
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the titanium-mediated catalytic enantioselective oxidation of aryl benzyl sulfides containing heterocyclic groups†
Maria Annunziata M. Capozzi *a,
Angel Alvarez-Larenab,
Joan F. Piniella Febrerc and
Cosimo Cardellicchio*d
*a,
Angel Alvarez-Larenab,
Joan F. Piniella Febrerc and
Cosimo Cardellicchio*d
aDipartimento di Chimica, Università di Bari, via Orabona 4, 70125 Bari, Italy
bServei de Difracció de Raigs X, Universitat Autònoma de Barcelona, 08193 Bellaterra, Cerdanyola del Vallès, Barcelona, Spain
cDepartament de Geologia, Universitat Autònoma de Barcelona, 08193 Bellaterra, Cerdanyola del Vallès, Barcelona, Spain
dCNR ICCOM, Dipartimento di Chimica, Università di Bari, via Orabona 4, 70125 Bari, Italy. E-mail: cardellicchio@ba.iccom.cnr.it
First published on 4th November 2024
Abstract
Our enantioselective oxidation protocol, based upon hydroperoxides in the presence of a titanium/(S,S)-hydrobenzoin catalyst, was tested for the first time with aryl benzyl sulfides containing heterocyclic moieties (2-thienyl, 2-pyridyl and benzimidazolyl), two of them being connected with the blockbuster omeprazole drug. Good yields of enantiopure sulfoxides were obtained in most cases. Two exceptions of unsatisfactory enantioselectivity in the oxidation of benzimidazolyl sulfides are reported. However, one of them was solved by crystallization of an enantio-enriched mixture. The present work was supported also by X-ray diffraction analysis of some synthesized sulfoxides and by energetic calculation of the crystal structures. The unexpected result is that the crystal structures of the racemic mixture of the two problematic benzimidazolyl sulfoxides are composed of separate enantiomers (a conglomerate), an interesting result that could be exploited in the future for the separation of the enantiomers of these sulfoxides.
Introduction
The asymmetric oxidation1 of sulfides to yield enantiopure sulfoxides2–5 is a crucial topic of investigation, because these strategic materials are relevant chiral building blocks,6 chiral ligands3,4 and some of them are also bioactive compounds.7 Among these compounds, a family of substituted benzimidazolyl 2-pyridylmethyl sulfoxides emerged as proton pump inhibitors, thus used for treating gastric diseases.7 (S)-omeprazol, one of the worldwide blockbusters drugs, is the best known molecule in this family.7–11The industrial process leading to this successful drug is based upon the hydroperoxide oxidation of the corresponding sulfide in the presence of a complex between titanium and diethyl tartrate.8–11 Many alternative procedures have been reported so far. Limiting only to recent examples, Nakamura et al. obtained (S)-omeprazole with an iron catalyzed asymmetric oxidation.12 Limiting only to titanium-based processes, Bryliakov et al. synthesised molecules of the omeprazole scaffold with an asymmetric oxidation in the presence of titanium–salalen13 or titanium–salan14 complexes. In both these cases, the employed complexes were not commercially available. The titanium–tartrate protocol was recently performed in continuous flow.15
However, the reaction conditions applied in the titanium–tartrate industrial process are in a sharp contrast with the original formulations of Kagan16,17 and Modena,18 who reported the first sulfides oxidation with hydroperoxides with an analogous complex. To say few, Kagan reported the oxidation of aryl alkyl sulfides with tert-butyl hydroperoxide (TBHP) or cumene hydroperoxide (CHP) acting as the oxidants in the presence of a titanium complex obtained by mixing titanium i-propoxide, 2 equivalents of diethyl tartrate and 1 equivalent of water in methylene chloride, at −20 °C.16,17 On the other hand, the Modena complex was prepared by mixing titanium i-propoxide with 4 equivalents of diethyl tartrate and no water.18 In a paper of us,19 followed by a similar report,20 we found that a complex formed with reduced amounts of water works better on our substrates. Other adjustments to the original recipe were reported for the oxidation of other sulfides.21 Finally, Kagan reported a new formulation for the catalyst based upon a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:4 complex between titanium i-propoxide/diethyl tartrate/i-propanol and molecular sieves.22 This variability of the experimental protocol is a serious issue of this process, because it seems that the reaction conditions should be tuned before each oxidation.
:2:4 complex between titanium i-propoxide/diethyl tartrate/i-propanol and molecular sieves.22 This variability of the experimental protocol is a serious issue of this process, because it seems that the reaction conditions should be tuned before each oxidation.
In the industrial process for the synthesis of (S)-omeprazole,8–11 the acting complex was prepared first by heating (54 °C) water, titanium i-propoxide and diethyl tartrate in 1:2.33:4.75 ratio in toluene, in the presence of the pro-chiral sulfide. Thus, the amount of water was more similar to our report.19,20 Later, N,N-di-i-propylethylamine and CHP were added, and the reaction temperature was adjusted to 30 °C.
In the last two decades, we have been involved in the enantioselective oxidation of sulfides by using hydroperoxides as the oxidant in the presence of catalytic amounts of a 1:2 complex between titanium and (S,S)-hydrobenzoin, a not expensive commercially available chiral ligand.1,23–31 The reaction is usually performed at room temperature with a simple “mix and wait” protocol and the purification steps are simple and effective.23–31
Among other results, we found that this reaction protocol oxidises with excellent efficiency the aryl benzyl sulfides, even in the presence of many different substituents.24,25 We synthesized23–31 with the same experimental protocol more than 60 aryl benzyl sulfoxides in high enantiomeric purity (ee > 90%), with the large majority of them being in an enantiopure form. From a stereochemical point of view, the (R)-sulfoxide is invariantly obtained, when the (S,S)-hydrobenzoin is used as a ligand of the titanium. The only exception of a lower enantioselectivity was connected to the oxidation of pentafluorobenzyl pentafluorophenyl sulfide (and similar compounds).30,31 However, a switch of the oxidant, from TBHP to CHP, leads again to the enantiopure sulfoxide.30,31
Our work was supported also with DFT calculations.24,27,30 We investigated the mono-metallic octahedral complexes between the titanium, two molecules of (S,S)-hydrobenzoin, the substrate and the hydroperoxide before the oxygen transfer. We calculated the diastereomeric paths deriving from these complexes and leading to the (R)- or (S)-sulfoxides. We found that the path leading to the (R)-sulfoxide (in accordance with the experiments) is energetically more stable, due to weak interactions involving the aryl groups.24,27,30
The oxidation of aryl pentafluorobenzyl sulfides with our procedure deserves a special attention. The very high enantioselectivities that were observed were explained with the model depicted in Fig. 1, obtained with DFT calculations.27 The crucial complex leading to the (R)-sulfoxide is clearly stabilised by π–π interactions between the pentafluorophenyl moiety and one aryl group of the (S,S)-hydrobenzoin coordinated to the titanium.
