
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical investigation of ozone adsorption on metal free element doped boron nitride monolayers
Zara Wang and
Xuan Luo*
and
Xuan Luo*
National Graphene Research and Development Center, Springfield, Virginia 22151, USA. E-mail: xluo@ngrd.org
First published on 15th November 2024
Abstract
Ozone in the troposphere poses significant environmental and health risks, contributing to global warming and being linked to respiratory diseases, making it critical to find effective methods to remove ozone from the atmosphere. This study investigates the adsorption of ozone on boron nitride (BN) monolayers doped with metal-free elements, specifically carbon, silicon, oxygen, and phosphorus, using first-principles calculations based on Density Functional Theory (DFT). Our results showed that ozone adsorbed on boron nitride doped with carbon exhibited physisorption and had an adsorption energy of −0.272 eV. Ozone adsorbed on silicon-doped boron nitride dissociated into an oxygen atom and an O2 molecule and showed chemisorption with an energy of −8.074 eV. Notably, phosphorus-doped boron nitride dissociated ozone, leading to the release of O2 and bonding of a single oxygen atom to the boron nitride monolayer. These findings highlight how carbon and silicon-doped boron nitride can be useful in removing ozone from the troposphere and the potential of phosphorus-doped boron nitride to adsorb ozone and release a much safer O2 molecule.
I. Introduction
Global warming is one of the most pressing challenges facing humanity today, characterized by the long-term increase in Earth's average surface temperature due to human activities, particularly the burning of fossil fuels, deforestation, and industrial processes.1 This phenomenon is primarily driven by the accumulation of greenhouse gasses in the atmosphere, including carbon dioxide (CO2), methane (CH4), nitrous oxide (N2O) and ozone (O3), which trap heat and disrupt the planet's natural climate systems.2,3 According to previous research, the rate of warming has increased each year with multiple locations on Earth breaking temperature records.4 Ozone is a crucial component of the Earth's atmosphere, playing a vital role in absorbing harmful ultraviolet radiation from the sun.5 However, at ground level, ozone becomes a significant air pollutant, contributing to respiratory problems, environmental damage, and climate change.6 Since 1995, free tropospheric ozone has increased by 2–12 percent per decade depending on the region of the world.7 This is especially harmful as ozone is known to cause bronchial inflammation and airway hyper-responsiveness via oxidative injury and inflammation.8Efforts to capture and mitigate tropospheric ozone began in the mid-20th century with the rise of industrialization and vehicle emissions. Early research focused on reducing the precursors of ozone formation such as nitrogen oxides and volatile organic compounds rather than directly capturing ozone itself.9 Later, ozone capture technologies began to emerge, relying on metal oxides, catalytic surfaces, and metal–organic frameworks to break down ozone into oxygen.10 More recently, research has shifted towards non-metal alternatives including carbon-based nanomaterials, and two-dimensional materials like boron nitride and graphene.11,12 These materials aim to adsorb and decompose ozone more efficiently, while also being sustainable and environmental friendly.
Since the discovery of graphene,13 two-dimensional metals have been studied extensively for their large surface area which provides accessible active sites, and a planar structure, which facilitates material modification to improve their properties.14 Many of these studies involve materials that either contain metal oxides, are doped with metals, or are inherently metallic. These include monolayers such as MoS2, Pt-decorated graphene, stanene, CaO, SnS monolayers, and iron-doped graphene.15,16 These studies showed substantial improvements in interaction strength, sensing capabilities, and the potential for ozone dissociation.17 However, a challenge with using transition metals is effectively controlling their electrical properties, which motivated the choice of nonmetals in our study.18
Recent research also highlights doped carbonaceous materials and its derivatives as promising catalysts for advanced oxidation processes due to their cost-effectiveness, environmental benefits, and high activity.19 Previous studies have found that adding structural or foreign-atom defects can serve as effective catalysts for ozone decomposition.20 Specifically, boron, nitrogen, silicon, and phosphorus-doped graphene have shown significant performance in organic degradation.21 Experimental studies have also demonstrated that carbon doped boron nitride nanotube exhibits an enhanced response to molecular oxygen, leading to increased reactivity of boron nitride and stable chemisorption.22 Due to the structural similarity between boron nitride and graphene,23 they have comparable ozone adsorption potentials. Given the success of graphene doped with silicon and phosphorus for ozone, and boron nitride doped with carbon for oxygen, these same dopants are expected to modify the boron nitride monolayer in a similar manner, enhancing its reactivity and adsorption capabilities for ozone.
We aim to investigate new boron nitride monolayers doped with carbon, oxygen, phosphorus, and silicon which could change properties and electronic structure to adsorb and potentially dissociate ozone. These dopants were chosen based on success found for other monolayers. We used DFT to calculate the optimized structures of pure boron nitride and boron nitride doped with carbon, oxygen, silicon, and phosphorus. We then calculated adsorption energy as well as band structure, charge transfer, total density of states, and projected density of states to analyze the level of adsorption and interaction between the monolayer and ozone.
