
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAg@Mg12@Ag20: a three-layer matryoshka structure with S6 symmetry†
Peng-Bo Liua,
Jing-Jing Guoa,
Yi-Sha Chena,
Hui-Yan Zhao*a,
Jing Wang a and
Ying Liu*ab
a and
Ying Liu*ab
aDepartment of Physics and Hebei Advanced Thin Film Laboratory, Hebei Normal University, Shijiazhuang 050024, Hebei, China. E-mail: hyzhao@hebtu.edu.cn; yliu@hebtu.edu.cn
bNational Key Laboratory for Materials Simulation and Design, Beijing 100083, China
First published on 6th November 2024
Abstract
The C60 fullerene, renowned for its soccer ball-like high-symmetry configuration, has attracted extensive interest. As research on C60 progresses, the synthesis of diverse C60 derivatives and the exploration of embedding varying numbers of atoms within the carbon cage, ranging from singular atoms to entire molecules, have emerged. This trend has prompted investigations into potential high-symmetry structures formed by incorporating main group or transition metal elements. This study presents a detailed analysis of a three-layer Ag@Mg12@Ag20 structure, featuring a Mg12 icosahedron enclosed within an Ag20 dodecahedron with a singular Ag atom at its core. Employing density-functional theory, the structure underwent comprehensive scrutiny, including energy minimization resulting in the adoption of a S6 symmetry, and subsequent evaluation of stability via vibrational frequency analysis and molecular dynamics simulations. The electronic structures and bonding characteristics of this three-layer Ag@Mg12@Ag20 architecture were explored through electron density analysis, density of states, and adaptive natural density partitioning analysis. Considering structural stability, the proposed three-layer Ag@Mg12@Ag20 structure exhibits promise as a novel constituent in the construction of other nano-materials.
Introduction
Highly connected and symmetric molecular cages, such as famous fullerene C60, have captivated the attention of chemists due to their unique bonding characteristics, as well as their potential to exhibit exceptional stability and reactivity. The incorporation of endohedral doping into these molecular cages has given rise to a family of endohedral clusters with remarkable properties.1 For instance, Health et al. unveiled the stable endohedral cluster La@C60, which belongs to the metallofullerene family.2 In the endohedral fullerenes, often with 60 or more carbon atoms, the doped atom does not interact strongly with the carbon cage. However, for smaller metallofullerenes of carbon, specifically M@Cn with n ≤ 28, metal doping has been employed to enhance the stability of the carbon fullerene cages. This instability arises due to the increased curvature of these carbon fullerene cages. T. Guo et al. experimentally observed U@C28 through the mass spectral analysis of U@Cn clusters.3Inspired by the idea of stabilizing the C28 fullerene cage by endohedral metal atom, metal doping has been used to stabilize cage clusters of other elements that are generally not stable. This idea has led to the prediction and subsequent experimental confirmation of novel, symmetric and highly stable Zr@Si16 fullerene4,5 and Zr doping within a Ge16 fullerene cage, forming a Frank–Kasper polyhedron Zr@Ge16 with remarkable stability.6 Similarly, the doping of Ti in Si16 has yielded a more stable Ti@Si16 Frank–Kasper polyhedron compared to the corresponding fullerene structure, suggesting the potential for designing novel clusters by tuning both endohedral and cage atoms.7–10 This concept of endohedral doping has broadened the scope to encompass various endohedral atoms, forming a diverse category of doped cage clusters.1
Among the intriguing doped cage clusters, a fascinating family known as “matryoshka clusters” or “onion-skin clusters” has garnered substantial attention. These clusters can be described as “endohedral cluster of endohedral cluster”, featuring a cage within a cage, rather than an atom inside a cage.11–15 In 2003, the [As@Ni12@As20]3− ion, an icosahedral [As@Ni12]3− fragment enclosed within an As20 dodecahedral cage to create an onion-skin-like structure, was successfully synthesized from As3−7 and Ni(COD)2 in ethylenediamine solutions.11 Following the discovery of the [As@Ni12@As20]3−, King and Zhao conducted an analysis of the chemical bonding in [As@Ni12@As20]3−, uncovering previously unrecognized principles governing the stability of large spherical metal clusters, notably the presence of 108 valence electrons excluding the filled d10 shells of metal atoms in the intermediate icosahedral layer.12 Subsequently, inspired by the matryoshka [As@Ni12@As20]3− cluster, extensive theoretical and experimental researches have been conducted to explore the structures and properties of matryoshka clusters.16–19 In 2011, a matryoshka cluster analogous to the icosahedral matryoshka doll, [Sn@Cu12@Sn20]12−, and its corresponding crystal structure in ternary A12Cu12Sn21 (A = Na to Cs) were successfully synthesized.16 Subsequently, Damianos et al. proposed the matryoshka structure as the lowest energy configuration of the Mg20Ni13 cluster.17 A consistent bonding model has been provided for understanding the two isoelectronic matryoshka clusters of [As@Ni12@As20]3− and [Sn@Cu12@Sn20]12−.18 Recent studies have delved into the magnetic response and spherical aromaticity of matryoshka-like clusters, such as [Sn@Cu12@Sn20]12−, revealing their potential for exhibiting aromatic behavior within multilayer structures.20 Furthermore, a series of icosahedral matryoshka clusters of A@B12@A20 (A = Sn, Pb; B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Mg, Zn, Cd) with significant HOMO–LUMO gaps and low formation energies has been proposed. Notably, Sn@Mn12@Sn20 and Pb@Mn12@Pb20 have been identified as ideal building blocks in novel magnetic materials and devices.21 Additionally, 33-atom intermetalloid clusters [TM13@Bi20]− (TM = 3d, 4d) in a three-shell icosahedral matryoshka structure have been proposed, with their potential to act as superatoms systematically explored using density functional theory (DFT) calculations.22 The compound [K([2.2.2]crypt)]4[In8Sb13], containing a 1
Mg, Zn, Cd) with significant HOMO–LUMO gaps and low formation energies has been proposed. Notably, Sn@Mn12@Sn20 and Pb@Mn12@Pb20 have been identified as ideal building blocks in novel magnetic materials and devices.21 Additionally, 33-atom intermetalloid clusters [TM13@Bi20]− (TM = 3d, 4d) in a three-shell icosahedral matryoshka structure have been proposed, with their potential to act as superatoms systematically explored using density functional theory (DFT) calculations.22 The compound [K([2.2.2]crypt)]4[In8Sb13], containing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of [Sb@In8Sb12]3− and [Sb@In8Sb12]5−, has been reported, with the [Sb@In8Sb12]3− exhibiting perfect Th symmetry.23 A recent study has proposed several ternary matryoshka clusters TM@Mn12@Au20 (TM = Fe, Co, Ni, Ru, Rh, Pd and Pt), demonstrating that Fe@Mn12@Au20 maintains the original spin direction of its atoms and forms ferromagnetic coupling.24 In a more recent development, Patzer et al. conceived some onion-like/matryoshka clusters comprising late main group and transition metal elements.25 Their study illuminates the presence of two energetically stable minima characterized by Ih and Th symmetries. This observation underscores the continued relevance and broad applicability of the underlying principles governing the construction of onion-like/matryoshka structures, representing an area that warrants further exploration. Sun et al. detailed the synthesis of an all-metal fullerene cluster, [K@Au12Sb20]5−, employing a wet-chemistry method, and further delineated the distinctive aromatic characteristics exhibited by this particular cluster.26
:1 mixture of [Sb@In8Sb12]3− and [Sb@In8Sb12]5−, has been reported, with the [Sb@In8Sb12]3− exhibiting perfect Th symmetry.23 A recent study has proposed several ternary matryoshka clusters TM@Mn12@Au20 (TM = Fe, Co, Ni, Ru, Rh, Pd and Pt), demonstrating that Fe@Mn12@Au20 maintains the original spin direction of its atoms and forms ferromagnetic coupling.24 In a more recent development, Patzer et al. conceived some onion-like/matryoshka clusters comprising late main group and transition metal elements.25 Their study illuminates the presence of two energetically stable minima characterized by Ih and Th symmetries. This observation underscores the continued relevance and broad applicability of the underlying principles governing the construction of onion-like/matryoshka structures, representing an area that warrants further exploration. Sun et al. detailed the synthesis of an all-metal fullerene cluster, [K@Au12Sb20]5−, employing a wet-chemistry method, and further delineated the distinctive aromatic characteristics exhibited by this particular cluster.26
Motivated by the captivating stability and diverse applications of matryoshka clusters, this work introduces a new stable Ag@Mg12@Ag20 matryoshka structure. In this paper, we systematically investigate its stabilities, electronic structures, and bonding characteristics using DFT calculations.
Methods
The three-layer Ag@Mg12@Ag20 structure was subjected to a full geometry optimization using DFT methods, employing the DMol3 software package.27 The structure was relaxed at the GGA-PBE (Generalized Gradient Approximation-Perdew–Burke–Ernzerhof) level of theory,28 with due consideration of relativistic effects through the utilization of a DFT-based semi-core pseudopotential (DSPP).29 The hybrid semi empirical dispersion-correction method proposed by Tkatchenko and Scheffler (TS) was adopted to account for dispersion effects. The geometry optimization employed a double numerical plus polarization (DNP)30 basis set without imposing any symmetry constraints and was conducted under spin-unrestricted conditions. The convergence criteria for the structural relaxation were set as follows: 10−5 Hartree for the energy charge, 0.002 Hartree Å−1 for the maximum force, 0.005 Å for the maximum displacement, and 10−6 Hartree for the self-consistent field tolerance.To verify the structural stability after post-geometry optimization, vibrational frequency analysis and ab initio molecular dynamics (AIMD) simulations were conducted using the DMol3 package. AIMD simulations were carried out in the NVT ensemble, employing a simple Nosé–Hoover thermostat,31 with a total simulation time of 5 ps and a time step of 1 fs.
In-depth exploration of the electronic structure and chemical bonding involved electron density analysis, partial density of states calculations, and the application of Adaptive Natural Density Partitioning (AdNDP) techniques. Specifically, the original data of AdNDP analysis were generated using Gaussian 09 package32 and the visualization of AdNDP analysis results was achieved through the utilization of the Multiwfn software.33
Results and discussions
As illustrated in Fig. 1, the optimized configuration of the Ag@Mg12@Ag20 cluster, achieved through unconstrained symmetry, exhibits a three-layer matryoshka structure characterized by S6 symmetry. The elucidated architecture of Ag@Mg12@Ag20 is predicated on the icosahedron-dodecahedron duality, wherein both the internal inner Mg12 icosahedron and the external Ag20 dodecahedron share an equivalent number of edges. Detailed insights into the construction of the three-layer Ag@Mg12@Ag20 are additionally provided in Fig. S1.† The mean Ag–Mg distance between the central Ag atom and its surrounding 12 Mg atoms measures 2.97 Å, the average Ag–Mg distance between Mg12 and Ag20 is 2.76 Å, the mean Mg–Mg distance within Mg12 is 3.12 Å, and the average Ag–Ag distance within Ag20 is 3.03 Å.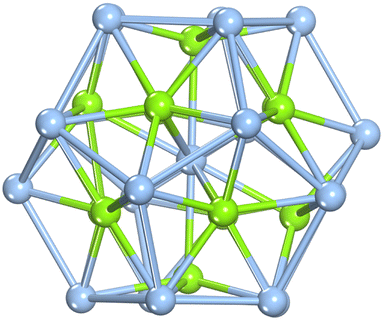 | ||
| Fig. 1 Configuration of the Ag@Mg12@Ag20 cluster, revealing three concentric structural layers as depicted below. Here, baby blue balls are for Ag atoms and green balls are for Mg atoms, respectively. | ||
The average binding energy per atom 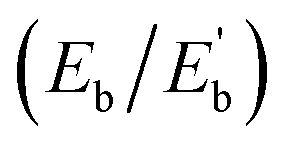 of Ag@Mg12 cluster and Ag@Mg12@Ag20 is calculated using the following formulas:
of Ag@Mg12 cluster and Ag@Mg12@Ag20 is calculated using the following formulas:
for Ag@Mg12,
| Eb = [E(Ag) + 12E(Mg) − E(Ag@Mg12)]/13 |
for Ag@Mg12@Ag20
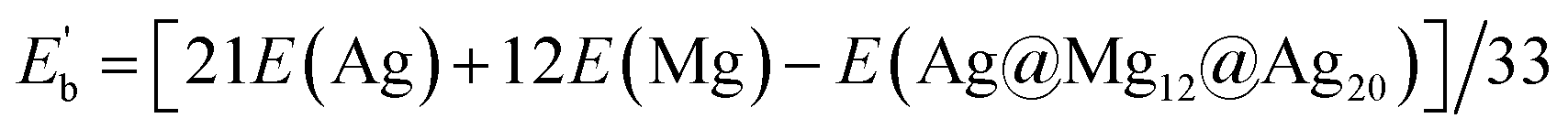
The results indicate that Eb of Ag@Mg12 is 0.72 eV/atom, while 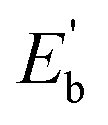 of Ag@Mg12@Ag20 is elevated to 1.84 eV/atom. A comparative analysis of
of Ag@Mg12@Ag20 is elevated to 1.84 eV/atom. A comparative analysis of 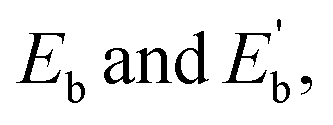 the average binding energy per atom has been largely increased when coated with Ag20.
the average binding energy per atom has been largely increased when coated with Ag20.
The embedding energy (De) of Ag@Mg12@Ag20 has been introduced to identify whether the Ag@Mg12 cluster can be encapsulated into Ag20 easily. Here, the De represents the gain in energy as Ag@Mg12 encapsulated into Ag20 and can be calculated using the following formula:
| De = E(Ag20) + E(Ag@Mg12) − E(Ag@Mg12@Ag20). |
The calculated De of Ag@Mg12@Ag20 is 27.93 eV, and the positive De means that the Ag@Mg12 cluster is suitable to be encapsulated into Ag20 cage to form stable three-layer structure.
The structural stability of Ag@Mg12@Ag20 has been further confirmed by comparing it with several isomers obtained through swaps of Ag–Mg pairs. All fifteen resulting structures exhibit higher binding energies than the ideal three-layer Ag@Mg12@Ag20 structure, as shown in Fig. S2.† Among them, the optimized structure resulting from the complete swap of Ag–Mg pairs, Mg@Ag12@Mg20, has the highest binding energy of −47.90 eV and displays a drum-like configuration, as shown in Fig. S2.† This comparison indicates that our proposed three-layer Ag@Mg12@Ag20 structure is relatively stable among the low-energy structures considered.
The investigation of the structural kinetic stability of the three-layer Ag@Mg12@Ag20 was conducted through a comprehensive vibrational frequency analysis as seen from Fig. 2. The vibrational frequencies of the Ag@Mg12@Ag20 span a range from 23.27 cm−1 to 319.66 cm−1, demonstrating the absence of any imaginary frequencies, thereby substantiating its kinetic stability. Furthermore, Fig. S3† presents the IR spectrum for three-layer Ag@Mg12@Ag20 with several typical peaks marked and corresponding vibration modes are presented in Fig. S4.† Notably, the lowest frequency at 23.27 cm−1 predominantly correspond to the stretching vibration modes associated with Ag–Ag bonds and the highest frequency at 319.66 cm−1 predominantly correspond to the stretching vibration modes associated with Mg–Mg bonds. While, peaks observed at frequency of 109.27 cm−1 emanate primarily from the vibration modes of central Ag atom, and the rest peaks observed at frequencies of 126.27 cm−1, 171.27 cm−1, and 259.27 cm−1 emanate primarily from the vibration modes of Mg12 as a whole.
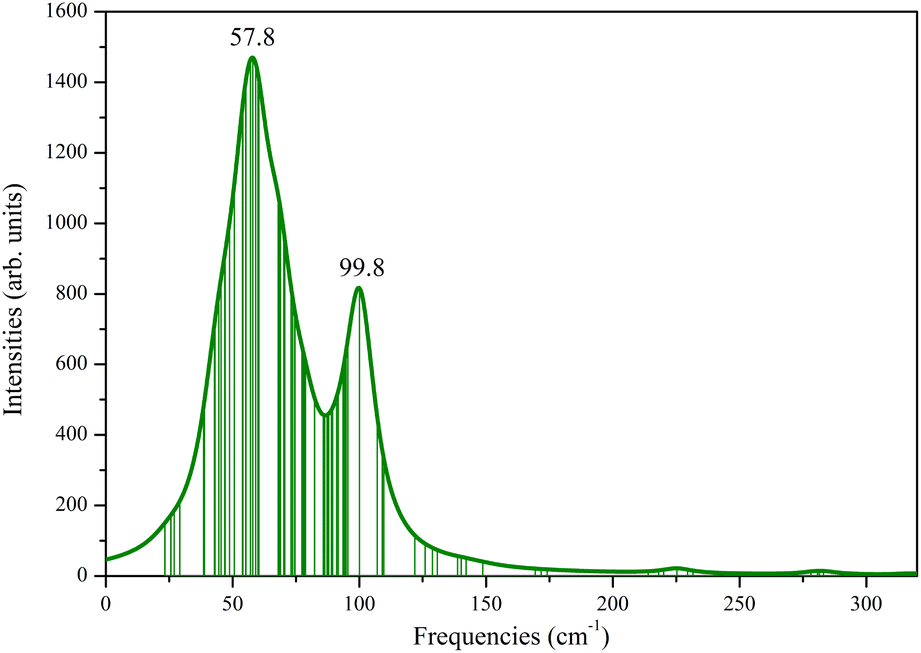 | ||
| Fig. 2 Simulated Raman spectrum for Ag@Mg12@Ag20 structure with the Lorentzian smearing of 20 cm−1, the assumed temperature of 300 K, and the assumed incident light wavelength of 488.00 nm, respectively. | ||
To assess the dynamic stability of the three-layer Ag@Mg12@Ag20 structure, ab into molecular dynamics simulations (AIMD) were conducted over a 5 ps duration at temperatures of 500 K, 600 K and 700 K. Corresponding snapshots at distinct time steps are presented in Fig. 3. At 500 K, the structural integrity of the Ag@Mg12@Ag20 configuration remains well-preserved, with only a discernible structural distortion observed in the Ag20 shell throughout the 5 ps simulations. The results of MD simulations at 500 K has been further confirmed by a longer MD simulations (10 ps), and the Ag@Mg12@Ag20 can basically maintain its original three-layer configuration even undergoing 10 ps MD simulations at 500 K. More details can be found in Fig. S6 and S7 in the ESI.† As shown in Fig. S7,† the coordinate root-mean-square deviation (RMSD) remains stable at approximately 0.4 Å, indicating that the structure is stable throughout the simulation. The deformed final configuration of 10 ps MD simulations at 500 K successfully reverts to its original S6 symmetry through energy minimization. Upon escalating the temperature to 600 K, a more substantial structural distortion becomes apparent in the Ag20 shell after the 5 ps simulations. One Ag atom in Ag20 shell forms chemical bond with the inner Ag atom (bond length: 2.66 Å), and this structural distortion cannot be resolved by geometrical optimization. However, at 700 K, the three-layer Ag@Mg12@Ag20 structure undergoes substantial degradation, characterized by a significant displacement of inner Mg atoms following the 5 ps simulations. Nevertheless, remnants of Ag5Mg units persist within the deformed Ag@Mg12@Ag20 structure. These results collectively underscore the robust dynamic stability exhibited by the Ag@Mg12@Ag20 configuration.