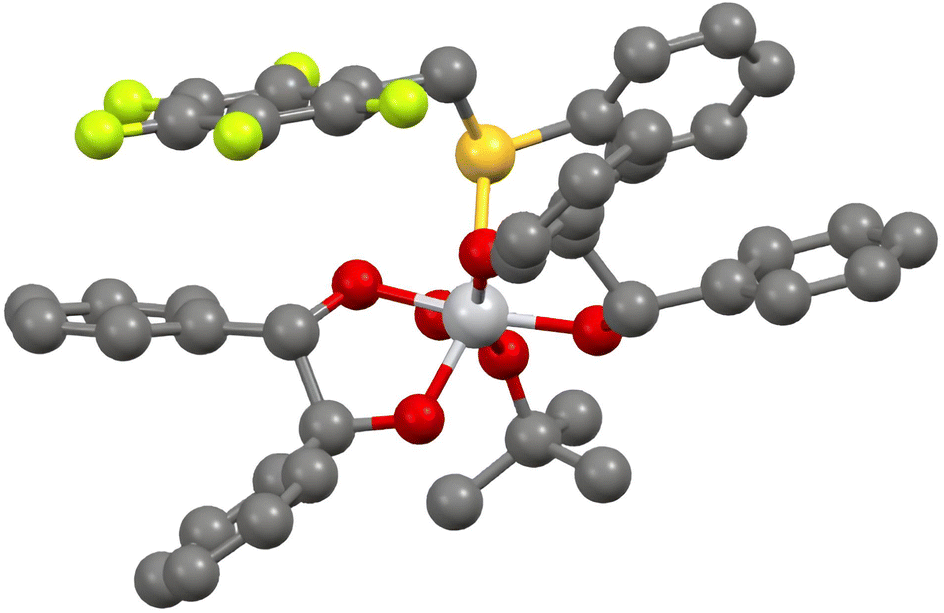 | ||
| Fig. 1 Pro-(R) titanium complex in the oxidation of pentafluorobenzyl phenyl sulfoxide. Hydrogen atoms are omitted for clarity. See ref. 27. | ||
The crucial role performed by electron-poor arene moieties was confirmed in the following experimental paper,31 in which groups different from the pentafluophenyl one perform in a similar way.
At this stage, we decided to extend our investigation to the oxidation of aryl benzyl sulfides containing heterocyclic moieties with hydroperoxides, in the presence of the complex between titanium and (S,S)-hydrobenzoin. The oxidation of sulfides connected with the omeprazole scaffold in the presence of a different complex between titanium and modified hydrobenzoins was once investigated,32 but those results could not benefit of the large mechanistic and computations framework collected in our work in the following years.
Results and discussion
The results of the present investigation are collected and summarised in Table 1.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Ar1 | Ar2 | Sulfide | Oxidant | Solvent | Catalyst (M/L/i-PrOH)a | Sulfoxide | Yieldb (%) | eec (%) |
| a The standard catalyst is a 1:2 mixture between titanium i-propoxide and (S,S)-hydrobenzoin. If i-PrOH is added, the ratios between reactants are reported.b Yields refer to pure isolated products.c Determined by HPLC (see Text).d Data reported in ref. 26.e After recrystallisation.f This product was reported in ref. 23.g This product was reported in ref. 27, 30 and 31. |
|||||||||
| 1 | 2-Thienyl | Ph | 1a | TBHP | n-Hexane | Standard | 1b | 85 | 85 |
| 2 | 2-Thienyl | C6F5 | 2a | TBHP | n-Hexane | Standard | 2b | 83 | >98 |
| 3 | 2-Py | Ph | 3a d | TBHP | n-Hexane | Standard | 3b d | 92 | 78 |
| 4 | 4-Br-C6H4 | 2-Py | 4a | TBHP | n-Hexane | Standard | 4b | 64 | 93 |
| 5 | BzIm | Ph | 5a | TBHP | Toluene | Standard | 5b | 97 | 31 |
| 6 | BzIm | Ph | 5a | CHP | Toluene | Standard | 5b | 66 | 50 |
| 7 | BzIm | Ph | 5a | CHP | Toluene | 1:2:2 |
5b | 57 | 66 |
| 8 | BzIm | Ph | 5a | CHP | Toluene | 1:2:4 |
5b | 55 | 76 (>98)e |
| 9 | BzIm | Ph | 5a | CHP | Toluene | 1:2:6 |
5b | 22 | 31 |
| 10 | 4-Br-C6H4 | Ph | 6a f | TBHP | Ethyl acetate | Standard | 6bf | 61 | 53 |
| 11 | C6F5 | C6F5 | 7a g | CHP | Ethyl acetate | Standard | 7bg | 71 | 65 |
| 12 | BzIm | Ph | 5a | CHP | Ethyl acetate | Standard | 5b | 51 | 45 (63)e |
| 13 | BzIm | Ph | 5a | TBHP | Ethyl acetate | Standard | 5b | 70 | 48 |
| 14 | BzIm | Ph | 5a | CHP | Ethyl acetate | 1:2:4 |
5b | 34 | 11 |
| 15 | BzIm | 2-Py | 8a | CHP | Toluene | Standard | 8b | 64 | 49 |
| 16 | BzIm | 2-Py | 8a | CHP | Toluene | 1:2:4 |
8b | 74 | 74 |
| 17 | BzIm | C6F5 | 9a | TBHP | n-Hexane | Standard | 9b | 89 | >98 |
| 18 | BzIm | C6F5 | 9a | TBHP | Ethyl acetate | Standard | 9b | 69 | >98 |
| 19 | BzIm/Me | Ph | 10a | TBHP | n-Hexane | Standard | 10b | 19 | 6 |
| 20 | BzIm/Me | Ph | 10a | TBHP | Ethyl acetate | Standard | 10b | 37 | 8 |
In a first series of reactions, we investigated the behaviour of 2-thienyl containing sulfides, since the thienyl moiety is the most similar to the phenyl group. We oxidised the benzyl 2-thienyl sulfide 1a33 with TBHP in n-hexane in the presence of 5% of the catalyst between the titanium and (S,S)-hydrobenzoin according to our procedure to yield the corresponding sulfoxide 1b34 (Table 1, entry 1, 85% yield; 85% ee). The results of this oxidation are almost identical with the results obtained with the analogous benzyl phenyl sulfides.29 Moreover, we oxidised the pentafluorobenzyl 2-thienyl sulfide 2a with the same protocol to obtain the corresponding sulfoxide 2b (Table 1, entry 2, 83% yield; >98% ee), thus showing a close similarity with the enantioselectivity obtained with the analogous pentafluorobenzyl phenyl sulfide.27 In our oxidation system, the exchange between phenyl and 2-thienyl moieties does not affect the stereochemical profile.
Then, we investigated the oxidation of aryl 2-pyridylmethyl sulfides. In sulfides in which the 2-pyridyl group is directly connected to the sulfur atom, we had reported26 satisfactory enantioselectivity and good yields (Table 1, entry 3). The same holds when the 2-pyridyl group is a part of the benzyl moiety. In fact, the oxidation of the 4-bromophenyl 2-pyridylmethyl sulfide 4a35 was accomplished with high enantioselectivity (Table 1, entry 4, 93% ee) with our protocol. Therefore, in these cases, the presence of the pyridyl moiety does not affect the usual favourable stereochemical path.
At this stage, we considered of interest to test benzimidazolyl benzyl sulfides, because the benzimidazolyl moiety is a part of omeprazole and other bioactive compounds.7
The oxidation of benzimidazolyl benzyl sulfide 5a36 with hydroperoxides, in the presence of the complex between titanium and (S,S)-hydrobenzoin to yield sulfoxide 5b37 has a special focus in this work (see Table 1). Sulfide 5a was almost insoluble in n-hexane and the reaction did not start at all, when this solvent is used. We tried a screening of alternative solvents, such as carbonates, methylene chloride and cyclopentyl methyl ether (ESI, Table S1†) in the oxidation of 5a with hydroperoxides according to our protocol. However, low values of enantioselectivity were recorded, with 20–22% ee values as the best results (ESI, Table S1†).
At this stage, we focused on toluene and ethyl acetate as solvents, as it occurs in the industrial process.8–11,32 The first oxidation of benzimidazolyl benzyl sulfide 5a with TBHP in toluene gave a 31% ee value of sulfoxide 5b (Table 1, entry 5). Better results were obtained by using CHP as the oxidant (Table 1, entry 6, 50% ee value).
