
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mild and convenient protocol for the synthesis of quinoxalin-2(1H)-ones and benzimidazoles†
Zhenbiao Luo *,
Mingyuan Wang,
Guidong Jiang,
Xinye Wang,
Liang Zhao,
Zhihui Hu,
Honghe Li and
Qing Ji
*,
Mingyuan Wang,
Guidong Jiang,
Xinye Wang,
Liang Zhao,
Zhihui Hu,
Honghe Li and
Qing Ji
Department of Brewing Engineering, Moutai Institute, Guizhou, Renhuai 564507, China. E-mail: Luozhenbiao@mtxy.edu.cn
First published on 5th November 2024
Abstract
We present a mild and simple method for the cyclization of N-protected o-phenylenediamines with carbonyl compounds in the presence of trifluoroacetic acid. This method reliably provides various substrates of benzimidazoles and quinoxalin-2(1H)-ones, with all reactions conducted at room temperature, demonstrating excellent substrate adaptability and a broad substrate scope.
Introduction
Nitrogen-containing heterocyclic compounds form the foundational structures of many bioactive natural products and synthetic molecules. Benzimidazoles are extensively utilized in various pharmaceuticals due to their notable anti-inflammatory,1 antibacterial, antiviral2 and anticancer properties,3 as well as their roles as antihypertensive agents, exemplified by drugs such as telmisartan4 and bendazol.5 Similarly, quinoxalines not only exhibit comparable therapeutic effects6 but also display potential as antiparasitic7 and anti-HIV agents.8 For instance, caroverine is recognized for its antioxidant and vasodilatory activities.9 Additionally, quinoxalines are used in various industrial materials, including organic semiconductors10 and efficient electro-luminescent materials11 (Fig. 1). Consequently, enhancing the synthesis methods for benzimidazoles and quinoxalines