II. Methods
A. Computational details
All first-principles calculations were based on Density Functional Theory (DFT) using the Generalized Gradient Approximation (GGA) in the format of Perdew–Burke–Ernzerhof (PBE) implemented in the ABINIT24 code. The projected augmented wave (PAW) method25 was used to generate pseudopotentials with the ATOMPAW code.26 Table 1 shows the different elements used in this study and their corresponding electron configurations and their radius cutoffs.| Element | Electron configuration | Radius cutoff (bohr) |
|---|---|---|
| Boron (B) | [He] 2s2 2p1 | 1.70 |
| Nitrogen (N) | [He] 2s2 2p3 | 1.20 |
| Oxygen (O) | [He] 2s2 2p4 | 1.41 |
| Carbon (C) | [He] 2s2 2p2 | 1.51 |
| Phosphorus (P) | [Ne] 3s2 3p3 | 1.91 |
| Silicon (Si) | [Ne] 3s2 3p2 | 1.91 |
B. Convergence details
To ensure the accuracy of the calculations, values for kinetic energy cutoff, k-point mesh, and vacuum were converged. The total energy self-consistent field (SCF) calculations were performed with a convergence criterion of 1.0 × 10−10 hartree. The dataset convergence was determined when the difference between two consecutive total energy was less than 0.0001 hartree (0.003 eV). Geometry optimizations were carried out with a convergence threshold for the maximum force set to 2.0 × 10−4 hartree per bohr (0.01 eV Å−1).C. Atomic structures
To isolate the ozone molecule, we utilized a large supercell and achieved convergence for both the kinetic energy cutoff and cell size. The optimized atomic structure is shown in Fig. 1(a) and the bond length is shown in Table 2.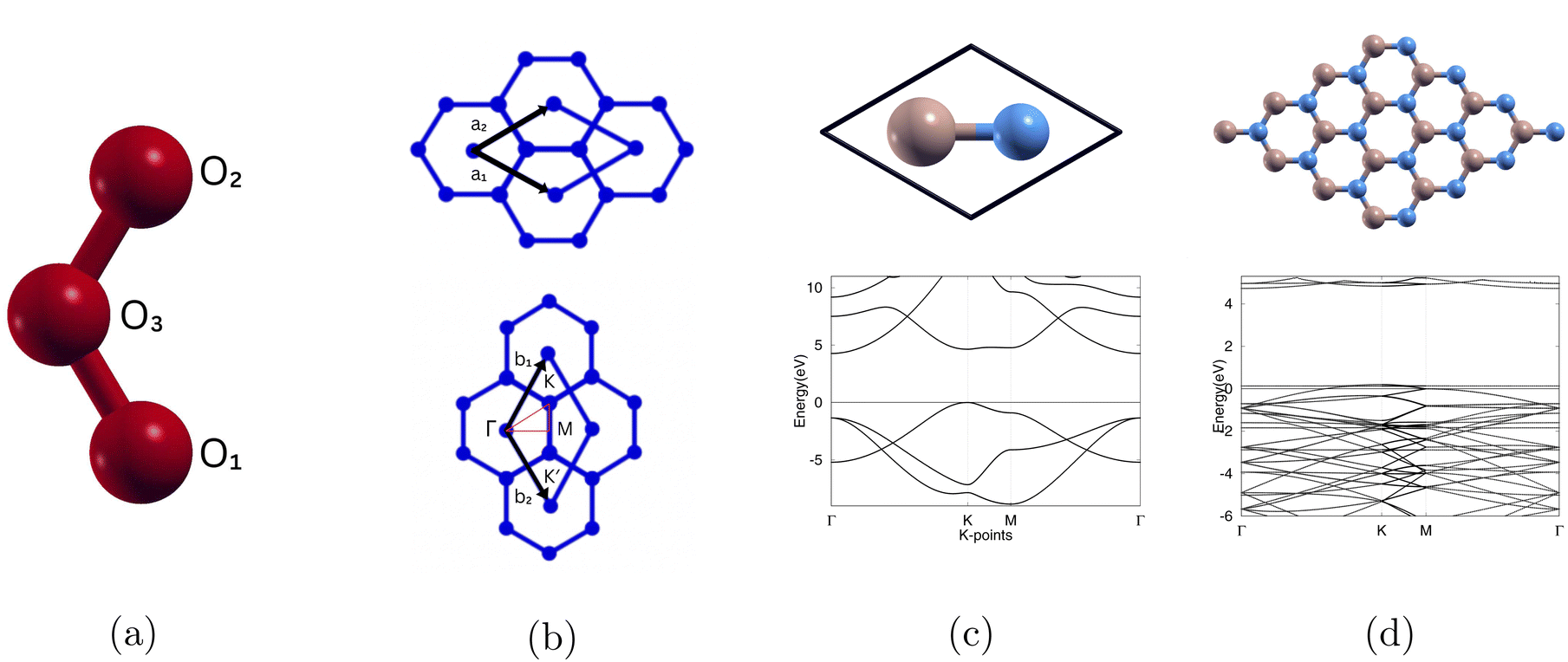 | ||
| Fig. 1 (a) Atomic structure of O3, (b) the Bravais lattice (a1, a2) and reciprocal lattice (b1, b2) of boron nitride monolayer. (c) The atomic and band structure of (1 × 1)-boron nitride monolayer, (d) the atomic and band structure of (4 × 4)-boron nitride monolayer. 0 eV is the Fermi level. Boron atoms and nitrogen atoms are beige and blue respectively. High symmetry k points in the first Brillouin zone are Γ (0,0,0), K (1/3,2/3,0), M (1/2,1/2,0). | ||
| Current | Other | Experimental | Error (%) | |
|---|---|---|---|---|
| d13 (Å) | 1.271 | 1.282 | 1.278 | 0.548 |
| d23 (Å) | 1.271 | 1.282 | 1.278 | 0.548 |
| ∠132 | 118.370 | 116.850 | 116.800 | 1.344 |
For the (1 × 1)-boron nitride (11BN) monolayer calculation, we utilized a primitive cell consisting of two atoms, one boron and one nitrogen, to represent the simplest possible configuration of the boron nitride structure. This small-scale model serves as a reference for calculating various properties such as lattice constants, bond lengths, and electronic structure, which can then be scaled up for larger systems.
For the (4 × 4)-boron nitride (44BN) calculation, we used a 4 × 4 × 1 supercell with 32 atoms. In four separate instances, we modified the monolayer with substituting a boron and nitrogen pair with 2 carbon (C), 2 oxygen (O), 2 silicon (Si), and 2 phosphorus (P). The defect formation energy is given in eqn (1)
| Ef = EmL − E44BN − 2Edopant + E11BN | (1) |
D. Adsorption calculations
The adsorption energy (Ead) between ozone and the monolayer is needed to assess whether physisorption or chemisorption will occur. We placed ozone on top of pure and doped boron nitride with a vacuum of 10.58 Å. We calculated the appropriate vacuum spacing based on the sum of the lattice parameters of the boron nitride monolayer and the ozone molecule. This gave a minimum spacing of 19 bohr, but to ensure no interactions, we increased the vacuum spacing to 20 bohr. We also tested 14 bohr and 24 bohr and found that the energy difference was less than 0.001 hartree for both, showing that our vacuum distance is effective. The adsorption energy was calculated using eqn (2)| Ead = Emol+mL − EmL − Emol | (2) |
E. Electronic structures
| Δρ = ρmol/mL − ρmL − ρmol | (3) |
III. Results and discussion
We calculated the atomic and band structures, adsorption energy, density of states, and charge transfer for ozone adsorbed on pure and doped boron nitride monolayers.A. Pure materials
We first calculated the optimized atomic structure, bond lengths, and bond angle for O3. From our current calculations, we observed the bond length between O1 and O2, bond length between O2 and O3, and bond angle of ozone to be 1.271 (Å), 1.271 (Å), and 118.370° respectively as shown in Table 2. This is comparable to other calculations27 and experimental data.28The converged lattice constant for (4 × 4)-boron nitride can be found in Table 3. The plotted band structures for (1 × 1)-boron nitride and (4 × 4)-boron nitride are shown in Fig. 1(c) and (d) and the calculated band gap is shown in Table 3. We used the same high symmetry points shown in Fig. 1(b).
