DOI:
10.1039/D4RA07259F
(Paper)
RSC Adv., 2024,
14, 36602-36609
Selectively electrolyzing CO2 to ethylene by a Cu–Cu2O/rGO catalyst derived from copper hydroxide nanostrands/graphene oxide nanosheets†
Received
9th October 2024
, Accepted 30th October 2024
First published on 15th November 2024
Abstract
Electrolyzing CO2 into ethylene (C2H4) is a promising strategy for CO2 utilization and carbon neutrality since C2H4 is an important industrial feedstock. However, selectively converting CO2 into C2H4 via the CO2 electro-reduction reaction (CO2 ERR) is still a great challenge. Herein, Cu–Cu2O nanoparticles anchored on reduced graphene oxide nanosheets (Cu–Cu2O/rGO) were prepared from copper hydroxide nanostrands (CHNs) and graphene oxide (GO) nanosheets via in situ electrochemical reduction. Cu–Cu2O nanoparticles with diameter less than 10 nm were formed on the surface of rGO nanosheets. After assembling the Cu–Cu2O/rGO catalyst into a flow cell, it demonstrated high Faraday efficiencies (FEs) of 55.4%, 37.6%, and 6.7% for C2H4, C2H6, and H2, respectively, and a total 93% FE for C2 at −1.3 V vs. the standard hydrogen electrode (SHE). Moreover, its FE was 68.2% for C2H4, 10.2% for C2H6, and 20.5% for H2 at −1.4 (vs. SHE). Besides, no liquid carbon product was detected. This high selectivity is attributed to the synergistic effect arising from the small diameter of Cu–Cu2O NPs with the combination of Cu0–Cu+ and rGO nanosheets, which promotes the activation of CO2 molecules, facilitates C–C coupling, and enhances stability. This may provide a facile way for designing an efficient catalyst for selectively electrolyzing CO2 into valuable C2 chemicals.
Introduction
CO2 electro-reduction into valuable chemicals is a desirable way for carbon neutralization.1 Tremendous attention has been paid to design novel catalysts to produce ethylene (C2H4), an important chemical feedstock, from CO2 via the CO2 electro-reduction reaction (ERR).2–4 Among them, the most promising are Cu-based catalysts owing to their unique electronic structures, which exhibit high CO2 activation ability and moderate CO adsorption affinity and facilitate the formation of C2H4.5–7 It has been found that copper crystal facets,8 copper oxidation states,9 crystal size,10 interaction between nanocrystals,11 grain boundaries,12 coordination environments,13 electrolytes14 and so forth could significantly affect the performance for electrolyzing CO2 to C2H4. Some researchers have confirmed that the Cu+ state in the Cu/Cu2O catalyst plays a critical role in the selective formation of C2H4.3,5,9–12 For example, Zhang et al. prepared Cu/Cu2O convex spheres with an adjustable Cu0–Cu+ interface to improve C2 production by modulating hydrothermal synthesis time.11 Gong's group also found that the synergism of Cu0–Cu+ pairs could enhance CO2 ERR activity. Cu0 activates CO2 and facilitates electron transfer, while Cu+ forces *CO adsorption and boosts C–C coupling.12 Most Cu0–Cu+ catalysts were prepared via the hydrothermal process from copper salts with a reducing agent.11,12 The hydrothermal process may be energy intensive like a black box and not easily controllable. In addition, the preservation of the oxidation of Cu+ in the Cu0–Cu+ catalyst during the CO2 EER process is still a challenge.13–17 Recently, it has been proposed that carbon-based materials,13–17 including C60,14,15 graphene-related materials16 and carbon nanotubes (CNTs),17 could enhance the stability of the Cu+ state of Cu–Cu+ during the CO2 EER process. Meanwhile, it is well known that the smaller the catalyst size, the higher the activity.2–5
In this work, we prepared Cu0–Cu+ (Cu–Cu2O) nanoparticles with a diameter less than 10 nm anchored on reduced graphene oxide (rGO) nanosheets, named as Cu–Cu2O/rGO, via in situ electrochemical reduction from a composite composed of 2 nm copper hydroxide nanostrands/graphene oxide nanosheets (CHNs/GO). It has been reported that the CHNs are highly positively charged.18 In addition, it is well known that GO nanosheets are negatively charged.19 Therefore, due to the strong electrostatic interaction, the CHNs and GO were well assembled together to form the CHNs/GO composite, similar to those reported.19 After drop-casting CHNs/GO with Nafion on a gas diffusion electrode, it was in situ electrochemical reduced to Cu–Cu2O/rGO in a flow cell. It demonstrated high selectivity and stable performance for CO2 ERR. The Faraday efficiency (FE) of C2H4 was 55.4% and 68.2% at −1.3 V and −1.4 V vs. standard hydrogen electrode (SHE), respectively, while it is 37.6% and 10.2% for C2H6. No liquid product was detected. This holds potential for the synthesis of efficient Cu-based catalysts for the electrolysis of CO2 to C2H4.
Experimental
Chemicals and materials
Copper nitrate (Cu(NO3)2·3H2O), 2-aminoethanol (NH2–CH2CH2OH, AE), and potassium bicarbonate (KHCO3) were purchased from ACROS Chemicals. Ethanol and isopropanol were purchased from Sinopharm Chemical Reagent Co. Ltd. Graphene oxide nanosheets dispersion with diameter of about 1 micrometer was purchased from GaoxiTech Co. Ltd. Nafion was purchased from Sigma-Aldrich. Ultrapure water with 18.2 MΩ cm, resistivity produced by a Millipore direct-Q system (Millipore), was used throughout the experiments. All materials used in this work were used without further purification.
Synthesis of CHNs
The CHNs were prepared by mixing equal volume of 4 mM Cu(NO3)2 and 1.6 mM AE aqueous solution under stirring for 5 min, then aged for 2 days at room temperature, as described elsewhere.18
Synthesis of the CHNs/GO composite
The CHNs/GO composite was typically synthesized by mixing 2 mL 2 mg mL−1 GO aqueous solution (zeta potential − 46 mV) with a certain volume of CHNs solution under stirring for 10 min following the process reported elsewhere.19 Then, the gel-like precipitate was collected by centrifugation at 8000 rpm and dried at room temperature overnight. CHNs/GO with various volume ratios of CHNs to GO were prepared, as listed in Table S1.†
Synthesis of the Cu–Cu2O/rGO composite
The dried CHNs/GO 10 mg powders, 0.02 mL Nafion solution, 0.96 mL isopropanol, and 1 mL water were mixed together and ground for 30 min. Then, this mixture was cast on the gas diffusion electrode (or porous carbon paper) with a loading amount of 2 mg cm−2 and dried under infrared light (0.1 W cm−2) for 3 hours. After drying, the electrode loaded with CHNs/GO was electrochemically reduced to Cu–Cu2O/rGO in 0.5 M KHCO3 solution, with CO2 flow rate of 20 mL min−1, under −3 V vs. SHE for 1 hour in the flow cell. The counter electrode was a Pt foil. The reference electrode was the Ag/AgCl electrode.
