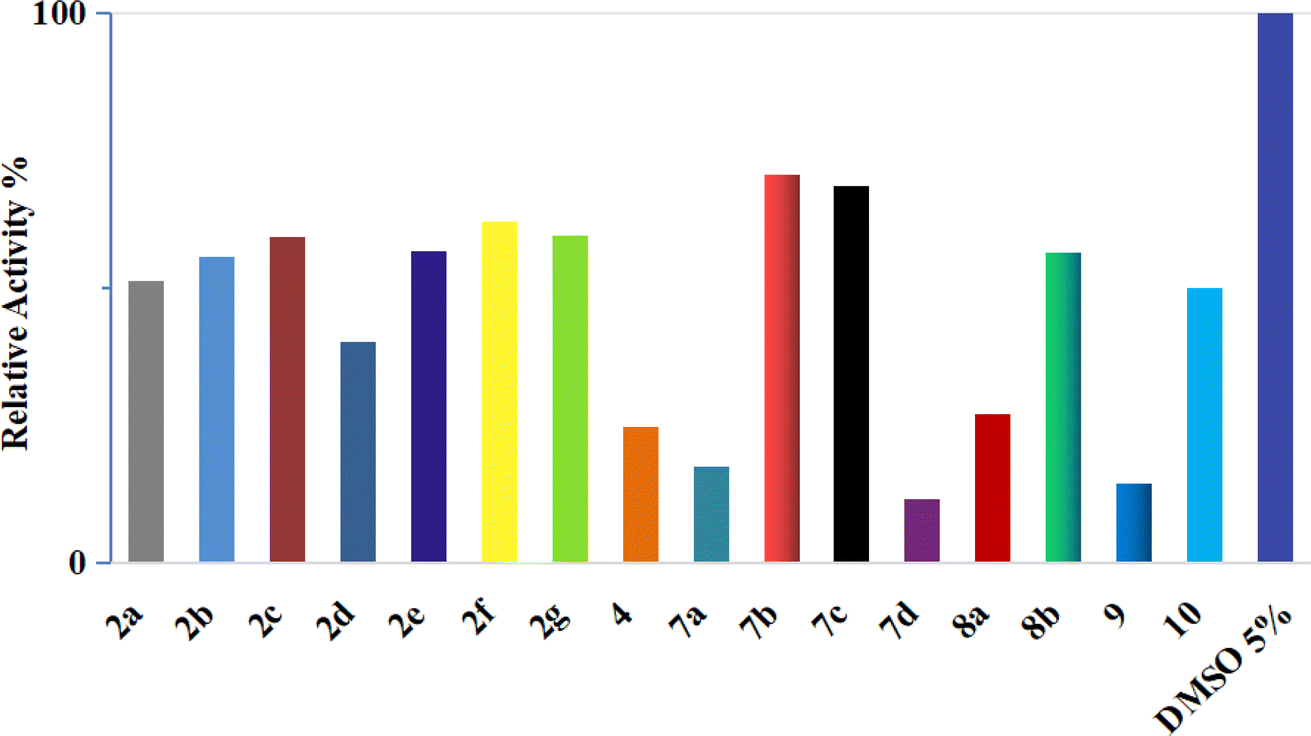 | ||
| Fig. 4 Inhibition of CDK2/cyclin A2 by compounds 2a–g, 4, 7a–d, 8a and b, 9, and 10 at a concentration of 50 μM. | ||
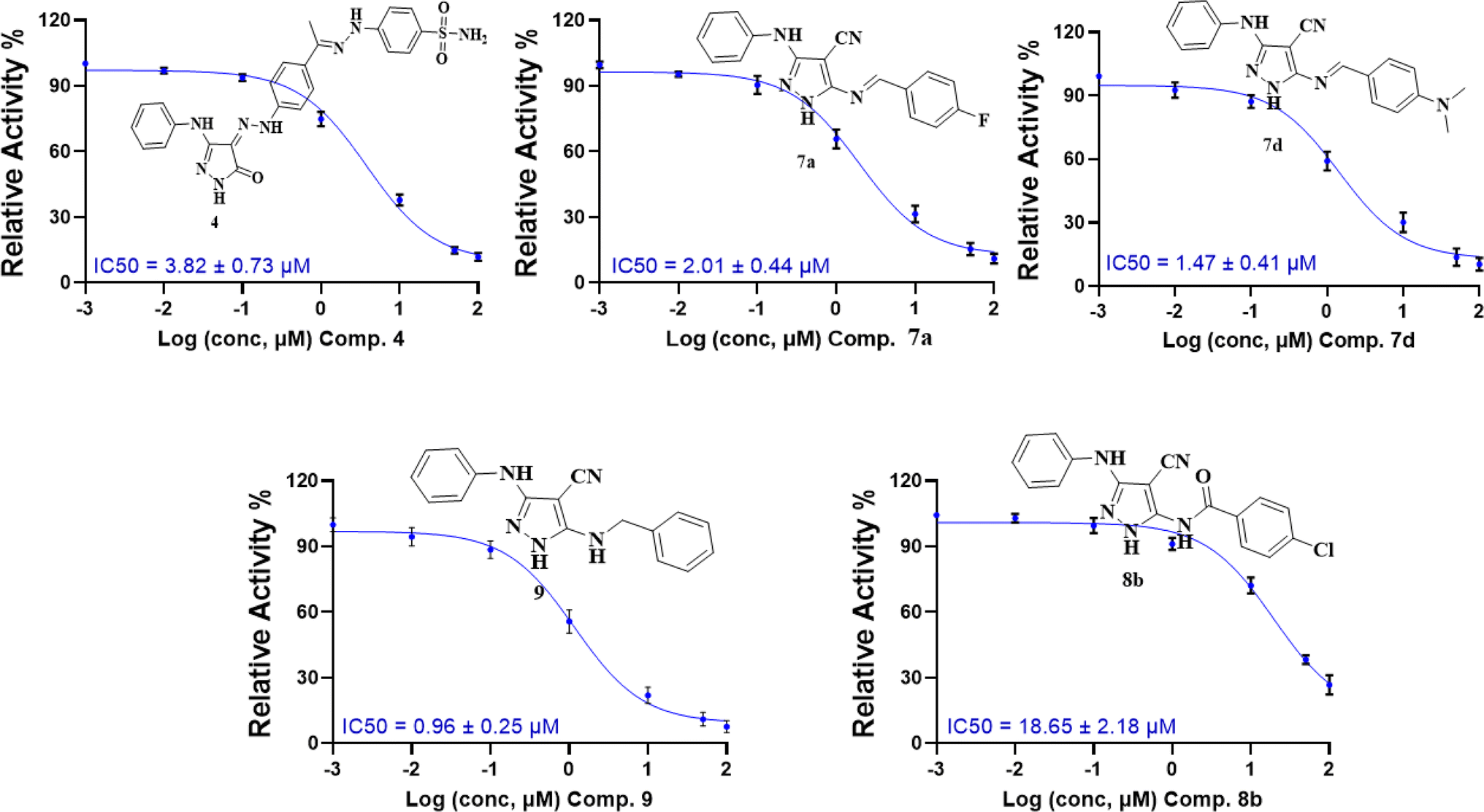 | ||
| Fig. 5 Dose–response curves with the inhibitory concentration at 50% (IC50) of compounds 4, 7a, 7d, 8b, and 9 using the CDK2/cyclin A2 protein kinase activity assay. | ||
These results are consistent with the recent CDK2 findings,10–13 confirming the anticipated inhibitory potential of the selected pyrazole derivatives. The observed IC50 values for compounds 9, 7a, 7d, and 4, particularly their strong activity against CDK2/cyclin A2, validate the structural modifications introduced during the design phase, further supporting their promising role as CDK2 inhibitors.
| Panel line | Cell line | GI% compound 4 | GI% compound 9 |
|---|---|---|---|
| Leukemia | CCRF-CEM | 96.41 | 81.65 |
| RPMI-8226 | 96.53 | — | |
| Melanoma cell lines | UACC-257 | 94.37 | — |
| SK-MEL-2 | — | 79.33 | |
| MALME-3M | — | 78.10 | |
| Breast cell lines | T-47D | — | 82.76 |
| Non-small cell lung cancer cell lines | NCI-H23 | 99.80 | — |
| NCI-H322M | 90.78 | — | |
| NCI-H460 | 91.13 | — | |
| Colon cancer cell lines | HT29 | 95.46 | — |
| KM12 | 96.31 | — | |
| HCT-116 | 99.70 | — | |
| CNS cancer cell lines | SF-268 | 90.67 | — |
| SF-295 | 94.88 | — |
A deep look at the results revealed that some derivatives of 2a–g revealed pronounced anticancer activity against some breast cancer cell lines such as compounds 2a, 2b and 2c, which showed activity against MCF7 with GI% of 73.8%, 86.1% and 77.1%, respectively. Furthermore, analog 2b exhibited comparable anticancer activity against MDA-MB-468 with GI% of 72.25%. Alternatively, compound 7c demonstrated remarkable activity against the MCF7 breast cancer cell line with GI% of 79.41%. The achieved results revealed the influence of electronic characteristics of the side chain substituents on the anticancer activity of sulfonamide candidate 4. Therefore, the electron-withdrawing-substituted derivatives 2b and 2c and unsubstituted analog 2a exhibited pronounced anticancer activity against the breast cancer cell line. Alternatively, changing the electronic environment of the substituents to electron-donating groups led to a decrease in the anticancer activity, as seen in compounds 2d, 2e, and 2f with GI% mean of 27.12%, 6.87% and 10.24%, respectively.