is crucial for lowering costs in pharmaceutical and industrial production. This ongoing demand makes it imperative for chemists to devise innovative synthetic pathways for the preparation of benzimidazole and quinoxaline derivatives.12 Traditional methods for synthesizing benzimidazoles and quinoxalin-2(1H)-ones typically involve the reaction of aniline derivatives with carbonyl compounds under harsh conditions, often necessitating photocatalysis,13 microwave irradiation,14 high temperatures,15 strong acids16 or metal catalysts.17 While numerous synthetic strategies have been established, we now present a straightforward and convenient approach that facilitates synthesis in an open flask at room temperature (Scheme 1).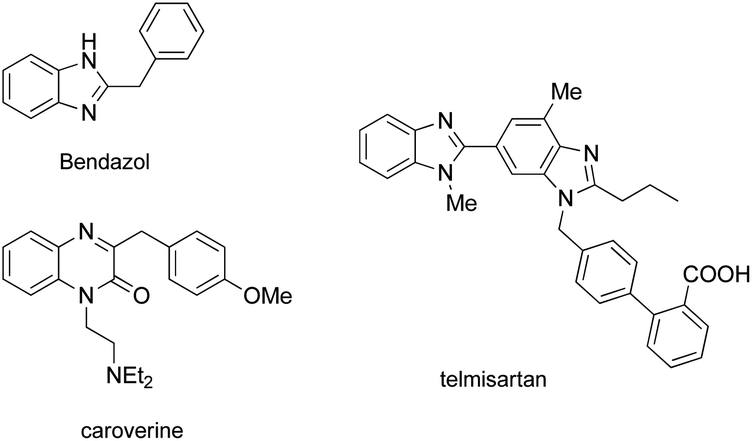 | ||
| Fig. 1 Examples of bioactive quinoxalin-2(1H)-one and benzimidazole derivative molecules. | ||
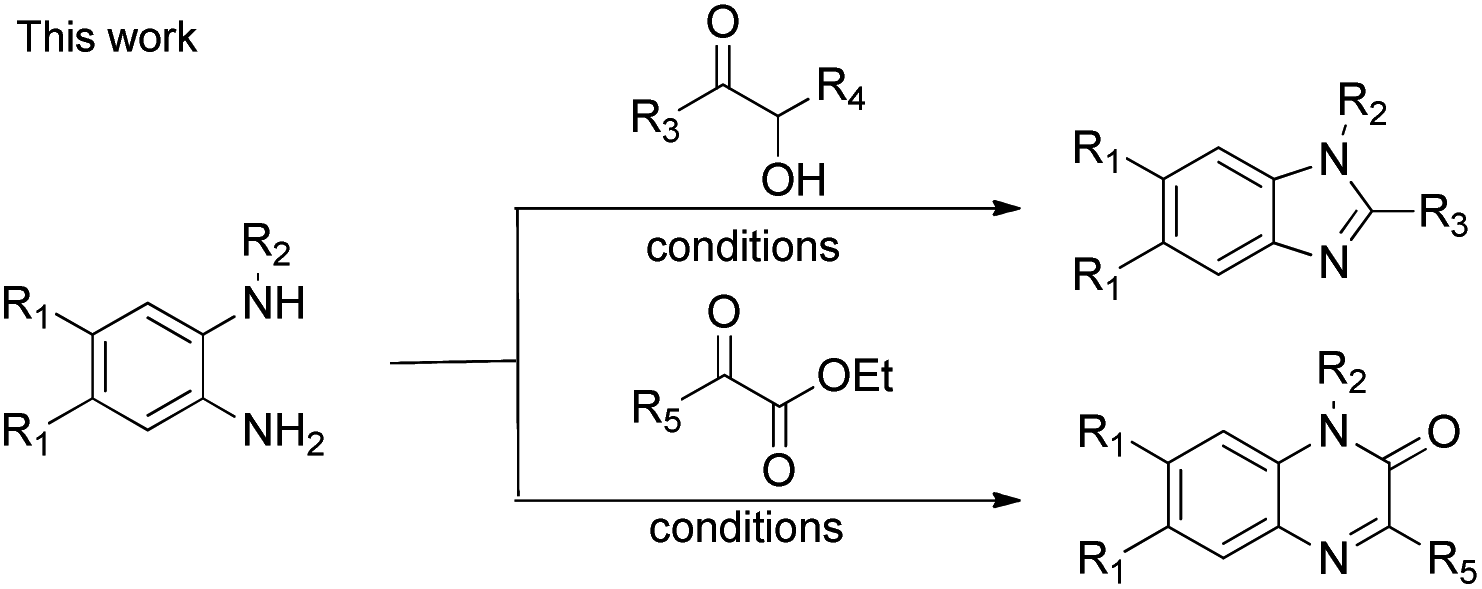 | ||
| Scheme 1 Synthesis of quinoxalin-2(1H)-ones and benzimidazoles. | ||
Results and discussion
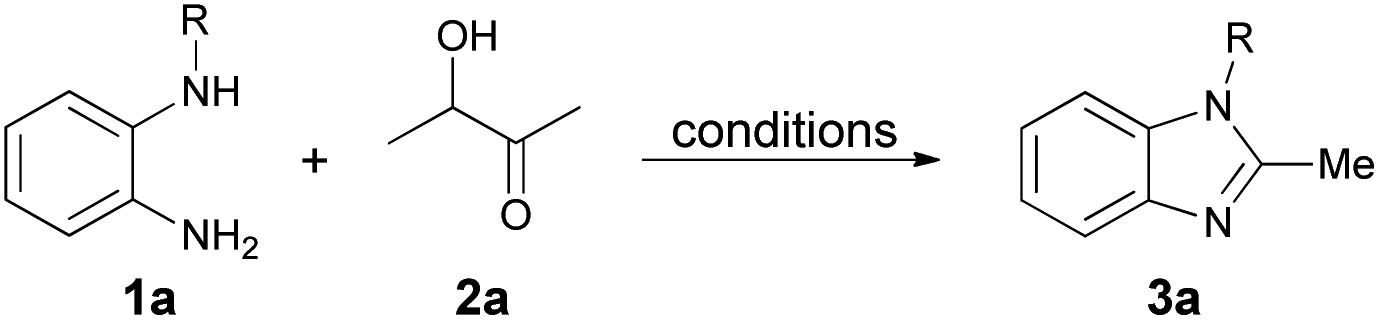
We initiated the model cyclization by reacting N-benzyl-o-phenylenediamine (1a) with acetoin (2a) in the presence of 2 M hydrochloric acid in dichloromethane (DCM) at room temperature (Table 1, entry 1). The reaction completed within two hours, as confirmed by TLC, yielding 78% of 1-benzyl-2-methyl-1H-benzo[d]imidazole (3a). To optimize the reaction conditions, we explored the necessity of an oxygen atmosphere. Reactions conducted under nitrogen and oxygen atmospheres, while maintaining the same experimental conditions, showed that oxygen significantly improved the yield to 83%. In contrast, the reaction failed under nitrogen, even at 40 °C, with only minimal decomposition of N-benzyl-o-phenylenediamine (1a).