Characterization
The crystalline data were collected on an X-ray powder diffractometer (SmartLab, 5°–70°, Cu Kα). Scanning electron microscopy (SEM, Ultra 55) with X-ray energy-dispersive spectroscopy (EDS) and transmission electron microscopy (TEM, HT7700) were used. The X-ray photoluminescence spectra (XPS) were recorded employing an XPS AXIS SUPRA (Kratos, UK). Liquid-phase products were detected by 1H nuclear magnetic resonance (NMR) spectroscopy (Bruker AVANCE AV III 500 MHz). 400 μL catholyte was added in 50 μL dimethyl sulfoxide (DMSO) (6 mM) solution as the internal standard and 50 μL D2O as the identification and quantification of liquid products. The spectra were measured with water suppression by excitation sculpting. Online gas chromatography (GC, FuLi 9790II) equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD) was employed to quantify the gas products.
CO2 ERR test
The CO2 ERR test was carried out in home-made flow cell assembled with an anion exchange membrane on an electrochemical workstation CHI 760E, with Pt foil counter electrode, Ag/AgCl reference electrode, and the gas diffusion electrode loaded with the catalyst working electrode. The electrolyte was 1 M KHCO3 aqueous solution (pH 8.4) for CO2 ERR, while it was 0.5 M KHCO3 aqueous solution (pH 8.2) for CHNs/GO reduction and activation. CO2 gas (99.99%) with 20 mL min−1 flow rate was filled. The effective area of the working electrode was 1 cm2.
Linear sweep voltammetry (LSV) was recorded by a flow cell with 20 mL min−1 flow rate of CO2 and 1 M KOH. The electrolyte (1 M KOH) was circulated at a flow rate of 10 mL min−1 by a pump. The potential was converted to the SHE without internal resistance correction.
In situ FTIR and Raman measurement
The in situ attenuated total reflection-surface enhanced infrared absorption spectroscopy (ATR-SEIRS) were recorded by an infrared spectrometer (Nicolet 6700) equipped with a liquid nitrogen cooled mercury–cadmium–telluride (MCT-A) detector. A homemade cell was assembled on top of the Si prism. First, an Au layer was chemically deposited on the Si prism. Then, the catalysts were loaded on the Au-coated Si prism and used as the working electrode. A Pt wire and Ag/AgCl served as the counter electrode and reference electrode, respectively. 0.1 M KOH was used as the electrolyte. After running for 100 s, the FTIR spectra were collected with a resolution of 8 cm−1 in the range from 1000 to 4000 cm−1 under a certain potential. In order to monitor the CHNs of the CNHs/GO conversion in the Cu–Cu2O process, in situ Raman spectra were recorded by a LabRAM Odyssey™ (HORIBA France SAS) using the same homemade cell with 0.5 M KHCO3 solution and CO2 flow rate of 20 mL min−1 at −3 V vs. SHE.
Results and discussion
Fabrication and characterization of Cu–Cu2O/rGO
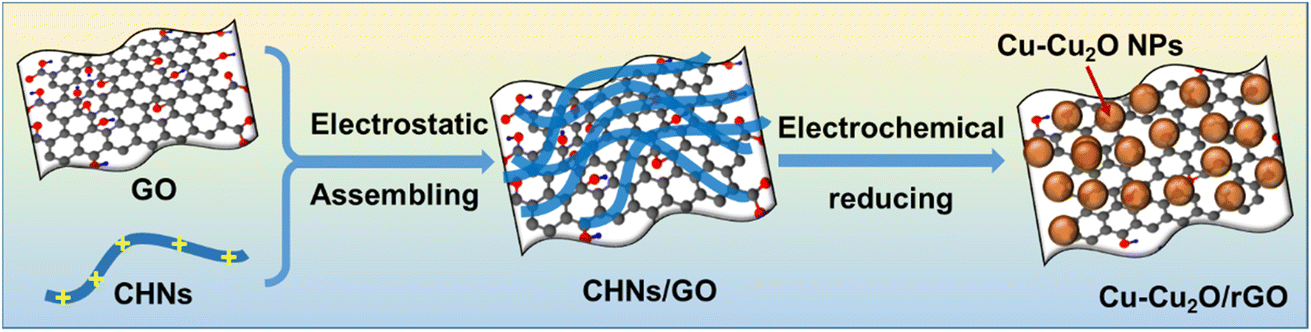
The typical synthesis process of Cu–Cu2O/rGO is illustrated in Scheme 1. The highly positively charged CHNs were prepared by adding AE solution into Cu(NO3)2 under stirring following the method described by Prof. Ichinose.18 They confirmed that half of the surface copper atoms of the CHNs are positively charged by adsorbing negatively charged dyes. The SEM image (Fig. S1a†) and TEM image (Fig. 1d) indicate that the linear structure of CHNs with diameter of about 2 nm is successfully formed, as reported elsewhere.18 The CHNs/GO composites were formed by mixing CHNs with GO solution via electrostatic interaction as described by Prof. Peng for molecular separation membrane.19 The microstructures of the CHNs/GO prepared from 125 mL CHNs and 1 mL GO solution (2 mg mL−1) are presented in Fig. 1a and e. Due to the highly charged surface of CHNs18 and negatively charged surface of the GO nanosheet, it is obvious that the ultrafine fibrous CHNs and GO nanosheets are uniformly assembled together via electrostatic interaction. Similar morphology was observed for the CHNs/GO with a volume ratio of CHNs to GO of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 150:1 (Fig. S1b and c†), respectively. The CHNs/GO was further mixed with Nafion and drop casted on a gas diffusion electrode (or porous carbon paper). An electrochemical reduction was applied to convert CHNs/GO to Cu–Cu2O/rGO in 0.5 M KHCO3 electrolyte under −3 V vs. SHE for 1 hour. The samples prepared from various volume ratios of CHNs solution and GO solution (2 mg mL−1) were named as S1 to S11 (see details in Table S1†). The SEM images of the S10 Cu–Cu2O/rGO catalyst (prepared from 125 CHNs and 1 mL 2 mg mL−1 GO) are shown in Fig. 1b (high magnification), Fig. 1c and S2† (large scale view), respectively. After electrochemical reduction, the fibrous CHNs disappeared, while nanoparticles with uniform size of less 10 nm were formed and anchored on the surface of the wrinkled rGO sheets. The wrinkled surface with grooves is rough and resulted in a high active surface area for the electrochemical reaction. The TEM image (Fig. 1f) of the Cu–Cu2O/rGO (S10) further confirms that the size of the nanoparticles is several nanometers. The corresponding SEM EDS element mapping images (Fig. 2) indicate Cu, C, and O are uniformly distributed in the sample. It also shows the overlapping part of Cu and O (Fig. 2b and c). All of these mean that the particles are mostly Cu and Cu–O species.