| Panel line | Cell line | GI50 (μM) | TGI (μM) | LC50 (μM) |
|---|---|---|---|---|
| Leukemia | CCRF-CEM | 2.63 | 8.92 | >100 |
| HL-60(TB) | 2.86 | 0.171 | 60.5 | |
| K-562 | 2.29 | 6.37 | 22.8 | |
| MOLT-4 | 1.64 | 5.24 | 34.9 | |
| RPMI-8226 | 3.36 | 28.2 | >100 | |
| SR | 1.74 | 5.33 | 23.6 | |
| Non-small cell lung cancer | A549/ATCC | 19.4 | >100 | >100 |
| EKVX | 5.39 | 26.9 | >100 | |
| HOP-62 | 2.05 | 6 | 30.3 | |
| HOP-92 | 1.3 | 6.55 | 30.7 | |
| NCI-H226 | 3.31 | 12.3 | 35.1 | |
| NCI-H23 | 2.54 | 6.53 | 23.10 | |
| NCI-H322M | 3.72 | 15.2 | 41.9 | |
| NCI-H460 | 3.21 | 11.10 | 45.3 | |
| NCI-H522 | 2.17 | 5.25 | 74.8 | |
| Colon cancer | COLO 205 | 3.28 | 12 | 34.6 |
| HCC-2998 | 2.29 | 6.08 | 21.3 | |
| HCT-116 | 3.04 | 10.3 | 32 | |
| HCT-15 | 5.29 | 18.4 | 42.9 | |
| HT29 | 3.58 | 11.6 | 34 | |
| KM12 | 2.66 | 8.95 | 29.9 | |
| SW-620 | 3.18 | 12.6 | 58.8 | |
| CNS cancer | SF-268 | 2.22 | 6.23 | 31.5 |
| SF-295 | 3.67 | 13.6 | 38.7 | |
| SF-539 | 1.58 | 3.68 | 8.56 | |
| SNB-19 | 2.59 | 8.76 | 29.6 | |
| U251 | 1.88 | 3.76 | 7.51 | |
| Melanoma | LOX IMVI | 2.13 | 4.33 | 8.81 |
| MALME-3M | 1.98 | 3.86 | 7.49 | |
| M14 | 2.83 | 7.55 | 35.1 | |
| MDA-MB-435 | 2.07 | 5.03 | 17.8 | |
| SK-MEL-2 | 2.11 | 4.38 | 9.1 | |
| SK-MEL-28 | 1.91 | 3.65 | 6.99 | |
| SK-MEL-5 | 3.32 | 12.8 | 35.8 | |
| UACC-257 | 3.97 | 16.7 | 75.5 | |
| UACC-62 | 1.96 | 4.34 | 9.6 | |
| Ovarian cancer | IGROV1 | 4.5 | 17.6 | 43.2 |
| OVCAR-3 | 1.87 | 4.54 | 12.5 | |
| OVCAR-4 | 2.84 | 9.26 | 40.7 | |
| OVCAR-5 | 11 | 24.4 | 54.2 | |
| OVCAR-8 | 2.59 | 7.81 | 29 | |
| NCI/ADR-RES | >100 | >100 | >100 | |
| SK-OV-3 | 2.34 | 6.47 | 23.7 | |
| Renal cancer | 786-0 | 2.14 | 6.27 | 24.7 |
| A498 | 11.4 | 32.8 | 94.7 | |
| ACHN | 5.9 | 18.5 | 43 | |
| CAKI-1 | 12.5 | 25.1 | 50.1 | |
| RXF 393 | 1.1 | 3.88 | 21.1 | |
| SN12C | 2.38 | 6.58 | 23.5 | |
| TK-10 | 18.6 | 56.8 | >100 | |
| UO-31 | 19.4 | >100 | >100 | |
| Prostate cancer | PC-3 | 3.06 | 11.3 | 35.8 |
| DU-145 | 3.21 | 10.4 | 32.2 | |
| Breast cancer | MCF7 | 2.4 | 7.34 | 27.4 |
| MDA-MB-231/ATCC | 2 | 4.07 | 8.28 | |
| HS 578T | 3.31 | 19.4 | >100 | |
| BT-549 | 2.83 | 9.66 | 31.1 | |
| T-47D | 2.22 | 5.92 | 23.9 | |
| MDA-MB-468 | 1.4 | 3 | 6.41 |
In addition, the mean graph midpoints (MG-MID) for the subpanel and full panel were calculated for the GI50 to demonstrate the average activity parameter for each compound. Commonly, compound 4 revealed obvious anticancer activity against almost the whole panel of cancer cell lines with GI50 values in the range of 1.1–5.9 μM. Remarkably, compound 4 displayed noticeable cytostatic activity against the following cell lines: leukemia (HL-60(TB); TGI 0.171 μM), breast cancer (MDA-MB-468; TGI 3 μM), melanoma (SK MEL-28; TGI 3.65 μM, MALME-3M: TGI 3.86 μM), CNS cancer (SF-539; TGI 3.68 μM and U251; TGI 3.76 μM), and renal cancer (RXF 393; TGI 3.88 μM).
Compound 4 exerted obvious antiproliferative activity against almost the whole NCI panel with a full panel GI50 (MGMID) value of 3.81 μM and subpanel GI50 (MG-MID) range of 2.6–9.17 μM. Among the tested cancer subpanels, breast cancer, CNS, leukemia, melanoma cancer, prostate cancer and colon cancer were the most susceptible subpanels to compound 4 with GI50 (MG-MID) values of 2.36, 2.388, 2.42, 2.47, 3.13 and 3.33 μM, respectively, as shown in Table 3.
| Subpanel type | MG-MID | Selectivity indexc |
|---|---|---|
| a MG-MID is the average activity parameter over individual subpanels/tested compound.b Full panel MG-MID is the average sensitivity over all cell lines (full panel)/tested compound.c Selectivity index was attained by dividing the full panel MG-MID (μM) for each compound by its individual subpanel MG-MID (μM). | ||
| Leukemia | 2.42 | 1.574 |
| Non-small cell lung cancer | 4.787778 | 0.795 |
| Colon cancer | 3.331429 | 1.143 |
| CNS cancer | 2.388 | 1.595 |
| Melanoma | 2.475556 | 1.539 |
| Ovarian cancer | 4.19 | 0.909 |
| Renal cancer | 9.1775 | 0.415 |
| Prostate cancer | 3.135 | 1.215 |
| Breast cancer | 2.36 | 1.614 |
| Full panel MG-MIDb = 3.81 | ||
| Comp. | DNA content results | ||||
|---|---|---|---|---|---|
| Conc. μM | %G0/G1 | %S | %G2/M | Results | |
| Control | 7.022 | 59.37 | 33.81 | 6.82 | — |
| Comp. 4 | 1.806 | 73.16 | 23.05 | 3.79 | Cell growth arrest at G1 |
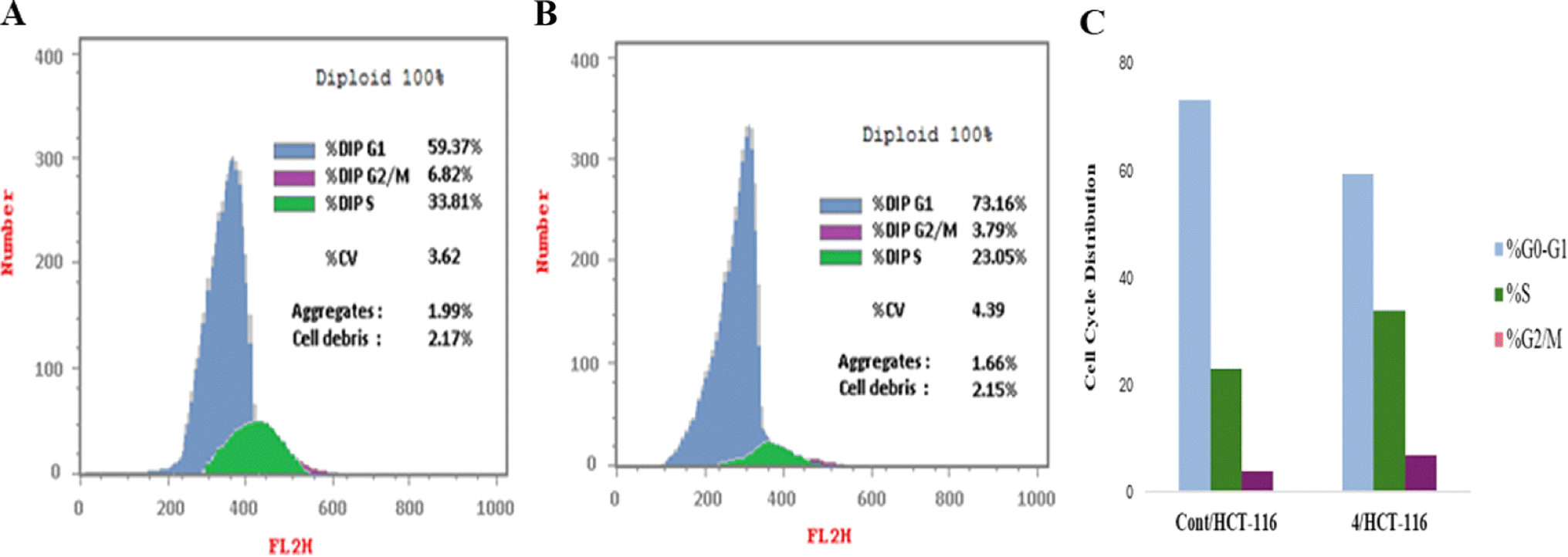 | ||
| Fig. 6 Flow cytometric analysis for cell cycle distribution. (A) Control HCT-116, (B) compound 4, and (C) graphical representation for cell cycle distribution analysis among differently treated cells. | ||
| Comp | Apoptosis | ||||
|---|---|---|---|---|---|
| Conc. μM | Total | Early | Late | Necrosis | |
| Control | 7.022 | 2.06 | 0.35 | 0.13 | 1.58 |
| Comp. 4 | 1.806 | 27.65 | 4.63 | 15.81 | 7.21 |
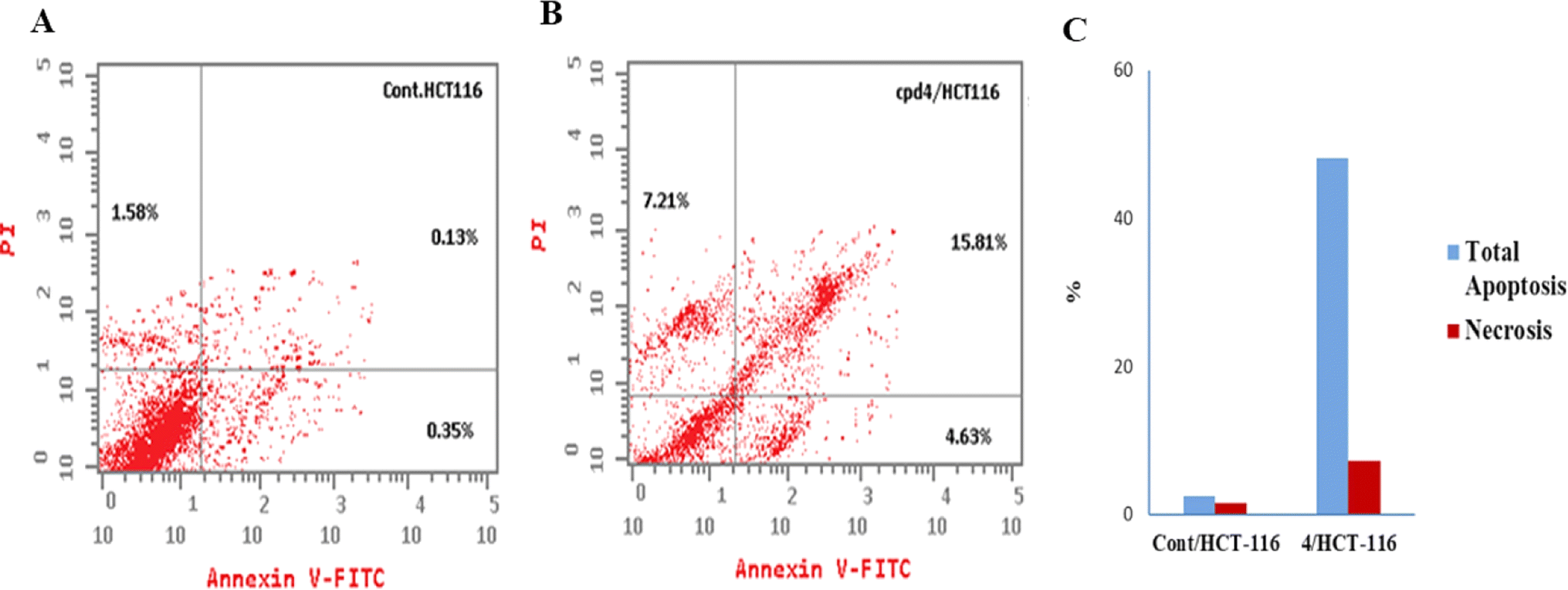 | ||
| Fig. 7 Flow cytometric analysis of apoptosis among treated cells. (A) Control HCT-116, (B) compound 4, and (C) graphical illustration of apoptosis% among differently treated cells. | ||
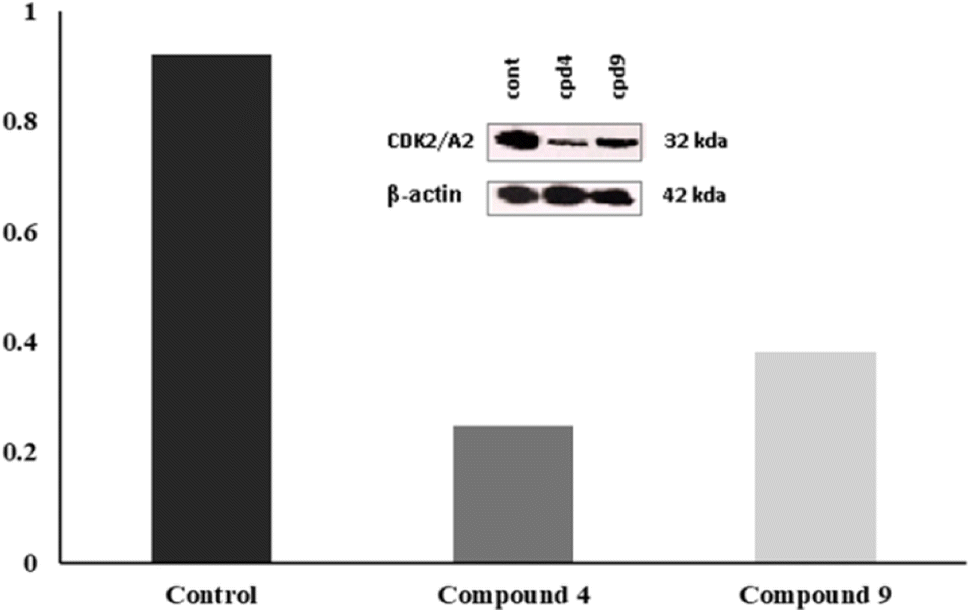 | ||
| Fig. 8 Western blot assay for compounds 4 and 9. | ||
2.3. In silico studies
The initial step of docking protocol was re-docking of the co-crystallized ligand to assess validation parameters as RMSD = 0.5227 Å, CDOCKER energy = – 12.8047 kcal mol−1. Furthermore, the following step was the identification of binding mode of the native ligand which confirmed the validity of the applied docking protocol. Two H bonds with the essential Leu83 residue were mediated via the pyrazole core N atom and side chain NH. In addition to an extra hydrogen bond formed between His84 residue and NH piperidine. The 2,6-dichlorobenzamide moiety occupied almost all of the ATP binding region, whereby the pyrazole moiety accommodated the adenine region, as shown in Fig. 9.
