
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of a novel curcumin–combretastatin A4 molecular skeleton: two pharmacophores†
Pravinkamaraj Ponraj and
Saravanakumar Rajendran *
*
Dept. of Chemistry, School of Advanced Sciences, Vellore Institute of Technology, Chennai Campus, Vandalur-Kelambakkam Road, Chennai 600127, Tamil Nadu, India. E-mail: Saravanakumar.r@vit.ac.in; Fax: +91 44 3993 2555; Tel: +91 44 3993 1479
First published on 20th November 2024
Abstract
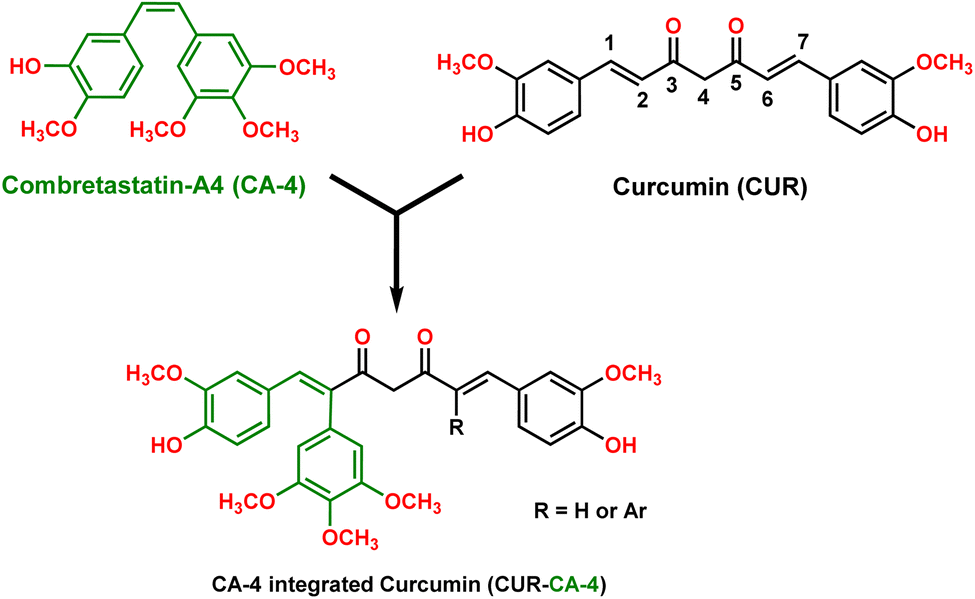
The logical design and synthesis of a novel compound combretastatin A-4-integrated curcumin is presented. Claisen condensation of phenylacetone with ethyl acetates formed 1,5-diphenylpentane-2,4-dione. Condensation of the dione with benzaldehyde via a modified Pabon procedure formed combretastatin A-4-integrated curcumin. The single-crystal X-ray structure of one of the CA-4 integrated CURs was established as a representative example. Curcumin (CUR) and combretastatin A-4 (CA-4) are well-known bioactive natural products; however, their poor pharmacokinetic profiles and cis–trans isomerization under in vivo conditions, respectively, have limited their biological applications. Herein, coupling of an aryl group at the olefinic C2 and/or C6 position of CUR integrates a CA-4-like structure with cis-configuration locked to CUR. At the same time, aryl coupling created steric hindrance around the olefinic bond and could resist the reductive metabolism of CUR and contribute to a better pharmacokinetic profile. Remarkably, this modification did not disturb the functional groups in both the natural products (CUR and CA-4), which is promising for their therapeutic effects. Thus, the synthesized CA-4-integrated CUR molecular architecture offers a new molecular skeleton to be explored for bio-application.
Introduction
The design and development of anticancer drug molecules with promising efficacy and reduced toxicity remain challenging in modern scientific research. Natural products and their variants are, by and large, preferable choices owing to their fewer or no side effects with high selectivity and efficacy. In this context, curcumin, an active principle of the plant Curcuma longa, commonly known as turmeric, is a well-known and extensively investigated natural product. Curcumin and its variants have been established to possess a broad spectrum of biological activities, including anticancer activity.1–4 Several studies have shown that CUR modulates/regulates multiple cellular and molecular targets, such as transcription factors, growth factors and their receptors, nuclear factors, and protein kinases, which may account for its broad spectrum of biological activity3,5–8 and fewer or no side effects.2,9 