
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel pyridine and pyrazolyl pyridine conjugates with potent cytotoxicity against HepG2 cancer cells as PIM-1 kinase inhibitors and caspase activators†
Mohamed S. Nafie *ab,
Ahmed Hamzabc,
Ahmed H. Moustafa*d,
Hassan A. El-Sayed*de,
Samir M. El Rayesb,
Hesham A. Morsyf and
Ahmed Aboelmagedb
*ab,
Ahmed Hamzabc,
Ahmed H. Moustafa*d,
Hassan A. El-Sayed*de,
Samir M. El Rayesb,
Hesham A. Morsyf and
Ahmed Aboelmagedb
aDepartment of Chemistry, College of Sciences, University of Sharjah, P.O. 27272, Sharjah, United Arab Emirates. E-mail: mohamed.elsayed@sharjah.ac.ae
bDepartment of Chemistry, Faculty of Science, Suez Canal University, Ismailia, 41522, Egypt. E-mail: mohamed_nafie@science.suez.edu.eg
cDepartment of Community Health, Community Health, Diwaniyah Technical Institute, Al-Furat Al-Awsat Technical University, Kufa, Iraq
dDepartment of Chemistry, Faculty of Science, Zagazig University, Zagazig, 44519, Egypt. E-mail: hasanneg@gmail.com; ah_hu_mostafa@yahoo.com
eAlzahrawi University College, Karbala, Iraq
fHigher Institution of Engineering & Modern Technology, El Marg, Cairo, Egypt
First published on 13th December 2024
Abstract
A novel series of nicotinonitrile and pyrazolyl nicotinonitrile were synthesized, and their PIM-1 kinase inhibitors and caspase activators were investigated. New Manich bases 6–8 were synthesized via reaction of pyridine 4 with piperidine, dimethyl amine, and morpholine in the presence of formalin. On the other hand, the pyrazolyl analogues 10–12 were synthesized via heterocyclization of acetohydrazide derivative 9 with acetylacetone, malononitrile, and ethyl cyanoacetate, respectively, in ethanol. The cytotoxic activity of compound 9 against MCF-7 and HepG2 cells was particularly noteworthy, with IC50 values of 0.34 μM and 0.18 μM, respectively, among these derivatives. Compared to staurosporine with potent PIM-1 kinase inhibition, which had an IC50 value of 16.7 nM and an inhibition of 95.6%, compound 9 had a strong inhibitory effect, with IC50 values of 20.4 nM and 93.8%. It induced apoptosis activity in HepG2 cancer cells. Accordingly, compound 9 was proven to be an effective chemotherapeutic drug that targets PIM-1 in treating liver cancer.
1 Introduction
Cancer is a dreadful disease that causes significant mortality worldwide. According to rough estimates, in 2024, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 001140 new cancer cases and 611720 cancer deaths are projected to occur in the United States. Liver cancer, one of the leading causes of cancer-related deaths, has around 34000 mortalities in the year 2024, and the numbers are still increasing.1
001140 new cancer cases and 611720 cancer deaths are projected to occur in the United States. Liver cancer, one of the leading causes of cancer-related deaths, has around 34000 mortalities in the year 2024, and the numbers are still increasing.1
PIM-1, a member of the calcium/calmodulin-dependent kinase (CaMK) family, is a serine/threonine kinase with a unique αC-helix-in/αC-helix-out equilibrium and a catalytic domain structure that includes an activation loop, a C-terminal extension, and a P+1-loop. One of three isoforms (PIM1-3) was first seen after retrovirus gene transformation; the most researched isoform is PIM-1.2 The 44 kDa cytosolic protein encoded by PIM-1 is known to serve many roles in various solid and hematological malignancies that exhibit elevated enzyme expression. Moreover, PIM-1 phosphorylates integrin-linked kinase (ILK), which subsequently plays a role in invading human osteosarcoma cells. Additionally, PIM-1 kinase reduces ROS production in hepatocellular cancer and may be an important target for starting cytotoxicity.3
Many biological processes rely on PIM kinases, including apoptosis, cell proliferation, differentiation, and signaling pathways linked to cancer.4 Multiple studies have linked high PIM expression levels to human hematologic and epithelial malignancies, making these genes attractive therapeutic targets.5 The unique active location of PIM-1 kinase makes it easier to design tiny molecules that block the enzyme.6 Evidence shows that increases in PIM-1 are associated with carcinogenesis and metastasis.7 Several tumor types, including pancreatic, breast, prostate, hepatic, and colon cancers, showed elevated expression of PIM-1 kinase.8 This suggests that targeting PIM-1 with small-molecule medicines could be an effective way to halt cancer progression.9 Liver cancer is among the leading causes of cancer mortality, emphasizing the need for novel therapeutic agents. PIM-1 kinase, known for its role in apoptosis and cell survival, has emerged as a promising target due to its overexpression in hepatic malignancies. Pyridine and pyrazolyl derivatives, widely recognized for their biological versatility, offer an excellent platform for developing kinase inhibitors.