|
|||||
|---|---|---|---|---|---|
| Entry | R | Acid | Solvent | T/°C | Yield (%) |
| a Reaction conditions: 1a (0.6 mmol), 2a (0.6 mmol), acid (1.0 eq.), solvent (2.0 mL) in open flask, 2 h.b O2 balloon (1.0 atm).c N2 balloon (1.0 atm). | |||||
| 1 | Bn | HCl | DCM | r.t. | 78 |
| 2 | Bn | HCl | DCM | r.t. | 83b |
| 3 | Bn | HCl | DCM | r.t. | 0c |
| 4 | BOC | HCl | DCM | r.t. | 0 |
| 5 | Ts | HCl | DCM | r.t. | 0 |
| 6 | Ac | HCl | DCM | r.t. | 0 |
| 7 | Bn | H2SO4 | DCM | r.t. | 55 |
| 8 | Bn | CF3COOH | DCM | r.t. | 81 |
| 9 | Bn | CF3SO3H | DCM | r.t. | 65 |
| 10 | Bn | CF3COOH | THF | r.t. | 83 |
| 11 | Bn | CF3COOH | EtOH | r.t. | 82 |
| 12 | Bn | CF3COOH | DMF | r.t. | 79 |
| 13 | Bn | CF3COOH | MeCN | r.t. | 91 |
| 14 | Bn | CF3COOH | MeCN | −15 | 41 |
| 15 | Bn | CF3COOH | MeCN | 0 | 93 |
| 16 | Bn | CF3COOH | MeCN | 40 | 57 |
| 17 | Bn | CF3COOH (2.0 eq.) | MeCN | r.t. | 72 |
| 18 | Bn | CF3COOH (1.5 eq.) | MeCN | r.t. | 83 |
| 19 | Bn | CF3COOH (0.5 eq.) | MeCN | r.t. | 47 |
| 20 | Bn | CF3COOH (0.0 eq.) | MeCN | r.t. | 0 |
This confirmed that the reaction requires oxygen (Table 1, entries 2 and 3). With this critical information, we continued to further optimize the reaction conditions. Unfortunately, some N-protected o-phenylene-diamines did not yield products, likely because electron-withdrawing substituents hindered the cyclization reaction (Table 1, entries 4–6). We treated 1a with 2a in the presence of several acids in dichloromethane at room temperature (Table 1, entries 7–9). Although each acid was examined and the target product 3a was obtained, trifluoroacetic acid demonstrated superior yields compared to other acids. Moreover, a much higher yield was achieved from the reaction conducted in acetonitrile tested respectively under oxygen atmospheres (Table 1, entries 10–13). Regarding temperature, we noted that lower temperatures generally enhanced the yield, although the effect was not substantial (Table 1, entries 14–16). Conversely, at elevated temperatures, the reaction became complex, leading to an increase in by-products and a consequent decrease in the desired product yield. Finally, we assessed the influence of varying amounts of CF3COOH on the reaction. Our findings revealed that the optimal quantity of CF3COOH was 1.0 equivalent (Table 1, entries 17–20). Notably, increasing the amount of acid resulted in shortened reaction times but lower yields, while reducing the acid amount inhibited the reaction's progression. Thus, we established concise and efficient conditions for this cyclization: N-protected o-phenylenediamines (1a, 1.0 eq.) and acyloin (2a, 1.0 eq.) in the presence of CF3COOH (1.0 eq.) in acetonitrile, conducted in an open flask at room temperature (Table 1, entry 13).
With the optimal reaction conditions established, we first explored the substrate scope of N-protected o-phenylene-diamines, and the results were summarized in Table 2. A variety of N-benzyl-1,2-diaminobenzenes, featuring both electron-donating and electron-withdrawing substituents, successfully underwent cyclization with 2a in an open flask, yielding modest to good amounts of the corresponding benzimidazoles (3a–c). Next, we evaluated the scope of α-hydroxy ketones under the established conditions. These substrates delivered high yields, including 1-hydroxycyclohexyl phenyl ketone (2c), despite bearing bulky substituent, 3d–i still achieved moderate yields. Similarly, various N-methyl-1,2-diaminobenzenes with different electron-donating and electron-withdrawing substituents underwent smooth cyclization with a range of α-hydroxy ketones, resulting in moderate to good yields (3j–r). Furthermore, the reaction of N1-phenyl-1,2-diaminobenzene (1g) with α-hydroxy ketones (2a–c) successfully afforded the target products in good yields (3s–u).
|
|||
|---|---|---|---|
| Entry | 1, R1, R2 | 2, R3, R4 | 3, yield (%) |
| a Reaction conditions: a mixture of 1 (0.6 mmol), 2 (0.6 mmol) and CF3COOH (0.6 mmol) in MeCN (2.0 mmol) while stirring in open flask at room temperature. | |||
| 1 | 1a, H, Bn | 2a, Me, Me | 3a, 91 |
| 2 | 1b, Me, Bn | 2a, Me, Me | 3b, 81 |
| 3 | 1c, F, Bn | 2a, Me, Me | 3c, 83 |
| 4 | 1a, H, Bn | 2b, Et, Et | 3d, 89 |
| 5 | 1b, Me, Bn | 2b, Et, Et | 3e, 78 |
| 6 | 1c, F, Bn | 2b, Et, Et | 3f, 82 |
| 7 | 1a, H, Bn | 2c, Ph, Cy | 3g, 74 |
| 8 | 1b, Me, Bn | 2c, Ph, Cy | 3h, 59 |
| 9 | 1c, F, Bn | 2c, Ph, Cy | 3i, 63 |
| 10 | 1d, H, Me | 2a, Me, Me | 3j, 89 |
| 11 | 1e, Me, Me | 2a, Me, Me | 3k, 69 |
| 12 | 1f, F, Me | 2a, Me, Me | 3l, 68 |
| 13 | 1d, H, Me | 2b, Et, Et | 3m, 86 |
| 14 | 1e, Me, Me | 2b, Et, Et | 3n, 64 |
| 15 | 1f, F, Me | 2b, Et, Et | 3o, 62 |
| 16 | 1d, H, Me | 2c, Ph, Cy | 3p, 73 |
| 17 | 1e, Me, Me | 2c, Ph, Cy | 3q, 58 |
| 18 | 1f, F, Me | 2c, Ph, Cy | 3r, 60 |
| 19 | 1g, H, Ph | 2a, Me, Me | 3s, 88 |
| 20 | 1g, H, Ph | 2b, Et, Et | 3t, 85 |
| 21 | 1g, H, Ph | 2c, Ph, Cy | 3u, 73 |
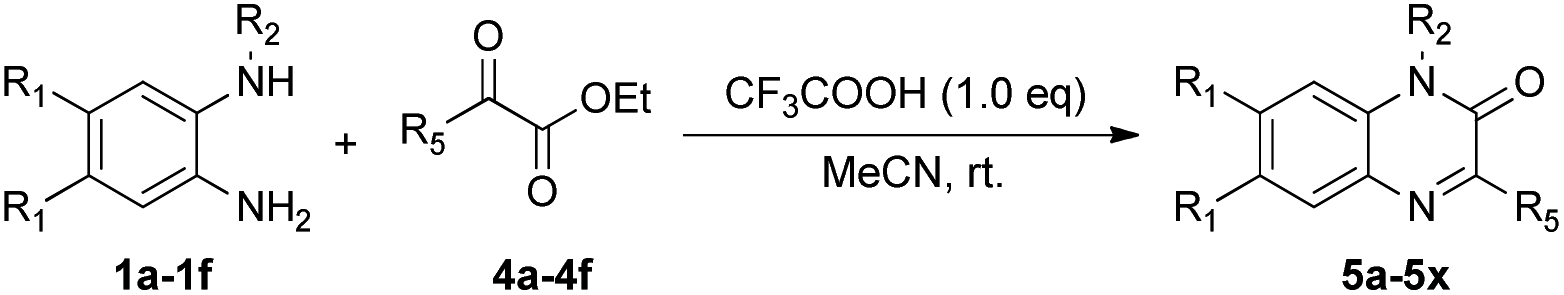
We then investigated the reactions of various N-protected o-phenylenediamines (1) with α-ketoesters (4) under the optimized conditions, as detailed in Table 3. Although the yields were slightly lower when using an open flask, this method was proven relatively straightforward and was therefore preferred (Table 3, entries 1 and 2). The reactions of various N-protected o-phenylenediamines containing electron-donating or electron-withdrawing substituents with α-ketoesters proceeded smoothly, furnishing the corresponding quinoxaline derivatives in moderate yields (5b–i, 5m–u). Notably, the reaction showed good tolerance to a range of functional groups, and ethyl 2-cyclohexyl-2-oxoacetates were successfully introduced into the desired products (5j–l, 5v–x).
