
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExperimental verification of halomethyl carbinol synthesis from carbonyl compounds using a TiCl4–Mg bimetallic complex promoter†
Olga Michalak *a,
Sylwia Żurawickaa,
Marek Kubiszewskib,
Piotr Krzeczyńskia,
Andrzej Leśc,
Sławomir Filipekd and
Marcin Cybulskia
*a,
Sylwia Żurawickaa,
Marek Kubiszewskib,
Piotr Krzeczyńskia,
Andrzej Leśc,
Sławomir Filipekd and
Marcin Cybulskia
aPharmacy, Cosmetic Chemistry and Biotechnology Research Group, Łukasiewicz Research Network-Industrial Chemistry Institute, Rydygiera 8, 01-793 Warsaw, Poland. E-mail: olga.michalak@ichp.lukasiewicz.gov.pl; Tel: +48 453 056 175
bPharmaceutical Analysis Laboratory, Łukasiewicz Research Network-Industrial Chemistry Institute, Rydygiera 8, 01-793 Warsaw, Poland
cFaculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
dBiological and Chemical Research Centre, Faculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
First published on 16th December 2024
Abstract
A critical evaluation of the feasibility of a previously published method for synthesising halomethyl carbinols from carbonyl compounds and CH2Br2 or CH2Cl2 using a bimetallic TiCl4–Mg complex is presented. The synthesis of compounds lacking the –CH2– group in their structure was achieved by following the procedures proposed in the reference literature or by introducing modifications to selected process parameters. These compounds were not identified as expected β-halohydrins but as products of reductive dimerisation or subsequent pinacolic rearrangement of carbonyl substrates. This paper proposes a formation mechanism of vicinal 1,2-diols in the presence of a TiCl4–Mg system, supported by experimental data and theoretical DFT calculations (DFT/B3LYP).
1 Introduction
β-Halohydrins, bearing a hydroxyl and halide functional group, are privileged building blocks used in organic synthesis to obtain compounds with biological activity.1 To date, halohydrins could be prepared by halohydroxylation of olefins,2 chemical or biotechnological reduction of haloketones,3–6 ring-opening reaction of epoxides,7–9 nucleophilic substitution of benzyl halides,10 oxidative β-halogenation of alcohols11 or gold catalysed regioselective hydration of haloalkynes.12 An important method for obtaining β-halohydrins uses carbonyl compounds as the initial reactants, followed by their treatment with in situ generated mono- and polyhalomethyl anions. Tarhouni et al. obtained monohalomethyllithium from the corresponding dihalogenomethane (XCH2Br; X = Br, Cl) by bromine–lithium exchange. The reactions were carried out in the presence of sec-butyl lithium (sec-BuLi) and one lithium bromide equivalent (LiBr) at a low temperature of −110 °C.13 The temperature of the lithium/halogen exchange was revealed as a critical parameter in the use of such carbenoid species. Relatively elevated temperatures promoted undesirable metal-assisted α-elimination with the formation of carbene-like species.14–17 For enantiomerically enriched monodeuterated chloromethyllithium the temperature −78 °C resulted in its rapid decomposition.18–20Nishimura et al. synthesised chlorohydrins in good yields from aldehydes and lithium magnesium carbenoid ClCH2MgCl–LiCl through the iodine/magnesium exchange reaction of the “turbo Grignard” i-PrMgCl–LiCl with chloroiodomethane.21 Pace et al. reviewed monofluoromethylation reactions using fluoroiodomethane to generate a fluoromethyl group. Depending on the reaction conditions adopted, the reagent serves as a precursor for differently functionalised CHF fragments.22 The preparations of iodomethyl carbinol using an metallic samarium and diiodomethane were also reported.23–25 The instability of lithium halocarbenoids can be overcome by the in situ trapping of previously introduced carbonyl compounds in flow chemistry processes.26 For example, direct nucleophilic monofluoroalkylation using flow microreactor technology allowed the use of short-lived intermediates, such as 1-fluoro-2-phenylethyllithium, 1-fluoro-3-phenylpropyllithium, generated by the lithium/iodine exchange reaction.27,28
Many synthetic applications have been found using lithium carbenoids.29 These reagents have been applied to the large-scale preparation of aminoepoxide intermediates of HIV protease inhibitors,30 to the total synthesis of fumagillin, an angiogenesis inhibitor,31 and the synthesis of fluconazole.26 Recently, Yan et al. described the synthesis of bromomethyl or chloromethyl carbinols by coupling various aldehydes and ketones with CH2Br2 or CH2Cl2, promoted by bimetallic TiCl4–Mg complex.32 Earlier studies by the Yan group proved that the TiCl4–Mg/CH2Cl2 system can also be successfully applied to the direct methylenation of various ketones and aldehydes.33 However, the reaction conditions from both papers32,33 differed essentially only in the TiCl4–Mg ratio.
In one of our current projects, there was a need to obtain a specific halohydrin from a particular carbonyl compound. For this reason, we started to prepare bromomethyl or chloromethyl carbinols using Yan's reference protocols.32
The present paper describes the results of our synthetic studies on Yan's halomethyl carbinols synthesis from aldehydes and ketones with CH2Br2 and CH2Cl2 in the presence of TiCl4–Mg bimetallic catalyst. We have also attempted to explain the mechanisms of formation of the identified products, which are different from the expected bromomethyl or chloromethyl carbinols, by analysing the experimental data and the results of numerous DFT calculations.
2 Results and discussion
2.1 Chemistry
Yan's32 method involves the treatment of an aldehyde or ketone with dibromomethane in the presence of Mg, TiCl4 and an Electron Pair Donor (EPD), at a relatively mild (0 °C) temperature when compared to the minimum temperatures for the stability of lithium halocarbenoids. The authors consider dimethoxy ethane (DME) to be the best EPD due to the reaction efficiency. However, according to the proposed mechanism (Fig. 1), the reaction also begins immediately after the addition of THF.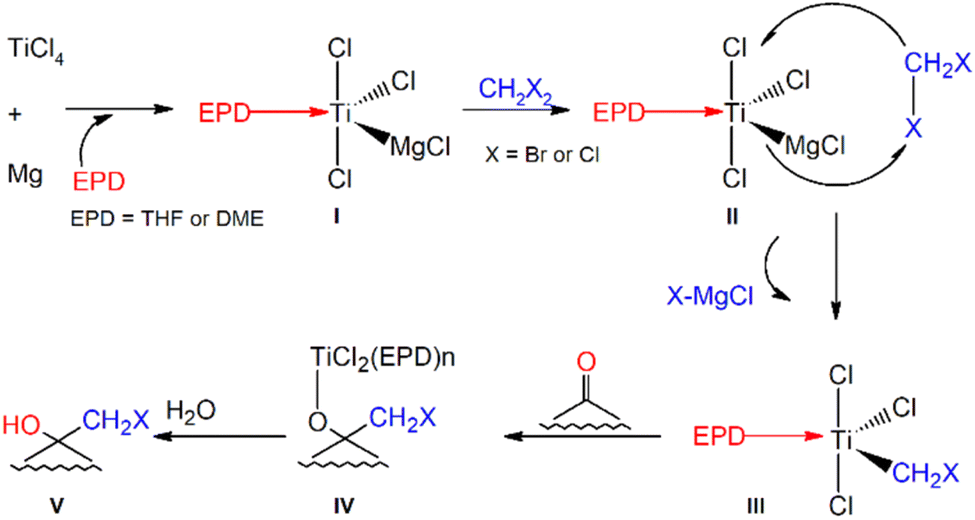 | ||
| Fig. 1 The mechanism for TiCl4–Mg mediated halomethylcarbinol formation proposed by Yan et al.32 | ||
Therefore, in the first series of our experiments, we used the given reaction conditions for the formylcyclohexane and dibromomethane substrates. Unfortunately, the procedure involving Mg, TiCl4 and DME did not yield the expected α-bromomethylcarbinol derivative, despite repeating the reaction and changing the temperature conditions. The results of the 1H and 13C NMR analyses indicated the absence of the –CH2– group in the chemical structure of the isolated products (ESI S.11†). These negative observations prompted us to investigate the reaction mechanism in detail. Thus, several different carbonyl compounds were subjected to the same reaction procedure to assess whether or not other substrates also led to products without the –CH2– group. We carefully repeated Yan's32 reference experimental protocol by dissolving the carbonyl compounds in 1,2-dichloroethane, followed by their addition to the suspension of Mg and TiCl4 in CH2Br2. Finally, after 5 minutes, DME (used here as EPD) was added to the reaction mixtures. This was done in two different ways: by fast addition in one portion, which led to a drastic increase of the reaction temperature up to 80 °C (procedure A2, Table 1) or by dropwise addition, while the temperature was maintained at 0 °C (procedure A1, Table 1).