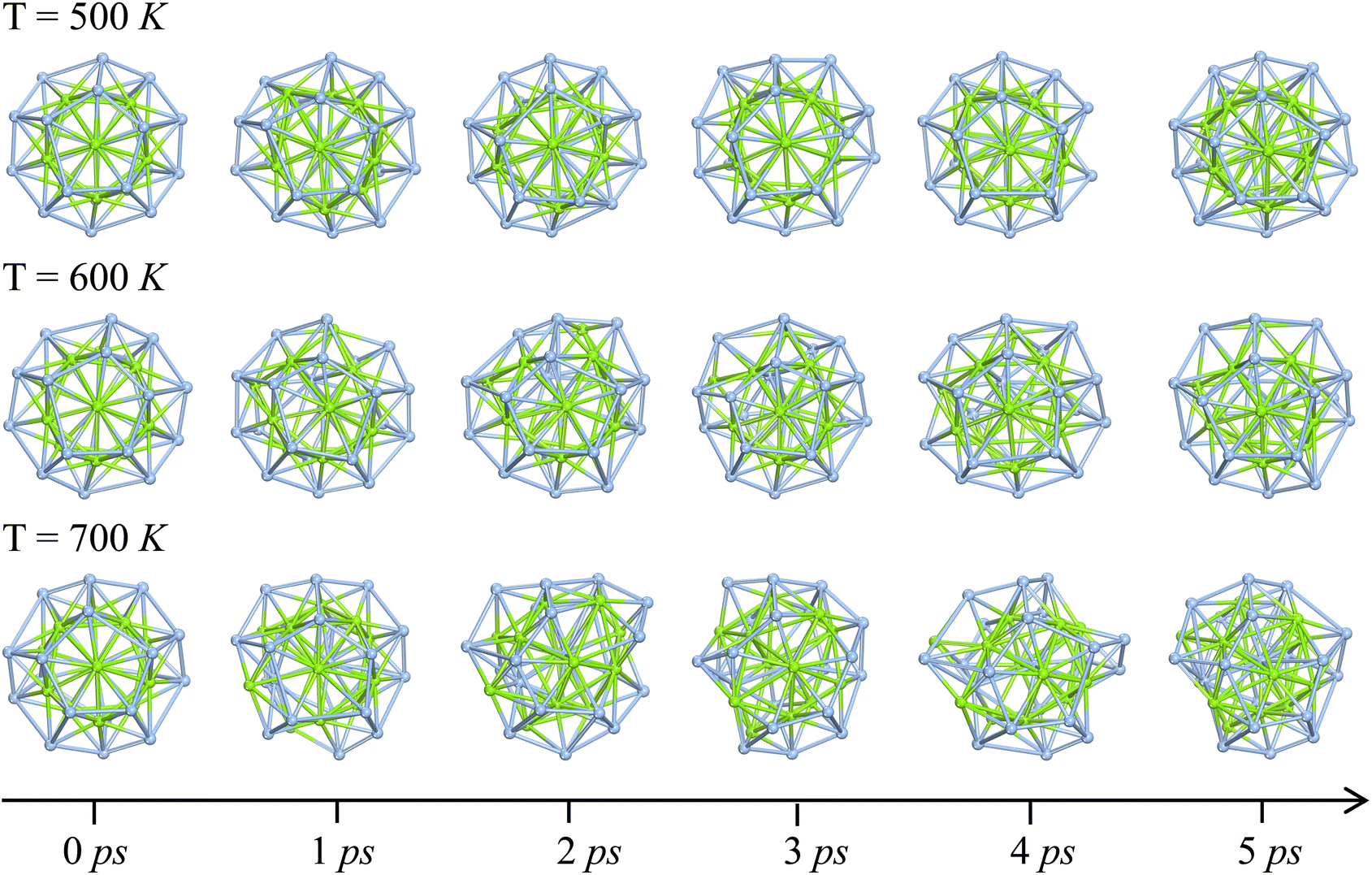 | ||
| Fig. 3 Ab into molecular dynamics simulations of the Ag@Mg12@Ag20 structure at 500 K, 600 K, and 700 K; insets depicting snapshots at 0 ps, 1 ps, 2 ps, 3 ps, 4 ps, and 5 ps, maintaining consistent three-layer orientation across species. | ||
Fig. 4 illustrates the electron density, encompassing both deformation electron density and total electron density for the three-layer Ag@Mg12@Ag20 structure. Fig. 4(a) shows an excess of electron charge density between the Ag atoms in the Ag20 shell and the Mg atoms in the Mg12 shell. Fig. 4(b) demonstrates electron charge density excess mainly around the inner Ag atom. These may suggest that there are strong bonding interaction between the Ag atoms in the Ag20 shell and the Mg atoms in the Mg12 shell, while the interaction between the inner Ag atom and the neighboring Mg atoms is weaker. The natural population charge analysis (NPA) presented in Table S1† shows that the inner Ag atom has a charge state of −0.72 e, the Mg atoms exhibit charge states ranging from −0.46 e to −0.51 e, and the 20 Ag atoms in shell show charge states ranging from 0.21 e to 0.39 e. The similar charge state between inner Ag atom and shell Mg atoms results that there are not typical covalent interactions between them. Furthermore, the weak interactions between the internal Ag atom and the surrounding Mg atoms can be confirmed by the reduced density gradient (RDG) for Ag@Mg12@Ag20 as shown in Fig. S8.† The RDG values between the inner Ag atom and surrounding Mg atoms are all lower than 0.5, indicating the presence of weak non-covalent interactions. The presentation of the total electron density in Fig. 4(c) underscores the distribution of charge across the entirety of the cluster, emphasizing its pivotal role in maintaining the structural integrity of the entire assembly.