At this point, we applied the Kagan modification,22 to our oxidation system. The reaction was performed by using toluene as the solvent, CHP as the oxidant and the titanium/(S,S)-hydrobenzoin catalyst was modified by adding molecular sieves 4 Å and variable amount of i-propanol (Table 1, entries 7–9). In a first attempt (Table 1, entry 7), the acting catalyst was formed by 1 equivalent of titanium i-propoxide, 2 equivalent of (S,S)-hydrobenzoin and 2 equivalents of i-propanol. We recorded an increase of the enantioselectivity (66% ee). When the i-propanol amount was increased to obtain a ratio between the reactants of 1:2:4 (Table 1, entry 8), a further increase was observed, with the formation of benzimidazolyl benzyl sulfoxide 5b in a 76% ee value. In this case, we were able to recrystallise this sulfoxide to obtain a sample of the enantiopure sulfoxide 5b. A further increase of i-propanol (Table 1, entry 9, 1:2:6 ratio) was detrimental and the ee value of the sulfoxide 5b decreases.
The role of i-propanol in the formation of the catalyst for this oxidation is not clear. Kagan hypothesized22 that an excess of this species can favour the formation of a more performing catalyst, blocking parallel less performing processes. However, up to now, nobody has investigated again this topic.
Our research continued by testing ethyl acetate as the solvent. Benzyl 4-bromophenyl sulfoxide 6b23 and pentafluorobenzyl pentafluorophenyl sulfoxide 7b30,31 were obtained in an enantiopure form, when n-hexane is the reaction solvent. As a preliminary test, we oxidised sulfides 6a and 7a with our protocol but using ethyl acetate as the solvent (Table 1, entries 10 and 11). We recorded a decrease of enantioselectivity (Table 1, entries 10 and 11) in comparison with the optimal results obtained with n-hexane.23,30,31
At this stage, we oxidized the benzimidazolyl benzyl sulfide 5a with our protocol using ethyl acetate as a solvent. We recorded a 45 and 48% ee values of sulfoxide 5b when CHP and TBHP were used as the oxidants (Table 1, entries 12–13). The attempt to improve these results with the modified catalyst of entry 8 was not successful (Table 1, entry 14). In summary, the reaction protocol described in Table 1, entry 8 (CHP as the oxidant, toluene as the solvent and a 5% catalyst formed by 1 eq. of titanium i-propoxide, 2 eq. of (S,S)-hydrobenzoin and 4 eq. of i-propanol and molecular sieves) is the optimal set up to obtain sulfoxide 5b.
Later, we investigated the oxidation of benzimidazolyl 2 pyridyl sulfide 8a,38 to sulfoxide 8b39 in which we combined the two previously investigated heterocyclic moieties. The reaction was performed according to the conditions of Table 1, entry 6. We observed that the corresponding benzimidazolyl 2 pyridyl sulfoxide 8b (Table 1, entry 15) was obtained with a 49% ee, a value that is very similar to the 50% ee value described in entry 6.
The oxidation reaction of sulfide 8a was repeated with the improved conditions reported in entry 8, that is the addition of molecular sieves 4 Å and i-propanol during the formation of the acting catalyst. Sulfoxide 8b was obtained (Table 1, entry 16) with an ee value of 74%, that is very similar to the values obtained for sulfoxide 5b (Table 1, entry 8). These experiments confirm that the 2-pyridyl moiety does not affect particularly the enantioselectivities of our protocol, when it is a part of the benzyl position of the aryl benzyl sulfides (Table 1, entries 4, 15 and 16).
At this stage, we studied the enantioselective oxidation of benzimidazolyl 2,3,4,5,6-pentafluorobenzyl sulfide 9a. Surprisingly, we observed that the oxidation of 9a with TBHP according to our protocol occurred successfully in n-hexane, yielding the sulfoxide 9b in high yield (89%, Table 1, entry 17) and in an enantiopure form (>98% ee). Moreover, when the reaction was repeated in a low performing solvent, such ethyl acetate (Table 1, Entry 18), the sulfoxide 9b was obtained in a lower yield (69%), but also in an enantiopure form (>98% ee). The highly beneficial effect of the pentafluorobenzyl group on the enantioselectivity is confirmed also in this case, in which the problematic benzimidazolyl moiety is present. The mechanistic considerations depicted in Figure 124,27,30 can be reasonably extended to the present work.
According to previous investigations,11 we investigated also the influence of the acidic benzimidazolyl hydrogen atom on the enantioselectivity. We synthesized the N-methyl-benzimidazolyl benzyl sulfide 10a40 and we oxidised it to sulfoxide 10b41 with our protocol (Table 1, entries 19–20). Also in this case, the solubility in n-hexane was low and the yields and the ee values were not satisfactory (Table 1, entry 19). Although a better solubility was observed in ethyl acetate (Table 1, entry 20), the results were only slightly better (37% yield; 8% ee). The investigation on the oxidation of 10a was not continued, due to the crystallographic results that will be discussed hereinafter.