| Monolayer | a (Å) | Eg (eV) | Gap type | Ef (Ha) |
|---|---|---|---|---|
| 44BN | 9.997 | 4.252 | Indirect | N/A |
| C-BN | 10.047 | 3.177 | Direct | 0.072 |
| O-BN | 10.118 | 1.805 | Indirect | 0.207 |
| Si-BN | 10.400 | 1.532 | Indirect | 0.911 |
| P-BN | 10.346 | 0.692 | Direct | 0.746 |
B. Doped monolayers
To investigate the effects of various dopants on the electronic properties of boron nitride, we substituted a pair of 2C, 2O, 2Si, 2P for a boron and nitrogen pair. The optimized atomic structures and band structures are shown in Fig. 2 where Fig. 2(a) shows C2 sub (4 × 4)-BN monolayer (C-BN), Fig. 2(b) shows Si2 sub (4 × 4)-BN monolayer (Si-BN), Fig. 2(c) shows P2 sub (4 × 4)-BN monolayer (P-BN), and Fig. 2(d) shows O2 sub (4 × 4)-BN monolayer (O-BN). The lattice constants are shown in Table 3 with Si-BN having the largest lattice constant of 10.400 (Å) and C-BN having the smallest lattice constant of 10.047 (Å).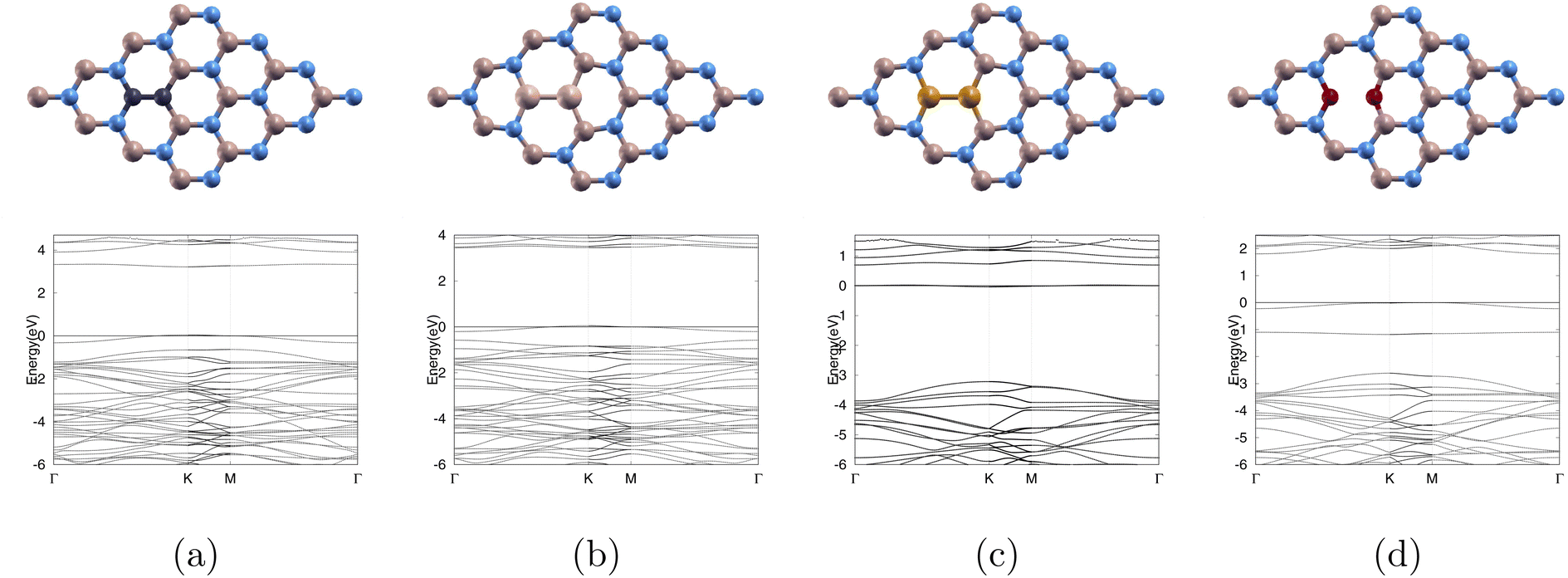 | ||
| Fig. 2 Top row represents the atomic structure of optimized monolayers and bottom row represents the band structure for (a) C-BN, (b) Si-BN, (c) P-BN, and (d) O-BN. 0 eV is the Fermi level. Color code as following: boron atoms, nitrogen atoms, carbon atoms, silicon atoms, phosphorus atoms, and oxygen atoms are colored beige, blue, black, white, yellow, and red. | ||
We observe that Si-BN has the largest band gap of 3.393 eV while P-BN has the smallest band gap of 0.692 eV as shown in Table 3.
In order to get defect formation energy, we calculated the total energies of our doped monolayer, pure (4 × 4)-BN monolayer, dopant, and (1 × 1)-BN monolayer. Using eqn (1) we were able to calculate the exact values as listed in Table 3. We observed that C-BN had the smallest Ef of 0.072 Ha indicating that it requires the least energy to obtain our doped monolayer, while Si-BN had the largest Ef of 0.911 Ha.