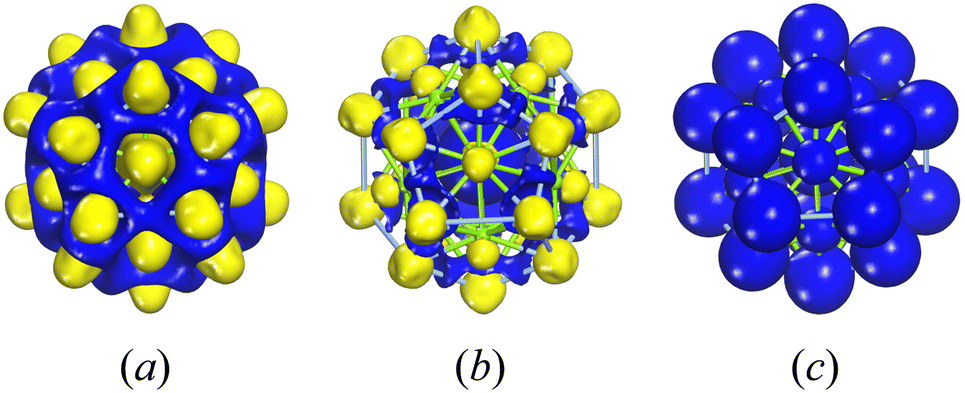 | ||
| Fig. 4 Electron density for the three-layer Ag@Mg12@Ag20. Part (a) and part (b) show the deformation electron density with isosurface values of 0.01 e Å−3 and 0.03 e Å−3 respectively. Part (c) shows the total electron density with isosurface values of 0.2 e Å−3. | ||
Fig. 5 depicts the partial density of states (PDOS) of the three-layer Ag@Mg12@Ag20 structure. Examining the PDOS diagram reveals that the Fermi level is predominantly occupied by s electrons, with a minor presence of p and d electrons. This observation aligns with the characteristics of the highest occupied molecular orbital (HOMO) orbitals, as illustrated in Fig. S3.† The proportion of p electrons increases inversely with energy, a trend consistent with the features of the HOMO−2 orbital. Regarding the lowest unoccupied molecular orbital (LUMO), it predominantly exhibits s-p-d hybridization, with discernible d-like traits on the Ag atoms, as depicted in Fig. S9.† With increasing energy, the proportion of p electrons rises, manifesting evident p-like orbital characteristics on both inner and outer Ag atoms in the LUMO+3 orbital. A HOMO–LUMO gap of 0.10 eV can be found in three-layer Ag@Mg12@Ag20 structure.
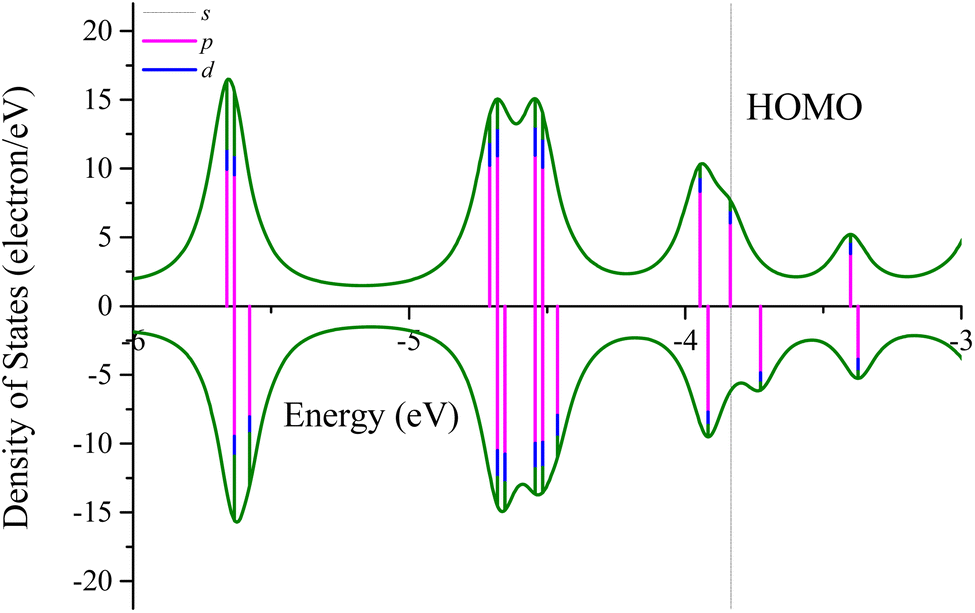 | ||
| Fig. 5 Partial density of states of the three-layer Ag@Mg12@Ag20 structure. Here, the spin up and spin down states are denoted by the positive and negative DOSs, with the lengths of each vertical line color-coded in pink, blue, and olive to signify the proportion of s, p, and d electrons, respectively. The HOMO level are shown by dashed lines. | ||
The investigation of the electronic configuration within the three-layer Ag@Mg12@Ag20 structure has been further delved into using the AdNDP method, with corresponding findings depicted in Fig. 6. The function of Ag in serving as a template support for the overall cluster stability mirrors that of the potassium ion within the core of [K@Au12Sb20]5−, as recently synthesized by the Sun group.24 This suggests the potential for the synthesis of the Ag@Mg12@Ag20 structure in a manner akin to the all-metal fullerene [K@Au12Sb20]5−. Consequently, the subsequent AdNDP analysis concentrates primarily on delineating the bonding characteristics inherent to the Mg12@Ag20 structure. With a total of 44 valence electrons and 100 d-type lone pairs within the Mg12@Ag20 configuration, individual Ag atoms exhibit five d-type lone pairs, as exemplified in Fig. S10.