|
|||
|---|---|---|---|
| Entry | 1, R1, R2 | 4, R5 | 5, yielda (%) |
| a Reaction conditions: a mixture of 1 (0.6 mmol), 4 (0.6 mmol) and CF3COOH (0.6 mmol) in MeCN (2.0 mmol) while stirring in open flask at room temperature.b N2 balloon (1.0 atm). | |||
| 1 | 1a, H, Bn | 4a, i-Pr | 5a, 78 |
| 2 | 1a, H, Bn | 4a, i-Pr | 5a, 81b |
| 3 | 1b, Me, Bn | 4a, i-Pr | 5b, 57 |
| 4 | 1c, F, Bn | 4a, i-Pr | 5c, 61 |
| 5 | 1a, H, Bn | 4b, n-Pr | 5d, 71 |
| 6 | 1b, Me, Bn | 4b, n-Pr | 5e, 55 |
| 7 | 1c, F, Bn | 4b, n-Pr | 5f, 57 |
| 8 | 1a, H, Bn | 4c, n-Bu | 5g, 55 |
| 9 | 1b, Me, Bn | 4c, n-Bu | 5h, 56 |
| 10 | 1c, F, Bn | 4c, n-Bu | 5i, 57 |
| 11 | 1a, H, Bn | 4d, Cy | 5j, 71 |
| 12 | 1b, Me, Bn | 4d, Cy | 5k, 55 |
| 13 | 1c, F, Bn | 4d, Cy | 5l, 51 |
| 14 | 1d, H, Me | 4a, i-Pr | 5m, 71 |
| 15 | 1e, Me, Me | 4a, i-Pr | 5n, 52 |
| 16 | 1f, F, Me | 4a, i-Pr | 5o, 56 |
| 17 | 1d, H, Me | 4b, n-Pr | 5p, 67 |
| 18 | 1e, Me, Me | 4b, n-Pr | 5q, 65 |
| 19 | 1f, F, Me | 4b, n-Pr | 5r, 71 |
| 20 | 1d, H, Me | 4c, n-Bu | 5s, 66 |
| 21 | 1e, Me, Me | 4c, n-Bu | 5t, 61 |
| 22 | 1f, F, Me | 4c, n-Bu | 5u, 63 |
| 23 | 1d, H, Me | 4d, Cy | 5v, 53 |
| 24 | 1e, Me, Me | 4d, Cy | 5w, 54 |
| 25 | 1f, F, Me | 4d, Cy | 5x, 51 |
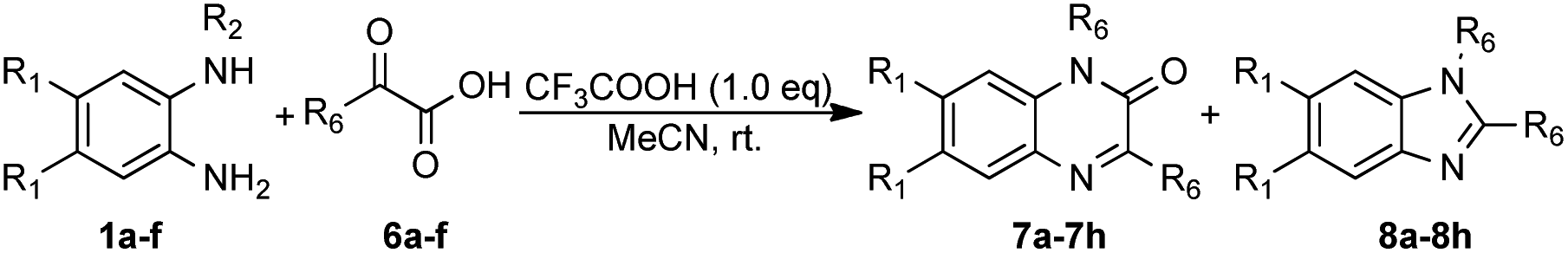
To enhance the practicality of this methodology, the optimized conditions were utilized to synthesize a series of benzimidazole and quinoxaline derivatives. We next examined the reaction of N-phenyl-o-phenylenediamine (1a) with phenylpyruvic acid (6a) under the optimized conditions (Table 4). The reaction successfully produced the target product, 1,3-dibenzyl quinoxalin-2(1H)-one (7a), with a yield of 9%, along with 1,2-dibenzyl-1H-benzo[d]imidazole (8a) at 76% yield, which was a precursor to the antihypertensive drug bendazol. Subsequently, the reaction of 1a with 6a was scaled up to 10 mmol, resulting in a yield of 76% for 8a. N-Phenyl-o-phenylenediamines (1a, 1c) bearing diverse substitutions reacted with trimethylpyruvic acid, yielding benzimidazoles (8b, 8c) in 73% and 71%, respectively, along with 7b and 7c in 11% and 12%. In sharp contrast, the reaction of N1-benzyl-4,5-dimethylbenzene-1,2-diamine (1b) was performed under the standard conditions to isolate 7d. Similarly, the reaction of N1-methyl-benzene-1,2-diamine (1d) with phenylpyruvic acid (6a) afforded 3-benzyl-1-methylquinoxalin-2(1H)-one (7e) but failed to isolate benzimidazoles. Next, we explored the reaction of N-methyl-o-phenylenediamines with various α-ketoesters; however, this obtained only quinoxalines (7f–i). Benzimidazoles were not isolated even in the presence of oxygen. Clearly, substituents played a directing role in the reaction.
