
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRepurposing of Indomethacin and Naproxen as anticancer agents: progress from 2017 to present
Asmaa E. Kassab *a and
Ehab M. Gedawyab
*a and
Ehab M. Gedawyab
aDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Cairo University, Kasr El-Aini Street, P. O. Box 11562, Cairo, Egypt. E-mail: asmaa.kassab@pharma.cu.edu.eg; Fax: +2023635140; Tel: +2023639307
bDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy and Pharmaceutical Industries, Badr University in Cairo (BUC), Badr City, P. O. Box 11829, Cairo, Egypt
First published on 23rd December 2024
Abstract
Inflammation is strongly linked to cancer and is essential for the growth and development of tumors. Targeting inflammation and the mediators involved in the inflammatory process could therefore provide a suitable method for cancer prevention and therapy. Numerous studies have shown that inflammation can predispose tumors. Non-steroidal anti-inflammatory drugs (NSAIDs) can affect the tumor microenvironment through increasing apoptosis and chemo-sensitivity while decreasing cell migration. Since the development of novel drugs requires a significant amount of money and time and poses a significant challenge for drug discovery, there has been a recent increase in interest in drug repositioning or repurposing. The growing body of research suggests that drug repurposing is essential for the quicker and less expensive development of anticancer therapies. In order to set the course for potential future repositioning of NSAIDs for clinical deployment in the treatment of cancer, the antiproliferative activity of derivatives of Indomethacin and Naproxen as well as their mechanism of action and structural activity relationships (SARs) published in the time frame from 2017 to 2024 are summarized in this review.
1. Introduction
The most fatal non-infectious disease is cancer, which is brought on by unchecked cell multiplication. The incidence of human cancers is constantly increasing, emerging as one of the greatest challenges in medical healthcare. Invasive cancer is the leading cause of death in developed countries and the second leading cause of death in developing countries. In 2020, 19.3 million cases of cancer and 10 million deaths were recorded.1 Despite the enhanced accessibility of many anticancer drugs, high mortality has persisted in recent decades.2 According to projections, there will be 26 million new instances of cancer worldwide in 2030.3,4 There is a significant gap between the supply and demand of new anticancer drugs. Currently, available anticancer drugs are associated with difficult follow-up care side effects because many of them are not selective, not distinguishing between cancer and healthy cells. The development of resistance to existing anticancer medications is a second obstacle.5 Hence invention and development of new and selective anticancer drugs with the fewest side effects and multiple treatment modalities is still an ongoing research area.2. Inflammation and cancer
The relationship between inflammation and cancer was first reported after the discovery of the presence of leukocytes in tumor cells.6 A microenvironment that is abundant in inflammatory cells, growth factors, and DNA-damage-inducing compounds promotes neoplastic risk by promoting prolonged and increased cell proliferation and survival.7 Presently, it is a recognized truth that chronic inflammation comes about an expanded hazard of cancer.8 During inflammation different growth factors, such as epidermal growth factor (EGF) and fibroblast growth factors (FG), are released stimulating cell proliferation and, hence, inducing cancer.9 The cyclooxygenase (COX) enzyme is a crucial catalyst in the production of prostaglandins and plays a significant role in the pathophysiology of inflammation. It exists in three isoforms, namely COX-1, COX-2, and COX-3. COX-1 is present in healthy tissues, regulating prostaglandin synthesis and maintaining normal physiological function. On the other hand, COX-2 is synthesized during inflammation and cancer development.10 The third and most recently identified cyclooxygenase isozyme is COX-3 which is not active in humans.11,123. NSAIDs and cancer
Non-steroidal anti-inflammatory drugs (NSAIDs) impede the activity of COX-1 and COX-2 enzymes to varying degrees, leading to a decrease in the production of prostaglandins.13 The obstruction of COX-1 results in various complications, with the most frequent being gastrointestinal ulceration.14 COX-2 is the principal inflammatory agent and its production is increased during inflammation.15–17 As a result, COX-2 has been the focus of anti-inflammatory drugs for numerous years.18 COX-2 plays a crucial role in the onset of cancer as it obstructs programmed cell death and commences the progression of blood vessel formation. A rise in COX-2 concentration has been noted in various types of cancer such as colorectal, breast, pancreatic, esophageal, lung cancer, and melanoma.19–21NSAIDs are commonly employed for the treatment of pain and inflammation associated with cancer. There was a minimal prevalence of primary or recurring tumors in patients on long-term NSAID therapy. Furthermore, mortality in cancer patients was significantly decreased following NSAIDs combination therapy.22 The blocking of COX-2 by certain NSAIDs has resulted in the suppression of tumor growth and the promotion of metastasis.23 As a result, COX-2 has been identified as a potential focal point for novel anti-neoplastic medications, prompting escalated exploration in this regard.24 Celecoxib, a selective COX-2 inhibitor has demonstrated anti-tumor characteristics for certain types of cancer, such as ovarian cancer and adenomas.25 Moreover, COX-2 inhibitors have demonstrated the ability to overcome multidrug resistance by decreasing the expression of efflux pumps, thereby revealing an alternative domain.26 One of the following can mediate the mechanism of action of NSAIDs' anticancer activity.
3.1. COX-dependent anticancer activity
The overexpression of COX-2 in a variety of solid tumor types is one of the compelling evidence for the involvement of COX enzymes in cancer.27–30 Initiation of tumors in the mammary gland epithelial cells was also linked to COX-2 overexpression.31 The overexpression of COX-2 was associated with the development of resistance to the induced apoptosis in the intestinal epithelial cells.32 Additionally, overexpression of COX-2 inhibits apoptosis,33 activates pro-carcinogen,34 and promotes metastasis and angiogenesis.35,36 Accordingly, metastasis,37 angiogenesis,38 and nuclear factor kappa B (NF-kB)39 may be inhibited by COX inhibitors, which in turn may mediate their anticancer effect. There is evidence that both COX-1 and COX-2 genetic disruption reduces carcinogenesis. Additionally, several studies in favor of COX-1's involvement in cancer were reported. A compelling evidence for the significance of COX-1 in malignancies was made by the antitumor effect of selective COX-1 inhibitors.40–423.2. COX-independent anticancer activity
Numerous studies disprove the idea that NSAIDs' chemo-preventive effects are exclusively brought on by COX inhibition by showing that these effects can be at least partially mediated by COX-independent mechanisms. For instance, in vitro research has shown that NSAIDs can suppress proliferation and/or trigger apoptosis in a variety of tumor cell lines from various origins, regardless of how much COX-1 or COX-2 is expressed in the cells.43–46 Even though they lack COX inhibitory action, several NSAIDs have shown antitumor efficacy.47 Several NSAIDs have shown antiproliferative effects on cancer cells that are COX-2 positive or negative.46 Certain NSAIDs show similar COX inhibitory action, even though they have distinct anticancer effects against the same cell lines.48 NSAIDs decrease cancer cell proliferation at concentrations greater than those needed to inhibit COX.494. Drug repurposing
In the last ten years, there has been an exponential rise in “drug repurposing”. This phrase refers to a drug discovery technique, whose main objective is to uncover novel applications for already FDA-approved medications.50 The economic gain continues to be the main driving force behind this phenomenon, notwithstanding the possibility of other contributing variables. This strategy gives prolonged patents to pharmaceutical corporations as a way to get around the challenges caused by the so-called productivity problem. Saving time, resources, and money, and allowing for direct admission into clinical trials because approved drugs have previously undergone toxicity and safety profiles is another significant benefit of drug repositioning. For a variety of reasons, drug reprofiling is more cost-effective than novel drug discovery and development because it provides one of the best risk-to-reward trade-offs among the various drug development methodologies.50,51 Several chemotherapeutic approaches to treat human health issues have used drug repurposing such as cancer,52–57 neurodegenerative diseases,58,59 autoimmune diseases,60 asthma61 and systemic lupus erythematosus.625. The limitations and challenges of drug repurposing in cancer treatment
In oncology, epidemiological data can be used to support drug repurposing; for example, it may be observed that using a non-anticancer medication such as Aspirin and Metformin significantly lowers the incidence, severity, or death of certain cancer forms. Despite compelling results from randomized trials and supporting evidence, Aspirin is still not advised in the most important clinical guidelines (NCCN or ESMO) even though high-level evidence supports its use following curative therapy of colorectal cancer to prevent new adenomas.63,64 There are general obstacles faced by the drug repurposing community, such as organizational challenges, regulatory issues, and patent concerns.65Drug repurposing in cancer treatment has had minimal success due to several issues. A combined therapy approach is more likely to be clinically beneficial for oncology patients since the pathogenesis of cancer involves several, rather than single, cellular pathways. In investigator-initiated studies of repurposed drugs, one drug is often employed, so carrying out clinical trials of repurposed drugs targeting cancer is much more difficult.66
Additionally, there are several financial causes of this failure such as a lack of funding incentives for medication developers and a lack of experience developing drugs in the nonprofit sector. Trials intended to examine repurposed anticancer drugs are expensive and prone to failure, like any experiment. No financial incentives presently exist for repurposing generic medications outside of the treatment of uncommon and/or pediatric tumors, making a return on investment all but impossible. Most non-profit organizations cannot afford to undertake phase III clinical studies of repurposed pharmaceuticals. Investigator-initiated trials are typically one-off studies with no foresight into future developments and/or strategies. The outcomes of these trials are frequently not widely disseminated, which limits their impact.66 To take advantage of drug repurposing for the benefit of patients and society worldwide, significant efforts and research are urgently needed.
In the next sections, as a continuation of our previous work,67 we'll go over the most current studies including derivatives of Indomethacin and Naproxen as they are among the most widely refashioned NSAIDs as anticancer candidates. Their potential antiproliferative activity, mechanism of action, and structural activity relationships (SARs) will be discussed. Analysis of the anticancer activity of these compounds may contribute to repositioning them for cheaper and safer cancer therapeutics.
6. Anticancer activity of Indomethacin derivatives
A Pt(IV) prodrug 1 (Fig. 1) was developed that could accumulate in cancer cells more so than in healthy cells and be activated by endogenous reducing molecules to release cisplatin and Indomethacin moieties simultaneously to suppress tumor progression synergistically. Human normal liver cells LO-2 and several human cancer cell lines HCT-116, HepG-2, PC-3, SGC7901, and SGC7901/CDDP (cisplatin) were used in vitro assays to determine the effectiveness of Indomethacin derivative 1.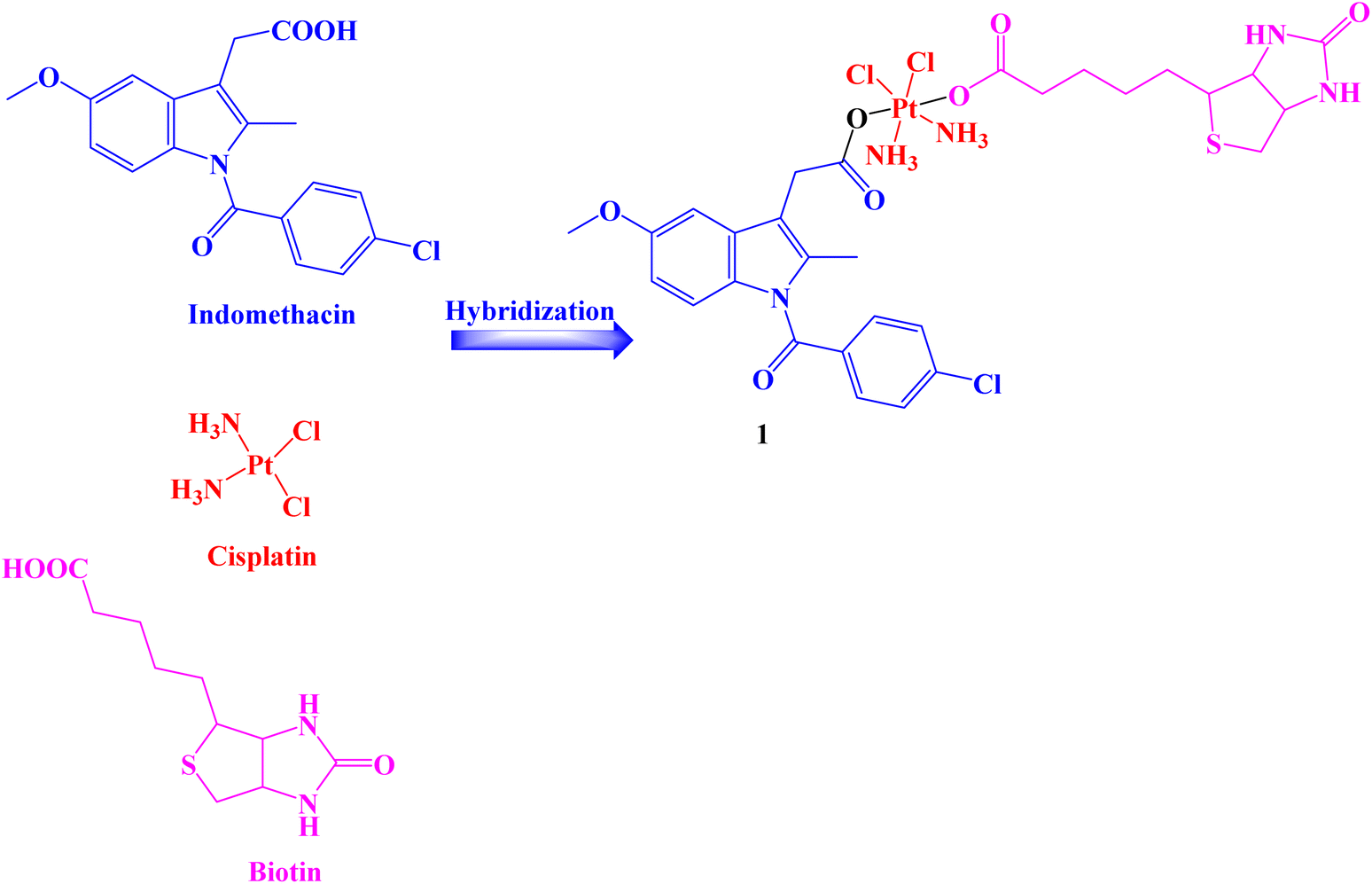 | ||
| Fig. 1 The structure of Indomethacin–Pt(IV) prodrug 1. | ||
The results showed that compound 1 significantly inhibited the tested cancer cell lines and was selective to cancer cells that were resistant to cisplatin (IC50 range: 0.91 to 59.64 μM). Additionally, prodrug 1 demonstrated cyclooxygenase inhibition properties to lessen tumor-associated inflammation, decreased the invasiveness of the highly aggressive PC-3 cells and interfered with the creation of capillary-like tubes in human umbilical vein endothelial EA.hy926 cells. In comparison to negative and positive controls, compound 1 treatment of PC-3 cells caused an increase in the population of cells in the G2/M phase over time, showing that the cell cycle was arrested in the G2/M phases. Therefore, it may be concluded that hybrid 1 prevented the proliferation of PC-3 cells by first preventing DNA synthesis and then, as time goes on, having an impact on PC-3 cells' karyokinesis.
The anti-apoptotic protein BCL-2 was found to be less abundant in PC-3 cells after treatment with compound 1. In parallel, the prodrug enhanced the level of the pro-apoptotic protein Bax in the cells under the same circumstances as compared to the cells that were not treated with the complex. Both early and late apoptotic cells were present in equal amounts 23.35% and 25.01%, respectively. Only 3.53% and 2.97% of early and late apoptotic cells, respectively, were discovered in the untreated control. These findings suggested that cisplatin-related resistance in tumor cells might be almost eliminated by the biotin-Pt(IV)–Indomethacin combination.68
Amide bond was used to create the proposed Indomethacin–methotrexate hybrid 2 (Fig. 2). By using the MTT assay, the cytotoxic effects of the synthesized hybrid against the cancer cells HeLa and MCF-7 were assessed. The findings demonstrated that hybrid 2 was more cytotoxic than Indomethacin (IC50 values of 100 and 94 μM against HeLa and MCF-7 cancer cells, respectively) and methotrexate alone and had IC50 values of 16 and 10 μM, respectively, against HeLa and MCF-7 cancer cells.69
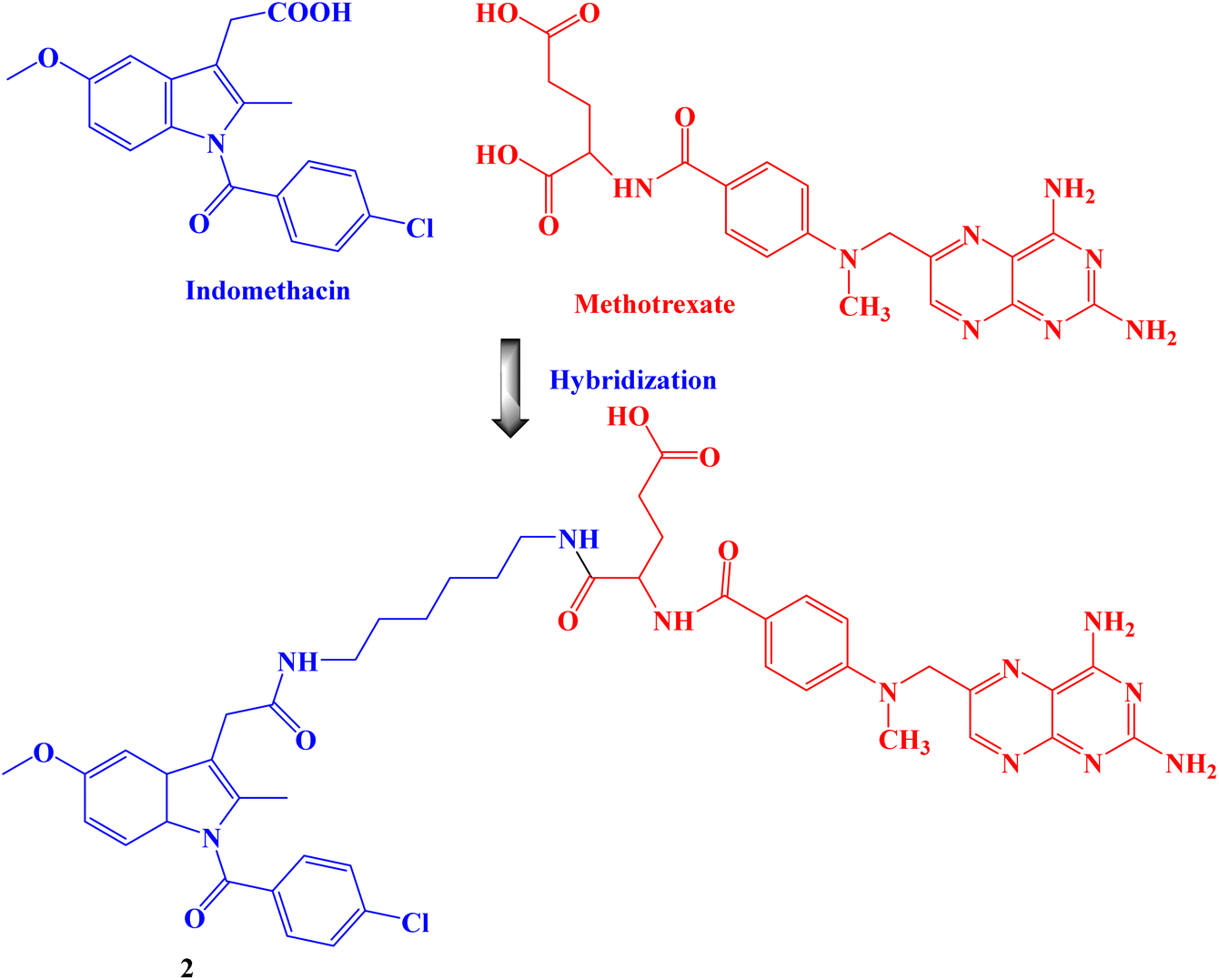 | ||
| Fig. 2 The structure of Indomethacin–methotrexate hybrid 2. | ||
Novel amide analogs were developed using the molecular structure of Indomethacin as a starting point, and their effects on the growth of human cancer cells were assessed using the colon cancer cell lines HCT-116, CACO-2, and HT-29. The novel compounds showed noticeably higher activity when compared to Indomethacin (IC50 values of 50, 53, and 30 μg mL−1 against HCT-116, HT-29, and CACO-2 cancer cells, respectively). With IC50 values ranging from 0.055 to 4.0 μg mL−1 compared to 0.7–5.45 μg mL−1 for 5-fluorouracil (5-FU), two Indomethacin analogs, 3a and 3b (Fig. 3), intriguingly demonstrated strong growth inhibitory activity in the nano- to micro-molar range against all three human colon cancer cell lines. While the diamide derivative 3b had strong growth inhibitory activity for all the tested cancer cell lines with the IC50 values smaller than 1 μg mL−1, the most powerful molecule 3a had 99 and 7.5-fold more cytotoxic activities against CACO-2 and HT-29 cell lines, respectively, than 5-FU.
 | ||
| Fig. 3 The structure of Indomethacin derivatives 3a,b. | ||
Additionally, the cytotoxic effect of the intriguing compounds 3a and 3b on the HT-29 and HCT-116 cell lines, respectively, was investigated as well as possible mechanisms. According to the findings, Indomethacin derivatives 3a and 3b might cause apoptosis by arresting the cell cycle at the G1/S and G0/G1 phases in HT-29 and HCT-116 cells, respectively. At IC50 values, the kinase inhibitory activity of 3a and 3b was assessed against CDK-2A. According to the profiling results, the action of 3a on HT-29 resulted in a 55 and 79% inhibition of CDK-2A activities after 24 and 48 hours, respectively, as compared to the control cell. Contrarily, 3b demonstrated strong enzyme inhibition at its IC50 concentration; the CDK-2A activities were reduced by 93 and 90%, respectively, in comparison to control.70
Hepatocellular carcinoma treatment is significantly hampered by multidrug resistance (MDR) to chemotherapeutic drugs. The development of novel anti-MDR antineoplastic drugs is an efficient strategy to combat cancer resistance. The antiproliferative properties of novel podophyllotoxin-NSAID conjugates were assessed in vitro against drug-sensitive human hepatocellular carcinoma cells Bel-7402, drug-resistant human hepatocellular carcinoma cells Bel-7402/5-FU and normal human liver cells L-O2 adopting CCK-8 assay. The Indomethacin conjugate 4 (Fig. 4), with an IC50 value of 2.15 μM and a reduced resistant factor value of 0.36, demonstrated selective cytotoxicity against resistant Bel-7402/5-FU cells. It was much less toxic to L-O2 cells (IC50 = 1.85 μM).
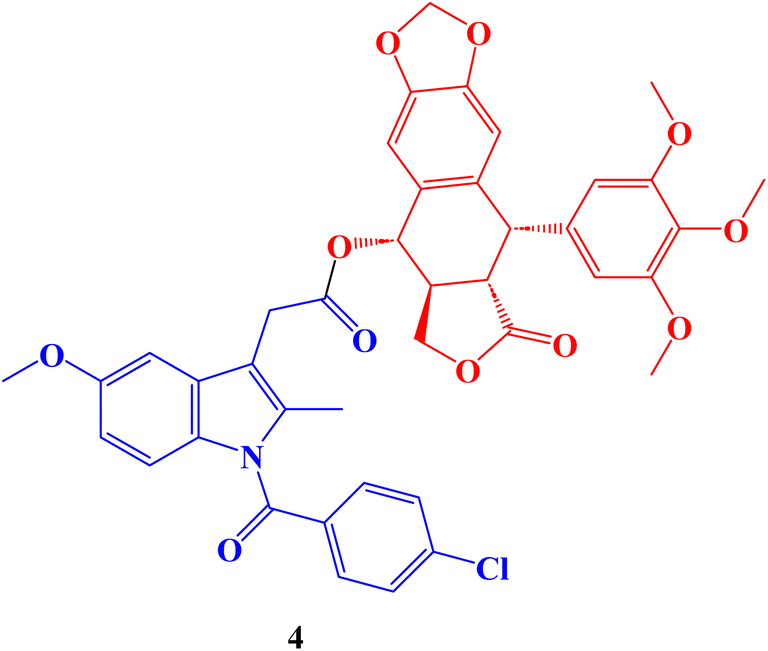 | ||
| Fig. 4 The structure of Indomethacin podophyllotoxin conjugate 4. | ||
The migration of Bel-7402/5-FU cells was also stopped by all conjugate compounds, which also induced apoptosis, harmed the microtubule network, and caused cell cycle arrest at the S/G2 phase. Finally, in Bel-7402/5-FU cells, these conjugates controlled the amounts of proteins linked to cell cycle arrest, apoptosis, migration, and inflammation. In general, better selective efficacy of the conjugates for resistant Bel-7402/5-FU cells and less toxicity to regular L-O2 cells were accomplished when NSAIDs were conjugated to the C-4 OH of the podophyllotoxin nucleus.71
Using the pharmacophore hybridization principle, dual-acting hybrid antioxidant/anti-inflammatory agents were developed. Stable antioxidant nitroxides were combined with traditional NSAIDs to create hybrid agents. On two Non-Small Cell Lung Cancer (NSCLC) cells (A549 and NCI-H1299) and in alleviating oxidative stress caused in 661W retinal cells, some of the hybrid nitroxide-NSAID conjugates showed promising antioxidant and anti-inflammatory properties. The effects of oxidative stress on 661W retinal neurons were greatly reduced by a nitroxide associated with the anti-inflammatory drug Indomethacin 5 (Fig. 5), with efficacies comparable to or greater than those of the antioxidant Lutein. With an IC50 value of 151 μM, Indomethacin derivative 5 demonstrated moderate cell inhibitory efficacy against NSCLC A549 cells.72
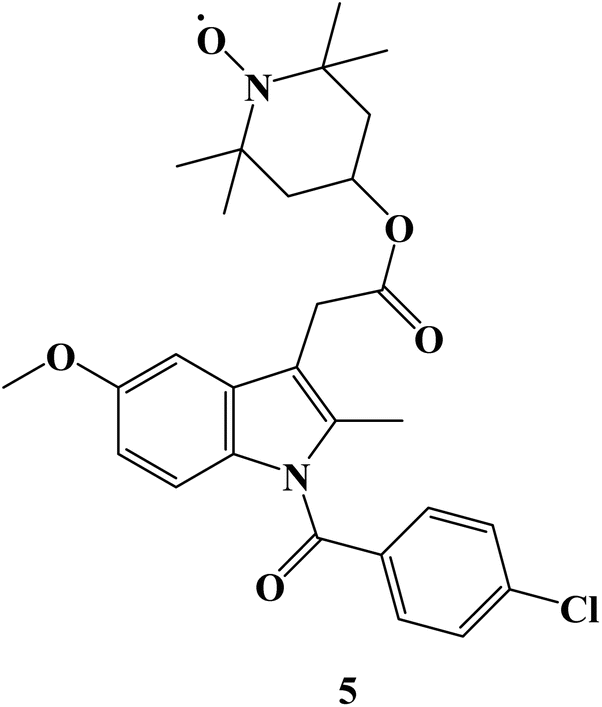 | ||
| Fig. 5 The structure of Indomethacin nitroxide hybrid 5. | ||
Rundstadler et al.73 reported the design and synthesis of a series of zinc(II)–phenanthroline–Indomethacin complexes 6a–d (Fig. 6) with equivalent potency (in the sub-micro- or micromolar range) for the killing of breast cancer stem cells (CSCs) (HMLER-shEcad) and bulk breast cancer cells (HMLER). The complete tumor population may theoretically be eradicated with a single dose of the zinc(II) complexes. Salinomycin, a drug that is anti-breast CSC, had an IC50 value of 4.2 μM. The potency of compounds 6a–d (with IC50 values in the range 0.7 to 1.1 μM) against CSC-like HMLER-shEcad cells was up to 6-fold higher.
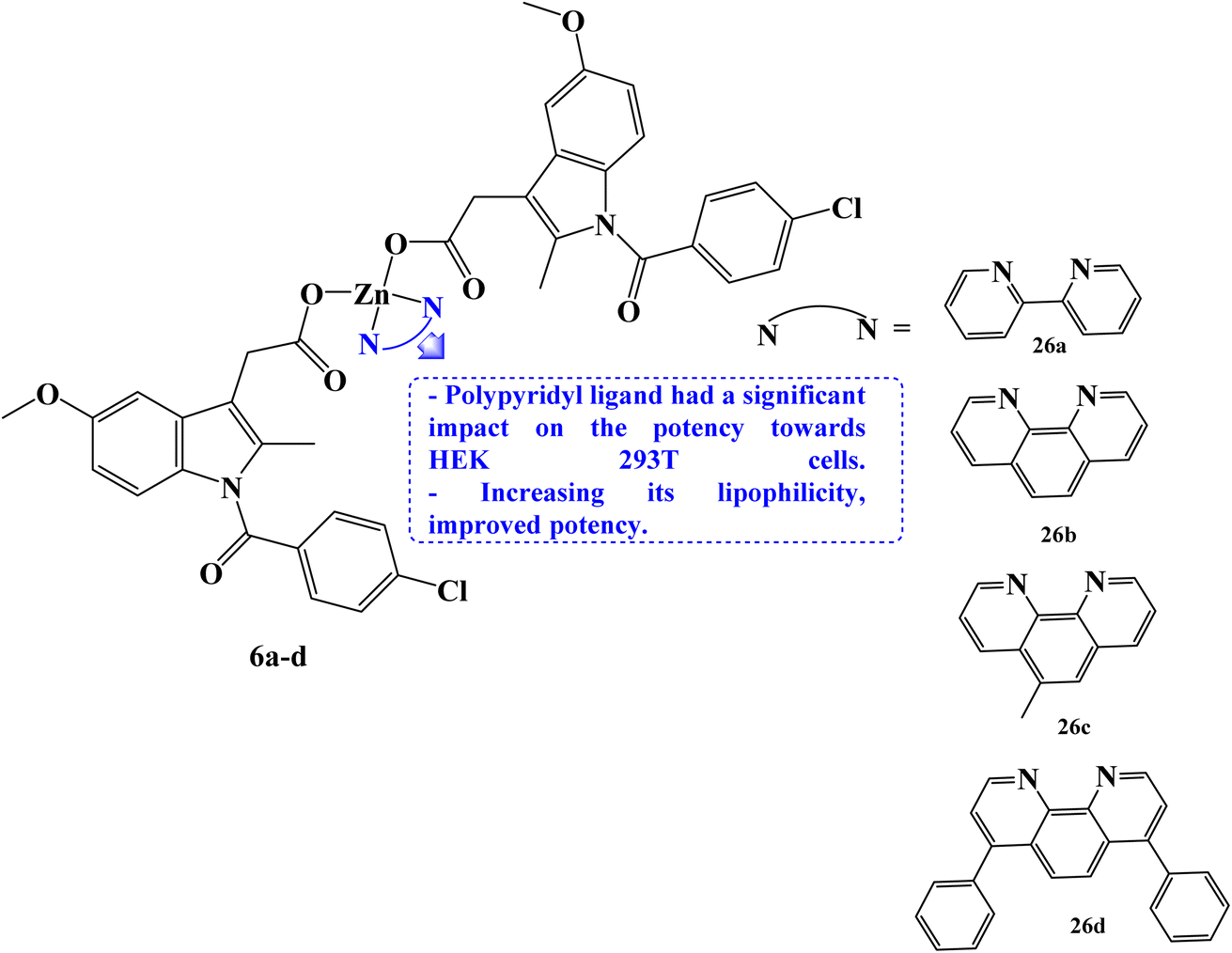 | ||
| Fig. 6 The structure of zinc(II)–phenanthroline–Indomethacin complexes 6a–d. | ||
The efficacy of Indomethacin derivatives 6a–d against human embryonic kidney (HEK 293T) cells was examined to gauge its therapeutic potential. The polypyridyl ligand has a significant impact on the potency towards HEK 293T cells, which rises in the order 6a< 6b < 6c < 6d. The 2,2′-bipyridine- and 1,10-phenanthroline-bearing complexes, 6b and 6c, showed promisingly preferred toxicity towards breast cancer cells (bulk and CSC-like cells) over healthy cells, being up to 17-fold less potent towards HEK 293T cells than HMLER and HMLER-shEcad cells. Several of the zinc(II) complexes inhibited mammosphere growth and viability to an equivalent or greater extent than salinomycin. Under the same circumstances, compound 6d containing 4,7-diphenyl-1,10-phenanthroline had an IC50 value of 2.7 μM and was 7 times more potent than salinomycin, which had an IC50 value of 18.5 μM.
Se derivatives of NSAIDs, such as selenocyanates and diselenides, were synthesized and characterized. The anticancer activities of these derivatives were evaluated against human tumor cell lines derived from various human cancer types-SW480 (human colon adenocarcinoma cells), HeLa (human cervical cancer cells), A549 (human lung carcinoma cells), and HepG2 (human hepatic carcinoma cells). Most of the novel compounds were effective in lowering the viability of various cancer cells. With IC50 values ranging from 7.9 to 39.5 μM against four cancer cell lines, Indomethacin derivative 7a (Fig. 7) demonstrated strong anticancer potential, notably to HepG2 with an IC50 value of 7.9 μM.
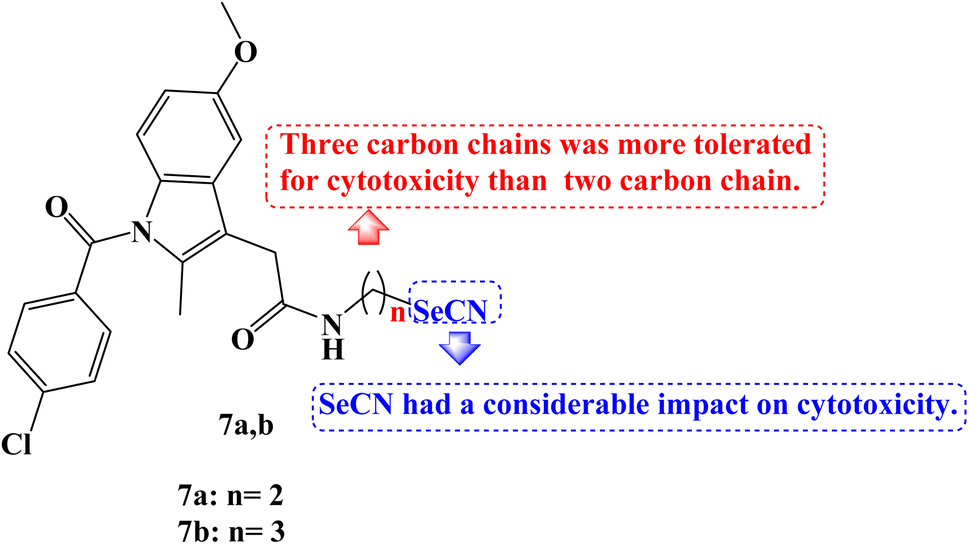 | ||
| Fig. 7 The structure of Indomethacin-Se derivatives 7a,b. | ||
The hybrid Se derivatives' IC50 values demonstrated that adding the SeCN moiety to the respective parent NSAIDs had a considerable impact on cytotoxicity. Except for HepG2 cells, compound 7b (Fig. 7) was moderately cytotoxic to all cell lines. It was interesting to note that compounds with three carbon chains between NSAIDs and SeCN moiety had stronger anticancer activity than the corresponding two carbon chain derivatives; perhaps this is due to the enhanced lipophilicity of these NSAIDs-Se derivatives.74
The MTT assay was used to test a new series of NSAIDs thioesters for their in vitro anticancer effects against a panel of four human tumor cell lines, including HepG2, MCF-7, HCT-116, and Caco-2. Compounds 8a,b (Fig. 8) outperformed the reference drugs 5-FU, afatinib, and celecoxib in terms of their potent, broad-spectrum anticancer activity against the selected tumor cell lines. The thioesters 8a,b showed strong anticancer activity concerning selectivity against the hepatocellular carcinoma cell line (HepG2), with IC50 values of 7.86 and 14.91 μM, respectively. With IC50 values of 9.65 and 17.10 μM, respectively, the thioesters 8a,b also demonstrated a significant sensitivity to the MCF-7 cell line. Additionally, the colon cancer cell line (HCT-116) was very susceptible to thioester 8a, with an IC50 value of 14.58 μM, but only moderately susceptible to thioester 8b, with an IC50 value of 34.05 μM. The thioester 8a also showed potent anticancer activity against the Caco-2 colorectal cancer cell line, with an IC50 value of 18.13 μM.
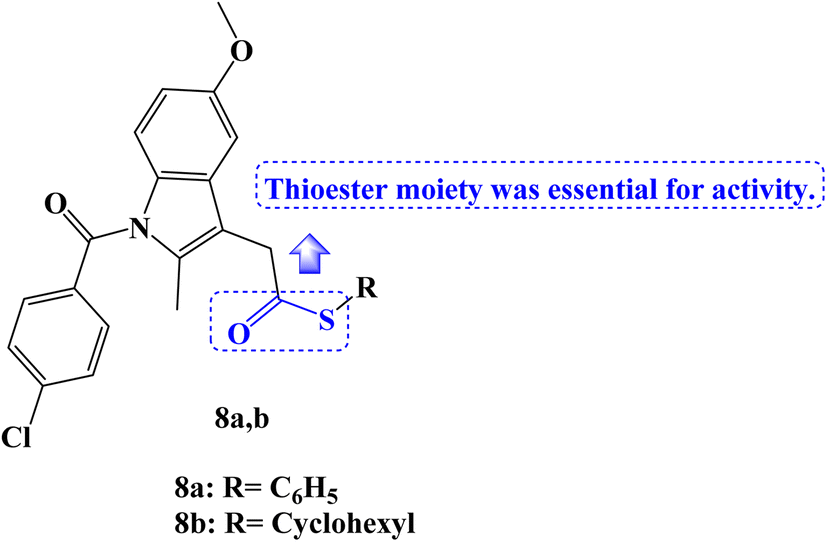 | ||
| Fig. 8 The structure of Indomethacin thioesters 8a,b. | ||
These derivatives were therefore chosen for mechanistic research on COX inhibition and kinase tests. When compared to celecoxib (IC50 = 0.16 μM, COX-2 SI: >312.5), the results of the in vitro COX-1/COX-2 enzyme inhibition assay showed that compounds 8a,b selectively inhibited the COX-2 enzyme (IC50 values of 0.22 and 0.49 μM, respectively), with SI values of 227 and 102, respectively. However, the COX-1 enzyme was not inhibited by the investigated derivatives (IC50 > 50 μM). On the other hand, 10 μM concentrations were used to examine EGFR, HER2, HER4, and cSrc kinase inhibition experiments. The chosen candidates showed only modest activity against the various kinases that were examined. The results of the molecular docking investigation demonstrated the significance of the thioester moiety for the compound's interaction with the amino acids in the COX-2 active site.75
As novel BCL-2 family protein inhibitors, several Indomethacin derivatives were developed by Chen et al.76 By including a benzyl scaffold, potent compounds were produced. Two benzyl groups were included in several compounds, which demonstrated more favorable inhibitory activity and even performed 2–3 times better than WL-276 and 5–6 times better than UMI-77. Meta-substitution proved to be more effective than para- and ortho-substitution in the benzyl scaffold. The addition of electron-withdrawing or electron-donating groups also had no discernible effect on the inhibitory activity. The 3-NO2-4-Cl substituted benzene sulfonamide moiety exerted greater efficacy than the 4-Br substituted counterparts.
Due to their strong binding affinity for both BCL-2 and Mcl-1, compounds 9a–f (Fig. 9) were chosen to be tested for their antiproliferative abilities in vitro. As members of the leukemia cancer cell line, Jurkat (acute T-cell leukemia cell) and K562 (chronic myelogenous leukemia cell) displayed high levels of BCL-2 and Mcl-1 expression, while PC-3 (prostate cancer cell), a member of the prostate cancer cell line, displayed high levels of Mcl-1 expression but low levels of BCL-2 expression.
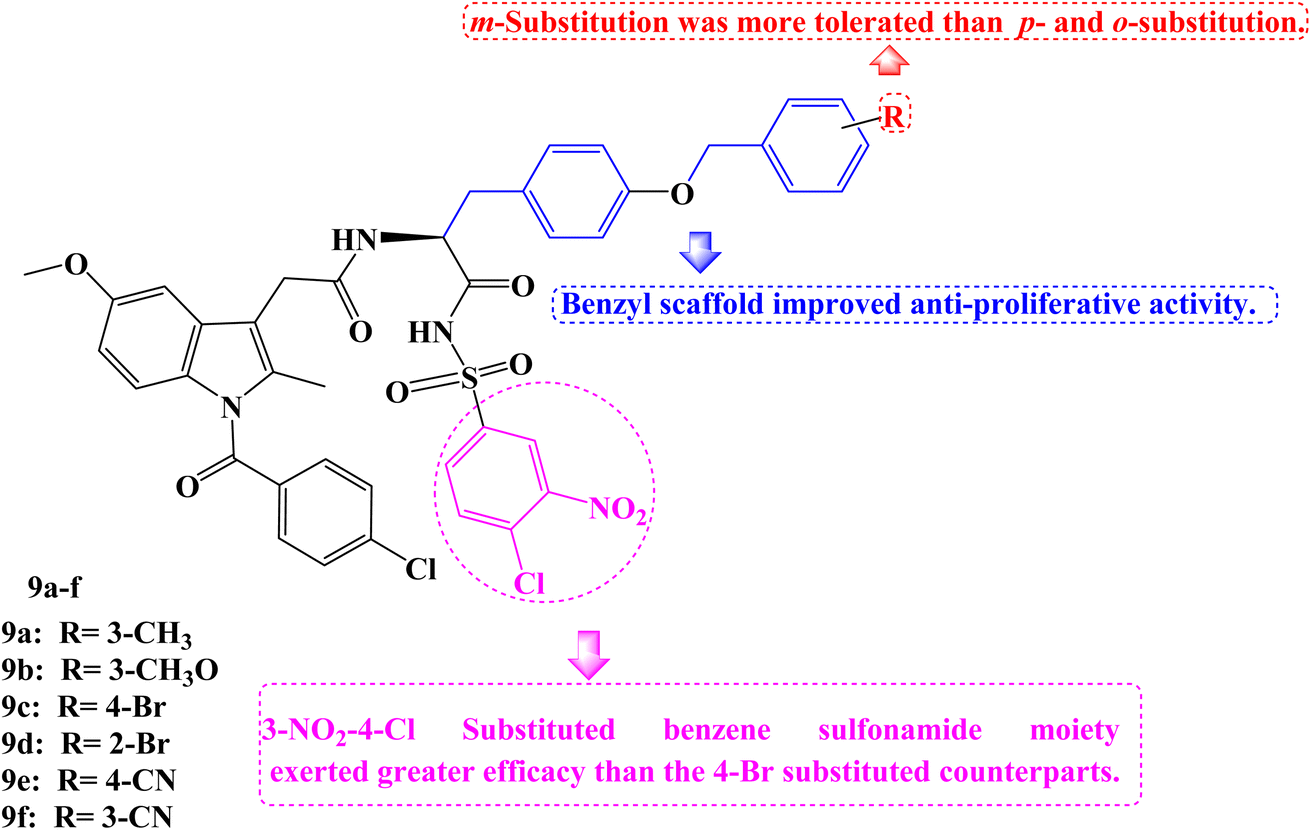 | ||
| Fig. 9 The structure of Indomethacin derivatives 9a–f. | ||
Then the MTT assay was used to examine Jurkat, K562, and PC-3. Most of the compounds under test had better antiproliferative actions against the three tumor cell lines compared to WL-276. The PC-3, Jurkat, and K562 cell lines were the most sensitive for compound 9c (IC50 = 25.79, 24.69, and 35.90 μM, respectively). Particularly, WL-276 performs two times worse than Jurkat cell lines in terms of antiproliferative activity. The growth of PC-3, Jurkat, and K562 could be greatly inhibited, and apoptosis could be induced by the most active Indomethacin derivative, 9c (BCL-2 Ki = 0.44 μM and Mcl-1 Ki = 0.44 μM). The elevated expression of the proteins Mcl-1 and BCL-2 in these cell lines might be the cause of 9c's potent anti-tumor effects. Using flow cytometry, annexin-V and propidium iodide (PI) double labelling was done to assess compound 9c's capacity to cause apoptosis in the Jurkat cell line. Compound 9c-induced apoptosis in a dose-dependent manner. Additionally, exposing the Jurkat cell line to 20 μM and 40 μM of 9c for 48 hours results in 43.1% and 56.8% of early and late cell apoptosis, respectively, as opposed to 13.3% in the DMSO control. In the Jurkat cell line, compound 9c was more effective at inducing apoptosis than WL-276, which causes 29.1% and 34.2% of cells to die after being exposed to those concentrations for 48 hours.
Low toxicity and noninvasive properties make photodynamic therapy (PDT) a viable cancer treatment strategy. Several metalloporphyrin–Indomethacin conjugates attached to poly(ethylene glycol) (PEG) chains were developed and characterized (Fig. 10). Using the 2′,7′-dichlorofluorescin (DCFH) method, the conjugates' singlet oxygen generation was assessed. The singlet oxygen (1O2) quantum yield of the metal porphyrin complexes was greater than that of the free base porphyrin due to the heavy atom effect. The synthesized porphyrins produced 1O2 in the following order: PtPor > PdPor > ZnPor > Por. The MTT assay using HeLa cells confirmed the low cytotoxicity of dark-activated porphyrin–Indomethacin conjugates. The platinated porphyrin (PtPor), among these conjugates, demonstrated the strongest therapeutic effectiveness after radiation exposure, most likely because of its high 1O2 production efficiency. Using a confocal laser scanning microscope, the conjugates' cellular uptake and subcellular localization were further assessed. According to the findings, HeLa cell lysosomes were where the conjugates were mostly found.77
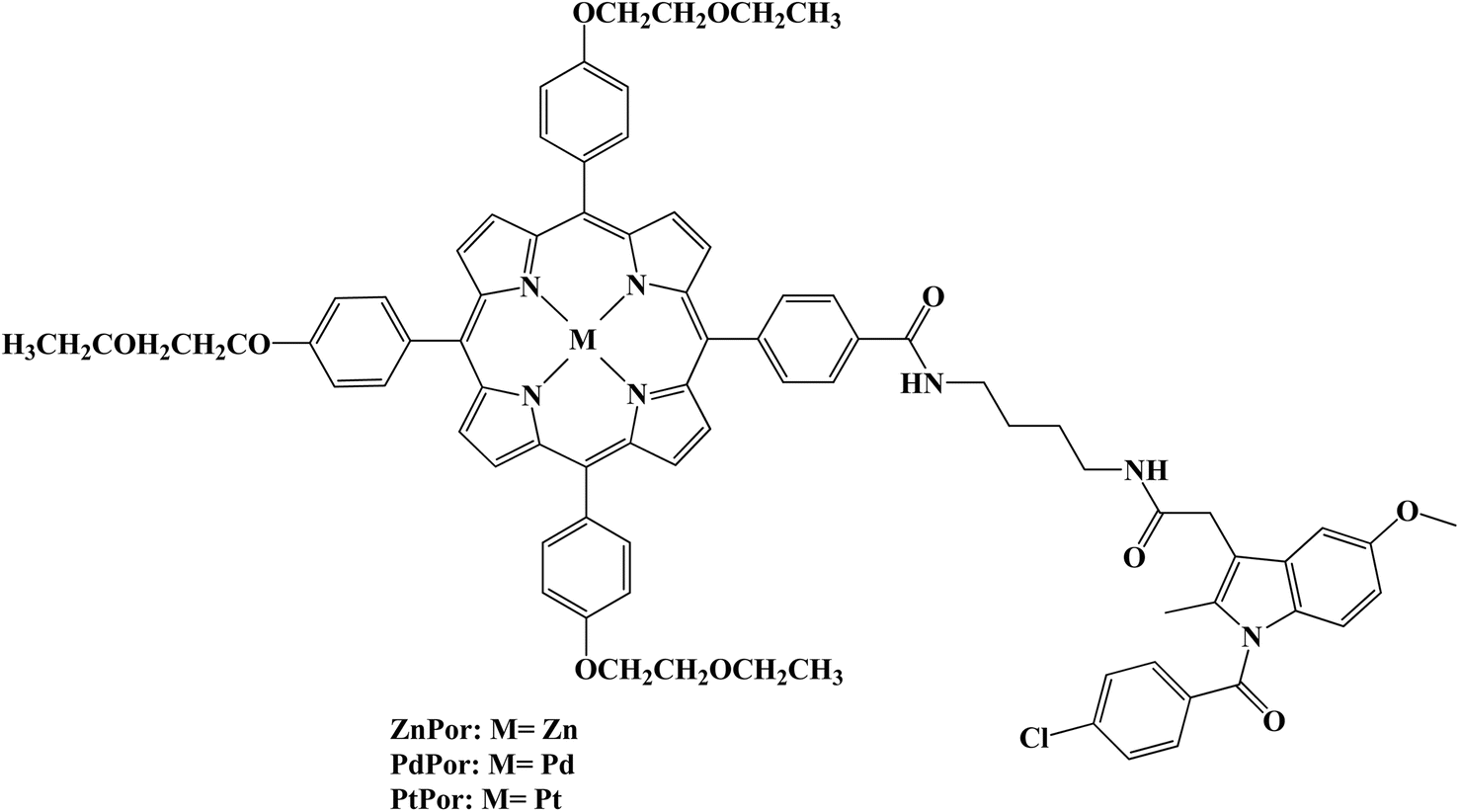 | ||
| Fig. 10 The structure of metalloporphyrin–Indomethacin conjugates PtPor, PdPor, and ZnPor. | ||
Cancers caused by inflammation are significantly influenced by Nrf2 and NF-kB. Due to the downregulation of these pathways, combining anti-inflammatory drugs with oleanolic acid oxime (OAO) may increase their therapeutic potential. THLE-2 immortalized normal hepatocytes and HepG2 hepatoma cells were used to investigate the effects of novel OAO compounds conjugated with Indomethacin on Nrf2 and NF-kB activation and expression concerning cell cycle arrest and apoptosis. In HepG2 cells, treatment with OAO-Indomethacin conjugates decreased Nrf2 and NF-kB activation and the expression of their active forms, but in healthy hepatocytes, Nrf2 activation was raised, and NF-kB activation was decreased.
The most potent modulator of Nrf2 translocation was found to be conjugate 10a (Fig. 11). The most effective cytotoxic compounds were 10a, 3-Indomethacinoxyiminoolean-12-en-28-oic acid morpholide (IC50 values of 42.5 and 38.5 μM against THLE-2 and HepG2 cell lines, respectively), and 10b, 3-Indomethacinoxyiminoolean-12-en-28-oic acid benzyl ester (Fig. 11) (IC50 values of 41.5 and 40 μM against THLE-2 and HepG2 cell lines, respectively). SOD-1 and NQO1 expression were dramatically increased in THLE-2 cells after treatment with these conjugates as opposed to HepG2 cells. In both cell lines, COX-2 expression was reduced. OAO-Indomethacin derivatives induced the cell cycle arrest in G2/M, which boosted apoptosis and increased the amount of HepG2 cells that were resting.78
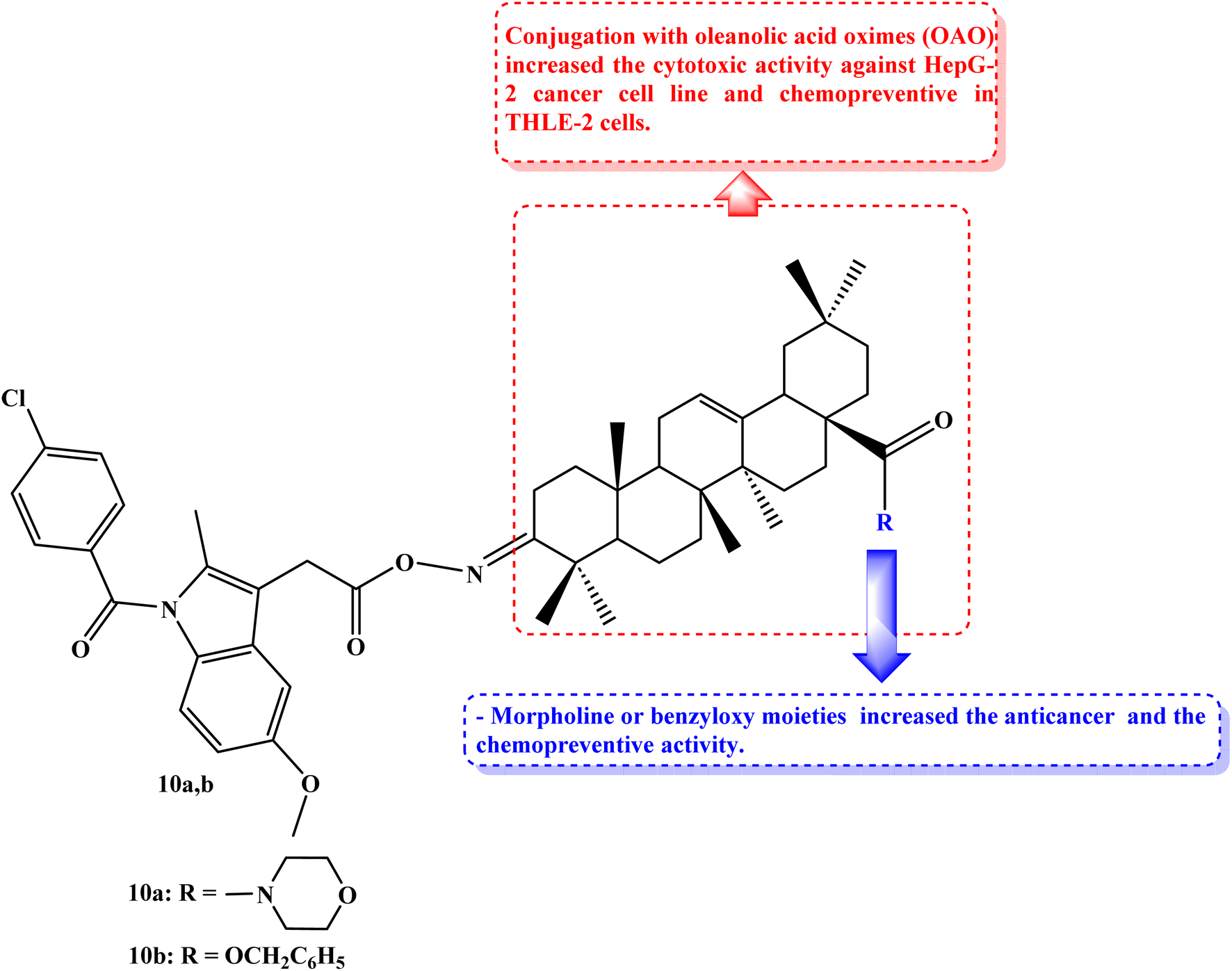 | ||
| Fig. 11 The structure of OAO-Indomethacin conjugates 10a,b. | ||
The particular COX-2 binding and intrinsic fluorescence features of 6- or 7-substituted coumarin-Indomethacin hybrids have been assayed. The best hybrid in this series was hybrid 11 (Fig. 12), which demonstrated strong binding to COX-2 and displayed higher fluorescence intensity in cancer cells than in normal cells. All of the cells were exposed to different concentrations of hybrid 11 (0–50 μM) for 24 hours before the MTT assay was used to determine the viability of the cells. At concentrations up to 10 μM, all of the cells maintained more than 80% vitality, and at concentrations as high as 50 μM, viability gradually decreased to around 50%. Next, it was investigated how selective compound 11 was for cancer cells that overexpressed COX-2. First, it was established that cellular absorption was time-dependent. All cells treated with compound 11 for 0, 0.5, 1, and 3 hours showed higher blue fluorescence signals with longer incubation times. Additionally, it was discovered that hybrid 11's fluorescence in cancer cells was dose-dependent. Consequently, in light of drug development tools, this unique hybrid system may be a promising targeted probe for detecting cancer cells and inflammation with COX-2 overexpression.79
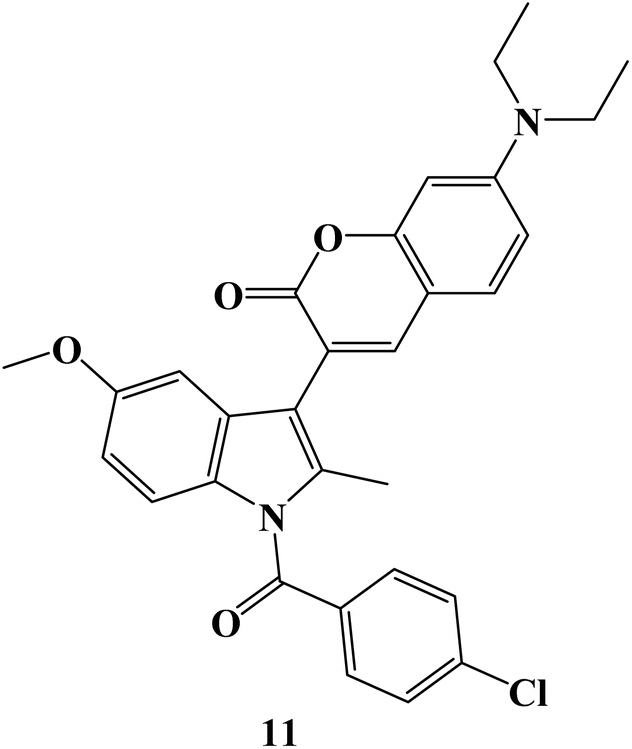 | ||
| Fig. 12 The structure of coumarin–Indomethacin hybrid 11. | ||
Five different human cancer cell lines (colon cancer cell lines HCT-116, HT-29, and Caco-2, hepatic cell line HepG-2, and breast cell line MCF-7) were used to test the cytotoxic effects of a new series of structurally related Indomethacin analogs. When tested against the three colon cancer cell lines, most of the derivatives showed strong anti-cancer activity. Indomethacin derivative 12a (Fig. 13) showed the strongest cytotoxic effect of all investigated derivatives when compared to the reference drugs 5-FU and Indomethacin, with IC50 values ranging from 0.83 to 1.54 μM. Cell cycle arrest during the G2/M phase was discovered by a mechanistic analysis of the most effective derivative 12a against the HCT-116, HT-29, and Caco-2 cell lines. It was discovered that compound 12a induced apoptosis by up-regulating Bax and p53 by 7.4 and 8.5 folds, respectively, and down-regulating BCL-2 by 3.2 folds when compared to the control.
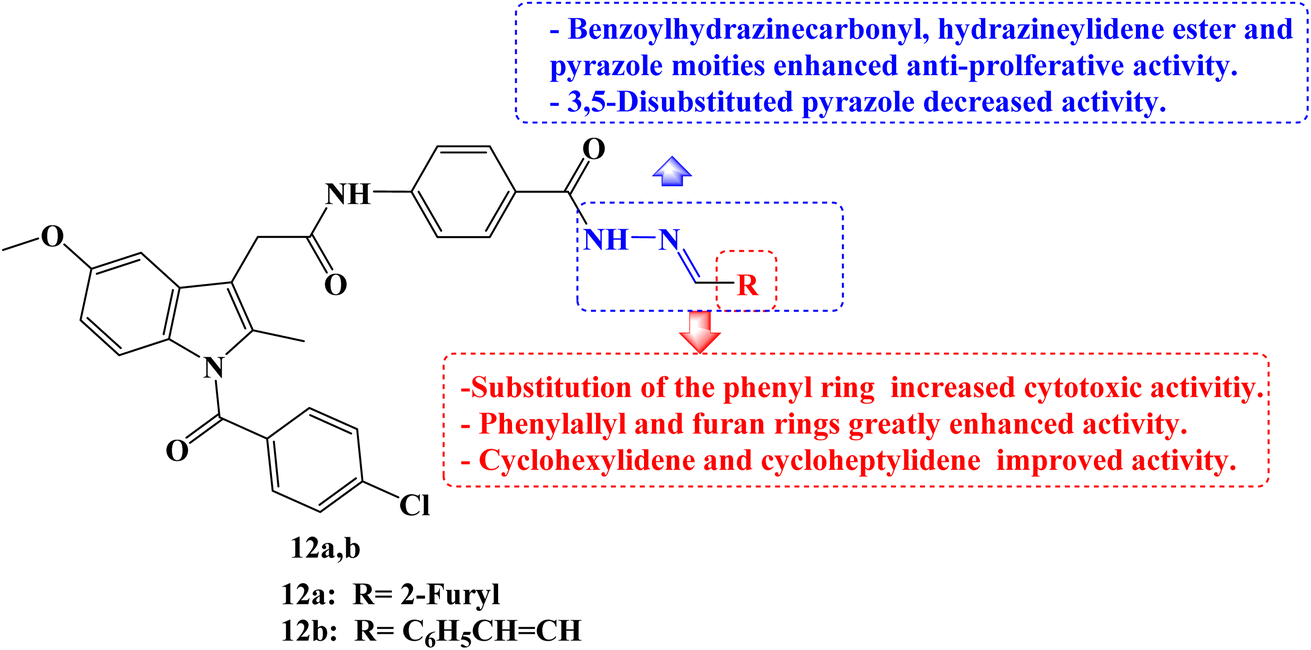 | ||
| Fig. 13 The structure of Indomethacin derivatives 12a,b. | ||
On HCT-116 cells, a western blot experiment was conducted, and the results showed a substantial reduction of CDK1 and BCL-2 expression as well as a concentration-dependent rise in the expression of caspase-3, Bax, and p53. The chemo-informatic features of compounds 12a and 12b (Fig. 13) were predicted, and results showed that they were orally accessible with no blood–brain barrier permeability. SARs studies revealed that the cytotoxic activities increased when the phenyl ring was substituted. It was interesting to note that Indomethacin derivatives with phenylallyl and furan rings, respectively, had higher potencies than phenyl and substituted phenyl derivatives. With IC50 values ranging from 2.8 to 9.16 μM, the cyclohexylidene and cycloheptylidene derivatives showed strong and broad-spectrum anticancer activity. In comparison to 5-FU, the benzoylhydrazinecarbonyl derivatives were more toxic to HCT-116, Caco-2, and HepG-2 cells while being marginally less effective against HT-29. The derivative containing the hydrazineylidene ester group and the pyrazole derivative both displayed strong and versatile antiproliferative properties. However, the activity of the derivative with the 3,5-disubstituted pyrazole moiety was slightly lower.80
New Indomethacin derivatives have been developed. Cytotoxicity evaluation utilizing the MTT assay was conducted after adopting molecular docking of derivatives to estimate their inhibitory effect on platelet-derived growth factor-α (PDGFR-α). The Indomethacin derivative 13 (Fig. 14) was chosen for the in vivo investigation because it had the greatest inhibitory activity for PDGFR-α in the docking study and had a good cytotoxic effect on the HepG2 cell line (IC50 = 2.13 μM). Treatment with derivative 13 induced a statistically significant decrease in body weight gain, the number of nodules, and the liver weight to body weight ratio. Compound 13 had a hepatoprotective effect on tumor-specific indicators such as α-fetoprotein, carcinoembryonic antigen, and PDGF levels. Treatments were observed to lower liver indicators such as ALT, ALP, AST, LDH, and total bilirubin levels. Additionally, the protective effect of derivative 13 was seen during a histological analysis. The immune-histochemical study conducted after treatment revealed elevated P53 expression as well as compound 13's upregulation of the VEGF gene.81
 | ||
| Fig. 14 The structure of Indomethacin derivative 13. | ||
As possible photosensitizers (PSs) for the non-invasive photodynamic treatment (PDT) of cancer, porphyrins have drawn a lot of attention. The design of the Indomethacin–porphyrin and –chlorin conjugates (P2-Ind and C2-Ind) (Fig. 15) was reported. Both conjugates were tested for in vitro cellular toxicity and singlet oxygen production capability. The 1,3-diphenylisobenzofuran singlet oxygen trap method was used to evaluate the singlet oxygen-generating properties, and the results demonstrated that C2-Ind is the best singlet oxygen photosensitizer.
 | ||
| Fig. 15 The structure of Indomethacin–porphyrin and –chlorin conjugates (P2-Ind and C2-Ind). | ||
Additionally, it was discovered that Indomethacin did not affect the formation of singlet oxygen by either chlorin or porphyrin. Porphyrin– and chlorin–Indomethacin conjugates had similar dark cytotoxicity, according to investigations of the conjugate's cytotoxicity in human HEp2 cells, however, chlorin C2 was found to be the most phototoxic. Chlorin C2 demonstrated a more widespread localization in HEp2 cells after 24 hours, although having a lower cellular absorption than C2-Ind, which could only be seen as tiny aggregates. DFT calculations were carried out to examine the relative stability of different isomers in solution and to offer light on the reaction energy involved in the production of the Indomethacin conjugates. Additionally, their suitability as photosensitizers to produce singlet oxygen for PDT was confirmed by the predicted energy of their initial excited triplet state structures.82
A number of novel Indomethacin derivatives were synthesized through the Pd(II)-catalyzed synthesis of substituted N-benzoylindole. Castration-resistant prostate cancer (CRPC) was used to assess the anticancer activity of these new Indomethacin derivatives. These compounds were demonstrated to be highly effective against CRPC tumor growth in vitro using CCK-8 cell viability and colony formation assays. The most effective derivative against CRPC cell survival was 14 (Fig. 16), with an IC50 value of 1.088 μM. It caused increased protein levels of cleaved Caspase-7 and PARP1 and upregulated Caspase-3/7 activity. Cell cycle arrest at the G2/M phase and a decline in the proportion of cells in the G1 and S phases were the outcomes.
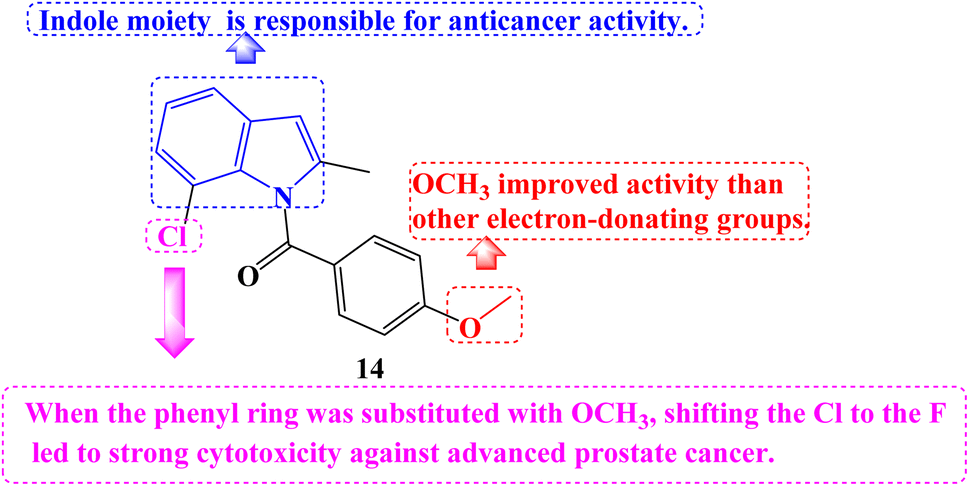 | ||
| Fig. 16 The structure of Indomethacin derivative 14. | ||
By degrading the androgen receptor (AR) and its variants, compound 14 dramatically reduced the expression of the gene networks that the AR targets. It consistently greatly reduced tumor growth in vivo in xenograft and PDX models based on CRPC cell lines. According to SARs studies, the indole moiety at the compound's core is responsible for the anticancer properties of Indomethacin and its derivatives. Among the derivatives with the identical indole core substitution, those with the phenyl ring substituted with OCH3 had higher activity than derivatives with different electron-donating groups. On the other hand, when the phenyl ring was substituted with OCH3, shifting the Cl to the F atom in the indole core led to strong cytotoxicity against advanced prostate cancer.83
Twenty-five new carboxylic acid, methyl ester, methyl amide, and cyano NSAID derivatives containing selenium in the chemical form of selenoester were described. When compared to the parent NSAID scaffolds (Aspirin, Salicylic acid, Naproxen, Indomethacin, and Ketoprofen), twenty Se-NSAID analogs showed a higher cytotoxic potency. The top five analogs were also submitted to the DTP program of the NCI's panel of 60 cancer cell lines to further investigate their cytotoxicity in a larger panel of cancer cells. With IC50 values below 10 μM against two breast cancer cells (T-47D and MDA-MB-231) and/or two lung (H1299 and A549) cancer cells and a selectivity index more than 5 in breast cancer cells, compounds 15a and 15b (Fig. 17) stood out.
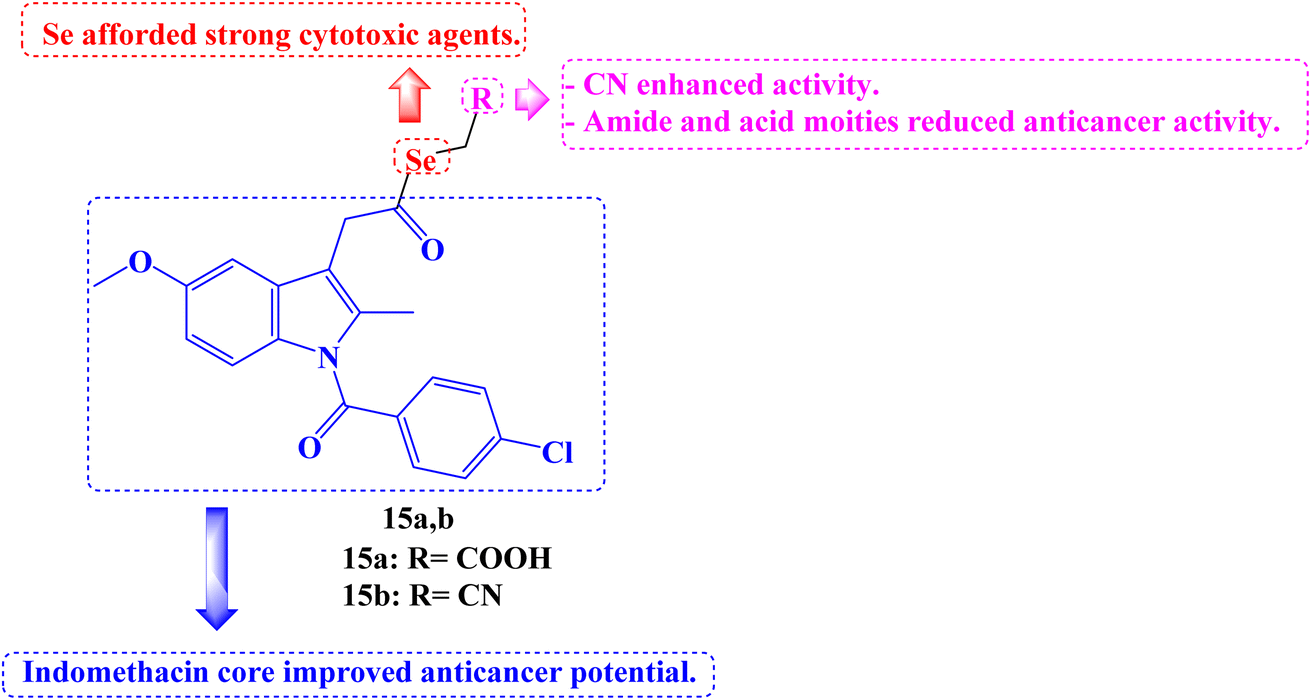 | ||
| Fig. 17 The structure of Se-Indomethacin analogs 15a,b. | ||
Surprisingly, it was discovered that analog 15b induced apoptosis, which inhibited cell development, particularly in two breast cancer cell lines, and that it is metabolized to release both the parent NSAID and the Se fragment. The following SARs can be inferred. The incorporation of the Se atom as selenoester into NSAID derivatives is a legitimate method for producing strong cytotoxic agents. Inactive derivatives were produced when an amide moiety was present in the selenoesters generated from carboxylic acids. In every cancer cell line examined, the Se-NSAID compounds that contained a nitrile moiety with either one or two methylene groups demonstrated a potent suppression of cell proliferation and even cell death at 10 μM. Exhibiting cell growth below 20% in the four tested lines, compounds containing an ester group linked to the selenoester displayed the best activity among the carboxylic derivatives. Less active molecules were produced because of the acid group's presence. The most active and selective NSAID derivatives were produced by Indomethacin.84
Due to the role that mitochondrion plays in the growth and development of cancer; it has become a crucial therapeutic target for anticancer strategies. For mitochondrial localization, a small molecule library of Indomethacin derivatives with triarylphosphonium moiety 16a–d (Fig. 18) was developed. When compared to Indomethacin, compound 16a–d showed significantly better MCF-7 cell-killing ability, with compound 16a having the lowest IC50 value of 1 μM.
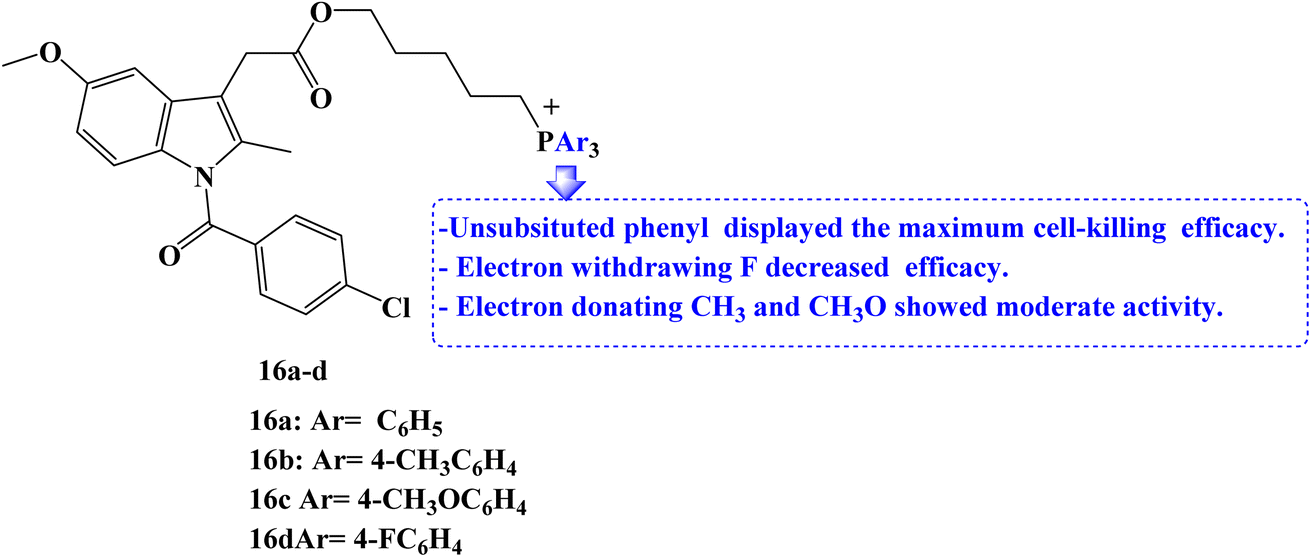 | ||
| Fig. 18 The structure of triarylphosphonium–Indomethacin derivatives 16a–d. | ||
To examine the effects of the NSAIDs derivatives on non-cancerous cells, RPE-1 human retinal epithelial cells were treated with the library members in a dose-dependent manner for 24 hours. Cell viability was then determined using the MTT assay. All of the library's members displayed high IC50 values with minimal cell death. The results showed that NSAID derivatives accelerated the death of cancer cells relative to non-cancerous cells. The ability of two library members to cause MCF-7 breast cancer cells to undergo exceptional apoptosis through the induction of ROS and mitochondrial damage through outer membrane permeabilization (MOMP) has been determined.
Western blot analysis was used to assess the expression of the apoptosis markers executioner caspase-3 and the anti-apoptotic protein BCL-2 to better understand the apoptosis mechanism. According to the western blot images, the expression of BCL-2 was reduced, and the expression of caspase-3 was increased as expected compared to the control cells as well as Indomethacin-treated cells. It was interesting to note that the cell-killing efficacy of the electron-withdrawing p-fluorophenyl-phosphonium derivatives was the lowest. On the other hand, triphenylphosphonium compounds displayed maximum efficacy, whereas p-methyl and p-methoxyphenyl-phosphonium derivatives had only moderate activity.85
There has been a lot of interest in photodynamic therapy, in which harmless photosensitizers can be photoactivated to produce cytotoxic ROS. However, the therapeutic effectiveness is constrained by the potent aggregation and poor tumor targeting of photosensitizers, as well as the limited lifetime and action radius of ROS. In order to address these three issues sequentially, Indomethacin, a COX-2 inhibitor, was introduced to conjugate with zinc phthalocyanines.
The proposed photosensitizer 17 (Fig. 19) could attach to COX-2 with reduced aggregation due to the Indomethacin moiety, and docking calculations and fluorescence enhancement were used to show how this binding occurred. The Indomethacin moiety thus helped compound 17 target only COX-2-expressing tumor cells and then accumulate in the Golgi apparatus. Results showed that compound 17 had improved intracellular ROS production and boosted anticancer effectiveness. Overall, such a new photosensitizer is a viable candidate for enhancing treatment efficacy due to its “three-in-one” COX-2 driven dual targeting and aggregation inhibition.86
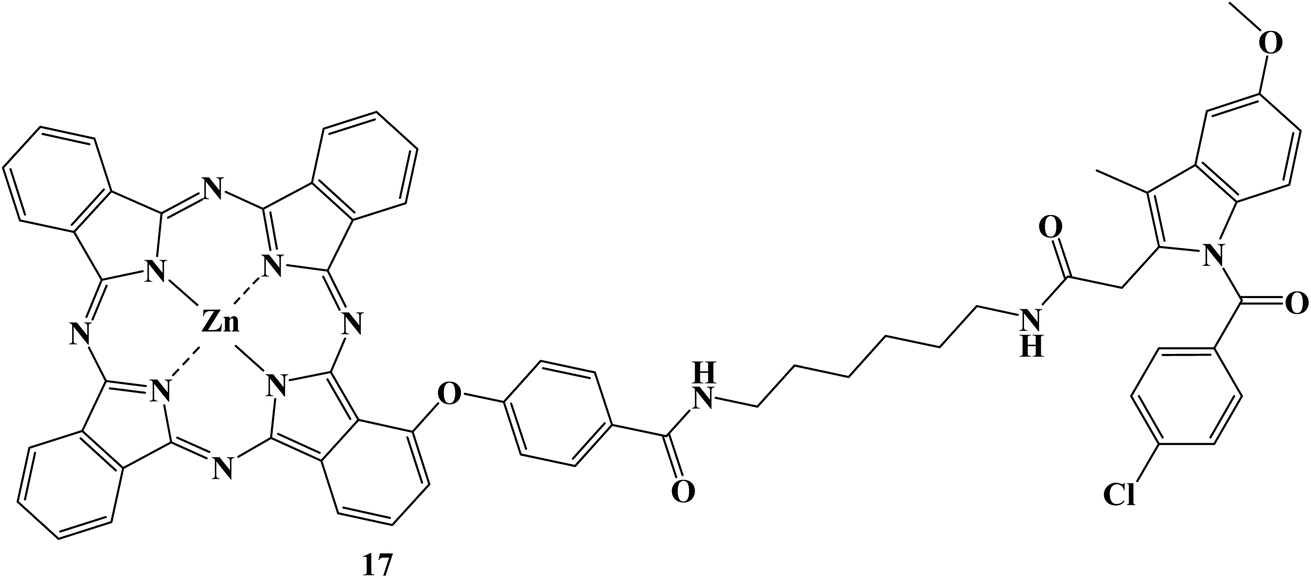 | ||
| Fig. 19 The structure of Indomethacin derivative 17. | ||
In 2023, several new indole compounds were designed, synthesized, and evaluated for their ability to bind to BCL-2 family proteins and to inhibit the proliferation of three different cancer cell lines (PC-3, Jurkat, and MDA-MB-231). Compounds 18a–c (Fig. 20) showed potential inhibitory effects against BCL-2 protein with IC50 values of 7.63, 29.6, and 34.3 μM, respectively. Additionally, they demonstrated strong binding affinity for the MCL-1 protein with IC50 values of 1.53, 15.3, and 37.1 μM, respectively. It was interesting to note that none of these three derivatives interacted with BCL-XL protein while exclusively binding with BCL-2 protein.
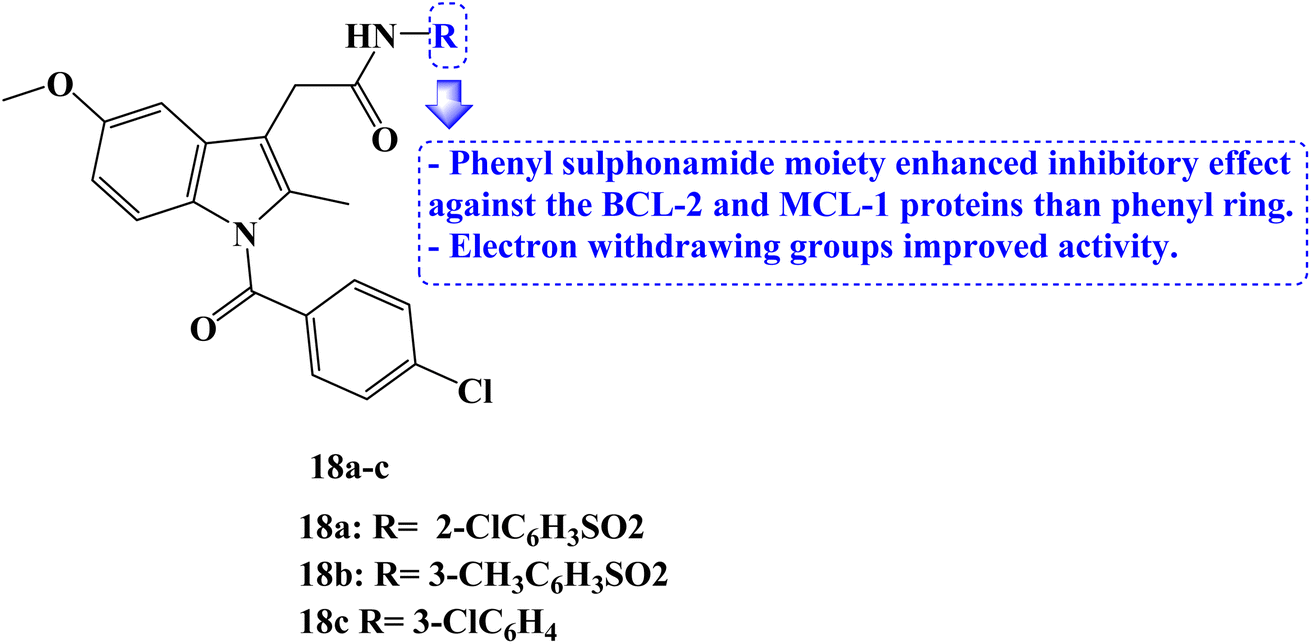 | ||
| Fig. 20 The structure of Indomethacin derivatives 18a–c. | ||
The compounds' inhibitory effect against the BCL-2 and MCL-1 proteins was favored by the electron-withdrawing group. The CCK8 test and AT-101 as the positive control were then used to evaluate the anticancer activity of the compounds 18a–c. With an IC50 value of 39 μM, compound 18c demonstrated the strongest antitumor efficacy against Jurkat cells. It could bind to the active pocket of the BCL-2 and MCL-1 proteins through hydrogen bonds and van der Waals forces, according to docking studies.87
In 2024, the anticancer activity of the synthesized Indomethacin derivative 19 (Fig. 21) was evaluated against colon HCT-116, HT-29, and pancreatic BxPC-3 cancer cell lines besides the normal cell line, MRC-5. It showed significant anticancer potential with IC50 values of 4.97, 12.78, and 9.78 μM, against HCT-116, HT-29, and BxPC-3 cells, respectively. A wound-healing assay employing the SW620 cell line was used to evaluate the anti-migratory properties of Indomethacin derivative 19. It exhibited a potent anti-migratory effect; its relative wound closure was 3.20% after 24 hours and 5.08 percent after 48 hours.88
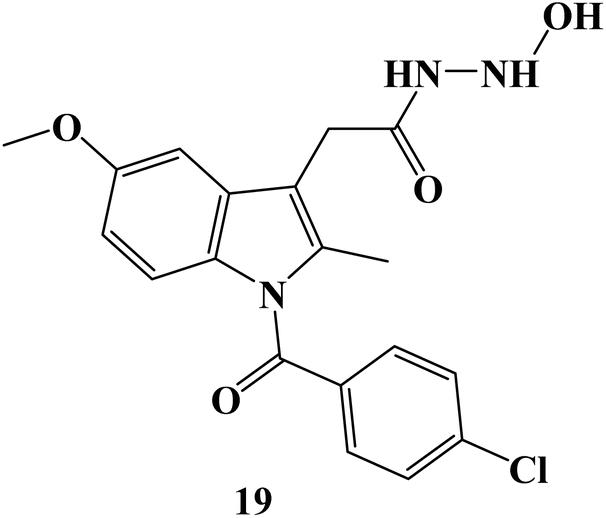 | ||
| Fig. 21 The structure of Indomethacin derivative 19. | ||
7. Anticancer activity of Naproxen derivatives
A total of four copper(II) complexes, 20a–d (Fig. 22), were developed, each comprising the NSAID Naproxen and regioisomeric vanillin Schiff base derivatives. In cell-free systems, every complex successfully breaks down DNA, with 20d having the highest nuclease activity. According to DNA binding, the derivative 20d attached to DNA in the grooves before causing oxidative DNA cleavage (1.3 ± 0.3 × 105 M−1). At micromolar concentrations, three of the complexes (20a, 20c, and 20d) killed cancer stem cell (CSC)-enriched cells (HMLER-shEcad) and bulk cancer cells (HMLER). The most efficient compound, 20d, had similar effects to salinomycin, a known CSC-potent substance, in that it decreased mammosphere production and size (38% decrease after 5 days of therapy at IC20 value). Micromolar toxicity of complex 20d was shown to kill both bulk breast cancer cells and breast CSCs (IC50 values of 37.6 and 36.0 μM, respectively).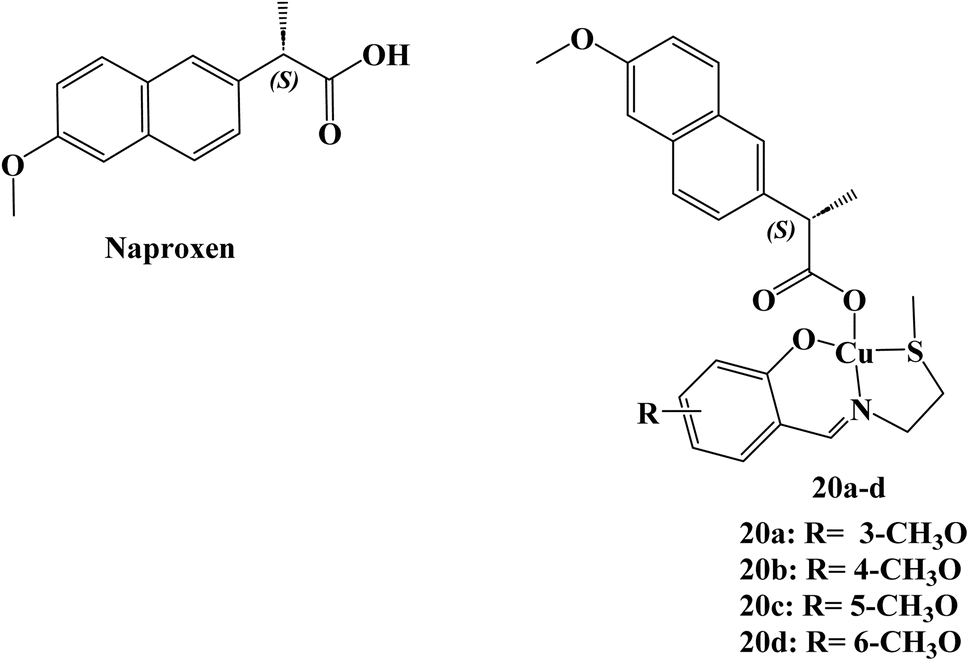 | ||
| Fig. 22 The structure of copper(II)–Naproxen complexes 20a–d. | ||
Studies on the mechanics of 20d's action revealed that it rapidly entered CSCs, increased intracellular ROS levels, damaged DNA, and triggered caspase-dependent death. Furthermore, 20d prevented the expression of COX-2 and resulted in CSC mortality that is dependent on COX-2. Derivative 20d could eradicate whole tumor populations (bulk cancer cells and CSCs) with a single dose, which makes it superior to treatments that are selective for bulk cancer cells or CSCs.89
The design and synthesis of five Naproxen derivatives, 21a–e (Fig. 23), were reported. The impact of electron-withdrawing groups (NO2 and Br) and electron-donating groups (CH3 and OCH3) on SARs, quantitative structure–activity relationships (QSAR), and frontier molecular orbitals (FMOs), global reactivity descriptors, and molecular electrostatic potentials (MEP) has been thoroughly investigated.
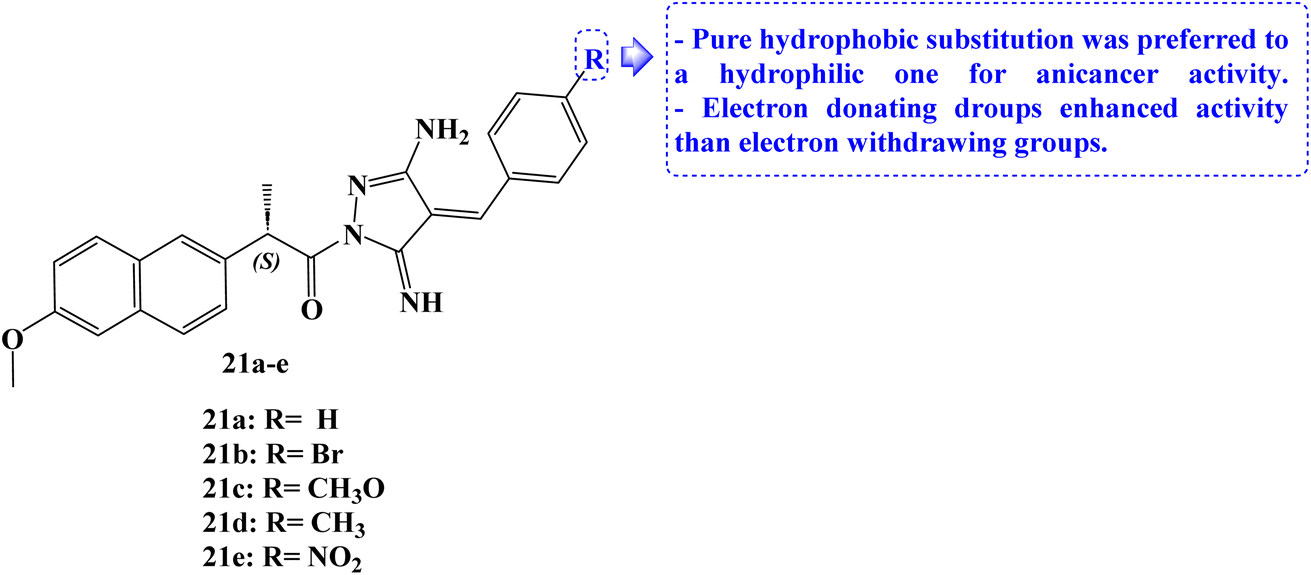 | ||
| Fig. 23 The structure Naproxen derivatives 21a–e. | ||
According to molecular docking studies, a pure hydrophobic substitution at position 4 of the aldehyde part is preferred to a hydrophilic one. With an IC50 value of 1.49 μM, compound 21c demonstrated potent antiproliferative activity against MCF-7 cells. Compounds 21d and 21e had moderate and weak antiproliferative activity, respectively, against the same cell line, with IC50 values of 17.64 and 23.28 μM. The docking studies showed that compound 21c's regular alkane chain was enhancing its biological activity, supporting its endorsement to bury well in the active site and enhancing hydrophobic interactions. In comparison to Naproxen, newly synthesized compounds demonstrated greater antiproliferative efficacy against the evaluated cell lines.90
New non-carboxylic analogs of Naproxen were developed, including the oxadiazoles 22a–c and 23a–c, cycloalkanes 24a–d, cyclic imides 25a–c, and triazoles 26–28 and 29a–c (Fig. 24). Additionally, the target compounds' cyclooxygenase isozymes (COX-1/COX-2) inhibition assay and in vitro anticancer efficacy were investigated. With an IC50 range of 4.83–14.49 μM, the antitumor activity assay results showed that compounds 24b, 23c, 29b, and 29c had the strongest antitumor effects against the tested cell lines MCF-7, MDA-231, HeLa, and HCT-116. Doxorubicin, afatinib, and celecoxib, the reference drugs, showed IC50 values of 3.18–26.79, 6.20–11.40, and 22.79–42.74 μM, respectively.
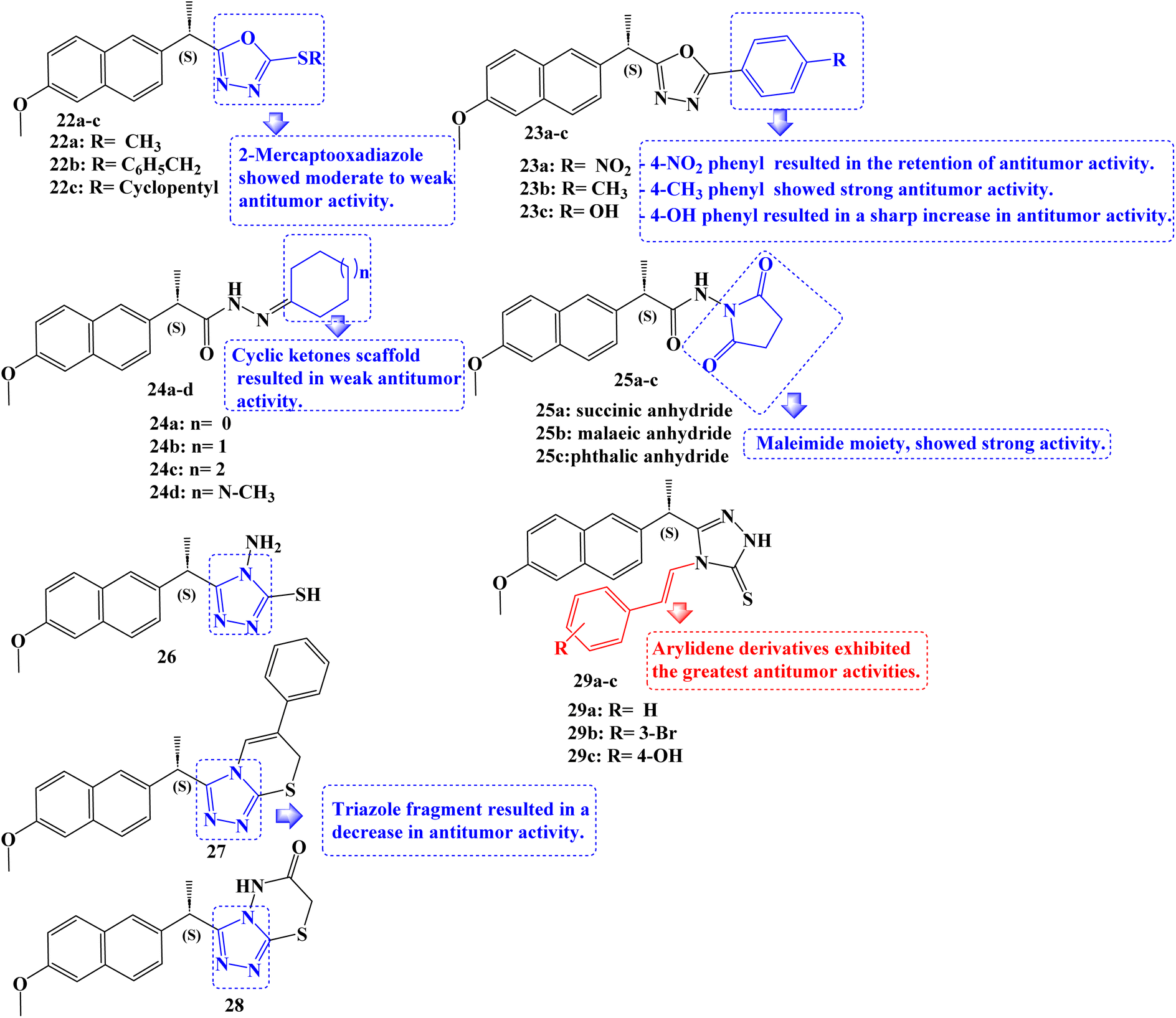 | ||
| Fig. 24 The structure of Naproxen derivatives 22–29. | ||
Additionally, using celecoxib as a reference standard (IC50 = 0.11 μM; COX-2 SI: >227.20), in vitro COX-1/COX-2 inhibition tests revealed that the compounds 24b, 23c, 29b, and 29c exhibited effective COX-2 inhibition, with IC50 values of 0.40–1.20 μM and selectivity index (SI) values of >62.50–20.83. The COX-2 binding site was docked with compounds 24b and 23c, which were powerful COX-2 inhibitors. Here, these compounds displayed substantial interactions.
The SARs for antitumor activities revealed, in accordance with these findings, that the Naproxen scaffold containing 2-mercapto oxadiazole derivatives displayed moderate to weak antitumor activity against the examined cell lines (IC50 values of roughly 23.19 to >100 μM), and that replacement of the mercapto moiety with a 4-nitrophenyl fragment at the same position resulted in the retention of antitumor activity (IC50 values of roughly 35.1 to >100 μM) against the five cell lines studied. The addition of a 4-methylphenyl moiety to the oxadiazole ring revealed strong anticancer activity against the cell lines MDA-231 and HeLa with IC50 values of 16.16 and 19.45 μM, respectively. Replacing the 4-nitrophenyl moiety with a 4-hydroxyphenyl fragment at the same position significantly increased the antitumor activity against all of the tested cell lines, including MCF-7, MDA-231, HeLa, HCT-116, and Caco-2, with IC50 values of 5.89, 7.29, 6.15, 10.50, and 33.94 μM, respectively.
Additionally, the Naproxen scaffold including certain cyclic ketones was shown to have mild anticancer activity as evidenced by their IC50 values, which ranged from about 51.57 to >100 μM. With IC50 values of 12.11, 9.65, 13.00, 14.49, and 18.81 μM against MCF-7, MDA-231, HeLa, HCT-116, and Caco-2 cancer cell lines, respectively, similar derivatives replaced with a cyclohexanone fragment, demonstrated a marked increase in antitumor activity. Only compound 25b, which incorporated a maleimide moiety in the Naproxen scaffold's terminal fragment and had IC50 values of 11.08 and 15.12 μM, demonstrated potent activity against the MDA-231 and HeLa cancer cell lines. Except for the HeLa cell line, which displayed an IC50 value of 20.71 μM with derivative 26, the antitumor activity against all of the examined cancer cell lines decreased as a result of the replacement of the oxadiazole ring system with a triazole fragment. Surprisingly, among the developed compounds, arylidene derivatives showed the strongest anticancer activity. The MCF-7, MDA-231, HeLa, and HCT-116 cancer cell lines were particularly sensitive to compound 29c's anticancer effects (IC50 values ranged from 4.83 to 9.03 μM).91
Han et al.92 developed a new series of 1,2,4-triazoles from (S)-Naproxen 30a–f (Fig. 25). The human methionine aminopeptidase 2 (MetAP2) was used to study the molecular modelling of these derivatives. Using the 3(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2(4-sulfophenyl)-2H-tetrazolium colorimetric technique, all synthesized compounds were tested for anticancer activity against three prostate cancer cell lines (PC-3, DU145, and LNCaP). With IC50 values of 26.0, 34.5, and 48.8 μM for the PC-3, DU145, and LNCaP cancer cell lines, respectively, compound 30a demonstrated the best activity. The IC50 values for compounds 30b, 30c, and 30d against the cancer cell lines PC-3 and DU-145 were 43.0, 36.5, 29.3 μM and 49.8, 49.1, 31.6 μM, respectively.
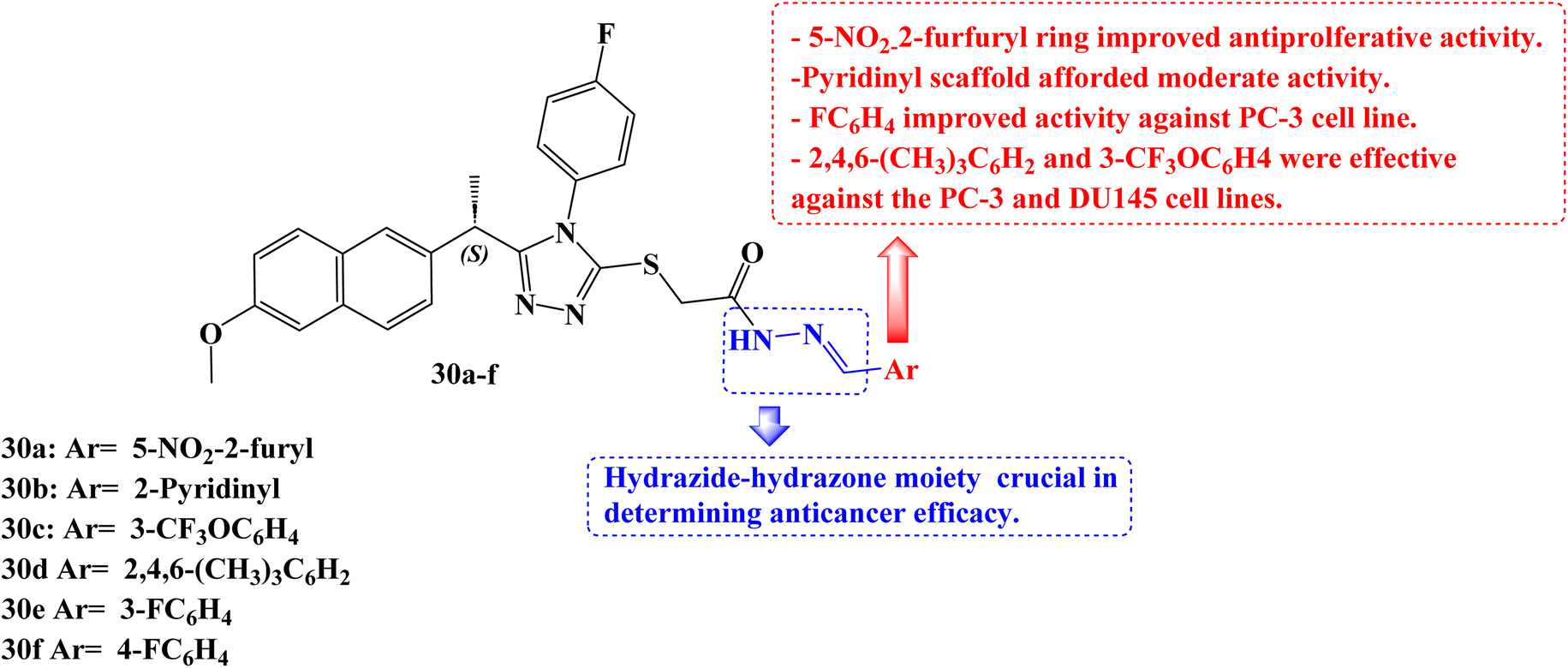 | ||
| Fig. 25 The structure Naproxen derivatives 30a–f. | ||
To evaluate the biodistribution of IRDye800 in mice, either 100 μM of free dye or compound 30a labelled with dye was administered intravenously into the mice's tails. The in vivo imaging system spectrum equipment was used to capture images 60, 120, 180, 240, 300, and 360 minutes after injection. After 360 minutes, in vivo, tests were conducted to see where the dye had accumulated in the urogenital system. Compound 30a might have promise for the treatment of prostate cancer, according to in vivo experiments that revealed a larger concentration of the compound in the prostate than the free dye.
It was anticipated that these derivatives would significantly reduce MetAP2 activity at nanomolar concentrations. The maximum inhibitory activity against the enzyme was determined to be at a concentration of compound 30a (inhibition constant, Ki = 24.76 nM), which is consistent with experimental findings.
To clarify the role of Akt and phosphoinositide 3 kinase (PI3K) in the PC-3 cell line's response to derivative 30a, at a concentration of 40 μM, 30a was exposed to PC-3 cells for 40, 60, 90, and 120 minutes. The Naproxen derivative had a notable impact on PI3K and Akt phosphorylation as well as a considerable reduction in the phosphorylation of the EGFR. Bax, BCL-2, caspase-3, and caspase-9 messenger RNA (mRNA) expression study was carried out after PC-3 cells were exposed to 30a (40 μM) for 0, 6, and 12 hours. In the 30a treated PC-3 cells compared to the control, there was a time-dependent increase in the expression levels of Bax, caspase-3, and caspase-9, according to a real-time polymerase chain reaction (RT-PCR) study. In addition to these findings, BCL-2 mRNA levels in 30a-treated PC-3 cells were considerably lower. All of these findings demonstrated that compound 30a significantly increased apoptotic markers while decreasing antiapoptotic markers.
Regarding cytotoxic activity in this class of compounds, the presence of the 5-nitro-2-furfuryl ring is necessary. The activity of the 2-pyridinyl, 3-pyridinyl, and 4-pyridinyl substituents were modest. Only the PC-3 cell line showed good activity against the 3- and 4-fluorophenyl rings. Both the 2,4,6-trimethylphenyl and 3-trifluoromethoxyphenyl rings were effective against the PC-3 and DU145 cell lines. For many of the compounds, the activity was significantly improved when the hydrazide scaffolds took the place of all substituents in comparison to the equivalent analogs. According to this finding, the hydrazide-hydrazone moiety may be crucial in determining anticancer efficacy.
Tabares et al.93 reported the development of novel Ru(II) organometallics and evaluated their anticancer potential. Organometallic 31 (Fig. 26) was found to decrease cell growth in both NCI-H460 and A549 lung cancer cell lines (IC50 values of 161 and 145.3 μM against A549 and NCI-H460 cell lines, respectively). It was determined that the structural alteration of Naproxen via the addition of the pyridine amide moiety was essential for the anticancer potential. The mitochondrial membrane potential (MMP) was not significantly affected by compound 31 at all.
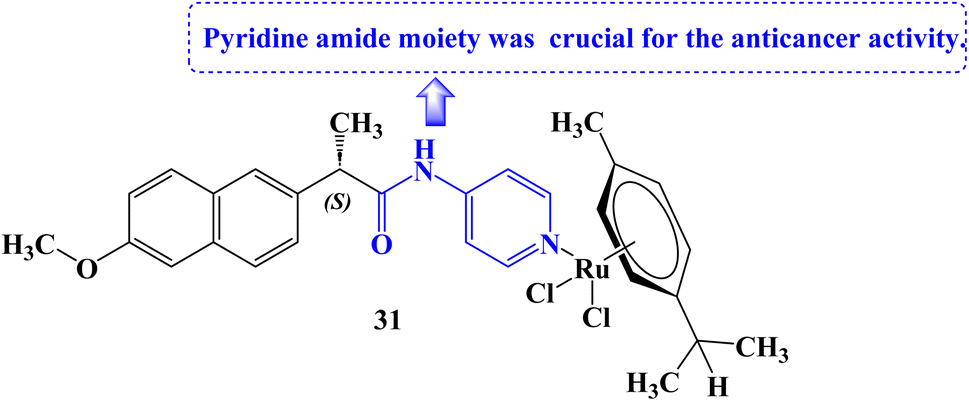 | ||
| Fig. 26 The structure Naproxen organometallic derivative 31. | ||
The development of novel anti-inflammatory medications known as H2S-releasing non-steroidal anti-inflammatory drugs (H2S-NSAIDs), showing remarkable promise for chemoprevention in malignancies, had taken advantage of the positive effects of H2S-release and COXs-inhibition. Naproxen-4-hydroxybenzodithioate (Naproxen-HBTA) 32 (Fig. 27), a novel Naproxen derivative that releases H2S, was tested by Ercolano et al.94 to see how well it reduced the characteristics of metastatic melanoma both in vitro and in vivo.
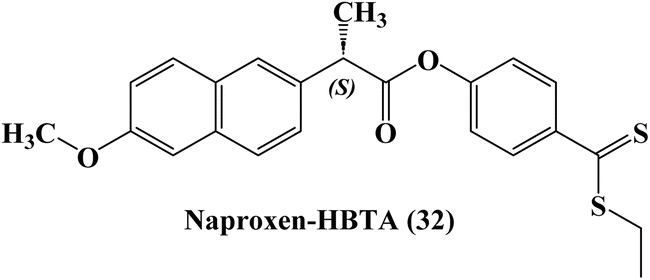 | ||
| Fig. 27 The structure of Naproxen-HBTA (32). | ||
In a cutaneous melanoma model, the impact of Naproxen-HBTA on a variety of metastatic features of human melanoma cells, including proliferation, migration, invasion, and colony formation, was investigated both in vitro and in vivo. Studies in cell culture showed that Naproxen-HBTA decreased motility, invasiveness, and focus formation while inducing caspase-3-mediated death. Finally, daily oral administration of Naproxen-HBTA dramatically slowed the growth and spread of melanoma in mice. With this dual strategy, it was concluded that the COX-2 and H2S pathways could be viewed as novel therapeutic targets to develop new “combination therapy”-based treatment options for melanoma.
Compounds 33a–g (Fig. 28) which are Naproxen triazole-thioether hybrid molecules were developed. Using the MTT assay method, they were assessed against the PC-3, DU-145, and LNCaP cell lines of androgen-dependent and androgen-independent prostate cancer. The anticancer activity of compounds 33a, 33b, 33c, and 33d was 14.2, 5.8, 10.8, and 8.4 μM against PC-3 cell lines, respectively. Compounds 33d, 33e, and 33g showed anticancer activity against DU-145 cell lines 18.8, 12.25, and 10.2 μM, and Naproxen hybrids 33e, 33f and 33g demonstrated anticancer activity against LNCaP cell lines (IC50 values of 12.25, 22.76 and 2.21 μM, respectively). The most potent activity against androgen-dependent and independent prostate cancer cell lines was demonstrated by compounds 33d and 33g, suggesting that these compounds might be effective small molecules against prostate cancer.
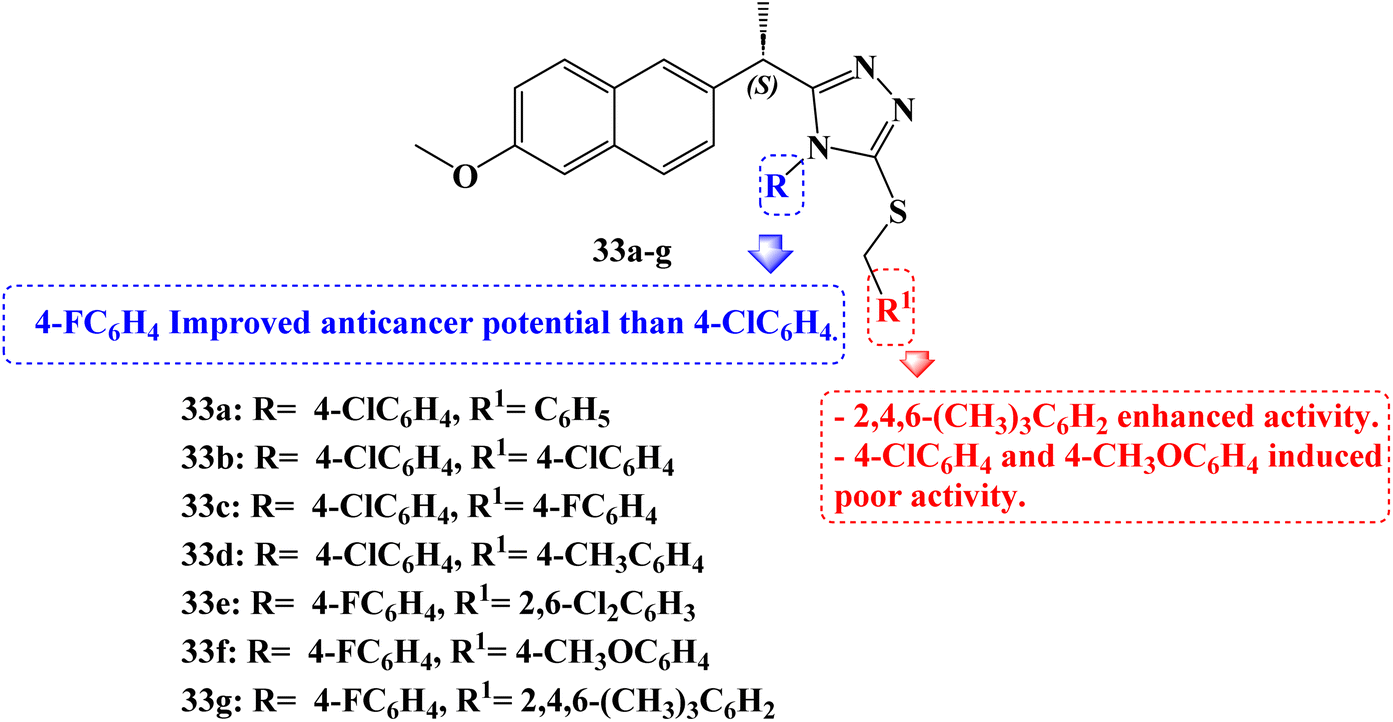 | ||
| Fig. 28 The structure of Naproxen triazole–thioether hybrids 33a–g. | ||
Furthermore, using the western blot method, it was determined whether hybrid 33g activated the androgen receptor, the protein kinase B (AKT) pathway, and the mitogen-activated protein kinase (MAPK) pathway in LNCaP cells. Real-time PCR analysis was used to evaluate the effect of compound 33g on mRNA expression analyses of the Bax, BCL-2, caspase-3, and caspase-9 genes. Real-time PCR analysis revealed a time-dependent increase in proapoptotic Bax, caspase-3, and caspase-9 expression in the compound 33g treated LNCaP cells compared to the control. In addition to these findings, compound 33g treatment of LNCaP resulted in a considerable drop in anti-apoptotic BCL-2 mRNA levels.
All of these findings demonstrated that treatment with compound 33g significantly increased apoptotic indicators while significantly lowering anti-apoptotic markers. Comparatively, to the control group, nude male mice with cancer received compound 33g. In contrast to the control group, compound 33g was observed to reverse the malignant phenotype in the nude male mouse models of prostate cancer. When compared to the control, an analysis of some blood parameters in the study revealed that they were within normal ranges. Animals treated in accordance with the control group's blood values likewise showed compliance with the blood limit values.
Its potential action was explained by molecular docking and dynamics simulation of compound 33g binding to MetAP2 enzyme. Additionally, the MetAP2 enzyme inhibition assay of compound 33g was assessed. Within the investigated dosage (12.50–200 μM), compound 33g could inhibit the enzymatic function by up to 42–77.7%. By raising the concentration of compound 33g, the potency of the inhibition was improved.
Several substituents on the triazole ring or the S-alkyl side chain carried on the triazole scaffold gave several new Naproxen 1,2,4-triazole-thioether hybrid compounds their unique anticancer properties. In nearly all cell lines, 4-fluorophenyl groups exhibit better activity than 4-chlorophenyl groups when substituents on the triazole ring are compared. When substituents on the alkyl side chain carried on a triazole scaffold, were compared, not only the 4-methoxyphenyl group but also the 4-chlorophenyl group exhibited poor activity against the DU-145 and LNCaP cell lines. However, an improved anticancer impact was seen when the 2,4,6-trimethylphenyl group was introduced instead of the 4-methoxyphenyl and 4-chlorophenyl groups. The activity also was diminished when both sides of a substituent were substituted with a substituent containing at least one electronegative atom.95
Several organoselenium compounds were developed, characterized, and tested against four different cancer cell lines: SW480 (human colon adenocarcinoma cells), HeLa (human cervical cancer cells), A549 (human lung carcinoma cells), and MCF-7 (human breast adenocarcinoma cells). These compounds were based on the hybridization of NSAID scaffolds and Se functionalities (–SeCN and –SeCF3). It was interesting to note that the majority of the tested organoselenium compounds demonstrated activity in lowering the viability of several cancer cell lines.
The IC50 values for Naproxen derivative 34a (Fig. 29) against the SW480, HeLa, A549, and MCF-7 cell lines were 12.0, 21.5, 32.3, and 28.3 μM, respectively. The IC50 values for the Naproxen–SeCF3 derivative 34b (Fig. 29) against the SW480, HeLa, A549, and MCF-7 cell lines were 10, 16.4, 28.4, and 19.3 μM, respectively. NSAIDs–SeCF3 compounds had better anticancer activity than their comparable NSAIDs–SeCN derivatives. Perhaps this was because of their increased lipophilicity.96
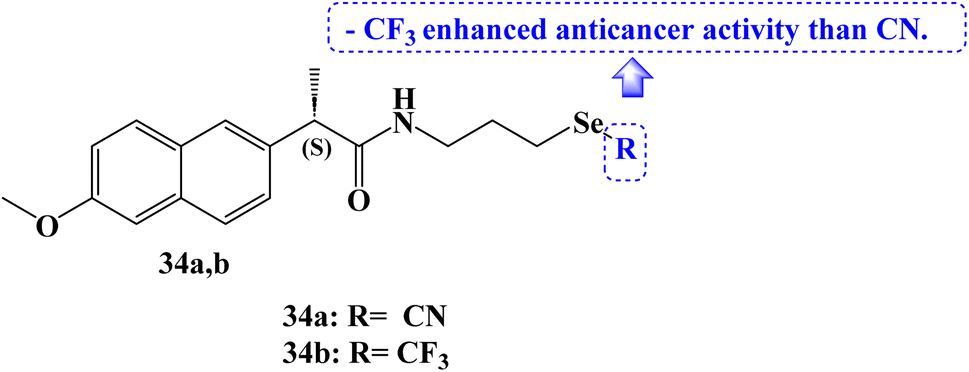 | ||
| Fig. 29 The structure of Naproxen organoselenium compounds 34a,b. | ||
As cytotoxic agents, a number of novel tryptophan-1,2,3-triazole Naproxen derivatives based on peptides were investigated. By using an MTT assay, the compounds' cytotoxicity was evaluated against the human lung carcinoma A549 cancer cell line. In contrast to Naproxen derivative 35 (Fig. 30), which had the lowest IC50 value (9.07 μM), several of these compounds demonstrated potential activity with IC50 values in the 9.07 to 47.1 μM range.97
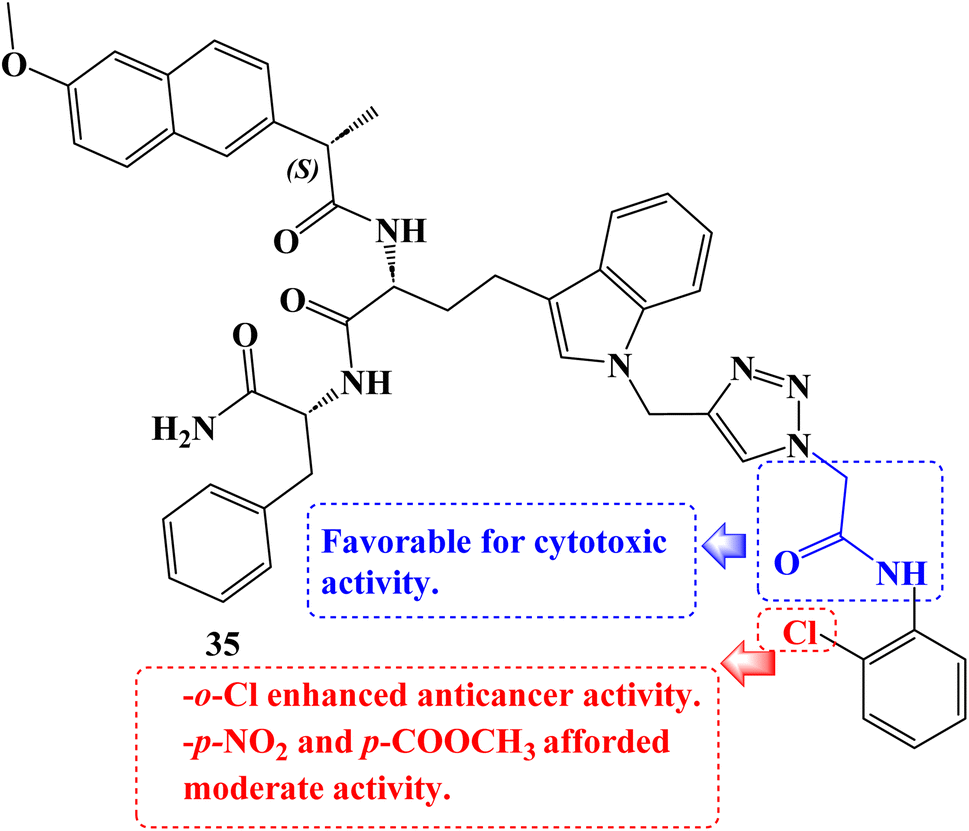 | ||
| Fig. 30 The structure of tryptophan-1,2,3-triazole Naproxen derivative 35. | ||
The anticancer effects of novel selenocyanate and diselenide compounds against the human cancer cell lines Caco2, BGC-823, MCF-7, and PC-3 were identified. It was interesting to note that the majority of the novel compounds were effective at lowering the viability of several cancer cell lines. Naproxen derivative 36 (Fig. 31) showed IC50 values of 15.5, 13.7, 8.3, and 8.3 μM against Caco2, BGC-823, MCF-7, and PC-3 cancer cell lines, respectively at 72 hours.
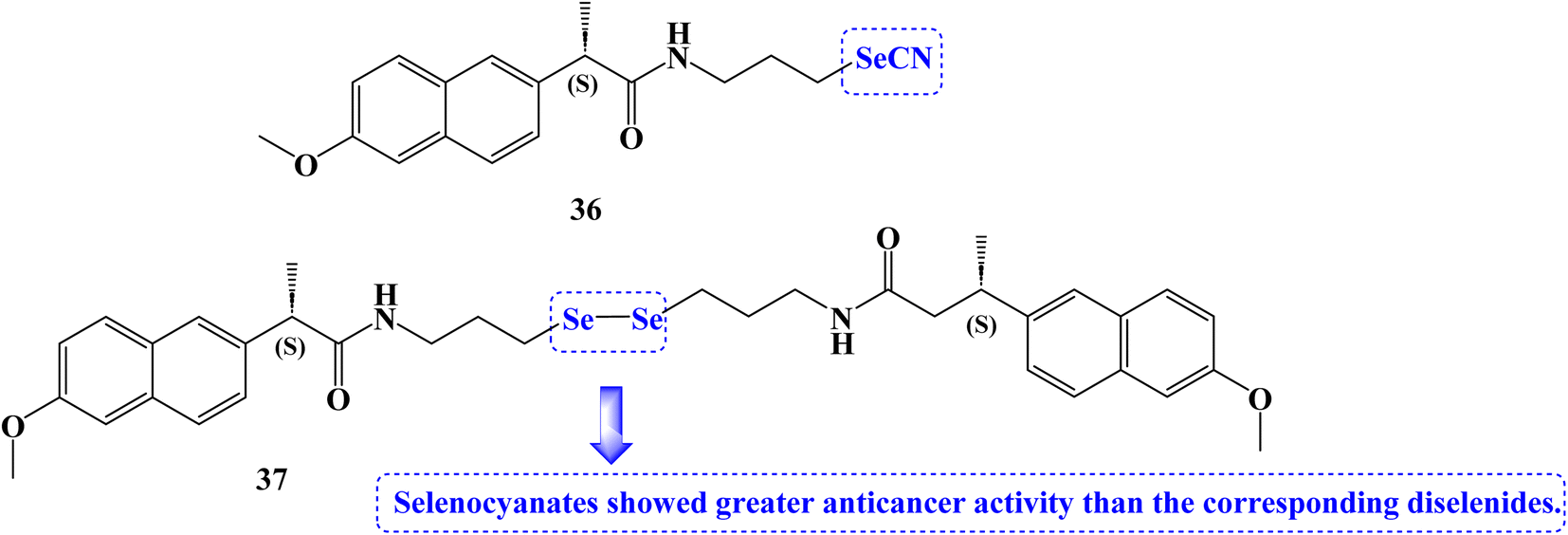 | ||
| Fig. 31 The structure of Naproxen selenocyanate and diselenide compounds 36 and 37. | ||
Compound 37 (Fig. 31) showed IC50 values of 8.3 and 22.4 μM against Caco2 and PC-3 cancer cell lines, respectively at 72 hours. Selenocyanates showed greater anticancer activity than the corresponding diselenides. The reduction in absorbance at 340 nm caused by the oxidation of NADPH to NADP+ served as an indicator of the synthesized compounds' glutathione peroxidase (GPx) activity. The positive control was ebselen. Due to their GPx-like activity that was superior to that of other derivatives, diselenide 37 seemed to be of interest. Compound 36 was up to two times more active than the GPx mimic ebselen in this assay.98
To develop novel anticancer agents, a hybrid of three different structural types-tryptophan, Naproxen, and triazole-has been investigated. When tested against the A549 cancer cell line, analogs with a ((4-chlorophenyl)amino)-2-oxoethyl moiety 38a (Fig. 32) and a ((4-nitrophenyl) amino)-2-oxoethyl moiety 38b (Fig. 32) linked to the triazole ring demonstrated cytotoxic potential (IC50 = 39.35 and 28.52 μg mL−1, respectively).
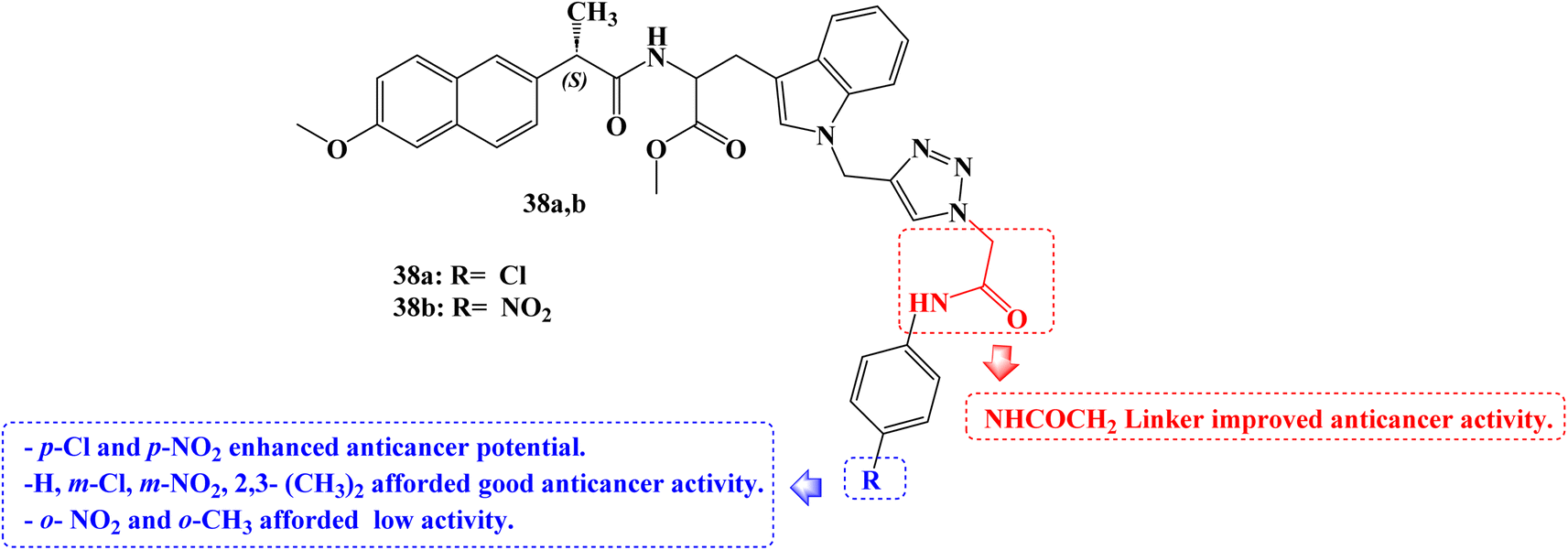 | ||
| Fig. 32 The structure of tryptophan-Naproxen-triazole hybrids 38a,b. | ||
The SARs suggested that the kind and position of the aromatic ring group attached to the triazole moiety through the NHCOCH2 linker directly affect the activities. p-Cl and p-NO2 groups enhanced anticancer potential. Good activity was seen in the m-Cl or m-NO2 groups, however o-NO2 or o-CH3 group reduced the anticancer activity.99
Novel (S)-Naproxen derivatives with hydrazide-hydrazone moiety were designed, synthesized, and their anticancer potential was assessed. All synthesized derivatives were tested for anticancer efficacy against the MDA-MB-231 and MCF-7 cell lines, two different types of human breast cancer. The most effective anticancer action against both cancer cell lines with good selectivity was demonstrated by (S)-2-(6-methoxynaphthalen-2-yl)-N′-{(E)-[2-(trifluoromethoxy)phenyl]methylidene}propanehydrazide (39) (Fig. 33). The IC50 values for this compound were 22.42 and 59.81 μM, respectively.
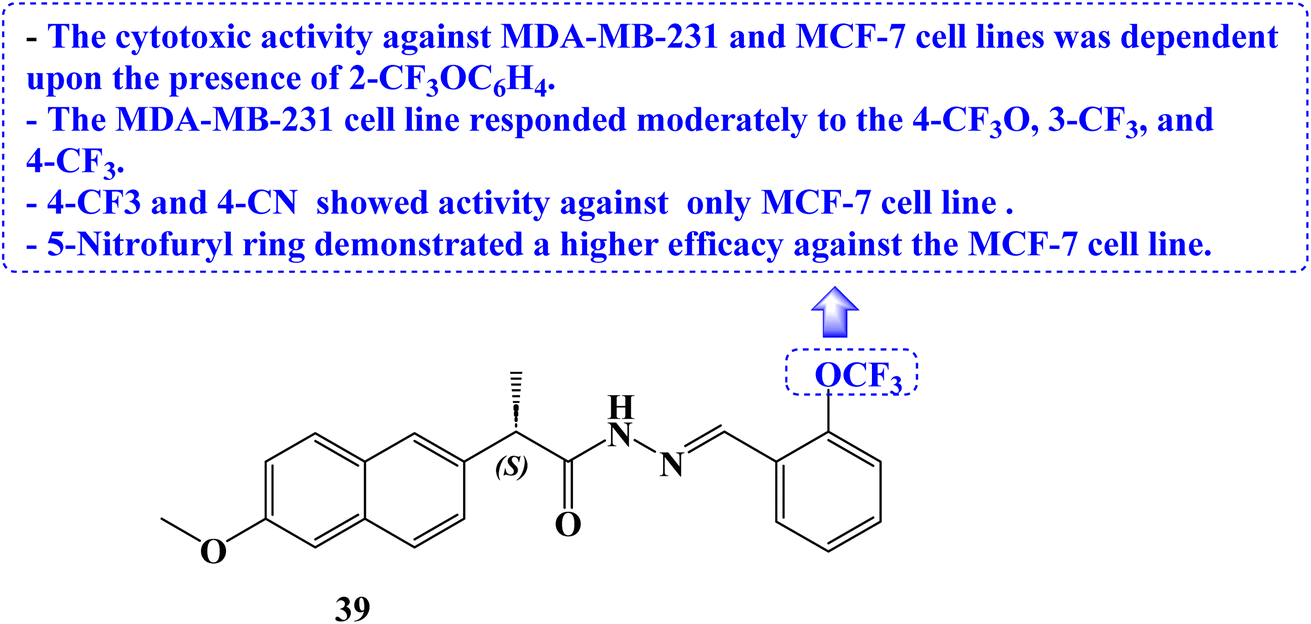 | ||
| Fig. 33 The structure of Naproxen derivative 39. | ||
Additionally, VEGFR-2 was used to study the molecular modelling of these compounds. The interaction between the Naproxen derivatives and the VEGFR-2/VEGF-A complex structure was clear. Western blotting was used to examine how compound 39 treated MDA-MB-231 cells' inhibition of VEGFR-2 and the apoptotic protein BCL-2. In the compound 39-treated group, anti-apoptotic BCL-2 protein expression was down-regulated, and VEGFR-2 protein expression was decreased.
Additionally, DAPI labelling was used in fluorescence microscopy to identify apoptosis. A substantial increase in the S and M phases was found in flow cytometry analysis of cell cycle phases compared to untreated control cells, demonstrating the cancer cell cycle arrest. Treatment with compound 39 at the IC50 concentration caused nuclear condensation and DNA fragmentation in the cells, whereas no such morphological change was seen in cells treated with Naproxen. Compound 39 caused cell death with a variety of morphological changes.
The Ehrlich acid tumor model, a reliable in vivo ectopic breast cancer model in mice, was used to examine compound 39's anticancer properties. In mice, Naproxen derivative 39 displayed anticancer action and reduced tumor volume at low (60 mg kg−1) and high (120 mg kg−1) doses, according to the results. Using two separate healthy epithelial cells, the cytotoxic potential of compound 39 was evaluated. Both the healthy ovarian surface epithelial cell line (OSE) and the embryonic kidney cell line HEK293T were employed. Compound 39's IC50 was 162.2 and 49 μM, respectively. Both IC50 values were higher than the IC50 values of the examined cancer cells, proving that compound 39 is less hazardous to healthy cells.
The cytotoxic activity against the two cell lines was dependent upon the presence of 2-trifluoromethoxyphenyl. The MDA-MB-231 cell line responded moderately to the 4-trifluoromethoxy, 3-trifluoromethyl, and 4-trifluoromethyl substituents. The 5-nitrofuryl ring displayed a better efficacy against the MCF-7 cell line than the 4-trifluoromethyl and 4-cyano substituents, which solely showed action against the MCF-7 cell line.100
New 1,3,4-oxadiazole compounds based on Naproxen have been developed. Utilizing a sulforhodamine assay, the antiproliferative efficacy of the target compounds was evaluated against the MCF-7, HepG2, and HCT-116 cancer cell lines. One of the synthetic derivatives was 2-(4-((5-((S)-1-(2-methoxynaphthalen-6-yl)ethyl)-1,3,4-oxadiazol-2yl-thio) methyl)-1H-1,2,3-triazol-1-yl)phenol (40a) (Fig. 34) with IC50 values of 2.13 and 1.63 μg mL−1 for MCF-7 and HepG2 cancer cells, respectively, was the molecule with the greatest potency against these cancer cells. It was also equivalent to doxorubicin, which had an IC50 value of 1.62 μg mL−1 for HepG2.
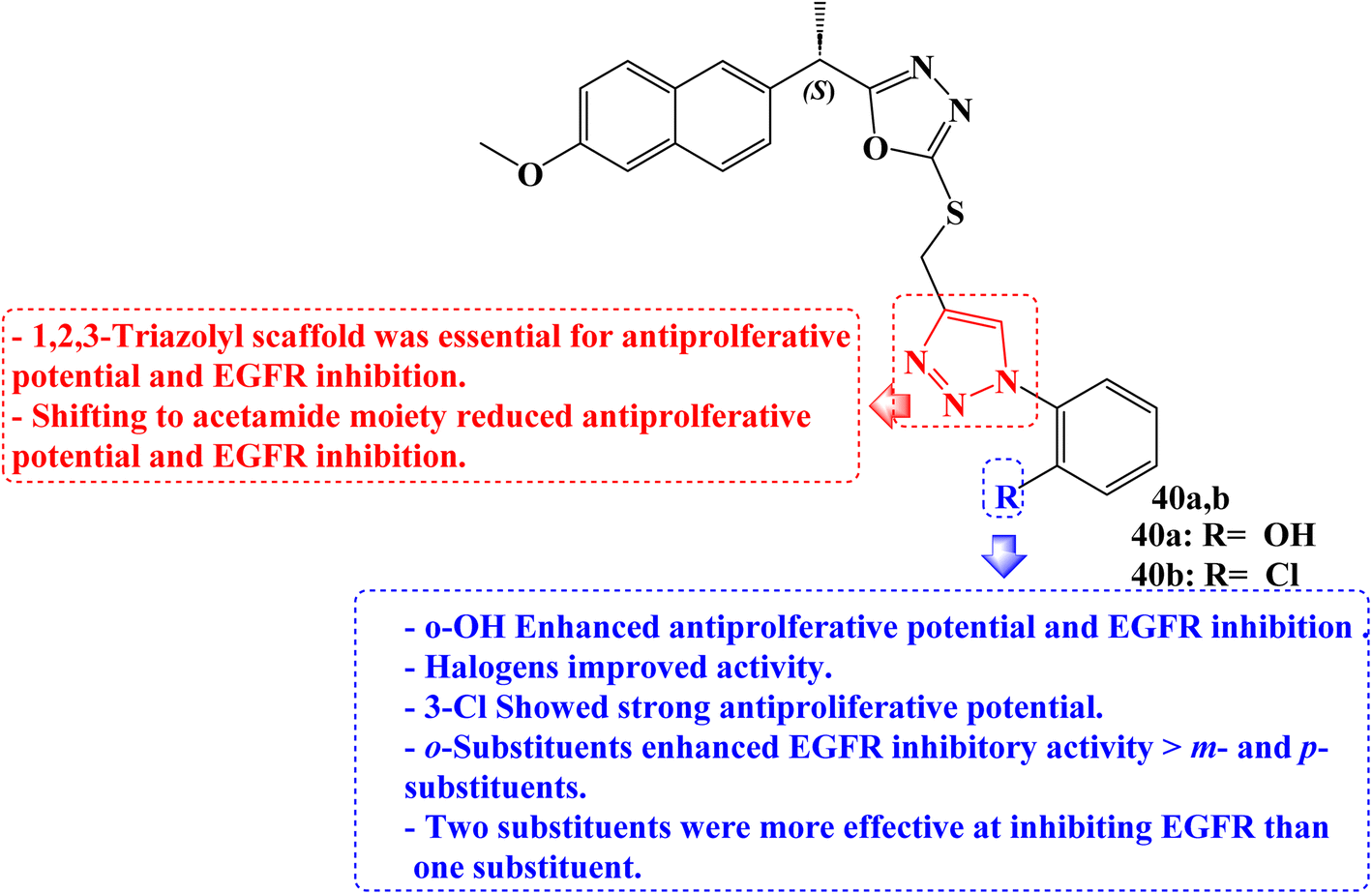 | ||
| Fig. 34 The structure of Naproxen-1,3,4-oxadiazole derivatives 40a,b. | ||
Furthermore, when compared to the reference drug erlotinib (IC50 = 0.30 μM), two compounds, 40a and 40b (Fig. 34), were the most effective at suppressing EGFR kinase with IC50 values of 0.41 μM and 0.67 μM, respectively. The active compound 40a induced a significant percentage of necrosis in the MCF-7, HePG2, and HCT 116 cells.
The biological data was also supported by the docking studies, DFT, and MEP. The most effective hybrid against MCF-7 and HepG2 cancer cells was a Naproxen derivative having an ortho position hydroxyl group on the triazolyl ring. The IC50 for the halogen-substituted 1,2,3-triazolyl analogs ranged from 5.05 to 15.84 μg mL−1 for MCF-7 cells and 3.53 to 25.49 μg mL−1 for HepG2. The anticancer impact of Naproxen derivatives on the examined cell lines was less potent when it contained oxadiazole and acetamide moieties. Against colorectal HCT-116 cells, a Naproxen derivative containing a 3-Cl group showed strong antiproliferative potential (IC50 = 1.24 μg mL−1) which was twofold the potency of doxorubicin (IC50 = 2.11 μg mL−1).
In terms of EGFR inhibitory efficacy, all hybrids showed significant inhibition of EGFR with IC50 values ranging from 0.41 to 7.31 μM. Hybrids of Naproxen and 1,2,3-triazole were more effective at inhibiting EGFR than those of acetamide. The IC50 values for Naproxen hybrids containing acetamide groups were 0.82 μM and 1.08 μM, respectively, and showed excellent inhibition.
The position of the substituent on the phenyl ring connected to the 1,2,3-triazole was found to be crucial in EGFR inhibition. A compound with OH and Cl substituents at the ortho position significantly inhibited the activity of the EGFR compared to hybrids with meta and para substituents. The ortho-substituted group's tight attachment to the receptor may be the cause of this activity trend. Additionally, it was found that hybrids with two substituents were more effective at inhibiting EGFR than those with one substituent. The EGFR activity was increased when the bromo group was replaced with the COOH group.101
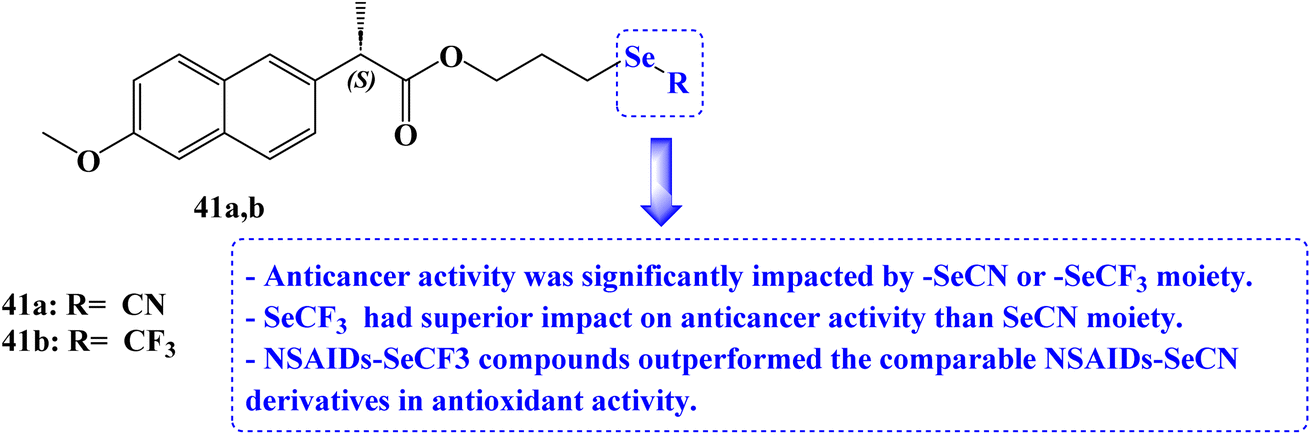
He et al.102 reported the synthesis of organoselenium compounds based on the hybridization of NSAIDs skeleton and organoselenium scaffold (–SeCN and –SeCF3). Human colon adenocarcinoma cell line Caco-2, human gastric cancer cell line BGC-823, human breast cancer cell line MCF-7, and human prostatic cancer cell line PC-3 were used to test the anticancer activities. The IC50 values for the Naproxen derivative 41a (Fig. 35) against the Caco2, BGC-823, MCF-7, and PC-3 cancer cell lines were 21.5, 17.3, 32.8, and 22 μM, respectively. The IC50 values for the Naproxen derivative 41b (Fig. 35) against the Caco2, BGC-823, MCF-7, and PC-3 cancer cell lines were 16.3, 10.8, 12.4, and 18.4 μM, respectively.
 | ||
| Fig. 35 The structure of Naproxen organoselenium derivatives 41a,b. | ||
It was intriguing to note that cancer cell lines are significantly impacted by the parent NSAIDs that correlate to the –SeCN or –SeCF3 moiety. Considering the lipophilicity and electron-withdrawing effect, NSAIDs with trifluoromethyl selenides moiety had superior anticancer activity than equivalent NSAIDs with selenocyanates moiety. By using bleomycin-dependent DNA damage, glutathione peroxidase (GPx)-like assays, 2, 2-didiphenyl-1-picrylhydrazyl (DPPH), and other tests, the redox characteristics of the NSAIDs-Se compounds were examined. In comparison to vitamin C (96.4% antioxidant activity), Naproxen derivatives 41a and 41b (24.6 and 37% antioxidant activity) showed good free-radical scavenging activity. NSAIDs–SeCF3 compounds outperformed the comparable NSAIDs–SeCN derivatives in this assay.
New (S)-Naproxen hybrids with thiosemicarbazide/1,2,4-triazole moiety were developed, and tested for their ability to inhibit the growth of the human breast cancer cell line MDA-MB-231. (S)-4-(2,4-Dichlorophenyl)-5-[1-(6-methoxynaphthalen-2-yl)ethyl]-4H-1,2,4-triazole-3-thione (42) (Fig. 36) exhibited the most potent anticancer activity with a good selectivity (IC50 = 9.89 μM). The IC50 value for compound 42 was greater (749 μM) as compared to the NIH-3T3 healthy control cell line.
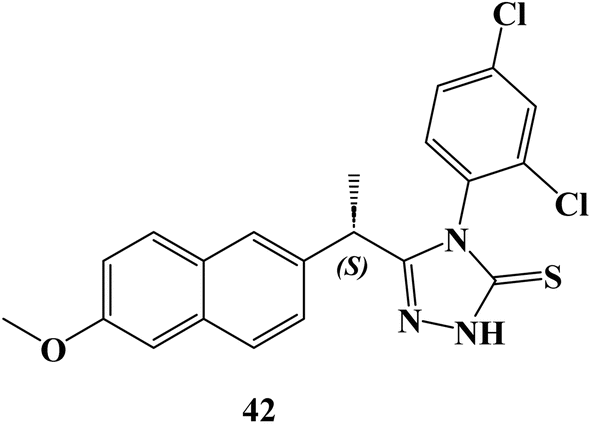 | ||
| Fig. 36 The structure of Naproxen-1,2,4-triazole hybrid 42. | ||
AO/EB staining of MDA-MB-231 cells revealed that live control cells had big green nuclei, indicating that their cell membranes were still intact. Naproxen triazole compound 42, on the other hand, drastically decreased the number of cells with large green nuclei and caused nearly all cells to undergo nuclear condensation and form apoptotic bodies, which discolored the cells orange. When MDA-MB-231 cells were treated with Naproxen triazole compound 42 at the IC50 concentration compared to the untreated control group, it was found that the number of apoptotic cells increased considerably. JC-1 monomers increased in comparison to the control group when Naproxen triazole compound 42 was incubated with MDA-MB-231 cells at the IC50 concentration, and the ratio of aggregate to monomer (red/green) in the cells dramatically decreased.
Caspase-3 enzyme activity was also activated, which showed apoptosis. The flow cytometric study revealed that compound 42, in a dose-dependent manner, increased the number of cells in the S phase and decreased the number of cells in the G2/M phase. Approximately 60% of BCL-2 protein expression was reduced by compound 42. It restricted the BCL-2 protein, which caused apoptosis. Compound 42 demonstrated anticancer effect and decreased the tumor volume at both low (60 mg kg−1) and high (120 mg kg−1) doses in mice when used in the Ehrlich acid tumor model, a well-validated in vivo ectopic breast cancer model.103
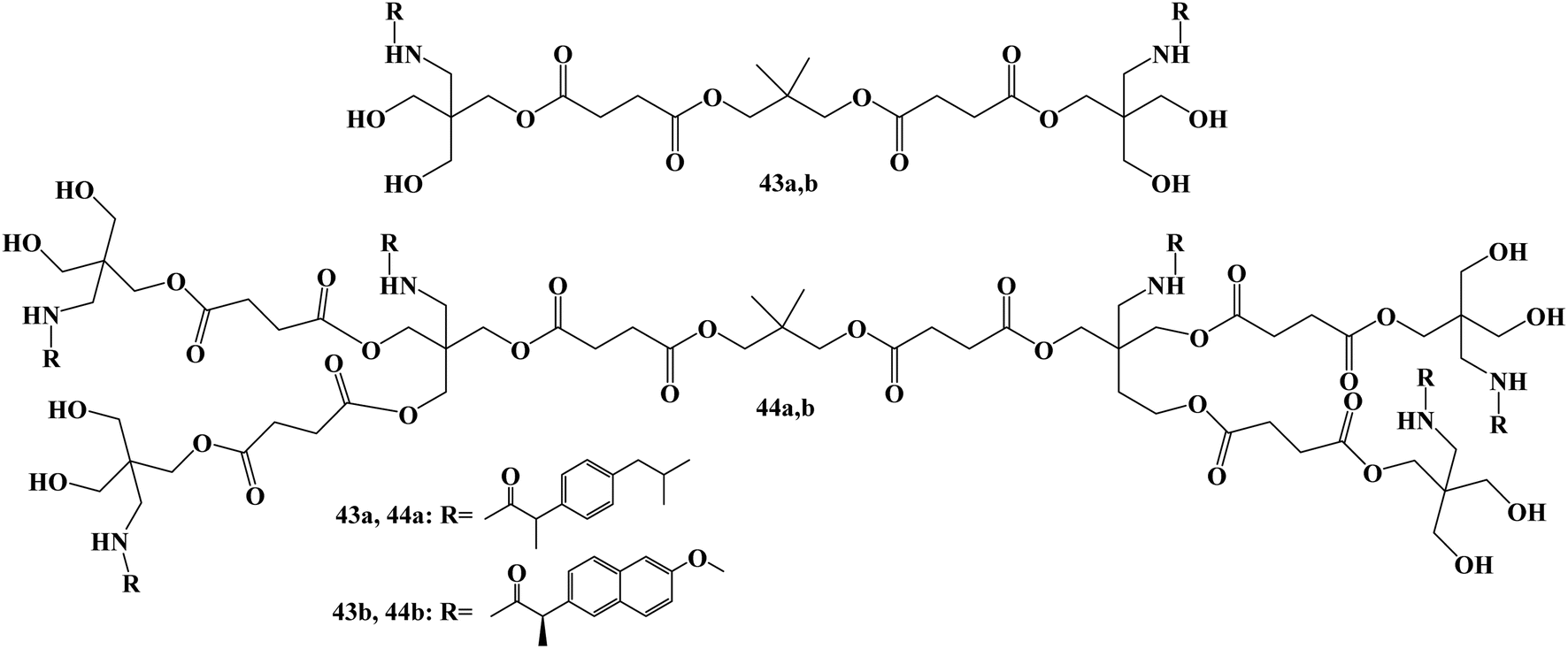
Bifunctional dendrimers 43 and 44 (Fig. 37) were developed via synthesizing tris(hydroxymethyl)-aminomethane (Tris) conjugates with Ibuprofen or Naproxen in the inner and hydroxy groups on the outside. Although less toxic to healthy COS-7 cells than cisplatin, the first-generation combination with Naproxen demonstrated strong anticancer efficacy. For all cell lines (U251, PC-3, K562, MCF-7, SKLU-1, and COS7), the inhibitory concentration (IC50) values ranged from 0.1 to 5 μM in cancer cells.
 | ||
| Fig. 37 The structure of bifunctional Ibuprofen and Naproxen dendrimers 43 and 44. | ||
The in vitro assays generally showed that the examined derivatives had good efficacy. With IC50 values of 9.9 and 10.6 μM, respectively, the first-generation compound with Ibuprofen 43a (with OH) demonstrated strong potential against human prostate cancer (PC-3) and human lung adenocarcinoma (SKLU-1). With IC50 values of 10.9 and 11.9 μM, respectively, the second-generation compound with Ibuprofen 44a (with OH) demonstrated good selectivity against two lines, PC-3 and SKLU-1. All of the cell lines under investigation were effectively inhibited by the first-generation conjugate of Naproxen 43b (with OH). With IC50 values ranging from 6 to 15.5 μM, the conjugate 44b (with OH) demonstrated good cytotoxic activity against five cell lines.104
Derived from (S)-Naproxen, new thiosemicarbazides, 1,2,4-triazoles, and thioethers were synthesized. The derivatives' molecular binding to MetAP-2 was carried out. All compounds were found to be active, however, compounds 45a and 45b (Fig. 38) stood out for their strong ability to bind to the MetAP2 enzyme with free energies of binding −11.4 and −11.75 kcal mol−1, respectively. The anticancer effects of the synthesized compounds were examined using the MTT assay method on the MCF-7 (which contains estrogen and progesterone receptors) and MDA-MB-231 (which lacks estrogen and progesterone receptors) adenocarcinoma cell lines for 24 hours at doses of 0, 10, 25, 50, 75, and 100 μM. On the MCF-7 breast cancer cell line and the MDA-MB-231 cell lines, the IC50 values of new (S)-Naproxen derivatives ranged from 3.3 to 100 μM.
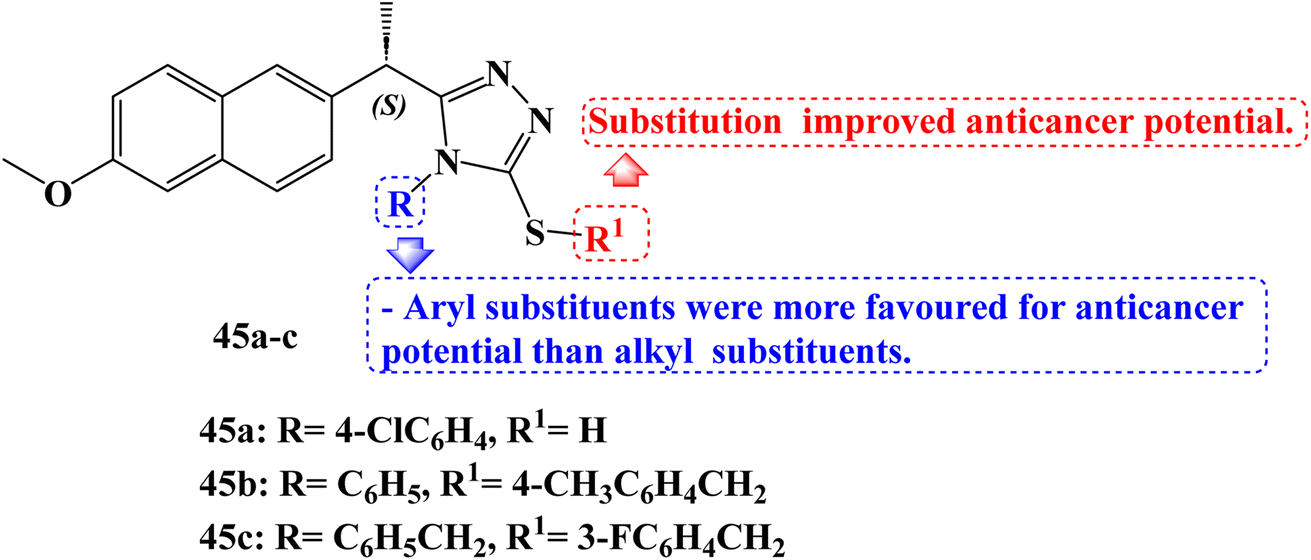 | ||
| Fig. 38 The structure of Naproxen-1,2,4-triazole hybrid 45a–c. | ||
Using the Tali Image-Based Cytometer, the apoptotic activity of compounds 45a and 45b was initially measured by annexin-V staining. The results of apoptosis were determined to be quite significant. A 17.6% increase in MCF-7 and a 20% increase in MDA-MB-231 cells was noticed after adding 50 μM of compound 45b.
Mitochondrial membrane potential variations in MCF-7 and MDA-MB-231 cells after JC-1 staining for compounds 45a and 45b were measured in a fluorescent plate reader. The effect of these derivatives on the cell viability of the 4T1 triple-negative (ER, PR, and HER2 negative) mouse breast cancer cell line was examined using the WST-1 cell viability and proliferation assay (IC50 × 1, IC50 × 2, IC50 × 3, IC50 × 4, and IC50 × 5). Next, a WST-1 cell survival and proliferation experiment with 4T1 was carried out to ascertain the toxicity of compound 45c (Fig. 38) and docetaxel together. The average viability of 4T1 cells was reduced by 43% by compound 45c at a concentration of 3 × IC50, 60% at a concentration of 4 × IC50, and 69% at a concentration of 5 × IC50.105
Recently, in 2023, Alsenani106 described the synthesis of new 1,2,4-triazole-Schiff base derivatives of Naproxen which were evaluated for their anticancer activity against MCF-7, Huh-7, and A-549 cancer cell lines cell line. Compound 46a (Fig. 39) revealed potent anticancer activity with IC50 values 4.72, 1.91, and 3.71 μM against the examined cell lines respectively, which represented 1.48 folds more potent than the used reference standard doxorubicin against the A-549 lung cancer cell line. In addition, compound 46b (Fig. 39) showed comparable activity against the hepatic Huh-7 cell line with IC50 (3.33 μM).
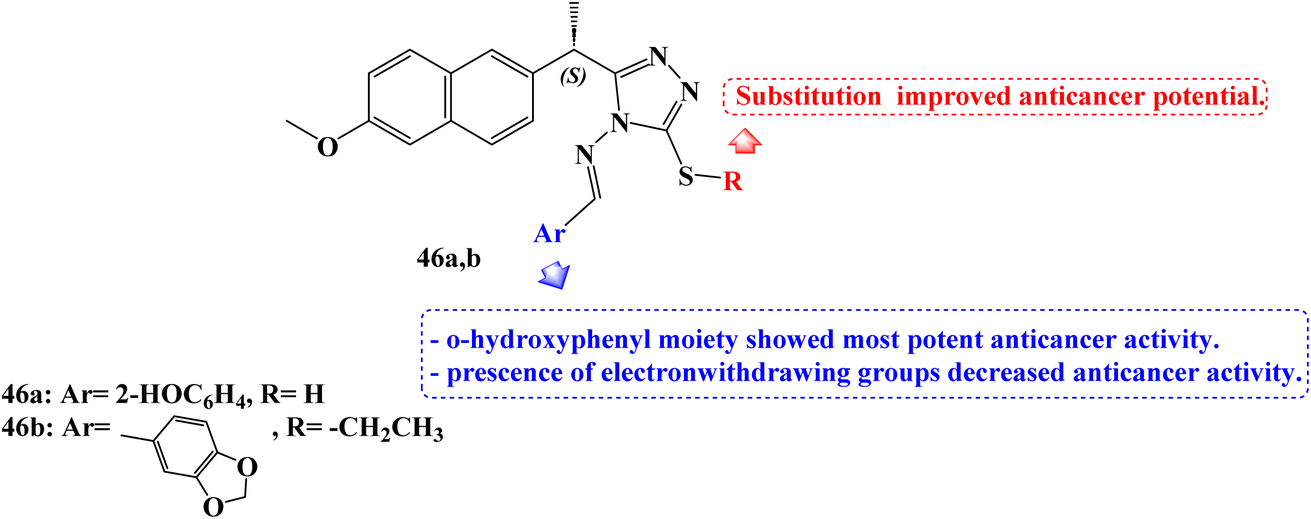 | ||
| Fig. 39 The structure of Naproxen-1,2,4-triazole-Schiff base derivatives 46a,b. | ||
In the same year, Nedeljković et al.107 reported the synthesis of new thiourea derivatives of (S)-Naproxen which were evaluated for their anticancer activity against the MRC5, MDAMB-231, HCT 116, and HeLa cells.108 Most of the synthesized Naproxen derivatives exhibited insignificant to weak anticancer activity towards both fibroblast MRC5 and breast MDAMB-231 cell lines. Moreover, compounds 47a and 47b (Fig. 40) showed moderate activity against colorectal HCT-116 cell lines with IC50 52.90 and 33.30 μM.
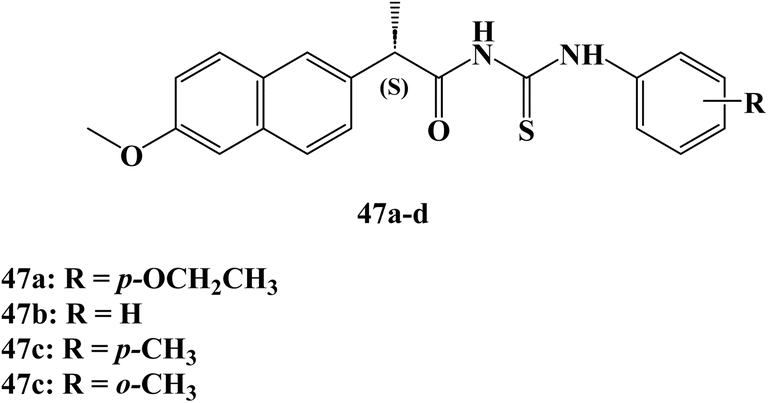 | ||
| Fig. 40 The structure of Naproxen thiourea derivatives 47a–d. | ||
On the other hand, Naproxen derivatives 47c and 47d (Fig. 40) showed potent anticancer activity against HeLa cervical cancer cells with IC50 11.70 and 9.10 μM. In addition, the results of cell cycle analysis of the HeLa cell line indicate that both compounds 47c, and 47d effectively induce G0/G1 phase arrest in HeLa cells, which could attribute the significant anticancer activity against HeLa cells through mechanisms involving the extrinsic pathway of apoptosis and cell cycle arrest.
In 2024, Bošković et al.88 examined the cytotoxicity of several synthesized derivatives of NSAID drugs, among the studied derivatives was the Naproxen hydroxamic acid derivative 48 (Fig. 41). The anticancer activity of the synthesized compounds was evaluated against colon HCT-116, HT-29, and pancreatic BxPC-3 cancer cell lines besides the normal cell line, MRC-5. The Naproxen hydroxamic acids derivative 48 exhibited cytotoxicity against the cell lines evaluated with IC50 values more than 100 μM.
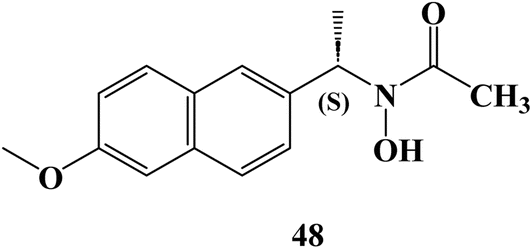 | ||
| Fig. 41 The structure of Naproxen hydroxamic acid derivative 48. | ||
Saez and coworkers109 synthesized and studied the anticancer activity of novel Naproxen–phosphane gold(I) complexes 49a,b (Fig. 42) against colon cancer. Specifically, the synthesized complexes. The synthesized complexes were found to have more potent anticancer activity than the reference standard auranofin with IC50 values against Caco2-/TC7 cell line 0.98, 0.24, and 1.80 μM respectively. Moreover, complexes 49a and 49b revealed significant selectivity towards cancer cells with selectivity index values of 7.47 and 33.10, respectively. Besides, Naproxen gold complexes induced G1-phase arrest in the tested colon cancer cells that resulted in an elevation in the percentage of cells in the G1 phase and a marked decrease percentage in the S and G2/M phases, indicating a specific disruption in DNA replication.
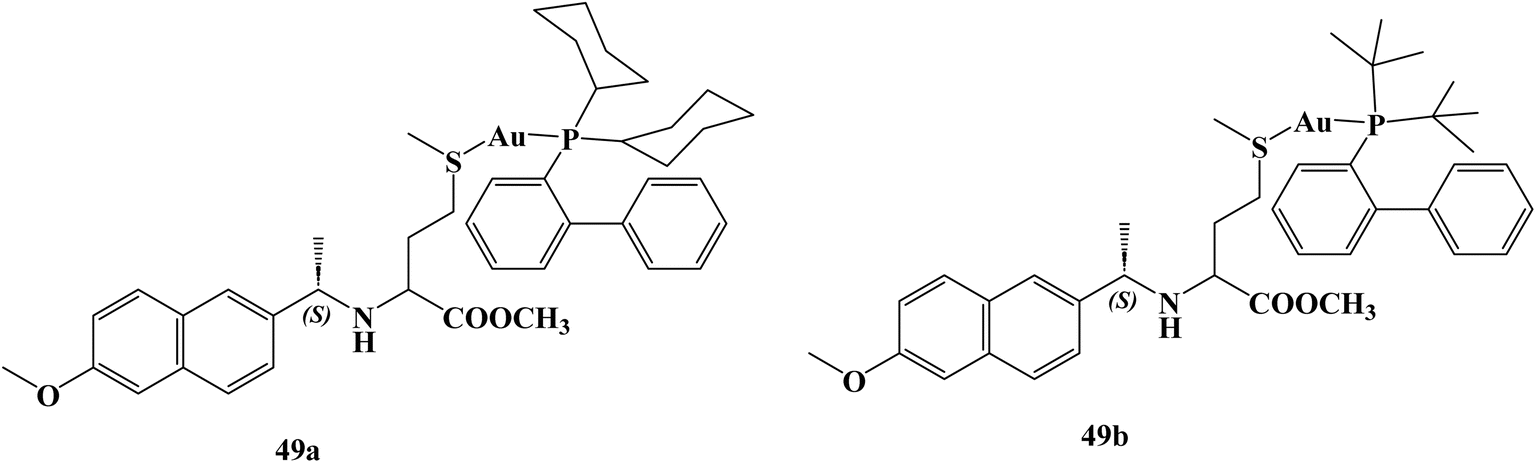 | ||
| Fig. 42 The structure of Naproxen–phosphane gold(I) complexes 49a,b. | ||
8. Conclusion
The exceedingly varied character of human cancers necessitates a variety of therapeutic approaches. There has been substantial improvement in the last 20 years in finding new treatments for various malignancies. However, a sizable portion of cancer patients remain incurable due to acquired resistance to treatment options, which continues to be a serious issue for researchers and medical professionals. Due to the exorbitant expense of the existing anticancer drugs, the majority of countries throughout the world are currently unable to provide for the medical demands of cancer patients. An efficient method for creating new therapeutic choices for the treatment of cancer patients at a reasonable cost in clinics is the repurposing of FDA-approved non-cancerous medications.Inflammation has a close connection to cancer and is crucial to the growth and development of tumors. Chronic inflammation encourages the development of cancer by triggering angiogenesis, metastasis, and proliferation while diminishing the body's ability to respond to chemotherapeutic drugs and the immune system. In preclinical and clinical investigations, NSAIDs and their derivatives demonstrated potential anticancer action by controlling several important molecular mechanisms and oncogenic pathways in human malignancies which may be COX-dependent pathways or non-COX targets as the apoptosis induction, metastasis inhibition, or induction of cell cycle arrest.
This review emphasizes the most recent antiproliferative activity of Indomethacin and Naproxen derivatives between 2017 and 2024. The investigation of Indomethacin and Naproxen derivatives with various modes of action as effective anticancer agents, based on SARs, has been effectively addressed. Several derivatives of Indomethacin and Naproxen showed strong, broad-spectrum antiproliferative activity against various human tumor cell lines. They showed IC50 values in the nano- to micro-molar range.
Strong cytotoxic candidates were developed when the Indomethacin and Naproxen were hybridized with other scaffolds such as N-acylhydrazone, oxadiazole, stable antioxidant nitroxides, coumarin or triazole that are known for their antiproliferative properties. Hybridization of Indomethacin and Naproxen derivatives with anticancer drugs such as podophyllotoxin, cisplatin, or methotrexate also produced synergistic anticancer effects. Selenium-containing analogs of Indomethacin and Naproxen exhibited potent antiproliferative potential. The Indomethacin and Naproxen derivatives' metal complexes showed significant cytotoxic action (Fig. 43).
 | ||
| Fig. 43 The summary of SARs of Indomethacin and Naproxen derivatives as antiproliferative agents. | ||
The meta-analysis of Indomethacin and Naproxen derivatives indicated their potential anticancer activity through various modes of action. A number of these derivatives are multitargeted potent anticancer agents, and it is hoped that they may soon be approved for the treatment of cancer. These compilations may therefore serve as a guide for medicinal chemists to develop bioactive Indomethacin and Naproxen derivatives that can be employed as lead candidates for the development of potent and safe anticancer drugs that specifically target tumors and could eventually be used in clinical settings. Drug repurposing brings up a new area for the study of already-approved medications and may offer higher chances for prompt treatment. It may open up new possibilities for the faster and less expensive discovery of novel anticancer drugs.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
The authors are thankful to the Faculty of Pharmacy, Cairo University.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- H. Maeda and M. Khatami, Clin. Transl. Med., 2018, 7, 1–20 CrossRef PubMed.
- A. Jazieh, O. B. Da’ar, M. Alkaiyat, Y. Zaatreh, A. A. Saad, R. Bustami, M. Alrujaib and K. Alkattan, Cancer Manage. Res., 2019, 11, 9665–9674 CrossRef PubMed.
- D. C. Whiteman and L. F. Wilson, Cancer Epidemiol., 2016, 44, 203–221 CrossRef.
- T. Stanković, J. Dinić, A. Podolski-Renić, L. Musso, S. S. Burić, S. Dallavalle and M. Pešić, Curr. Med. Chem., 2019, 26, 6074–6106 CrossRef.
- S. Sano, K. S. Chan, S. Carbajal, J. Clifford, M. Peavey, K. Kiguchi, S. Itami, B. J. Nickoloff and J. DiGiovanni, Nat. Med., 2005, 11, 43–49 CrossRef PubMed.
- M. Philip, D. A. Rowley and H. Schreiber, Semin. Cancer Biol., 2004, 14, 433–439 CrossRef.
- A. Mantovani, Nature, 2009, 457, 36–37 CrossRef PubMed.
- H. Achiwa, Y. Yatabe, T. Hida, T. Kuroishi, K. Kozaki, S. Nakamura, M. Ogawa, T. Sugiura, T. Mitsudomi and T. Takahashir, Clin. Cancer Res., 1999, 5, 1001–1005 Search PubMed.
- L. Y. Pang, E. A. Hurst and D. J. Argyle, Stem Cells Int., 2016, 2016, 2048731 CrossRef.
- R. Botting, Thromb. Res., 2003, 110, 269–272 CrossRef PubMed.
- N. V. Chandrasekharan, H. Dai, K. L. T. Roos, N. K. Evanson, J. Tomsik, T. S. Elton and D. L. Simmons, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 13926–13931 CrossRef PubMed.
- J. Steinmeyer, Arthritis Res. Ther., 2000, 2, 1–7 Search PubMed.
- K. Brune and P. Patrignani, J. Pain Res., 2015, 105–118 CrossRef PubMed.
- D. Wang and R. N. DuBois, Oncogene, 2010, 29, 781–788 CrossRef.
- A. A. Khan, M. Iadarola, H.-Y. T. Yang and R. A. Dionne, J. Pain, 2007, 8, 349–354 CrossRef PubMed.
- Y. N. Ye, W. K. K. Wu, V. Y. Shin, I. C. Bruce, B. C. Y. Wong and C. H. Cho, Carcinogenesis, 2005, 26, 827–834 CrossRef.
- L. Minghetti, J. Neuropathol. Exp. Neurol., 2004, 63, 901–910 CrossRef PubMed.
- E. R. Greene, S. Huang, C. N. Serhan and D. Panigrahy, Prostaglandins Other Lipid Mediators, 2011, 96, 27–36 CrossRef PubMed.
- B. Zhong, X. Cai, S. Chennamaneni, X. Yi, L. Liu, J. J. Pink, A. Dowlati, Y. Xu, A. Zhou and B. Su, Eur. J. Med. Chem., 2012, 47, 432–444 CrossRef CAS PubMed.
- F. H. Sarkar, S. Adsule, Y. Li and S. Padhye, Mini-Rev. Med. Chem., 2007, 7, 599–608 CrossRef CAS PubMed.
- E. R. Rayburn, S. J. Ezell and R. Zhang, Mol. Cell. Pharmacol., 2009, 1, 29–43 CrossRef.
- A.-M. Alaa, A. Angeli, A. S. El-Azab, M. E. A. Hammouda, M. A. El-Sherbeny and C. T. Supuran, Bioorg. Chem., 2019, 84, 260–268 CrossRef.
- M. Vosooghi and M. Amini, Expert Opin. Drug Discovery, 2014, 9, 255–267 CrossRef.
- S. N. Kang, S.-S. Hong, M.-K. Lee and S.-J. Lim, Int. J. Pharm., 2012, 428, 76–81 CrossRef.
- H.-B. Xu, F.-M. Shen and Q.-Z. Lv, Eur. J. Pharmacol., 2015, 769, 1–7 CrossRef.
- U. C. Nzeako, M. E. Guicciardi, J.-H. Yoon, S. F. Bronk and G. J. Gores, Hepatology, 2002, 35, 552–559 CrossRef.
- T. Fujita, M. Matsui, K. Takaku, H. Uetake, W. Ichikawa, M. M. Taketo and K. Sugihara, Cancer Res., 1998, 58, 4823–4826 Search PubMed.
- K. C. Zimmermann, M. Sarbia, A. A. Weber, F. Borchard, H. E. Gabbert and K. Schrör, Cancer Res., 1999, 59, 198–204 Search PubMed.
- A. T. Koki and J. L. Masferrer, Cancer Control, 2002, 9, 28–35 CrossRef PubMed.
- C. H. Liu, S. H. Chang, K. Narko, O. C. Trifan, M. T. Wu, E. Smith, C. Haudenschild, T. F. Lane and T. Hla, J. Biol. Chem., 2001, 276, 18563–18569 CrossRef PubMed.
- M. Tsujii and R. N. DuBois, Cell, 1995, 83, 493–501 CrossRef.
- D. E. A. Francés, P. I. Ingaramo, R. Mayoral, P. Través, M. Casado, Á. M. Valverde, P. Martín-Sanz and C. E. Carnovale, J. Cell. Biochem., 2013, 114, 669–680 CrossRef PubMed.
- J. P. Plastaras, F. P. Guengerich, D. W. Nebert and L. J. Marnett, J. Biol. Chem., 2000, 275, 11784–11790 CrossRef PubMed.
- L. Qu and B. Liu, Cancer Cell Int., 2015, 15, 69 CrossRef PubMed.
- H. Hu, T. Han, M. Zhuo, L.-L. Wu, C. Yuan, L. Wu, W. Lei, F. Jiao and L.-W. Wang, Sci. Rep., 2017, 7, 470 CrossRef PubMed.
- N. Kundu and A. M. Fulton, Cancer Res., 2002, 62, 2343–2346 Search PubMed.
- C. Rosas, M. Sinning, A. Ferreira, M. Fuenzalida and D. Lemus, Biol. Res., 2014, 47, 27 CrossRef PubMed.
- Y. Yamamoto and R. B. Gaynor, J. Clin. Invest., 2001, 107, 135–142 CrossRef PubMed.
- A. Pannunzio and M. Coluccia, Pharmaceuticals, 2018, 11(4), 101 CrossRef PubMed.
- T. Kitamura, T. Kawamori, N. Uchiya, M. Itoh, T. Noda, M. Matsuura, T. Sugimura and K. Wakabayashi, Carcinogenesis, 2002, 23, 1463–1466 CrossRef PubMed.
- N. Niho, T. Kitamura, M. Takahashi, M. Mutoh, H. Sato, M. Matsuura, T. Sugimura and K. Wakabayashi, Cancer Sci., 2006, 97, 1011–1014 CrossRef PubMed.
- D. J. Elder, D. E. Halton, A. Hague and C. Paraskeva, Clin. Cancer Res., 1997, 3, 1679–1683 Search PubMed.
- S. Aggarwal, N. Taneja, L. Lin, M. B. Orringer, A. Rehemtulla and D. G. Beer, Neoplasia, 2000, 2, 346–356 CrossRef PubMed.
- T. Vogt, M. McClelland, B. Jung, S. Popova, T. Bogenrieder, B. Becker, G. Rumpler, M. Landthaler and W. Stolz, Melanoma Res., 2001, 11, 587–599 CrossRef PubMed.
- M. L. Smith, G. Hawcroft and M. A. Hull, Eur. J. Cancer, 2000, 36, 664–674 CrossRef.
- S. Zhang, A. Suvannasankha, C. D. Crean, V. L. White, A. Johnson, C.-S. Chen and S. S. Farag, Clin. Cancer Res., 2007, 13, 4750–4758 CrossRef PubMed.
- T. Wu, J. Leng, C. Han and A. J. Demetris, Mol. Cancer Ther., 2004, 3, 299–307 CrossRef PubMed.
- T. C. He, T. A. Chan, B. Vogelstein and K. W. Kinzler, Cell, 1999, 99, 335–345 CrossRef.
- S. Pushpakom, F. Iorio, P. A. Eyers, K. J. Escott, S. Hopper, A. Wells, A. Doig, T. Guilliams, J. Latimer and C. McNamee, Nat. Rev. Drug Discovery, 2019, 18, 41–58 CrossRef.
- T. T. Ashburn and K. B. Thor, Nat. Rev. Drug Discovery, 2004, 3, 673–683 CrossRef PubMed.
- M. Antoszczak, A. Markowska, J. Markowska and A. Huczyński, Eur. J. Pharmacol., 2020, 866, 172784 CrossRef.
- R. G. Armando, D. L. Mengual Gómez and D. E. Gomez, Int. J. Oncol., 2020, 56, 651–684 Search PubMed.
- T. Masuda, Y. Tsuruda, Y. Matsumoto, H. Uchida, K. I. Nakayama and K. Mimori, Cancer Sci., 2020, 111, 1039–1046 CrossRef PubMed.
- G. Mudduluru, W. Walther, D. Kobelt, M. Dahlmann, C. Treese, Y. G. Assaraf and U. Stein, Drug Resistance Updates, 2016, 26, 10–27 CrossRef PubMed.
- P. Nowak-Sliwinska, L. Scapozza and A. R. i Altaba, Biochim. Biophys. Acta, Rev. Cancer, 2019, 1871, 434–454 CrossRef.
- M. B. Serafin, A. Bottega, T. F. da Rosa, C. S. Machado, V. S. Foletto, S. S. Coelho, A. D. da Mota and R. Hörner, Am. J. Ther., 2021, 28, e111–e117 CrossRef PubMed.
- A. Corbett, G. Williams and C. Ballard, Front. Biosci., Scholar Ed., 2015, 7, 184–188 CrossRef PubMed.
- A. A. de Castro, E. F. F. da Cunha, A. F. Pereira, F. V Soares, D. H. S. Leal, K. Kuca and T. C. Ramalho, Curr. Alzheimer Res., 2018, 15, 1161–1178 CrossRef.
- A. C. Grammer and P. E. Lipsky, Rheum. Dis. Clin. N. Am., 2017, 43, 467–480 CrossRef.
- Y. Huo and H.-Y. Zhang, Genes, 2018, 9(5), 237 CrossRef PubMed.
- A. C. Grammer, M. M. Ryals, S. E. Heuer, R. D. Robl, S. Madamanchi, L. S. Davis, B. Lauwerys, M. D. Catalina and P. E. Lipsky, Lupus, 2016, 25, 1150–1170 CrossRef PubMed.
- B. F. Cole, R. F. Logan, S. Halabi, R. Benamouzig, R. S. Sandler, M. J. Grainge, S. Chaussade and J. A. Baron, JNCI, J. Natl. Cancer Inst., 2009, 101, 256–266 CrossRef.
- P. M. Rothwell, M. Wilson, J. F. Price, J. F. F. Belch, T. W. Meade and Z. Mehta, Lancet, 2012, 379, 1591–1601 CrossRef CAS.
- S. Pushpakom, F. Iorio, P. A. Eyers, K. J. Escott, S. Hopper, A. Wells, A. Doig, T. Guilliams, J. Latimer, C. McNamee, A. Norris, P. Sanseau, D. Cavalla and M. Pirmohamed, Nat. Rev. Drug Discovery, 2019, 18, 41–58 CrossRef PubMed.
- F. Bertolini, V. P. Sukhatme and G. Bouche, Nat. Rev. Clin. Oncol., 2015, 12(12), 732–742 CrossRef PubMed.
- A. E. Kassab and E. M. Gedawy, Curr. Pharm. Des., 2024, 30, 1217–1239 CrossRef.
- W. Hu, L. Fang, W. Hua and S. Gou, J. Inorg. Biochem., 2017, 175, 47–57 CrossRef PubMed.
- F. Yazdi, M. Abbasi and H. Sadeghi-Aliabadi, Iran. J. Pharmacol. Ther., 2017, 15(1), 1–7 Search PubMed.
- S. I. Eissa, Med. Chem. Res., 2017, 26, 2205–2220 CrossRef.
- L. Zhang, L. Liu, C. Zheng, Y. Wang, X. Nie, D. Shi, Y. Chen, G. Wei and J. Wang, Eur. J. Med. Chem., 2017, 131, 81–91 CrossRef PubMed.
- K. Thomas, T. W. Moody, R. T. Jensen, J. Tong, C. L. Rayner, N. L. Barnett, K. E. Fairfull-Smith, L. A. Ridnour, D. A. Wink and S. E. Bottle, Eur. J. Med. Chem., 2018, 147, 34–47 CrossRef.
- T. K. Rundstadler, A. Eskandari, S. M. Norman and K. Suntharalingam, Molecules, 2018, 23(9), 2253 CrossRef.
- L. Liu, S. Li, X. Li, M. Zhong, Y. Lu, Y. Jiajie, Z. Yongmin and X. He, Med. Chem. Res., 2018, 27, 2071–2078 CrossRef.
- A. S. El-Azab, A. A. M. Abdel-Aziz, L. A. Abou-Zeid, W. M. El-Husseiny, A. M. ElMorsy, M. A. El-Gendy and M. A. A. El-Sayed, J. Enzyme Inhib. Med. Chem., 2018, 33, 989–998 CrossRef.
- C. Chen, Y. Nie, G. Xu, X. Yang, H. Fang and X. Hou, Bioorg. Med. Chem., 2019, 27, 2771–2783 CrossRef PubMed.
- F. Wu, M. Yang, J. Zhang, S. Zhu, M. Shi and K. Wang, J. Biol. Inorg. Chem., 2019, 24, 53–60 CrossRef PubMed.
- M. Narożna, V. Krajka-Kuźniak, B. Bednarczyk-Cwynar, R. Kleszcz and W. Baer-Dubowska, Pharmaceuticals, 2020, 14, 32 CrossRef PubMed.
- S. Wet-osot, T. Pewklang, K. Chansaenpak, N. Chudapongse, R. Y. Lai and A. Kamkaew, ChemMedChem, 2021, 16, 1660–1666 CrossRef PubMed.
- M. F. Harras, R. Sabour, Y. A. Ammar, A. B. M. Mehany, A. M. Farrag and S. I. Eissa, J. Mol. Struct., 2021, 1228, 129455 CrossRef.
- S. Patel, P. Nanavati, J. Sharma, V. Chavda and J. Savjani, Arch. Med. Res., 2021, 52, 483–493 CrossRef PubMed.
- J. Almeida, G. Zhang, M. Wang, C. Queirós, A. F. R. Cerqueira, A. C. Tomé, G. Barone, M. G. H. Vicente, E. Hey-Hawkins, A. M. G. Silva and M. Rangel, Org. Biomol. Chem., 2021, 19, 6501–6512 RSC.
- H. Wang, Z. Chang, G. di Cai, P. Yang, J. he Chen, S. shu Yang, Y. feng Guo, M. yu Wang, X. hua Zheng, J. ping Lei, P. qing Liu, D. peng Zhao and J. jian Wang, Acta Pharmacol. Sin., 2022, 43, 1024–1032 CrossRef PubMed.
- S. Ramos-Inza, I. Encío, A. Raza, A. K. Sharma, C. Sanmartín and D. Plano, Eur. J. Med. Chem., 2022, 244, 114839 CrossRef PubMed.
- A. Bajpai, Deepshikha, D. Chhabria, T. Mishra, S. Kirubakaran and S. Basu, Bioorg. Med. Chem., 2022, 64, 116759 CrossRef PubMed.
- K. Huang, H. Zhang, M. Yan, J. Xue and J. Chen, Dyes Pigm., 2022, 198, 109997 CrossRef.
- Y. Liu, J. Li, G. Zhou, J. Zhang, Y. Teng, Z. Bai and T. Liu, Med. Chem. Res., 2023, 32, 99–108 CrossRef.
- J. Bošković, V. Dobričić, O. Keta, L. Korićanac, J. Žakula, J. Dinić, S. Jovanović Stojanov, A. Pavić and O. Čudina, Pharmaceutics, 2024, 16(6), 826 CrossRef PubMed.
- C. Lu, A. Eskandari, P. B. Cressey and K. Suntharalingam, Chem.—Eur. J., 2017, 23, 11366–11374 CrossRef PubMed.
- A. G. Al-Sehemi, A. Irfan, M. Alfaifi, A. M. Fouda, T. Ma'mon El-Gogary and D. A. Ibrahim, J. King Saud Univ., Sci., 2017, 29, 311–319 CrossRef.
- W. M. El-Husseiny, M. A. A. El-Sayed, N. I. Abdel-Aziz, A. S. El-Azab, Y. A. Asiri and A. A. M. Abdel-Aziz, Eur. J. Med. Chem., 2018, 158, 134–143 CrossRef PubMed.
- M. İ. Han, H. Bekçi, A. I. Uba, Y. Yıldırım, E. Karasulu, A. Cumaoğlu, H. Y. Karasulu, K. Yelekçi, Ö. Yılmaz and Ş. G. Küçükgüzel, Arch. Pharm., 2019, 352, 1800365 CrossRef.
- J. P. G. Tabares, R. L. S. R. Santos, J. L. Cassiano, M. H. Zaim, J. Honorato, A. A. Batista, S. F. Teixeira, A. K. Ferreira, R. B. Viana, S. Q. Martínez, A. C. Stábile and D. de Oliveira Silva, Inorg. Chim. Acta, 2019, 489, 27–38 CrossRef.
- G. Ercolano, P. De Cicco, F. Frecentese, I. Saccone, A. Corvino, F. Giordano, E. Magli, F. Fiorino, B. Severino, V. Calderone, V. Citi, G. Cirino and A. Ianaro, Front. Pharmacol, 2019, 10, 1–11 CrossRef.
- K. Birgül, Y. Yıldırım, H. Y. Karasulu, E. Karasulu, A. I. Uba, K. Yelekçi, H. Bekçi, A. Cumaoğlu, L. Kabasakal, Ö. Yılmaz and Ş. G. Küçükgüzel, Eur. J. Med. Chem., 2020, 208, 112841 CrossRef.
- X. He, M. Zhong, S. Li, X. Li, Y. Li, Z. Li, Y. Gao, F. Ding, D. Wen, Y. Lei and Y. Zhang, Eur. J. Med. Chem., 2020, 208, 112864 CrossRef PubMed.
- S. Srinivas, P. Neeraja, K. Naveen, V. Banothu, P. K. Dubey, K. Mukkanti and S. Pal, ChemistrySelect, 2020, 5, 6786–6791 CrossRef.
- Y. Nie, M. Zhong, S. Li, X. Li, Y. Zhang, Y. Zhang and X. He, Chem. Biodiversity, 2020, 17(5), 1900603 CrossRef.
- S. Srinivas, P. Neeraja, V. Banothu, P. Kumar Dubey, K. Mukkanti and S. Pal, ChemistrySelect, 2020, 5, 14741–14746 CrossRef.
- M. İ. Han, P. Atalay, C. Ü. Tunç, G. Ünal, S. Dayan, Ö. Aydın and Ş. G. Küçükgüzel, Bioorg. Med. Chem., 2021, 37, 116097 CrossRef PubMed.
- M. M. Alam, S. Nazreen, A. S. A. Almalki, A. A. Elhenawy, N. I. Alsenani, S. E. I. Elbehairi, A. M. Malebari, M. Y. Alfaifi, M. A. Alsharif and S. Y. M. Alfaifi, Pharmaceuticals, 2021, 14(9), 870 CrossRef.
- X. He, Y. Nie, M. Zhong, S. Li, X. Li, Y. Guo, Z. Liu, Y. Gao, F. Ding, D. Wen and Y. Zhang, Eur. J. Med. Chem., 2021, 218, 113384 CrossRef PubMed.
- M. İ. Han, C. Ü. Tunç, P. Atalay, Ö. Erdoğan, G. Ünal, M. Bozkurt, Ö. Aydın, Ö. Çevik and G. Küçükgüzel, New J. Chem., 2022, 46, 6046–6059 RSC.
- L. D. Pedro-Hernández, T. Ramirez-Ápan and M. Martínez-García, ChemistrySelect, 2022, 7(27), e202201335 CrossRef.
- K. Birgül, A. I. Uba, O. Çuhadar, S. K. Sevinç, S. Tiryaki, P. M. Tiber, O. Orun, D. Telci, Ö. Yılmaz, K. Yelekçi and Ş. Güniz Küçükgüzel, J. Mol. Struct., 2022, 1259, 132739 CrossRef.
- N. I. Alsenani, Journal of Umm Al-Qura University for Applied Sciences, 2023, 9, 294–303 CrossRef.
- N. Nedeljković, V. Dobričić, J. Bošković, M. Vesović, J. Bradić, M. Anđić, A. Kočović, N. Jeremić, J. Novaković, V. Jakovljević, Z. Vujić and M. Nikolić, Pharmaceuticals, 2023, 16(5), 666 CrossRef PubMed.
- N. Nedeljković, M. Nikolić, P. Čanović, M. Zarić, R. Živković Zarić, J. Bošković, M. Vesović, J. Bradić, M. Anđić, A. Kočović, M. Nikolić, V. Jakovljević, Z. Vujić and V. Dobričić, Pharmaceutics, 2023, 16(1), 1 CrossRef.
- J. Saez, J. Quero, M. J. Rodriguez-Yoldi, M. C. Gimeno and E. Cerrada, Inorg. Chem., 2024, 63, 19769–19782 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |