
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface-modified spinel high entropy oxide with hybrid coating-layer for enhanced cycle stability and lithium-ion storage performance†
Feiyue Zhaiabc,
Shengji Gaobcd,
Wenfeng Zhangbc,
Gaoping Caobc,
Huimin Zhang bc,
Yalan Xinga,
Yu Xiang*bc and
Shichao Zhanga
bc,
Yalan Xinga,
Yu Xiang*bc and
Shichao Zhanga
aSchool of Materials Science and Engineering, Beihang University, Beijing 100191, China
bChemical Defense Institute, Beijing, 100191, PR China. E-mail: drxiangyu2016@126.com
cBeijing Key Laboratory of Advanced Chemical Energy Storage Technology and Materials, Beijing 100191, China
dHebei University of Technology, Tianjin 100131, China
First published on 21st October 2024
Abstract
High-entropy oxide (HEO) has emerged as a promising anode material for high-energy lithium-ion batteries (LIBs) due to its high theoretical specific capacity. However, the further application of HEO is restricted by its complicated interface problems and inevitable expansion effect. In this work, a simple approach to coat spinel HEO (FeCoNiCrMn)3O4 with a hybrid layer of lithium titanate (LTO) and carbon is presented. The coating is applied through a solution-chemistry method followed by calcination under an inert atmosphere. This hybrid layer significantly improves the electrochemical kinetics and stability at the electrode/electrolyte interface. Additionally, the diffusion of Ti4+ into the HEO bulk during synthesis provides an inactive metal skeleton, potentially improving cycle stability. Electrochemical test results show that the HEO@LTO/C achieved a reversible specific capacity of 1090 mA h g−1 at 0.5 A g−1 and remained stable after 800 cycles. Moreover, the first-coulomb efficiency was increased from 63.7% to 72.8%, and rate performance has improved by at least 100 mA h g−1. This work demonstrates that hybrid surface-modifying of HEO is an effective measure to improve and stabilize its electrochemical properties.
Introduction
Since they were first proposed in 2018, HEOs have shown promising potential as anodes for high-energy lithium-ion batteries (LIBs).1–4 For example, rock salt type HEO (MgCoNiCuZn)O could display a reversible capacity of about 650 mA h g−1 at a current density of 200 mA g−1 for 300 cycles.5 This demonstrated superior cycle stability compared to conventional transition metal oxide (TMO) anodes.6–8 It was found that the inactive MgO component could act as a stable framework and alleviate the volume expansion of HEO anode during the long cycling, realizing a relatively stable performance. The synergistic effect of multiple elements can promote more electrons to participate in the redox reaction, so the high entropy effect can increase the specific capacity of the material.9 For example, spinel HEOs(SHEOs) have been designed based on the tunability of the components, such as (MgTiZnCuFe)3O4,10 (NiCoCuFeMg)3O4,11 and (CrMnFeNiZn)3O4.12 The metal ions of SHEOs are located at two Wyckoff sites, which are tetrahedral 8a and octahedral 16d sites, respectively. The random distribution of cations at these two sites results in higher hybrid valence states compared to rock type HEOs, leading to significantly higher theoretical capacities.13 Huang et al. synthesized non-equimolar SHEO (CrMnFeCoNi)3O4 with the atom ratio of Cr, Mn, Fe, Co, Ni being 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:2:8:4 via a surfactant-assisted hydrothermal method.14 The first reversible capacity was about 1250 mA h g−1 at 20 mA g−1 with a specific capacity of 750 mA h g−1 after 200 cycles at 0.5 A g−1. This is a high level that can be achieved with SHEO anodes, demonstrating a higher specific capacity than other conventional TMO anodes.
:8:2:8:4 via a surfactant-assisted hydrothermal method.14 The first reversible capacity was about 1250 mA h g−1 at 20 mA g−1 with a specific capacity of 750 mA h g−1 after 200 cycles at 0.5 A g−1. This is a high level that can be achieved with SHEO anodes, demonstrating a higher specific capacity than other conventional TMO anodes.
Although SHEOs have displayed obvious advantages in the applications of high-energy LIBs over TMOs, there are still three issues limiting their performance enhancement. Firstly, the interfaces of SHEOs are not very steady during the life-cycle span, resulting in the continuous consumption of electrolytes. This phenomenon leads to an exhaustion of electrolytes and a sharp collapsed capacity, which has been reported in our previous work.15 Secondly, all metal ions in SHEOs have electrochemical activity to provide capacity in most cases. Hence, the cycle stabilities of SHEOs are usually inferior to rock-salt HEOs due to the lack of inactive metal skeleton. Thirdly, the poor electrical conductivity of SHEOs also limits the electrochemical performance. Hence, it is necessary to solve the above-mentioned three shortcomings to promote its application process.
Interface engineering modifies the surface of SHEOs through chemical modification, thereby stabilizing the interface with electrolytes and enhancing the electrochemical performance of active materials. During the intercalation/deintercalation, SHEO particles continuously break into nanocrystals which decreases the diffusion resistance of lithium ions, increases the contact interface, and improves the kinetics of Li+ penetration. However, the fresh surfaces of nanocrystal fragmentations lead to constant consumption of electrolytes for (Solid Electrolyte Interphase) SEI information. Therefore, coating electrochemically stable compounds has been used to modify the interfacial reactivity of materials.16–18 In LIB research, lithium titanate (LTO) is one of the most common coating materials for interface modification.19–21
Firstly, LTO has high Li+ conductivity, which is beneficial for enhancing rate performance. Secondly, LTO is chemically inert that could effectively isolate the direct contact between active material and electrolyte. This is useful to reduce the electrolyte's corrosion and improve the cycle stability. Abundant studies on LTO coating for improving the performance of cathode materials have been reported previously.22–24 Some scholars have also shown that LTO coating on graphite has effectively improved the ion diffusion ability and structural stability of the anode.18
In this work, we initially synthesized SHEO anode (FeCoNiCrMn)3O4 by a conventional solid-phase method. The hybrid coated layer of LTO and amorphous carbon on HEO (HEO@LTO/C) was then achieved using hydrothermal and one-step calcination methods. In the hybrid coating layer, LTO enhances Li+ diffusion and amorphous carbon improves electron transport. This reduces the interfacial electrochemical reaction resistance and improves the performance of SHEO. Meanwhile, the coating layer protects the inner active material and reduces its direct contact with the electrolyte, thus improving the stability and service life. Furthermore, a tiny amount of Ti4+ is diffused into the bulk of SHEO in the synthesis process, which could serve as inactive metal skeleton and is beneficial to enhance the cycle stability. The as-prepared HEO@LTO/C has shown significant improvements in initial coulombic efficiency, cycling specific capacity, and stability. This provides an effective way to enhance the electrochemical performance of other SHEO anodes.
Experimental
Materials preparation
Original SHEO was synthesized with solid-phase reaction using five metal oxides Fe2O3, Co3O4, NiO, Cr2O3, and MnO2 as raw materials. Equimolar amounts of the five metal oxide powders were grinded via a planetary ball-milling process at 350 rpm for 5 h. Then, the mixture was calcined at 900 °C with a heating rate of 6 °C min−1 under an Ar atmosphere for 12 h. The SHEO product was finally obtained after naturally cooled and high-energy ball milled for 2 h.The modified SHEO was prepared with the following procedures. The synthesized SHEO powder was dispersed homogeneously into amount of anhydrous ethanol to obtain suspension. Quantitative Ti (OC4H9)4 was dissolved in anhydrous ethanol with 0.1 vol% iced ethylic acid and then was completely added into the HEO suspension by drops. After stirring continuously for another 2 h, the reacted solution was filtered to collect solid powder. Afterwards, the obtained powder was dried for 1 hour at 80 °C, and then ball-milled with specific ratios of CH3COOLi at 200 rpm for 30 min. The grinded mixture was treated at 800 °C under Ar atmosphere for 2 h with a heating rate of 5 °C min−1 to obtain the final product, which was noted as HEO@LTO/C.
Material characterizations
A 10 kV scanning electron microscope (SEM, ZEISS Gemini SEM 300) and transmission electron microscopy (TEM, JEOL JEM-F200) were utilized to analyze the morphology and elemental distribution of active materials or electrodes. X-ray diffraction (XRD, Bruker D8 Advance) was applied to study the crystal structure with a scanning range of 5–90° and a rate of 1° min−1. X-ray Photoelectron Spectroscopy (XPS, Thermo Scientific K-Alpha) was used to measure the valences of metallic elements.Electrochemical measurements
To get electrode slurries, HEO or HEO/LTO@C powder, polyvinylidene fluoride binder (PVDF), and carbon black (Super P) were distributed uniformly with a weight ratio of 8:1:1 in N-methyl pyrrolidone (NMP). After stirring for 0.5 h, the slurry was coated onto a Cu foil. The coated copper foil was subsequently dried in a vacuum oven at 120 °C for 10 h and then cut into Φ 12 mm disks. The active mass loading of these disks was about 1 mg cm−2. To test the electrochemical performance of active material, half-cells were assembled in an argon-filled glovebox, with metallic lithium foil as the counter electrode, polypropylene as the separator, and 1 M LiPF6 EC (ethylene carbonate)/DEC (diethyl carbonate) (with a volume ratio of 1:1) as the electrolyte.
Galvanostatic performances were tested using the LAND CT3002A multi-channel battery test system at room temperature with a voltage range from 0.01 V to 3.00 V (vs. Li/Li+). Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) experiments were carried out with a solartron electrochemical workstation. The CV tests involved a potential window of 0.01–3.00 V at various scan rates ranging from 0.05 to 2.0 mV s−1. EIS measurements were measured with a frequency range from 106 Hz to 0.01 Hz and a 5 mV AC perturbation.
Results and discussion
Composition, structural, and morphological analysis
Fig. 1 shows the synthesis process of HEO@LTO/C. Firstly, HEO is prepared with solid phase reaction method. Then, Ti(OC4H9)4 is hydrolyzed in situ at the surface of HEO particles, resulting in a –(Ti–O–Ti)–n·xH2O pre-coating layer. Subsequently, the pre-coating layer is mixed with CH3COOLi via ball-milling treatment. Under the sintering process in Ar circumstance, two transformation reactions have emerged between the pre-coating layer and HEO particle to obtain HEO@LTO/C. One is that a small quantity of Ti elements in the pre-coating layer have migrated into the HEO bulk. The other is that the residual components have also transformed into rock salt type LiTiO2 and amorphous carbon to form a hybrid coating-layer. The formation mechanism of rock salt type LiTiO2 has been reported in previous articles.19 The inorganic hybrid coating-layer could hinder the corrosion of harmful products originated from the decomposition of electrolytes and increase the steadiness of the particle surfaces. The binary coating layer consisting of ionic conductor rock salt type LiTiO2 and amorphous carbon electron conductor is able to improve kinetic of interface electrochemical reaction, which is very beneficial to achieve better performances. Furthermore, the doped Ti element in the HEO bulk is electrochemical inertness and could serve as stable metal skeleton to enhance the long-cycle performance. Hence, it is inferred that as-prepared HEO@LTO/C is probably displays better electrochemical properties than original HEO.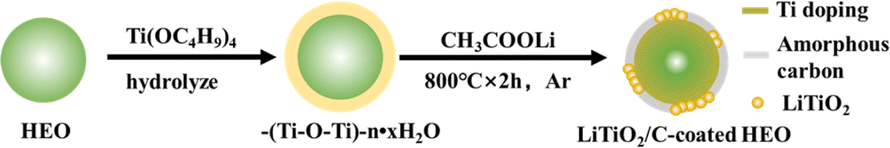 | ||
| Fig. 1 Schematic illustration of the synthetic process of HEO@LTO/C. | ||
Comparing the SEM images of original HEO (Fig. 2a) with modified HEO@LTO/C particles (Fig. 2b), it is presented that the surface of HEO@LTO/C is relatively rougher and a coating-layer exists in the prepared HEO@LTO/C anode material. According to the XRD results in Fig. 2c, it is found that the dominated phases in HEO@LTO/C and HEO are both attributed to spinel structure, representing the (FeCoNiCrMn)3O4 bulks. In HEO@LTO/C, it is also additionally observed the characteristic broad peak of amorphous carbon at around 25° and the represent diffraction peaks of the rock salt-type LiTiO2 at 37.6°, 43.9° and 63.6°. The TEM image in Fig. 2d and e displays that the coating-layer in HEO@LTO/C is about 2–5 nm thickness. It is also confirmed (311) face of spinel (FeCoNiCrMn)3O4 in the bulk (200) face of rock-salt type LiTiO2, and amorphous carbon in the coating-layer. Considering LiTiO2 and amorphous carbon are separately Li+ and electron conductor, it is inferred that the hybrid coating-layer is useful to improve the dynamics of interface electrochemical reactions, resulting in the performance improvement. Meanwhile, the inorganic character of the hybrid coating layer could also reduce the electrolyte corrosion effect and increase the interface stability.18
 | ||
| Fig. 2 SEM images of HEO (a) and HEO@LTO/C (b) and (c) XRD patterns of HEO and HEO@LTO/C, (d) and (e) TEM images of HEO@LTO/C, (f) FIB-SEM image of HEO@LTO/C, (g)–(l) Energy Dispersive Spectrum (EDS) with Fe, Co, Ni, Cr, Mn and Ti intensity maps of the corresponding HEO@LTO/C section shown in (f). | ||
Focused Ion Beam Scanning Electron Microscopy (FIB-SEM) method is applied to further investigate the element distribution in HEO@LTO/C. The cut section is shown in Fig. 2f and relative EDS analysis results are shown in Fig. 2g–l. It is revealed that Fe, Co, Ni, Cr, and Mn are uniformly distributed in the bulk without evident element segregation, presenting the successful preparation of SHEO. Notably, it is also found a slight amount of Ti element also homogenously disperses in the bulk, proving Ti element indeed migrates from the coating layer into the bulk. Unlike the other five metal elements in HEO, Ti3+/4+ ions are difficulty reduced to metal Ti. Hence, Ti3+/4+ could serve as a metallic skeleton to enhance the cycle stability.
The valence states of all metal elements in HEO@LTO/C are investigated from the XPS characterization in Fig. 3. The Fe 2p spectrum in Fig. 3a shows two major peaks of Fe 2p3/2 and Fe 2p1/2, plus three satellite peaks at 715.2, 718.8, and 733.0 eV. The co-existence of Fe2+/Fe3+ is shown from the deconvolution of Fe 2p spectrum, where Fe2+ is located at 710.9/724.2 eV and Fe3+ at 713.7/726.6 eV. The concentration ratio of Fe2+/Fe3+ is 53.63/46.37.25,26 Two spin–orbit peaks of Co 2p3/2 and Co 2p1/2 are located separately at 780.2 and 796.1 eV in Fig. 3b. The Co 2p3/2 can be split into two peaks, referring to Co2+ (782.6 eV) and Co3+ (780.4 eV). The Co 2p1/2 could be assigned to Co2+ (797.0 eV) and Co3+ (795.1 eV). The concentration ratio of Co2+/Co3+ is 44.20/55.80.25,26 In Fig. 3c, the Ni 2p spectrum displays two satellite peaks at 861.7 and 880.0 eV. Simultaneously, the spin–orbit split Ni 2p3/2 and Ni 2p1/2 peaks are detected at 854.9 and 872.3 eV, respectively. The deconvolution of the Ni 2p3/2 and Ni 2p1/2 peaks reveals the presence of Ni2+ and Ni3+ at different energies. The Ni2+ species is indexed to peaks at 854.5 and 872.1 eV, comprising 59.48% of the total Ni content. The Ni3+ species is ascribed to peaks at 856.2 and 873.8 eV, with a concentration of 40.52%.25 The deconvolution of the Cr 2p spectrum attributed the Cr3+ species at 575.8, 577.0, and 585.8 eV, with a dominant concentration of 73.89%. Meanwhile, the Cr6+ species was assigned to peaks at 579.1 and 588.1 eV, occupying a concentration of 26.11%.25,27 The Mn 2p spectrum exhibits two prominent peaks corresponding to the Mn 2p3/2 and Mn 2p1/2, along with a satellite peak at 643.4 eV. Deconvolution of the Mn 2p spectrum displays the co-existence of Mn2+ and Mn4+ species, with Mn2+ identified at 640.2/652.1 eV and Mn4+ at 641.6/653.7 eV. The ratio of Mn2+ to Mn4+ is determined to be 29.88/70.12.26,27 According to the XPS analysis, all metal elements in HEO@LTO/C show mixed valence states. The existence of mixed valence states means that the metal elements in the material have different oxidation states, which provides more possibilities for electron transfer and storage.26,28,29 Moreover, the higher content of high valence states in the metals contributes to a larger specific capacity.
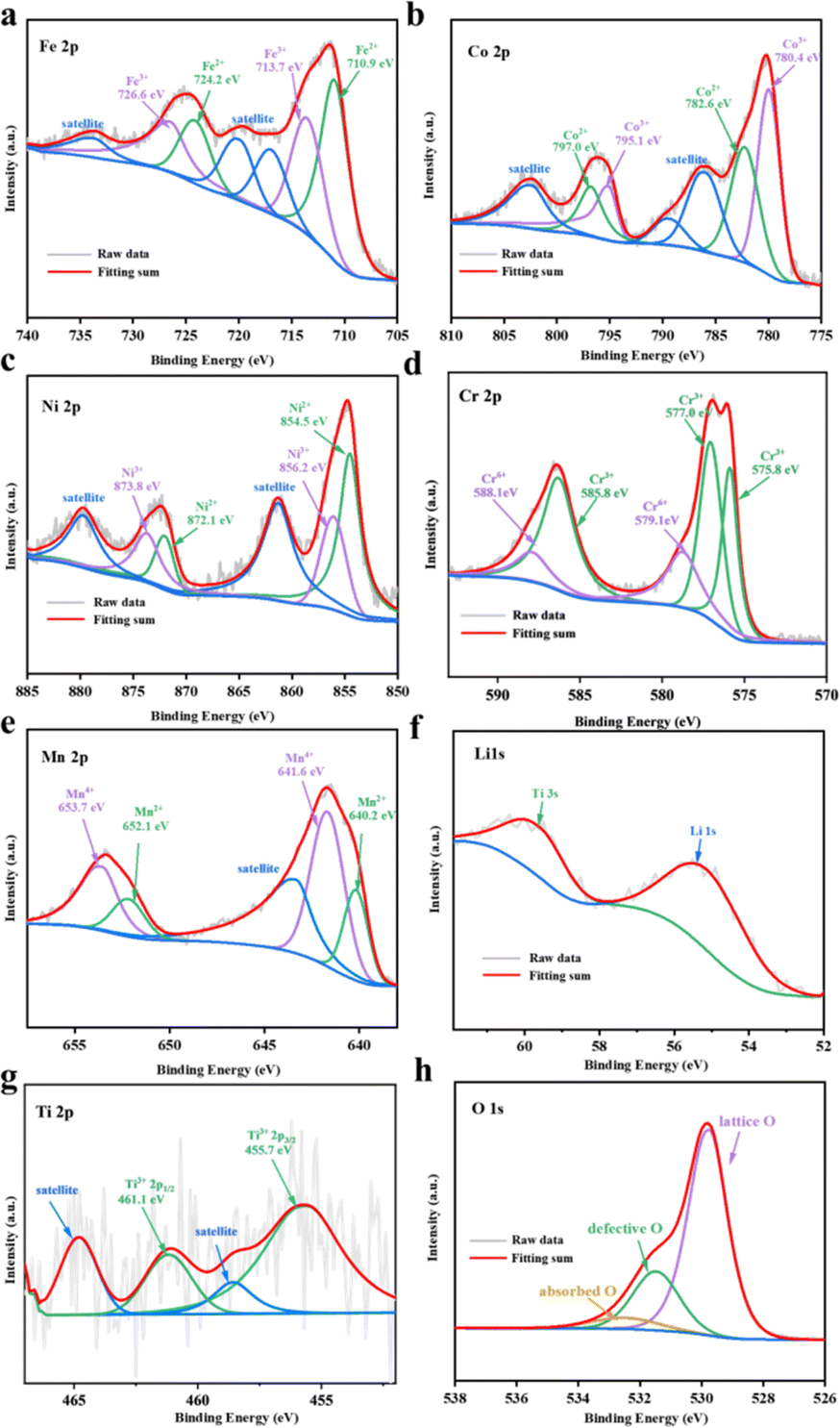 | ||
| Fig. 3 XPS spectra and fitting results of (a) Fe 2p, (b) Co 2p, (c) Ni 2p, (d) Cr 2p, (e) Mn 2p, (f) Li 1s, (g) Ti 2p, (h) O 1s. | ||
The spectra of Li and Ti elements are performed to verify the presence of LiTiO2. As shown in Fig. 3f, the Li 1s spectrum located at 55.11 eV is the characteristic peak of Li–O bond, and the characteristic peak of Ti 3s can also be observed, which provides evidence for the presence of Ti.20 Due to the relatively low concentration of Ti, the Ti 2p XPS spectrum exhibits a poor signal-to-noise ratio, but it is possible to find clear evidence of the presence of Ti3+ at 455.7 eV and 461.1 eV. The peaks at 458.6 eV and 464.4 eV are satellite peaks of Ti3+.30,31 The O 1s spectra in Fig. 3h show three forms of oxygen state. The lattice oxygen content at 529.76 eV is 70.50%, the relative concentration of oxygen vacancies at 531.48 eV is 22.82%, and the surface chemisorbed oxygen content is 6.68%.
Lithium storage performance
The CV curves of HEO and HEO@LTO/C from the 1st to 3rd cycles are presented in Fig. 4a and b, respectively. In the first cycle of HEO, there is an obvious cathodic peak around 0.54 V corresponding to the reduction of metal cations and the formation of SEI. The anodic peak at 1.64 V is identified as the oxidation of metal nanoparticles and the decomposition of Li2O. In the subsequent two cycles, the cathodic peak drifted to about 1.0 V. In Fig. 4b, the peak around 0.05 V in the 1st cathodic scan is usually interpreted as the decomposition potential of the electrolyte.32 The response peaks at this potential become very sharp, which may be related to the significant increase in the specific surface area of the coated particles. Another proof is that the sharp anodic peak in the first cycle nearly disappears in the subsequent two cycle curves. It can be seen that there are two more faint reduction peaks located around 0.8 V in the first scan and 0.5 V in the latter cycles, and one small peak around 2.0 V in the cathodic scan. This is the characteristic of the participation of LiTiO2 in the electrochemical reaction.30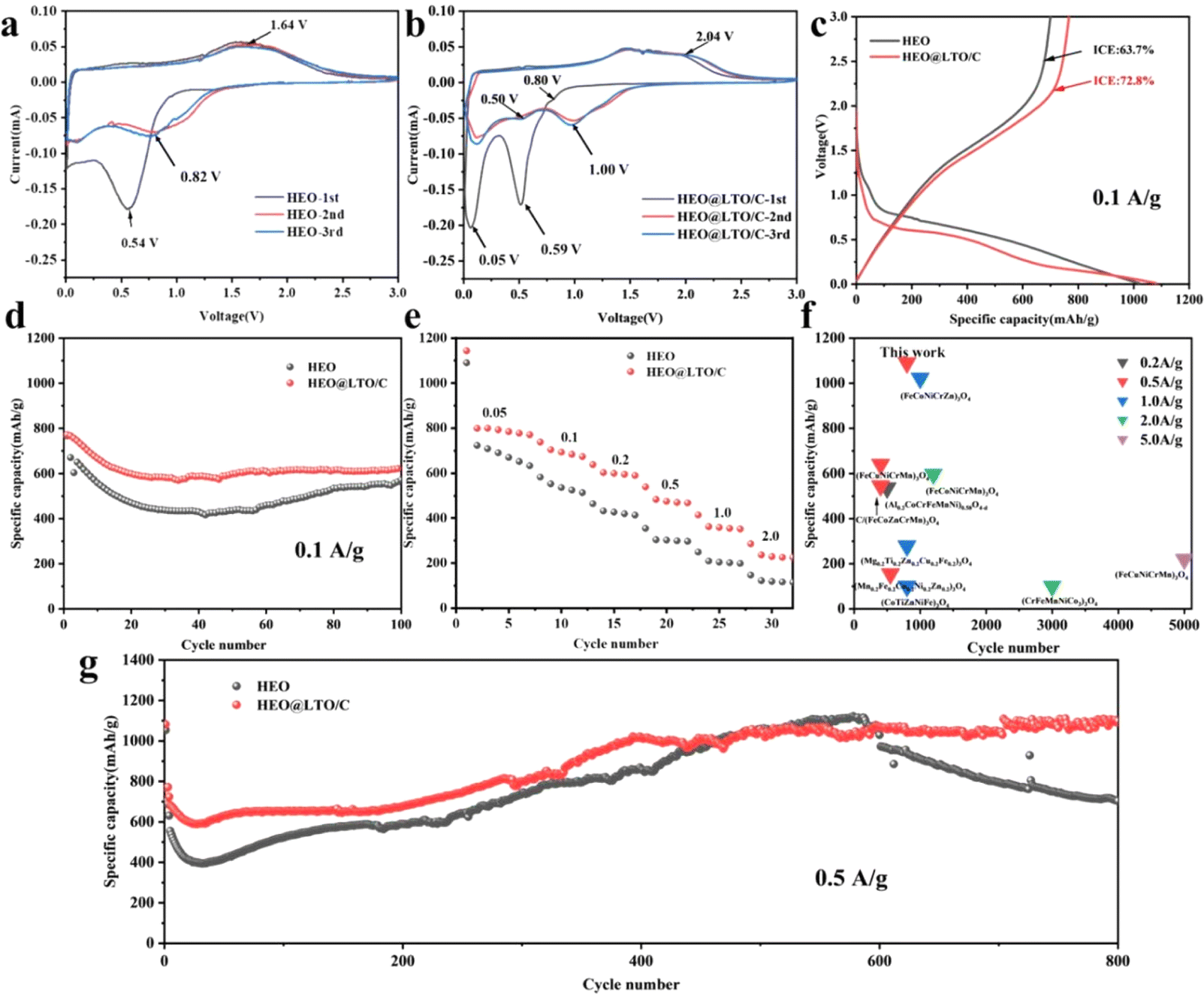 | ||
| Fig. 4 CV curves of HEO (a) and HEO@LTO/C (b) at 0.05 mV s−1 from 1st to 3rd cycles. The results of initial charge–discharge curves (c) and 100 cycles performance (d) at 0.1 A g−1 for HEO and HEO@LTO/C. (e) The rate performances of HEO and HEO@LTO/C from 0.05 to 2 A g−1. (f) Comparison of long-term cycle performance between this work and relative literature. (g) The long-cycle performance of HEO and HEO@LTO/C at 0.5 A g−1. | ||
In Fig. 4c, the charge–discharge curves of both samples roughly show similar shapes, indicating that the fundamental reaction mechanisms remain unchanged. However, the HEO@LTO/C sample displays slightly lower polarization, as evidenced by the smaller voltage gap between charge and discharge curves. This reduced polarization aligns with the CV results and further confirms the improved conductivity and charge transfer kinetics provided by the modified strategy. Moreover, the initial coulombic efficiencies of HEO and HEO@LTO/C are 63.7% and 72.8%, respectively. The initial capacity loss is generated from the irreversible transformation of Li2O and the formation of SEI, which is a common phenomenon in TMO anodes.3,33 The increased initial efficiency of HEO@LTO/C indicates that the irreversible reaction of Li2O transformation and SEI information have been alleviated, demonstrating the positive influence of the hybrid coating layer.
The cycling performances of HEO and HEO@LTO/C at 0.1 A g−1 are displayed in Fig. 4d. The capacity decay of HEO is much more obvious than HEO@LTO/C, showing a lower stability. Especially at the lowest point of the cycling curve (at about 40 cycles), the specific capacity of HEO@LTO/C is 150 mA h g−1 higher than that of HEO. According to the structure character of HEO@LTO/C, it is proved that Ti doping and the inorganic coating-layer could enhance the cycle stability. The rate performances at 0.05 A g−1, 0.1 A g−1, 0.2 A g−1, 0.5 A g−1, 1.0 A g−1, and 2.0 A g−1 are presented in Fig. 4e. It is clearly that the reversible capacities of HEO@LTO/C are higher than HEO at different current densities. At 2.0 A g−1, HEO@LTO/C displays above 280 mA h g−1, but HEO only exhibits about 100 mA h g−1. It is demonstrated that HEO@LTO/C owns much better dynamics.
To further investigate the cycle stability, the performance comparison between HEO and HEO@LTO/C within 1000 cycles at 0.5 A g−1 is shown in Fig. 4g. As the cycle goes on, the reversible capacity starts to show a rising trend, which is caused by the fragmented crystals. The fresh interface along the fragmented crystals could bring on persistent electrolyte decomposition.34 Hence, the specific capacity of HEO starts to collapse at about the 600th cycle due to the exhaustion of electrolytes. However, the capacity of HEO@LTO/C keeps increasing in the whole cycle-span test, indicating a hindered interface irreversible reaction. As shown in Fig. 4f, the cycle performance of this work is the best among existing SHEOs with similar compositions.3,10,28,35–40
To further characterize the kinetic properties, the EIS and CV curves at different scan speeds were tested. The CV curves of HEO and HEO@LTO/C at scanning rates of 0.1, 0.2, 0.5, and 1.0 mV s−1 are shown in Fig. 5a and b. As the scanning rate increases, the positions of the oxidation and reduction peaks are shifted. But the curve of HEO@LTO/C shows less variation at different scanning rates, indicating better stability at the electrode/electrolyte interface brought by the hybrid layer. The CV curves of HEO show obvious peak shifts at high scanning rates, suggesting that the diffusion control process is more significant. And the peak shift of HEO@LTO/C is smaller, indicating that its diffusion resistance is lower because of the LTO coating.41,42 What's more, the CV curves of HEO@LTO/C are more symmetrical at different scanning rates, which suggests that the kinetics of the electrochemical reaction have been improved.43 The hybrid coating layer can provide HEO@LTO/C with a larger active surface and more defects, so the pseudocapacitive effect of the electrode cannot be ignored.44 According to the pseudo capacitance share plot in Fig. 5c, it can be seen that the capacitance contribution of HEO@LTO/C is higher than that of HEO, indicating that the coating layer enhances the electron transfer in the conversion reaction. This result is supported by the impedance test in Fig. 5d. The impedance results measured before cycling often correspond to the properties of the material itself. The charge transfer resistance Rct of HEO@LTO/C is significantly smaller than that of HEO, representing an increase in electronic conductivity thanks to the carbon coating. The linear relationship between Z′ and ω−0.5 is related to the diffusion process with electrolyte ions on the electrode surface. The slope of HEO@LTO/C is 367.33, lower than that of HEO at 433.41. Therefore, HEO@LTO/C has a superior ionic diffusion rate than HEO (Fig. S1†).
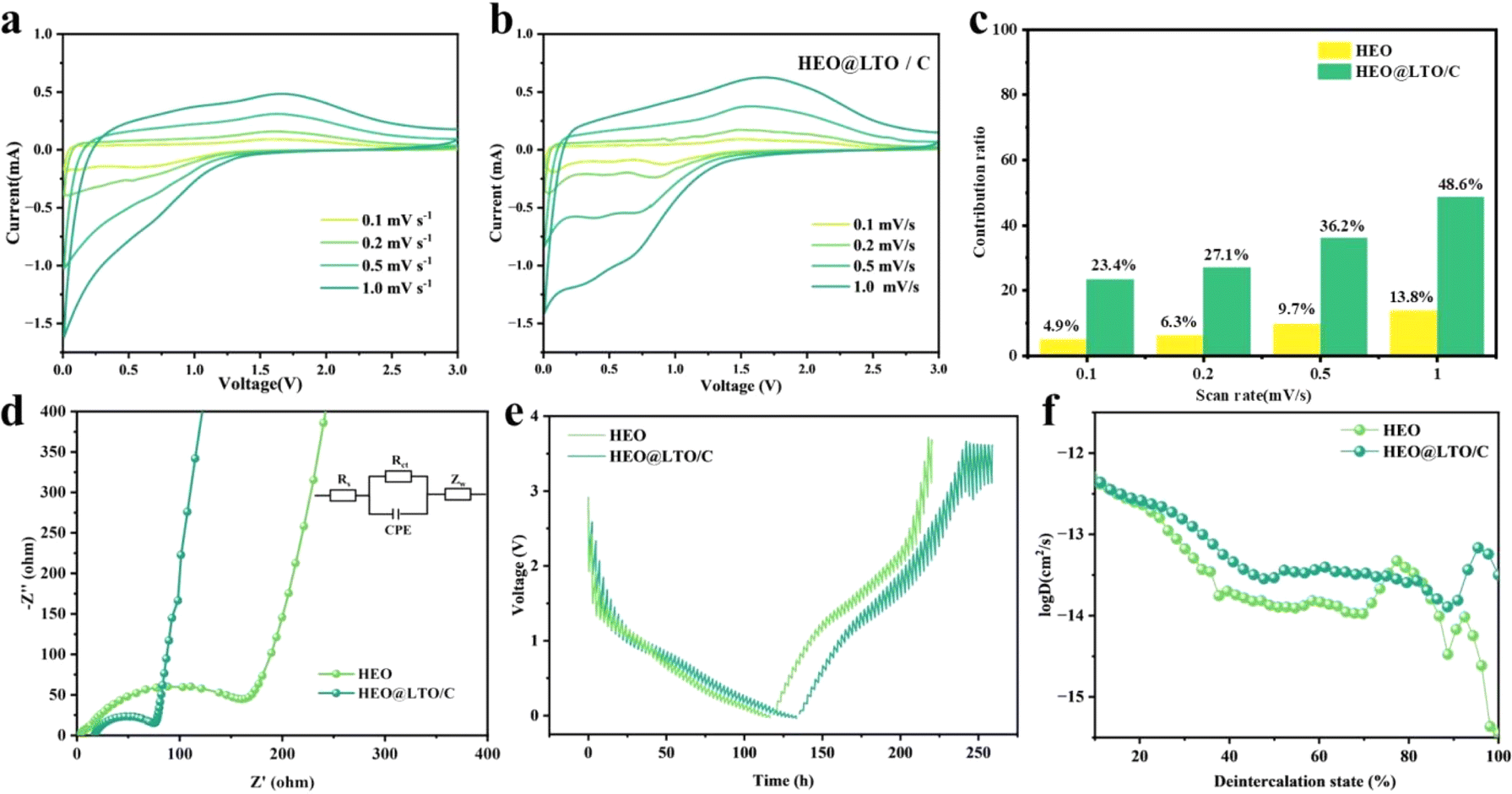 | ||
| Fig. 5 The comparison of HEO and HEO@LTO/C in electrochemical tests: CV curves of (a) HEO and (b) HEO@LTO/C at different scan rates, (c) the ratio of capacitance contribution at different scan rates, (d) Nyquist plots of the electrodes before the cycle, (e) GITT curves, (f) the DLi+ at different deintercalation state. | ||
The galvanostatic interval titration technique (GITT) is performed on HEO and HEO@LTO/C to further characterize lithium ion diffusion behavior. Based on the data collected from the tests and the formula below, the specific lithium-ion diffusion rate can be obtained.
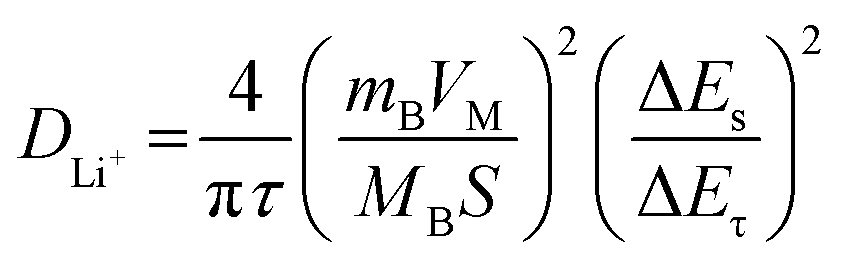 | (1) |
As shown in Fig. 5e, the GITT curves indicate that the main difference between the coated and uncoated samples lies in the later lithium extraction process. Although there are some fluctuations, the overall charge–discharge curve shape of HEO@LTO/C is similar to that of HEO, indicating that the basic reaction mechanism has not undergone significant changes. By presenting the variation of Li-ion diffusion coefficient with the degree of lithium extraction (Fig. 5f), it can be clearly seen that the Li-ion diffusion coefficient of HEO@LTO/C is significantly higher than that of HEO.
Stability enhancement
Having a stable and uniform electrode–electrolyte interface is crucial for rechargeable batteries with high energy density and long cycle life.45 XPS was used to evaluate the chemical stability of the electrode surface in the surface composition analysis of the electrodes after 40 cycles. As shown in Fig. 6a and b, C 1s spectra were mainly used to evaluate the decomposition of carbonate solvents, which may decompose during battery charging and discharging to form side products and affect the battery performance. And F 1s spectra are usually used to evaluate the decomposition of LiPF6 salt,46 which is a commonly used electrolyte salt in LIBs. Its decomposition also negatively affects the battery performance. By comparing Fig. 6a and b, it is evident that the overall content of C in the C 1s spectra decreases at the same test intensity. This phenomenon indicates that the decomposition of the carbonate solvent is reduced during the cycling process. This may be due to the protective effect of the coating layer on the surface of HEO@LTO/C. The uncoated HEO underwent obvious electrolyte decomposition. These changes can be attributed to the structural reconstruction of the SEI film, with an outer layer of organic compounds. Accompanying the continuous electrochemical reduction and decomposition of the electrolyte into main products like Li2CO3 and organic carbonates.47 The coating layer reduces the direct contact between the electrolyte and the electrode material, thus inhibiting the decomposition of the carbonate solvent. Similarly, in Fig. 6c and d, the relative content of LixPOyFz is significantly increased, implying the reduction of decomposition during the cycling process.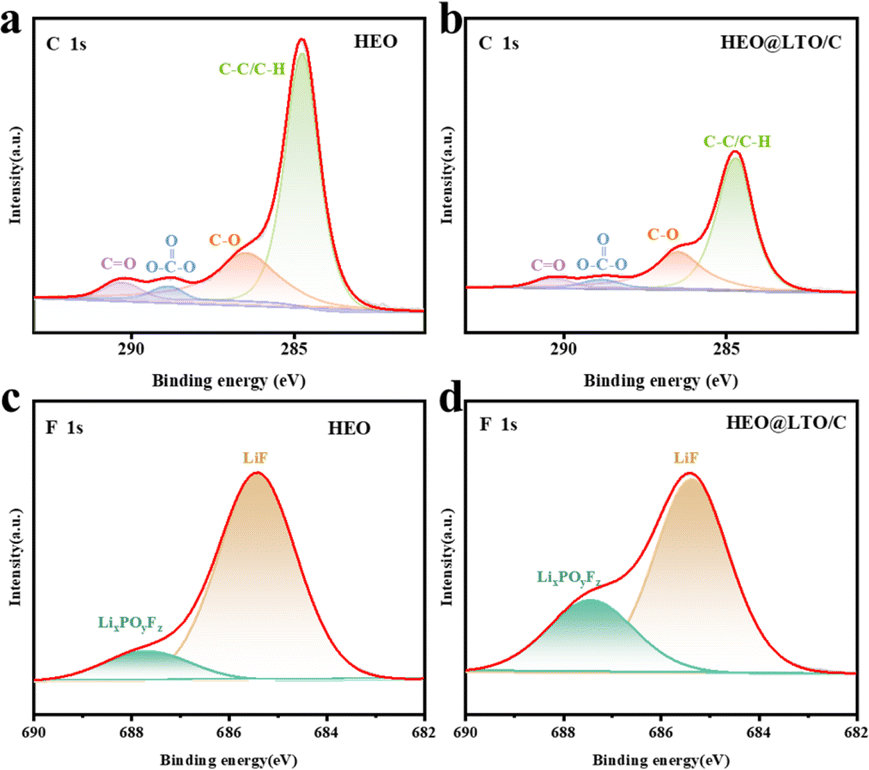 | ||
| Fig. 6 XPS spectra and fitting results of C 1s (a) and (b) and F 1s (c) and (d) after 40 cycles in HEO and HEO@LTO/C. | ||
Based on these results, it can be concluded that the coating layer on the surface of the electrode can effectively inhibit the decomposition of the main components in the electrolyte. This inhibition helps to maintain the chemical stability of the electrode material and reduces the occurrence of side reactions, thus improving the cycling stability.
In order to compare the structural stability of the electrode plates before and after coating, SEM was used in the detailed surface and cross-section morphology observation of the electrode plate after 40 cycles.
First, the observation of the surface morphology is performed in Fig. 7a and b. At a magnification of 1000, the size of the active substance particles is relatively uniform and larger particles are largely invisible after 40 cycles in the uncoated sample. The particles underwent significant fragmentation during the electrochemical reaction. This may be due to the volume change caused by the extraction and insertion of lithium ions, increasing the mechanical stress of the particles. In contrast, more large particles appeared on the surface of the coated sample. This observation visually demonstrates the inhibiting effect of the coating layer on particle fragmentation and the protection of the structural integrity of the electrode material.
 | ||
| Fig. 7 SEM images of the surface and cross-section of the electrode after 40 cycles: (a) surface of the HEO electrode; (b) surface of the HEO@LTO/C electrode; (c) cross section of the HEO electrode; (d) cross section of the HEO@LTO/C electrode. | ||
The cross-sectional thickness of the electrode sheet is an important parameter to characterize the volume expansion of the electrode material, especially in materials with large changes in the volume of active substance particles.48 In Fig. 7c, the thickness of the electrode plate in the uncoated sample HEO can reach 46 μm after 40 cycles. However, in Fig. 7d, the thickness of the electrode plate in HEO@LTO/C is only 27 μm, indicating that the coating significantly inhibits vertical volume expansion.
Conclusions
In summary, as-prepared HEO@LTO/C owns a hybrid coating-layer consisting of ionic and electron conductors plus slight Ti-doping. When applied to the anode of LIB, the reversible specific capacity of HEO@LTO/C reaches 1090 mA h g−1 and remains about 1000 mA h g−1 after 800 cycles at 0.5 A g−1. Compared to original HEO without any surface coating-layer, HEO@LTO/C's initial coulombic efficiency increases from 63.7% to 72.8% and the rate capacities are always at least 100 mA h g−1 higher at 0.5–2 A g−1. Series of characterizations have confirmed that the enhanced performances of HEO@LTO/C originate from the following merits: (1) the hybrid coating-layer acts as an efficient electron and ion diffusion channel to improve the electrochemical dynamics at the electrolyte/electrode interface; (2) the inorganic hybrid coating-layer could hinder the corrosion from organic electrolyte to improve interface stability; (3) doped Ti elements serve as steady metallic framework to enhance structure stability. Therefore, it provides a simple surface-modified strategy to improve the lithium storage and cycle stability of HEO in this work. Building on these findings, future research could explore the optimization of the hybrid coating layer to further enhance the modified effect. This may involve fine-tuning the composition and thickness of the coating to achieve even better electrochemical performance and stability. Additionally, investigating the scalability of this surface modification technique for large-scale production could pave the way for its commercial application, potentially revolutionizing the development of high-performance anode materials for next-generation LIBs.Data availability
The data are available from the corresponding author on reasonable request.Author contributions
Feiyue Zhai: conceptualization, investigation, data curation, formal analysis, writing – original draft. Shengji Gao: data curation, formal analysis. Wenfeng Zhang: conceptualization, investigation, data curation, formal analysis. Gaoping Cao: conceptualization, investigation, resources. Huimin Zhang: conceptualization, investigation, data curation, formal analysis. Yalan Xing: validation, supervision, writing – review & editing. Yu Xiang: conceptualization, investigation, validation, supervision, writing – review & editing, funding acquisition. Shichao Zhang: validation, supervision, writing – review & editing.Conflicts of interest
The authors have no conflicts to disclose.Acknowledgements
This work is supported by the National Natural Science Foundation of China (NSFC 22109176).References
- L. Su, J. Ren, T. Lu, K. Chen, J. Ouyang, Y. Zhang, X. Zhu, L. Wang, H. Min, W. Luo, Z. Sun, Q. Zhang, Y. Wu, L. Sun, L. Mai and F. Xu, Adv. Mater., 2023, 35, e2205751 CrossRef PubMed.
- N. Qiu, H. Chen, Z. Yang, S. Sun, Y. Wang and Y. Cui, J. Alloys Compd., 2019, 777, 767–774 CrossRef CAS.
- B. Xiao, G. Wu, T. Wang, Z. Wei, Y. Sui, B. Shen, J. Qi, F. Wei and J. Zheng, Nano Energy, 2022, 95, 106962 CrossRef CAS.
- X. Liu, X. Li, Y. Li, H. Zhang, Q. Jia, S. Zhang and W. Lei, EcoMat, 2022, 4, e12261 CrossRef CAS.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kübel, T. Brezesinski, S. S. Bhattacharya, H. Hahn and B. Breitung, Nat. Commun., 2018, 9, 3400 CrossRef.
- D. Gu, W. Li, F. Wang, H. Bongard, B. Spliethoff, W. Schmidt, C. Weidenthaler, Y. Xia, D. Zhao and F. Schüth, Angew. Chem., Int. Ed., 2015, 54, 7060–7064 CrossRef CAS.
- S. H. Yu, S. H. Lee, D. J. Lee, Y. E. Sung and T. Hyeon, Small, 2015, 12, 2146–2172 CrossRef PubMed.
- S. L. P. Poizot, S. Grugeon, L. Dupont and J.-M. Tarascon, Nature, 2000, 407, 496–499 CrossRef.
- N. Zhang, X. Dong, Q. Yan, J. Wang, F. Jin, J. Liu, D. Wang, H. Liu, B. Wang and S. Dou, Energy Storage Mater., 2024, 72, 103734 CrossRef.
- H. Chen, N. Qiu, B. Wu, Z. Yang, S. Sun and Y. Wang, RSC Adv., 2020, 10, 9736–9744 RSC.
- X. L. Wang, E. M. Kim, T. G. Senthamaraikannan, D.-H. Lim and S. M. Jeong, Chem. Eng. J., 2024, 484, 149509 CrossRef CAS.
- T. X. Nguyen, J. Patra, C. C. Tsai, W. Y. Xuan, H. Y. T. Chen, M. S. Dyer, O. Clemens, J. Li, S. B. Majumder, J. K. Chang and J. M. Ting, Adv. Funct. Mater., 2023, 33, 2307923 Search PubMed.
- J. Zhao, X. Yang, Y. Huang, F. Du and Y. Zeng, ACS Appl. Mater. Interfaces, 2021, 13, 58674–58681 CrossRef CAS PubMed.
- C.-Y. Huang, C.-W. Huang, M.-C. Wu, J. Patra, T. Xuyen Nguyen, M.-T. Chang, O. Clemens, J.-M. Ting, J. Li, J.-K. Chang and W.-W. Wu, Chem. Eng. J., 2021, 420, 129838 CrossRef CAS.
- F. Zhai, X. Zhu, W. Zhang, G. Cao, H. Zhang, Y. Xing, Y. Xiang and S. Zhang, J. Power Sources, 2024, 603, 234418 CrossRef CAS.
- J.-Z. Yen, Y.-C. Yang and H.-Y. Tuan, Chem. Eng. J., 2022, 450, 137924 CrossRef CAS.
- C. Jin, Y. Wang, Y. Wei, R. Nan, Z. Jian, Z. Yang and Q. Ding, J. Power Sources, 2024, 613, 234926 CrossRef CAS.
- H. Guo, J. Shen, T. Wang, C. Cheng, H. Yao, X. Han and Q. Zheng, Ceram. Int., 2022, 48, 3344–3350 CrossRef CAS.
- Z. Wang, S. Huang, B. Chen, H. Wu and Y. Zhang, J. Mater. Chem. A, 2014, 2, 19983–19987 RSC.
- Y. Sun, C. Wang, W. Huang, G. Zhao, L. Duan, Q. Liu, S. Wang, A. Fraser, H. Guo and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202300962 CrossRef CAS PubMed.
- J.-Y. Eom, Y.-H. Cho, S.-I. Kim, D. Han and D. Sohn, J. Alloys Compd., 2017, 723, 456–461 CrossRef CAS.
- Y.-D. Xu, W. Xiang, Z.-G. Wu, C.-L. Xu, Y.-C. Li, X.-D. Guo, G.-P. Lv, X. Peng and B.-H. Zhong, Electrochim. Acta, 2018, 268, 358–365 CrossRef CAS.
- T.-F. Yi, J. Shu, Y. Wang, J. Xue, J. Meng, C.-B. Yue and R.-S. Zhu, Surf. Coat. Technol., 2011, 205, 3885–3889 CrossRef CAS.
- P. Liu, L. Xiao, Y. Chen and H. Chen, Ceram. Int., 2019, 45, 18398–18405 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- S. Hou, L. Su, S. Wang, Y. Cui, J. Cao, H. Min, J. Bao, Y. Shen, Q. Zhang, Z. Sun, C. Zhu, J. Chen, Q. Zhang and F. Xu, Adv. Funct. Mater., 2023, 34, 202307923 Search PubMed.
- B. Xiao, G. Wu, T. Wang, Z. Wei, Z. Xie, Y. Sui, J. Qi, F. Wei, X. Zhang, L.-b. Tang and J.-c. Zheng, ACS Appl. Mater. Interfaces, 2023, 15, 2792–2803 CrossRef CAS PubMed.
- J. Patra, T. X. Nguyen, C. C. Tsai, O. Clemens, J. Li, P. Pal, W. K. Chan, C. H. Lee, H. Y. T. Chen, J. M. Ting and J. K. Chang, Adv. Funct. Mater., 2022, 32, 2110992 CrossRef CAS.
- F. Liu, Z. Chen, G. Fang, Z. Wang, Y. Cai, B. Tang, J. Zhou and S. Liang, Nano-Micro Lett., 2019, 11, 25 CrossRef CAS PubMed.
- C. Lu, R. Fang, Y. Gan, X. He, Z. Xiao, H. Huang, J. Zhang, X. Xia, W. Zhang and Y. Xia, ACS Appl. Mater. Interfaces, 2023, 16, 898–906 CrossRef.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- A. Ponrouch, P.-L. Taberna, P. Simon and M. R. Palacín, Electrochim. Acta, 2012, 61, 13–18 CrossRef CAS.
- D. Wang, S. Jiang, C. Duan, J. Mao, Y. Dong, K. Dong, Z. Wang, S. Luo, Y. Liu and X. Qi, J. Alloys Compd., 2020, 844, 156158 CrossRef CAS.
- H. Kim, W. Choi, J. Yoon, J. H. Um, W. Lee, J. Kim, J. Cabana and W. S. Yoon, Chem. Rev., 2020, 120, 6934–6976 CrossRef CAS.
- H.-Z. Xiang, H.-X. Xie, Y.-X. Chen, H. Zhang, A. Mao and C.-H. Zheng, J. Mater. Sci., 2021, 56, 8127–8142 CrossRef CAS.
- B. Petrovičovà, W. Xu, M. G. Musolino, F. Pantò, S. Patanè, N. Pinna, S. Santangelo and C. Triolo, Appl. Sci., 2022, 12, 5969 CrossRef.
- X. Yang, H. Wang, Y. Song, K. Liu, T. Huang, X. Wang, C. Zhang and J. Li, ACS Appl. Mater. Interfaces, 2022, 14, 2c07576 Search PubMed.
- C. Triolo, M. Maisuradze, M. Li, Y. Liu, A. Ponti, G. Pagot, V. Di Noto, G. Aquilanti, N. Pinna, M. Giorgetti and S. Santangelo, Small, 2023, 19, e2304585 CrossRef PubMed.
- C. Liu, J. Bi, L. Xie, X. Gao and L. Meng, Mater. Today Commun., 2023, 35, 106315 CrossRef CAS.
- C. Liu, J. Bi, L. Xie, X. Gao and J. Rong, J. Energy Storage, 2023, 71, 108211 CrossRef.
- W. Pholauyphon, P. Charoen-amornkitt, T. Suzuki and S. Tsushima, Electrochem. Commun., 2024, 159, 107654 CrossRef CAS.
- F. Wang, B. Wang, Z. Yu, C. Zhu, P. Liu, J. Li, B. Wang, Y. Zhou, D. Wang, H. K. Liu and S. Dou, Cell Rep. Phys. Sci., 2022, 3, 100872 CrossRef CAS.
- Q. Wang, G. Yu, B. Luo, W. Ji, Z. Liu, M. Li, Y. Nong, Y. Tian, X. Wang, J. Zhang, C. L. Chen, C. K. Chang, Z. Sang, Z. Zhao, R. Zhao and J. Liang, ACS Nano, 2024, 18, 18622–18634 CrossRef CAS PubMed.
- N. Zhang, Q. Yan, X. Dong, J. Wang, F. Jin, J. Liu, D. Wang, H. Liu, B. Wang and S. Dou, Chin. Chem. Lett., 2024, 110328 CrossRef.
- N. Zhang, B. Wang, F. Jin, Y. Chen, Y. Jiang, C. Bao, J. Tian, J. Wang, R. Xu, Y. Li, Q. Lv, H. Ren, D. Wang, H. Liu, S. Dou and X. Hong, Cell Rep. Phys. Sci., 2022, 3, 101197 CrossRef CAS.
- G. Yang, K. Pan, F. Lai, Z. Wang, Y. Chu, S. Yang, J. Han, H. Wang, X. Zhang and Q. Li, Chem. Eng. J., 2021, 421, 129964 CrossRef CAS.
- Q. Lv, Y. Jing, B. Wang, B. Wu, S. Wang, C. Li, L. Wang, L. Xiao, D. Wang, H. Liu and S. Dou, Energy Storage Mater., 2024, 65, 103122 CrossRef.
- C. Bao, J. Wang, B. Wang, J. Sun, L. He, Z. Pan, Y. Jiang, D. Wang, X. Liu, S. X. Dou and J. Wang, ACS Nano, 2022, 16, 17197–17209 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06878e |
| This journal is © The Royal Society of Chemistry 2024 |