|
||||
|---|---|---|---|---|
| Entry | 1, R1, R2 | 6, R6 | 7, yield (%) | 8, yield (%) |
| a Reaction conditions: a mixture of 1 (0.6 mmol), 6 (0.6 mmol) and CF3COOH (0.6 mmol) in MeCN (2.0 mmol) while stirring in open flask at room temperature. | ||||
| 1 | 1a, H, Bn | 6a, Bn | 7a, 9 | 8a, 76 |
| 2 | 1a, H, Bn | 6b, t-Bu | 7b, 11 | 8b, 73 |
| 3 | 1c, F, Bn | 6b, t-Bu | 7c, 12 | 8c, 71 |
| 4 | 1b, Me, Bn | 6b, t-Bu | 7d, 79 | Trace |
| 5 | 1d, H, Me | 6a, Bn | 7e, 69 | Trace |
| 6 | 1d, H, Me | 6b, t-Bu | 7f, 71 | 0 |
| 7 | 1e, Me, Me | 6b, t-Bu | 7h, 73 | 0 |
| 8 | 1f, F, Me | 6b, t-Bu | 7i, 59 | 0 |
Based on the above results and previous reports,18–21 a probable mechanism is proposed. N-protected o-phenylenediamines 1 with α-hydroxy ketone 2 afforded intermediate A in the presence of acid, subsequently, intermediate A undergone the favored 5-exo-tet cyclization to generated intermediate B. Under oxidative conditions, intermediate B was converted into benzimidazole (3), accompanied by the generation of aldehydes or ketones (for details, see the ESI†). In addition, N-protected o-phenylene-diamines 1 with α-ketoesters 4 in the presence of acid to give intermediate C, which subsequently undergone 6-exo-trig cyclization to afford intermediate D. Finally, after the elimination of ethanol, quinoxalin-2(1H)-ones (5) was obtained. The reaction mechanisms for the formation of compounds 7 and 8 was similar to the aforementioned. However, we have not yet been able to provide a satisfactory explanation for the different ratios of 7 and 8. We will continue to investigate this reaction mechanism in our laboratory (Scheme 2).
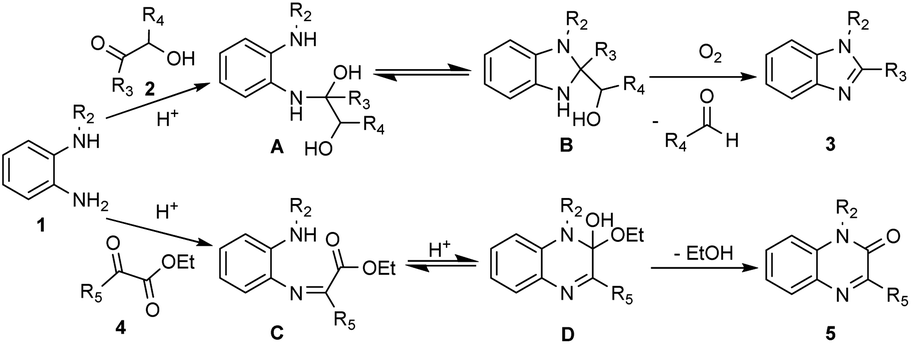 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Conclusions
We have established a concise and efficient protocol for the synthesis of benzimidazoles and quinoxalin-2(1H)-ones in an open flask at room temperature. The method accommodated substrates with a variety of substitutions, obtaining moderate to good yields under mild reaction conditions. On going investigations in our laboratory aim to further explore the applications of this cyclization protocol for the synthesis of additional benzimidazole and quinoxaline derivatives.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the NSFC (No. 22161027); Scientific Research Foundation of Moutai Institute (No. mygccrc[2022]016, mygccrc[2022]004, mygccrc[2022]023, mygccrc[2022]027, mygccrc[2022]031); Joint Project of Zunyi Science & Technology Bureau and Moutai Institute (No. ZunShiKeHe HZ zi[2021]307).Notes and references
- A. F. Mohammed, S. G. Abdel-Moty, M. A. Hussein and A. M. Abdel-Alim, Arch. Pharmacal Res., 2013, 36, 1465–1479 CrossRef CAS PubMed.