In addition, curcumin has been shown to be extremely safe even at high doses, 12 g per day, in various animal models and human studies.2,10 Despite its promising therapeutic properties and safety profile, curcumin is not approved as a therapeutic agent owing to its poor absorption, low bioavailability, rapid metabolism (via reduction and conjugation pathways in the liver and intestinal wall)11,12 and rapid systemic clearance of what little is absorbed.5,9,12,13 Curcumin's olefinic bonds, active methylene carbon, phenolic-OH and β-diketone have been established as primary reaction sites for its weak pharmacokinetic profile.10,12 At the same time, SAR studies revealed that these functional groups are important for curcumin's therapeutic effect.5 Thus, modifications altering these sites modify curcumin's pharmacokinetic profile and biological action. In earlier reports, modification of curcumin was primarily focused on aryl substitution, β-diketone, and active methylene carbon.5,6 Studies pertaining to modification at the olefinic bond are limited and have been scarcely explored. For example, Koo et al. (2015) reported a set of curcumin derivatives with a methyl group introduced at olefinic bonds at the C2 and C6 positions or the C2 position only of CUR. The methyl substitution created steric hindrance and thus prevented reductive metabolism of CUR as well as contributed to the enhancement of the bioavailability of CUR. Moreover, these compounds showed enhanced antiangiogenesis and antitumor activity against glioma, U87MG, in comparison with the parent compound, CUR.14On the other hand, combretastatin A-4 (CA-4), a cis-stilbenoid, isolated from the bark of the South African bush willow tree Combretum caffrum is a well-known microtubule depolymerizing agent.15–17 Several preclinical studies have shown that CA-4 is a potent anticancer agent.16,18,19 It binds to the colchicine site of tubulin and inhibits tubulin polymerization and arrests the cell cycle in the metaphase and triggers apoptosis.16,20 Also, it was found to be a potent vascular disrupting agent.16,18,20 Structure–activity relationship (SAR) studies suggested that trimethoxy substitution on the ring A and cis-configuration of the olefinic bond are crucial for its bioactivity (Fig. 1).16,18–20 Despite its high efficacy, CA-4 is vulnerable to undergoing transformation from the active cis-form to the inactive trans form under in vivo conditions.16,18,19 Several variants of CA-4 have been synthesized to improve its water solubility, pharmacological properties, and stability.16,19 One of the strategies that has been widely adopted is locking the cis-configuration of the olefinic bond into heterocyclic rings, such as imidazole, pyrazole, β-lactam, and