† Of the 44 valence electrons, 20 are allocated to form four-center two-electron (4c-2e) σ bonds amid three inner Mg atoms and one outer Ag atom, while two twelve-center two-electron (12c-2e) σ bonds emerge amidst twelve inner Mg atoms. These σ bonds are distributed throughout the entirety of the three-layer structure, postulated to underlie the structural stability of the three-layer arrangement.
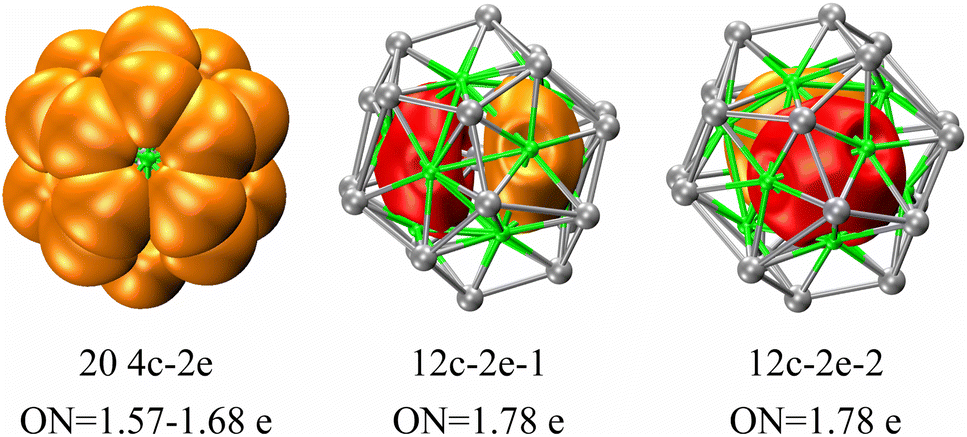 | ||
| Fig. 6 AdNDP analysis for the three-layer Ag@Mg12@Ag20 structure. Occupation numbers (ON) are listed under each structure. Of all 22 multi-center two-center σ bonds, 20 four-center two-electron (4c-2e) σ bonds are observed among 3 inner Mg atoms and 1 outer Ag atom; 2 twelve-center two-electron (12c-2e) σ bonds are observed among 12 Mg atoms. | ||
Conclusions
In summary, a stable three-layer Ag@Mg12@Ag20 structure with S6 symmetry has been comprehensively studied based on the first-principles calculations. The structural stability has been further explored by the vibrational frequency analysis and the molecular dynamics simulations. There are no imaginary frequencies for three-layer Ag@Mg12@Ag20 structure and the core–shell topology configuration can be maintained well after 5 ps MD simulations at 500 K. Furthermore, the AdNDP analysis shows that there are 22 σ bonds distributing throughout the structure. The proposed three-layer Ag@Mg12@Ag20 structure may open a way to new findings of endohedral clusters showing some intriguing properties as the novel superatoms.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Peng-Bo Liu: conceptualization, methodology, writing-original draft, writing-review & editing. Jing-Jing Guo: writing-reviewing and editing. Yi-Sha Chen: writing-reviewing and editing. Hui-Yan Zhao: conceptualization, methodology, validation, investigation, writing-review & editing. Jing Wang: writing-review & editing, funding acquisition. Ying Liu: conceptualization, methodology, validation, funding acquisition, investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 12174084), the Natural Science Foundation of Hebei Province (Grant No. A2021205024), the Doctoral Research Start-up Foundation of Hebei Normal University (Grant No. L2023B08), and the Key Program of Scientific and Technological Foundation of Hebei Province (Grant No. ZD2021065).References
- J. Zhao, Q. Du, S. Zhou and V. Kumar, Chem. Rev., 2020, 120, 43 Search PubMed.