C. Ozone adsorbed on monolayers
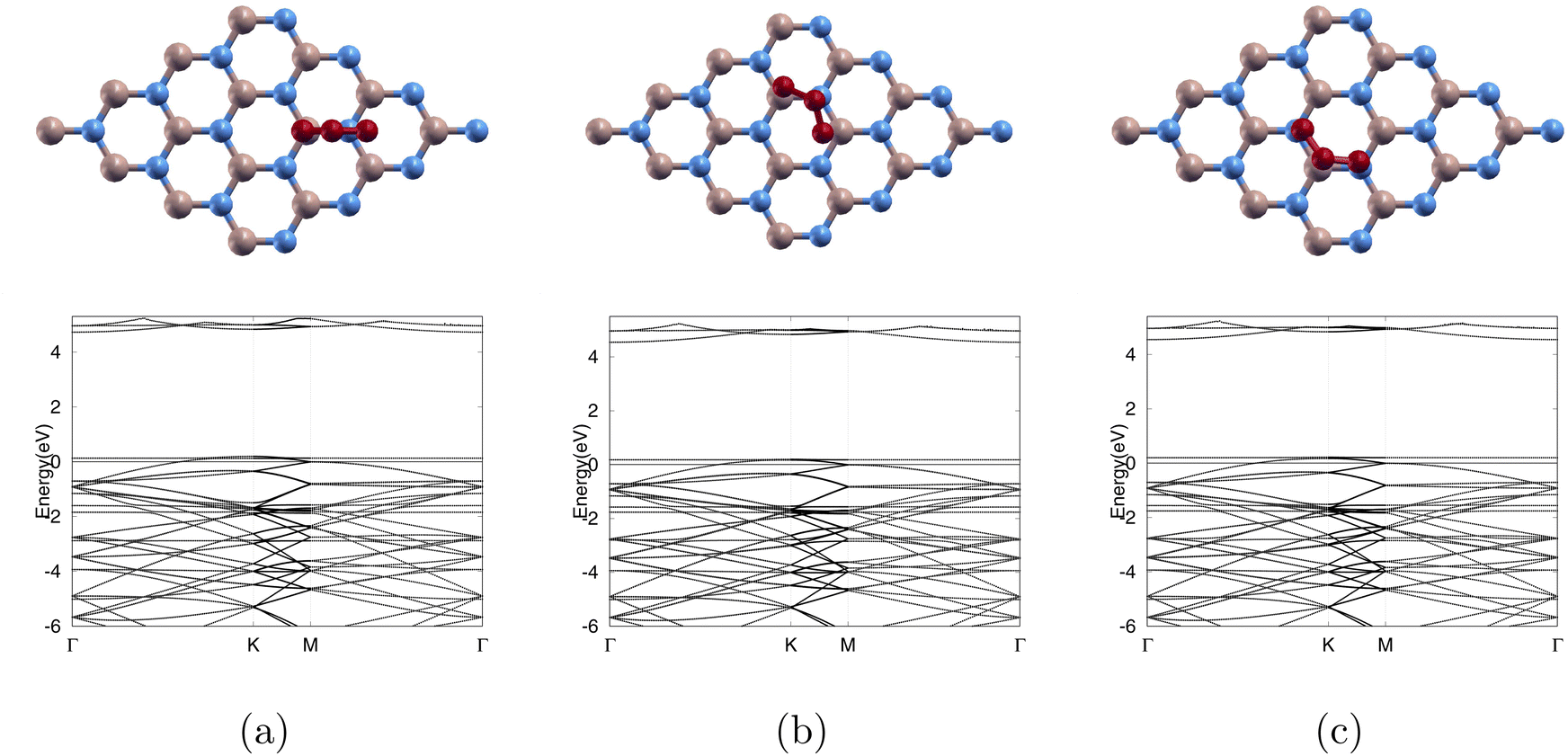 | ||
| Fig. 3 Ozone adsorbed on pure monolayers in 3 different positions: top row shows atomic structures and bottom row shows band structures for (a) BN-TBN, (b) BN-TBB, and (c) BN-TNN. 0 eV is the Fermi level. Color code as following: boron atoms, nitrogen atoms, and oxygen atoms are beige, blue, and red respectively. | ||
In Fig. 4, we depict the optimized atomic structures of our doped (4 × 4)-BN monolayers. The top row and bottom row represent the atomic structure before and after optimization. Fig. 4(a) C2 sub (4 × 4)-BN (O3/C-BN). Fig. 4(b) Si2 sub (4 × 4)-BN (O3/Si-BN). Fig. 4(c) P2 sub (4 × 4)-BN (O3/P-BN). Fig. 4(d) O2 sub (4 × 4)-BN (O3/O-BN). In Fig. 4(a), O3 bonded to the monolayer. In Fig. 4(b), O3 bonded to the monolayer and was dissociated into O and O2. In Fig. 4(c), O3 was dissociated into an O atom bonded to the monolayer and a released O2 molecule. In Fig. 4(d), O3 showed no adsorption. We successfully identified the converged state for (O3/C-BN) and (O3/Si-BN). However, we were unable to achieve convergence for (O3/P-BN) and (O3/O-BN). As a result, we proceeded with the following calculations (density of states, band structure, and charge transfer) for only (O3/C-BN) and (O3/Si-BN).
