
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and application of intramolecular arylogous nitroaldol condensation to access 2-aryl-benzofuran and -indole derivatives and formal synthesis of saprisartan†
Manyam Subbi Reddy ab,
Killari Satyamab and
Surisetti Suresh*ab
ab,
Killari Satyamab and
Surisetti Suresh*ab
aDepartment of Organic Synthesis and Process Chemistry, CSIR-Indian Institute of Chemical Technology (CSIR-IICT), Hyderabad 500 007, India. E-mail: surisetti@iict.res.in; suresh.surisetti@yahoo.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
First published on 20th November 2024
Abstract
This study presents the design and application of intramolecular arylogous nitroaldol (Henry) condensation. A transition metal-free, base-mediated reaction of ortho-heteroatom-substituted aryl aldehydes/ketones and 2-nitrobenzyl (pseudo)halides has been developed to access a wide range of 2-(2-nitroaryl)benzofuran/2-(2-nitroaryl)indole derivatives in high yields. The reaction appears to proceed through O-/N-benzylation and intramolecular arylogous nitroaldol condensation. The application potential of the presented strategy has been demonstrated in the formal synthesis of anti-hypertensive agent saprisartan through the efficient synthesis of an advanced key intermediate, which has been accomplished in a ten-fold high overall yield by using resourceful, inexpensive and less-toxic reagents compared to the known methods.
Benzofurans and indoles are important and privileged heterocycles because of their diverse biological activities and relevance in drug development. 2-Arylbenzofuran and 2-arylindole derivatives have attracted significant interest as biologically valuable compounds and pharmaceutical agents.1 Their potential to target a wide range of diseases and conditions continue to make them promising candidates for future drug development. In particular, 2-(2-arylamino)benzofuran/indole derivatives have received considerable attention in medicinal chemistry and drug discovery research programs. For example, saprisartan, an approved drug belonging to the sartan family that is devoid of the diphenyl moiety, functions as an angiotensin II receptor antagonist and helps to treat anti-hypertensive symptoms as well as heart failure.2 Compound A displays strong bladder relaxant effects, as well as high bladder (versus aorta) selectivity.3a,b Furthermore, 2-(2-nitroaryl)benzofuran derivatives serve as key starting materials for the synthesis of targeted products that have exhibited light emitting properties.3c,d Paullones possess excellent inhibitory profiles against cyclin dependent kinases (CDKs) and glycogen synthase kinase 3 (GSK3) (Fig. 1).4 The vinylogy principle has emerged as one of the important tools in organic chemistry enabling the synthesis of complex and structurally varied molecules including natural products and pharmaceutical compounds.5 It continues to drive advancements in materials science, drug discovery, and other areas of chemical research. On the other hand, 2-aryl-benzofurans/indoles are a class of compounds that have gained significant attention in the field of organic/medicinal chemistry. In continuation of our research efforts in vinylogous transformations6 and considering their importance as potential building blocks, we envisaged to develop arylogous nitroaldol condensation7 to access 2-(2-nitroaryl)benzofuran and 2-(2-nitroaryl)indole derivatives where the nitro group can serve as a synthetic handle.
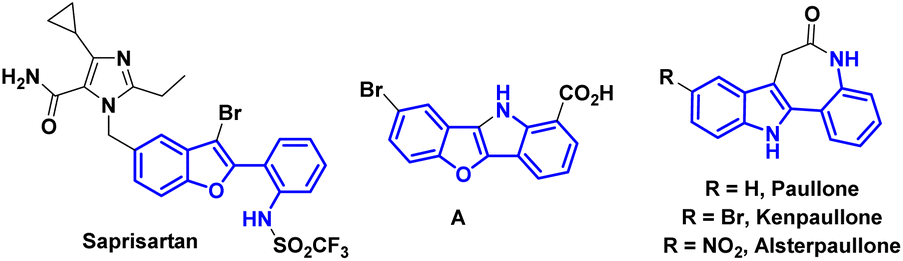 | ||
| Fig. 1 Bioactive molecules containing 2-arylaminobenzofuran and indole cores. | ||
To the best of our knowledge, transition metal catalysts are generally employed to access 2-(2-nitroaryl)benzofurans/2-(2-nitroaryl)indoles. Palladium-catalyzed cross-coupling reactions of 2-substituted benzofurans/indoles and 2-halonitroarenes were employed to obtain the aforementioned derivatives (Scheme 1a).8 Pd-catalyzed decarboxylative/desulfitative cross-coupling reactions of the respective benzofuran carboxylic acids and 2-nitrobenzoic acid/2-nitrobenzenesulfonyl chloride were also reported to access 2-(2-nitroaryl)benzofurans (Scheme 1b).9 In 2018, Patil and coworkers reported the synthesis of 2-(2-nitroaryl)benzofurans using desilylative C(sp2)–C(sp2) cross-coupling reaction of 2-benzofuranylsilane with 2-nitrophenyldiazonium salts, catalyzed by Au–Ru–Cu/photoredox system (Scheme 1c).10 Transition metal-free base-mediated reactions were also reported for the synthesis of a derivative of 2-(2-nitroaryl)benzofuran in four stages (Scheme 1d).11 Although, these methods are elegant, they suffer from the use of expensive transition metal catalysts, lengthy synthetic steps and use of not-readily-accessible metalated/substituted benzofuran/indole derivatives as starting materials. Herein, we report the development of a base-mediated intramolecular arylogous nitroaldol condensation to construct the benzofuran/indole nuclei enabling the synthesis of 2-(2-nitroaryl)benzofuran and 2-(2-nitroaryl)indole derivatives using commercially available, inexpensive and less-toxic starting materials/reagents obviating transition metals (Scheme 1e).