The heterocyclic molecules are highly valued in drug design and development due to their natural origin and the presence of a versatile pharmacophore that exhibits biological activity. Notably, approximately 90% of bioactive pharmaceuticals contain a heterocyclic moiety,10 and over 75% of FDA-approved marketed drugs feature a heterocyclic analog with nitrogen in their ring structure11–13 Pyridine is a vital class of compounds characterized by a six-membered ring structure containing one nitrogen atom. Pyridine derivatives are the most common and significant heterocyclic compounds, which show their fundamental characteristics to various pharmaceutical agents and natural products.14,15 It also serves as an important structural unit in numerous pharmaceuticals,16,17 and the pyridine moiety is associated with a wide range of biological activities, including anti-cancer, antiviral, analgesic, and antioxidant properties.8–10 Interestingly, pyridine-based structures (V–VIII), including nicotinonitrile were able to induce PIM-1 kinase inhibition and exhibited promising anti-cancer activities (Fig. 1).18–20 Pyrazole-based structures (I–IV), belonging to the class of azoles, are highly effective heterocycles that play a crucial role in drug chemistry, as well as in biological and photochemical properties. Moreover, the pyrazole scaffold is highly versatile and is utilized in a wide range of drugs that exhibit a broad spectrum of pharmacological activities, including anti-infective anti-cancer through PIM-1 kinase inhibition (Fig. 1).25,26 As seen in Fig. 1B with molecular visualization of PIM-1 kinase protein of the co-crystallized ligand making analyses of lipophilic and hydrophobic moieties to highlight the importance of cyanopyridine-based compounds.
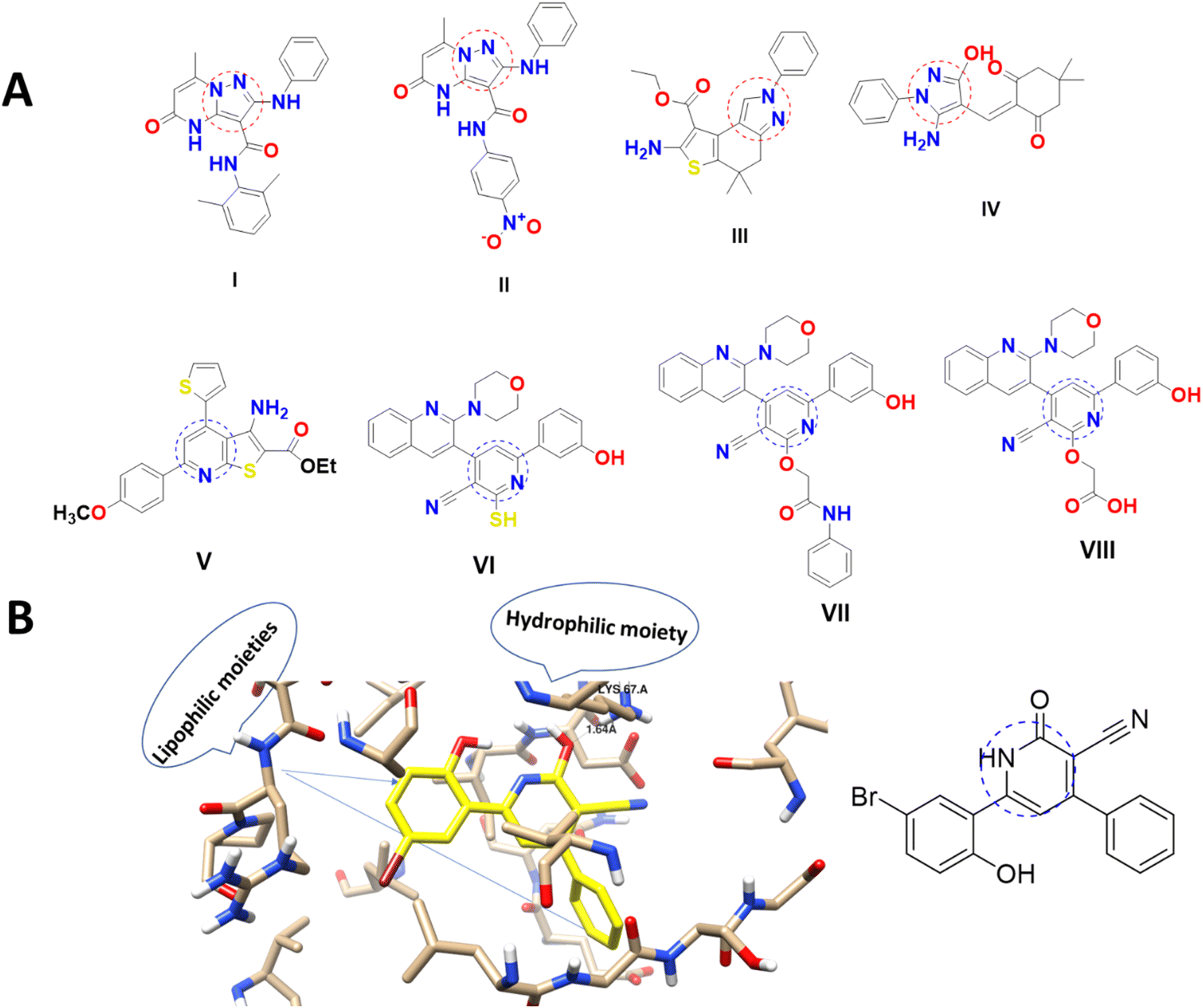 | ||
| Fig. 1 (A) Pyrazole and cyanopyridine derivatives as PIM-1 kinase inhibitors18–23 and (B) molecular visualization of the PIM-1 active site (PDB ID = 2OBJ) with cyanopyridine inhibitor.24 | ||
Apoptosis and cell cycle progression are two of the most important biological processes, and PIM kinases play an essential role in regulating these proteins. Notably, Pim kinases are overexpressed in liver cancer. Therefore, they are seen as promising targets for the development of new therapies that target the liver.4,27
One useful method in medicinal chemistry is the Mannich reaction, which is used to synthesize novel chemical compounds with important biological features. Pharmacological activity, bioavailability, and effectiveness are all affected by the physicochemical properties of prospective drug candidates, which can be altered using this process. Pharmaceutics and medicinal chemistry are persistently interested in Mannich bases because of their helpful biological activities and techniques of synthesis.28 Given all this, the pharmacological properties of the pyridine scaffold can be improved by incorporating pyrazolyl, Mannich bases, and acetyl hydrazide at C![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) center of the pyridine nucleus. Hydrazide moiety was reported to enhance hydrogen bonding potential with the kinase active site and modulate the pharmacokinetics of drugs.29
center of the pyridine nucleus. Hydrazide moiety was reported to enhance hydrogen bonding potential with the kinase active site and modulate the pharmacokinetics of drugs.29
The performed structural analysis, together with the results of the biological tests, show that cyanopyridine could potentially be an efficient liver cancer therapeutic compound. Focusing on the molecular mechanism of action of cyanopyridine specifically for liver cancer provides insight into the possible therapeutic effect of this compound. Furthermore, the obtained data can be used for further development of cyanopyridine as a safe and effective liver cancer therapeutic strategy. In continuation of our research line, our current research interests aim at developing novel pyridine and pyrazolyl pyridine conjugates with potent cytotoxicity against liver cancer cells and identifying the molecular target and effective cell death mechanism.30–33
2 Results chemistry
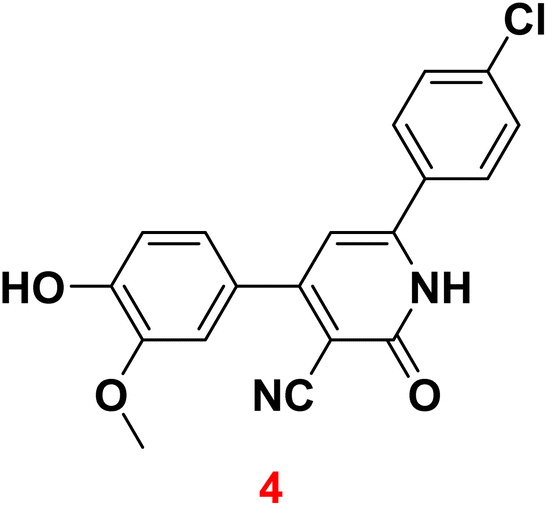
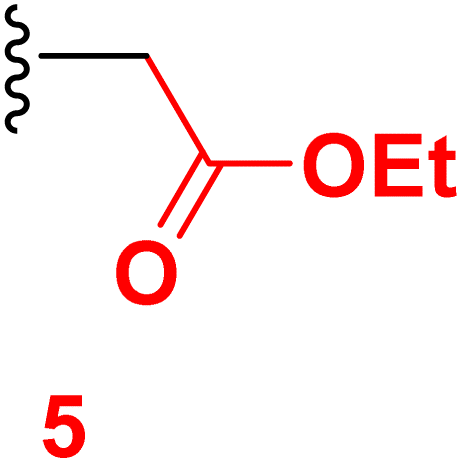
The present work aims to prepare a new series of 2-oxo-nicotinonitrile derivatives such as N-alkylated, Mannich bases, and pyrazolyl analogues. The preparation of 2-pyridone derivative 4 is the main point in the work, where it is synthesized via one-pot condensation of vanillin, 4-chloroacetophenone, ethyl cyanoacetate in the presence of ammonium acetate11,12,34 (Scheme 1). The reaction proceeded at reflux temperature in acetic acid for 6 hours, and the product's structure agreed with its spectral data (IR, 1H, and 13C NMR). Nicotinonitrile 4 was subjected to an alkylation reaction with ethyl bromo acetate in the presence of potassium carbonate to afford the respective N-alkylated analogue. The reaction smoothly proceeds at room temperature for two hours and gives a good yield (92%). The presence of lactam carbonyl (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 1645 cm−1 confirmed the formation of an N-alkylated isomer.
O) at 1645 cm−1 confirmed the formation of an N-alkylated isomer.
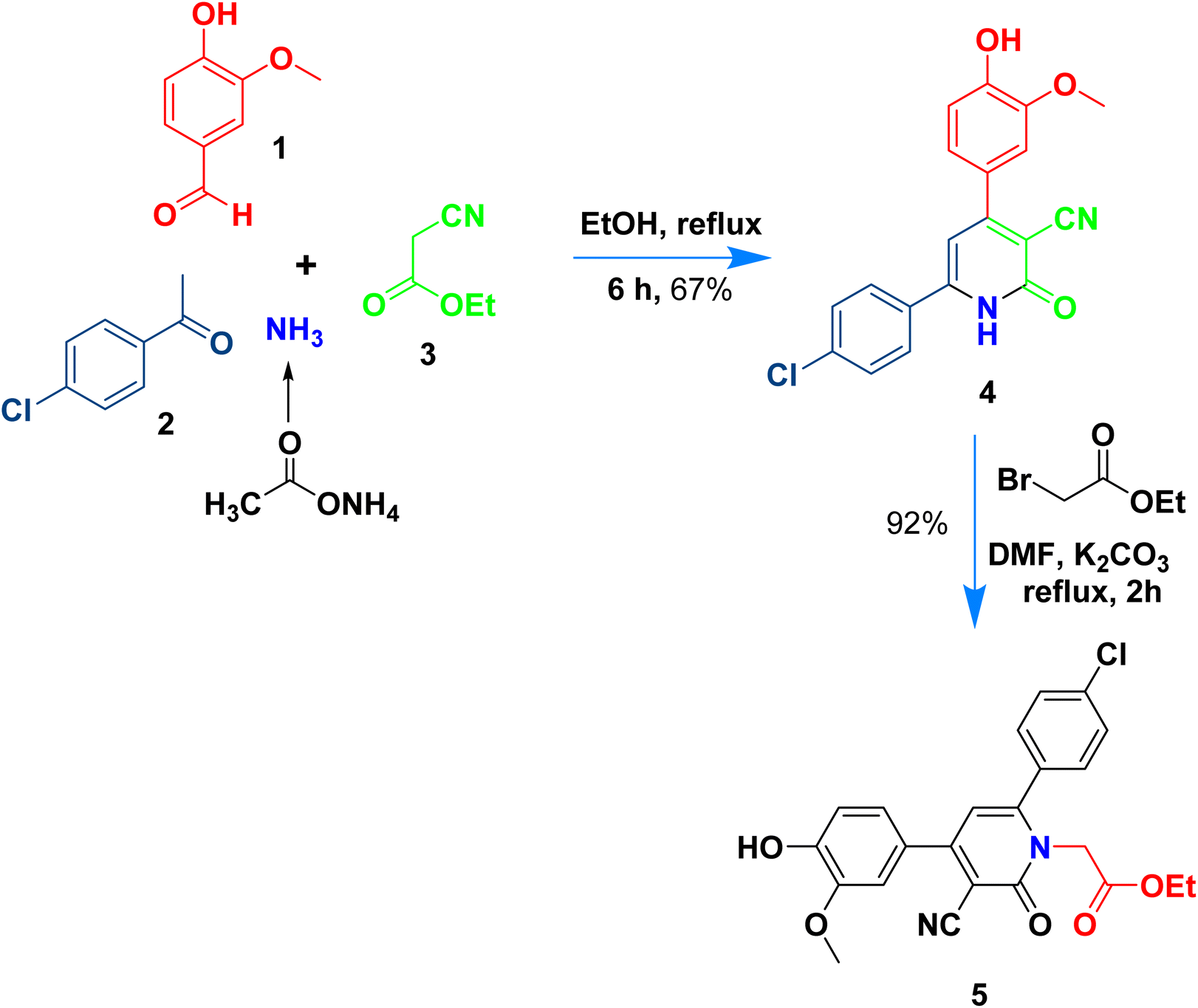 | ||
| Scheme 1 Synthetic route of N-alkylated nicotinonitrile 5. | ||
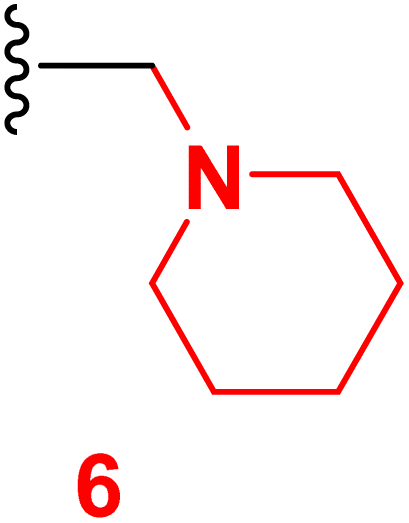
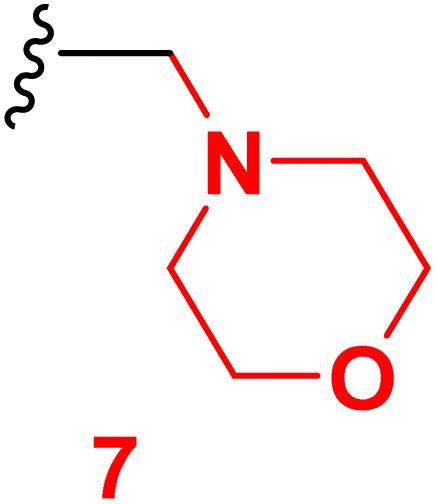
Scheme 2 outlined the Mannich reaction of nicotinonitrile 4 with piperidine, morpholine, and dimethyl amine in formaldehyde. The Mannich compounds are formed under heating for 8 hours and isolated in good yields (85 and 86%). Compound 8 contains bands at 3484, 2196, and 1646 cm−1 for OH, CN, and CO functions. Its 1H NMR confirmed the presence of NCH2N– protons at 3.60 ppm as a singlet peak, and the piperidine protons appeared at δH 1.20, 1.50, and 1.60 ppm as two multiplet and triplet.
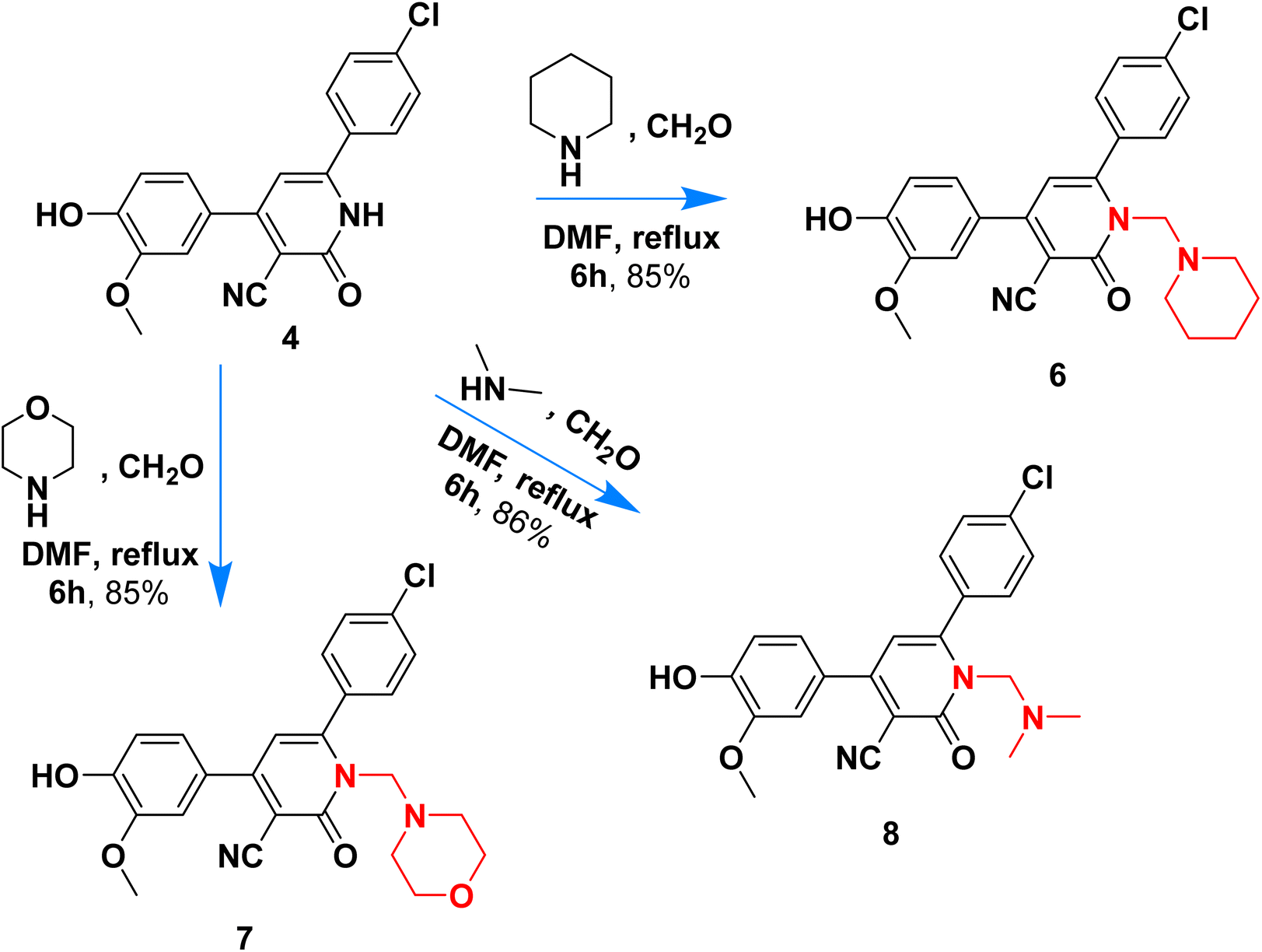 | ||
| Scheme 2 Mannich type reaction of nicotinonitrile 4. | ||
Its 13C NMR spectrum accounted for 19 signals at δC 18.55, 28.20, 53.13, 55.81, 56.02, 106.4, 112.5, 115.5, 117.0, 121.8, 126.5, 128.8, 129.6, 135.8, 147.5, 149.1, 149.7, 159.4, 162.3 for sp3, sp2, sp carbons.
Hydrazinolysis of ester 5 with hydrazine hydrate followed by heterocyclization with acetyl acetone, ethyl acetoacetate, and ethyl cyanoacetate afforded the respective pyrazolyl 10–12 in 75–88% yield (Scheme 3). The formation of pyrazolyl derivatives was performed under reflux for 6 hours in ethanol, and the products were obtained in high yield (above 75%). Hydrazide 6 contains two adjacent nucleophilic centers (NH–NH2) that enable such compound to act as a key precursor for the synthesis of different five-membered rings via its reaction with 1,3-dielectrophiles.
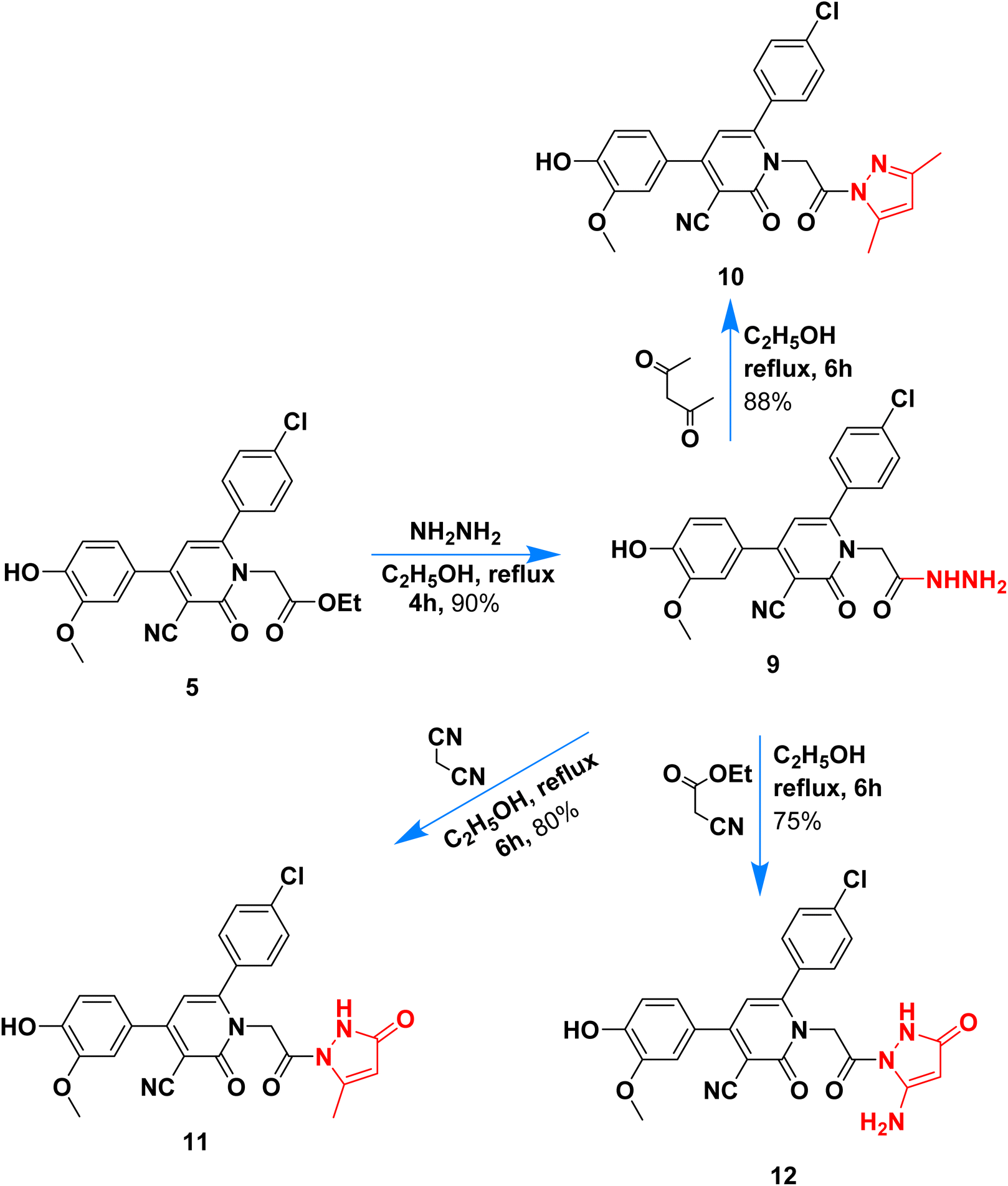 | ||
| Scheme 3 Synthetic route of pyrazolyl nicotinonitriles 10–12. | ||
The chemical structure of pyrazolyl derivatives 10–12 was confirmed from spectral analysis, for example, the IR spectrum of 6-(4-chlorophenyl)-1-(2-(3,5-dimethyl-1H-pyrazol-1-yl)-2-oxoethyl)-4-(4-hydroxy-3-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (10) showed main bands at 3337, 2210, 1686 cm−1 for OH, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and CO. Its 1H NMR spectrum, has a signal at 1.72, 2.05 ppm as two singlets for two methyl-attached pyrazole, two singlets at 3.83 and 5.14 for OCH3 and NCH2CO, and the pyridine pyrazole and aromatic protons appeared at the expected field at 6.93–6.95, 6.96–9.98, 7.23–7.25, 7.33–7.38, 7.59–7.61, and 7.91–7.93 ppm, in addition to phenolic hydroxide group appears at 9.73 ppm. The 13C NMR data assigned 23 peaks at 13.68, 14.16, 51.81, 55.73, 90.55, 111.4, 113.7, 114.1, 116.2, 120.4, 122.0, 128.7, 128.9, 129.0, 129.2, 134.0, 135.4, 137.6, 147.3, 147.7, 155.0, 163.1, 168.3 for sp3, sp2, sp carbons.
N and CO. Its 1H NMR spectrum, has a signal at 1.72, 2.05 ppm as two singlets for two methyl-attached pyrazole, two singlets at 3.83 and 5.14 for OCH3 and NCH2CO, and the pyridine pyrazole and aromatic protons appeared at the expected field at 6.93–6.95, 6.96–9.98, 7.23–7.25, 7.33–7.38, 7.59–7.61, and 7.91–7.93 ppm, in addition to phenolic hydroxide group appears at 9.73 ppm. The 13C NMR data assigned 23 peaks at 13.68, 14.16, 51.81, 55.73, 90.55, 111.4, 113.7, 114.1, 116.2, 120.4, 122.0, 128.7, 128.9, 129.0, 129.2, 134.0, 135.4, 137.6, 147.3, 147.7, 155.0, 163.1, 168.3 for sp3, sp2, sp carbons.
2.1 Biological investigations
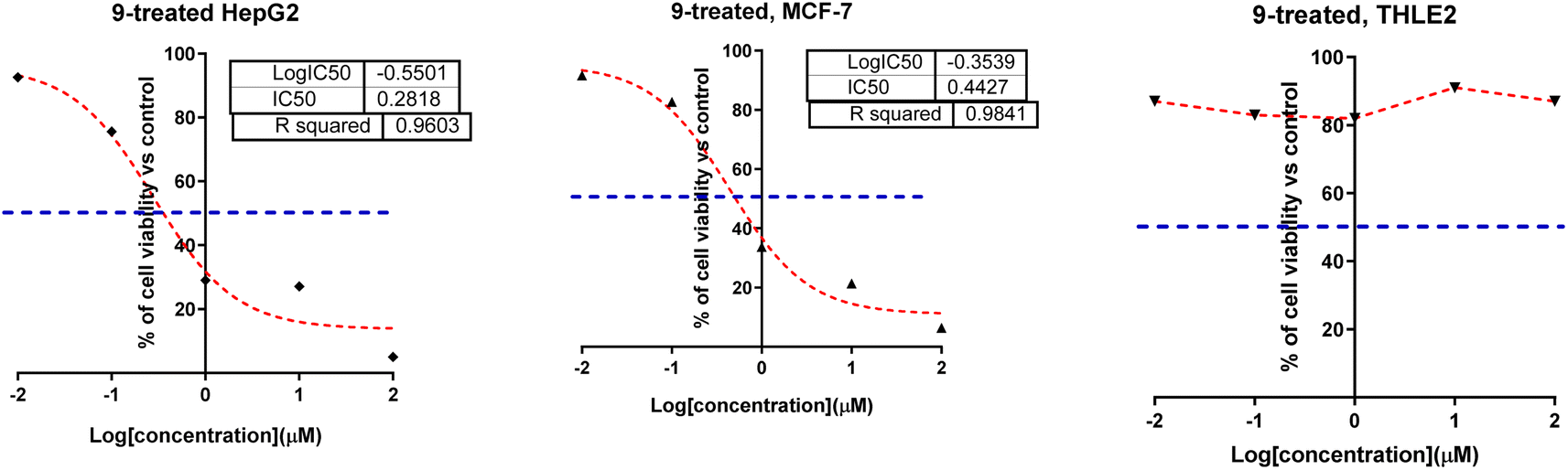 | ||
| Fig. 2 Dose–response non-linear regression curve fitting the percentage of cell viability of HepG2, MCF-7, and THLE2 cells vs. log[con. μM], R square ≈1 using the GraphPad prism. | ||
Correlating the compounds' differential activity patterns to their corresponding structural features has been considered helpful in highlighting key structural requirements for cytotoxic activity. It is worth noting that the cytotoxic activity against the human breast (MCF-7) cancerous cell line favors polar functionality rather than hydrophobic ones. The latter has been demonstrated for introducing ethoxyacetyl, dimethyl amino, and hydrazide functionality at the compound structural diversity site. Compared to the unsubstituted lead compound (4), the activity patterns have increased from micromolar concentration (6.17 ± 0.32) to lower micromolar range for compounds 5 and 8 (4.15 ± 0.21 and 2.14 ± 0.1, respectively) till reaching a sub-micromolar concentration for compound 9 (0.44 ± 0.01). Contrarily, the cytotoxic activity at MCF-7 was found intolerant to more hydrophobic substituents such as the cyclic aliphatic moieties (piperidine; 6 or morpholine; 7) as well as the heteroaromatic scaffold (pyrazole ring; 10). The MCF-7 depicted activity patterns have also been consistent with those at the hepatic (HepG2) cancerous cell line where top activity IC50s were assigned to the compounds with many polar substitutions (compounds 5, 8, and 9). The later structure–cytotoxic relationship is quite in line with the current literature, where the introduction of polar scaffolds with relevant hydrogen bond potentiality (hydrogen bond donor and/or acceptor) has been associated with improved pharmacokinetic properties, particularly the dissolution and distribution patterns.35 Moreover, polar scaffolds have been reported with the merits of mediating key polar contacts with target sites resulting in better binding affinity and improved pharmacodynamic profiles.36
To better acknowledge the polar potentiality of the synthesized compounds, we have deduced more relevant bioactivity indicators that consider both compounds' polarity and lipophilicity indices. Physiochemical properties of compounds like hydrogen bond acceptor/donor (number of heteroatoms) and structural-related lipophilic characteristics (logP) are best accounted within the estimation of ligand's efficiencies (LEs) and lipophilic efficiencies (LLEs) bioactivity indices.37 Typically, compound with promising activity profiles have been reported with LE and LLE values above the 0.2 and 2, respectively.38,39 In the case of our synthesized compounds, compounds with incorporated higher polar functionality were assigned with the relevant LE and LLE indices (Fig. 3). Notably, compounds 5 and 8 harboring the ethoxyacetyl and dimethyl amino groups showed LE; LLE of 0.29; 2.65 and 0.27; 2.97 at MCF-7 and 0.31; 2.93 and 0.26; 2.76 at HepG2 respectively, whereas compound 9 was estimated with the best LE; LLE values (0.29; 5.41 at MCF-7 and 0.30; 5.60 at HepG2). The later efficiency indices further highlight the advent of incorporating the polar functionalities for cytotoxic activity of these synthesized pyridine and pyrazolyl pyridine conjugates. Fig. 4 summarizes the process of generating the suggested SAR model. In counting for the advent of polar functionality for improving the compound's pharmacokinetic and pharmacodynamics profile has been also proceeded throughout the following sections. An investigation for tampered pharmacokinetics was evaluated through the in silico solubility profile prediction while as molecular docking simulation was adopted to assess the potential ligand binding affinity.
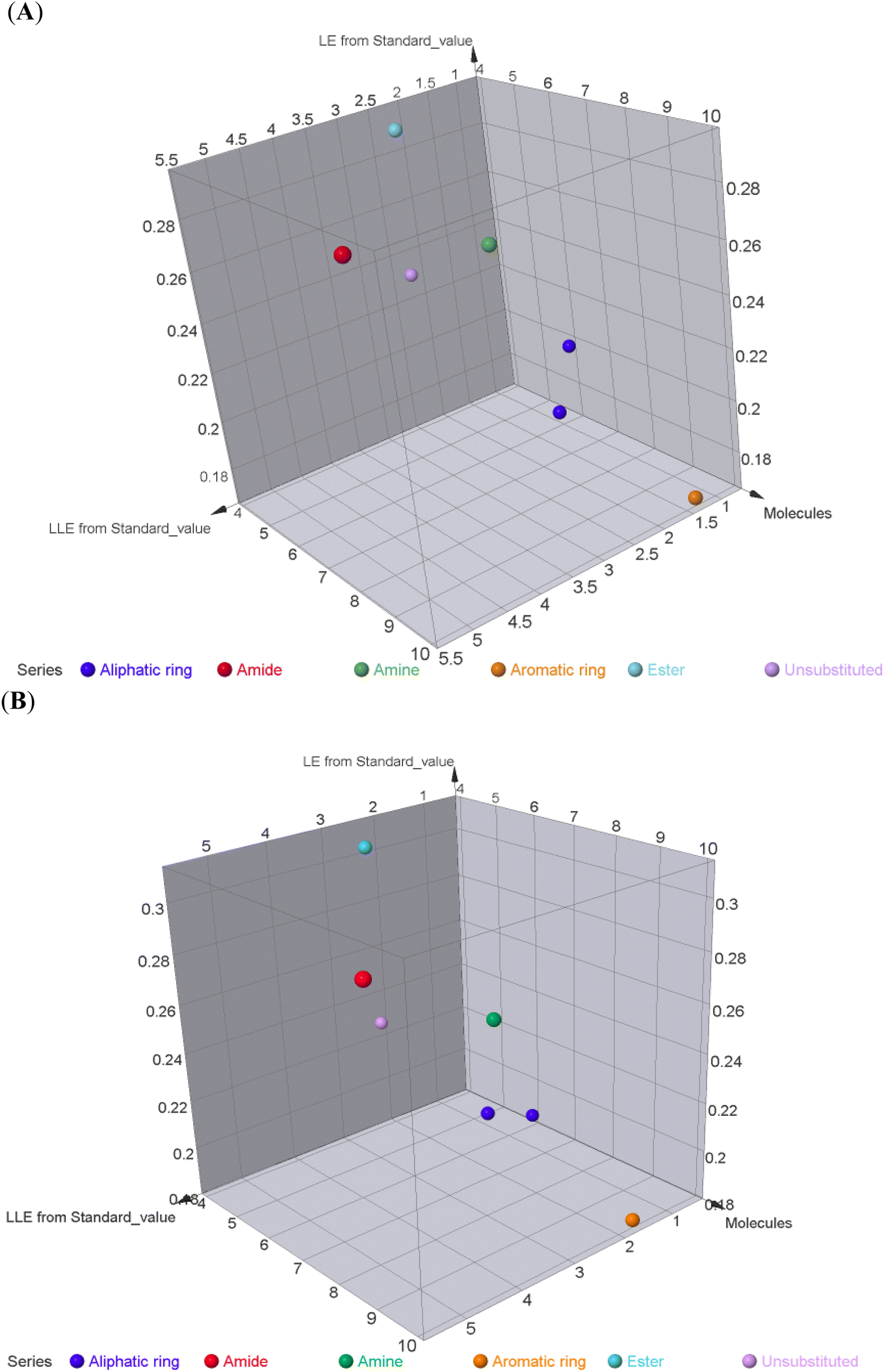 | ||
| Fig. 3 Three-dimensional charts for estimated compounds' LE and LLE indices being calculated from incorporating the compounds' polarity and lipophilicity characteristics within the obtained cytotoxicity IC50 values against MCF-7 (A) and HepG2 (B) cancerous cell lines. Charts are constructed via DataWarriors V6.2.5 software. | ||
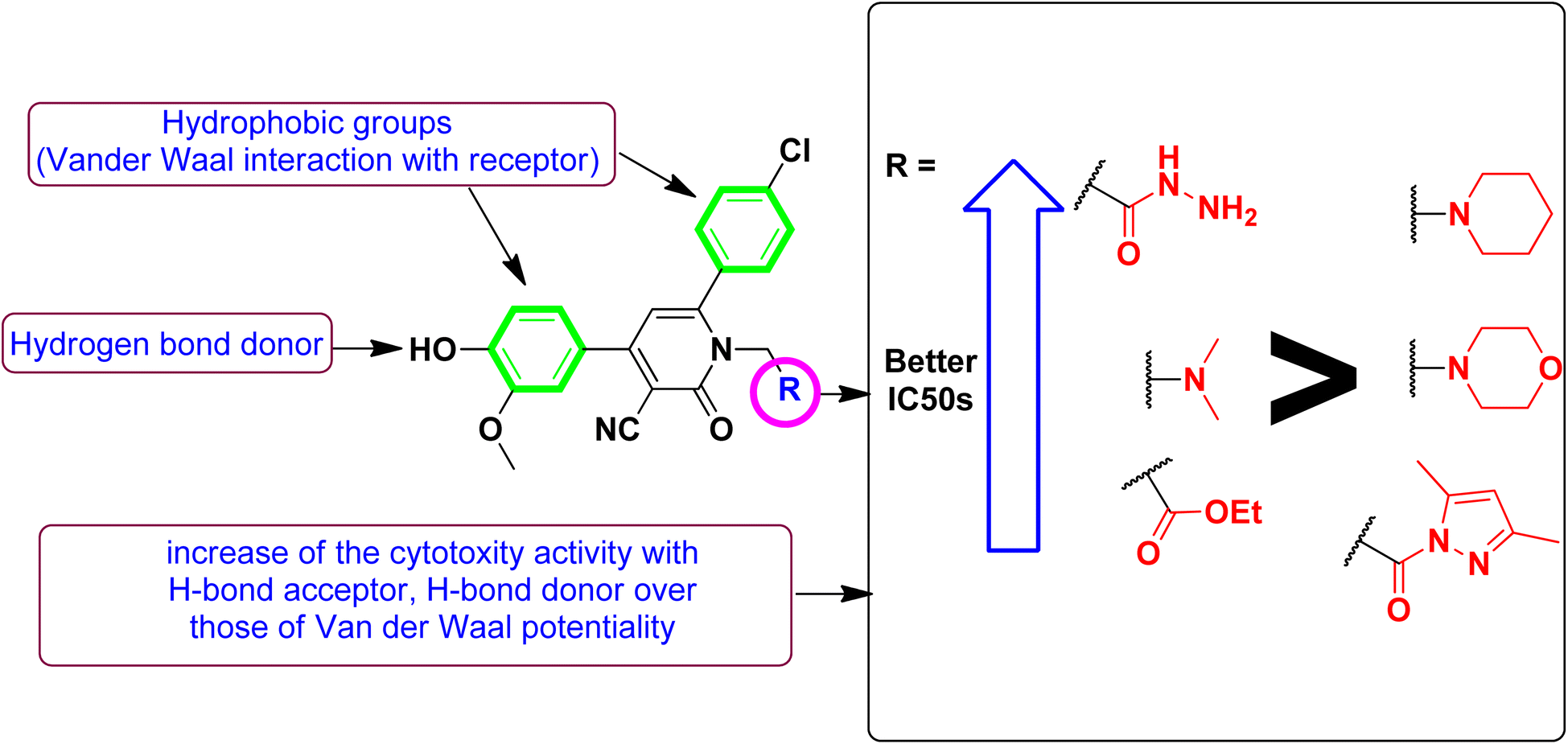 | ||
| Fig. 4 Substituents anchored on the main scaffold with cytotoxic activities of the designed derivatives. | ||
| Compound | % of PIM-1 inhibition | IC50 ± SDa (nM) |
|---|---|---|
| a Values are expressed as an average of three independent replicates. “IC50 values were calculated using sigmoidal non-linear regression curve fit of percentage inhibition against five concentrations of each compound”. | ||
| 5 | 83.4 ± 2.3 | 64.6 ± 0.2 |
| 9 | 98.8 ± 1.9 | 8.4 ± 0.2 |
| 10 | 87.6 ± 3.2 | 34.6 ± 0.12 |
| Pyrido[2,3-d] pyrimidine-6-carbonitrile | 97.8 ± 1.8 | 11.4 ± 0.13 (ref. 40) |
| Staurosporine | 95.6 ± 2.4 | 16.7 ± 0.32 |
| Compound | Concentrationa [ng mL−1] | ||
|---|---|---|---|
| Caspase 3 | Caspase 8 | Caspase 9 | |
| a Values are expressed as mean ± SD of three independent trials.b (P ≤ 0.05) significantly different between treated and treated groups using GraphPad prism. | |||
| 9-Treated HepG2 | 9.8b ± 0.9 | 74.4b ± 2.2 | 112.6b ± 2.9 |
| Control | 4.01 ± 0.21 | 55.01 ± 1.19 | 43.33 ± 0.49 |
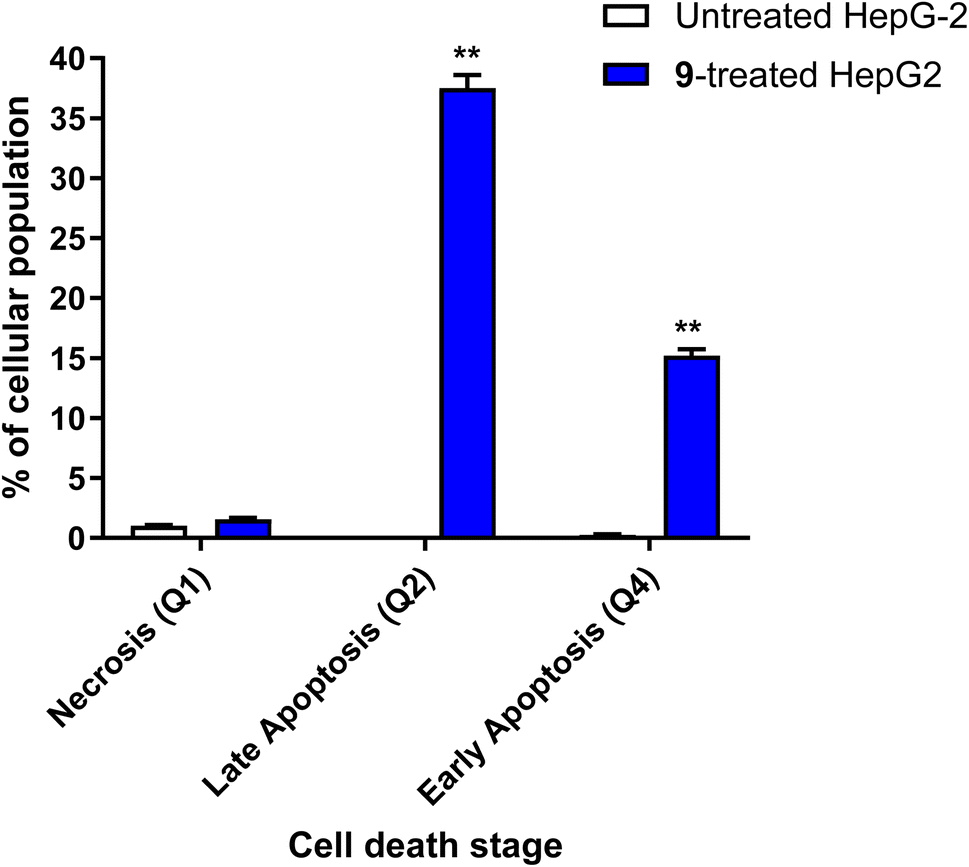 | ||
| Fig. 5 Bar chart of apoptosis and necrosis assessment using Annexin V/PI staining of untreated and 9-treated HepG2 cancer cells. **(P ≤ 0.01) significantly different between untreated and treated cells using the unpaired t-test using GraphPad prism. Annexin V/PI histograms are supported in the ESI.† | ||
It was also reported45–48 that caspase activation plays a central role in the execution of apoptosis as programmed cell death. Caspases 3, 8 and 9 are mainly incorporated in extrinsic and intrinsic pathways. So, Apoptotic cell death can be defined as caspase cascade activators.
Taken together, cyanopyridine–pyrazole conjugates exhibited potent anti-cancer activity in vitro against liver cancers. A target identification study revealed a potential protein tyrosine kinase (PTK) as the mode of action of cyanopyridine. Furthermore, the compound directly exhibited potent PIM-1 kinase inhibition, leading to apoptosis of liver cancer cells. This study concludes that cyanopyridine, as an anti-cancer small molecule, is a PIM-1 kinase inhibitor and can be considered as a promising lead for the development of more potent anti-cancer compounds.
2.2 In silico studies
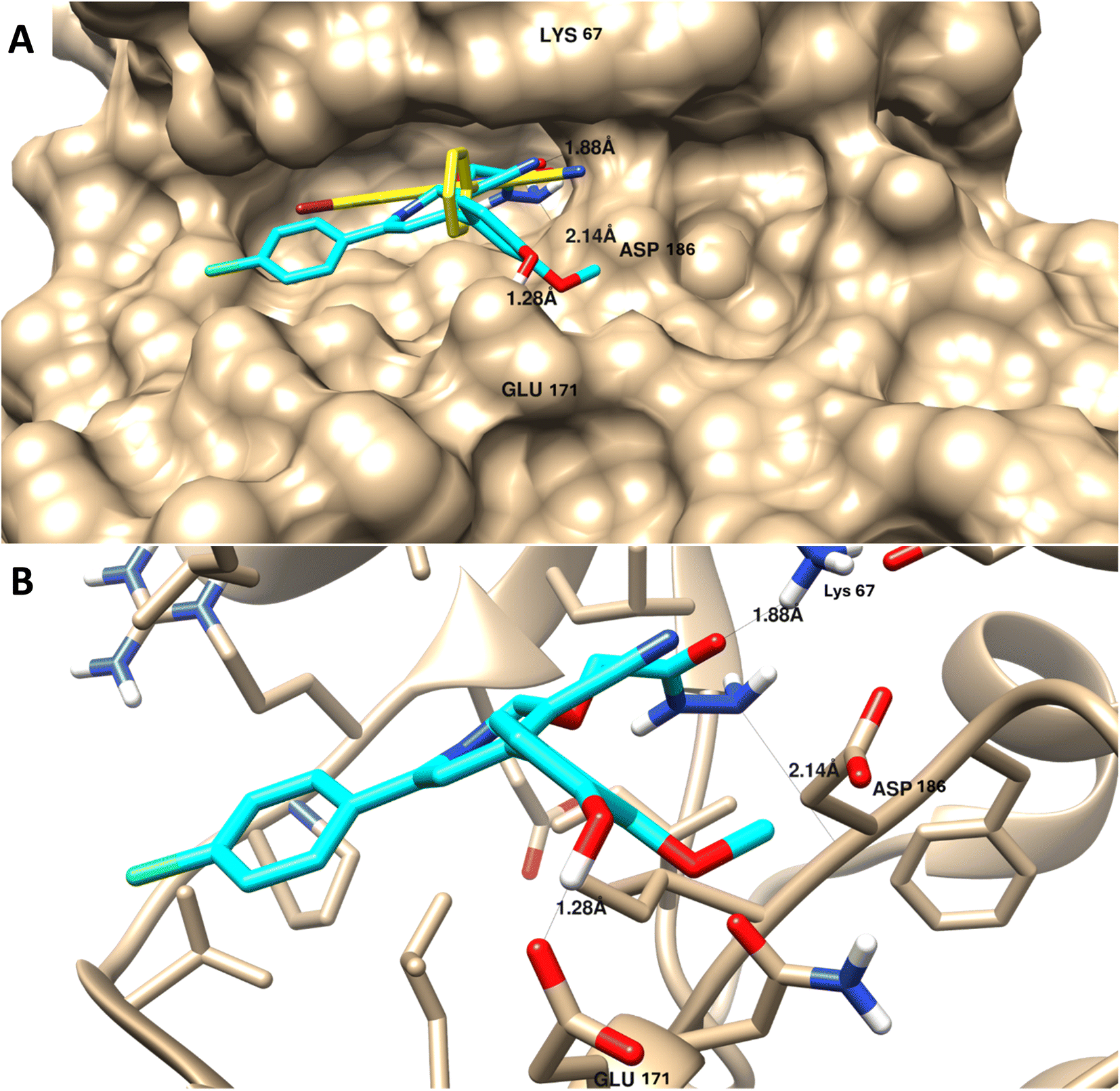 | ||
| Fig. 6 Molecular docking of 9 towards PIM-1 protein (PDB = 2OBJ). (A) Surface disposition of the co-crystallized ligand (yellow-cored) and the docked compound (cyan-colored) and (B) interactive view of the compound with the highlighted key amino acids. 3D images were generated by Chimera-UCSF software. | ||
As summarized in Table 4, in contrast with other compounds that exhibited less binding affinities with lower binding energies ranging from −9.34 kcal mol−1 to −12.76 kcal mol−1 and less interactions with the key amino acids of protein active sites, in agreement with experimental results of PIM-1 kinase inhibition.
| Compounds | Binding affinitiesa | |
|---|---|---|
| Binding energy (kcal mol−1) | Binding interactions | |
| a Binding affinities were calculated from docking results by AutoDock Vina with Chimera. | ||
| Co-crystallized ligand | −19.57 | 1 H-bond with Lys 67 |
| 4 | −9.34 | 1 H-bond with Lys 67 |
| 5 | −10.12 | 1 H-bond with Lys 67 |
| 6 | −11.25 | 1 H-bond with Asp 196 |
| 7 | −12.23 | 1 H-bond with Glu 171 |
| 8 | −12.76 | 1 H-bond with Lys 67 |
| 9 | −20.84 | 3 H-bond with Lys 67, Asp 196, and Glu 171 |
| 10 | −11.98 | 1 H-bond with Lys 67 |
It appears that the efficient target for apoptosis-mediated cell death is PIM-1 kinase inhibition, and the experimental enzyme target activity was supported by the molecular docking investigation.
| # | Molsoft | SwissADME | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HBA | HBD | Solubility (mg L−1) | Drug score | M. wt (D) | MV (A3) | PSA (A2) | logp |
BBB score | Nviolations | Drug likeness (Lipinski Pfizer filter) | |
| a M. wt: molecular weight, MV: molecular volume, PAS: polar surface area, logp: logP: octanol–water partition coefficient, nrotb: number of rotatable bonds, nviolations: number of violations, HBA: hydrogen bond acceptor, HBD: hydrogen bond donor, drug-likeness score, compounds having negative, or zero value should not be considered as drug-like”. Drug likeness (Lipinski Pfizer filter)/“Yes, drug-like” MW ≤ 500, logp ≤ 4.25, HBA ≤ 10 and HBD ≤ 5.40 |
|||||||||||
| 5 | 6 | 1 | 72.27 | −0.09 | 438.10 | 464.16 | 76.96 | 3.65 | 3.17 | 0 | Yes |
| 9 | 6 | 5 | 1036.43 | 0.78 | 426.11 | 432.93 | 108.99 | 1.69 | 2.22 | 0 | Yes |
| 10 | 5 | 1 | 126.13 | 0.46 | 409.12 | 443.85 | 62.11 | 3.45 | 4.4 | 0 | Yes |
3 Experimental
Melting points were detected using an Electro-thermal IA 9100 apparatus. IR spectra (KBr) were recorded on a Nexus 670 FTIR Nicolet, Fourier transform infrared spectrometer. The 1H and 13C NMR spectra were measured in DMSO-d6 with a JEOL-JNM-LA 400 MHz spectrometer and 100 MHz for 13C NMR. The chemical shifts are expressed on the δ (ppm) scale using TMS as the standard reference. TLC was performed on Merck Silica Gel 60F254 and detected by UV light. Elemental analysis measured on a PerkinElmer 240 (microanalysis), Microanalysis Center, Cairo University.3.1 6-(4-Chlorophenyl)-4-(4-hydroxy-3-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (4)
A mixture of 4-hydroxy-3-methoxybenzaldehyde (vanillin) (1) (0.02 mole) and 4-chloroacetophenone (2) (0.02 mole) and ethyl cyanoacetate (3) (0.02 mole) in the presence of ammonium acetate (0.16 mole) with acetic acid (20 mL) was heated under reflux (6 hours). After cooling, the resulting solid was filtered off and dried. Pale yellow solid (AcOH), mp = 340–342 °C, yield: 67%. IR spectrum, ν, cm−1: 3483 (OH and NH), 2226 (CN), 1645 (CO), 822 (C–Cl). 1H NMR spectrum, δ, ppm: 3.85 (s, 3H, OCH3), 6.85 (s, 1H, Hpyridine), 6.92–6.94 (d, 1H, 3J = 8.0 Hz, Haryl), 7.22–7.24 (d, H, 3J = 6.40 Hz, Haryl), 7.25 (s, 1H, Haryl), 7.57–7.59 (d, 2H, 3J = 8.8 Hz, Haryl), 7.89–7.91 (d, 2H, 3J = 8.80 Hz, Haryl), 9.75 (s, 1H, OH), 12.66 (s 1H, NH). 13C NMR spectrum, δC, ppm: 104.9, 112.1, 115.3, 116.8, 120.2, 122.7, 126.2, 128.7, 128.9, 135.5, 147.1, 149.3, 157.4, 159.4, 160.2 (sp3, sp2, sp carbons), found, %: C, 64.78; H, 3.66; N, 8.00. Calculated, C19H13ClN2O3 (352.77) %: C, 64.69; H, 3.71; N, 7.94.
3.2 Ethyl 2-(6-(4-chlorophenyl)-3-cyano-4-(4-hydroxy-3-methoxyphenyl)-2-oxo pyridin-1(2H)-yl)acetate (5)
A mixture of 6-(4-chlorophenyl)-4-(4-hydroxy-3-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (1) (0.01 mole) and ethyl cyanoacetate (0.01 mole) in the presence of potassium carbonate in DMF (20 mL) was stirring at room temperature for 2 hours. The reaction mixture was diluted with HCl, and the formed solid was filtered off. Pale yellow solid (EtOH), mp = 292–294 °C, yield: 92%. IR spectrum, ν, cm−1: 3485 (OH), 2222 (CN), 1750, 1683 (2CO). 1H NMR spectrum, δ, ppm: 1.16–1.20 (t, 3H, Hethyl), 3.88 (s, 3H, OCH3), 4.15–4.20 (q, 2H, Hethyl), 5.13 (s, 2H, CH2), 6.95–6.97 (d, 1H, 3J = 8.0, Hz, Haryl), 7.23–7.25 (d, 1H, 3J = 8.0, Hz, Haryl), 7.36 (s, 1H, Haryl), 7.57–7.59 (d, 2H, 3J = 8.80, Hz, Haryl), 7.86 (s, 1H, Hpyridine), 8.16–8.18 (d, 2H, 3J = 8.80, Hz, Haryl), 9.72 (s, 1H, OH). Found, %: C, 62.95; H, 4.36; N, 6.38. Calculated, C23H19ClN2O5 (438.86) %: C, 62.95; H, 4.36; N, 6.38.
3.3 6-(4-Chlorophenyl)-4-(4-hydroxy-3-methoxyphenyl)-2-oxo-1-(piperidin-1-ylmethyl)-1,2-dihydropyridine-3-carbonitrile (6)
A mixture of 6-(4-chlorophenyl)-4-(4-hydroxy-3-methoxyphenyl)-2-oxo-1,2-dihydro pyridine-3-carbonitrile (4) (0.01 mole), (2 mL) formaldehyde (0.01 mole) and piperidine (0.01 mole) in DMF (20 mL) was heated under reflux (8 hours). After cooling, the resulting solid was filtered off and dried. Pale yellow solid (EtOH), mp = 310–312 °C, yield: 85%. IR spectrum, ν, cm−1: 3484 (OH), 2196 (CN), 1646 (CO). 1H NMR spectrum, δ, ppm: 1.20 (m, 2H, Hpip), 1.50 (m, 4H, Hpip), 1.60 (t, 2H, Hpip), 3.85 (s, 3H, OCH3), 3.75 (s, NCH2N), 6.86 (s, 1H, Hpyridine), 6.92–9.94 (d, 1H, 3J = 8.0, Hz, Haryl), 7.23–7.25 (dd, 1H, 2J = 2.0, 3J = 8.40, Hz, Haryl), 7.33–7.34 (d, 1H, 2J = 2.0 Hz, Haryl), 7.58–7.60 (dd, 2H, 2J = 1.60, 3J = 6.80, Hz, Haryl), 7.90–7.93 (d, 2H, 3J = 8.80, Hz, Haryl), 9.73 (s, 1H, OH). 13C NMR spectrum, δC, ppm: 18.55, 28.20, 53.13, 55.81, 56.02, 106.4, 112.5, 115.5, 117.0, 121.8, 126.5, 128.8, 129.6, 135.8, 147.5, 149.1, 149.7, 159.4, 162.3 (sp3, sp2, sp carbons). Found, %: C, 66.66; H, 5.35; N, 9.40 and calculated, C25H24ClN3O3 (449.94) %: C, 66.74; H, 5.38; N, 9.34.
3.4 6-(4-Chlorophenyl)-4-(4-hydroxy-3-methoxyphenyl)-1-(morpholinomethyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (7)
A mixture of 6-(4-chlorophenyl)-4-(4-hydroxy-3-methoxyphenyl)-2-oxo-1,2-dihydro pyridine-3-carbonitrile (4) (0.01 mole), (2 mL) formaldehyde (0.01 mole) and morpholine (0.01 mole) in DMF (20 mL) was heated under reflux (8 hours). After cooling, the resulted solid was filtered off and dried. Pale yellow solid (EtOH), mp = 332–334 °C, yield: 85%. IR spectrum, ν, cm−1: 3484 (OH), 2201 (CN), 1645 (CO). 1H NMR spectrum, δ, ppm: 2.54 (d, 4H, Hmorph), 3.40 (d, 4H, Hmorph), 3.76 (s, NCH2N), 3.85 (s, 3H, OCH3), 6.86 (s, 1H, Hpyridine), 6.92–9.94 (d, 1H, 3J = 8.40, Hz, Haryl), 7.23–7.25 (dd, 1H, 2J = 2.0, 3J = 8.40, Hz, Haryl), 7.33–7.34 (d, 1H, 2J = 2.0 Hz, Haryl), 7.58–7.59 (dd, 2H, 2J = 2.40, 3J = 8.40, Hz, Haryl), 7.90–7.92 (d, 2H, 3J = 8.40, Hz, Haryl), 9.72 (s, 1H, OH). 13C NMR spectrum, δC, ppm: 36.0, 42.3, 48.0, 55.82, 106.2, 112.5, 115.5, 116.9, 121.8, 126.5, 128.8, 129.8, 129.6, 131.4, 135.8, 147.5, 149.1, 159.4, 162.3 (sp3, sp2, sp carbons). Found, %: C, 63.88; H, 494; N, 9.27, calculated, C24H22ClN3O4 (451.91) %: C, 63.79; H, 4.91; N, 9.30.
3.5 6-(4-Chlorophenyl)-1-((dimethylamino)methyl)-4-(4-hydroxy-3-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (8)
A mixture of 6-(4-chlorophenyl)-4-(4-hydroxy-3-methoxyphenyl)-2-oxo-1,2-dihydro pyridine-3-carbonitrile (4) (0.01 mole), (2 mL) formaldehyde (0.01 mole) and dimethylamine (0.01 mole) in DMF (20 mL) was heated under reflux (8 hours). After cooling, the resulting solid was filtered off and dried. Pale yellow solid (EtOH), mp = 302–304 °C, yield: 86%. IR spectrum, ν, cm−1: 3485 (OH), 2210 (CN), 1646 (CO). 1H NMR spectrum, δ, ppm: 1.90 (s, 6H, Hmethyl), 3.60 (s, NCH2N), 3.85 (s, 3H, OCH3), 6.87 (s, 1H, Hpyridine), 6.92–9.94 (d, 1H, 3J = 8.40, Hz, Haryl), 7.23–7.25 (dd, 1H, 2J = 2.40, 3J = 8.40, Hz, Haryl), 7.33–7.34 (d, 1H, 2J = 2.0 Hz, Haryl), 7.59–7.61 (dd, 2H, 2J = 2.0, 3J = 6.80, Hz, Haryl), 7.91–7.93 (d, 2H, 3J = 8.40, Hz, Haryl), 9.72 (s, 1H, OH). 13C NMR spectrum, δC, ppm: 18.55, 21.04, 48.59, 56.02, 92.01, 108.1, 112.5, 115.5, 116.9, 121.8, 126.5, 128.8, 129.6, 135.8, 147.5, 149.1, 159.5, 162.2 (sp3, sp2, sp carbons). Found, %: C, 64.36; H, 4.89; N, 10.29, calculated, C22H20ClN3O3 (409.87) %: C, 64.47; H, 4.92; N, 10.25.
3.6 2-(6-(4-Chlorophenyl)-3-cyano-4-(4-hydroxy-3-methoxyphenyl)-2-oxopyridin-1(2H)-yl)acetohydrazide (9)
A mixture of ethyl 2-(6-(4-chlorophenyl)-3-cyano-4-(3-hydroxy-4-methoxyphenyl)-2-oxo pyridin-1(2H)-yl) acetate (4) (0.01 mole) and hydrazine hydrate (0.01 mole) with ethanol (20 mL) was heated under reflux (4 hours). After cooling, the resulting solid was filtered off and dried. Pale yellow solid (EtOH), mp = 226–228 °C, yield: 90%. IR spectrum, ν, cm−1: 3286, 3203, 3130 (OH, NH, NH2), 2212 (CN), 1663 (CO). 1H NMR spectrum, δ, ppm: 3.88 (s, 3H, OCH3), 4.50 (bs, 1H, NH2), 4.99 (s, 2H, CH2), 6.97–6.99 (d, 1H, 3J = 8.40, Hz, Haryl), 7.25–7.27 (d, 1H, 3J = 8.40, Hz, Haryl), 7.36 (s, 1H, Haryl), 7.57–7.59 (d, 2H, 3J = 8.40, Hz, Haryl), 7.84 (s, 1H, Hpyridine), 8.22–8.24 (d, 2H, 3J = 8.40, Hz, Haryl), 9.40 (s, 1H, OH), 9.70 (bs, 1H, NH). 13C NMR spectrum, δC, ppm: 55.93, 64.31, 92.28, 112.8, 113.9, 115.5, 121.8, 126.3, 128.8, 129.2, 129.5, 135.4, 148.8, 155.2, 156.5, 163.4 and 166.5 (sp3, sp2, sp carbons). Found, %: C, 59.29; H, 4.00; N, 13.25. Calculated, C21H17ClN4O4 (424.84) %: C, 59.37; H, 4.03; N, 13.19.
3.7 6-(4-Chlorophenyl)-1-(2-(3,5-dimethyl-1H-pyrazol-1-yl)-2-oxoethyl)-4-(4-hydroxy-3-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (10)
A mixture of 2-(6-(4-chlorophenyl)-3-cyano-4-(4-hydroxy-3-methoxyphenyl)-2-oxopyridin-1(2H)-yl) acetohydrazide (6) (0.01 mole) and acetyl acetone (0.01 mole) with ethanol (20 mL) absolute was heated under reflux (6 hours). After cooling, the reaction mixture was diluted with water, and the formed precipitate was filtered off. Pale yellow solid (EtOH), mp = 206–208 °C, yield: 88%. IR spectrum, ν, cm−1: 3337 (OH), 2210 (CN), 1686 (CO). 1H NMR spectrum, δ, ppm: 1.72, 2.05 (2s, 6H, Hpyrazole), 3.83 (s, 3H, OCH3), 5.14 (s, NCH2CO), 6.93–6.95 (m, 2H, Hpyridine and pyrazole), 6.96–9.98 (d, 1H, 3J = 8.0, Hz, Haryl), 7.23–7.25 (dd, 1H, 2J = 2.0, 3J = 8.0, Hz, Haryl), 7.33–7.38 (d, 1H, 2J = 2.0 Hz, Haryl), 7.59–7.61 (dd, 2H, 2J = 2.0, 3J = 8.40, Hz, Haryl), 7.91–7.93 (d, 2H, 3J = 8.40, Hz, Haryl), 9.73 (s, 1H, OH). 13C NMR spectrum, δC, ppm: 13.68, 14.16, 51.81, 55.73, 90.55, 111.4, 113.7, 114.1, 116.2, 120.4, 122.0, 128.7, 128.9, 129.0, 129.2, 134.0, 135.4, 137.6, 147.3, 147.7, 155.0, 163.1, 168.3 (sp3, sp2, sp carbons). Found, %: C, 64.36; H, 4.89; N, 10.29, calculated, C22H20ClN3O3 (409.87) %: C, 64.47; H, 4.92; N, 10.25.
3.8 6-(4-Chlorophenyl)-4-(4-hydroxy-3-methoxyphenyl)-1-(2-(5-methyl-3-oxo-2,3-dihydro-1H-pyrazol-1-yl)-2-oxoethyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (11)
A mixture of 2-(6-(4-chlorophenyl)-3-cyano-4-(4-hydroxy-3-methoxyphenyl)-2-oxopyridin-1(2H)-yl) acetohydrazide (6) (0.01 mole) and ethyl acetoacetate (0.01 mole) with ethanol (20 mL) absolute was heated under reflux (6 hours). After cooling, the reaction mixture was diluted with water, and the formed precipitate was filtered off. Pale yellow solid (EtOH), mp = 206–208 °C, yield: 80%. IR spectrum, ν, cm−1: 3321 (OH), 2224 (CN), 1692, 1671 (CO). 1H NMR spectrum, δ, ppm: 2.02 (s, 3H, Hmethyl), 3.87 (s, 3H, OCH3), 5.14 (s, NCH2CO), 6.92–6.95 (m, 2H, Hpyridine and pyrazole), 6.96–6.98 (d, 1H, 3J = 8.0, Hz, Haryl), 7.23–7.25 (dd, 1H, 2J = 2.0, 3J = 8.0, Hz, Haryl), 7.33–7.38 (d, 1H, 2J = 2.0 Hz, Haryl), 7.59–7.61 (dd, 2H, 2J = 2.0, 3J = 8.40, Hz, Haryl), 7.91–7.93 (d, 2H, 3J = 8.40, Hz, Haryl), 9.73 (s, 1H, OH), 10.68 (s, 1H, NH). 13C NMR spectrum, δC, ppm: 16.35, 55.86, 60.77, 91.86, 112.5, 114.1, 115.7, 121.9, 126.1, 126.7, 128.7, 128.9, 129.5, 135.2, 135.6, 147.5, 148.9, 155.1, 156.8, 163.1, 168.3, 169.5 (sp3, sp2, sp carbons). Found, %: C, 64.36; H, 4.89; N, 10.29. Calculated, C22H20ClN3O3 (409.87) %: C, 64.47; H, 4.92; N, 10.25. Found, %: C, 61.09; H, 3.87; N, 11.47. Calculated, C25H19ClN4O5 (490.90) %: C, 61.17; H, 3.90; N, 11.41.
3.9 1-(2-(5-Amino-3-oxo-2,3-dihydro-1H-pyrazol-1-yl)-2-oxoethyl)-6-(4-chlorophenyl)-4-(4-hydroxy-3-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (12)
A mixture of 2-(6-(4-chlorophenyl)-3-cyano-4-(4-hydroxy-3-methoxyphenyl)-2-oxopyridin-1(2H)-yl) acetohydrazide (6) (0.01 mole) and ethyl cyanoacetate (0.01 mole) with ethanol (20 mL) absolute was heated under reflux (6 hours). After cooling, the reaction mixture was diluted with water, and the formed precipitate was filtered off. Pale yellow solid (EtOH), mp = 180–182 °C, yield: 75%. IR spectrum, ν, cm−1: 3321 (OH), 2223 (CN), 1692, 1652 (CO). 1H NMR spectrum, δ, ppm: 3.85 (bs, 2H, NH2), 3.89 (s, 3H, OCH3), 5.15 (s, NCH2CO), 6.84 (s, 1H, Hpyrazole), 6.92–6.94 (m, 2H, Hpyridine and aryl), 6.96–6.99 (d, 1H, 3J = 8.0, Hz, Haryl), 7.23–7.25 (d, 1H, 3J = 8.0, Hz, Haryl), 7.57–7.59 (d, 2H, 3J = 8.40, Hz, Haryl), 7.91–7.93 (d, 2H, 3J = 8.40, Hz, Haryl), 9.70 (s, 1H, OH), 9.80 (s, 1H, NH). Found, %: C, 58.51; H, 3.73; N, 14.18, calculated, C24H18ClN5O5 (491.89) %: C, 58.60; H, 3.69; N, 14.24.
3.10 Biology
:100) and propidium iodide (PI)” at a concentration equals 10 μg mL−1 in the dark for 30 min. The flow cytometric data were analyzed using FlowJo version-10 software (TreeStar, Ashland, OR, USA).54–564 Conclusion
Nicotinonitrile and pyrazolyl nicotinonitrile were synthesized, and their PIM-1 kinase inhibitors and caspase activators were investigated. A new Manich bases 6–8 were synthesized via reaction of pyridine 4 with piperidine, dimethyl amine, and morpholine in the presence of formalin. On the other hand, the pyrazolyl analogues 10–12 were synthesized via heterocyclization of acetohydrazide derivative 9 with acetylacetone, malononitrile, and ethyl cyanoacetate, respectively, in ethanol. Compound 9 showed exceptional cytotoxic effects on MCF-7 and HepG2 cells among these derivatives, with IC50 values of 0.34 μM and 0.18 μM, respectively. Curiously, compound 9 exhibited potent inhibition of PIM-1 kinase activity, with IC50 values of 20.4 nM and 93.8% inhibition, respectively, compared to staurosporine, which had an IC50 value of 16.7 nM and 95.6% inhibition. In addition, the molecular docking investigation showed that compound 9 bound to the PIM-1 kinase site similarly to a co-crystallized ligand, demonstrating a strong binding affinity.Data availability
All data associated with this manuscript will be available upon reasonable request from the corresponding authors.Author contributions
A. Hamza, A. Aboelmaged, H. A. Morsy synthesized the entire series of derivatives under the supervision of A. H. Moustafa, H. A. El-Sayed, S. M. El Rayes, and M.S. Nafie with the characterization of structure elucidation. At the same time, M. S. Nafie initiated the idea and design of the biology part by carrying out in vitro cytotoxic screening, enzyme targeting flow cytometry, and in silico studies with linguistic revision and manuscript finalizing. All authors contributed to data analysis and manuscript writing original drafts in their corresponding parts. All authors validated it in the final submitted form.Conflicts of interest
The authors declare no conflict of interest.References
- R. L. Siegel, A. N. Giaquinto and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 12–49 CrossRef.
- M. Bachmann and T. Möröy, Int. J. Biochem. Cell Biol., 2005, 37, 726–730 CrossRef CAS.
- X. Zhang, M. Song, J. K. Kundu, M.-H. Lee and Z.-Z. Liu, J. Cancer Prev., 2018, 23, 109–116 CrossRef.
- X. Zhang, M. Song, J. K. Kundu, M.-H. Lee and Z.-Z. Liu, J. Cancer Prev., 2018, 23, 109 CrossRef PubMed.
- L. S. Chen, S. Redkar, P. Taverna, J. E. Cortes and V. Gandhi, Blood, 2011, 118, 693–702 CrossRef CAS PubMed.
- M. E. Abdelaziz, M. M. M. El-Miligy, S. M. Fahmy, M. A. Mahran and A. A. Hazzaa, Bioorg. Chem., 2018, 80, 674–692 CrossRef CAS PubMed.
- O. H. Rizk, M. Teleb, M. M. Abu-Serie and O. G. Shaaban, Bioorg. Chem., 2019, 92, 103189 CrossRef CAS.
- M. M. F. Ismail, A. M. Farrag, M. F. Harras, M. H. Ibrahim and A. B. M. Mehany, Bioorg. Chem., 2020, 94, 103481 CrossRef CAS.
- N. S. Magnuson, Z. Wang, G. Ding and R. Reeves, Future Oncol., 2010, 6, 1461–1478 CrossRef CAS.
- E. Kabir and M. Uzzaman, Results Chem., 2022, 4, 100606 CrossRef CAS.
- E. S. Tantawy, M. S. Nafie, H. A. Morsy, H. A. El-Sayed, A. H. Moustafa and S. M. Mohammed, RSC Adv., 2024, 14, 11098–11111 RSC.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS.
- A. Hamza, H. A. El-Sayed, A. H. Moustafa, S. M. El Rayes, A. Aboelmaged and R. A. Haggam, J. Heterocycl. Chem., 2023, 60, 803–813 CrossRef CAS.
- M. Alrooqi, S. Khan, F. A. Alhumaydhi, S. A. Asiri, M. Alshamrani, M. M. Mashraqi, A. Alzamami, A. M. Alshahrani and A. A. Aldahish, http://www.eurekaselect.com.
- S. De, A. K. SK, S. K. Shah, S. Kazi, N. Sarkar, S. Banerjee and S. Dey, RSC Adv., 2022, 12, 15385 RSC.
- H. A. El-Sayed, A. H. Moustafa, A. A. Fadda and K. E. Abd El-Rahman, Russ. J. Gen. Chem., 2019, 89, 339–347 CrossRef CAS.
- T. Tahir, M. Ashfaq, M. Saleem, M. Rafiq, M. I. Shahzad, K. Kotwica-Mojzych and M. Mojzych, Molecules, 2021, 26, 4872 CrossRef CAS.
- M. M. M. El-Miligy, M. E. Abdelaziz, S. M. Fahmy, T. M. Ibrahim, M. M. Abu-Serie, M. A. Mahran and A. A. Hazzaa, J. Enzyme Inhib. Med. Chem., 2023, 38, 2152810 CrossRef PubMed.
- A. M. Farrag, M. H. Ibrahim, A. B. M. Mehany and M. M. F. Ismail, Bioorg. Chem., 2020, 105, 104378 CrossRef CAS.
- N. Y. Megally Abdo, E. M. Samir and R. M. Mohareb, J. Heterocycl. Chem., 2020, 57, 1993–2009 CrossRef CAS.
- A. E. M. Mekky, S. M. H. Sanad, A. Y. Said and M. A. A. Elneairy, Synth. Commun., 2020, 50, 2376–2389 CrossRef CAS.
- R. M. Mohareb, F. M. Manhi, M. A. A. Mahmoud and A. Abdelwahab, Med. Chem. Res., 2020, 29, 1536–1551 CrossRef CAS.
- R. M. Mohareb, F. M. Manhi, M. A. A. Mahmoud and A. Abdelwahab, Med. Chem. Res., 2020, 29, 1536–1551 CrossRef CAS.
- I. W. Cheney, S. Yan, T. Appleby, H. Walker, T. Vo, N. Yao, R. Hamatake, Z. Hong and J. Z. Wu, Bioorg. Med. Chem. Lett., 2007, 17, 1679–1683 CrossRef CAS PubMed.
- Y. Zhang, C. Wu, N. Zhang, R. Fan, Y. Ye and J. Xu, Int. J. Mol. Sci., 2023, 24, 12724 CrossRef CAS PubMed.
- G. Li, Y. Cheng, C. Han, C. Song, N. Huang and Y. Du, RSC Med. Chem., 2022, 13, 1300–1321 RSC.
- N. A. Warfel and A. S. Kraft, Pharmacol. Ther., 2015, 151, 41–49 CrossRef CAS.
- G. Roman, Eur. J. Med. Chem., 2015, 89, 743–816 CrossRef CAS PubMed.
- S. Murugappan, S. Dastari, K. Jungare, N. M. Barve and N. Shankaraiah, J. Mol. Struct., 2024, 1307, 138012 CrossRef CAS.
- H. A. El-Sayed, S. A. Said and A. M. Abd El-Hamid, Synth. Commun., 2021, 51, 3116–3124 CrossRef CAS.
- A. A. Mohamed, B. H. Asghar, A. H. Moustafa, H. A. El-Sayed, D. El-Sayed, A. S. A. Mohamed and K. A. Asla, Russ. J. Gen. Chem., 2021, 91, 2564–2580 CrossRef CAS.
- S. M. Mohammed, A. H. Moustafa, H. A. El-Sayed, A. Ayman and A. S. A. Mohamed, Russ. J. Gen. Chem., 2021, 91, S84–S88 CrossRef CAS.
- A. H. Moustafa, H. A. El-Sayed, A. E.-F. Z. Haikal and E. S. H. El Ashry, Nucleos Nucleot. Nucleic Acids, 2011, 30, 340–352 CrossRef CAS.
- S. Chaudhuri, A. Ghosh and S. K. Chattopadhyay, Green Synth. Approaches Biol. Relevant Heterocycl., 2021, 617–653 CAS.
- P. W. Kenny, J. Med. Chem., 2022, 65, 14261–14275 CrossRef CAS.
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug Discovery, 2014, 13, 105–121 CrossRef CAS PubMed.
- D. Hadži, J. Kidrič, J. Koller and J. Mavri, J. Mol. Struct., 1990, 237, 139–150 CrossRef.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS.
- S. Schultes, C. de Graaf, E. E. J. Haaksma, I. J. P. de Esch, R. Leurs and O. Krämer, Drug Discov. Today Technol., 2010, 7, e157–e162 CrossRef CAS.
- E. S. Tantawy, M. S. Nafie, H. A. Morsy, H. A. El-Sayed, A. H. Moustafa and S. M. Mohammed, RSC Adv., 2024, 14, 11098–11111 RSC.
- F. G. Abdulrahman, R. Sabour, S. M. A. El-Gilil, A. B. M. Mehany and E. A. Taha, J. Mol. Struct., 2024, 1295, 136811 CrossRef CAS.
- R. M. Mohareb, N. Y. M. Abdo and W. W. Wardakhan, Med. Chem. Res., 2017, 26, 2520–2537 CrossRef CAS.
- M. S. Nafie, A. M. Amer, A. K. Mohamed and E. S. Tantawy, Bioorg. Med. Chem., 2020, 28, 115828 CrossRef CAS PubMed.
- S. Mohamady, A. F. Khalil, B. H. Naguib, M. S. Nafie, H. O. Tawfik and M. A. Shaldam, Bioorg. Chem., 2024, 143, 106988 CrossRef CAS PubMed.
- Y. Shi, Mol. Cell, 2002, 9, 459–470 CrossRef CAS PubMed.
- S. E. Logue and S. J. Martin, Biochem. Soc. Trans., 2008, 36, 1–9 CAS.
- S. Kumar, Cell Death Differ., 1999, 6, 1060–1066 CrossRef CAS PubMed.
- I. Budihardjo, H. Oliver, M. Lutter, X. Luo and X. Wang, Annu. Rev. Cell Dev. Biol., 1999, 15, 269–290 CrossRef CAS PubMed.
- E. S. Tantawy, M. S. Nafie, H. A. Morsy, H. A. El-Sayed, A. H. Moustafa and S. M. Mohammed, RSC Adv., 2024, 14, 11098–11111 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- E. S. Tantawy, A. M. Amer, E. K. Mohamed, M. M. Abd Alla and M. S. Nafie, J. Mol. Struct., 2020, 1210, 128013 CrossRef CAS.
- M. E. Abdelaziz, M. M. M. El-Miligy, S. M. Fahmy, M. A. Mahran and A. A. Hazzaa, Bioorg. Chem., 2018, 80, 674–692 CrossRef CAS.
- CST, HTScan® Pim-1 Kinase Assay Kit, http://www.cellsignal.com/products/cellular-assay-kits/htscan-pim-1-kinase-assay-kit/7573, accessed July 17, 2020.
- M. S. Nafie, K. Arafa, N. K. Sedky, A. A. Alakhdar and R. K. Arafa, Chem. Biol. Interact., 2020, 324, 109087 CrossRef CAS.
- M. S. Nafie, A. M. Amer, A. K. Mohamed and E. S. Tantawy, Bioorg. Med. Chem., 2020, 28, 115828 CrossRef CAS PubMed.
- E. M. Gad, M. S. Nafie, E. H. Eltamany, M. S. A. G. Hammad, A. Barakat and A. T. A. Boraei, Molecules, 2020, 25, 2523 CrossRef CAS.
- M. S. Nafie, M. A. Tantawy and G. A. Elmgeed, Steroids, 2019, 152, 108485 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic characterizations of the synthesized compounds are provided as ESI. See DOI: https://doi.org/10.1039/d4ra07963a |
| This journal is © The Royal Society of Chemistry 2024 |