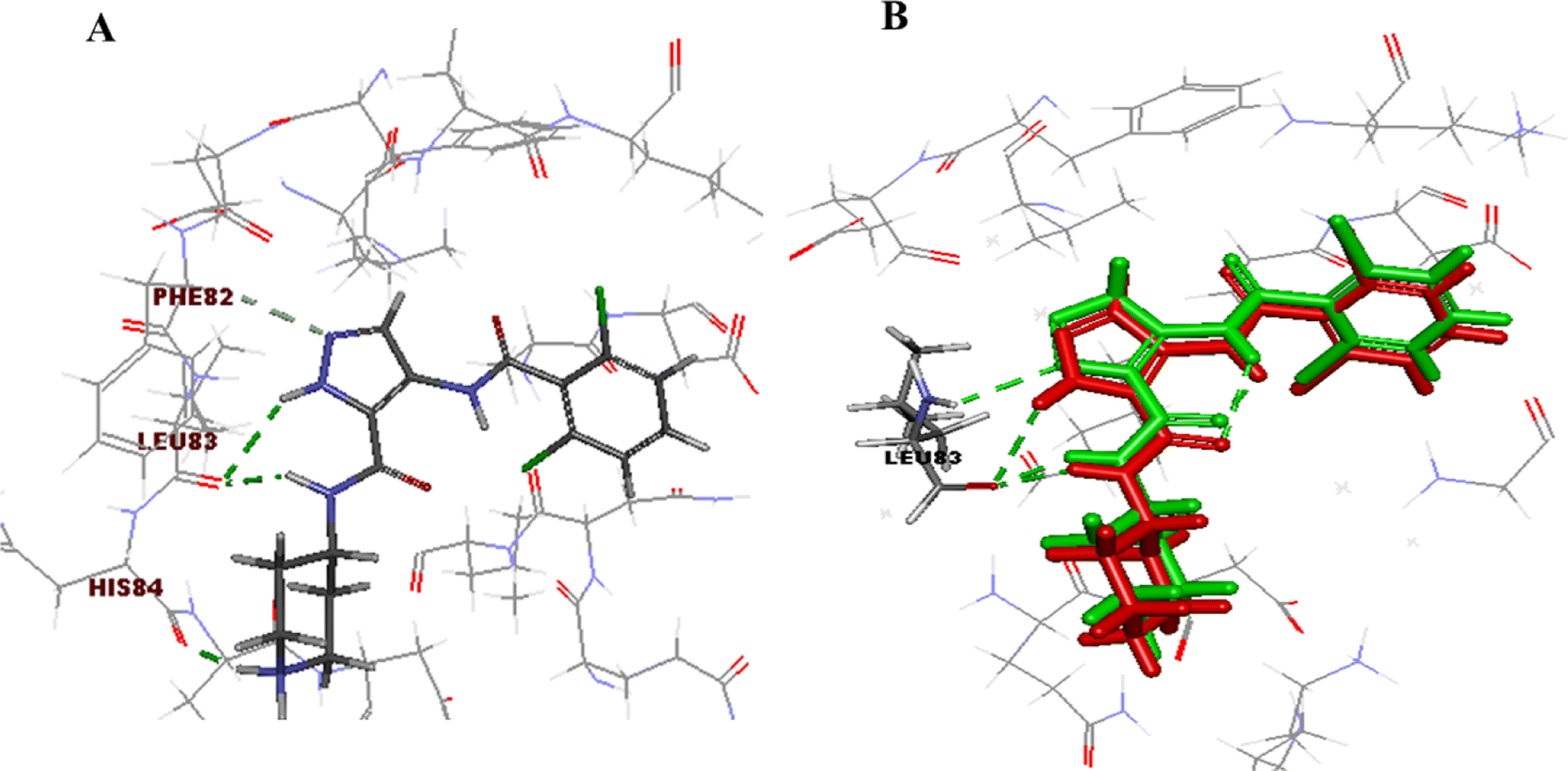 | ||
| Fig. 9 2D ligand AT7519 (I) interaction diagram (A) and 3D representation (B) of superimposition of the native ligand (green) and its re-docked pose (red) in CDK2 ATP binding site. | ||
Successively, we applied the same docking protocol for our compounds of interest. Most of the docked compounds presented a similar binding pattern to that of the reference AT7519 (I).
As shown in Fig. 10, the most potent CDK2 inhibitors 4 (IC50 of 4.2 μM), 7a (IC50 of 3.98 μM), and 9 (IC50 of 0.97 μM) could mediate the essential interactions with the enzyme binding site, in particular the H-bonding with the Leu83 residue via the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group, phenylamino NH groups and the pyrazole NH; an observation that can rationalize their obtained activity. In contrast, docking of compound 4 (IC50 of 4.2 μM) was significantly different from the reference AT7519 (I), given that it demonstrated H-bonding between Lys20 and NH2SO2. The aforementioned binding pattern for compound 4 can explain its observed inhibitory activity.
N group, phenylamino NH groups and the pyrazole NH; an observation that can rationalize their obtained activity. In contrast, docking of compound 4 (IC50 of 4.2 μM) was significantly different from the reference AT7519 (I), given that it demonstrated H-bonding between Lys20 and NH2SO2. The aforementioned binding pattern for compound 4 can explain its observed inhibitory activity.
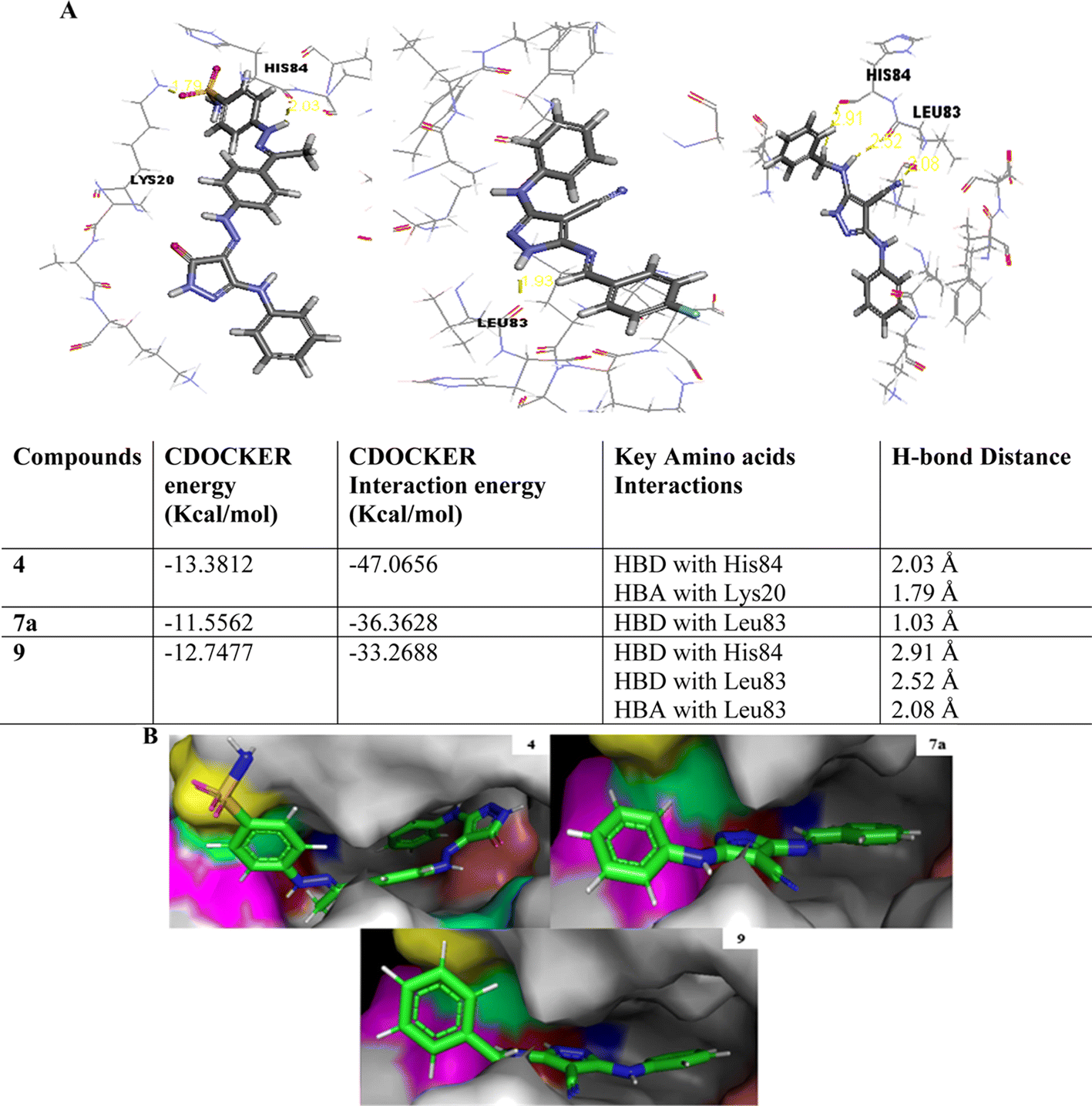 | ||
| Fig. 10 Interaction diagrams presenting the best synthesized compounds 4, 7a and 9 in the binding site of the CDK2. (A) 2D representation showing H-bonding and bond distance with CDOCKER and CDOCKER interaction energies values and (B) 3D representation where yellow: Lys20, blue: Glu82, red: Leu83, pink: His84, green: Phe82, cyan: Gln131, and salmon: Asp145. | ||
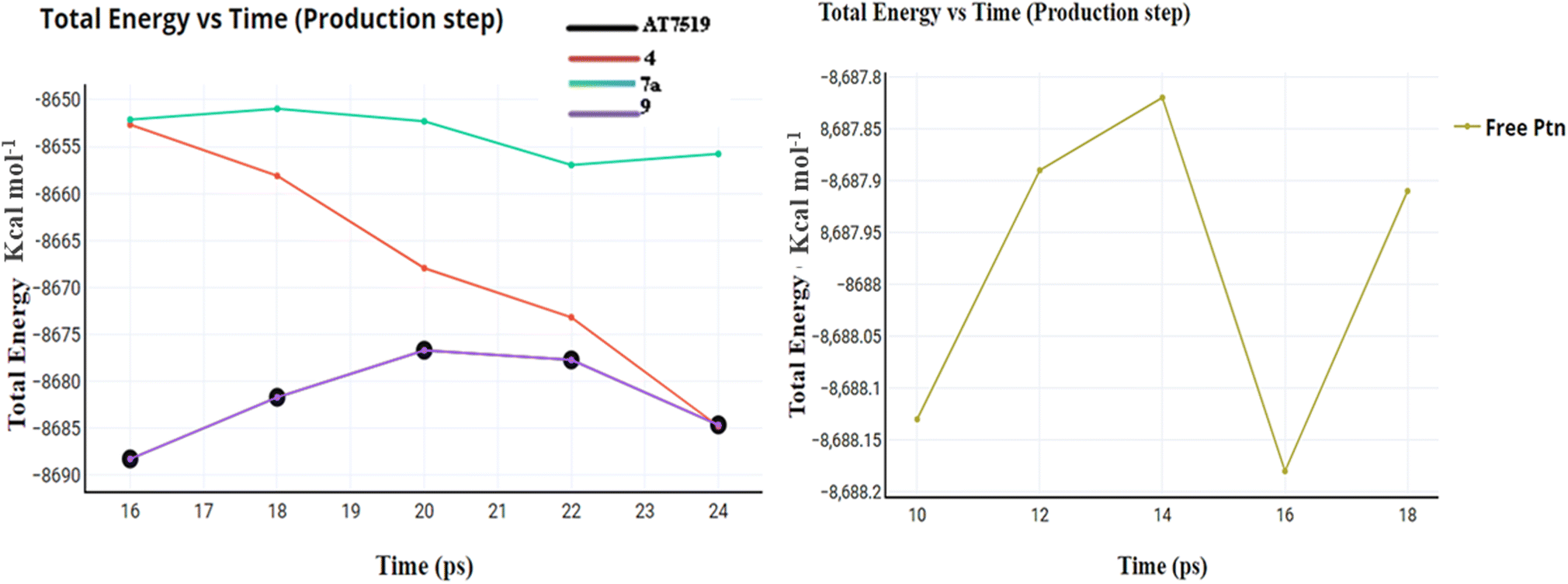 | ||
| Fig. 11 Total energy versus time for compounds AT7519 (I), 4, 7a and 9 and the free protein itself. | ||
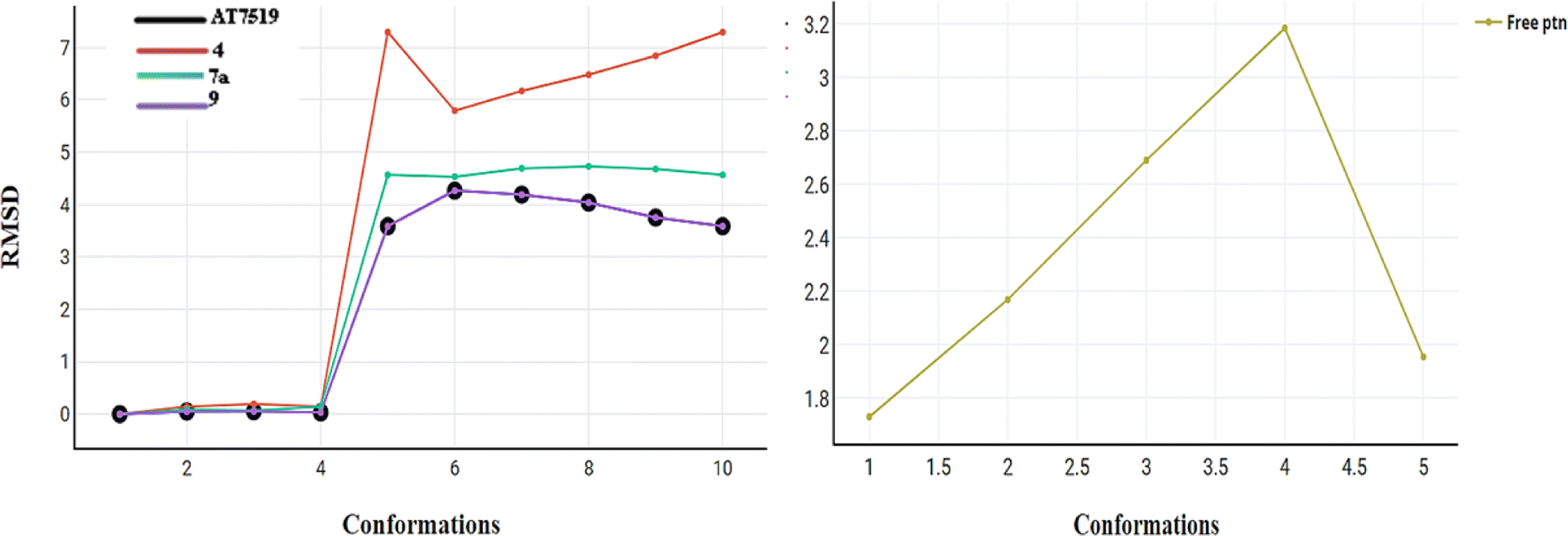 | ||
| Fig. 12 RMSD for compounds AT7519 (I), 4, 7a and 9 and the free protein itself. | ||
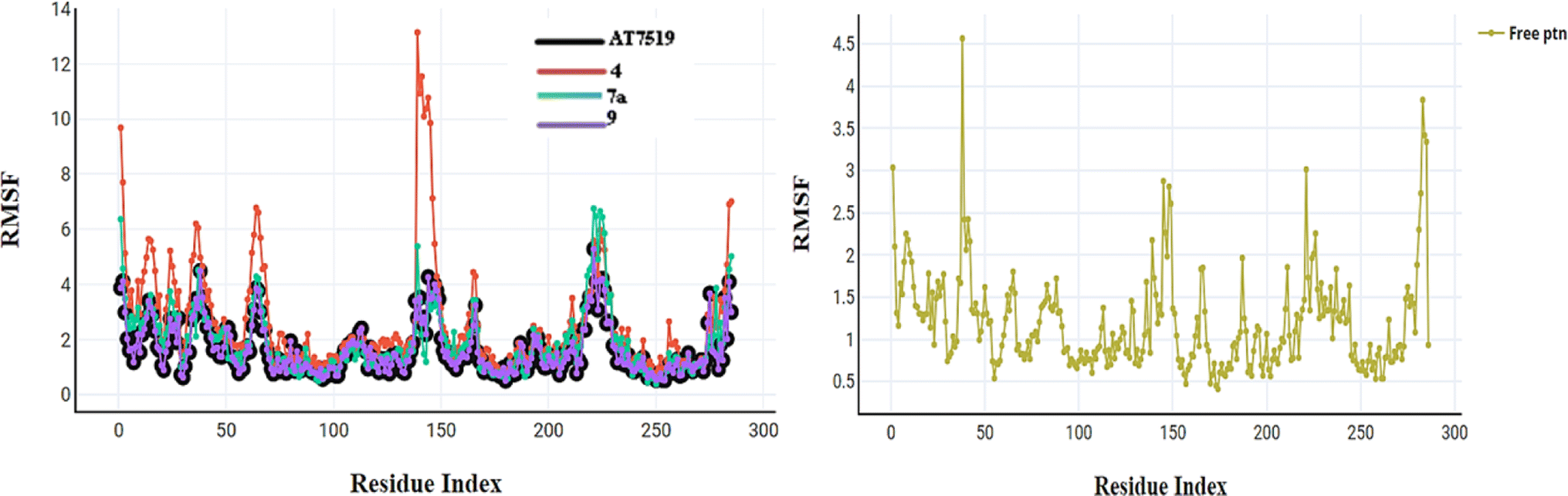 | ||
| Fig. 13 RMSF for compounds AT7519 (I), 4, 7a, and 9 and the free protein itself. | ||
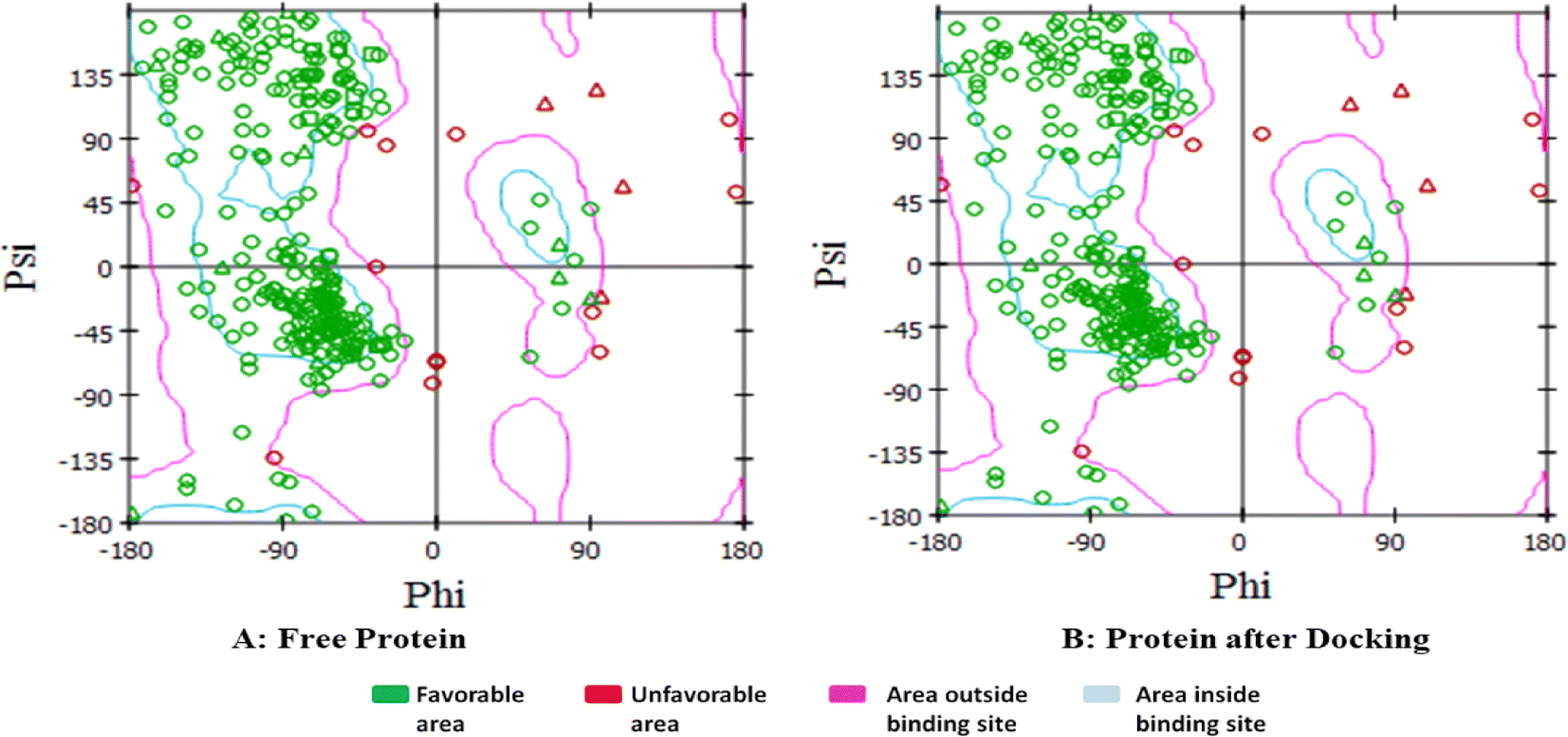 | ||
| Fig. 14 Ramachandran plot presenting the torsional energy conformations for interaction between the newly synthesized molecules and CDK2, where (A) represents the free protein before docking while (B) represents the protein after docking. | ||
2.3.4.1 Pharmacokinetic and drug-likeness prediction. Herein, we used the SwissADME web server (http://www.swissadme.ch/) to predict the drug-likeness and pharmacokinetics parameters of the novel compounds 2a–g, 7a–d, 8a and b, 9 and 10. In this work, we aimed to define the relationship between the chemical structure of our compounds of interest and definite parameters including blood–brain barrier (BBB) permeation, human gastrointestinal absorption (HIA), substrate or non-substrate for the permeability glycoprotein (P-gp), log
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kp and interaction of molecules with cytochrome P450 isomers (CYP). The results are presented using the “BOILED-EGG chart (2D plot between the calculated TPSA and logP properties of the target molecule). Thus, the GIT passive absorption probability, BBB penetration, effluxing by P-gp (PGP+) and no effluxing via P-gp (PGP−) are indicated as white area, yellow region, blue dots, and red dot, respectively.37
kp and interaction of molecules with cytochrome P450 isomers (CYP). The results are presented using the “BOILED-EGG chart (2D plot between the calculated TPSA and logP properties of the target molecule). Thus, the GIT passive absorption probability, BBB penetration, effluxing by P-gp (PGP+) and no effluxing via P-gp (PGP−) are indicated as white area, yellow region, blue dots, and red dot, respectively.37Most of the target compounds are predicted to have no drug–drug interactions upon administration with no activity on cytochrome P450 isomers (CYP3A2 and CYP2D6).38 The BOILED-Egg charts of the target compounds exhibited minimal CNS adverse effects, GIT passive absorption probability and no BBB permeability predicting. In addition, there is no possibility of tumor cell lines resistance through the efflux mechanism to the target compounds given that they may not be substrates for P-gp (PGP−).39 Additionally, the target molecules are expected to have good bioavailability with score = 0.55 according to their five rule-based filters compliance40 including Lipinski,41 Ghose,42 Veber,43 Egan44 and Muegge rules,45 as shown in Fig. 15.
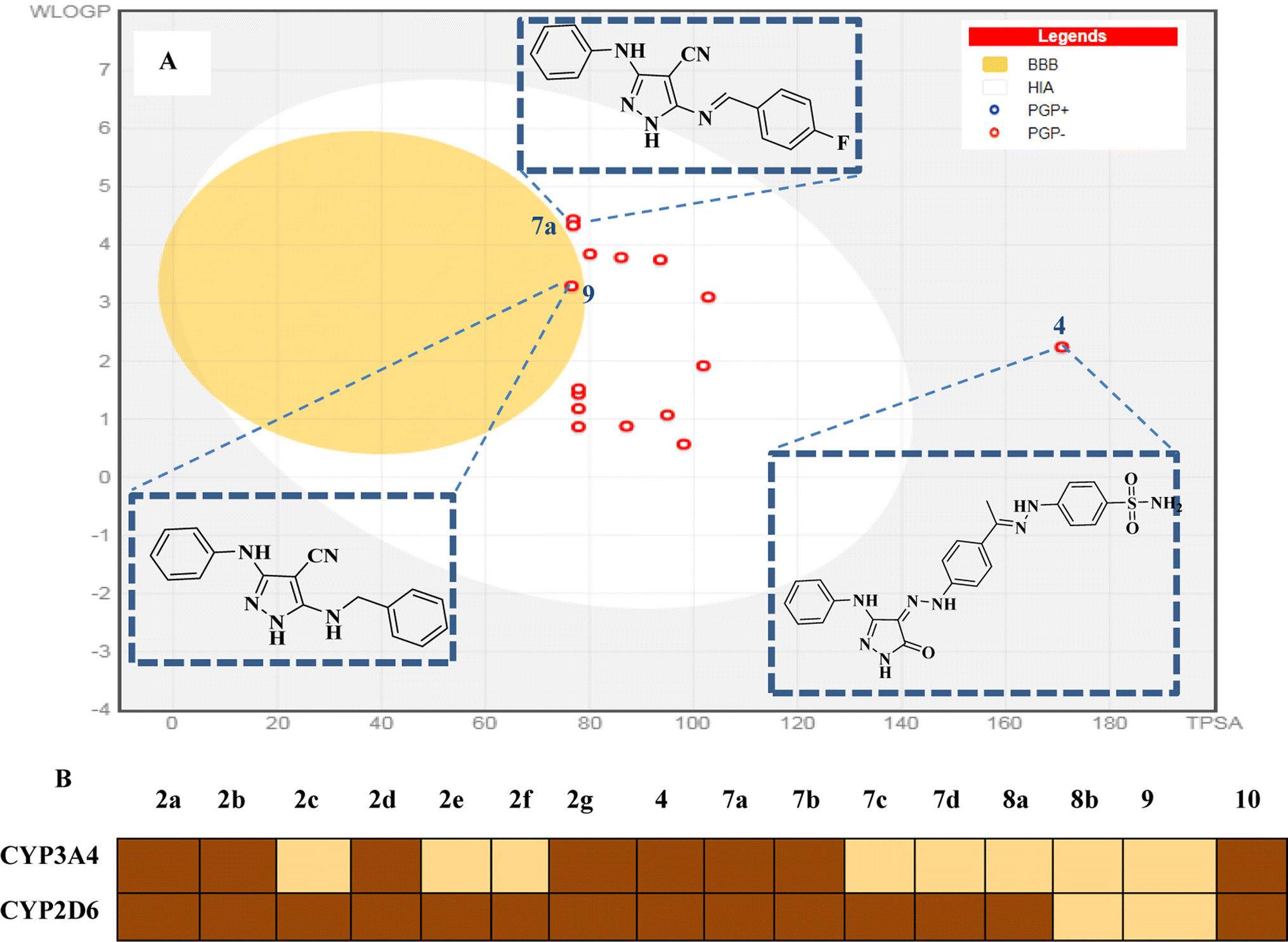 | ||
| Fig. 15 Pharmacokinetic profile. (A) Boiled-egg diagram of all the synthesized compounds. White region: high probability of passive absorption by the gastrointestinal tract; yellow region (yolk): high probability of brain penetration. Yolk and the white region are not mutually exclusive. Bluepoint: P-gp substrate. Redpoint: no P-gp substrate. (B) Inhibitory capacity on the set of CYP450 enzymes. | ||
2.3.4.2 Toxicity prediction. The probable toxicities such as mutagenicity, carcinogenicity, tumorigenicity and teratogenicity were predicted by subjecting our compounds of interest to another virtual filter (Osiris Property Explorer (http://www.organic-chemistry.org/prog/peo/)). This online program compares the target compounds with in vitro and in vivo studied compounds within its database based on their functional group similarity. The color-coded results are red, green and yellow. Red color predicts high probability of toxicity, while yellow indicates moderate toxicity and green color means low toxic potential, as shown in Table 6.46,47 The results exhibited that some of the studied compounds are predicted to be safe and displayed low or no toxicity concerning tumorigenicity, mutagenicity, irritant effect and effect on the reproductive system.
| Comp. | Probable toxicities |
|---|---|
| 2a | High risk for irritant effect |
| High risk of mutagenicity | |
| Moderate toxic effects on the reproductive system | |
| 2b | High risk for irritant effect |
| Moderate toxic effects on the reproductive system | |
| 2c | High risk for irritant effect |
| Moderate toxic effects on the reproductive system | |
| 2d | High risk for irritant effect |
| Moderate toxic effects on the reproductive system | |
| 2e | High risk for irritant effect |
| Moderate toxic effects on the reproductive system | |
| 2f | High risk for irritant effect |
| Moderate tumorigenicity | |
| Moderate toxic effects on the reproductive system | |
| 2g | High risk of mutagenicity |
| High risk for irritant effect | |
| Moderate toxic effects on the reproductive system | |
| 4 | High risk for irritant effect |
| Moderate toxic effects on the reproductive system | |
| 7a | Moderate risk of mutagenicity |
| 7b | Moderate risk of mutagenicity |
| 7c | Moderate risk of mutagenicity |
| 7d | Moderate risk of mutagenicity |
| High tumorigenicity | |
| 8a | Moderate risk of mutagenicity |
| 8b | Moderate risk of mutagenicity |
| 9 | Moderate risk of mutagenicity |
| 10 | Moderate risk of mutagenicity |
| Moderate tumorigenicity |
3. Conclusion
The newly synthesized pyrazole derivatives 2a–g, 7a–d, 8a and b, 9, and 10 were assessed for their CDK2 inhibitory activity, and among them compounds 4, 7a, and 9 were found to be promising CDK2 inhibitors (IC50 of 4.2, 3.98, and 0.97 μM, respectively). All the compounds were subjected to full panel screening for their anticancer activity against 60 cancer cell lines by NCI/USA. Two compounds, 4 and 9, displayed promising growth inhibitory activity with mean GI% of 96.47 and 65.90, respectively. The NCI five dose assay of compound 4 exhibited its remarkable anticancer activity against almost the full panel (GI50 range: 1.1–5.9 μM) and (full panel GI50 (MG-MID) of 3.81 μM). Comparable binding interactions with the co-crystallized ligand AT7519 (I) (PDB code: 2VU3) were noticed for the docked compounds in the CDK2 ATP binding site, especially binding with the Leu83 residue. Finally, the synthesized compounds showed good ADMET properties and toxicity profiles. As a future plan, these proposed compounds will serve as targeted scaffolds that require further optimization to enhance their therapeutic potential, including improving their selectivity, efficacy, and pharmacokinetic properties.4. Experimental
4.1. Chemistry
The starting materials, reagents and solvents were purchased from Sigma-Aldrich (USA) or Alfa-Aesar Organics and used as received without further purification. The reactions were monitored by analytical thin layer chromatography (TLC), which was performed on pre-coated (0.25 mm) silica gel GF254 plates (E. Merck, Germany), and the compounds were detected with 254 nm UV lamp. Silica gel (60–230 mesh) was employed for routine column chromatography separation. Melting points (°C) were determined using the open capillary tube method on a Stuart SMP30 apparatus and are uncorrected. Mass spectroscopy was carried out on a direct inlet part to the mass analyzer in a Thermo Scientific GCMS model ISQ at the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University, Nasr City, Cairo. 1H NMR and 13C NMR spectra were recorded using a Bruker AVANCE III 400 MHz High-performance Digital FT-NMR spectrometer (Bruker Corporation, Germany) at the Microanalytical Unit, Faculty of Pharmacy, Cairo University and Mansoura University. 1H/13C NMR spectra were run at 400/100 MHz, respectively, in DMSO-d6 as the solvent, and chemical shifts are quoted in δ as parts per million (ppm) downfield from tetramethylsilane (TMS) as the internal standard. Infrared spectra were determined using a Shimadzu Fourier-transform infrared spectrometer (IR-470, Shimadzu, Kyoto, Japan). All spectra were expressed as v cm−1. Compounds 1,22 5,25,26 3 (ref. 48) and 6 (ref. 27) were synthesized according to the reported methods.4.1.1.1 5-(Phenylamino)-4-(2-phenylhydrazono)-2,4-dihydro-3H-pyrazol-3-one 2a. Dark-brick red powder; yield (83%); M. P.: 215–217 °C; IR: (v max, cm−1): 3460 (NH), 3150 (NH), 3050 (NH), 1598 (CO); 1H-NMR (400 MHz, DMSO-d6) δ: 6.91 (t, 1H, J = 8, Ar–H), 7.17 (t, 1H, J = 8, Ar–H), 7.29 (t, 2H, J = 6, Ar–H), 7.44 (t, 2H, J = 8, Ar–H), 7.69 (d, 2H, J = 8, Ar–H), 7.76 (d, 2H, J = 8, Ar–H), 8.81 (s, 1H, NH, exchangeable by D2O), 11.10 (s, 1H, NH, exchangeable by D2O), 13.06 (s, 1H, NH, exchangeable by D2O); MS: (M. wt: 279.30): m/z, 279.11 (M+, [100%]); anal. calcd for C15H13N5O; C, 64.51; H, 4.69; N, 25.07; found: C, 64.21; H, 4.75; N, 25.3.
4.1.1.2 4-(2-(4-Fluorophenyl)hydrazono)-5-(phenylamino)-2,4-dihydro-3H-pyrazol-3-one 2b. Dark-brown powder; yield (82%); M. P.: 239–240 °C; IR: (v max, cm−1): 3417 (NH), 3178 (NH), 3163 (NH), 1662 (CO); 1H-NMR (400 MHz, DMSO-d6) δ: 6.92 (t, 1H, J = 6, Ar–H), 7.30 (dd, 4H, J = 6.6, Ar–H), 7.73–7.78 (m, 4H, Ar–H), 8.80 (s, 1H, NH, exchangeable by D2O), 11.08 (s, 1H, NH, exchangeable by D2O), 13.05 (s, 1H, NH, exchangeable by D2O); MS: (M. wt: 297.29): m/z, 297.1 (M+, [100%]); anal. calcd for C15H12FN5O; C, 60.60; H, 4.07; N, 23.56; found: C, 60.43; H, 4.29; N, 23.72.
4.1.1.3 4-(2-(4-Chlorophenyl)hydrazono)-5-(phenylamino)-2,4-dihydro-3H-pyrazol-3-one 2c. Dark-brick red powder; yield (86%); M. P.: 249–251 °C; IR: (v max, cm−1): 3450 (NH), 3250 (NH), 3190 (NH), 1672 (CO); 1H-NMR (400 MHz, DMSO-d6) δ: 6.93 (t, 1H, J = 8, Ar–H), 7.31 (t, 2H, J = 6, Ar–H), 7.49 (d, 2H, J = 12, Ar–H), 7.75 (d, 4H, J = 8, Ar–H), 8.82 (s, 1H, NH, exchangeable by D2O), 11.10 (s, 1H, NH, exchangeable by D2O), 13.00 (s, 1H, NH, exchangeable by D2O); MS: (M. wt: 313.74): m/z, 313.39 (M+, [25.3%]); anal. calcd for C15H12ClN5O; C, 57.42; H, 3.86; N, 17.91; found: C, 57.60; H, 3.97; N, 22.18.
4.1.1.4 4-(2-(4-Hydroxyphenyl)hydrazono)-5-(phenylamino)-2,4-dihydro-3H-pyrazol-3-one 2d. Dark-red powder; yield (83%); M. P.: 257–259 °C; IR: (v max, cm−1): 3479 (OH), 3417 (NH), 3259 (NH), 3147 (NH), 1666 (CO); 1H-NMR (400 MHz, DMSO-d6) δ: 6.83 (d, 2H, J = 8, Ar–H), 6.89 (t, 1H, J = 6, Ar–H), 7.28 (t, 2H, J = 8, Ar–H), 7.52 (d, 2H, J = 12, Ar–H), 7.73 (d, 2H, J = 8, Ar–H), 8.71 (s, 2H, NH, exchangeable by D2O), 9.59 (s, 1H, OH, exchangeable by D2O), 11.00 (s, 1H, NH, exchangeable by D2O); MS: (M. wt: 295.30): m/z, 295.88 (M+, [104.6%]); anal. calcd for C15H13N5O2; C, 61.01; H, 4.44; N, 23.72; found: C, 60.89; H, 4.67; N, 23.9.
4.1.1.5 5-(Phenylamino)-4-(2-(p-tolyl)hydrazono)-2,4-dihydro-3H-pyrazol-3-one 2e. Reddish-brown powder; yield (86%); M. P.: 251–253 °C; IR: (v max, cm−1): 3302 (NH), 3151 (NH), 3024 (NH), 1670 (CO);1H-NMR (400 MHz, DMSO-d6) δ: 2.31 (s, 3H, CH3), 6.89 (d, 1H, J = 3.86, Ar–H), 7.23–7.30 (m, 3H, Ar–H), 7.49 (d, 1H, J = 7.86, Ar–H), 7.58 (d, 1H, J = 8, Ar–H), 7.75 (d, 1H, J = 8, Ar–H), 8.78 (s, 1H, Ar–H), 9.17 (s, 1H, Ar–H), 10.37 (s, 1H, NH, exchangeable by D2O), 11.07 (s, 1H, NH, exchangeable by D2O), 13.08 (s, 1H, NH, exchangeable by D2O); MS: (M. wt: 293.32): m/z, 293.13 (M+, [100%]); anal. calcd for C16H15N5O; C, 65.52; H, 5.15; N, 23.88; found: C, 65.7; H, 5.34; N, 24.09.
4.1.1.6 4-(2-(4-Methoxyphenyl)hydrazono)-5-(phenylamino)-2,4-dihydro-3H-pyrazol-3-one 2f. Dark-brick red powder; yield (86%); M. P.: 240–242 °C; IR: (v max, cm−1): 3302 (NH), 3143 (NH), 3055 (NH), 1666 (CO); 1H-NMR (400 MHz, DMSO-d6) δ: 3.78 (s, 3H, OCH3), 6.90 (t, 1H, J = 8, Ar–H), 7.02 (d, 2H, J = 8, Ar–H), 7.28 (t, 2H, J = 6, Ar–H), 7.65 (d, 2H, J = 8, Ar–H), 7.75 (d, 2H, J = 8, Ar–H), 8.75 (s, 1H, NH, exchangeable by D2O), 11.03 (s, 2H, NH, exchangeable by D2O); 13C-NMR (100 MHz, DMSO-d6) δ: 55.86, 115.12, 115.45, 117.53, 117.61, 117.91, 118.15, 120.93, 121.69, 129.1, 129.35, 135.76, 141.33, 146.22, 157.25, 158.71; MS: (M. wt:309.32): m/z, 309.12 (M+, [100%]); anal. calcd for C16H15N5O2; C, 65.3; H, 4.79; N, 19.04; found: C, 62.35; H, 5.01; N, 22.87.
4.1.1.7 4-(2-(4-Acetylphenyl)hydrazono)-5-(phenylamino)-2,4-dihydro-3H-pyrazol-3-one 2g. Dark-reddish brown powder; yield (85%); M. P.: 265–267 °C; IR: (v max, cm−1): 3363 (NH), 3163 (NH), 3055 (NH), 1666 (CO), 1566 (CO);1H-NMR (400 MHz, DMSO-d6) δ: 2.58 (s, 3H, Ar–COCH3), 6.91 (t, 1H, J = 6, Ar–H), 7.17 (t, 2H, J = 8, Ar–H), 7.29 (t, 2H, J = 6, Ar–H), 7.44 (t, 2H, J = 8, Ar–H), 7.69 (d, 1H, J = 8, Ar–H), 7.76 (d, 1H, J = 8, Ar–H), 8.91 (s, 1H, NH, exchangeable by D2O), 11.18 (s, 1H, NH, exchangeable by D2O), 13.06 (s, 1H, NH, exchangeable by D2O);13C-NMR (100 MHz, DMSO-d6) δ: 27.06, 115.63, 115.82, 118.15, 118.65, 121.31, 125.4, 125.82, 129.17, 129.5, 130.41, 133.02, 141.01, 146.05, 146.14, 158.15, 197; MS: (M. wt: 321.33): m/z, 321.72 (M+, [21.8%]); anal. calcd for C17H15N5O2; C, 63.54; H, 4.71; N, 21.79; found: C, 63.72; H, 4.89; N, 22.04.
Dark-brick red powder; yield (86%); M. P.: 249–251 °C; IR: (v max, cm−1): 3420 (NH), 3320 (NH), 3190 (NH), 1663 (CO), 1177 and 1338 (SO2); 1H-NMR (400 MHz, DMSO-d6) δ: 4.39 (t, 1H, J = 4.8, CH3), 6.93 (t, 1H, Ar–H), 6.94 (s, 2H, NH2, exchangeable by D2O), 7.30 (d, 2H, J = 8.4, Ar–H), 7.34 (d, 2H, J = 5.6, Ar–H), 7.69 (d, 2H, J = 8.8, Ar–H), 7.73 (d, 2H, J = 8.8, Ar–H), 7.77 (d, 2H, J = 8, Ar–H), 7.90 (d, 2H, J = 8.8, Ar–H), 8.84 (s, 1H, NH, exchangeable by D2O), 9.77 (s, 1H, NH, exchangeable by D2O), 11.12 (s, 1H, NH, exchangeable by D2O), 13.14 (s, 1H, NH, exchangeable by D2O); 13C-NMR (100 MHz, DMSO-d6) δ:13.56, 112.45, 115.97, 118.05, 121.13, 123.48, 127.03, 127.75, 129.15, 134.03, 135.58, 141.21, 141.98, 143.19, 146.15, 149.00, 158.54; MS: (M. wt: 490.54): m/z, 490.13 (M+, [50%]); anal. calcd for C23H22N8O3S; C, 56.31; H, 4.52; N, 22.84; S, 6.54; found: C, 56.47; H, 4.65; N, 23.19; S, 6.62.
4.1.3.1 5-((4-Fluorobenzylidene)amino)-3-(phenylamino)-1H-pyrazole-4-carbonitrile 7a. Dark-yellow powder; yield (81%); M. P.: 265–267 °C; IR: (v max, cm−1): 3317 (NH), 3190 (NH), 2222 (CN); 1H-NMR (400 MHz, DMSO-d6) δ: 6.88–6.90 (m, 1H, Ar–H), 7.27 (t, 3H, J = 8, Ar–H), 7.43 (t, 3H, J = 8, Ar–H), 8.06–8.09 (m, 2H, Ar–H), 9.00 (s, 1H, imine-CH), 13.13 (s, 2H, NH, exchangeable by D2O); 13C-NMR (75 MHz, DMSO-d6) δ:114.66, 116.73, 116.95, 117.1, 120.96, 129.37, 132.24, 132.33, 142.24, 164.05, 166.55, 217.59; MS: (M. wt: 305.31): m/z, 305.11 (M+, [100%]); anal. calcd for C17H12FN5; C, 66.88; H, 3.96; N, 22.94; found: C, 66.71; H, 4.15; N, 23.17.