:1 and 150:1 (Fig. S1b and c†), respectively. The CHNs/GO was further mixed with Nafion and drop casted on a gas diffusion electrode (or porous carbon paper). An electrochemical reduction was applied to convert CHNs/GO to Cu–Cu2O/rGO in 0.5 M KHCO3 electrolyte under −3 V vs. SHE for 1 hour. The samples prepared from various volume ratios of CHNs solution and GO solution (2 mg mL−1) were named as S1 to S11 (see details in Table S1†). The SEM images of the S10 Cu–Cu2O/rGO catalyst (prepared from 125 CHNs and 1 mL 2 mg mL−1 GO) are shown in Fig. 1b (high magnification), Fig. 1c and S2† (large scale view), respectively. After electrochemical reduction, the fibrous CHNs disappeared, while nanoparticles with uniform size of less 10 nm were formed and anchored on the surface of the wrinkled rGO sheets. The wrinkled surface with grooves is rough and resulted in a high active surface area for the electrochemical reaction. The TEM image (Fig. 1f) of the Cu–Cu2O/rGO (S10) further confirms that the size of the nanoparticles is several nanometers. The corresponding SEM EDS element mapping images (Fig. 2) indicate Cu, C, and O are uniformly distributed in the sample. It also shows the overlapping part of Cu and O (Fig. 2b and c). All of these mean that the particles are mostly Cu and Cu–O species.
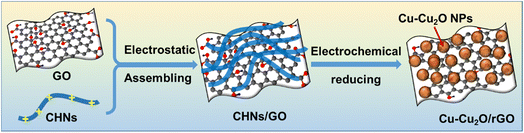 |
| | Scheme 1 Illustration of the synthesis process of Cu–Cu2O/rGO from CHNs/GO via in situ electrochemical deposition. | |
 |
| | Fig. 1 (a) SEM image of CHNs/GO prepared from 125 mL CHNs and 1 mL 2 mg mL−1 GO. (b and c) SEM images of the sample S10 Cu–Cu2O/rGO after in situ electrochemical reduction. (d) The TEM image of CHNs. (e) TEM image of CHNs/GO. (f) TEM image of Cu–Cu2O/rGO. | |
 |
| | Fig. 2 (a) SEM image of S10 Cu–Cu2O/rGO with EDS mapping. The corresponding EDS mapping images; (b) overall elements combined together; (c) enlarged section of (b); (d) Cu, (e) oxygen and (f) carbon. | |
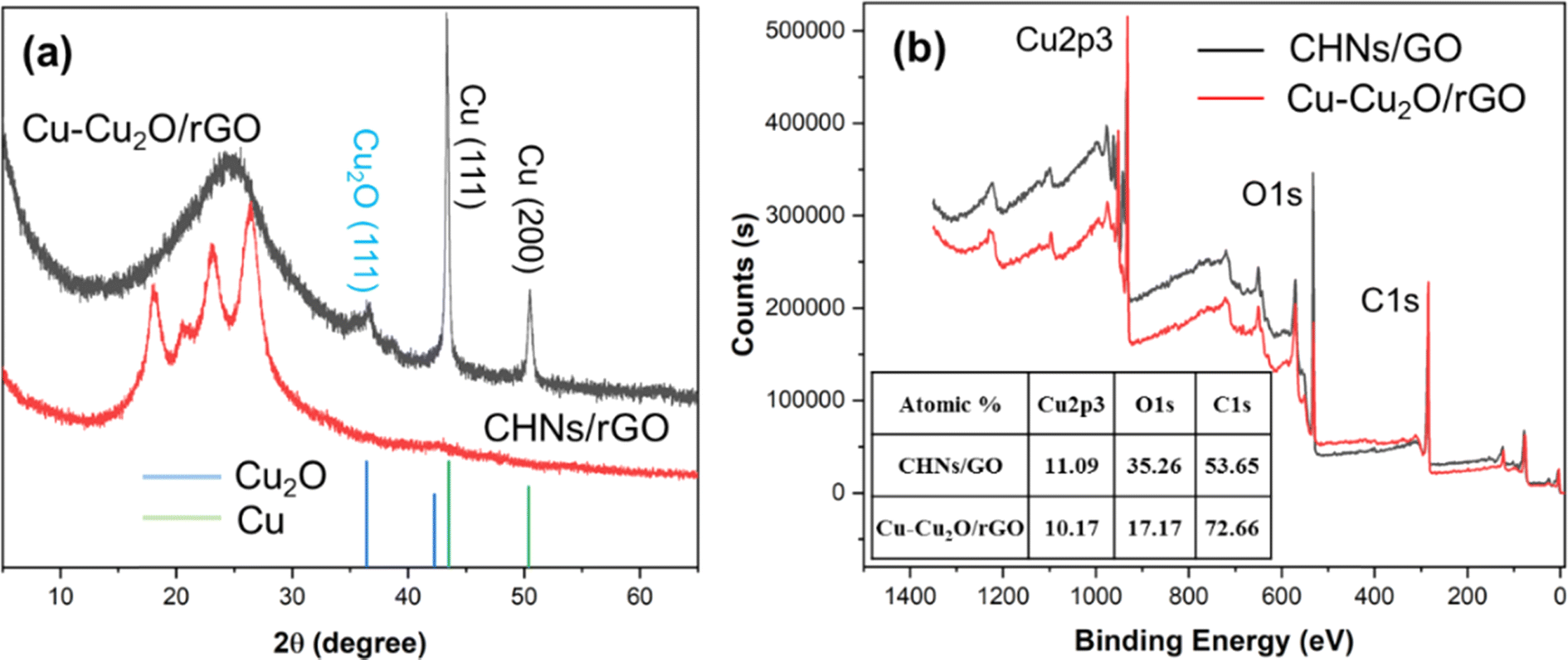
The XRD patterns of the S10 Cu–Cu2O/rGO sample before and after electrochemical reduction are shown in Fig. 3a. Two strong peaks at 43.3° and 50.4° are indexed to the (111) and (200) planes of Cu, respectively (JCPDS 04-0836).7,11 The small peak located at 36.4° is assigned to the (111) plane of Cu2O (JCPDS 005-0667).7,11 No peaks for crystalline CuO were observed. The broad peak at 25° is indexed to rGO.20 These results indicate that the major products of the electrochemical reduction of CHNs are Cu with minor Cu2O. Similar results for S9 and S11 were achieved, as shown in Fig. S3.† In order to monitor the CHNs electrochemical reduction process, the in situ Raman spectra (Fig. S4†) in the range from 200 to 700 cm−1 were recorded during the electrochemical reduction of CHNs/GO. The characteristic peaks at about 292 and 470 cm−1 assigned to Cu(OH)2 (ref. 21 and 22) continuously decline and finally disappear after 1 hour, while the peak at about 525 cm−1 indexed to Cu2O21,22 is clearly observed after 15 min and up to 1 hour. These results indicate the Cu2O phase is really formed during the electrochemical reduction. The formation of Cu–Cu2O is further supported by the XPS results (Fig. 3d and 4) recorded for S10 before and after electrochemical reduction. Fig. 3d is the corresponding survey scan XPS spectra. It clearly demonstrates the Cu 2p, O 1s and C 1s peaks.7,9,11 The atomic percent of these elements are 11.09 at% for Cu, 35.26 at% for O, and 53.65 at% for C in CHNs/GO. After electrochemical reduction, Cu is almost the same while oxygen significantly deceases to 17.17 at% and C dramatically increases to 72.66 at% (inset table in Fig. 3b). These mean that the reduction of CHNs and GO occurs. Fig. 4a, b and d–f are the corresponding high resolution Cu 2p, O 1s and C 1s XPS spectra of CHNs/GO and Cu–Cu2O/rGO, respectively.
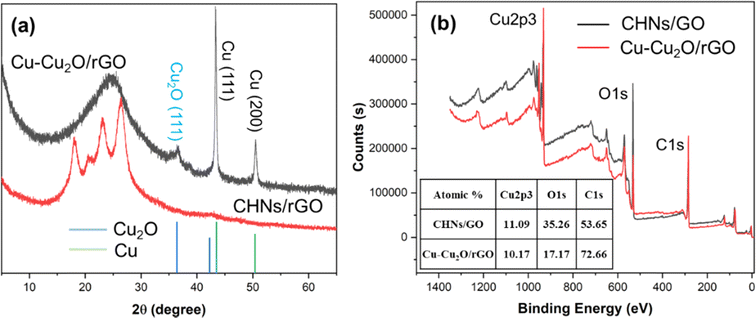 |
| | Fig. 3 (a) The XRD patterns and (b) survey scan XPS spectra of S10 before (CHNs/GO) and after electro-reduction (Cu–Cu2O/rGO). The inset table in (b) lists the atomic ratio of Cu, O, and C of S10 before and after electro-reduction, calculated from the XPS results. | |
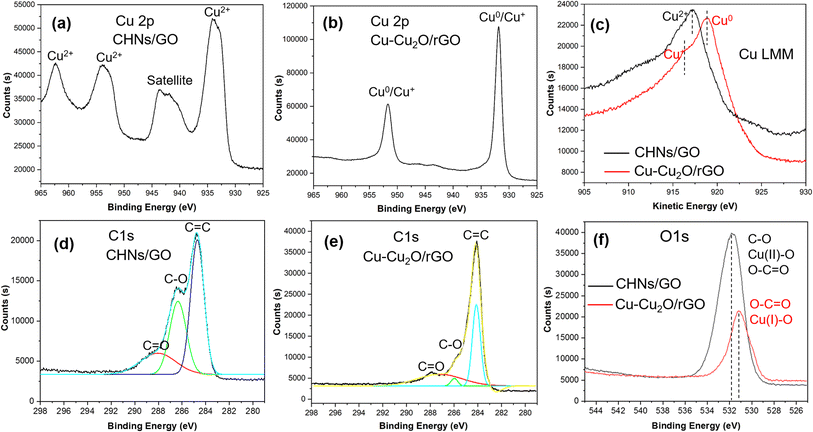 |
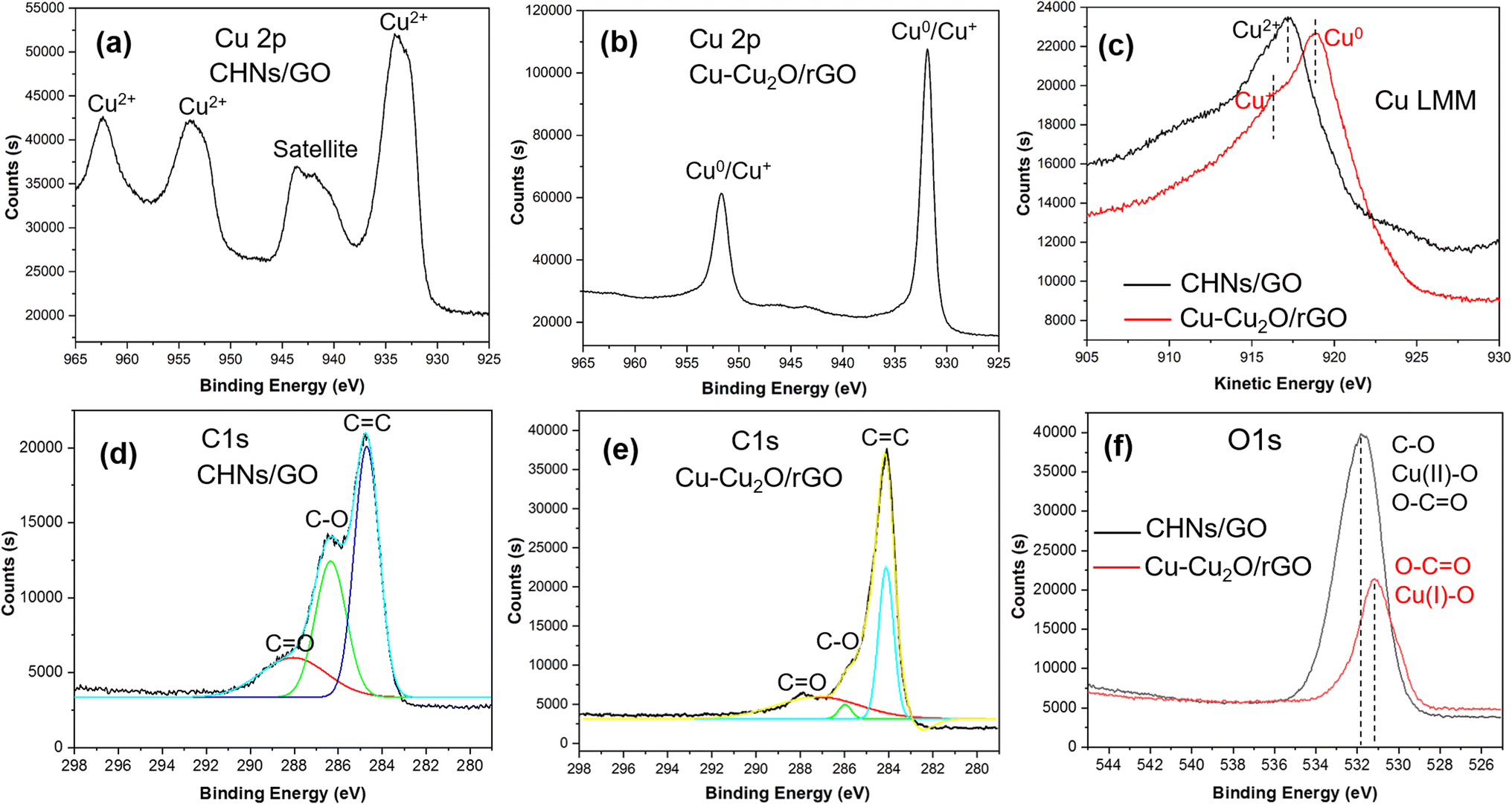
| | Fig. 4 High resolution XPS spectra of (a and b) Cu 2p, (c) Cu LMM, (d and e) C 1s; (f) O 1s of S10 before (CHNS/GO) and after (Cu–Cu2O/rGO) electro-reduction. | |
It is clearly seen that before electrochemical reduction, the Cu valent state is Cu2+ with typical XPS spectrum having two obvious satellite peaks at 942 and 945 eV (Fig. 4a) in CHNs/GO, while, it is Cu0 and Cu+ with overlapped two strong peaks and small satellite peaks after reduction in Cu–Cu2O/rGO (Fig. 4b).9,11,23–30 To more clearly distinguish the valent sate of Cu0 and Cu+, the Cu LMM spectra (Fig. 4c) were recorded. The typical XPS LMM Cu2+ peak of CHNs/GO is observed, while a strong Cu0 peak with a weak shoulder Cu+ peak in the XPS LMM spectrum of Cu–Cu2O/rGO is seen.9,11,24,27 Comparing the C 1s spectra of CHNs/GO (Fig. 4d) with that of Cu–Cu2O/rGO (Fig. 4e), the C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peaks significantly decrease. Similar decrease trends of C–O and Cu(II)O peaks are also observed in the O 1s spectra (Fig. 4f).31,32 The intensity of O 1s from the Cu2+O and C–O peaks dramatically declines.9,11,32,33 In addition, the total intensity of O 1s is significantly decreased after reduction. These mean that both CHNs and GO are really reduced. Based on the weak Cu2O peak in the XRD pattern (Fig. 3a) and small shoulder peak of Cu+ in the Cu LMM spectrum (Fig. 4f), the atomic ratio of Cu+ to Cu in the Cu–Cu2O/rGO (S10) calculated by Gauss-fitting is about 10%.
O peaks significantly decrease. Similar decrease trends of C–O and Cu(II)O peaks are also observed in the O 1s spectra (Fig. 4f).31,32 The intensity of O 1s from the Cu2+O and C–O peaks dramatically declines.9,11,32,33 In addition, the total intensity of O 1s is significantly decreased after reduction. These mean that both CHNs and GO are really reduced. Based on the weak Cu2O peak in the XRD pattern (Fig. 3a) and small shoulder peak of Cu+ in the Cu LMM spectrum (Fig. 4f), the atomic ratio of Cu+ to Cu in the Cu–Cu2O/rGO (S10) calculated by Gauss-fitting is about 10%.
CO2 ERR performance
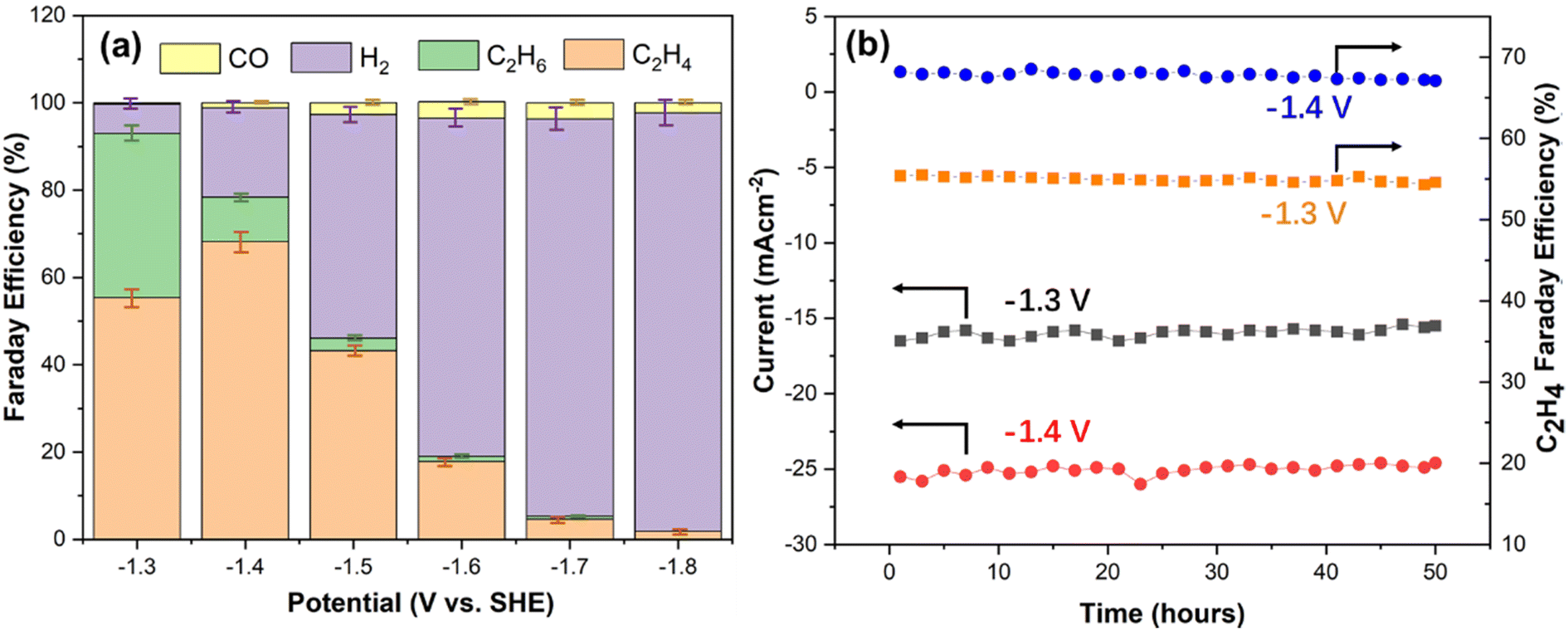