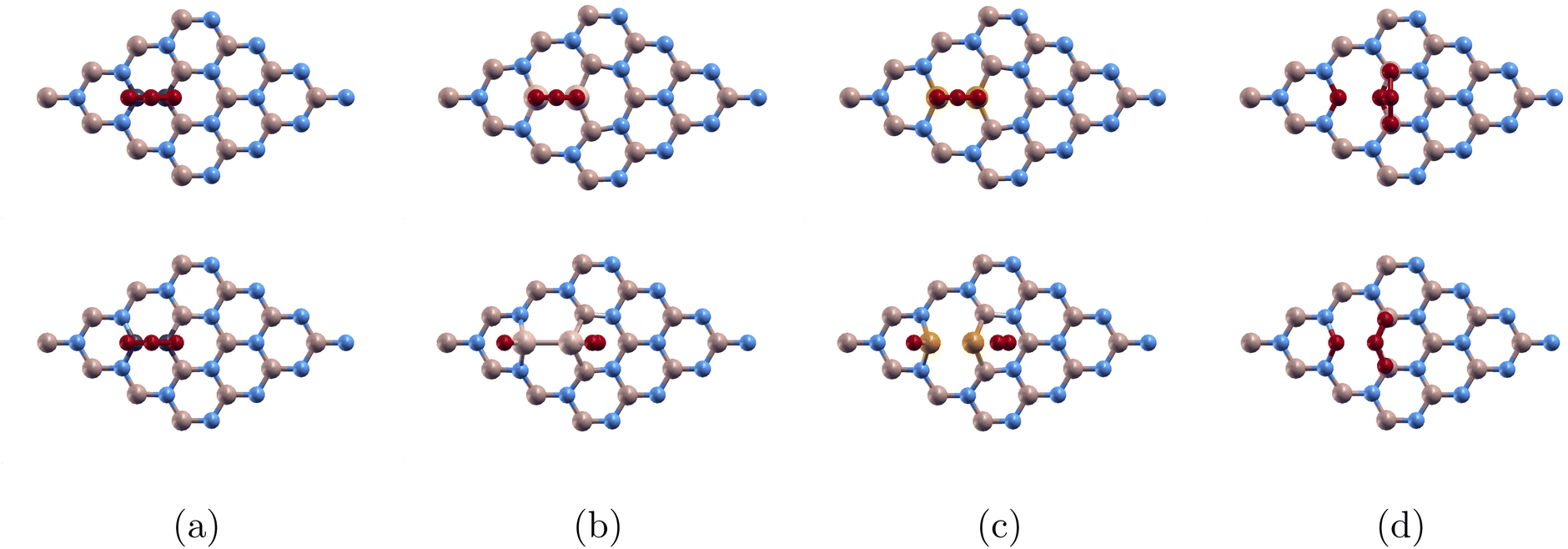 | ||
| Fig. 4 Ozone adsorbed on doped monolayers before and after optimization: top row and bottom row shows atomic structure before and after optimization respectively for (a) O3/C-BN, (b) O3/Si-BN, (c) O3/P-BN, and (d) O3/O-BN. Color code as following: boron atoms, nitrogen atoms, carbon atoms, silicon atoms, phosphorus atoms, and oxygen atoms are colored beige, blue, black, white, yellow and red respectively. | ||
| Monolayer | Ead (eV) | h (Å) | Eg (eV) | Gap type |
|---|---|---|---|---|
| BN-TBB | −0.155 | 3.18 | 0 | Metallic |
| BN-TBN | −0.155 | 3.26 | 0 | Metallic |
| BN-TNN | −0.155 | 3.27 | 0 | Metallic |
| C-BN | −0.272 | 1.98 | 2.314 | Indirect |
| Si-BN | −8.074 | 2.57 | 3.393 | Indirect |
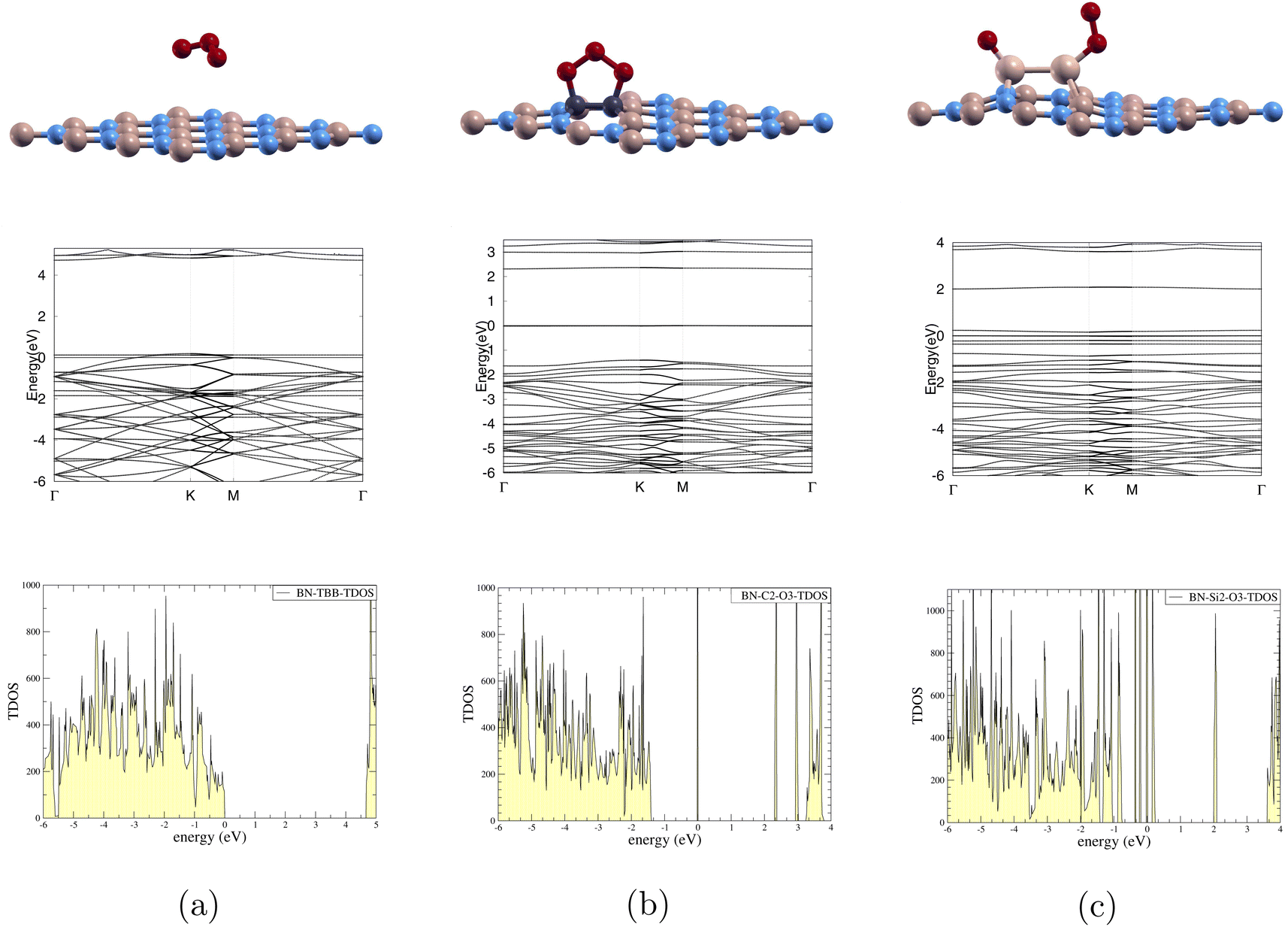 | ||
| Fig. 5 Ozone adsorbed on monolayers: top row shows side view of atomic structures, middle row shows band structure, and bottom row shows total density of states. 0 eV is Fermi level. (a) BN-TBB, (b) O3/C-BN, (c) O3/Si-BN. | ||
In Fig. 5(a), the band gap is noted to decrease from 4.252 eV to 0 eV after ozone adsorption shown in Tables 3 and 4 respectively. This indicates metallic nature. Furthermore, the TDOS graph shows significant electronic states at the Fermi level 0 eV. This presence of states suggests that electrons can move freely through the conduction band, facilitating electrical conductivity.
In Fig. 5(b), the band gap is noted to decrease from 3.177 eV to 2.314 eV after ozone adsorption as shown in Tables 3 and 4 respectively. The TDOS graph reveals states around the set Fermi level 0 eV. The presence of these straight lines indicates flat bands, which suggest localized electronic states at these energies. The flat bands at these specific energies likely result from interactions with ozone and carbon dopants, and these localized states are evident in both the band structure and TDOS.
For Fig. 5(c), the band gap is noted to decrease from 3.393 eV to 1.532 eV after ozone adsorption as shown in Tables 3 and 4 respectively. We observe states around the Fermi level, set at 0 eV in the TDOS graph. This suggests that the electronic states introduced by the silicon dopants are more localized near the Fermi level and have a higher density of available electronic states. These closely packed states can enhance the likelihood of charge transfer or bonding interactions between the monolayer and the ozone, leading to higher Ead of −8.074 eV which can be seen in Table 4.
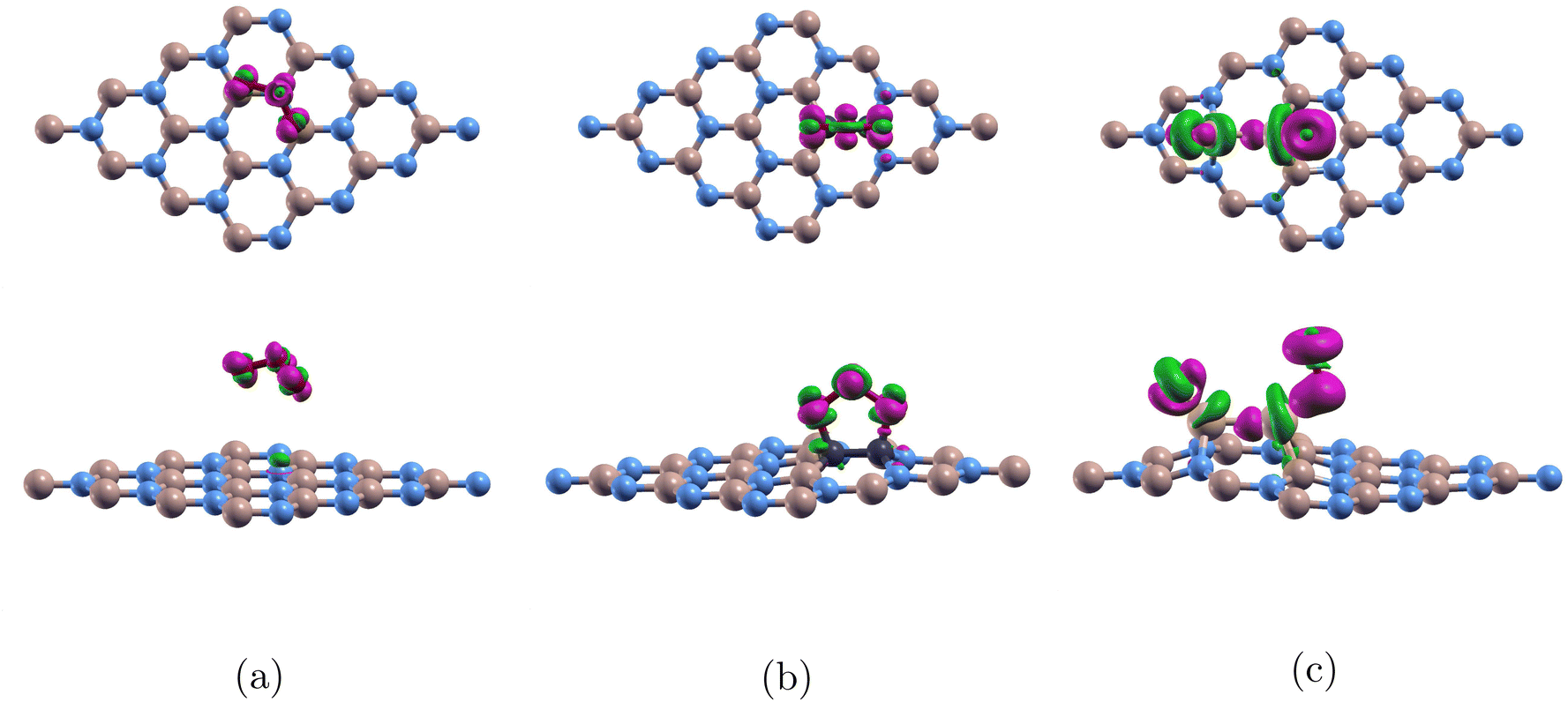 | ||
| Fig. 6 Top view and side view of charge transfer of ozone adsorbed on monolayers: (a) BN-TBB isovalue = 0.001 electron per bohr3, (b) O3/C-BN isovalue = 0.02 electron per bohr3, (c) O3/Si-BN isovalue = 0.01 electron per bohr3. Color code as following: boron atoms, nitrogen atoms, oxygen atoms, carbon atoms, silicon atoms are colored beige, blue, red, back, and white respectively. The pink and green regions represent charge accumulation and charge depletion respectively and charge is transferred from the green to the pink regions. | ||
For Fig. 6(a), the figure shows ozone detached from pure (4 × 4)-BN and analysis reveals no significant bonding or charge transfer between the ozone molecule and the monolayer. The projected density of states (PDOS) shows minimal interaction between the orbitals of the oxygen and boron atoms, and the charge transfer results confirm that no substantial electron redistribution occurs. As a result, there is no evidence of covalent bonding between ozone and the boron nitride surface. This can be explained by the large band gap of 4.252 eV in Table 3 and boron nitride's robust covalent bonds.31
Fig. 6(b) shows ozone adsorbed onto C-BN. For C2-doped (4 × 4)-BN, electrons are transferred within the ozone molecule. There is minimal interaction between the monolayer and the molecule. This is because carbon does not introduce states near the Fermi level that can strongly interact with the orbitals of ozone. As a result, there are no accessible energy states that would allow for significant electron exchange between the monolayer and the molecule. This finding is consistent from when we calculated for adsorption energy and confirms the weak Ead of −0.272 eV between the monolayer and the ozone molecule.