It is worth to say that high enantioselectivities were observed in the oxidation with our standard protocol (n-hexane as the solvent; TBHP as the oxidant) of the investigated sulfides 1a, 2a, 4a and 9a. In the case of sulfide 5a, a modification of the synthetic protocol (toluene, as the solvent; CHP as the oxidant; i-propanol and molecular sieves added in the formation of the catalyst) lead to higher enantioselectivity. It must be stressed that the two investigated aryl pentafluorobenzyl sulfides were oxidised with the usual high efficiency, regardless of the heterocyclic components of the substrates, as the main driving force of the enantioselection is connected to the interactions depicted in Fig. 1. In Chart 1, we draw the most successful products synthesised in this work.
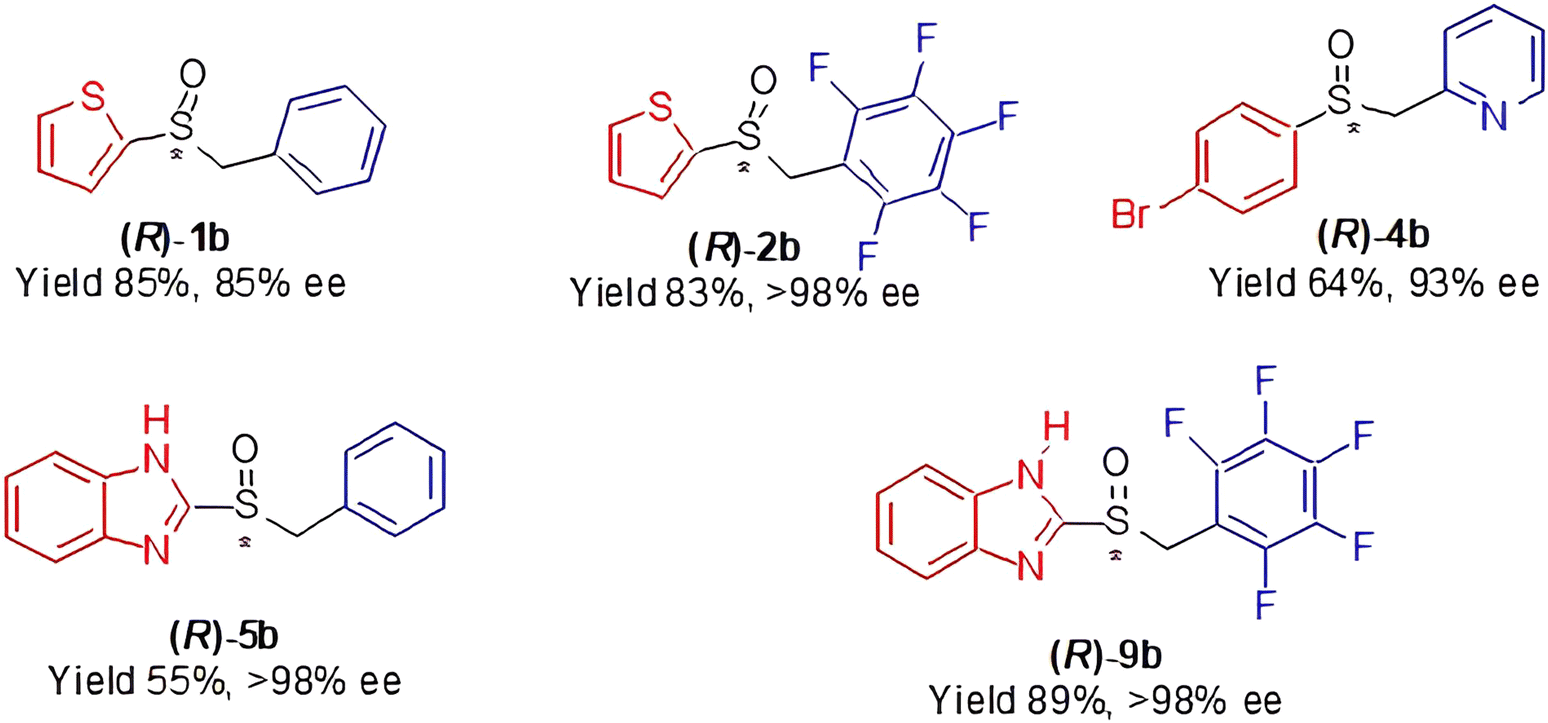 | ||
| Chart 1 A selection of products from Table 1. | ||
Single crystal X-ray diffraction studies
The X-ray diffraction experiments of the single crystals of sulfoxides 2b and 9b were used to determine their absolute configuration. This investigation was extended to sulfoxide 5b and 10b. In the ESI,† crystal data and structure refinements are collected in Tables S2–S5;† ORTEP plots and packing plots are depicted in Fig. S1–S8.† All these compounds have the (R)-configuration at the sulphur stereogenic center, thus confirming the empirical rule that this configuration was invariantly obtained, when the (S,S)-hydrobenzoin was employed in our oxidation system.Lattice energies can be estimated starting from the coordinates recorded in the crystallographic files with the aid of the Crystal Explorer 21 program.42 The procedure is based upon first building a 10 Å radius network of molecules around the central species and then evaluating the pairwise interactions of each molecule of the network with the central species. The interaction energy of each pairwise interaction was calculated as a weighted sum of four different contributions (electronic, polarization, dispersion and repulsion energies).42 The sum of these pairwise interactions is the estimation of the total structure energy. The calculations connected to the crystal structures 2b, 5b, 9b, and 10b are collected in the ESI Section (Tables S6–S9).†
Predominant contributions to the final energy derive mainly from electronic or dispersion energies. The contribution of each different type of energy can be depicted graphically by the Crystal Explorer 21 “Energy Frameworks”,42 or discussed according to our recent approach.43
In the case of the (R)-pentafluorobenzyl 2-thienyl sulfoxide 2b (Fig. 2), we observed a weak hydrogen bonding between one methylene hydrogen atom and the sulfinyl oxygen atom (CH⋯O distance 2.49 Å), a peculiar behaviour of aryl benzyl sulfoxides, that was reported previously.28 Moreover, this interaction favours a parallel displaced set up of the pentafluorobenzyl moieties, that also contributes to the stability of the crystal structure (distance between the planes of the pentafluophenyl moieties around 3 Å). The sulfoxide is in gauche-conformation (torsion angle 59°).28
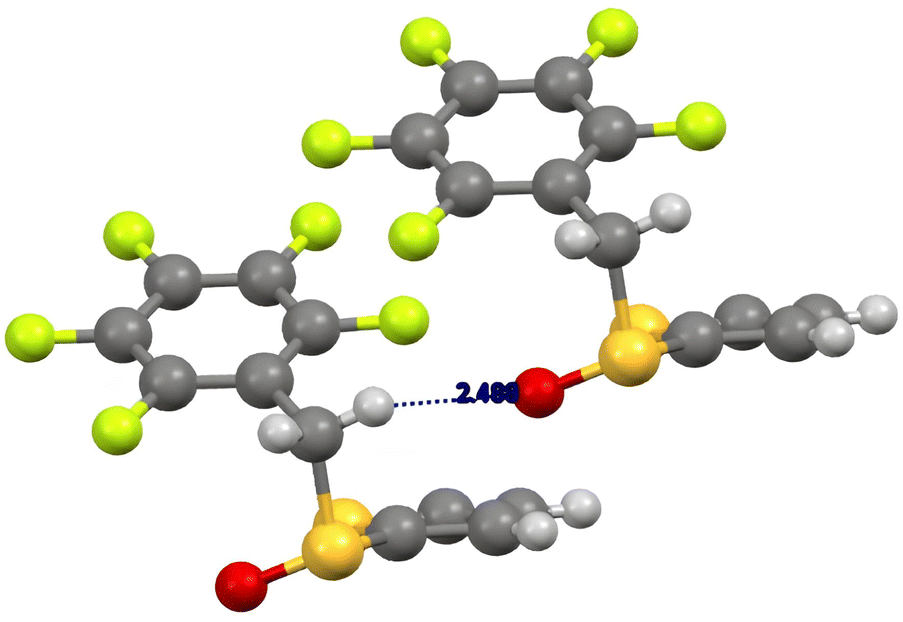 | ||
| Fig. 2 Crystal structure of (R)-2-(2,3,4,5,6-pentafluorobenzylsulfinyl)thiophene 2b. | ||
In the case of (R)-benzimidazolyl pentafluorobenzyl sulfoxide 9b (Fig. 3), the major contributions to the final energy are due both to electronic and dispersion energies (ESI, Table S8†). A relevant hydrogen bonding between the imidazolyl hydrogen atom and the sulfinyl oxygen atom (NH⋯O distance 2.05 Å) is present. Main geometric characteristic of hydrogen bondings are collected in ESI, Table S10.† Dispersion interactions connected to the pentafluophenyl moiety are also present. The sulfoxide is in anti-conformation (Caryl–sulfur–CMethylene–CAryl torsion angle 177°).28
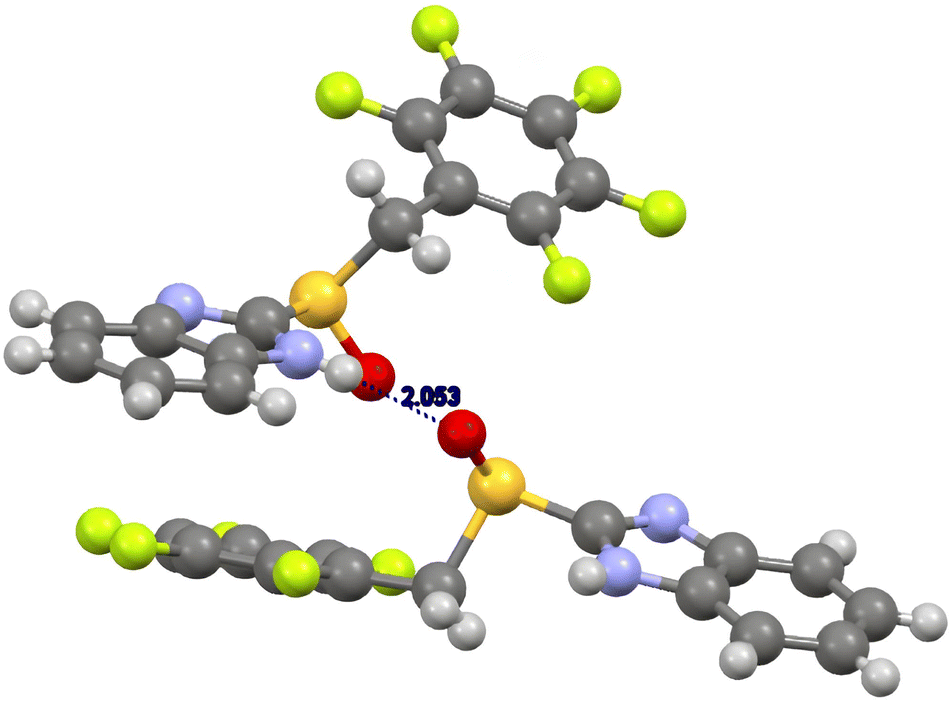 | ||
| Fig. 3 Crystal structure of (R)-2-(2,3,4,5,6-pentafluorobenzylsulfinyl)-1H-benzo[d]imidazole 9b. | ||
The (R)-benzimidazolyl benzyl sulfoxide 5b arranges in a gauche-conformation (torsion angle 62°)28 (Fig. 4). We observed that the most relevant interaction is the hydrogen bonding between the imidazolyl hydrogen atom with the nitrogen atom of another molecule (NH⋯N distance 2.07 Å). This interaction was accompanied by the interaction between the sulfinyl oxygen atom with one of the benzyl hydrogen atoms.28
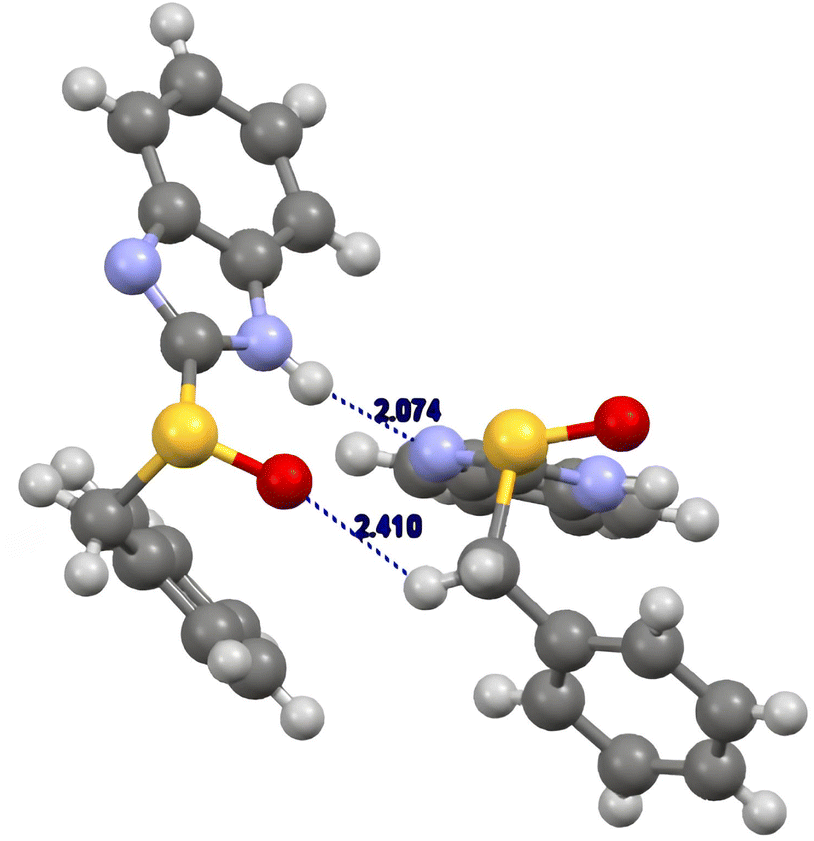 | ||
| Fig. 4 Crystal structure of (R)-2-benzylsulfinyl-1H-benzo[d]imidazole 5b. | ||
The crystal structure of the racemic benzimidazolyl benzyl sulfoxide 5b was already published, obtained by an achiral oxidation reaction.37 However, the reported data referred only to the (S)-enantiomer, solved in a Sohncke space group.37 Probably, the authors did not realise that they picked up a crystal of a single enantiomer. Therefore, it is likely that their “racemic” structure is a conglomerate, as it can be confirmed by overlaying the inverted structure of their “racemic” compound with the enantiopure (R)-5b structure solved by us (ESI, Fig. S9†).
Conglomerates are valuable compounds from an industrial point of view44 because, in principle, a protocol to separate the enantiomers could be set up without resorting to asymmetric synthesis. Recent papers45,46 designed a strategy for searching conglomerates in the crystallographic databases.
Stimulated from the previous observations, we analysed also the racemic (HPLC checked) N-methylated sulfoxide 10b. A single crystal of this compound was analysed with the X-ray diffraction experiment. We found that this crystal structure was formed only by (R)-enantiomer, solved in a Sohncke space group (Fig. 5).
 | ||
| Fig. 5 Crystal structure of 2-benzylsulfinyl-1-methyl-1H-benzo[d]imidazole 10b. | ||
Since the crystal structure of (S)-10b is available in the literature,41 a satisfactory overlay of our crystal structure, referring to a racemic compound, with the (R)-10b, obtained by inverting the literature report, is depicted in ESI (Fig. S10).† They are indeed the same crystal structure.
Energetic calculations (ESI, Table S9†) show that the interactions between the two closest anti-conformed (torsion 173°) (R)-configured molecules account for more than 2/3 (50.8 out of 74 kJ mol−1) of the overall energy. The nature of this interaction is mainly connected to dispersion phenomena, such as the stacking between the aryl groups. Probably, this very tight packing of sulfoxides of the same chirality causes the preferential crystallization of single enantiomers, and thus a conglomerate.
Conclusions
In the present work, we analysed further synthetic targets for our enantioselective oxidation of sulfides with hydroperoxides in the presence of a complex between titanium and (S,S)-hydrobenzoin. This consolidated and versatile oxidation protocol has been tested with aryl benzyl sulfides containing heterocyclic moieties, that can originate bioactive sulfoxides. The substitution of the phenyl with the 2-thienyl or 2-pyridyl groups does not alter a framework of high enantioselectivities, that is peculiar of our protocol. An exception of lower enantioselectivity was observed in the case of the benzimidazolyl benzyl sulfide, but it was recovered with a slight modification of the catalyst. It is worth to mention that pentafluorobenzyl sulfides are always oxidized successfully, even in the case of the problematic benzimidazolyl sulfide, according to our mechanism.However, our crystallographic analysis adds relevant information to this investigation. The possibility that conglomerates are present in the family of benzimidazolyl sulfoxides opens new perspectives. In fact, in principle, it should be possible to set up a separation procedure without resorting to asymmetric synthesis. Then, in our opinion, future synthetic investigation on these intermediates should be always accompanied by a crystallographic analysis, to verify the possibility of finding conglomerates, and thus new large-scale separations of enantiomers.
Experimental section
Synthesis and characterization of compounds
Chemicals were used as received. TBHP is a commercial 80% solution of tert-butyl hydroperoxide (in di-tert-butyl peroxide/water 3:2). CHP is a commercial 80% solution. High resolution Mass Spectra were determined with a HPLC-QTOF spectrometer via direct infusion of the samples, using methanol as the eluent. 1H-NMR spectra were recorded at 500 MHz or 400 MHz. 13C-NMR spectra were recorded at 125 MHz or 100 MHz. Only absolute values of the coupling constants were reported.
Synthesis of sulfides (1a)–(9a) Sulfides were synthesized on a 6 mmol scale by adding the commercially available thiol to an ethanol solution of potassium carbonate and the corresponding benzyl bromide.30,31 The mixture was reacted for 2 hours at room temperature. Usual work up30,31 gave a crude mixture that was purified by distillation.
2-(Benzylthio)thiophene (1a).33 Kugelrohr oven temp 120–125 °C, p = 0.1 torr. 59% yield. 1H-NMR (500 MHz, CDCl3) 7.38–7.26 (m, 4H), 7.25–7.19 (m, 2H), 7.02–6.94 (m, 2H), 4.01 (s, 2H). 13C-NMR (125 MHz, CDCl3) 137.5, 134.2, 133.4, 129.6, 128.9, 128.3, 127.3, 127.1, 43.7.
2-(2,3,4,5,6-Pentafluorobenzylthio)thiophene (2a). Kugelrohr oven temp 98–102 °C, p = 0.1 torr. 53% yield. 1H-NMR (400 MHz, CDCl3) 7.41 (dd, J = 5.3, J = 1.4 Hz, 1H), 7.02 (dd, J = 3.4, J = 1.4 Hz, 1H), 6.97 (dd, J = 5.3, J = 3.4 Hz, 1H), 3.97 (broad s, 2H). 13C-NMR (100 MHz, CDCl3) 145.0 (dm, J = 249 Hz), 140.3 (dm, J = 254 Hz), 137.3 (dm, J = 254 Hz), 136.1, 131.7, 131.0, 127.8, 112.1 (m), 30.2. HRMS (ESI-TOF), m/z calcd for C11H4F5S2 [M − H]+ 294.9674. Found [M − H]+ 294.9712.
2-(4-Bromophenylthiomethyl)pyridine (4a).35 Kugelrohr oven temp 145–150 °C, p = 0.1 torr. 81% yield. 1H-NMR (500 MHz, CDCl3) 8.55–8.50 (m, 1H), 7.72–7.66 (m, 1H), 7.39–7.32 (m, 3H), 7.25–7.16 (m, 3H), 4.30 (s, 2H). 13C-NMR (125 MHz, CDCl3) 156.8, 148.0, 138.0, 134.3, 132.0, 131.4, 123.5, 122.6, 120.6, 39.6.
2-Benzylthio-1H-benzo[d]imidazole (5a).36 mp 188–190 °C (lit.36 mp 184–185 °C). 95% yield. 1H-NMR (400 MHz, DMSO-d6) 12.61–12.52 (m, 1H), 7.58–7.34 (m, 4H), 7.33–7.21 (m, 3H), 7.14–7.10 (m, 2H), 4.56 (s, 2H). 13C-NMR (100 MHz, DMSO-d6) 149.6, 143.5, 137.6, 135.4, 128.8, 128.4, 127.2, 121.6, 121.1, 117.3, 35.0 ppm.
2-(2-Pyridylmethylthio)-1H-benzo[d]imidazole (8a)38 mp 95–97 °C (lit.38 mp 100–101 °C). 79% yield. 1H-NMR (500 MHz, CDCl3) 8.67 (d, J = 4.7 Hz, 1H), 7.77 (dt, J = 1.7 Hz, J = 7.7 Hz, 1H), 7.59–7.55 (m, 2H), 7.40 (d, J = 7.9 Hz, 1H), 7.34–7.31 (m, 1H), 7.24–7.18 (m, 2H), 4.39 (s, 2H). 13C-NMR (125 MHz, CDCl3) 158.1, 148.9, 138.2, 123.8, 123.1, 122.2, 111.1, 109.8, 37.8 ppm.
2-(2,3,4,5,6-Pentafluorobenzylthio)-1H-benzo[d]imidazole (9a). mp 203–204 °C. 91% yield. 1H-NMR (400 MHz, Acetone-d6) 12.06–11.40 (m, 1H), 7.60–7.40 (m, 2H), 7.20–7.13 (m, 2H), 4.71 (s, 2H). 13C-NMR (100 MHz, Acetone-d6) 147.5, 145.2 (dm, J = 248 Hz), 140.5 (dm, J = 251 Hz), 137.3 (dm, J = 249 Hz), 122.0–121.0 (m), 112.1 (m), 23.2 ppm. HRMS (ESI-TOF), m/z calcd for C14H8F5N2S 331.0329. Found [M + H]+ 331.0330.
2-Benzylthio-1 methyl-1H-benzo[d]imidazole (10a)40 was synthesized by methylation of sulfide 5a with methyl iodide and potassium carbonate in ethanol.30,31 mp 87–88 °C (lit40 70–71 °C). Yield 57%. 1H-NMR (500 MHz, CDCl3) 7.77–7.73 (m, 1H), 7.46–7.41 (m, 2H), 7.36–7.25 (m, 6H), 4.64 (s, 2H), 3.62 (s, 3H). 13C-NMR (125 MHz, CDCl3) 151.6, 143.3, 136.7, 136.6, 129.0, 128.6, 127.6, 122.0, 121.8, 118.3, 108.5, 37.2, 29.9 ppm.
Synthesis of racemic sulfoxides. Racemic sulfoxides 1b–10b were used in the optimisation of the conditions for the HPLC separations and were obtained by standard m-chloroperoxybenzoic acid oxidation of the corresponding sulfides 1a–10a.
Synthesis of sulfoxides (1b) – (10b) by enantioselective oxidation of the corresponding sulfides. Representative procedure
According to our previous work,23–31 under a nitrogen atmosphere, it was prepared a solution of (S,S)-hydrobenzoin (21 mg, 0.1 mmol) in 8 ml of the solvent reported in Table 1. If i-propanol and molecular sieves 4A are required (see Table 1), they were added to the solution at this stage. Then, a solution of Ti(O-i-Pr)4 99.999% (14 mg, 0.05 mmol) in 4 ml of the specified solvent was added. After 1 h stirring at room temperature, a solution of 1 mmol of the sulfide to be oxidised in 8 ml of the solvent was added. The stirring was continued for 30 minutes. After this time, 1.1 mmol of a commercial solution of TBHP or CHP (see Table 1) was added and the stirring was continued for two days. After this time, the solvent was evaporated and the residual was subjected to column chromatography (silica gel), followed by crystallisation. Ee values were measured with chiral HPLC (see later).(R)-2-(Benzylsulfinyl)thiophene (1b).34 mp 78–80 °C. [α]D25 = −94.3 (c = 0.65, CHCl3) for a sample having 85% ee. Lit.34 -86.7 (c = 0.85, CHCl3). 85% yield. The ee value was measured by HPLC (Column: Chiralcel OD-H. Eluent n-hexane/i-propanol 8:2. Flow rate 0.5 ml min−1. Separation factor α 1.39). 1H-NMR (500 MHz, CDCl3) 7.67–7.61 (m, 1H), 7.37–7.23 (m, 3H), 7.17–7.00 (m, 4H), 4.39 (d, J = 12.4 Hz, 1H), 4.16 (d, J = 12.4 Hz, 1H). 13C-NMR (125 MHz, CDCl3) 131.1, 130.2, 129.8, 129.1, 128.9, 128.7, 128.4, 127.1, 64.9.
(R)-2-(2,3,4,5,6-Pentafluorobenzylsulfinyl)thiophene (2b). Mp 125–127 °C. [α]D25 = +30.6 (c = 0.36, CHCl3). 83% yield. The ee value was measured by HPLC (Column: Chiralpak IA. Eluent n-hexane/i-propanol 7:3. Flow rate 0.5 ml min−1. Separation factor α 1,20). 1H-NMR (400 MHz, CDCl3) 7.71 (dd, J = 5.1, J = 1.4 Hz, 1H), 7.33 (dd, J = 3.8, J = 1.4 Hz, 1H), 7.13 (dd, J = 5.1, J = 3.8 Hz, 1H), 4.36 (d, J = 13.0 Hz, 1H), 4.31 (d, J = 13.0 Hz, 1H). 13C-NMR (100 MHz, CDCl3) 145.7 (dm, J = 251 Hz), 144.3, 141.3 (dm, J = 254 Hz), 137.5 (dm, J = 252 Hz), 132.0, 129.5, 127.5, 104.2 (m), 51.7 (broad). HRMS (ESI-TOF), m/z calcd for C11H5F5NaOS2 334.9599. Found [M + Na]+ 334.9595.
(R)-2-(4-Bromophenylmethylsulfinyl)pyridine (4b)35 Mp 82–83 °C. [α]D25 = +195.3 (c = 0.3, CH3CN) for a 93% ee value. 64% yield. The ee value was measured by HPLC (Column: Chiralcel OD-H. Eluent n-hexane/i-propanol 7:3. Flow rate 0.5 ml min−1. Separation factor α 1.22).1H-NMR (500 MHz, CDCl3) 8.57–8.52 (m, 1H), 8.39–8.33 (m, 1H), 7.94–7.89 (m, 1H), 7.85–7.78 (m, 1H), 7.69–7.57 (m, 4H), 4.96 (d, J = 13.2 Hz, 1H), 4.72 (d, J = 13.2 Hz, 1H). 13C-NMR (125 MHz, CDCl3) 146.6, 144.7, 141.0, 139.5, 132.8, 128.9, 126.6, 125.8, 125.7, 57.7.