4.1.3.2 5-((4-Chlorobenzylidene)amino)-3-(phenylamino)-1H-pyrazole-4-carbonitrile 7b. Orange powder; yield (80%); M. P.: 273–275 °C; IR: (v max, cm−1): 3344 (NH), 3190 (NH), 2218 (CN); 1H-NMR (400 MHz, DMSO-d6) δ: 6.89–6.91 (m, 1H, Ar–H), 7.27 (t, 3H, J = 6, Ar–H), 7.67 (d, 2H, J = 8, Ar–H), 8.02 (d, 2H, J = 8, Ar–H), 9.01 (s, 2H, imine-CH + Ar–H), 13.18 (s, 2H, NH, exchangeable by D2O); 13C-NMR (100 MHz, DMSO-d6) δ: 114.63, 117.12, 129.4, 129.81, 131.34, 131.68, 134.24; MS: (M. wt: 321.76): m/z, 321.08 (M+, [100%]), 323.08 (M+2, [33.9%]); anal. calcd for C17H12ClN5; C, 63.46; H, 3.76; N, 21.77; found: C, 63.19; H, 3.84; N, 21.98.
4.1.3.3 5-((4-Methoxybenzylidene)amino)-3-(phenylamino)-1H-pyrazole-4-carbonitrile 7c. Yellow powder; yield (85%); M. P.: 233–235 °C; IR: (v max, cm−1): 3332 (NH), 3190 (NH), 2214 (CN); 1H-NMR (400 MHz, DMSO-d6) δ: 3.88 (s, 3H, OCH3), 6.86–6.88 (m, 1H, Ar–H), 7.14 (d, 3H, J = 8, Ar–H), 7.26 (t, 2H, J = 8, Ar–H), 7.49 (s, 1H, NH, exchangeable by D2O), 7.95 (d, 2H, J = 8, Ar–H), 8.91 (s, 2H, imine-CH + Ar–H), 13.05 (s, 1H, NH, exchangeable by D2O); 13C-NMR (100 MHz, DMSO-d6) δ: 56.13, 114.94, 115.14, 115.73, 116.52, 116.98, 120.78, 127.97, 129.04, 129.37, 130.01, 131.9, 132.37, 142.24, 143.02, 163.66, 164.71, 192.02; MS: (M. wt: 317.34): m/z, 317.03 (M+, [11.3%]); anal. calcd for C18H15N5O; C, 68.13; H, 4.76; N, 22.07; found: C, 68.4; H, 4.91; N, 22.33.
4.1.3.4 5-((4-(Dimethylamino)benzylidene)amino)-3-(phenylamino)-1H-pyrazole-4-carbonitrile 7d. Pale yellow powder; yield (83%); M. P.: 284–285 °C; IR: (v max, cm−1): 3317 (NH), 3205 (NH), 2214 (CN); 1H-NMR (400 MHz, DMSO-d6) δ: 3.07 (s, 6H, 2CH3), 6.83 (d, 3H, J = 8, Ar–H), 7.23–7.25 (m, 2H, Ar–H, NH, exchangeable by D2O), 7.51–7.52 (m, 2H, Ar–H), 7.78 (d, 2H, J = 8, Ar–H), 8.71–8.75 (m, 1H, imine-CH + Ar–H), 12.85 (s, 1H, NH, exchangeable by D2O); 13C-NMR (100 MHz, DMSO-d6) δ: 70.42, 111.54, 112.04, 115.33, 116.57, 116.76, 120.00, 122.33, 128.93, 129.08, 131.88, 142.82, 152.93, 154.01, 154.97, 164.71; MS: (M. wt: 330.39): m/z, 330.16 (M+, [100%]); anal. calcd for C19H18N6; C, 69.07; H, 5.49; N, 25.44; found: C, 68.79; H, 5.62; N, 25.63.
4.1.4.1 4-Chloro-N-(4-cyano-3-(phenylamino)-1H-pyrazol-5-yl)benzamide 8a. Faint-yellow powder; yield (84%); M. P.: 169–171 °C; IR: (v max, cm−1): 3429 (NH), 3309 (NH), 3244 (NH), 2214 (CN), 1689 (CO); 1H-NMR (400 MHz, DMSO-d6) δ: 6.88 (t, 1H, J = 6, Ar–H), 7.22 (t, 2H, J = 8, Ar–H), 7.54 (d, 2H, J = 8, Ar–H), 7.65 (d, 2H, J = 8, Ar–H), 8.06 (d, 2H, J = 8, Ar–H), 8.19 (s, 2H, NH, exchangeable by D2O), 8.98 (s, 1H, NH, exchangeable by D2O); MS: (M. wt: 337.76): m/z, 337.07(M+, [100%]), 339.07 (M2+, [33.9%]); anal. calcd for C17H12ClN5O; C, 60.45; H, 3.58; N, 20.73; found: C, 60.74; H, 3.8; N, 20.99.
4.1.4.2 N-(4-Cyano-3-(phenylamino)-1H-pyrazol-5-yl)-4-methoxybenzamide 8b. Yellow powder; yield (85%); M. P.: 233–235 °C; IR: (v max, cm−1): 3390 (NH), 3302 (NH), 3244 (NH), 2214 (CN), 1666 (CO); 1H-NMR (400 MHz, DMSO-d6) δ: 3.89 (s, 3H, OCH3), 6.89 (t, 1H, J = 8, Ar–H), 7.10 (d, 2H, J = 8, Ar–H), 7.24 (d, 2H, J = 8, Ar–H), 7.59 (d, 2H, J = 8, Ar–H), 8.12–8.15 (m, 4H, Ar–H + 2NH, exchangeable by D2O), 8.96 (s, 1H, NH, exchangeable by D2O); 13C-NMR (100 MHz, DMSO-d6) δ: 56.06, 56.14, 65.06, 113.66, 113.86, 114.24, 116.57, 118, 121.22, 123.62, 124.95, 129.08, 129.43, 133.61, 134.13, 134.23, 141.45, 151.38, 156.73, 163.24, 168.47; MS: (M. wt: 333.34): m/z, 333.03 (M+, [68.9%]); anal. calcd for C18H15N5O2; C, 64.86; H, 4.54; N, 21.01; found: C, 64.7; H, 4.72; N, 21.25.
Dark-off white powder; yield (85%); M. P.: 136–137 °C; IR: (v max, cm−1): 3282 (NH), 3186 (NH), 3028 (NH), 2210 (CN); 1H-NMR (400 MHz, DMSO-d6) δ: 4.52 (s, 2H, CH2), 5.10 (s, 1H, NH, exchangeable by D2O), 6.84–6.90 (m, 2H, Ar–H), 7.05 (d, 1H, J = 8, Ar–H), 7.22–7.33 (m, 8H; 7Ar–H, 1H, NH, exchangeable by D2O), 8.75 (s, 1H, NH, exchangeable by D2O); 13C-NMR (100 MHz, DMSO-d6) δ: 51.2, 52.51, 71.29, 115.26, 116.54, 121.23, 127.47, 127.52, 127.84, 128.25, 128.81, 128.86, 129.67, 137.01, 138.48, 142.65, 147.15, 156.57, 217.59; MS: (M. wt: 289.33): m/z, 289.69 (M+, [106.6%]); anal. calcd for C17H15N5; C, 70.57; H, 5.23; N, 24.21; found: C, 70.68; H, 5.40; N, 24.39.
Off-white powder; yield (87%); M. P.: 173–175 °C; IR: (v max, cm−1): 3400 (NH), 3310 (NH), 2229 (CN), 1585 (CO), 1558 (CO); 1H-NMR (400 MHz, DMSO-d6) δ: 2.19 (s, 3H, CH3), 2.62 (s, 3H, CH3), 6.89–6.97 (m, 1H, Ar–H), 7.25–7.32 (m, 1H, Ar–H), 7.67 (d, 1H, J = 8, Ar–H), 7.99 (s, 1H, Ar–H), 8.92 (s, 1H, Ar–H), 9.12 (s, 1H, NH, exchangeable by D2O); 10.80 (s, 1H, NH, exchangeable by D2O); 13C-NMR (100 MHz, DMSO-d6) δ: 23.7, 24.1, 65.07, 115.26, 116.54, 121.23, 127.47, 127.52, 127.84, 128.25, 128.81, 128.86, 129.67, 137.01, 138.48, 142.65, 147.15, 156.57, 217.59; MS: (M. wt: 283.29): m/z, 283.93 (M+, [21.4%]); anal. calcd for C14H13N5O2; C, 59.36; H, 4.63; N, 24.72; found: C, 59.54; H, 4.75; N, 24.98.
4.2. Biological assays
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
All authors are appreciative of the efforts by the National Cancer Institute (NCI) Developmental Therapeutic Program (http://www.dtp.nci.nih.gov) for the anti-proliferative activity screening of the targeted compounds against 60 cancer cell lines.References
- H. K. Matthews, C. Bertoli and R. A. M. de Bruin, Cell cycle control in cancer, Nat. Rev. Mol. Cell Biol., 2022, 23, 74–88 Search PubMed.
- T. Otto and P. Sicinski, Cell cycle proteins as promising targets in cancer therapy, Nat. Rev. Cancer, 2017, 7, 93–115 CrossRef PubMed.
- U. Asghar, A. K. Witkiewicz, N. C. Turner and E. S. Knudsen, The history and future of targeting cyclin-dependent kinases in cancer therapy, Nat. Rev. Drug Discovery, 2015, 14, 130–146 CrossRef CAS PubMed.
- N. Chunder, L. Wang, C. Chen, W. W. Hancock and A. D. Wells, Cyclin-dependent kinase 2 controls peripheral immune tolerance, J. Immunol., 2012, 189, 5659–5666 CrossRef CAS PubMed.
- P. Saurus, S. Kuusela, V. Dumont, E. Lehtonen, C. L. Fogarty, M. I. Lassenius, C. Forsblom, M. Lehto, M. A. Saleem, P. H. Groop and S. Lehtonen, Cyclin-dependent kinase 2 protects podocytes from apoptosis, Sci. Rep., 2016, 6, 21664 CrossRef CAS PubMed.
- F. Granes, M. B. Roig, H. J. Brady and G. Gil-Gomez, Cdk2 activation acts upstream of the mitochondrion during glucocorticoid induced thymocyte apoptosis, Eur. J. Immunol., 2004, 34, 2781–2790 CrossRef CAS PubMed.
- A. J. Deans, K. K. Khanna, C. J. McNees, C. Mercurio, J. Heierhorst and G. A. McArthur, Cyclin-dependent kinase 2 functions in normal DNA repair and is a therapeutic target in BRCA1-deficient cancers, Cancer Res., 2006, 66, 8219–8226 Search PubMed.
- X. F. Yin, J. Yu, Y. Zhou, C. Y. Wang, Z. M. Jiao, Z. N. Qian, H. Sun and B. H. Chen, Identification of CDK2 as a novel target in treatment of prostate cancer, Future Oncol., 2018, 14, 709–718 Search PubMed.
- P. Marion, P. Camille, P. Morgan and C. M. May, Targeting Cyclin-Dependent Kinases in Human Cancers: From Small Molecules to Peptide Inhibitors, Cancers, 2015, 7, 179–237 CrossRef.
- A. M. Shaker, M. I. Shahin, A. M. AboulMagd, H. M. Abdel-Rahman and D. A. Abou El Ella, Design, synthesis, and molecular docking of novel 1, 3, 4-triaryl pyrazole derivatives bearing methylsulfonyl moiety with anticancer activity through dual targeting CDK2 and COX-2 enzymes, J. Mol. Struct., 2024, 1301, 137323 CrossRef CAS.