- M. Chen, S. Su, Q. Zhou, X. Tang, T. Liu, F. Peng, M. He, H. Luo and W. Xue, J. Saudi Chem. Soc., 2021, 25, 101194–101207 CrossRef CAS.
- O. Algul, A. Kaessler, Y. Apcin, A. Yilmaz and J. Jose, Molecules, 2008, 13, 736–748 CrossRef CAS.
- (a) M. Imenshahidi, A. Roohbakhsh and H. Hosseinzadeh, Biomed. Pharmacother., 2024, 171, 116169–116183 CrossRef CAS PubMed; (b) C. V. Rizos and M. S. Elisaf, Curr. Med. Res. Opin., 2016, 32, 1397–1398 CrossRef.
- J. Charton, S. Girault-Mizzi, M. Debreu-Fontaine, F. Foufelle, I. Hainault, J. Bizot-Espiard, D. Caignard and C. Sergheraert, Bioorg. Med. Chem., 2006, 14, 4490–4518 CrossRef CAS PubMed.
- (a) S. N. Khattab, S. A. H. Abdel Moneim, A. A. Bekhit, A. M. EI Massry, S. Y. Hassan, A. El-Faham, H. E. Ali Ahmed and A. Amer, Eur. J. Med. Chem., 2015, 93, 308–320 CrossRef CAS PubMed; (b) B. Wu, Y. Yang, X. Qin, S. Zhang, C. Jing, C. Zhu and B. Ma, ChemMedChem, 2013, 8, 1913–1917 CrossRef CAS PubMed; (c) R. Liu, Z. H. Huang, M. G. Murray, X. Y. Guo and G. Liu, J. Med. Chem., 2011, 54, 5747–5768 CrossRef CAS.
- J. Soto-Sánchez and J. Da. Ospina-Villa, Chem. Biol. Drug Des., 2021, 98, 683–699 CrossRef PubMed.
- J. Balzarini, A. Karlsson, C. Meichsner, A. Paessens, G. Riess, E. De Clercq and J. P. Kleim, J. Virol., 1994, 68, 7986–7992 CrossRef CAS.
- (a) N. Udilova, A. V. Kozlov, W. Bieberschulte, K. Frei, K. Ehrenberger and H. Nohl, Biochem. Pharmacol., 2003, 65, 59–65 CrossRef CAS PubMed; (b) Y. Ishida, H. Ozaki and S. Shibata, Br. J. Pharmacol., 1980, 71, 343–348 CrossRef CAS PubMed.
- S. Dailey, W. J. Feast, R. J. Peace, I. C. Sage, S. Tilla and E. L. Wood, J. Mater. Chem., 2001, 11, 2238–2243 RSC.
- K. R. J. Thomas, M. Velusamy, J. T. Lin, C. Chuen and Y.-T. Tao, Chem. Mater., 2005, 17, 1860–1866 CrossRef CAS.
- P. Pattanayak, D. Panigrahi, S. Kumari, H. N. Yadav, C. R. Ashby, S. Kerber, M. J. S. A. Shahwan, A. K. Tiwari and G. P. Mishra, J. Mol. Struct., 2024, 1295, 136716–136735 CrossRef CAS.