(R)-2-Benzylsulfinyl-1H-benzo[d]imidazole (5b). Mp 190–192 °C (lit.37 182.9–183.2 °C for a “racemic” sample – see Text). [α]D25 = +332 (c = 0.23, CHCl3). Lit.32 −39.9 (c = 1, CHCl3) for a 50% ee sample of the (S)-enantiomer. 55% yield. The ee value was measured by HPLC (Column: Chiralpak IA. Eluent n-hexane/i-propanol 7:3. Flow rate 0.5 ml min−1. Separation factor α 1.22). 1H-NMR (400 MHz, acetone-d6) 12.15–11.89 (m, 1H), 7.80–7.71 (m, 1H), 7.63–7.55 (m, 1H), 7.35–7.22 (m, 5H), 7.20–7.16 (m, 2H), 4.62 (d, J = 13.2 Hz, 1H). 4.39 (d, J = 13.2 Hz, 1H). 13C-NMR (100 MHz, Acetone-d6) 153.6, 144.0, 134.4, 130.3, 129.7, 128.1, 128.0, 123.5, 122.4, 119.7, 111.9, 60.4.
(R)-2-(2-Pyridylmethylsulfinyl)-1H-benzo[d]imidazole (8b)39 mp 145–147 °C (lit.39 mp 121.3–122.9 °C). [α]D25 = +101 (c = 0.15, CHCl3) for a sample of 74% ee. 74% yield. The ee value was measured by HPLC (Column: Chiralcel OD-H. Eluent n-hexane/i-propanol 7:3. Flow rate 0.5 ml min−1. Separation factor α 1.26). 1H-NMR (500 MHz, CDCl3) 8.54–8.49 (m, 1H), 7.74–7.53 (broad, 3H), 7.36–7.30 (m, 2H), 7.24–7.18 (m, 1H), 7.17–7.12 (m, 1H), 4.79 (d, J = 13 Hz, 1H), 4.59 (d, J = 13 Hz, 1H). 13C-NMR (125 MHz, CDCl3) 152.3, 150.0, 140.5, 136.8, 125.3, 123.8–123.7 (broad), 123.3, 109.6, 62.6 ppm.
(R)-2-(2,3,4,5,6-Pentafluorobenzylsulfinyl)-1H-benzo[d]imidazole (9b). Mp 190–192 °C. [α]D25 = +219.1 (c = 0.6, CHCl3). 69% yield. The ee value was measured by HPLC (Column: Chiralpak IA. Eluent n-hexane/i-propanol 7:3. Flow rate 0.5 ml min−1. Separation factor α 1.55). 1H-NMR (400 MHz, Acetone-d6) 12.29–12.14 (m, 1H), 7.78–7.73 (m, 1H), 7.65–7.59 (m, 1H), 7.40–7.29 (m, 2H), 4.84 (d, J = 13.7 Hz, 1H), 4.63 (d, J = 13.7 Hz, 1H). 13C-NMR (100 MHz, Acetone-d6) 152.6, 145.4 (d, J = 253 Hz), 144.0, 140.9 (d, J = 253 Hz), 137.2 (d, J = 250 Hz), 134.4, 124.0, 122.7, 119.9, 112.0, 104.4, 48.3 ppm. HRMS (ESI-TOF), m/z calcd for C14H5F5N2NaOS 369.0097. Found [M + H]+ 369.0113.
2-Benzylsulfinyl-1-methyl-1H-benzo[d]imidazole (10b)41 mp 133–134 °C. 37% yield. The ee value was measured by HPLC (Column: Chiralpak IA. Eluent n-hexane/i-propanol 7:3. Flow rate 0.5 ml min−1. Separation factor α 1.44). 1H-NMR (500 MHz, CDCl3) 7.86–7.83 (m, 1H), 7.39–7.35 (m, 2H), 7.33–7.27 (m, 2H), 7.25–7.21 (m, 2H), 7.06–7.03 (m, 2H), 4.65 (d, J = 12.8 Hz, 1H). 4.47 (d, J = 12.8 Hz, 1H), 3.47 (s, 3H). 13C-NMR (125 MHz, CDCl3) 150.0, 142.0, 136.5, 130.5, 128.8, 128.7, 128.6, 124.4, 123.4, 120.7, 109.6, 60.8, 29.8.
Absolute configuration of sulfoxides. The (R)-configuration of the sulfoxides was attributed on the basis of the X-ray diffraction experiments that were performed in this work for sulfoxides 2b, 5b and 9b or by comparison with known sample for sulfoxide 1b34 and for sulfoxide 8b.47 In our work,23–31 we have always observed that (R)-sulfoxides are obtained when (S,S)-hydrobenzoin is employed as a titanium ligand. The results of the present investigation confirm this trend, and can be used to attribute the configuration to sulfoxide (+)-(4-bromophenylsulfinilmethyl)pyridine 4b, which has not satisfactory crystals for an X-ray diffraction experiment. As a further confirmation, the (R)-configuration had been attributed to the analogous sulfoxide (+)-2-(4-tolylsulfinilmethyl)pyridine,48 differing only for the presence of the tolyl moiety instead of the bromine atom. The same considerations can be extended to sulfoxide 8b.
X-ray diffraction experiments
Data collection for single crystal X-ray diffraction experiments was performed by using Mo Kα radiation in a Bruker SMART-APEX diffractometer. An empirical absorption correction was applied (SADABS). Structures were solved by direct methods (SHELXS-86) and refined by full-matrix least-squares methods on F2 for all reflections (SHELXL-2016).49 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms bonded to carbon atoms were placed in calculated positions with isotropic displacement parameters fixed at 1.5 (CH3) or 1.2 (CH2 and CH) times the Ueq of the corresponding carbon atoms. The hydrogen atom bonded to a nitrogen atom was located in a difference Fourier map and refined with an isotropic displacement parameter. Crystal data and further refinement details are collected in the ESI (Tables S1–S5).†Data availability
The data underlying this study are available in the published article and in the ESI.† Crystallographic data were deposited at the Cambridge Crystallographic Data Centre with the following depository codes: 2328883 (10b), 2328884 (2b), 2328885 (5b), 2328886 (9b). Copies of available material can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-1223-336033 or e-mail: E-mail: deposit@ccdc.cam.ac.uk).cif files and checkcif files are also attached.Author contributions
M. A. M. Capozzi: synthesis and chemical characterization of the molecules; writing of the manuscript. A. Alvarez-Larena: X-ray diffraction experiments; resolution of the crystal structures; writing of the manuscript. J. F. Piniella: X-ray diffraction experiments; resolution of the crystal structures; writing of the manuscript. C. Cardellicchio: synthesis and chemical characterization of the molecules; theoretical calculations; writing of the manuscript; coordination of the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Italian Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR) is gratefully acknowledged for support. The projects “Fibre e tessuti intelligenti ed ECOsostenibili per l'abbigliamento TECnico e l'alta moda (ECOTEC)” and “Processi di EstRazione di bioprodotti da sCarti agroIndustriali e VALorizzazione in cascata PON ARS01_00869 PERCIVAL- Programma di ricerca n. 03.231- CUP: B95F21001900005” are gratefully acknowledged. Dr Agostina Capodilupo is gratefully acknowledged for technical support in NMR spectra recording.References
- H. Pellissier, Coord. Chem. Rev., 2022, 463, 214537 CrossRef.
- H. B. Kagan, Asymmetric Synthesis of Chiral Sulfoxides, in Organosulfur Chemistry in Asymmetric Synthesis, ed. T. Toru and C. Bolm, Wiley, Weinheim, 2008, pp 1–29 Search PubMed.
- E. Wojaczyńska and J. Wojaczyński, Chem. Rev., 2010, 110, 4303 CrossRef PubMed.
- E. Wojaczyńska and J. Wojaczyński, Chem. Rev., 2020, 120, 4578 CrossRef PubMed.
- J. Han, V. A. Soloshonok, K. D. Klika, J. Drabowicz and A. Wzorek, Chem. Soc. Rev., 2018, 47, 1307 RSC.
- M. A. M. Capozzi and C. Cardellicchio, Organosulfur Compounds as Chiral Building Blocks, in Chiral Building Blocks in Asymmetric Synthesis: Synthesis and Applications, ed. E. Wojaczyńska and J. Wojaczyński, John Wiley and Sons, Hoboken, 2022, pp 441–462 Search PubMed.
- A. S. Surur, L. Schulig and A. Link, Arch. Pharm., 2019, 352, e1800248 Search PubMed.
- H. Cotton, T. Elebring, M. Larsson, L. Li, H. Sorensen and S. von Unge, Tetrahedron: Asymmetry, 2000, 11, 3819 CrossRef.
- P. J. Hogan, P. A. Hopes, W. O. Moss, G. E. Robinson and I. Patel, Org. Process Res. Dev., 2002, 6, 225 CrossRef.
- M. Seenivasaperumal, H.-J. Federsel, A. Ertan and K. J. Szabó, Chem. Commun., 2007, 2187 RSC.
- M. Seenivasaperumal, H.-J. Federsel and K. J. Szabó, Adv. Synth. Catal., 2009, 351, 903 CrossRef.
- S. Nishiguchi, T. Izumi, T. Kouno, J. Sukegawa, L. Ilies and E. Nakamura, ACS Catal., 2018, 8, 9738 CrossRef.
- E. P. Talsi, T. V. Rybalova and K. P. Bryliakov, ACS Catal., 2015, 5, 4673 CrossRef.
- E. P. Talsi and K. P. Bryliakov, Catal. Today, 2017, 279, 84 CrossRef CAS.
- T. Gao, J. Zhao, H. Sun, J. Li, C. Ma and Q. Meng, Org. Process Res. Dev., 2023, 28, 1464 CrossRef.
- P. Pitchen, E. Duñach, M. N. Deshmukh and H. B. Kagan, J. Am. Chem. Soc., 1984, 106, 8188 CrossRef CAS.
- S. H. Zhao, O. Samuel and H. B. Kagan, Tetrahedron, 1987, 43, 5135 CrossRef CAS.
- F. Di Furia, G. Modena and R. Seraglia, Synthesis, 1984, 325 CrossRef CAS.
- C. Cardellicchio, G. Fracchiolla, F. Naso and P. Tortorella, Tetrahedron, 1999, 55, 525 CrossRef CAS.
- D. Brózda, A. Głuszyńska, A. Kościołowicz and M. D. Rozwadowska, Tetrahedron: Asymmetry, 2005, 16, 953 CrossRef.
- V. E. Aggarwal, B. N. Esquivel-Zamora, G. R. Evans and E. Jones, J. Org. Chem., 1998, 63, 7306 CrossRef CAS PubMed.
- J. M. Brunel and H. B. Kagan, Synlett, 1996, 404 CrossRef CAS.
- M. A. M. Capozzi, C. Cardellicchio, F. Naso and V. Rosito, J. Org. Chem., 2002, 67, 7289 CrossRef CAS PubMed.
- F. Naso, M. A. M. Capozzi, A. Bottoni, M. Calvaresi, V. Bertolasi, F. Capitelli and C. Cardellicchio, Chem. Eur. J., 2009, 15, 13417 CrossRef CAS.
- M. A. M. Capozzi, C. Centrone, G. Fracchiolla, F. Naso and C. Cardellicchio, Eur. J. Org Chem., 2011, 4327 CrossRef CAS.
- M. A. M. Capozzi, G. Fracchiolla and C. Cardellicchio, J. Sulf. Chem., 2013, 34, 646 CrossRef CAS.
- M. A. M. Capozzi, F. Capitelli, A. Bottoni, M. Calvaresi and C. Cardellicchio, ChemCatChem, 2013, 5, 210 CrossRef CAS.
- M. A. M. Capozzi, F. Capitelli and C. Cardellicchio, Cryst. Growth Des., 2014, 14, 5442 CrossRef.
- M. A. M. Capozzi, G. Terraneo, G. Cavallo and C. Cardellicchio, Tetrahedron, 2015, 71, 4810 CrossRef.
- M. A. M. Capozzi, A. Bottoni, M. Calvaresi and C. Cardellicchio, Tetrahedron, 2018, 74, 2041 CrossRef.
- M. A. M. Capozzi, V. Frascaro, G. Pescitelli and C. Cardellicchio, Tetrahedron, 2019, 75, 2406 CrossRef.
- B. Jiang, X.-L. Zhao, J.-J. Dong and W.-J. Wang, Eur. J. Org. Chem., 2009, 987 CrossRef.
- L. Liu, Y. Tang, K. Wang, T. Huang and T. Chen, J. Org. Chem., 2021, 86, 4159 CrossRef.
- L. Zong, X. Ban, C.-W. Kee and C.-H. Tan, Angew. Chem., Int. Ed., 2014, 53, 11849 CrossRef.
- F. Haviv, R. W. DeNet, R. J. Michaels, J. D. Ratajczyk, G. W. Carter and P. R. Young, J. Med. Chem., 1983, 26, 218 CrossRef.
- K. Bharami, M. M. Khodaei and M. S. Arabi, J. Org. Chem., 2010, 75, 6208 CrossRef PubMed.
- I. V. Loginova, K. S. Rodygin, S. A. Rubtsova, P. A. Slepukhin, A. V. Kuchin and V. A. Polukeev, Russ. J. Org. Chem., 2011, 47, 124 CrossRef.
- D. Y. Cruz-Gonzalez, R. González-Olvera, D. Angeles-Beltrán, G. E. Negrón-Silva and R. Santillan, Synthesis, 2013, 45, 3281 CrossRef.
- B. Hernández-Ochoa, S. Gómez-Mancho, A. Sánchez-Carrillo, J. Marcial-Quino, L. M. Rocha-Ramirez, A. Santos-Segura, E. J. Ramirez-Nava, R. Arreguin-Espinosa, M. Cuevas-Cruz, A. Méndez-Tenorio and E. Calderón-Jaimes, Molecules, 2020, 25, 3979 CrossRef.
- R. Wang, H. Xu, Y. Zhang, Y. Hu, Y. Wei, X. Du and H. Zhao, Org. Biomol. Chem., 2021, 19, 5899 RSC.
- X. Ye, A. M. P. Moeljadi, K. F. Chin, H. Hirao, L. Zong and C.-H. Tan, Angew Chem. Int. Ed. Engl., 2016, 55, 7101 CrossRef PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006 CrossRef PubMed.
- M. A. M. Capozzi, A. Alvarez-Larena, J. F. Piniella Febrer and C. Cardellicchio, Cryst. Res. Technol., 2024, 2400096 CrossRef.
- G. Coquerel, Preferential Crystallization, in Novel Optical Resolution Technologies, Topics in Current Chemistry, ed. K. Sakai, N. Hirayama and R. Tamura, Springer, Berlin, Heidelberg, 2006, vol. 269, pp. 1–51 Search PubMed.
- M. P. Walsh, J. A. Barclay, C. S. Begg, J. Xuan, N. T. Johnson, J. C. Cole and M. O. Kitching, JACS Au, 2022, 2, 2235 CrossRef.
- M. P. Walsh, J. A. Barclay, C. S. Begg, J. Xuan and M. O. Kitching, Cryst. Grow. Des., 2023, 23, 2837 CrossRef.
- J. Deng, J. Zhu, J. Liao and J. Zhu, Racemic Enantiomers Resoluting, WO Pat., WO2010/118575 A1, 2010 Search PubMed.
- K.-U. Baldenius and H. B. Kagan, Tetrahedron: Asymmetry, 1990, 1, 597 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2328883–2328886. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra07088g |
| This journal is © The Royal Society of Chemistry 2024 |