- B. K. Chagaleti, B. S. Kumar, R. Rajagopal, A. Alfarhan, J. Arockiaraj, K. M. Kumaradoss and S. K. R. Namasivayam, Targeting cyclin-dependent kinase 2 CDK2: Insights from Molecular Docking and Dynamics Simulation-A systematic computational approach to discover novel cancer therapeutics, Comput. Biol. Chem., 2024, 108134 CrossRef CAS.
- M. I. El-Gamal, S. O. Zaraei, M. M. Madkour and H. S. Anbar, Evaluation of substituted pyrazole-based kinase inhibitors in one decade (2011–2020): Current status and future prospects, Molecules, 2022, 27(1), 330 CrossRef CAS PubMed.
- S. D. Packiapalavesam, V. Saravanan, A. A. Mahajan, M. H. Almutairi, B. O. Almutairi, J. Arockiaraj and S. K. R. Namasivayam, Identification of novel CA IX inhibitor: Pharmacophore modeling, docking, DFT, and dynamic simulation, Comput. Biol. Chem., 2024, 110, 108073 CrossRef CAS PubMed.
- S. I. Elewa, E. Mansour, I. F. Nassar and A. A. I. Mekawey, Synthesis of some new pyrazoline-based thiazole derivatives and evaluation of their antimicrobial, antifungal, and anticancer activities, Russ. J. Bioorg. Chem., 2020, 46, 382–392 CrossRef.
- E. Eggert, R. C. Hillig, S. Koehr, D. Stockigt, J. Weiske, N. Barak, J. Mowat, T. Brumby, C. D. Christ, A. Ter Laak, T. Lang, A. E. Fernandez-Montalvan, V. Badock, H. Weinmann, I. V. Hartung, D. Barsyte-Lovejoy, M. Szewczyk, S. Kennedy, F. Li, M. Vedadi, P. J. Brown, V. Santhakumar, C. H. Arrowsmith, T. Stellfeld and C. Stresemann, Discovery and characterization of a highly potent and selective aminopyrazoline-based in vivo probe (BAY-598) for the protein lysine methyltransferase SMYD2, J. Med. Chem., 2016, 59, 4578–4600 CrossRef CAS PubMed.
- V. K. Mishra, M. Mishra, V. Kashaw and S. K. Kashaw, Synthesis of 1,3,5-trisubstituted pyrazolines as potential antimalarial and antimicrobial agents, Bioorg. Med. Chem., 2017, 25, 1949–1962 CrossRef CAS PubMed.
- M. A. Abdel-Sayed, S. M. Bayomi, M. A. El-Sherbeny, N. I. Abdel-Aziz, K. E. ElTahir, G. S. Shehatou and A. A. Abdel-Aziz, Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibition activities and molecular docking study of pyrazoline derivatives, Bioorg. Med. Chem., 2016, 24, 2032–2042 CrossRef CAS PubMed.
- P. G. Wyatt, A. J. Woodhead, V. Berdini, J. A. Boulstridge, M. G. Carr, D. M. Cross, D. J. Davis, L. A. Devine, T. R. Early, R. E. Feltell, E. J. Lewis, R. L. McMenamin, E. F. Navarro, M. A. O'Brien, M. O'Reilly, M. Reule, G. Saxty, L. C. A. Seavers, D.-M. Smith, M. S. Squires, G. Trewartha, M. T. Walker and A. J.-A. Woolford, Identification of N-(4-Piperidinyl)-4-(2,6dichlorobenzoylamino)-1H-pyrazole-3-carboxamide (AT7519), a Novel Cyclin Dependent Kinase Inhibitor Using Fragment-Based X-Ray Crystallography and Structure Based Drug Design, J. Med. Chem., 2008, 51, 4986–4999 CrossRef CAS.
- M. Anderson, D. M. Andrews, A. J. Barker, C. A. Brassington, J. Breed, K. F. Byth, J. D. Culshaw, M. R. V. Finlay, E. Fisher, H. H. J. McMiken, C. P. Green, D. W. Heaton, I. A. Nash, N. J. Newcombe, S. E. Oakes, R. A. Pauptit, A. Roberts, J. J. Stanway, A. P. Thomas, J. A. Tucker, M. Walker and H. M. Weir, Imidazoles: SAR and development of a potent class of cyclin-dependent kinase inhibitors, Bioorg. Med. Chem. Lett., 2008, 18, 5487–5549 CrossRef CAS PubMed.
- C. Effects and M. Paprska, 4-Arylazo-3, 5-diamino-1 H -pyrazole CDK Inhibitors: SAR Study, Crystal Structure in Complex, J. Med. Chem., 2006, 49, 6500–6509 CrossRef.
- T. Lin, J. Li, L. Liu, Y. Li, H. Jiang, K. Chen, P. Xu, C. Luo and B. Zhou, Design, synthesis, and biological evaluation of 4-benzoylamino-1Hpyrazole-3-carboxamide derivatives as potent CDK2 inhibitors, Eur. J. Med. Chem., 2021, 215, 113281 CrossRef CAS PubMed.
- H. D. P. Weissberger, Investigation of Pyrazole Compounds III: The Condensation of α Carbethoxyacetothioacetanilide with Hydrazines, J. Am. Chem. Soc., 1943, 65, 732–734 CrossRef.
- S. M. Abou-seri, W. M. Eldehna, M. M. Ali, D. A. Abou and E. Ella, Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: Synthesis and in vitro biological evaluation, Eur. J. Med. Chem., 2016, 215, 165–179 CrossRef.
- E. Z. Mohammed, W. R. Mahmoud, R. F. George, G. S. Hassan, F. A. Omar and H. H. Georgey, Synthesis, in vitro anticancer activity and in silico studies of certain pyrazole-based derivatives as potential inhibitors of cyclin dependent kinases (CDKs), Bioorg. Chem., 2021, 116, 105347 CrossRef CAS PubMed.
- S. Baumli, G. Lolli, E. D. Lowe, S. Troiani and L. A. N. Rusconi, The Structure of P-TEFb (CDK9/Cyclin T1), its Complex with Flavopiridol and Regulation by Phosphorylation, EMBO J., 2008, 27, 1907–1918 CrossRef CAS PubMed.
- S. K. Hanks and T. Hunter, Protein Kinases 6. The Eukaryotic Protein Kinase Superfamily: Kinase (Catalytic) Domain Structure and Classification, FASEB J., 1995, 9, 576–596 CrossRef CAS.
- H. L. De Bondt, J. Rosenblatt, J. Jancarik, H. D. Jones, D. O. Morgan and D. O. Kim, Crystal Structure of Cyclin-Dependent Kinase 2, Nature, 1993, 363, 595–602 CrossRef CAS PubMed.
- S. Baumli, A. J. Hole, L. Z. Wang, M. E. Noble and J. A. Endicott, The CDK9 Tail Determines the Reaction Pathway of Positive Transcription Elongation Factor B, Structure, 2012, 20, 1788–1795 CrossRef CAS PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Targeting cancer with small molecule kinase inhibitors, Nat. Rev, Cancer, 2009, 9, 28–39 CAS.
- S. Baumli, G. Lolli, E. D. Lowe, S. Troiani and L. A. N. Rusconi, The Structure of P-TEFb (CDK9/Cyclin T1), its Complex with Flavopiridol and Regulation by Phosphorylation, EMBO J., 2008, 27, 1907–1918 CrossRef CAS.
- K. M. Amin, M. M. Hanna, H. E. Abo-Youssef and R. F. George, Synthesis, analgesic and anti-inflammatory activities evaluation of some bi-, tri- and tetracyclic condensed pyrimidines, Eur. J. Med. Chem., 2009, 44, 4572–4584 CrossRef CAS.
- S.-H. Lu, P.-L. Liu and F. F. Wong, Vilsmeier reagent-mediated synthesis of 6- [(formyloxy)methyl]-pyrazolopyrimidines via a one-pot multiple tandem reaction, RSC Adv., 2015, 5, 47098–47107 RSC.
- J. Liu, H. Huang, X. Deng, R. Xiong, X. Cao, G. Tang, X. Wu, S. Xu and J. Peng, Design, synthesis and broad-spectrum Bcr-Abl inhibitory activity of novel thiazolamide–benzamide derivatives, RSC Adv., 2019, 9, 2092–2101 RSC.
- S. M. Abou-seri, W. M. Eldehna, M. M. Ali, D. A. Abou and E. Ella, 1Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: Synthesis and in vitro biological evaluation, Eur. J. Med. Chem., 2016, 107, 165–179 CrossRef CAS PubMed.
- E. M. Azmy, M. Hagras, M. A. Ewida, A. S. Doghish, E. G. Khidr, A. A. El-Husseiny, M. H. Gomaa, H. M. Refaat, N. S. Ismail, I. F. Nassar and W. H. Lashin, Development of pyrolo [2, 3-c] pyrazole, pyrolo [2, 3-d] pyrimidine and their bioisosteres as novel CDK2 inhibitors with potent in vitro apoptotic anti-proliferative activity: Synthesis, biological evaluation and molecular dynamics investigations, Bioorg. Med. Chem., 2023, 139, 106729 CrossRef CAS PubMed.
- J. Zhang, Y. Gan, H. Li, J. Yin, X. He, L. Lin, S. Xu, Z. Fang, B. W. Kim, L. Gao and L. Ding, Inhibition of the CDK2 and Cyclin A complex leads to autophagic degradation of CDK2 in cancer cells, Nat. Commun., 2022, 13, 2835 CrossRef CAS PubMed.
- A. Daina and V. Zoete, A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules, ChemMedChem, 2016, 11, 1117–1121 CrossRef CAS PubMed.
- T. Lynch and A. Price, The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects, Am. Fam. Physician, 2007, 76, 391–396 Search PubMed.
- M. C. O. Campos, D. B. Castro-Pinto, G. A. Ribeiro, M. M. Berredo-Pinho, L. H. F. Gomes, M. S. da Silva Bellieny, C. M. Goulart, A. Echevarria and L. L. Leon, P-glycoprotein efflux pump plays an important role in Trypanosoma cruzi drug resistance, Parasitol. Res., 2013, 112, 2341–2351 CrossRef.
- A. Daina, O. Michielin and V. Zoete, SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 17, 1–13 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. A Qualitative and Quantitative Characterization of Known Drug Databases, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS.
- W. J. Egan, K. M. J. Merz and J. J. Baldwin, Prediction of drug absorption using multivariate statistics, J. Med. Chem., 2000, 43, 3867–3877 CrossRef CAS.
- I. Muegge, S. L. Heald and D. Brittelli, Simple Selection Criteria for Drug-like Chemical Matter, J. Med. Chem., 2001, 44, 1841–1846 CrossRef CAS.
- T. Sander, Actelion's property explorer, Pharm. Ltd, Gewerbestrasse, 2001, vol. 16, p. 4123 Search PubMed.
- T. Sander, J. Freyss, M. von Korff, J. R. Reich and C. Rufener, OSIRIS, an entirely in-house developed drug discovery informatics system, J. Chem. Inf. Model., 2009, 49, 232–246 CrossRef CAS PubMed.
- K. S. Abdelrahman, H. A. Hassan, S. A. Abdel-Aziz, A. A. Marzouk, R. shams, M. Tajiri, M. A. Aziz and H. Konno, Design, Synthesis, Molecular Docking and Biological Evaluation of Novel 1,5- Diarylpyrazole-N,O-Dimethyl Hydroxamate Derivatives as Antiproliferative agents, J. Adv. Biomedical Pharm. Sci., 2021, 4, 214–225 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06500j |
| This journal is © The Royal Society of Chemistry 2024 |