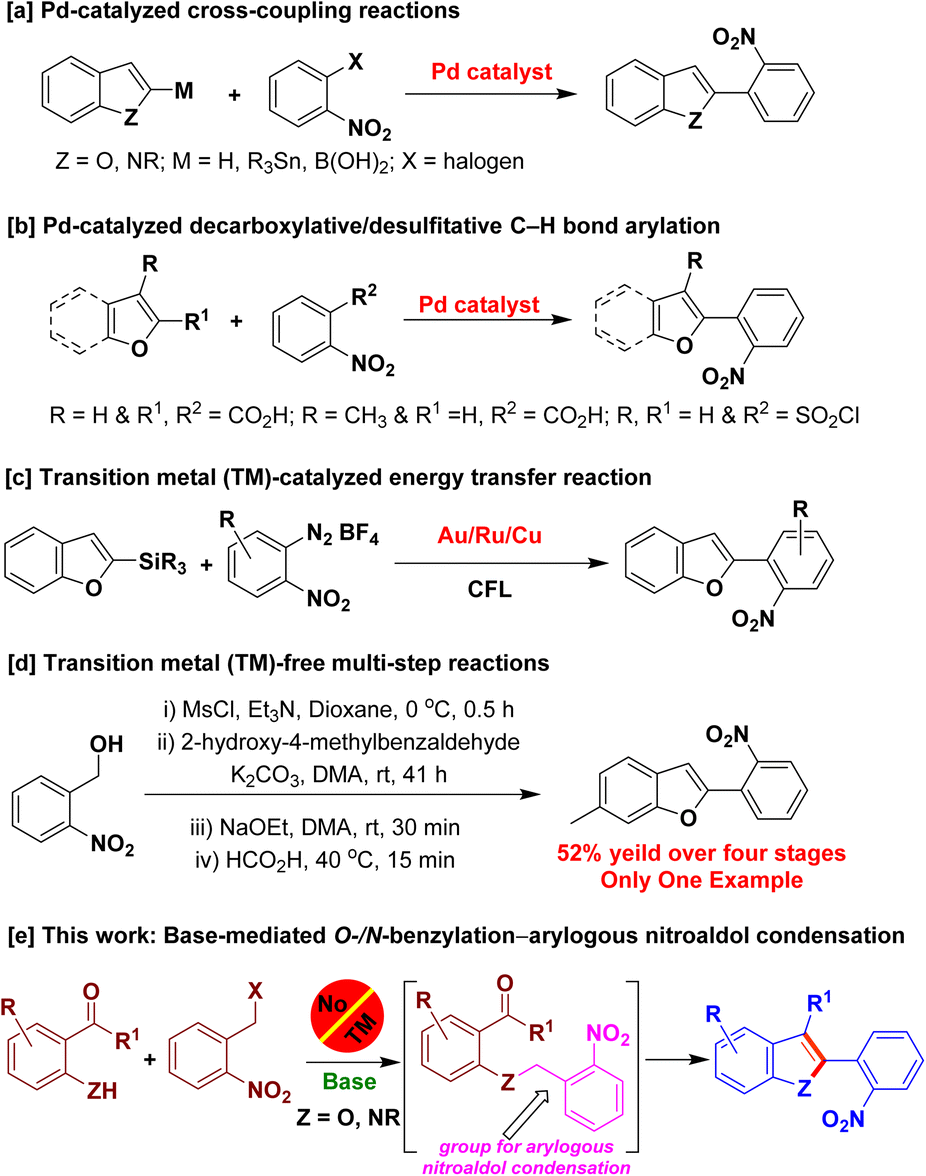 | ||
| Scheme 1 Construction of 2-(2-nitroaryl)benzofuran/indole derivatives and this work. | ||
We designed an easily accessible O-alkylated intermediate 3a integrating the arylogous nitroaldol donor moiety and electrophilic carbonyl moiety to validate our idea. Salicylaldehyde 1a and 1-(bromomethyl)-2-nitrobenzene 2a were used to generate the intended intermediate 3a. Following our approach, compound 3a was treated with base DBU in N,N-dimethylformamide for 3 h at 80 °C. We observed the formation of 2-(2-nirophenyl)benzofuran 4a in 55% yield. Subsequently, benzofuran 4a was produced in a higher yield of 65% using a one-pot process starting from the reaction of 1a and 2a (Scheme 2).
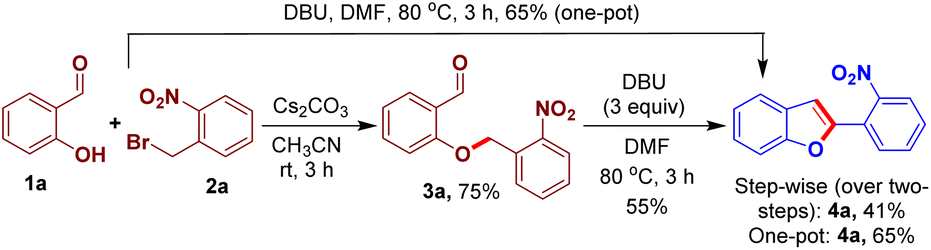 | ||
| Scheme 2 Step-wise and one-pot synthesis of 2-(2-nitrophenyl)benzofuran 4a. | ||
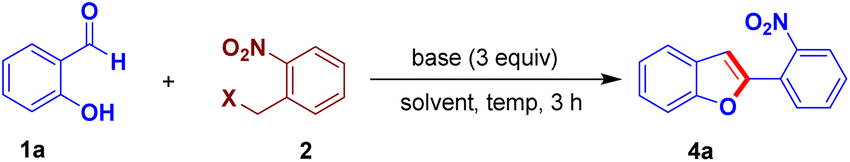
Then, we conducted an optimization study of the one-pot reaction by varying bases, solvents, and reaction conditions in order to obtain 4a in better yields. The use of bases such as DABCO, K2CO3, Cs2CO3 and KOtBu provided inferior results (Table 1, entries 2–5). Increasing the reaction temperature was not useful (Table 1, entry 6). To our delight, while screening the solvents, toluene was found to be the best solvent to give 4a in a very good yield (Table 1, entries 7 and 8). Then, we tested different leaving groups such as chloro, OMs and OTs in place of Br group in 2 and isolated 4a in slightly lower yields (Table 1, entries 9–11).
|
|||||
|---|---|---|---|---|---|
| Entry | Base | X in 2 | Solvent | Temp. °C | % yield of 4ab |
| a Reaction conditions: 1a (0.5 mmol), 2 (0.6 mmol), base (1.5 mmol), solvent (4 mL).b Isolated yields. | |||||
| 1 | DBU | Br | DMF | 80 | 65 |
| 2 | DABCO | Br | DMF | 80 | 21 |
| 3 | K2CO3 | Br | DMF | 80 | 38 |
| 4 | Cs2CO3 | Br | DMF | 80 | 46 |
| 5 | KOtBu | Br | DMF | 80 | 55 |
| 6 | DBU | Br | DMF | 120 | 58 |
| 7 | DBU | Br | DMSO | 80 | 45 |
| 8 | DBU | Br | Toluene | 80 | 85 |
| 9 | DBU | Cl | Toluene | 80 | 53 |
| 10 | DBU | OMs | Toluene | 80 | 80 |
| 11 | DBU | OTs | Toluene | 80 | 76 |
Having a set of optimization conditions, the substrate scope of the sequential O-alkylation–intramolecular arylogous nitroaldol condensation was explored as illustrated in the Scheme 3. Various substitutions on the salicylaldehyde partner were examined. The reaction of methyl-substituted-salicylaldehydes and 2a furnished the respective benzofurans 4b–4d in good yields. Noteworthy to mention we could obtain 6-methyl-2-(2-nitrophenyl)benzofuran 4c in 70% yield in one step using commercially available starting materials (Scheme 3) whereas the same was reported to have been synthesized in four stages with a low overall yield (Scheme 1d).11 5,7-Di-tert-butyl-2-(2-nitrophenyl)benzofuran 4e was obtained in 58% yield from the reaction of 3,5-di-tert-butylsalicylaldehyde 1e and 2a. The reaction of methoxy-substituted-salicylaldehydes and 2a provided the corresponding benzofuran derivatives 4f–4h in good yields. Naphthofuran derivative 4i was obtained in 80% yield from the reaction of 2-hydroxy-1-naphthaldehyde and 2a. The reaction of 2-hydroxy-5-(thiophen-2-yl)benzaldehyde 1j and 2a furnished the respective benzofuran 4j in 71% yield. Later, the sequential process of 2a and salicylaldehydes bearing halogen groups was examined. Accordingly, halogen-substituted-benzofuran derivatives 4k–4m were obtained in moderate to good yields. Furthermore, we used dihalogen-substituted-salicylaldehydes that furnished the respective benzofuran products 4n–4p in high yields. Salicylaldehyde bearing halogen and electron releasing group (ERG) was employed in this protocol to obtain the respective benzofuran derivative 4q in 71% yield. Strong electron-withdrawing groups (EWGs) such as nitro, nitrile and ester groups on salicylaldehyde were well tolerated and afforded the corresponding benzofuran derivatives 4r–4t in moderate to good yields. Furthermore, in place of aldehyde group we tested keto groups in the reaction partner 1. Accordingly, acetophenone and substituted benzophenones were engaged in this transformation furnishing the corresponding 2,3-disubstituted-benzofuran derivatives 4u, 4v and 4w in 68%, 73% and 48% yields, respectively.
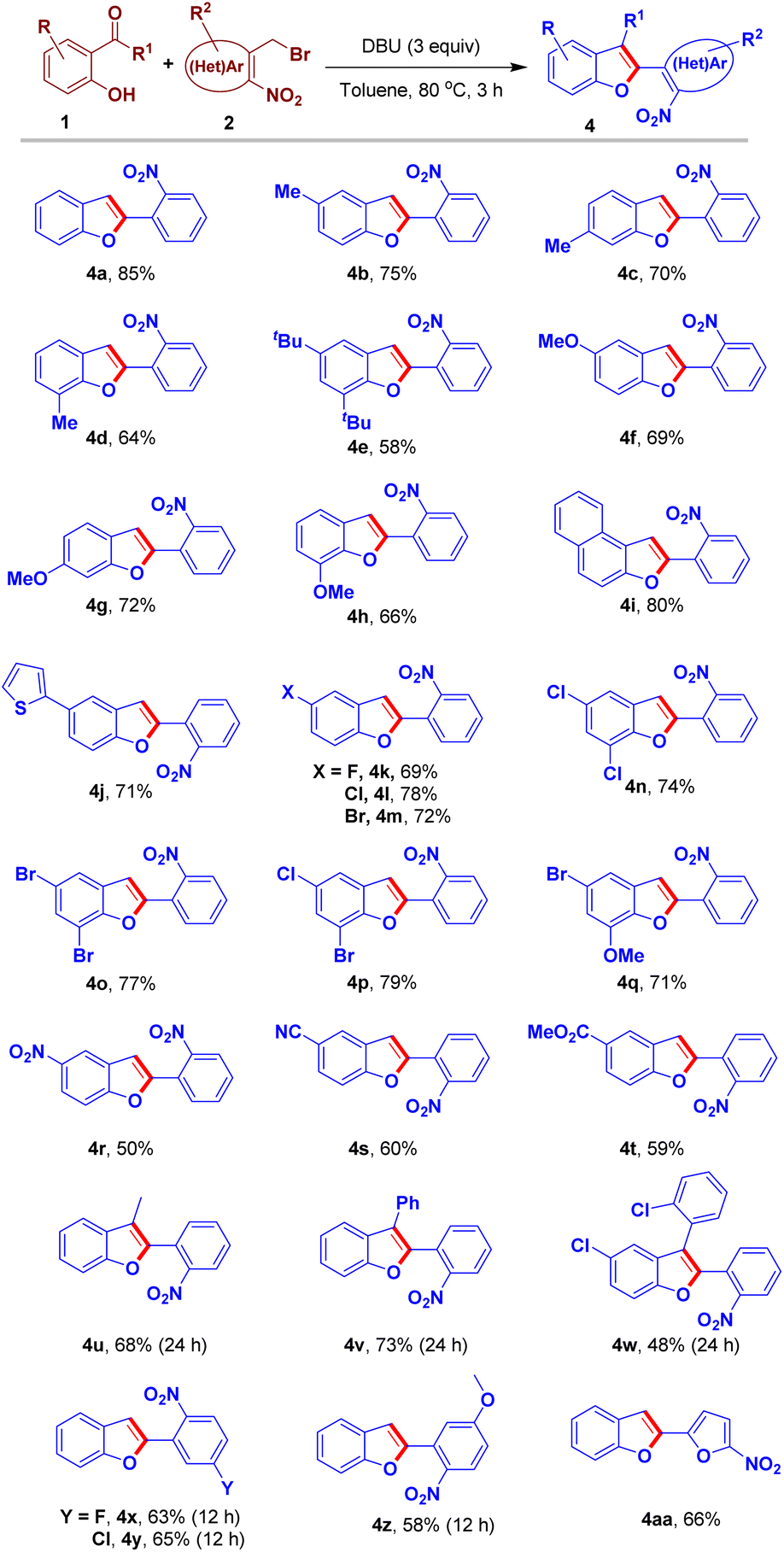 | ||
| Scheme 3 Scope of the one-pot reaction to access 2-(2-nitroaryl)benzofuran derivatives 4a–4aa. | ||
We also examined the scope of ortho-nitrobenzy/heteroarylmethyl bromides through a reaction of the respective substrate 2b–2e and salicylaldehyde 1a affording the corresponding benzofuran derivatives 4x-4aa in a good yields (Scheme 3).
Subsequently, we became interested to implement the optimized reaction conditions to develop an N-benzylation-intramolecular nitroaldol condensation process in order to obtain 2-(2-nitrophenyl)indole 7a, in one-pot, from the reaction of N-(2-formylphenyl)-4-methylbenzenesulfonamide 5a and 2a. However, the desired 7a product was isolated in lower yields, under the optimized conditions and even after screening different bases and solvents (see the ESI† for more details). An N-benzylated compound 6a was then prepared from the reaction of 5a and 2a. Delightfully, the intramolecular arylogous Henry condensation reaction of 6a, in the presence of DBU in toluene, afforded the desired indole derivative 7a in 78% yield with 69% yield over two-steps. Consequently, we carried out a sequential reaction of 5a and 2a, without the isolation of N-benzyl intermediate 6a, that efficiently produced 7a in 74% yield (Scheme 4).
 | ||
| Scheme 4 Step-wise and sequential reactions of N-benzylation followed by intramolecular arylogous nitroaldol condensation for the synthesis of 7a. | ||
By selecting the competitive reaction conditions, the scope of the sequential N-benzylation and arylogous nitroaldol condensation was investigated. Initially, we focused on the substitutions on benzene ring of 2-(N-tosylamido)benzaldehyde 5. Accordingly, the reactions of methyl-substituted 5 and 2a were performed to generate the corresponding 2-aryl-substituted-indole derivatives 7b–c in good yields. Benzo-fused indole derivative 7d was synthesized in 63% yield from the reaction of ortho-tosylamidonaphthaldehyde 5d and 2a. We also prepared the starting materials 5 having different N-sulfonylaryl groups and subsequently subjected them to the N-benzylation–intramolecular arylogous nitroaldol condensation process to provide the corresponding 2-(2-nitrophenyl)indole derivatives 7e–7g in good yields. The reaction of N-(2-formylphenyl)methanesulfonamide 5h, bearing an N-sulfonylalkyl group, and 2a gave the respective indole derivative 7h in 69% yield. The reaction of ketone such as benzophenone 5j and 2a were performed to generate the corresponding 2,3-diaryl-substituted indole derivative 7i in 77% yield. We also examined the scope of ortho-nitrobenzy bromide through a reaction of 2-(bromomethyl)-4-fluoro-1-nitrobenzene 2b and 5a affording the respective indole derivative 7j in 70% yield (Scheme 5).
 | ||
| Scheme 5 Scope of the sequential transformation to access 2-(2-nitrophenyl)indole derivatives 7a–7j. | ||
However, this transformation was not successful having an N-benzyl group in the place of sulfonyl group in 5 suggesting the requirement of a strong EWG that would enhance the acidity of the methylene group of the intermediate 6 to undergo intramolecular arylogous nitroaldol condensation. To establish the practicality of the present transformation, we conducted gram-scale reactions of 1a/5a and 2a, individually, under the optimized reaction conditions to successfully furnish the corresponding benzofuran/indole derivatives 4a/7a in 70% and 67% yield, respectively (Scheme 6).
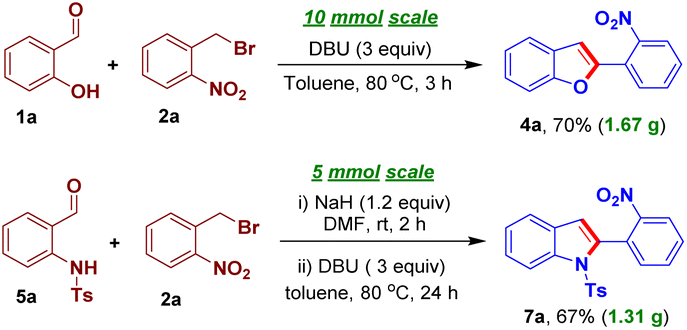 | ||
| Scheme 6 Gram-scale syntheses of the compounds 4a and 7a. | ||
The mechanism of the present O-/N-benzylation–intramolecular arylogous nitroaldol condensation process has been studied to know the requirement of nitro group and the involvement of the well-defined reaction intermediates. A reaction was performed using benzyl bromide 8, instead of 1-(bromomethyl)-2-nitrobenzene 2a, and salicylaldehyde; however, the corresponding benzofuran 4a′ was not observed and only O-alkylated product 9 was isolated in 80% yield (Scheme 7a). We then investigated alternative EWGs in place of NO2 group in 2a. Accordingly, the reactions of benzyl bromides 10/11 bearing nitrile (CN)/aldehyde (CHO) and salicylaldehyde were performed. In these reactions, the corresponding benzofuran derivatives were not observed while O-alkylated intermediates 12 and 13 were obtained in 88% and 83% of yield, respectively (Scheme 7b). These experiments suggest that a strong EWG such as nitro group is required that smoothly drive the later intramoleculararylogous condensation constructing the desired benzofuran derivative. We also conducted some control experiments to further know the intermediates of the reaction. The O-/N-alkylated intermediate 3a/6a was reacted with one equivalent of DBU to furnish the 3-hydroxy-dihydrobenzofuran 4ab/3-hydroxy-indoline 7aa in 82% and 90% yield, respectively, albeit with low diastereoselectivities while favouring the cis-isomer as determined by 2D-NMR analysis (see ESI† for more details). The dihydro compounds 4ab/7aa underwent dehydration reaction upon the treatment with 1.2 equiv. of DBU to give 4a/7a in high yields (Scheme 7c).
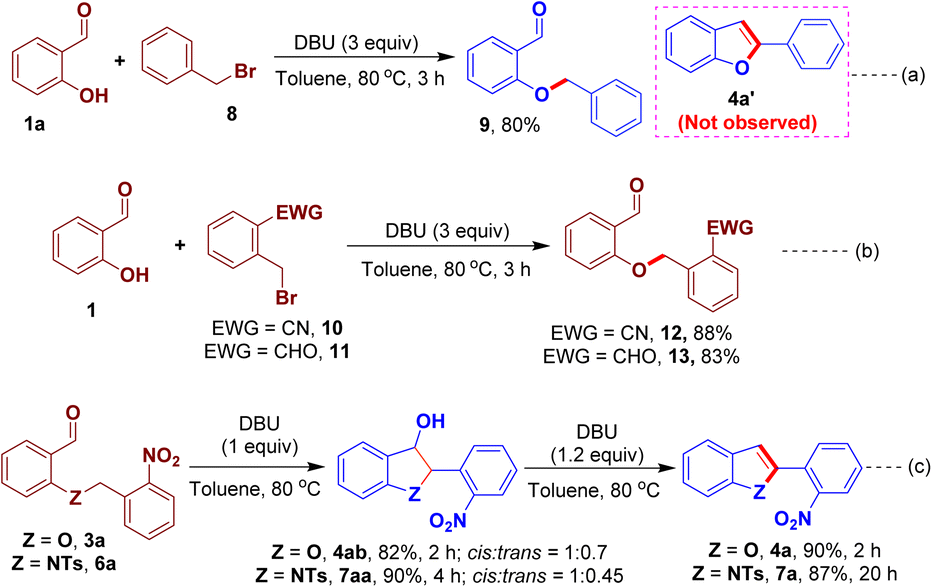 | ||
| Scheme 7 Control experiments. | ||
A mechanism for the present O-/N-benzylation followed by intramolecular arylogous nitroaldol condensation process is proposed based on the literature7d,e and control experiments. O-benzylation/N-benzylation of 1a/5a with 2a begins with a base-mediated reaction to give the intermediate 3a/6a, which had also been isolated (see Schemes 2 and 4). Base abstracts the acidic proton present on the benzylic CH2 group of 3a/6a, activated by the nitro group through conjugation, to provide intermediate I. The ensuing intermediate I undergoes intramolecular arylogous nitroaldol addition reaction to provide 3-hydroxy-substituted dihydrobenzofuran/indoline intermediate 4ab/7aa, which was also isolated (see Scheme 7). Base-mediated dehydration of 4ab/7aa delivers 2-(2-nitrophenyl)-benzofuran/indole derivative 4a/7a (Scheme 8).
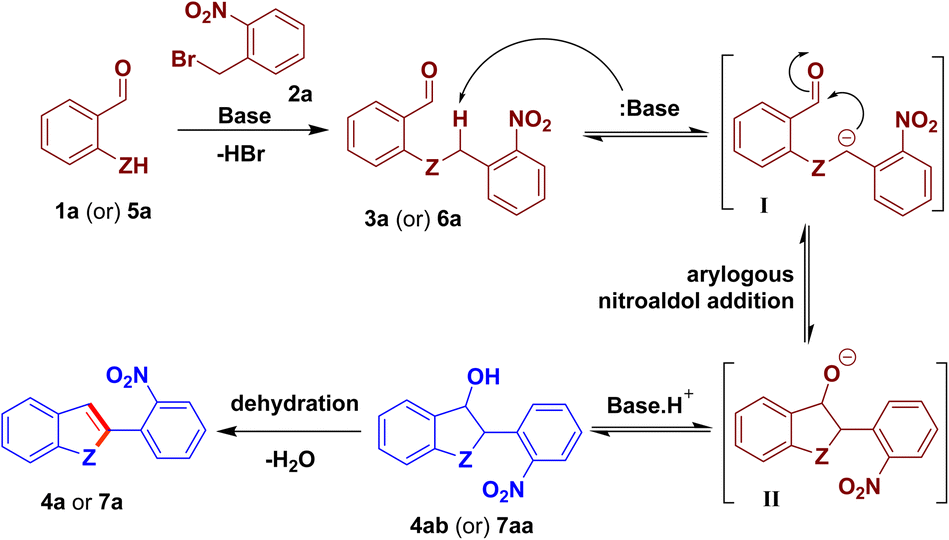 | ||
| Scheme 8 Plausible mechanism. | ||
A few post-synthetic transformations were performed using the representative benzofuran 4a and indole 7a. The nitro group of the compound 4a/7a was reduced in presence of Fe/NH4Cl to provide the corresponding 2-(benzofuran-2-yl)aniline 14 and 2-(1-tosyl-1H-indol-2-yl)aniline 15 in 92% and 90% yield, respectively. The compound 7a underwent detosylation to access the respective NH-indole derivative 16 in 92% yield12 (Scheme 9).
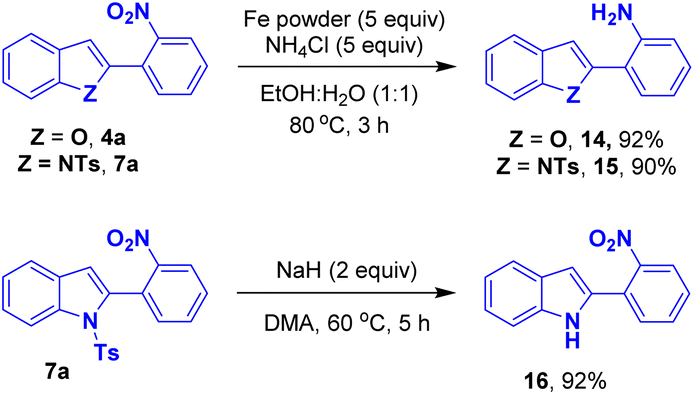 | ||
| Scheme 9 Post-synthetic transformations of benzofuran 4a and indole 7a. | ||
We became interested to demonstrate the potential application of our method in the synthesis of an approved anti-hypertensive drug, saprisartan bearing substituted-benzofuran moiety. According to our one-pot O-alkylation–arylogous nitroaldol condensation we could obtain 5-methyl-2-(2-nitrophenyl)benzofuran 4b from commercially available 5-methylsalicylaldehyde 1b and 2a(Scheme 3). The compound 4b underwent an NBS-mediated bromination at C-3 position of benzofuran followed by nitro reduction to yield the corresponding benzofuran derivative 17 in 63% yield over two steps. The compound 17 was treated with Boc anhydride, resulting in critical intermediate 18 in 72% yield. Compound 18 is an advanced key intermediate in the production of saprisartan, a medication approved to treat hypertension and heart failure. The known Pd-catalyzed method, starting from 5-methylbenzofuran, needed five consecutive steps to give compound 18 in only 3% overall yield.2b Furthermore, Noël and Wang, independently reported the palladium-catalyzed synthesis of an early intermediate of saprisartan, methyl 2-(5-methylbenzofuran-2-yl)benzoate.2c,e In sharp contrast, our strategy involving intramolecular arylogous nitroaldol condensation, using transition metal-free conditions, as a key step delivered the advanced intermediate 18 in four consecutive steps in a eleven-fold 34% overall yield (Scheme 10). Saprisartan can be synthesized from 18 using the reported literature, thus constituting the formal synthesis (see ESI† for details).
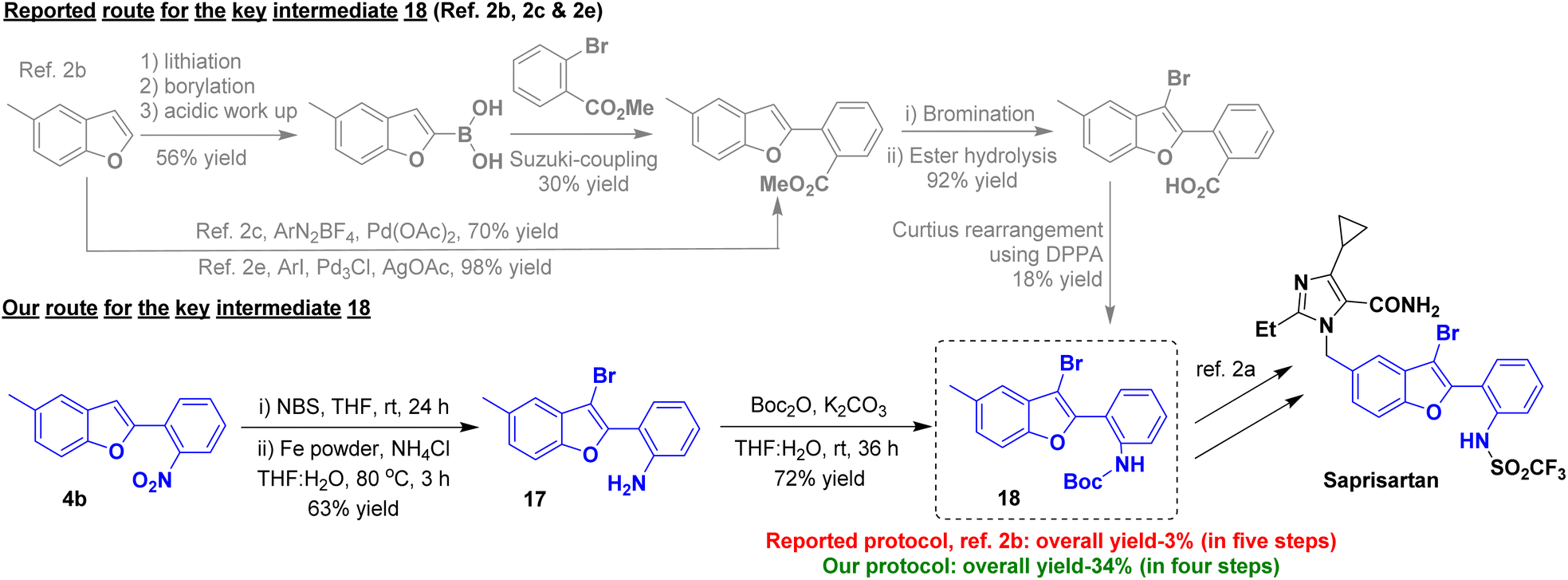 | ||
| Scheme 10 Formal synthesis of saprisartan. | ||
Conclusions
In summary, we have demonstrated a convenient, facile and transition metal-free protocol for the synthesis of diverse range of 2-(2-nitroaryl)benzofuran/indole derivatives via O-/N-alkylation followed by intramolecular arylogous nitroaldol condensation from the reaction of ortho-heteroatom-substituted aryl aldehydes/ketones and ortho-nitrobenzyl halides. Gram-scale reactions and post-synthetic modifications have been achieved proving the practicality of the developed method. Furthermore, the application of the developed protocol was successfully demonstrated in the synthesis of an advanced key intermediate and accomplished a formal synthesis of saprisartan.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Science & Engineering Research Board (SERB), Department of Science and Technology (DST), India for financial support in the form of a Core Research Grant (CRG) to SS (CRG/2022/001459). MSR thanks ICMR (File No. 45/10/2022-DDI/BMS), KS thanks CSIR for financial support in the form of fellowship. IICT communication No. IICT/Pubs./2024/408.Notes and references
- For selected papers on the chemistry and biology 2-aryl-substituted benzofuran and indoles, see: (a) S. Samosorn, J. B. Bremner, A. Ball and K. Lewis, Bioorg. Med. Chem., 2006, 14, 857–865 CrossRef CAS PubMed; (b) Y. Luo, Z. He and H. Li, Fitoterapia, 2007, 78, 211–214 CrossRef CAS PubMed; (c) L. D. Luca, G. Nieddu, A. Porcheddu and G. Giacomelli, Curr. Med. Chem., 2009, 16, 1–20 CrossRef PubMed; (d) Y. Jiang, B. Gao, W. Huang, Y. Liang, G. Huang and Y. Ma, Synth. Commun., 2009, 39, 197–204 CrossRef CAS; (e) M. T. Hovey, C. T. Check, A. F. Sipher and K. A. Scheidt, Angew. Chem., Int. Ed., 2014, 53, 9603–9607 CrossRef CAS PubMed; (f) K. Bera and C. Schneider, Chem.–Eur. J., 2016, 22, 7074–7078 CrossRef CAS PubMed; (g) Y. Yun, Y. Miao, X. Sun, J. Sun and X. Wang, J. Enzyme Inhib. Med. Chem., 2021, 36, 1345–1355 CrossRef CAS PubMed.
- (a) M. D. Dowle and D. B. Judd, US Pat., US005332831A, 1994; (b) D. B. Judd, M. D. Dowle, D. Middlemiss, D. I. C. Scopes, B. C. Ross, T. I. Jack, M. Pass, E. Tranquillini, J. E. Hobson, T. A. Panchal, P. G. Stuart, J. M. S. Paton, T. Hubbard, A. Hilditch, G. M. Drew, M. J. Robertson, K. L. Clark, A. Travers, A. A. E. Hunt, J. Polley, P. J. Eddershaw, M. K. Bayliss, G. R. Manchee, M. D. Donnelly, D. G. Walker and S. A. Richards, J. Med. Chem., 1994, 37, 3108–3120 CrossRef CAS PubMed; (c) H. P. L. Gemoets, I. Kalvet, A. V. Nyuchev, N. Erdmann, V. Hessel, F. Schoenebeck and T. Noël, Chem. Sci., 2017, 8, 1046–1055 RSC; (d) I. Balbuena-Rebolledo, I. I. Padilla-Martínez, M. C. Rosales-Hernández and M. Bello, Pharmaceuticals, 2021, 14, 791 CrossRef CAS PubMed; (e) J. Yao, L. Shao, X. Kang, M. Zhu, X. Huo and X. Wang, J. Org. Chem., 2024, 89, 1719–1726 CrossRef CAS PubMed.
- (a) J. A. Butera, S. A. Antane, B. Hirth, J. R. Lennox, J. H. Sheldon, N. W. Norton, D. Warga and T. M. Argentieri, Bioorg. Med. Chem. Lett., 2001, 11, 2093–2097 CrossRef CAS PubMed; (b) I. d. Peña and J. H. Cheong, J. Biomed. Biotechnol., 2011, 2011, 1 CrossRef PubMed; (c) J. S. Ha, Y. Cha, W. J. Cho, W. Lee, S. Geum and S. Kim, KO Pat., KR20230138209A, 2023 Search PubMed; (d) E. Lee, S.-B. Ko, S. Kim, E. Ahn, M. Jeon and J. Ju, US Pat., US2023240131A1, 2023 Search PubMed.
- (a) C. Schultz, A. Link, M. Leost, D. W. Zaharevitz, R. Gussio, E. A. Sausville, L. Meijer and C. Kunick, J. Med. Chem., 1999, 42, 2909–2919 CrossRef CAS PubMed; (b) W. F. Schmid, R. O. John, G. Mühlgassner, P. Heffeter, M. A. Jakupec, M. S. Galanski, W. Berger, V. B. Arion and B. K. Keppler, J. Med. Chem., 2007, 50, 6343–6355 CrossRef CAS PubMed; (c) B. Harish, M. Subbireddy and S. Suresh, Chem. Commun., 2017, 53, 3338–3341 RSC; (d) D. A. Aksenov, A. S. Akulova, E. A. Aleksandrova, N. A. Aksenov, A. V. Leontiev and A. V. Aksenov, Molecules, 2023, 28, 2324 CrossRef CAS PubMed.
- For recent excellent reviews on vinylogous transformations, see: (a) H. B. Hepburn, L. Dell'Amico and P. Melchiorre, Chem. Rec., 2016, 16, 1787–1806 CrossRef CAS PubMed; (b) C. Curti, L. Battistini, A. Sartori and F. Zanardi, Chem. Rev., 2020, 120, 2448–2612 CrossRef CAS PubMed and references cited therein..
- (a) B. Harish, M. Subbireddy, O. Obulesu and S. Suresh, Org. Lett., 2019, 21, 1823–1827 CrossRef CAS PubMed; (b) B. Harish, S. Yadav and S. Suresh, Chem. Commun., 2021, 57, 231–234 RSC; (c) L. Gummidi, A. Muddassar, G. V. M. Sharma, V. Murugesh and S. Suresh, Org. Biomol. Chem., 2022, 20, 773–777 RSC; (d) M. S. Reddy, J. B. Nanubolu and S. Suresh, Org. Biomol. Chem., 2023, 21, 5387–5397 RSC; (e) P. C. Behera, B. Anilkumar, J. B. Nanubolu and S. Suresh, Org. Lett., 2024, 26, 8654–8661 CrossRef CAS PubMed.
- For the selected papers on vinylogous/arylogousnitroaldol reactions, see: (a) M. F. A. Adamo and S. Suresh, Tetrahedron, 2009, 65, 990–997 CrossRef CAS; (b) E. Rajanarendar, S. R. Krishna, D. Nagaraju, K. G. Reddy, B. Kishore and Y. N. Reddy, Bioorg. Med. Chem. Lett., 2015, 25, 1630–1634 CrossRef CAS PubMed; (c) Y. Zhang, B.-W. Wei, H. Lin, L. Zhang, J.-X. Liu, H.-Q. Luo and X.-L. Fan, Green Chem., 2015, 17, 3266–3270 RSC; (d) O. Obulesu, J. B. Nanubolu and S. Suresh, Org. Biomol. Chem., 2015, 13, 8232–8240 RSC; (e) O. Obulesu, V. Murugesh, B. Harish and S. Suresh, J. Org. Chem., 2018, 83, 6454–6465 CrossRef CAS PubMed; (f) L. Yang, J. Zhao, X. Yang, M. Chena and Y. Xue, RSC Adv., 2019, 9, 4932–4941 RSC; (g) M. Ono, Y. Fuchi, T. Fuchigami, N. Kobashi, H. Kimura, M. Haratake, H. Saji and M. Nakayama, ACS Med. Chem. Lett., 2010, 1, 443–447 CrossRef CAS PubMed; (h) A. D’Amato and G. D. Sala, Catalysts, 2021, 11, 1545 CrossRef.
- (a) A. Ohta, Y. Akita, T. Ohkuwa, M. C. R. Fukunaga, A. Miyafuji, T. Nakata, N. Tani and Y. Aoyagi, Heterocycles, 1990, 31, 1951–1958 CrossRef CAS; (b) R. L. Hudkins, J. L. Diebold and F. D. Marsh, J. Org. Chem., 1995, 60, 6218–6220 CrossRef CAS; (c) A. Bourderioux, P. Kassis, J.-Y. Mérour and S. Routier, Tetrahedron, 2008, 64, 11012–11019 CrossRef CAS; (d) H. Srour, T.-H. Doan, E. D. Silva, R. J. Whitby and B. Witulski, J. Mater. Chem. C, 2016, 4, 6270–6279 RSC; (e) R. K. Konidena, K. h. Lee, J. Y. Lee and W. P. Hon, Org. Electron., 2019, 70, 211–218 CrossRef CAS.
- (a) P. Hu, Y. Shang and W. Su, Angew. Chem., Int. Ed., 2012, 51, 5945–5949 CrossRef CAS PubMed; (b) K. Pei, X. Jie, H. Zhao and W. Su, Eur. J. Org Chem., 2014, 4230–4233 CrossRef CAS; (c) A. Hfaiedh, K. Yuan, H. B. Ammar, B. B. Hassine, J.-F. Soulé and H. Doucet, ChemSusChem, 2015, 8, 1794–1804 CrossRef CAS PubMed.
- I. Chakrabarty, M. O. Akram, S. Biswas and N. T. Patil, Chem. Commun., 2018, 54, 7223–7226 RSC.
- H. R. Moon, H. Y. Chung, H. J. An, S. J. Son and D. H. Kim, EP Pat., EP3613735A1, 2020.
- W. Sun, X. Chen, Y. Hu, H. Geng, Y. Jiang, Y. Zhou, W. Zhu, M. Hu, H. Hu, X. Wang, X. Wang, S. Zhang and Y. Hu, Tetrahedron Lett., 2020, 61, 152442 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07741e |
| This journal is © The Royal Society of Chemistry 2024 |