- J. R. Heath, S. C. O'Brien, Q. Zhang, Y. Liu, R. F. Curl, F. K. Tittel and R. E. Smalley, J. Am. Chem. Soc., 1985, 107, 7779 CrossRef CAS.
- T. Guo, M. D. Diener, Y. Chai, M. J. Alford, R. E. Haufler, S. M. McClure, T. Ohno, J. H. Weaver, G. E. Scuseria and R. E. Smalley, Science, 1992, 257, 1661 CrossRef CAS PubMed.
- V. Kumar and Y. Kawazoe, Phys. Rev. Lett., 2001, 87, 045503 CrossRef CAS PubMed.
- V. Kumar and Y. Kawazoe, Phys. Rev. Lett., 2003, 91, 199901 CrossRef.
- V. Kumar and Y. Kawazoe, Phys. Rev. Lett., 2002, 88, 235504 CrossRef PubMed.
- M. Ohara, K. Koyasu, A. Nakajima and K. Kaya, Chem. Phys. Lett., 2003, 371, 490 CrossRef CAS.
- K. Koyasu, M. Akutsu, M. Mitsui and A. Nakajima, J. Am. Chem. Soc., 2005, 127, 4998 CrossRef CAS PubMed.
- S. Furuse, K. Koyasu, J. Atobe and A. Nakajima, J. Chem. Phys., 2008, 129, 064311 CrossRef PubMed.
- J. T. Lau, K. Hirsch, Ph. Klar, A. Langenberg, F. Lofink, R. Richter, J. Rittmann, M. Vogel, V. Zamudio-Bayer, T. Möller and B. V. Issendorff, Phys. Rev. A, 2009, 79, 053201 CrossRef.
- M. J. Moses, J. C. Fettinger and B. W. Eichhorn, Science, 2003, 300, 778 CrossRef CAS PubMed.
- R. B. King and J. Zhao, Chem. Commun., 2006, 4204 RSC.
- J. Zhao and R. H. Xie, Chem. Phys. Lett., 2004, 396, 161 CrossRef CAS.
- C. Chang, A. B. C. Patzer, E. Sedlmayr and D. Sülzle, Phys. Rev. B, 2005, 72, 235402 CrossRef.
- C. Chang, A. B. C. Patzer, E. Sedlmayr, D. Sülzle and T. Steinke, Comput. Mater. Sci., 2006, 35, 387 CrossRef CAS.
- S. Stegmaier and T. F. Fassler, J. Am. Chem. Soc., 2011, 133, 19758 CrossRef CAS PubMed.
- K. Damianos, P. Solokha and R. Ferrando, RSC Adv., 2013, 3, 9419 RSC.
- F. K. Sheong, W. J. Chen, H. Kim and Z. Lin, Dalton Trans., 2015, 44, 7251 RSC.
- L. Zhang, J. Huang, W. Wang, Q. Li and J. Yang, RSC Adv., 2017, 7, 12704 RSC.
- M. Kulichenko, N. Fedik, A. Boldyrev and A. Munoz-Castro, Chemistry, 2020, 26, 2263 CrossRef CAS PubMed.
- X. Huang, J. Zhao, Y. Su, Z. Chen and R. B. King, Sci. Rep., 2014, 4, 6915 CrossRef CAS PubMed.
- C. Y. Kou, L. Zhuang, G. Q. Wang, H. Cui, H. K. Yuan, C. L. Tian, J. Z. Wang and H. Chen, RSC Adv., 2015, 5, 92134 RSC.
- C. Liu, N. V. Tkachenko, I. A. Popov, N. Fedik, X. Min, C. Q. Xu, J. Li, J. E. McGrady, A. I. Boldyrev and Z. M. Sun, Angew. Chem., Int. Ed., 2019, 58, 8367 CrossRef CAS PubMed.
- X. Bai, J. Lv and H. S. Wu, Mol. Phys., 2020, 118, e1659434 CrossRef.
- A. B. C. Patzer, C. Chang, H. Bauer and D. Sülzle, The 228 valence electron rule for onion-like inorganic fullerenes X1@Y12@Z20 of Ih and Th symmetry, ChemRxiv, Cambridge Open Engage, Cambridge, 2023, This content is a preprint and has not been peer-reviewed Search PubMed.
- Y.-H. Xu, W.-J. Tian, A. Muñoz-Castro, G. Frenking and Z.-M. Sun, Science, 2023, 382, 4 Search PubMed.
- B. Delley, J. Chem. Phys., 2000, 113, 7756 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- B. Delley, Phys. Rev. B, 2002, 66, 155125 CrossRef.
- B. Delley, J. Chem. Phys., 1989, 92, 508 CrossRef.
- S. Nosé, Mol. Phys., 1984, 52, 255 CrossRef.
- G. W. Trucks, M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.-A. Petersson et al., Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: detailed construction, frequency modes, representative frontier molecular orbitals, natural population charge analysis for the three-layer Ag@Mg12@Ag20 matryoshka structure. See DOI: https://doi.org/10.1039/d4ra07046a |
| This journal is © The Royal Society of Chemistry 2024 |