triazole.16 However, many of the fused cyclic ring compounds displayed inferior antiproliferative activity than the parent compound, CA-4.16,19 Recently, Surendra R. Punganuru et al. (2016)21 adopted a different strategy to suppress the cis–trans isomerization. They realized that insertion of the aryl group to the olefinic bond (C-7 position) of piperlongumine (PL) (an active principle of long pepper) would create a stabilized CA-4-like structure locked in its cis-form while retaining the PL configuration. The new compounds exhibited potent antiproliferative activities against eight cancer cell lines, including the breast cancer cells MCF-7 and SKBR3. In another interesting study, a combination of curcumin (CUR) and combretastatin-A4 phosphate loaded in a nanodrug-delivery system (i.e. glycyrrhetinic acid (GA)-modified liposomes (GA LPs)) was studied for liver-targeted co-delivery. It was found to have higher cytotoxicity than the free drug molecules.22,23 The above study suggests a combination of curcumin and combretastatin-A4 works synergistically and has a better therapeutic effect. Keeping the above facts in mind, we designed and synthesized a new molecular skeleton that incorporates the structural features of both the natural products, curcumin and CA-4 (Fig. 1). Interestingly, the substitution of a phenyl group at the C2 and/or C6 positions of CUR integrated a CA-4 like structure to CUR. The new molecular skeleton now incorporated structural features of both CUR and CA-4 and has the following benefits:
 | ||
| Fig. 1 Schematic representation of novel CA-4-integrated curcumin. | ||
(a) Cis-configuration of CA-4 is locked to CUR, which is crucial for its anticancer activity. This was confirmed by single-crystal XRD structural analysis.
(b) Aryl substitution creates a steric barrier around the olefinic bond against reductive metabolizing enzymes and thus retards/prevents reductive metabolism, one of the major metabolic pathways leading to the low bioavailability of CUR.
(c) Key structural features responsible for the bioactivities of both CUR and CA-4 were retained, ensuring the innate activity of both CUR and CA-4 as well as their synergistic effect, offering improved efficacy, bioavailability, stability and selectivity.
Moreover, CUR derivatives with substitution at olefinic bonds (C2, and/or C6 positions) remain underexplored molecular architectures. Thus, our research represents an exciting step forward in exploring the medicinal chemistry and synergism of both natural products, CA-4 and CUR (Fig. 1).
Results and discussion