- (a) J. L. Falcó, M. Piqué, M. González, I. Buira, E. Méndez, J. Terencio, C. Pérez, M. Príncep, A. Palomer and A. Guglietta, Eur. J. Med. Chem., 2006, 41, 985–990 CrossRef PubMed; (b) Y. Qin, M. Hao, C. Xu and Z. Li, Green Chem., 2021, 23, 4161–4169 RSC; (c) M.-C. Wu, M.-Z. Li, J.-Y. Chen, J.-A. Xiao, H.-Y. Xiang, K. Chen and H. Yang, Chem. Commun., 2022, 58, 11591–11594 RSC; (d) C. Lu, J. Qu, C. Sun, S. Mao, D. Yu, M. Xue and X. Sun, J. Mol. Struct., 2024, 1312, 138496–138505 CrossRef CAS; (e) Y. Shiogai, M. Okaa and H. Iida, Org. Biomol. Chem., 2023, 21, 2081–2085 RSC; (f) S. Dadwal, M. Kumar and V. Bhalla, J. Org. Chem., 2020, 85, 13906–13919 CrossRef CAS PubMed; (g) R. Chen, Z. Jalili and R. Tayebee, RSC Adv., 2021, 11, 16359–16375 RSC; (h) H. Zhang, J. Xu, M. Zhou, J. Zhao, P. Zhang and W. Li, Org. Biomol. Chem., 2019, 17, 10201–10208 RSC; (i) S. Kumari, A. Joshi, I. Borthakur and S. Kundu, J. Org. Chem., 2023, 88, 11523–11533 CrossRef CAS PubMed.
- (a) S. Y. Kim, K. H. Park and Y. K. Chung, Chem. Commun., 2005, 1321–1323 RSC; (b) J. Gris, R. Glisoni, L. Fabian, B. Fernández and A. G. Moglioni, Tetrahedron Lett., 2008, 49, 1053–1056 CrossRef CAS.
- (a) S. Lin and L. Yang, Tetrahedron Lett., 2005, 46, 4315–4319 CrossRef CAS; (b) S. Kumar, R. Sivakumar., K. Padala and B. Maiti, ChemistrySelect, 2023, 8, e202303665 CrossRef CAS; (c) S. Kumar, K. Padala and B. Maiti, ACS Omega, 2023, 8, 33058–33068 CrossRef CAS.
- (a) A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, A. S. Bijieva, I. V. Aksenovaa and M. Rubin, Org. Biomol. Chem., 2015, 13, 4289–4295 RSC; (b) M. Radhakrishnan, K. D. Arghya, S. T. V. N. V. Tara, T. Sappanimuthu and D. Thangaraj, Chin. Chem. Lett., 2011, 22, 389–392 CrossRef; (c) B. Das, K. Venkateswarlu, K. Suneel and A. Majhi, Tetrahedron Lett., 2007, 48, 5371–5374 CrossRef CAS; (d) H. Huang, X. Lin, S. Yen and C. Liang, Org. Biomol. Chem., 2020, 18, 5726–5733 RSC; (e) M. N. Noolvi, H. M. Patel, V. Bhardwaj and A. Chauhan, Eur. J. Med. Chem., 2011, 46, 2327–2346 CrossRef CAS PubMed.
- (a) K. M. H. Nguyen and M. Largeron, Eur. J. Org Chem., 2016, 1025–1032 CrossRef CAS; (b) D. Xie, R. Tian, X. Zhang and S. Tian, Org. Biomol. Chem., 2022, 20, 4518–4521 RSC; (c) X. Zhu and Y. Wei, J. Chem. Res., 2013, 37, 119–121 CrossRef CAS; (d) S. Samanta, S. Mahato, R. Chatterjee, S. Santra, G. V. Zyryanov and A. Majee, Tetrahedron Lett., 2020, 61, 152177–152182 CrossRef CAS; (e) Z. Ke, B. Yu, H. Wang, J. Xiang, J. Han, Y. Wu, Z. Liu, P. Yang and Z. Liu, Green Chem., 2019, 21, 1695–1701 RSC; (f) E. M. Bonku, H. Qin, A. Odilov, S. Abduahadi, S. D. Guma, F. Yang, F. Zhu, H. A. Aisa and J. Shen, RSC Adv., 2024, 14, 6906–6916 RSC; (g) H. Wang and J. Huang, Adv. Synth. Catal., 2016, 358, 1975–1981 CrossRef CAS.
- Y.-S. Lee, Y.-H. Cho, S. Lee, J.-K. Bin, J. Yang, G. Chae and C.-H. Cheon, Tetrahedron, 2015, 71, 532–538 CrossRef CAS.
- Z. Yao, Z. Luo, Y. Pan, X. Zhang, B. Li, L. Xu, P. Wang and Q. Shi, Adv. Synth. Catal., 2022, 364, 658–664 CrossRef CAS.
- S. Lin and L. Yang, Tetrahedron Lett., 2005, 46, 4315–4319 CrossRef CAS.
- Y. Shiraishi, Y. Sugano, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2010, 49, 1656–1660 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and copies of NMR spectra for all compounds. See DOI: https://doi.org/10.1039/d4ra06887d |
| This journal is © The Royal Society of Chemistry 2024 |