The aforementioned XRD, in situ Raman and XPS results confirmed the formation of the Cu–Cu2O/rGO composite. It has been reported that the Cu0–Cu+ combination is desirable for selectively converting CO2 to C2H4 by the CO2 ERR process.3,5,7,9,11,12,14,17 It has also been confirmed that carbon-based supports for copper-based catalyst could enhance their stability during the CO2 ERR process.7,13–17 Therefore, the prepared Cu–Cu2O/rGO samples were examined for CO2 ERR. Linear sweep voltammetry (LSV) investigation was carried out using a flow cell under CO2 flow in 0.5 M KHCO3 electrolyte to study the activity of the resulting samples. A typical activation curve of S10 is shown in Fig. S5a.† It is clearly seen that the catalyst shows onset activity at −1.1 V vs. SHE. The chronoamperometry curves of S10 at different potentials is shown in Fig. S5b.† The current increases from −1.3 to −1.8 V vs. SHE. It is critical to suppress H2 evolution during the CO2 ERR.30–34 In order to find the appropriate sample with less H2 evolution, the FE of H2 by the catalysts prepared from different volume ratios of CHNs to GO, from S1 to S11, under −1.3 V vs. SHE were calculated from the gas products during the operation (Fig. S6†). Fig. S6† indicates that the FE of H2 increases from 8.2% to about 12% with the volume ratio of CHNs to GO in the range from 5:1 up to 20:1. Then, it declines to about 6.7% for the volume ratio of CHNs to GO at 125:1 (S10) and slightly increases when the volume ratio is 150:1 (S11). The FE calculation follows the methods described elsewhere.2–17,21–30,33–37 It is clear that S10 is the optimal catalyst, which has the lowest FE of H2. Therefore, S10 was chosen to investigate the CO2 ERR performance in detail. Fig. S5b† is the chronoamperometric curves of S10 under various potentials. The NMR results (Fig. S7–S12†) indicate that there are no liquid carbon products in the potential range from −1.3 to −1.8 V vs. SHE. The corresponding FE of the gas products are shown in Fig. 5a and Table S2.† The FE of C2H4, C2H6, H2 and CO are 55.4%, 37.6%, 6.7% and 0.3%, respectively, at −1.3 V vs. SHE. The total FE for C2H4 and C2H6 is 93%. When the potential is −1.4 V vs. SHE, the FE of C2H4, C2H6, H2 and CO are 68.2%, 10.2%, 20.5% and 1.1%, respectively, with the total FE for C2H4 and C2H6 of 78.4%. These FE of C2H4 and the total FE of C2 are competitive with those reported for Cu–Cu+ hybrids catalysts (Table S3†) with FE of C2H4 in the range from 10% to 59.3% and the FE of total C2 in the range from 61% to 90.5% in recent works (Table S3†).2,4,7,9,11,12,14,17,22,24,35 For example, Dinh et al. found that the FE of C2H4 and total C2+ were 70% and 83%, respectively, using the hydroxide-modified Cu nanocatalyst in 7 M KOH at −0.55 V vs. relative hydrogen electrode (RHE) by a flow cell electrolyzer.2 Wang et al. achieved 53% FE of C2H4 and 74% of total C2 using Cu–CuOx/carbon black catalyst in 0.1 M KHCO3 electrolyte at −1.3 V vs. RHE.4 A similar FE of 46% for C2H4 was observed in the case of CuO–Cu2O/carbon black in 0.1 M KHCO3 using a flow cell.7 The highest FE of 59.5% for C2H4 and 90.5% for total C2 were reported by Cuenya's group using Cux@Cu2O on carbon paper as the catalyst in an H cell filled with 0.1 M KHCO3 solution at −1.2 V vs. RHE.11 In our case, however, at a potential of −1.5 V vs. SHE, the FE of C2H4, C2H6, H2 and CO was 43.2%, 2.9%, 51.2% and 3.7%, respectively. The FE for C2 products dramatically declined to less than 10% when the potential was close to or less than −1.6 V vs. SHE. It is obvious that the more negative the potential, the more the formation of H2.
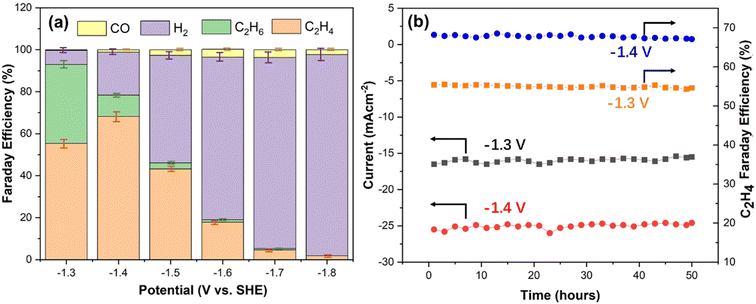 |
| | Fig. 5 (a) FE of the products generated by S10 at various potentials. (b) The FE of C2H4 and current density of the CO2 ERR using the S10 catalyst at −1.3 V and −1.4 V vs. SHE, respectively. | |
Considering the current density and FE efficiency of C2H4 products, the time dependence of the FE and current density of S10 for CO2 ERR were investigated to evaluate the stability at the potential of −1.3 V and −1.4 V, respectively. The results (Fig. 5b) indicate that during continuous operation for 50 hours, both the FE of C2H4 and current density of the CO2 ERR at these two potentials show almost no change. In addition, the SEM image of the catalyst after operating for 50 hours at −1.3 V vs. SHE is shown in Fig. S13.† It shows the similar nanoparticular structures on the rGO surface as those in the original catalyst (Fig. 2c). This result demonstrates the good stability of the Cu–Cu2O/rGO catalyst. It has been reported that fullerenes can stabilize the catalytic CO2 ERR performance of the Cu–Cu2O–C60 catalyst.14–16 Furthermore, it has been found that carbon-based materials, such as carbon layer,7 carbon black13 and CNTs,17 could stabilize the Cu+ species of the CuOx@C and Cu2O/CNTs since the carbon layer or CNTs can increase the electronic conductivity of Cu+ catalyst, which enables the electrons from the external circuit to transfer to the surface and facilitate the timely conversion of the reactant molecules, thus suppressing the reduction of Cu+ species.7,13,17 Similarly, in our case, the rGO layer will enhance the electronic conductivity of Cu2O, transfer the electrons to the catalyst surface and facilitate CO2 reduction, thus suppressing the reduction of Cu+ species.
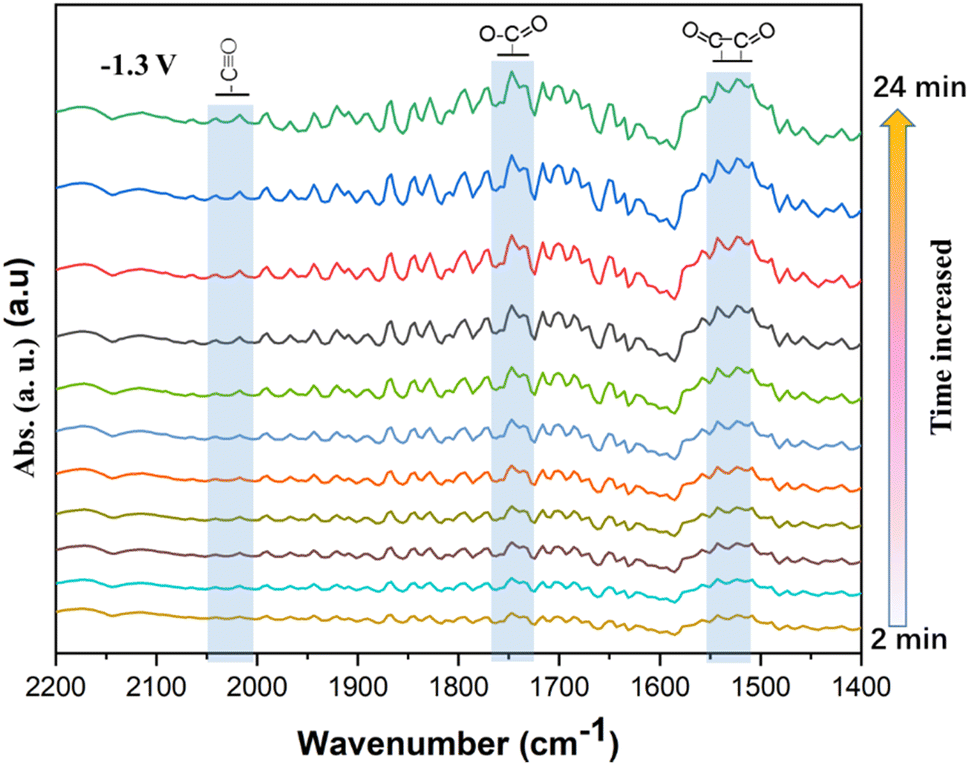
In situ ATR-SEIRS of the CO2 ERR
In order to understand the CO2 ERR process on the Cu–Cu2O/rGO catalyst, S10 was investigated by an in situ ATR-SEIRS setup (see details in the Experimental section). After running for 100 s, the FTIR spectra were recorded at −1.3 V vs. SHE. The interval for the spectra was 2 min. The spectra shown in Fig. 6 were recorded in the wavenumber range from 1400 to 2200 cm−1. The FTIR peak at about 2050 cm−1 is assigned to the adsorbed *CO.5,7,9 The peak at about 1750 cm−1 is indexed to O–CO intermediates.11–17 Also, the peak at about 1525 cm−1 comes from the OC–CO species, the key intermediate for the formation of C2H4 or other C2 products.24–30 It is obvious that the OC–CO dimers are the dominant species on the surface. Previous works11–17,24–30 indicate that the Cu0/Cu+ interfaces play a key role in the activation and CO dimerization, promoting the C2 selectivity. Similarly, the conversion of CO2 on Cu–Cu2O/rGO by CO2 ERR is achieved through the following steps: first, CO2 is activated by Cu and Cu+ to *CO and adsorbed on the catalyst surface. Two *CO species are coupled to form OC–CO and then further hydrated to C2H4 or C2H6. The rGO support enhances the electronic conductivity and protects the Cu+ species. Therefore, the synergistic effect of Cu0, Cu+ and rGO of the Cu–Cu2O/rGO catalyst contributes to its high selectivity and stability for the conversion of CO2 to C2H4.
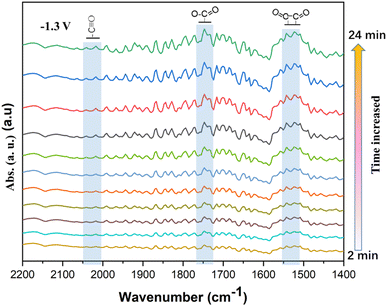 |
| | Fig. 6 Electrochemical in situ FTIR spectra of the S10 Cu–Cu2O/rGO at a potential of −1.3 V vs. SHE. All the spectra were subtracted from the initial one (recorded after activation of 100 s). | |
Conclusion
In summary, we prepared a Cu–Cu2O/rGO catalyst from the CHNs/GO composite via an in situ electrochemical reduction process in a CO2 ERR flow cell. The prepared Cu–Cu2O/rGO demonstrated ∼10 nm Cu–Cu2O nanoparticles with Cu–Cu+ combination anchored on the surface of rGO. These Cu–Cu+ species are desirable for the activation of CO2 and dimerization of C–C species, then selectively facilitating the formation of C2H4.5,7,9–17,24–30 Due to these advantages, this Cu–Cu2O/rGO catalyst presented highly selective CO2 ERR to convert CO2 to C2H4 in a flow-cell model. At −1.3 V vs. SHE, it shows an FE of 55.4% and 37.6% for C2H4 and C2H6, respectively, with a total C2 FE of 93%. At −1.4 V vs. SHE, it shows an FE of 68.2% and 10.2% for C2H4 and C2H6, respectively, with a total C2 FE of 78.4%. It also presents a good stability. This may provide a facile way to design Cu-based catalysts for selectively converting CO2 to C2H4 via CO2 ERR.
Data availability
The authors confirm that the data supporting the findings of this study are provided within the article and its ESI.† Raw data that support the findings of this study are available from the corresponding author, upon request.
Author contributions
Chenxiang Peng: primary investigation, methodology, formal analysis, and original draft writing and editing. Bing Yao: analysis, writing, reviewing and editing. Lei Wang: writing, reviewing and editing. Xinyi Wan: conceptualization, supervision, reviewing and editing, resources.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2023YFB3810900).
References
- C. P. O'Brien, R. K. Miao, A. S. Zeraati, G. Lee, E. H. Sargent and D. Sinton, Chem. Rev., 2024, 124, 3648 CrossRef PubMed.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. de Arquer, A. Kiani, J. P. Edwards, P. D. Luna, O. S. Bushuyer, C. Zuo, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783 CrossRef CAS PubMed.
- Y. Yang, Z. Tan and J. Zhang, Chem.–Asian J., 2022, 17, e202200893 CrossRef CAS.
- F. Yang, W. Fang, Q. Wang, P. Deng and B. Y. Xia, ACS Sustainable Chem. Eng., 2022, 10, 4677 CrossRef CAS.