| Substrate | Producta | Synthetic procedure and yield | |||||
|---|---|---|---|---|---|---|---|
| A1 | A2 | B1 | B2 | C1 | C2 | ||
| a Specific reaction conditions: A1: TiCl4–MgCH2Br2–C2H4Cl2–DME, added DME at 0 °C, then 0 °C; A2: TiCl4–MgCH2Br2–C2H4Cl2–DME, added DME without maintaining 0 °C, then 0 °C; B1: TiCl4–Mg–CH2Cl2–DME, added DME at 0 °C, then 0 °C; B2: TiCl4–Mg–CH2Cl2–THF, added THF at 0 °C, then 0 °C; C1: TiCl4–Mg–CH2Cl2–THF, added THF at rt, then rt, ultrasonication; C2: TiCl4–Mg–THF, added THF at rt, then rt, ultrasonication.b Numbers describing yield in [%]. | |||||||
 |
 |
22 | 12 | 28 | 30 | 9 | 3 |
 |
 |
17 | 12 | 21 | |||
 |
 |
4 | 1 | 19 | |||
 |
 |
18 | 2 | 22 | |||
 |
 |
23 | 1 | ||||
 |
 |
21 | 13 | 23 | |||
 |
 |
29 | 21 | 22 | |||
 |
 |
8 | 1 | ||||
 |
 |
6 | |||||
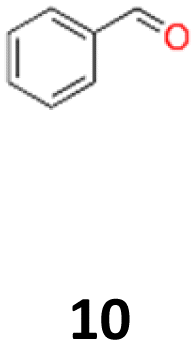 |
 |
10 | 12 | 4 | 51 | 21 | |
 |
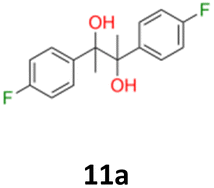 |
33 | 8 | ||||
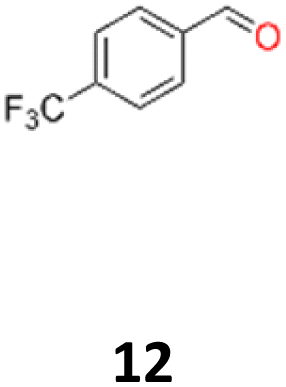 |
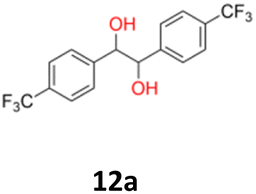 |
24 | 31 | ||||
After 3 hours, all reactions were quenched with potassium carbonate solution. The mixtures were filtrated through Celite pad and the organic layer was separated from the filtrates. The extracts were dried and evaporated to give crude products. The products were isolated by silica gel column chromatography, then characterised by NMR spectroscopy to determine the presence or absence of the –CH2– group. The analysis confirmed the lack of a methylene group for all obtained derivatives. The results also showed that the temperature increase associated with the fast addition of DME did not affect the reaction course, leading to the same type of products as under temperature-controlled conditions. However, maintaining the temperature at 0 °C resulted in a significant increase of reaction yields for isolated products.
Comparing the reaction conditions and reagents used in Yan's procedure to the important organic synthesis method known as “pinacol coupling”, it became clear that Yan's conditions may cause reductive dimerisation of substrates by an electron transfer process to form vicinal 1,2-diols. To date, several methods have been described for the radical activation of carbonyl derivatives, leading to their homodimerisation. Among others, various low-value titanium compounds have been shown to have efficiency in pinacol coupling. The described methods include coupling promoted by aqueous TiCl3 in basic media34 or ultrasound-supported coupling in acidic conditions.35 Similarly, the reaction easily occurs in anhydrous solvents, such as DCM/THF.36 The titanium(III) compounds were reported as readily accessible by in situ reduction of Ti(IV) precursors becoming very attractive catalysts for such reactions.37–41 The combination of TiCl3/Zn–Cu, introduced by McMurry and Rico was also successfully used for the intramolecular coupling of various aldehydes.41 Moreover, magnesium is the only alkaline earth metal, that found early application in the pinacol coupling reaction, even in an amalgamated form or as a Mg/MgI2 system.42 The conditions described by Li et al.43 for obtaining pinacols via the TiCl4–Mg system were most similar to those described by Yan et al.32 The transformation of benzaldehyde to the respective pinacol derivative occurred in the presence of THF and/or DME as primary ligands in DCM solvent with a yield of 68% (304![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d,l to meso).
:1 d,l to meso).
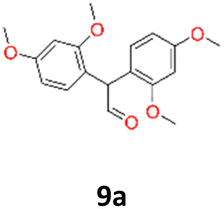
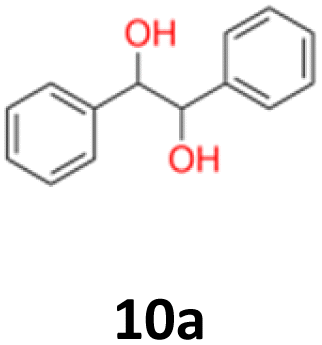
HRMS and EI analyses of our obtained reaction products confirmed the hypothesis (ESI S.12†). No characteristic signals of bromine isotopes were observed in the mass spectra. The isotopic mass distribution for the parent ions confirmed the presence of postulated vicinal diols as reaction products. Only for 2,4-dimethoxybenzaldehyde (9), in the presence of Mg/TiCl4/CH2Br2/C2H4Cl2/DME system, the isolated product (9a) was characterised as the product of pinacol rearrangement. The distinguishing features of this substrate from others was the presence of two strongly activating (EDG) methoxy substituents in the aromatic ring. This rearrangement type is well known and occurs mainly in the presence of acids.44,45 Normally, it can proceed either by a concerted mechanism without a carbocation intermediate or by a stepwise mechanism with a carbocation intermediate followed by a migration of the functional group in the presence of strong Brønsted or Lewis acids. However, when the migrating group is an aryl group, which facilitates the formation of a carbocation, the stepwise mechanism is predominantly favoured.46 The obtained compounds were characterised by NMR (1H and 13C) and mass spectroscopy (HRMS) (see structures in Table 1).
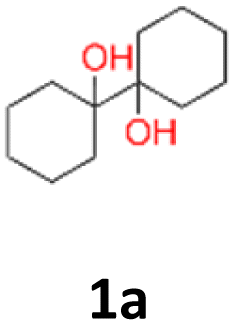
The effect of various reaction parameters on the types of products obtained was checked. Firstly, different EPDs were verified due to their importance in proposed Yan's mechanism. While carefully repeating the procedure for cyclohexanon (1) substrate with Mg, TiCl4, CH2Br2 and different EPD reagents i.e. THF, 1,4-dioxane, AcN or TEA, the reaction progress was observed only for THF to give small 3% amount of isolated vicinal 1,2-diol (1a). Changing the solvent from 1,2-dichloroethane to DCM also led to 1,2-diols instead of chloromethyl carbinols. In procedures B1 and B2, the corresponding vicinal 1,2-diols (1a) were obtained in similar yields (28% for DME or 30% for THF, respectively). In contrast, without the presence of TiCl4 or Mg in the reaction medium, no formation of products was observed. Therefore, the selected aldehydes/ketones were subjected to the next set of reaction conditions with THF additive and in DCM, which acted both as a solvent and halomethyl group donor (procedure B2). According to the literature, pinacols can also be obtained under ultrasound irradiation.41 Thus, two model substrates (benzaldehyde, cyclohexanone) were finally used to compare the effectiveness of 1,2-diol formation by TiCl4–Mg–CH2Br2–C2H4Cl2–DME Yan's32 system (A1) and TiCl4–Mg–CH2Cl2–THF (C1) ultrasound-assisted activation at room temperature for 0.5–1 h. For benzaldehyde substrate (10) the product was formed with a higher yield, i.e. 51%. However, for cyclohexanone, a higher yield i.e. 30% was obtained for the conditions not supported by ultrasound irradiation (12% yield).