Our group is actively involved in the synthesis of curcumin hybrid molecules. In this context, we designed and synthesized a novel molecular skeleton, combretastatin A4-integrated curcumin. Retrosynthesis analysis of the same is outlined in Scheme 1. The target molecules could be readily synthesized from diketones and benzaldehydes. The diketones in turn could be obtained from phenylacetone and ethyl acetates (Scheme 1).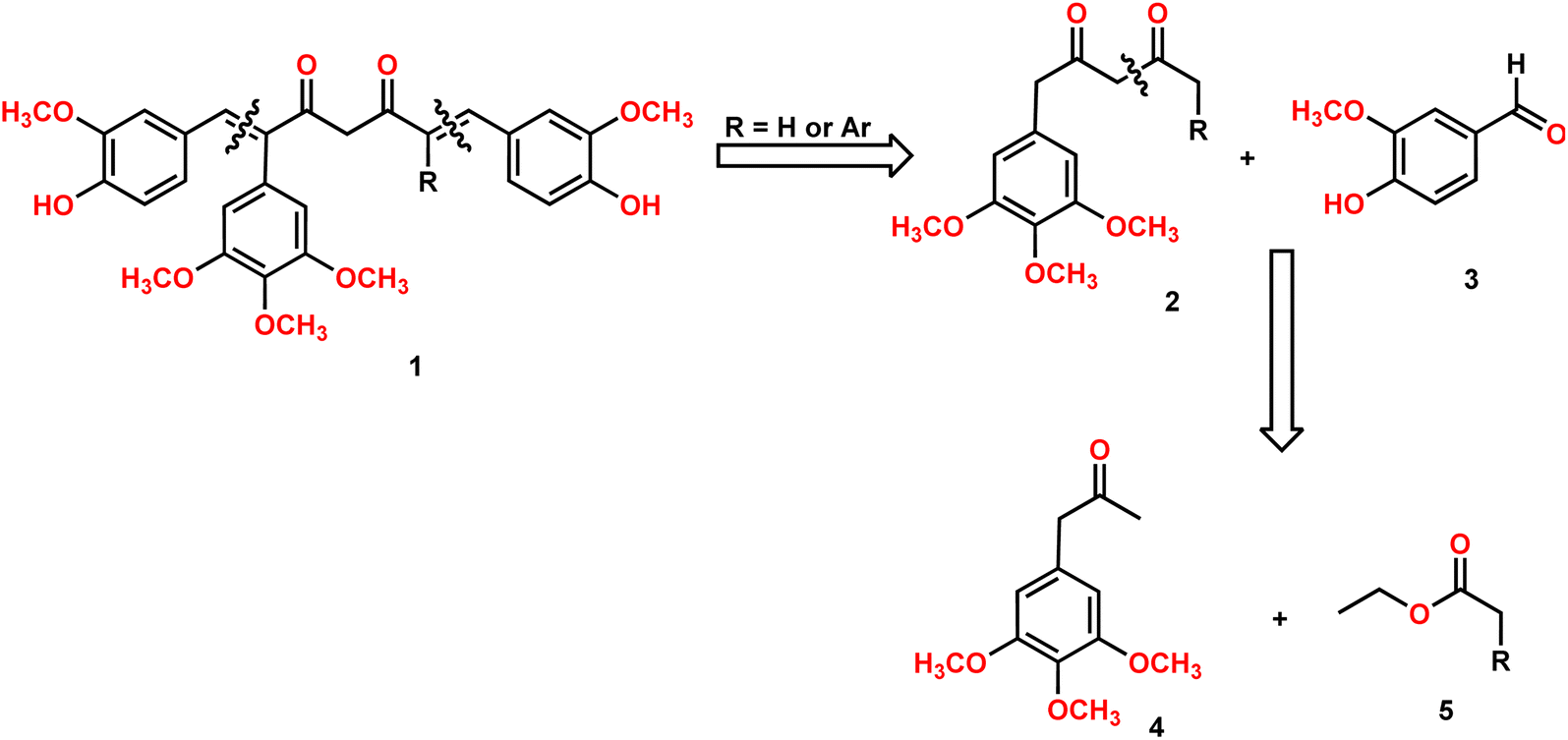 | ||
| Scheme 1 Retrosynthesis analysis of CA-4-integrated curcumin. | ||
Synthesis of diketones
Claisen condensation of phenyl acetones with esters (ethyl/ethyl phenyl acetate) under basic conditions yielded β-diketones in moderate yields (42–52%, Table 1). A literature-reported procedure was adopted for the synthesis of β-diketone.24 Initially, the reaction was attempted with various bases (NaH2, NaNH2, KOtBu3, KOH, K2CO3, and DBU) and solvents (THF, DMF, diethyl ether, toluene, methyl tertiary butyl ether (MTBE), and di-isopropyl ether (DIPE)). Sodamide and diethyl ether were found to be a suitable base and solvent to obtain the expected product in better yields. Phenyl 2,4-diones were prepared by reacting phenyl acetones with ethyl acetates (Table 1, Z = H), and di-phenyl 2,4-diones were prepared by reacting phenyl acetones with phenyl ethyl acetates (Table 1, Z = Ar). The synthesized diketones were characterized by 1H NMR, 13C NMR, and LC-MS (refer ESI†). The characterization data aligned with the structural attributes of diketones (refer to the ESI†).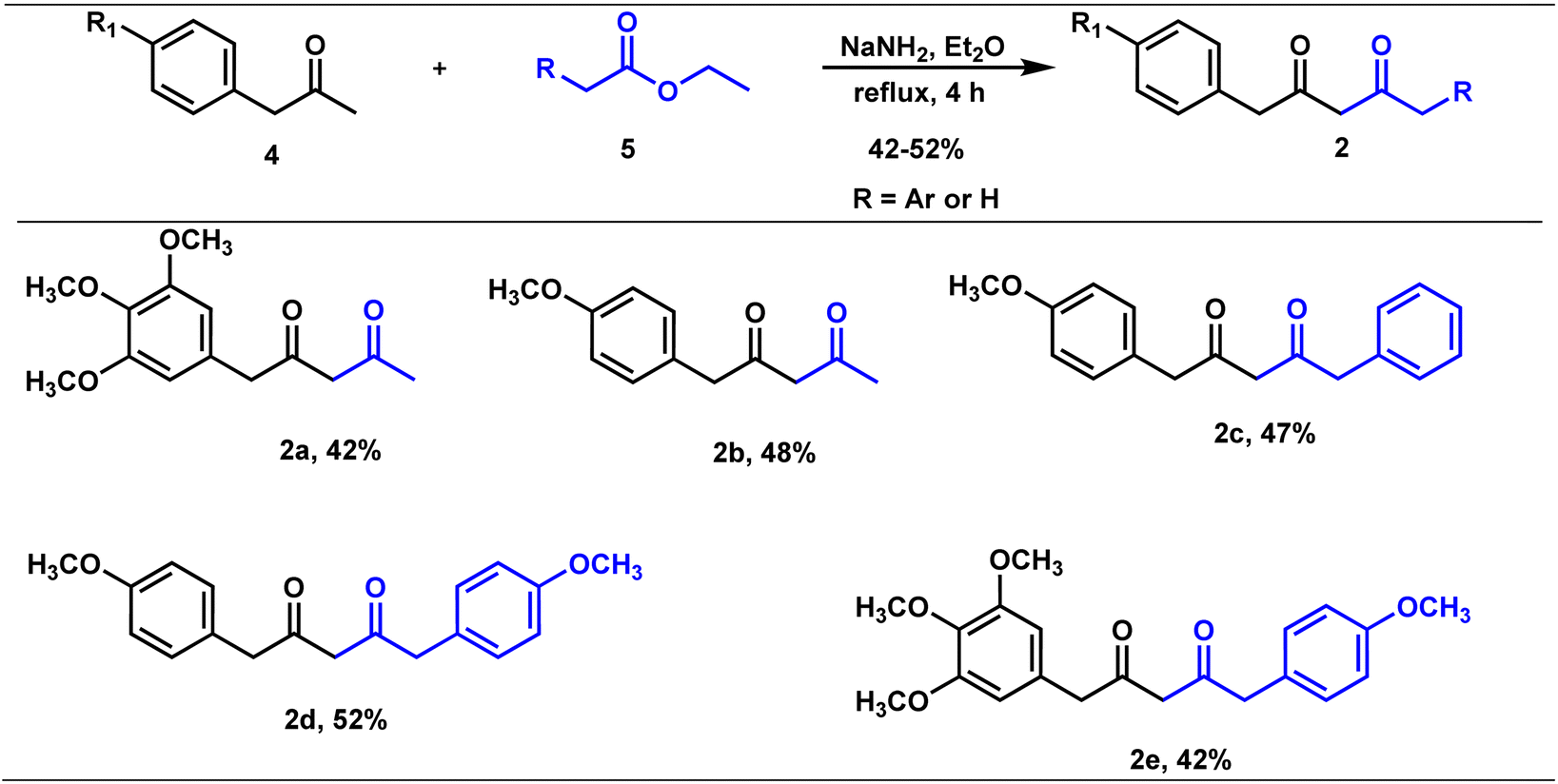 |
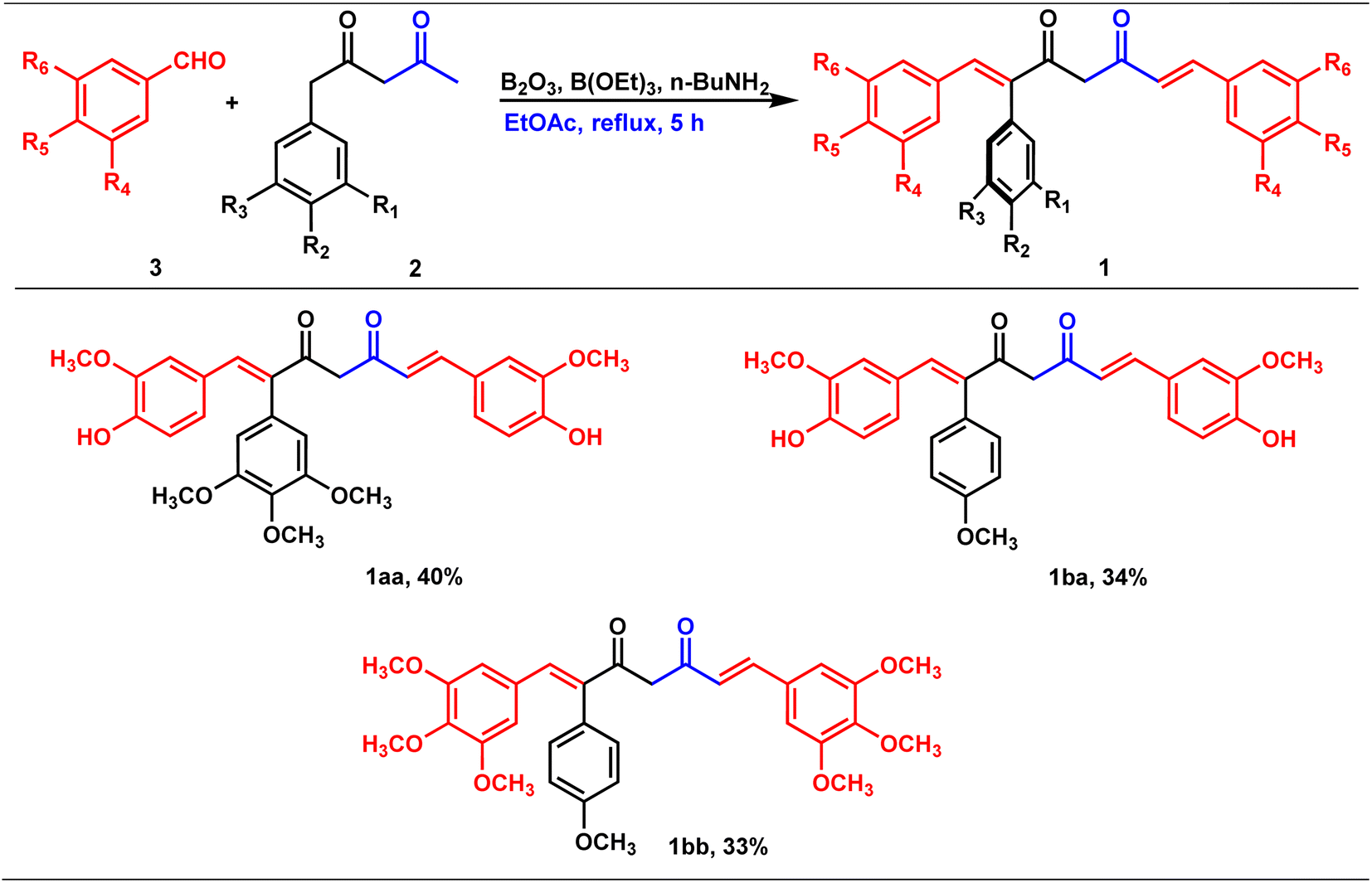
The precursors, phenyl 2,4-diones and di-phenyl 2,4-diones, synthesized were then condensed with vanillin/di or trimethoxy benzaldehydes to form mono- and di-combretastatin A4-integrated curcumins (Tables 2 and 3). Accordingly, phenyl 2,4-dione/di-phenyl 2,4-dione was reacted with benzaldehyde in the presence of boron trioxide (B2O3), trialkyl borate, and n-butylamine. A literature-reported4–6 synthetic protocol was adopted. In all of these methods, the initial step involved the formation of a 2,4-diketone–boron complex, which prevents Knoevenagel condensation between 2,4-diketone and benzaldehyde. A primary/secondary amine is used as a base to provide the necessary basicity for deprotonating the alkyl groups of the diketone. To eliminate the water generated during the condensation reaction, a scavenger, like alkyl borate, was used. Failure to remove the water can lead to its reaction with the diketone complex, thereby diminishing the yield of the target compounds. After completion, the reaction was acidified to break down the 2,4-diketone–boron complex and to form the expected product, mono/di-combretastatin A4-integrated curcumins in a 32–56% yield. The synthesis of mono- and di-combretastatin A4-integrated curcumins is outlined in Tables 2 and 3.
 |
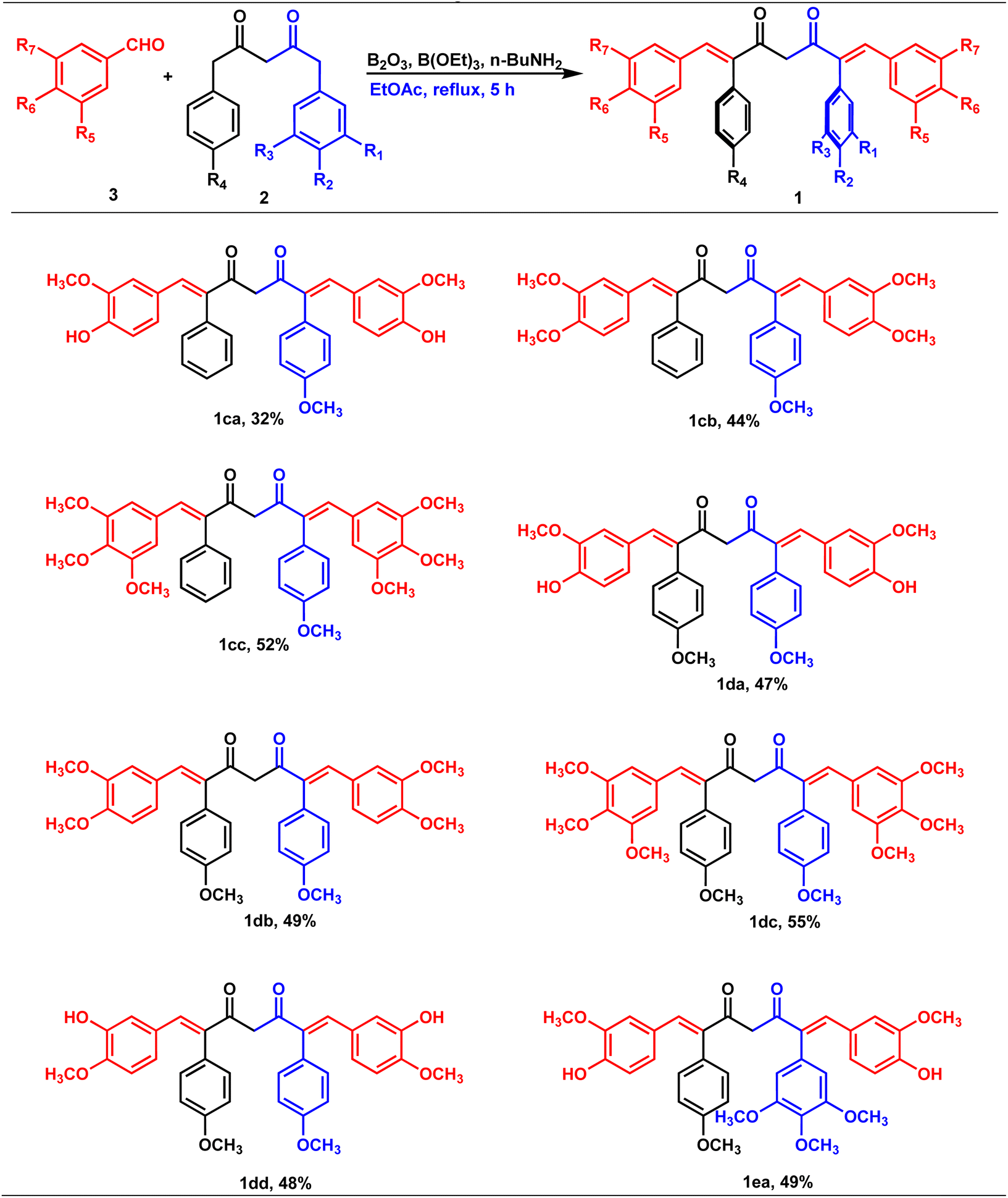 |
The formation of mono- and di-combretastatin A4-integrated curcumins was confirmed by 1H NMR, 13C NMR, FT-IR, and HR-MS. The 1H and 13C NMR spectral data were in accordance with the structural attributes of the mono- and di-combretastatin A4-integrated curcumins (refer to the ESI†). The structural elucidation of one of the di-combretastatin A4-integrated curcumins (1da) is described as a representative example (Fig. 2). NMR was recorded in DMSO-d6 at room temperature (25 °C). The broad singlet observed at 9.56 ppm was assigned to the phenolic –OH proton. The characteristic active methylene proton appeared as a singlet at 4.97 ppm. A distinct singlet at 3.76 ppm was assigned to the methoxy (–OCH3) proton adjacent to the phenolic group. The other methoxy proton peak merged with DMSO–H2O water and appeared as a broad singlet at 3.35 ppm. A representative AB-type doublet at 6.94 and 6.86 ppm was assigned to para-substituted phenyl protons. A doublet of doublet at 6.71 ppm corresponded to protons para to a methoxy group. A doublet at 6.63 ppm was designated to protons ortho to a phenolic group. An entwined doublet at 6.53 ppm was assigned to protons ortho to a methoxy group. 13C NMR showed a characteristic downfield signal for carbonyl carbon at 185.5 ppm. A distinct peak noticed at 159.2 ppm was assigned to β-olefinic carbon. Aromatic carbon signals were observed in the range of 148.8–114.0 ppm. A positive signal at 100.1 was assigned to active methylene carbon. Two peaks around 55.5 and 55.1 were designated to methoxy carbons. The FT-IR spectrum featured characteristic –OH stretching band around 3358 cm−1 and keto carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching band around 1591 cm−1. The high-resolution mass spectrum showed a molecular mass corresponding to [M + H] ion, which matched with the corresponding calculated mass. Further, the structure of one of the di-combretastatin A4-integrated curcumins (1ca) was unambiguously established by single-crystal X-ray diffraction analysis (Fig. 3).
O) stretching band around 1591 cm−1. The high-resolution mass spectrum showed a molecular mass corresponding to [M + H] ion, which matched with the corresponding calculated mass. Further, the structure of one of the di-combretastatin A4-integrated curcumins (1ca) was unambiguously established by single-crystal X-ray diffraction analysis (Fig. 3).
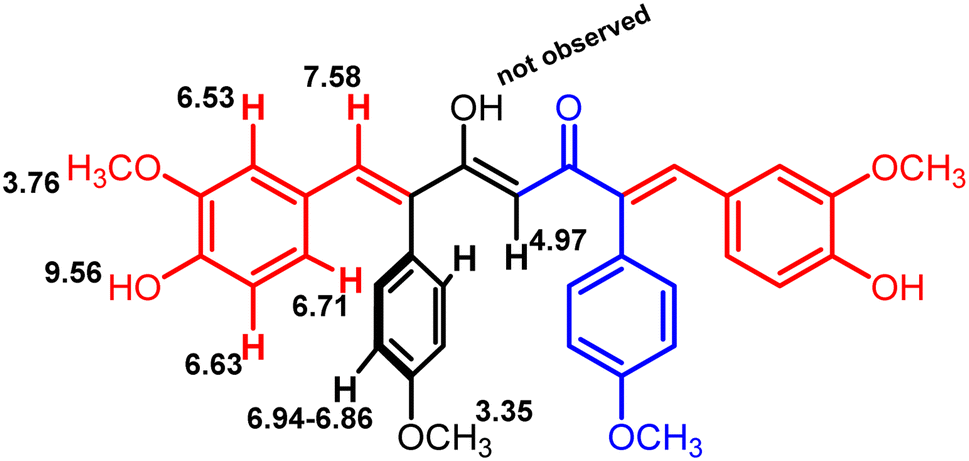 | ||
| Fig. 2 ChemDraw structure of compound 1da with 1H NMR peak values (in ppm) marked (refer ESI, Fig. 21†). | ||
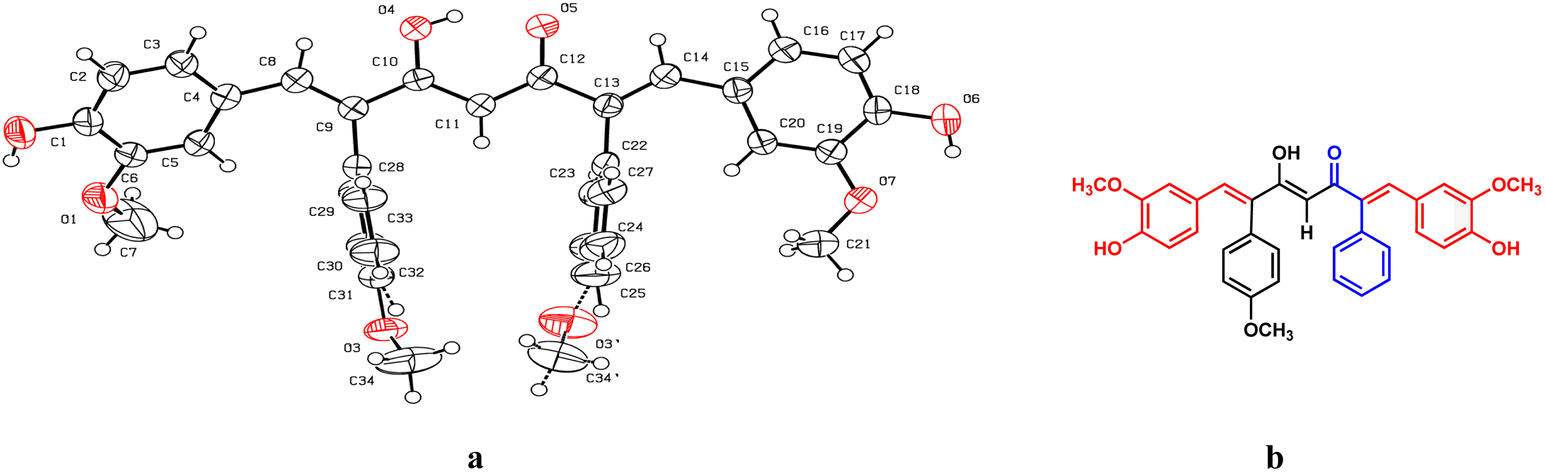 | ||
| Fig. 3 Single-crystal X-ray structure of combretastatin A4-integrated curcumin (1ca), (a) ORTEP diagram and (b) equivalent ChemDraw structure of 1ca. | ||
Single-crystal X-ray structure
A saturated solution of 1ca in toluene was allowed to evaporate slowly at room temperature. Pale yellow needle-shaped crystals were formed after a week. The quality of the crystals was found to be suitable for single-crystal X-ray diffraction studies. The compound crystallized in the triclinic system with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Fig. 3). From the crystal structure, it is clear that the CA-4 unit integrated into curcumin was locked in its cis-form, a crucial structural feature required for its biological activities. In the solid-state structure, combretastatin A4-integrated curcumin is in its enol form and the enol hydrogen is involved in intra- and intermolecular hydrogen bonding. Also, the enol oxygen is involved in aromatic C–H⋯O hydrogen bonds with adjacent molecules. The phenolic –OH groups are involved in hydrogen bonds with the phenolic –OH and –OCH3 of adjacent molecules and contribute to the stability of the solid-state structure (Fig. 3).
space group (Fig. 3). From the crystal structure, it is clear that the CA-4 unit integrated into curcumin was locked in its cis-form, a crucial structural feature required for its biological activities. In the solid-state structure, combretastatin A4-integrated curcumin is in its enol form and the enol hydrogen is involved in intra- and intermolecular hydrogen bonding. Also, the enol oxygen is involved in aromatic C–H⋯O hydrogen bonds with adjacent molecules. The phenolic –OH groups are involved in hydrogen bonds with the phenolic –OH and –OCH3 of adjacent molecules and contribute to the stability of the solid-state structure (Fig. 3).
Synthesis of pyrazole derivatives of combretastatin A-4-integrated curcumin
The therapeutic potential of curcumin is mainly hampered by its poor absorption, rapid metabolism (via reduction and conjugation pathways at the liver and intestinal wall),11,12 and rapid systemic clearance of what little is absorbed.13 Thus, variants of curcumin have been synthesized to improve its pharmacokinetic profile and bioactivities. One such class of derivative that is widely known and studied is curcumin pyrazoles, which possess improved pharmacological properties and metabolic stability compared to curcumin. For example, Jeong Kwon et al.25 showed that curcumin pyrazole could effectively inhibit the proliferation of bovine aortic endothelial cells (BAECs) at a very low concentration (IC50 = 520 nM) with no cell toxicity. They also found that curcumin pyrazole could be a potent anti-angiogenic agent. Similarly, in another report, Narlawar and co-workers26 found that curcumin pyrazole could inhibit the development of fibrillar Aβ42 and tau protein aggregation, with a 100-fold better activity than the parent compound, CU. Avadhesha Surolia et al.27 unveiled that curcumin pyrazoles are seven to nine times more potent than curcumin against chloroquine-sensitive (CQ-S) and chloroquine-resistant (CQ-R) Plasmodium falciparum. In this context, we intended to prepare combretastatin A4-integrated curcumin pyrazole derivatives (Table 4) with the hope of improving the pharmacological properties and aqueous solubility. Accordingly, the synthesized combretastatin A4-integrated curcumins (1db and 1dc) were reacted with hydrazine hydrate in acetic acid at 60 °C (oil bath) for 12 h to form their pyrazole derivatives (Table 4). A literature-reported procedure was adopted for the synthesis of pyrazole derivatives of curcumin. In principle, this simple modification should alter the compound's properties, including its pharmacological profile, solubility, and pharmacokinetic profile.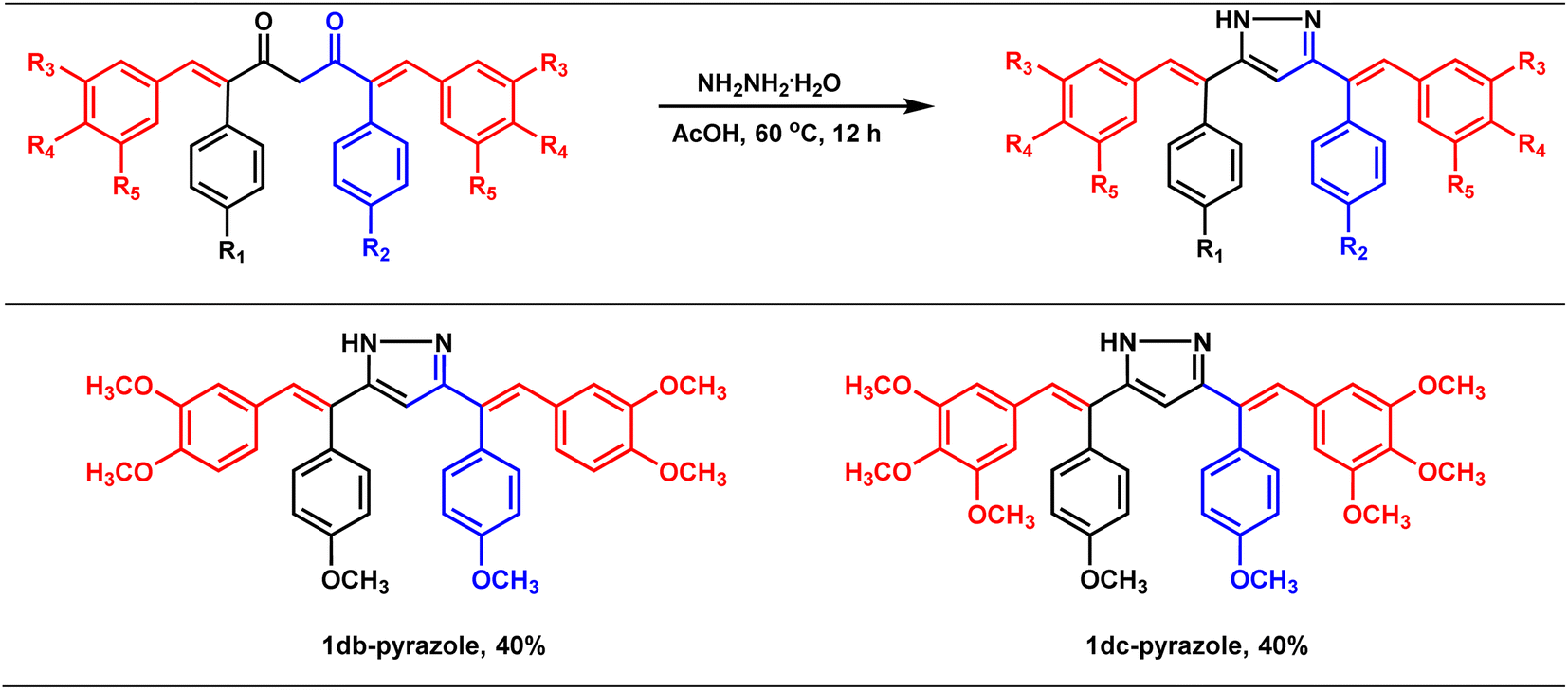 |
Conclusion
A logical design and synthesis of novel target compounds, combretastatin A4-integrated curcumins, was demonstrated. Coupling a phenyl group at the C2 and/or C6 position of curcumin integrated a combretastatin A4-like structure to curcumin to form the target compounds. A simple condensation of benzaldehyde with phenyl 2,4-dione/di-phenyl 2,4-dione via a modified Pabon procedure led to combretastatin A4-integrated curcumins in moderated yields. The target compounds were characterized by 1H, 13C NMR, and HR-MS. Further, the structure of one of the target compounds (1ca) was unambiguously established by single-crystal X-ray diffraction. The single-crystal X-ray diffraction study revealed that combretastatin A4 was locked in its cis-configuration, a crucial structural feature responsible for its pharmacological activity. To widen the scope, pyrazole derivatives of two of the combretastatin A4-integrated curcumins were synthesized and characterized. Thus, our findings unveiled a new molecular skeleton to be investigated with positive hopes for a better pharmacokinetic profile, and biological activity, with less or no side effects.Data availability
The full experimental data related to this article are given in the ESI,† including the synthetic procedure, FT-IR spectra of combretastatin A4-integrated curcumins, 1H NMR, and 13C NMR spectra of the combretastatin A4-integrated curcumins and their precursors. CIF data of combretastatin A4-integrated curcumin (1ca) are deposited in the Cambridge Crystallographic Data Centre (CCDC) and its CCDC number is 2383033.Conflicts of interest
Authors have no conflict of interest to declare.Acknowledgements
P. P. and R. S. K. thanks, Dept. of Chemistry and VIT Chennai, for providing infrastructure and instrumentation facilities (GC-MS FT-IR). SAIF, VIT Vellore is gratefully acknowledged for recording 1H and 13C NMR, single-crystal XRD and HR-MS.References
- G. Sa, T. Das, S. Banerjee and J. Chakraborty, Al Ameen J. Med. Sci., 2010, 3, 21–37 CAS.
- V. Soleimani, A. Sahebkar and H. Hosseinzadeh, Phytother. Res., 2018, 32, 985–995 CrossRef CAS PubMed.
- A. B. Kunnumakkara, D. Bordoloi, G. Padmavathi, J. Monisha, N. K. Roy, S. Prasad and B. B. Aggarwal, Br. J. Pharmacol., 2017, 174, 1325–1348 CrossRef CAS PubMed.
- F. Alibeiki, N. Jafari, M. Karimi and H. P. Dogaheh, Sci. Rep., 2017, 7, 2559 CrossRef PubMed.
- P. Anand, S. G. Thomas, A. B. Kunnumakkara, C. Sundaram, K. B. Harikumar, B. Sung, S. T. Tharakan, K. Misra, I. K. Priyadarsini, K. N. Rajasekharan and B. B. Aggarwal, Biochem. Pharmacol., 2008, 76, 1590–1611 CrossRef CAS PubMed.
- B. Aggarwal, L. Deb and S. Prasad, Molecules, 2014, 20, 185–205 CrossRef PubMed.
- P. Anand, C. Sundaram, S. Jhurani, A. B. Kunnumakkara and B. B. Aggarwal, Cancer Lett., 2008, 267, 133–164 CrossRef CAS PubMed.
- B. Kocaadam and N. Şanlier, Crit. Rev. Food Sci. Nutr., 2017, 57, 2889–2895 CrossRef CAS PubMed.
- P. Anand, A. B. Kunnumakkara, R. A. Newman and B. B. Aggarwal, Mol. Pharm., 2007, 4, 807–818 CrossRef CAS PubMed.
- G. Liang, L. Shao, Y. Wang, C. Zhao, Y. Chu, J. Xiao, Y. Zhao, X. Li and S. Yang, Bioorg. Med. Chem., 2009, 17, 2623–2631 CrossRef CAS PubMed.
- M. J. Yoon, Y. J. Kang, J. A. Lee, I. Y. Kim, M. A. Kim, Y. S. Lee, J. H. Park, B. Y. Lee, I. A. Kim, H. S. Kim, S.-A. Kim, A.-R. Yoon, C.-O. Yun, E.-Y. Kim, K. Lee and K. S. Choi, Cell Death Dis., 2014, 5, e1112 CrossRef CAS PubMed.
- M. H. Pan, T. M. Huang and J. K. Lin, Drug Metab. Dispos., 1999, 27, 486–494 CAS.
- C. Tamvakopoulos, K. Dimas, Z. D. Sofianos, S. Hatziantoniou, Z. Han, Z.-L. Liu, J. H. Wyche and P. Pantazis, Clin. Cancer Res., 2007, 13, 1269–1277 CrossRef CAS PubMed.
- H.-J. Koo, S. Shin, J. Y. Choi, K.-H. Lee, B.-T. Kim and Y. S. Choe, Sci. Rep., 2015, 5, 14205 CrossRef CAS PubMed.
- S. Aprile, E. Del Grosso, G. C. Tron and G. Grosa, Drug Metab. Dispos., 2007, 35, 2252–2261 CrossRef CAS PubMed.
- Z. S. Seddigi, M. S. Malik, A. P. Saraswati, S. A. Ahmed, A. O. Babalghith, H. A. Lamfon and A. Kamal, Med. Chem. Commun., 2017, 8, 1592–1603 RSC.
- K. G. Pinney, C. Jelinek, K. Edvardsen, D. J. Chaplin and G. Pettit, The discovery and development of the combretastatins, in Anticancer Agents from Natural Products, CRC Press, 2005 Search PubMed.
- G. C. Tron, T. Pirali, G. Sorba, F. Pagliai, S. Busacca and A. A. Genazzani, J. Med. Chem., 2006, 49, 3033–3044 CrossRef CAS PubMed.
- Q. Li and H. L. Sham, Expert Opin. Ther. Pat., 2002, 12, 1663–1702 CrossRef CAS.
- L. Li, S. Jiang, X. Li, Y. Liu, J. Su and J. Chen, Eur. J. Med. Chem., 2018, 151, 482–494 CrossRef CAS PubMed.
- S. R. Punganuru, H. R. Madala, S. N. Venugopal, R. Samala, C. Mikelis and K. S. Srivenugopal, Eur. J. Med. Chem., 2016, 107, 233–244 CrossRef CAS PubMed.
- H. Jiang, Z.-P. Li, G.-X. Tian, R.-Y. Pan, C.-M. Xu, B. Zhang and J. Wu, Int. J. Nanomed., 2019, 14, 1789–1804 CrossRef CAS PubMed.
- A. Reeves, E. Wickstrom and S. Vinogradov, Curcumin-Combretastatin Nanocells as Breast Cancer Cytotoxic and Antiangiogenic Agent, 2008, https://apps.dtic.mil/sti/tr/pdf/ADA494015.pdf Search PubMed.
- E. C. Y. Woon, P. T. Sunderland, H. A. Paine, M. D. Lloyd, A. S. Thompson and M. D. Threadgill, Bioorg. Med. Chem., 2013, 21, 5218–5227 CrossRef CAS PubMed.
- J. S. Shim, D. H. Kim, H. J. Jung, J. H. Kim, D. Lim, S.-K. Lee, K.-W. Kim, J. W. Ahn, J.-S. Yoo, J.-R. Rho, J. Shin and H. J. Kwon, Bioorg. Med. Chem., 2002, 10, 2987–2992 CrossRef CAS PubMed.
- R. Narlawar, M. Pickhardt, S. Leuchtenberger, K. Baumann, S. Krause, T. Dyrks, S. Weggen, E. Mandelkow and B. Schmidt, ChemMedChem, 2008, 3, 165–172 CrossRef CAS PubMed.
- S. Mishra, K. Karmodiya, N. Surolia and A. Surolia, Bioorg. Med. Chem., 2008, 16, 2894–2902 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2383033. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra06618a |
| This journal is © The Royal Society of Chemistry 2024 |