Fig. 6(c) shows ozone bonded to the Si2-doped (4 × 4)-BN. For Si2-doped (4 × 4)-BN, there is much more charge transfer between the monolayer and the two attached oxygens which accumulates in the Si–O bonds. This can be explained by the large electronegativity difference between Si (1.8) and O (3.5) which allows for the electrons to be pulled towards oxygen. This finding is uniform with our large Ead of −8.074 eV.
In Fig. 7(a), analysis revealed minimal interaction between the boron and oxygen orbitals. The p orbital plot for the two borons are similar, and the p orbital plot for the two oxygen atoms were also almost identical. This indicates that this configuration does not significantly alter the electronic structure of either the boron or oxygen atoms. This is due to the relatively stable and inert electronic structure of boron nitride, which has a wide band gap. The lack of interaction and hybridization supports why ozone doesn't bond to 44BN, and the material's properties remain largely unaffected by the presence of ozone in this scenario.
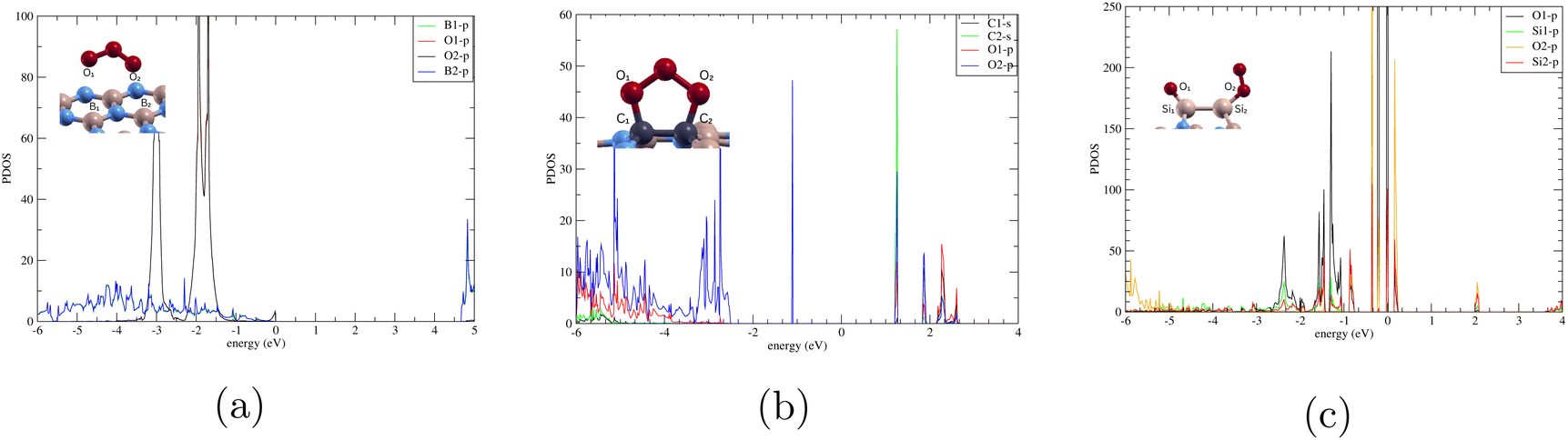 | ||
| Fig. 7 Projected density of states of (a) BN-TBB, (b) O3/C-BN, and (c) O3/Si-BN. The atomic orbitals in the right corner correspond to the atomic structure in the top left corner. | ||
In Fig. 7(b), we observe minimal overlapping between the s orbital from C1 and the p orbital from O2 suggesting some hybridization and bonding between the monolayer and the ozone molecule. This corresponds to the adsorption energy −0.272 eV that we calculated. Compared to Fig. 7(a), where there is no interaction and no adsorption, O3/C-BN's minimal interaction supports physisorption.
In Fig. 7(c), we observe strong interactions between the silicon atoms in the doped boron nitride and the oxygen atoms from the adsorbed ozone. The overlapping of the p orbital from O2 and p orbital of O1 suggests significant hybridization of their orbitals, leading to strong chemical bonding between silicon and oxygen. This corresponds to the large Ead −8.074 eV that we calculated. The proximity to the Fermi level highlights the high electronic activity associated with these atoms, implying that they contribute actively to the material's electronic structure. As a result, the system likely exhibits enhanced reactivity and possibly improved conductivity. The strong bonding interaction and electronic contribution from silicon and oxygen suggest that silicon-doped boron nitride could have promising applications in catalytic processes or chemical sensing, given its ability to effectively adsorb and interact with ozone. This observation matches the findings of Yadav who showed that Si doping significantly enhanced the sensing behaviour of BNNs compared to pristine BNNs.32
D. General results
We investigated the adsorption of ozone on both pure boron nitride and four doped monolayers: carbon, oxygen, silicon, and phosphorus. Among the tested monolayers, boron nitride doped with carbon exhibited the lowest formation energy at 0.072 hartree, indicating it is energetically favorable. After optimization, we found that ozone adsorbed on Si2 sub (4 × 4)-BN formed the most stable configuration, with an adsorption energy of −8.074 eV, highlighting strong interaction with ozone. Ozone adsorbed on P2 sub (4 × 4)-BN did not produce a stable configuration, but it led to the release of an oxygen molecule. Charge transfer and projected density of states (PDOS) analysis showed that silicon-doped boron nitride demonstrated the greatest charge transfer and the most hybridization with ozone. This suggests a strong interaction between ozone and the silicon atoms, making silicon doping particularly effective for ozone adsorption. The band structure calculations further revealed that ozone adsorbed on carbon-doped boron nitride monolayer exhibited a band gap of 2.314 eV, while the silicon-doped boron nitride monolayer had a slightly higher band gap of 2.57 eV, indicating both doped systems retain semiconducting properties while interacting with ozone.IV. Conclusion
Through first-principles calculations based on DFT, this study aimed to address the environmental and health challenges posed by ozone in the troposphere by exploring its adsorption on boron nitride (BN) monolayers doped with metal-free elements. Our findings reveal that different dopants result in varied behaviors of ozone adsorption and dissociation compared to pure boron nitride. Carbon-doped boron nitride exhibited physisorption with the entire ozone molecule weakly bonded to the monolayer. Silicon-doped boron nitride, showed a significant increase in adsorption energy, achieving chemisorption and effectively breaking the ozone molecule into O and O2, both of which remained bonded to the monolayer, making Si2 sub (4 × 4)-BN a promising candidate for ozone removal. Phosphorus-doped BN led to the dissociation of ozone, releasing O2 and bonding a single oxygen atom to the BN monolayer. Oxygen-doped BN demonstrated little interaction with ozone indicating it unfit for ozone capture. While this study is based on first-principles Density Functional Theory (DFT) calculations the energy values, lattice constants, and structural properties obtained in this study are consistent with previous theoretical works, which enhances confidence in the reliability of our computational model.These findings demonstrate the effectiveness of silicon-doped boron nitride in removing ozone from the troposphere. Given the high adsorption energy of the resulting oxygen species, it is reasonable to conclude that the surface of the doped boron nitride may be suitable for one-time use only. The strong chemisorption indicates that regeneration of the surface could be challenging. Therefore, the application of this material may be limited to specific single-use scenarios. Additionally, the results indicate the potential of phosphorus-doped boron nitride to adsorb ozone and release an oxygen molecule. However, further investigation is necessary to find the fully optimized structure and assess its capabilities in mitigating the harmful effects of tropospheric ozone on both the environment and human health. We hope this study will provide valuable insights on improving ozone adsorption technologies using doped boron nitride monolayers.