- X. Chen, Y. Zhao, J. Han and Y. Bu, ChemPlusChem, 2023, 88, e202200370 CrossRef CAS.
- E. Andreoli, Nat. Catal., 2021, 4, 8 CrossRef CAS.
- V. S. R. K. Tandava, M. C. Spadaro, J. Arbiol, S. Murcia-López and J. R. Morante, ChemSusChem, 2023, 16, e202300344 CrossRef CAS.
- B. Yin, C. Wang, S. Xie, J. Gu, H. Sheng, D.-X. Wang, J. Yao and C. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202405873 CrossRef CAS.
- D. Gao, I. Sinev, F. Scholten, R. M. Ará-Ais, N. J. Divins, K. Kvashina, J. Timoshenko and B. R. Cuenya, Angew. Chem., Int. Ed., 2019, 58, 17047 CrossRef CAS PubMed.
- R. Zhang, J. Zhang, S. Wang, Z. Tan, Y. Yang, Y. Song, M. Li, Y. Zhao, H. Wang, B. Han and R. Duan, Angew. Chem., Int. Ed., 2024, 63, e202405733 CrossRef CAS.
- H. Liu, C. Yang, T. Bian, H. Yu, Y. Zhou and Y. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202404123 CrossRef CAS.
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z.-J. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344 CrossRef CAS PubMed.
- Z. Wang, L. Xu, Y. Zhou, Y. Liang, J. Yang, D. Wu, S. Zhang, X. Han, X. Shi, J. Li, Y. Yuan, P. Deng and X. Tian, Chem. Soc. Rev., 2024, 53, 6295 RSC.
- B. Zhao, F. Chen, C. Cheng, L. Li and B. Zhang, Adv. Energy Mater., 2023, 13, 2204346 CrossRef CAS.
- J. Zheng, L. Huang, C.-H. Cui, Z.-C. Chen, X.-F. Liu, X. Duan, X.-Y. Cao, T.-Z. Yang, H. Zhu, K. Shi, P. Du, S.-W. Ying, C.-F. Zhu, Y.-G. Yao, G.-C. Guo, Y. Yuan, S.-Y. Xie and L.-S. Zheng, Science, 2022, 376, 288 CrossRef CAS PubMed.
- I. Shown, H.-C. Hsu, Y.-C. Chang, C.-H. Lin, P. K. Roy, A. Ganguly, C.-H. Wang, J.-K. Chang, C.-I. Wu, L.-C. Chen and K.-H. Chen, Nano Lett., 2014, 14, 6097 CrossRef CAS PubMed.
- X. Lv, Q. Liu, J. Wang, X. Wu, X. Li, Y. Wang, J. Yan, A. Wu and H. B. Wu, Appl. Catal., B, 2023, 324, 122272 CrossRef CAS.
- Y. Luo, J. Huang, J. Jin, X. Peng, W. Schmitt and I. Ichinose, Chem. Mater., 2006, 18, 1795 CrossRef CAS.
- H. Huang, Z. Song, N. Wei, L. Shi, Y. Mao, Y. Ying, L. Sun, Z. Xu and X. Peng, Nat. Commun., 2013, 4, 3979 Search PubMed.
- W. Lu, W. Sun, X. Tan, L. Gao and G. Zheng, Catal. Commun., 2019, 125, 98 CrossRef CAS.
- Y. Deng, A. D. Handoko, Y. Du, S. Xi and B. S. Yeo, ACS Catal., 2016, 6, 2473 CrossRef CAS.
- L. Wang, Z. Chen, Y. Xiao, L. Huang, X. Wang, H. Fruehwald, D. Akhmetzyankov, M. Hanson, Z. Chen, N. Chen, B. Billinghurst, R. D. L. Smith, C. V. Singh, Z. Tan and Y. A. Wu, Nat. Commun., 2024, 15, 7477 CrossRef CAS PubMed.
- M. Sookhakian, M. A. Teridi, G. B. Tong, P. M. Woi, M. Khalil and Y. Alias, ACS Appl. Nano Mater., 2021, 4, 12737 CrossRef CAS.
- Y. Zhang, Y. Chen, X. Wang, Y. Feng, Z. Dai, M. Cheng and G. Zhang, Nat. Commun., 2024, 15, 5172 CrossRef CAS PubMed.
- Y. Kim, S. Park, S.-J. Shin, W. Choi, B. K. Min, H. Kim, W. Kim and Y. J. Hwang, Energy Environ. Sci., 2020, 13, 4301 RSC.
- L. Zhang, Y. Men, B. Wu, Y. Feng, C. Song, S. Liu, J. Wang, W. An and T. T. Magkov, Top. Catal., 2023, 66, 1527 CrossRef CAS.
- W. Fang, R. Lu, F.-M. Li, C. He, D. Wu, K. Yue, Y. Mao, W. Guo, B. You, F. Song, T. Yao, Z. Wang and B. Y. Xia, Angew. Chem., Int. Ed., 2024, 63, e202319936 CrossRef CAS PubMed.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179 CrossRef CAS.
- M. Bisztyga-Szklaz, K. Mech, M. Mzrzec and R. Kalenarev, Materials, 2021, 14, 3171 CrossRef.
- Y. Zhao, X. Zu, R. Chen, X. Li, Y. Jiang, Z. Wang, S. Wang, Y. Wu, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2022, 144, 10446 CrossRef CAS PubMed.
- A. Salverda, S. Abner, E. Mena-Morcillo, A. Zimmer, A. Elsayed and A. Chen, J. Phys. Chem. C, 2023, 127, 7151 CrossRef CAS.
- R. Al-Gaashani, A. Najjar, Y. Zakaria, S. Mansour and M. A. Atieh, Ceram. Int., 2019, 45, 14439 CrossRef CAS.
- Q.-J. Wu, J. Liang, Y.-B. Huang and R. Cao, Acc. Chem. Res., 2022, 55, 2978 CrossRef CAS.
- H. S. Jeon, S. Kunze, F. Scholten and B. R. Cuenya, ACS Catal., 2018, 8, 531 CrossRef CAS.
- G.-Y. Duan, X.-Q. Li, G. R. Ding, L.-J. Han, B.-H. Xu and S.-J. Zhang, Angew. Chem., Int. Ed., 2023, 61, e202110657 CrossRef.
- B. Beslsa, L. Xia, V. Golovanova, B. Polesso, A. Pinilla-Sánchez, L. S. Martin, J. Ye, C. T. Dinh and F. P. G. de Arquer, Nat. Rev. Mater., 2024, 9, 535 CrossRef.
- Z. Chen, G. Zhang, H. Chen, J. Prakash, Y. Zheng and S. Sun, Renewable Sustainable Energy Rev., 2022, 155, 111922 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc
*bc