The majority of reductive coupling products displayed dl-diastereoselectivity.43,47 However, to date, few examples of meso-diastereoselectivity have been published. Aspinall et al. achieved diastereoselectivity of up to 95/5 (±/meso) for aliphatic aldehydes and, in opposition, up to 19/81 (±/meso) for aromatic aldehydes in samarium diiodide catalyzed diastereoselective pinacol couplings.48 Highly meso-diastereoselective pinacol coupling of aromatic aldehydes mediated, up to 1/99 (±/meso) promoted by aluminium powder/copper sulfate system in water, was also described.47 In the case of aromatic aldehydes, coupling involving TiCl3–Mg and under ultrasound irradiation, gave different dl/meso ratios depending on the substituents in the aromatic ring. For benzaldehyde and that with aromatic EDG d,l enantiomers were observed as major form, while for substrates bearing electron-withdrawing group (EWG) meso was predominantly measured.40
Similar to the reference results mentioned above, an excess of dl products (Table 2) was also evidenced in our experiments for cyclic aliphatic ketones (1) and (5) as substrates. While reacting flexible aliphatic ketone (8), the diastereoselectivity ratio was only ca. 1:1 (dl/meso). For substituted benzaldehydes, the dl mixture predominated over the meso form, ranging for procedures A1 and B1 with temperature control at 0 °C from ca. 9:1 (Table 2, 12a) to ca. 2:1 (Table 2, 11a). The impact of temperature on dl to meso ratio was observed in the reactions of aromatic aldehydes as substrates. Furthermore, in one example, an uncontrolled increase of temperature during the experiment led to the prevalence of the meso form of 3a. In reference to the mass balance issue, small amounts of other chromatographic fractions were also isolated and characterised by NMR as complicated mixtures of different substances. The peaks specific to halohydrins, pinacols or initial substrates were also not identified in these mixtures. Instead, spots of very high polarity (Rf = 0) were observed on TLC, even in polar eluents. This may be related to the formation of degradation products in the reaction medium and/or the formation of complexes with the metals involved in the process.
| Product | Synthetic procedure and dl/meso ratio* | |||||
|---|---|---|---|---|---|---|
| A1 | A2 | B1 | B2 | C1 | C2 | |
| a *The ratio of dl/meso was determined by: a 1H NMR or b 13C NMR analysis. | ||||||
 |
97:3 |
100:0 |
100:0 |
100:0 |
100:0 |
100:0 |
 |
86:14 |
69:31 |
— | 93:7 |
— | — |
 |
85:15 |
44:56 |
— | 79:21 |
— | — |
 |
75:25 |
76:24 |
— | 74:26 |
— | — |
 |
100:0 |
100:0 |
— | — | — | — |
 |
83:17 |
83:17 |
— | 68:32 |
— | — |
 |
88:12 |
86:14 |
— | 75:25 |
— | — |
 |
50:50 |
50:50 |
— | — | — | — |
 |
91:9 |
77:23 |
— | 90:10 |
87:13 |
79:21 |
 |
67:33 |
67:33 |
— | — | — | — |
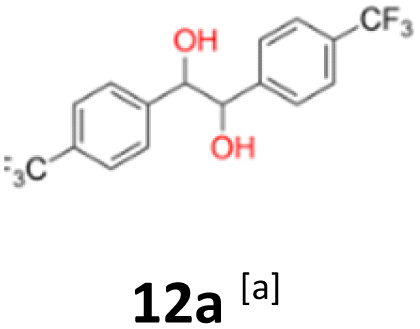 |
90:10 |
85:15 |
— | — | — | — |
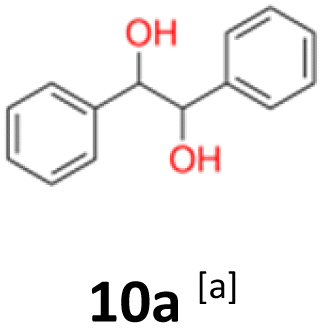
2.2 The quantum mechanical DFT calculations
Theoretical quantum mechanical calculation has been summarized in this section, with further detailed explanation provided in the ESI.† The aim of the theoretical modeling was to present our view on the subsequent steps for the synthesis of the selected pinacol compound, hydrobenzoin (10a). The confidence of the proposed stoichiometric reactions was estimated from the fact that the calculated Gibbs free energies change of the reactions was negative, indicating that the reaction should have occurred. The numerical values of the Gibbs free energy, enthalpy, and electron (+nuclear) energy were given in Table 3. The reaction barriers for the transition states were not considered in this approach. The magnesium powder used in the experimental part was modeled as a single Mg atom. The Mg reaction with CH2Br2 led to the formation of a complex (graphically labelled as “::”) which can be seen as composed of two radicals (species with an unpaired electron), i.e. [CH2Br](˙) and [MgBr](˙) (see eqn (E.1) below):| Mg + CH2Br2 → {[CH2Br](˙)::[MgBr](˙)} |
| ΔG = −52 kcal mol−1 | (E.1) |
| In this paper | ΔG | ΔH | ΔE | In ESI | Comment |
|---|---|---|---|---|---|
| (E.1) | −52 | −58 | −57 | (SE.1) | CH2Br2 split by Mg |
| (E.2) | −69 | −86 | −87 | (SE.3) | Ti/Mg catalyst formation |
| (E.3) | −19 | −28 | −29 | (SE.6.1) | Catalyst::benzaldehyde link |
| (E.4) | −15 | −31 | −34 | (SE.6.2) | (Catalyst::benzaldehyde)2 link |
| (E.5) | −59 | −104 | −108 | (SE.7.2) | Hydrobenzoin promoted by Ti/Mg catalyst |
| (E.6) | −88 | −157 | −167 | — | Hydrobenzoin, cumulative stoichiometric |
| (E.7) | −7 | −45 | −57 | (SE.8) | 1-(Bromomethyl)benzyl alcohol, cumulative stoichiometric |
Then, the addition of TiCl4 led to the formation of a bimetallic Mg/Ti catalyst:
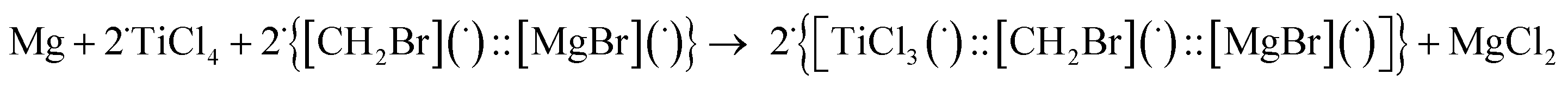
| ΔG = −69 kcal mol−1 | (E.2) |
Then we took the radical 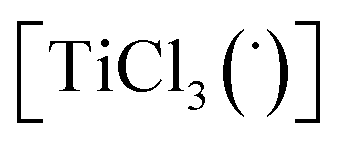 as a model for
as a model for 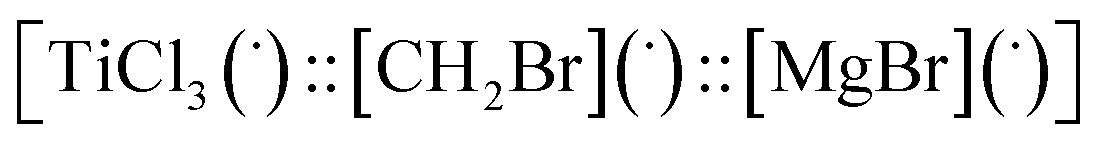 according to eqn (E.3).
according to eqn (E.3).
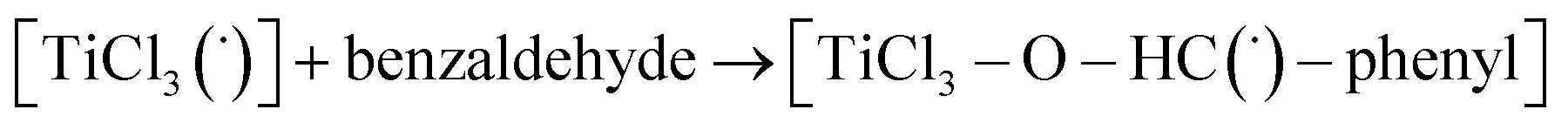
| ΔG = −19 kcal mol−1 | (E.3) |
In the reaction with benzaldehyde the following reactive complex was formed (Fig. 2):
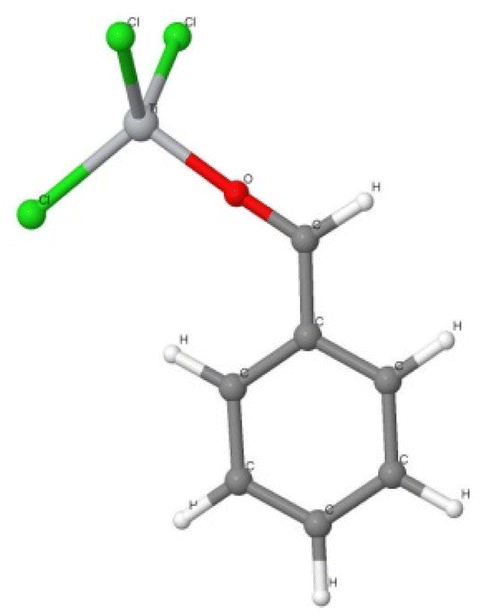 | ||
| Fig. 2 The [TiCl3–O–HC(˙)–phenyl] complex of TiCl3 and benzaldehyde. Colour codes: dark grey – C, light grey – H, red – O, grey – Ti, green – Cl. | ||
The dimerisation of this complex (according to eqn (E.4)) would form a pinacol-like structure (Fig. 3):
| 2[TiCl3–O–HC(˙)–phenyl] → [(TiCl3–O)–benzyl–benzyl–(O–TiCl3)] |
| ΔG = −15 kcal mol−1 | (E.4) |
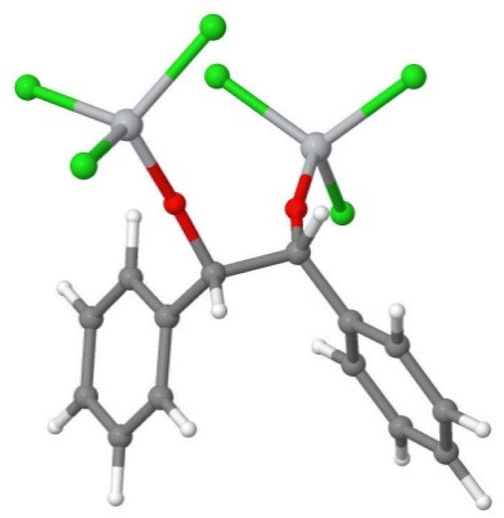 | ||
| Fig. 3 The molecular geometry of the {[TiCl3]–O–HC–(phenyl)—(phenyl)–CH–O–[TiCl3]} titanium-benzaldehyde (pinacol-like) hydrobenzoin complex. Titanium atoms were shown in light grey, and the oxygen atoms in red. | ||
Subsequent hydrolysis of such a pinacol-like structure (presented in Fig. 3) in KHCO3 aqueous solution should lead to hydrobenzoin according to eqn (E.5) (DBE = dibutyl ether as in a model of the Makino's for Ti/Mg bimetallic catalyst49):
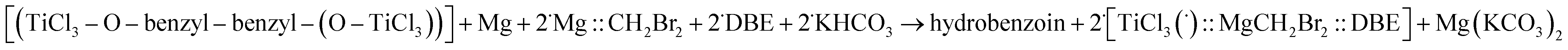
| ΔG = −59 kcal mol−1 | (E.5) |
A cumulative stoichiometric reaction leading to the formation of hydrobenzoin can be written in the form of eqn (E.6):
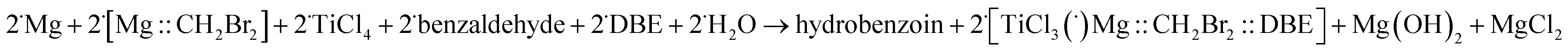
| ΔG = −88 kcal mol−1 | (E.6) |
The ΔG energy output of the reaction (E.6) can be even more negative when two radical Ti complexes couple to form a dimeric species. In (E.5) and (E.6), reaction a shorthand notation of Mg::CH2Br2 was used in place of {[CH2Br](˙)::[MgBr](˙)} as shown in (E.1). In reaction (E.6) one mole of hydrobenzoin requires the use of the Mg/Ti ratio (equiv.) of 2:1.
Let us consider the reaction (E.1) of Mg with CH2Br2, in the presence of TiCl4, benzaldehyde and water, i.e. the reaction (E.7):
| 4˙{[CH2Br](˙)::[MgBr](˙)} + TiCl4 + 4˙benzaldehyde + ˙H2O →→ 4˙{1-(bromomethyl)benzylalcohol} + TiO2 + 4˙MgClBr |
| ΔG = −7 kcal mol−1 | (E.7) |
In the reaction (E.7) one mole of {1-(bromomethyl)benzylalcohol} (presented in Fig. 4) requires the use of the Mg/Ti ratio (equiv.) of 4:1.
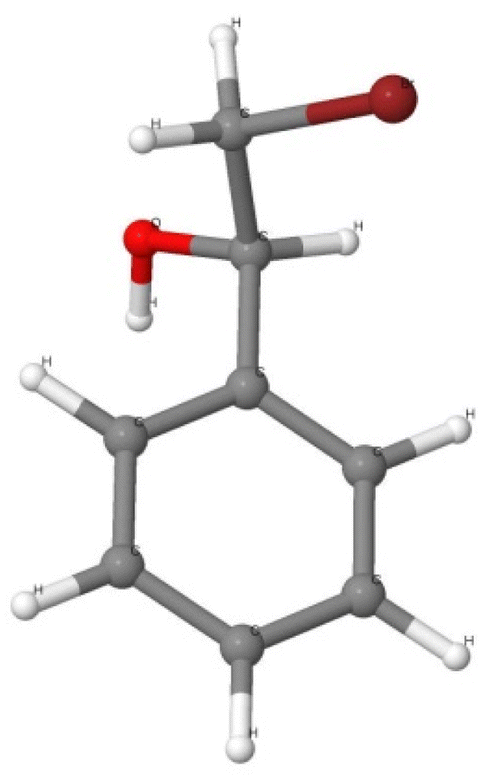 | ||
| Fig. 4 The optimized molecular geometry of 1-(bromomethyl) benzyl alcohol. Colour codes: the bromine atom was shown in brown, the oxygen atom in red, carbon atoms in dark grey and hydrogen atoms in light grey. | ||
In the reaction (E.7) one mole of {1-(bromomethyl)benzyl alcohol} requires the use of the Mg/Ti ratio (equiv.) of 4:1. The predicted energy output of (E.7) is relatively small by comparing the ΔG of hydrobenzoin formation (eqn (E.5) and (E.6)). Therefore, a possible formation of 1-(bromomethyl)benzylalcohol will likely not occur.
Apart from DBE, the influence of some other solvents on the reaction outputs was also modeled. No essential modifications comparable to the predicted DBE influence were noticed (details in the ESI†). In some preliminary studies, the model of magnesium powder in the form of Mg7 cluster was considered. It was interesting to see that the model reaction of Mg7 cluster with 1,2-dichloroethane suggested a split of the solvent molecule into ethylene and a few MgCl radicals. Details of the calculations are presented in Table 3 and ESI.†
3 Conclusion
2The feasibility of halomethyl carbinols synthesis from carbonyl compounds and CH2Br2 or CH2Cl2 using a bimetallic TiCl4–Mg complex was discussed. In all evaluated methods: A1, A2, B1, B2, C1 and C2 main products have been identified as the results of reductive dimerisation or further pinacol rearrangement of carbonyl substrates. In this paper, we have proposed a mechanism of example benzoin (10a) formation in the presence of TiCl4–Mg system. This was supported by experimental data and theoretical DFT calculations (DFT/B3LYP), as the result of Gibbs free energy (ΔG), enthalpy (ΔH) and electronic (+nuclear) energy (ΔE) outputs for model reaction. The hydrobenzoin (10a) synthesis became a complicated multistep reaction involving radicals as the intermediate species. The main source of radicals was the reaction of Mg powder with CH2Br2 being split into the [[CH2Br](˙)] and [[MgBr](˙)] radicals as well as with the components involved in the bimetallic Mg/Ti catalyst structure. Our suggested stoichiometric model supported several previous concepts about the formation of pinacol-like products. According to the DFT/B3LYP calculations performed, there was a rather low probability of obtaining 1-(bromomethyl)benzyl alcohol under the reaction conditions studied.4 Experimental
Reagents, solvents, and other materials were of commercial sources and used without additional operations. Reactions were monitored on silica gel TLC plates 60 F254 (Merck, Darmstadt, Germany). Visualizations were performed with UV light (254 and/or 365 nm) then with CeMo stain and subsequent charring. Melting points were determined using the MP70 Melting Point System (Mettler-Toledo, Greifensee, Switzerland). Solvents were evaporated under reduced pressure at 40 °C using the Büchi Rotavapor (BÜCHI Labortechnik AG, Flawil, Switzerland). Flash column chromatography was performed on silica gel (200–300 mesh). The 1H and 13C NMR spectra were acquired in CDCl3 solutions on Bruker AVANCE III HD 500 MHz spectrometer (Bruker Corporation, Billerica, MA, USA) at the temperature 298 K. To identify the structures of all isolated products, analysis of the results of 1D and 2D NMR experiments was performed. The 1H and 13C NMR chemical shifts are given relative to the TMS signal at δ = 0.0 ppm. Mass spectra were recorded on the MaldiSYNAPT G2-S HDMS (Waters Corporation, Milford, MA, USA) spectrometer via electrospray ionisation (ESI-MS). High-resolution mass spectrometry (HRMS) measurements were performed using the Synapt G2-Si mass spectrometer (Waters Corporation, Milford, MA, USA) equipped with an ESI source and a quadrupole-time-of-flight mass analyser. The results of the measurements were processed using the MassLynx 4.1 software (Waters Corporation, Milford, MA, USA).The DFT calculations were performed within the density functional theory with the B3LYP three-parameter functional and the 6-311++G(d,p) atomic basis sets. The thermodynamic parameters were obtained after calculation of the optimal geometries and all positive harmonic frequencies. We used the Gaussian G16 suite of programs implemented in the ICM Warsaw University Computer Centre within the computer grant G18-6. The ICM facilities are greatly acknowledged. The molecular structures of selected compounds are available in the ESI.†
4.1 Synthesis of compounds 1a–12a — general procedures
:hexane–AcOEt 20:1 to 5:1 v/v).(A2) The suspension of Mg (576 mg, 24 mmol) and TiCl4 (0.33 ml, 3 mmol) in 3 ml CH2Br2 were stirred for 1 min at 0 °C under nitrogen atmosphere, then a solution of aldehyde/ketone (2 mmol) in 8 ml of C2H4Cl2 was added. After being stirred for 5 min, DME (2 ml) was added without maintaining a temperature of 0 °C. The black-brown slurry was stirred for 3 h at 0 °C. Then the resulting suspension was quenched with 10 ml of saturated K2CO3 solution and filtered through a celite pad to remove the solid residues. The filtrate was extracted with C2H4Cl2 (1 × 15 ml). The combined organic layers were dried over anhydrous Na2SO4 and filtered. The solvent was evaporated under reduced pressure to give the crude product, which was separated by column chromatography on silica gel (eluent:hexane–AcOEt 20:1 to 5:1 v/v).
:hexane–AcOEt 20:1 to 5:1 v/v).(B2) The suspension of Mg (576 mg, 24 mmol) and TiCl4 (0.33 ml, 3 mmol) in 3 ml CH2Cl2 were stirred for 1 min at 0 °C under nitrogen atmosphere, then a solution of aldehyde/ketone (2 mmol) in 15 ml of C2H4Cl2 was added. After being stirred for 5 min, THF (2 ml) was added whilst maintaining the temperature at 0 °C. The black-brown slurry was stirred for 3 h at 0 °C. Then the resulting suspension was quenched with 10 ml of saturated K2CO3 solution and filtered through a celite pad to remove the solid residues. The filtrate was extracted with CH2Cl2 (1 × 15 ml). The combined organic layers were dried over anhydrous Na2SO4 and filtered. The solvent was evaporated under reduced pressure to give the crude product, which was separated by column chromatography on silica gel (eluent:hexane–AcOEt 20:1 to 5:1 v/v).
(C2) Mg (70 mg, 2.92 mmol) was added in one portion to the solution of TiCl4 (0.3 ml, 2.71 mmol) in THF (6 ml) under a nitrogen atmosphere at room temperature. The colour of the solution changed to green immediately. Then a solution of the aldehyde (1.1 mmol) was added in one portion. The mixture was irradiated in the water bath of the ultrasonic cleaner at room temperature for 1 h. Then the resulting suspension was quenched with 10 ml of 10% K2CO3 solution and filtered through a celite pad to remove the solid residues. The filtrate was extracted with ethyl acetate (3 × 15 ml). The combined organic layers were washed with saturated aqueous NaHCO3 and brine, dried over anhydrous Na2SO4 and filtered. The ethyl acetate was evaporated under reduced pressure to give the crude product, which was separated by filtration after maceration in hexane.
The following products were obtained according to the above procedures:
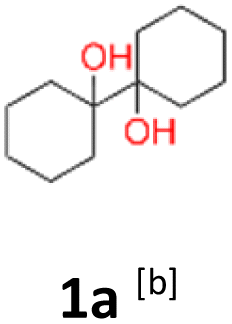
1a: yellow powder, yield: (A1: 86.2 mg, 22%, A2: 47.0 mg, 12%, B1: 108 mg, 28%, B2: 119.4 mg, 30%, C1: 17 mg, 9%, C2: 6.5 mg, 3%); mp: 127.3 °C (dec.); Rf: 0.32 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: 1.77, bs, 1H; 1.64–1.72, m, 3H; 1.52–1.63, m, 4H; 1.31–1.40, m, 2H; 1.04–1.16, m, 1H; 13C NMR (125 MHz, CDCl3), δ ppm: 75.6, 30.7, 25.9, 21.8; TOF MS AP-[M − H]− calculated for C12H21O2: 197.1542, found: 197.1541, TOF MS ES+ [M + Na]+ calculated for C12H22O2Na; 221.1517 found: 221.1513.
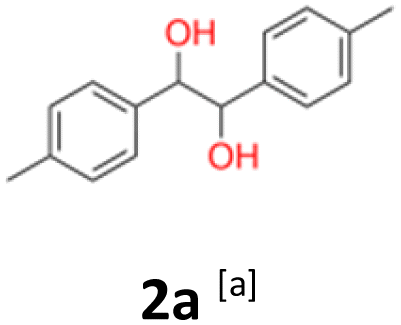
2a: white powder, yield: (A1: 103.0 mg, 17%, A2: 70.6 mg, 12%, B2: 130.1 mg, 21%); mp: 148.3 °C (dec.); Rf: 0.19 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: dl: 7.02–7.07, m, 4H; 4.67, s, 1H; 2.30, s, 3H; meso: 7.17–7.21, m, 2H; 7.12–7.16, m, 2H; 4.74, s, 1H; 2.35, s, 3H; 13C NMR (125 MHz, CDCl3), δ ppm: dl: 137.5, 137.0, 128.8, 126.8, 78.8, 21.1; meso: 137.8, 137.0, 129.0, 127.0, 78.1, 21.2; TOF MS ES+ [M + Na]+ calculated for C16H18O2Na: 265.1204, found: 265.1207; elem. anal.: C: 74.90:74.94%, H: 7.33:7.22%.
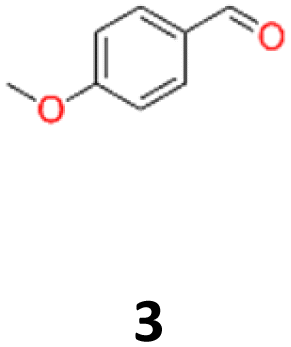
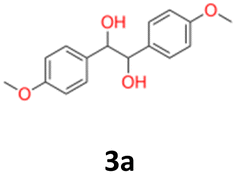
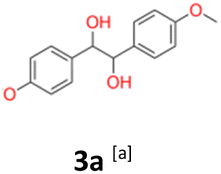
3a: white powder, yield: (A1: 61.0 mg, 4%, A2: 19 mg, 1%, B2: 129.0 mg, 19%); mp: 160.5 °C (dec.); Rf: 0.1 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: dl: 7.03, d (J = 8.7 Hz), 2H; 6.76, d (J = 8.7 Hz), 2H; 4.62, s, 1H; 3.76, s, 3H; meso: 7.20, d (J = 8.7 Hz), 2H; 6.85, d (J = 8.7 Hz), 2H; 4.73, s, 1H; 3.80, s, 3H; 13C NMR (125 MHz, CDCl3), δ ppm: dl: 159.2, 132.1, 128.2, 113.5, 78.8, 55.2, meso: 159.4, 132.0, 128.3, 113.7, 77.8, 55.3; TOF MS ES+ [M + Na]+ calculated for C16H18O4Na: 297.1103, found: 297.1104.
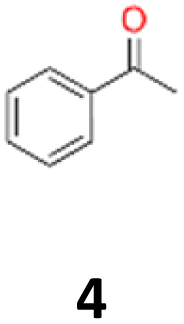
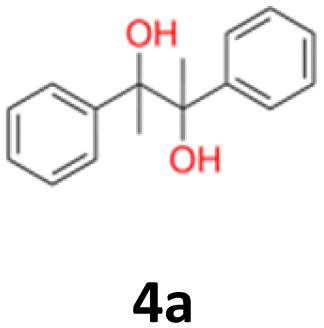
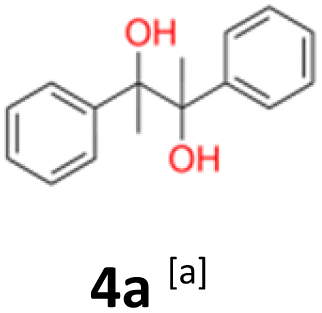
4a: yellow powder, yield: (A1: 87.2 mg, 18%, A2: 21.0 mg, 2%, B2: 259.0 mg, 22%); mp: 109.9 °C (dec.); Rf: 0.39 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: dl: 7.22–7.29, m, 3H; 7.18–7.22, m, 2H; 1.51, s, 3H, meso: 7.22–7.29, m, 5H; 1.59, s, 3H; 13C NMR (125 MHz, CDCl3), δ ppm: dl: 143.4, 127.3, 127.1, 127.0, 78.8, 25.0, meso: 143.8, 127.3, 126.9, 126.9, 78.6, 25.1; TOF MS ES-[M − H]− calculated for C16H17O2: 241.1229, found: 241.1228.
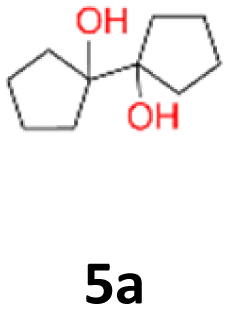
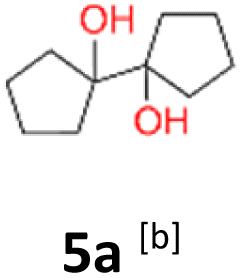
5a: yellow powder, yield: (A1: 192.0 mg, 23%, A2: 11.0 mg, 1%); mp: 110.8 °C (dec.); Rf: 0.125 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: 1.96, bs, 1H; 1.78–1.89, m, 2H; 1.68–1.78, m, 2H; 1.55–1.67, m, 4H; 13C NMR (125 MHz, CDCl3), δ ppm: 87.1, 36.4, 24.8; TOF MS ES+ [M + Na]+ calculated for C10H18O2Na: 193.1204, found: 193.1204; elem. anal.: C: 69.50:69.53%, H: 10.46:10.45%.
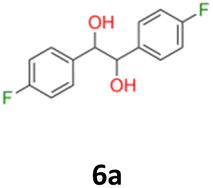
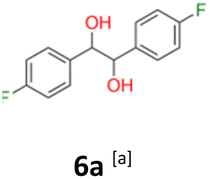
6a: white powder, yield: (A1: 410 mg, 21%, A2: 250 mg, 13%, B2: 467.9 mg, 23%); mp: 115.5 °C (dec.); Rf: 0.1 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: dl: 7.01–7.10, m, 2H; 6.87–6.95, m, 2H; 4.62, s, 1H; meso: 7.12–7.18, m, 2H; 6.94–7.01, m, 2H; 4.82, s, 1H; 13C NMR (125 MHz, CDCl3), δ ppm: dl: 162.4, d (J = 246.3 Hz); 135.3, d (J = 3.0 Hz); 128.6, d (J = 8.1 Hz); 115.1, d (J = 21.3 Hz); 78.7; meso: 162.5, d (J = 246.4 Hz); 135.2, d (J = 3.0 Hz); 128.7, d (J = 8.1 Hz); 115.1, d (J = 21.3 Hz); 77.3; TOF MS ES-[M − H]− calculated for C14H12F2O2: 249.0727, found: 249.0727.
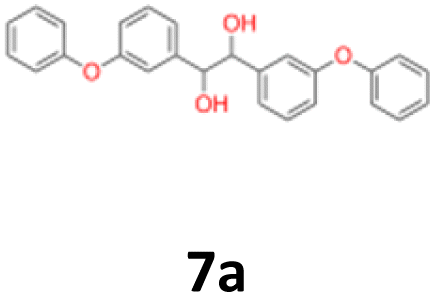
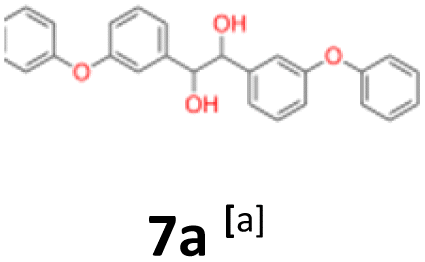
7a: yellow oil, yield: (A1: 227 mg, 29%, A2: 178 mg, 21%, B2: 224 mg, 22%); Rf: 0.13 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: dl: 7.27–7.34, m, 2H; 7.17–7.22, m, 2H; 7.07–7.13, m, 1H; 6.84–6.94, m, 4H; 6.70–6.74, m, 1H; 4.53, s, 1H; meso: 7.27–7.34, m, 2H; 7.19–7.24, m, 2H; 7.07–7.13, m, 1H; 6.84–6.94, m, 4H; 6.80–6.82, m, 1H; 4.79, s, 1H; 13C NMR (125 MHz, CDCl3), δ ppm: dl: 157.1, 156.8, 141.9, 129.8, 129.6, 123.2, 122.0, 118.7, 118.6, 117.9, 79.0, meso: 157.1, 156.8, 141.5, 129.8, 129.4, 123.2, 122.0, 118.7, 118.4, 117.7, 77.3; TOF MS ES-[M − H]− calculated for C26H21O4: 397.1440, found: 397.1446.
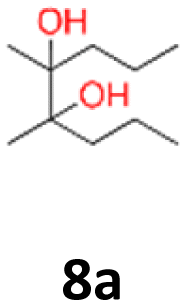
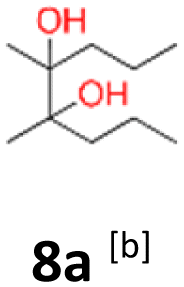
8a: brown solid, yield: (A1: 100 mg, 8%, A2: 8 mg, 1%); mp: 67.8 °C (dec.); Rf: 0.23 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: both dl and meso: 2.05, bs, 1H; 1.31–1.58, m, 4H; 1.13, s, 3H; 0.93, t (J = 7.8 Hz), 3H; 13C NMR (125 MHz, CDCl3), δ ppm: both dl and meso: 77.1, 17.0, 14.8, specific for dl or meso: 38.6, 20.6 (*), specific for meso or dl: 38.2, 21.0 (*); (*) signals cannot be assigned to specific structures; TOF MS ES+ [M + Na]+ calculated for C10H22O2Na: 197.1517, found: 197.1516.
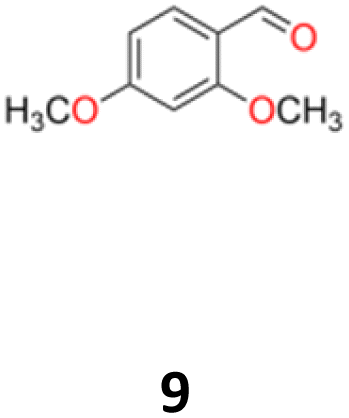
9a: brown oil, yield: (A1: 67 mg, 6%); Rf: 0.28 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: 9.79, d (J = 1.0 Hz), 1H; 6.90, d (J = 8.4 Hz), 2H; 6.52, d (J = 2.4 Hz), 2H; 6.45, dd (J = 8.4 Hz, J = 2.4 Hz), 2H; 5.16, s, 1H; 3.80, s, 3H; 3.79, s, 3H; 13C NMR (125 MHz, CDCl3), δ ppm: 200.9, 160.3, 158.2, 130.9, 118.1, 104.4, 98.8, 55.5, 55.4, 52.6; TOF MS ES+ [M + H]+ calculated for C18H20O5; 316.3700 found: 317.1392.
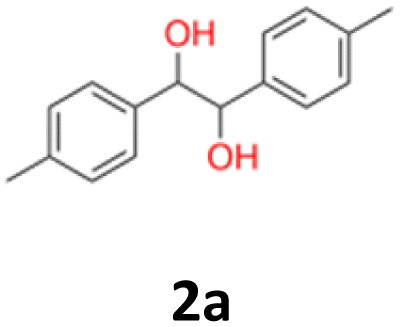
10a: white powder, yield: (A1: 98 mg, 10%, A2: 120 mg, 12%, B2: 33 mg, 4%, C1:117 mg, 51%, C2: 49 mg, 21%); mp: 116.0 °C (dec); Rf: 0.1 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: dl: 7.20–7.27, m, 3H; 7.09–7.15, m, 2H; 4.69, s, 1H; meso: 7.27–7.34, m, 3H; 7.22–7.27, m, 2H; 4.82, s, 1H; 13C NMR (125 MHz, CDCl3), δ ppm: dl: 139.8, 128.1, 127.9, 126.9, 79.1, meso: 139.7, 128.2, 128.1, 127.1, 78.1; TOF MS ES+ [M + Na]+ calculated for C14H14O2Na; 237.0891 found: 237.0886.
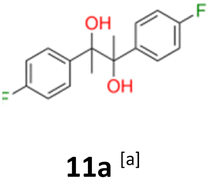
11a: brown solid, yield: (A1: 328 mg, 33%, A2: 79 mg, 8%); mp: 125.8 °C (dec.); Rf: 0.28 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: dl: 7.09–7.15, m, 2H; 6.89–6.95, m, 2H; 1.49, s, 3H; meso: 7.15–7.20, m, 2H; 6.88–6.94, m, 2H; 1.57, s, 3H; 13C NMR (125 MHz, CDCl3), δ ppm: dl: 162.0, d (J = 245.7 Hz); 139.0, d (J = 3.4 Hz); 129.1, d (J = 8.1 Hz); 113.9, d (J = 21.3 Hz); 78.6; 24.9; meso: 161.9, d (J = 245.9 Hz); 139.4, d (J = 3.1 Hz); 128.7, d (J = 8.0 Hz); 114.0, d (J = 21.2 Hz); 78.3; 25.2; TOF MS ES-[M − H]− calculated for C16H15F2O2; 277.1040 found: 277.1044.
12a: yellow powder, yield: (A1: 242 mg, 24%, A2: 309 mg, 31%); mp: 149.6 °C (dec.); Rf: 0.09 (SiO2, hexane–AcOEt 5:1 v/v); 1H NMR (500 MHz, CDCl3), δ ppm: dl: 7.41, d (J = 8.2 Hz), 2H; 7.13, d (J = 8.2 Hz), 2H; 4.61, s, 1H; meso: 7.44, d (J = 8.1 Hz), 2H; 7.18, d (J = 8.1 Hz), 2H; 4.85, s, 1H; 13C NMR (125 MHz, CDCl3), δ ppm: dl: 144.0; 129.9, q (J = 32.6 Hz); 127.3; 124.9, q (J = 3.9 Hz); 124.0, d (J = 272.2 Hz); 78.1; meso: 143.9; 129.7, q (J = 32.5 Hz); 127.3; 124.6, q (J = 3.9 Hz); 124.1, d (J = 271.8 Hz); 76.8; TOF MS ES-[M − H]− calculated for C16H11F6O2; 349.0663 found: 349.0672.
Data availability
The authors declare that the data supporting this study are available within the paper and in ESI file.† Any other data needed are available from the corresponding author upon reasonable request.Author contributions
Conceptualization, O. M. and M. C.; methodology, O. M., S. Ż., M. K., P. K., A. L. and S. F.; software, A. L., and M. K.; formal analysis, O. M., A. L and S. F.; investigation, O. M., S. Ż., M. K., P. K. and A. L.; resources, O. M., A. L. and S. F.; data curation, O. M., M. K. and A. L.; writing – original draft preparation, O. M., M. C., S. Ż and A. L.; writing – review and editing, M. C., O. M., S. F. and A. L.; visualization, O. M., M. C., P. K. and A. L.; supervision, O. M., S. F. and A. L.; project administration, O. M., A. L. and S. F.; funding acquisition, O. M., A. L. and S. F. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
Authors declare no conflict of interests.Acknowledgements
This research was funded by the Polish Ministry of Science and Higher Education under the framework of the Łukasiewicz–Industrial Chemistry Institute statutory project (grant no. 84134305). This research was also co-funded by the National Science Centre, Poland, grant OPUS-23 2022/45/B/NZ7/04246. The Interdisciplinary Centre of Mathematical and Computational Modeling (ICM) of Warsaw University is acknowledged for computer time and facilities within the G18-6 computer grant. We would like to thank Jamie Wojtasinski for consulting on the linguistic correctness of this publication.References
- D. A. Burnett, Curr. Med. Chem., 2004, 11(14), 1873 CrossRef CAS PubMed.
- R. E. Buckles and J. E. Maure, J. Org. Chem., 1953, 18(11), 1585 CrossRef.
- M. R. Naimi-Jamal, J. Mokhtari, M. G. Dekamin and G. Kaupp, Eur. J. Org Chem., 2009, 21, 3567 CrossRef.
- J. González-Rodríguez, J. Albarrán-Velo, R. L. G. Soengas, I. Lavandera, V. Gotor-Fernández and H. Rodríguez-Solla, Org. Lett., 2022, 24(39), 7082 CrossRef PubMed.
- L. C. Rocha, H. V. Ferreira, E. F. Pimenta, R. G. S. Berlinck, M. O. Oliveira Rezende, M. D. Landgraf and A. L. Meleiro Porto, Mar. Biotechnol., 2010, 12, 552 CrossRef CAS PubMed.
- W. Borzęcka, I. Lavandera and V. Gotor, J. Org. Chem., 2013, 78(14), 7312 CrossRef PubMed.
- H. J. Naeimi, J. Chin. Chem. Soc., 2008, 55(950), 1156 CrossRef CAS.
- L. Lelo, M. Miele, V. Pillari, R. Senatore, S. Mirabile, R. Gitto, W. Holzer, A. R. Alcántara and V. Pace, Org. Biomol. Chem., 2021, 19(9), 2038 RSC.
- W.-M. Ren, Y. Liu and X.-B. Lu, J. Org. Chem., 2014, 79(20), 9771 CrossRef CAS PubMed.
- S. S. Bhosale, P. L. Joshi and A. S. Rao, Org. Prep. Proced. Int., 1992, 24(6), 695 CrossRef CAS.
- L. Ai, W. Wang, J. Wei, Q. Li, S. Song and N. Jiao, Synlett, 2019, 30, 437 CrossRef CAS.
- S. González-Granda, L. Escot, I. Lavandera and V. Gotor-Fernández, ACS Catal., 2022, 12(4), 2552 CrossRef.
- R. Tarhouni, B. Kirschleger, M. Rambaud and J. Villieras, Tetrahedron Lett., 1984, 25(8), 835 CrossRef CAS.
- S. Monticelli, M. Rui, L. Castoldi, G. Missere and V. Pace, Monatsh. Chem., 2018, 149, 1285 CrossRef CAS PubMed.
- K. Okamoto, K. Muta, H. Yamada, R. Higuma, Y. Ashikari and A. Nagaki, React. Chem. Eng., 2024, 9, 1173 RSC.
- C. R. Emerson, L. N. Zakharov and P. R. Blakemore, Chem.–Eur. J., 2013, 19, 16342 CrossRef CAS PubMed.
- C. Kupper, S. Molitor and V. H. Gessner, Organometallics, 2014, 33(1), 347 CrossRef CAS.
- A. Wieczorek and F. Hammerschmidt, J. Org. Chem., 2012, 77(22), 10021 CrossRef CAS PubMed.
- D. C. Kapeller and F. Hammerschmidt, J. Am. Chem. Soc., 2008, 130(7), 2329 CrossRef CAS PubMed.
- V. Capriati and S. Florio, Chem.–Eur. J., 2010, 16, 4152 CrossRef CAS PubMed.
- R. H. V. Nishimura, F. T. Toledo, J. L. C. Lopes and G. C. Clososki, Tetrahedron Lett., 2013, 54(4), 287 CrossRef CAS.
- R. Senatore, M. Malik and V. Pace, Adv. Synth. Catal., 2022, 364, 2890 CrossRef CAS.
- T. Imamoto, T. Takeyama and H. Koto, Tetrahedron Lett., 1986, 27(28), 3243 CrossRef CAS.
- J. M. Concellón, P. L. Bernad and J. A. Pérez-Andrés, J. Org. Chem., 1997, 62(25), 8902 CrossRef.
- T. Tabuchi, J. Inanaga and M. Yamaguchi, Tetrahedron Lett., 1986, 27(33), 3891 CrossRef CAS.
- L. Degennaro, F. Fanelli, A. Giovine and R. Luisi, Adv. Synth. Catal., 2015, 357, 21 CrossRef CAS.
- M. Spennacchio, P. Natho, M. Andresini and M. Collela, J. Flow Chem., 2024, 14, 43 CrossRef CAS.
- M. Spennacchio, M. Colella, M. Andresini, R. S. Dibenedetto, E. Graziano, A. Aramini, L. Degennaro and R. Luisi, Chem. Commun., 2023, 59(10), 1373 RSC.
- R. H. V. Nishimura, V. E. Murie, R. A. Soldi, J. L. C. Lopes and G. C. Clososki, J. Braz. Chem. Soc., 2015, 26(11), 2175 CAS.
- P. L. Beaulieu, D. Wernic, J.-S. Duceppe and Y. Guindon, Tetrahedron Lett., 1995, 36(19), 3317 CrossRef CAS.
- M. Hutchings, D. Moffat and N. S. Simpkins, Synlett, 2001, 2001(5), 0661 CrossRef.
- T. H. Yan, B. Ananthan and S. H. Chang, Eur. J. Org Chem., 2019, 4, 778 CrossRef.
- T.-H. Yan, C.-C. Tsai, C.-T. Chien, C.-C. Cho and P.-C. Huang, Org. Lett., 2004, 6(26), 4961 CrossRef CAS PubMed.
- A. Clerici and O. Porta, Tetrahedron Lett., 1982, 23(34), 3517 CrossRef CAS.
- J.-T. Li, Z.-P. Lin and T.-S. Li, Sonochemistry, 2005, 12(5), 349 CrossRef CAS PubMed.
- A. Clerici, L. Clerici and O. Porta, Tetrahedron Lett., 1996, 17(22), 3035 CrossRef.
- A. Bensari, J.-L. Renaud and O. Riant, Org. Lett., 2001, 3(24), 3863 CrossRef CAS PubMed.
- S. Barroso, P. Adão, A. M. Coelho, J. Costa Pessoa and A. M. Martins, J. Mol. Catal. A: Chem., 2016, 412, 107 CrossRef CAS.
- S. M. Rele, S. K. Nayak and S. Chattopadhyay, Tetrahedron, 2008, 64(30–31), 7225 CrossRef CAS.
- J. T. Li, Y. X. Chen and T. S. Li, J. Chem. Res., 2005, 2005(6), 361 CrossRef.
- (a) K. Wang, S.-X. Wang, M.-Z. Gao and J.-T. Li, Synth. Commun., 2006, 36(10), 1391 CrossRef CAS; (b) A. Chatterjee and N. N. Joshi, Tetrahedron, 2006, 62(52), 12137 CrossRef CAS.
- E. Bergmann, J. Am. Chem. Soc., 1932, 54(9), 3773 CrossRef CAS.
- T.-Y. Li, L.-P. Wang, T. Zhang, Y.-X. Li and Z.-M. Wang, Chem. J. Chin. Univ., 2000, 21(9), 13911394 Search PubMed.
- P. Rashidi-Ranjbar and E. Kianmehr, Molecules, 2001, 6(5), 442 CrossRef CAS.
- H. Shi, C. Du, X. Zhang, F. Xie, X. Wang, S. Cui, X. Peng, M. Cheng, B. Lin and Y. Liu, J. Org. Chem., 2018, 83(3), 1312 CrossRef CAS PubMed.
- K. Nakamura and Y. Osamura, J. Am. Chem. Soc., 1993, 115(20), 9112 CrossRef CAS.
- Y. Mitoma, I. Hashimoto, C. Simion, M. Tashiro and N. Egashira, Synth. Commun., 2008, 38(19), 3243 CrossRef CAS.
- H. C. Aspinall, N. Greeves and C. Valla, Org. Lett., 2005, 7(10), 1919 CrossRef CAS PubMed.
- K. Makino, K. Tsuda and M. Takai, Polym. Bull., 1991, 26, 371 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: S1–S10: theoretical DFT calculations; S11: NMR spectra; S12: HRMS spectra. See DOI: https://doi.org/10.1039/d4ra07250b |
| This journal is © The Royal Society of Chemistry 2024 |