Data availability
All data for our study came from ABINIT calculations. All other data used can be found in the References section of the manuscript.Conflicts of interest
There are no conflicts to declare.References
- What is climate change?, https://science.nasa.gov/climate-change/what-is-climate-change/ Search PubMed.
- T. S. Ledley, E. T. Sundquist, S. E. Schwartz, D. K. Hall, J. D. Fellows and T. L. Killeen, EOS, Trans., Am. Geophys. Union, 2011, 80, 453 CrossRef.
- H. Tian, G. Chen, C. Lu, X. Xu, W. Ren, B. Zhang, K. Banger, B. Tao, S. Pan and M. Liu, et al., Ecosys. Health Sustain., 2017, 1, 1 Search PubMed.
- J. Blunden, G. Hartfield, D. S. Arndt, R. J. H. Dunn, M. R. Tye, S. Blenkinsop, M. Donat, I. Durre, M. Ziese and O. R. Cooper, et al., Am. Meteorol. Soc., 2018, 99, 1 CrossRef.
- L. O. Bjorn, Biol. Conserv., 2007, 135, 326 CrossRef.
- E. C. Filippidou and A. Koukouliata, Prog. Health Sci., 2011, 1, 144 Search PubMed.
- H. Wang, X. Lu, D. J. Jacob, O. R. Cooper, K.-L. Chang, K. Li, M. Gao, Y. Liu, B. Sheng and K. Wu, et al., Atmos. Chem. Phys., 2022, 22, 13753–13782 CrossRef CAS.
- S.-Y. Kim, E. Kim and W. J. Kim, Tuberc. Respir. Dis., 2020, 83, 56 Search PubMed.
- M. M. Fry, V. Naik, J. J. West, M. D. Schwarzkopf, A. M. Fiore, W. J. Collins, F. J. Dentener, D. T. Shindell, C. Atherton and D. Bergmann, et al., J. Geophys. Res., 2012, 117, D7 CrossRef.
- K. Erme and I. Jõgi, Environ. Sci. Technol., 2019, 53, 5266–5271 CrossRef CAS PubMed.
- D. Chen, J.-J. Yang, L.-H. Xie, G. Cui, W.-H. Fang and R.-J. Li, Nat. Commun., 2022, 13, 4991 CrossRef PubMed.
- G. Lee, B. Lee, J. Kim and K. Cho, J. Phys. Chem. C, 2009, 113, 14225–14229 CrossRef CAS.
- L. Patra, G. Sachdeva, R. Pandey and S. P. Karna, ACS Omega, 2021, 6, 19546 CrossRef CAS PubMed.
- P. Kumbhakar, J. S. Jayan, A. S. Madhavikutty, P. R. Sreeram, A. Saritha, T. Ito and C. S. Tiwary, iScience, 2023, 26, 106671 CrossRef CAS PubMed.
- A. Shokuhi Rad and D. Zareyee, Vacuum, 2016, 130, 113 CrossRef CAS.
- A. Abbasi and J. Jahanbin Sardroodi, Appl. Surf. Sci., 2019, 469, 781 CrossRef CAS.
- A. Shukla and N. K. Gaur, Phys. B, 2019, 572, 12 CrossRef CAS.
- D. Lee, M. Rabeel, Y. Han, H. Kim, M. Khan, D. Kim and H. Yoo, ACS Appl. Mater. Interfaces, 2023, 15(44), 51518–51526 CrossRef CAS.
- H. Zhang, J. Y. Lee and H. Liu, J. Phys. Chem. C, 2021, 125, 10948 CrossRef CAS.
- M. Wu, J. Sun, W. Xiang and S. Chen, J. Environ. Chem. Eng., 2022, 10, 108734 CrossRef CAS.
- G. Yu, Y. Xie, Q. Ge, Q. Dai, J. Xu and H. Cao, Chem. Eng. J., 2022, 430, 133114 CrossRef CAS.
- J. Zhang, K. P. Loh, J. Zheng, M. B. Sullivan and P. Wu, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 24 Search PubMed.
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 7698 CrossRef PubMed.
- X. Gonze, B. Amadon, P.-M. Anglade, J.-M. Beuken, F. Bottin, P. Boulanger, F. Bruneval, D. Caliste, R. Caracas and M. Cote, et al., Comput. Phys. Commun., 2009, 180, 2582 CrossRef CAS.
- P. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- N. A. W. Holzwarth, A. R. Tackett and G. E. Matthews, Comput. Phys. Commun., 2001, 135, 329 CrossRef CAS.
- L. Mainali, D. R. Mishra and M. M. Aryal, arXiv, 2011, preprint, arXiv:1102.1051, DOI:10.48550/arXiv.1102.1051.
- Experimental data for O3 (ozone), 2022, https://cccbdb.nist.gov/exp2x.asp?casno=10028156\%26charge=0 Search PubMed.
- D. Farmanzadeh and N. Askari Ardehjani, Appl. Surf. Sci., 2018, 444, 642 CrossRef CAS.
- D. García-Toral, R. Mendoza Báez, J. I. Sánchez, A. Flores-Riveros, G. H. Cocoletzi and J. F. Rivas-Silva, ACS Omega, 2021, 6, 14824–14837 CrossRef.
- G. R. Bhimanapati, N. R. Glavin and J. A. Robinson, Semicond. Semimetals, 2016, 95, 101 CAS.
- A. Yadav, Silicon, 2022, 15, 1847 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |