
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimizing CO2 methanation: effect of surface basicity and active phase reducibility on Ni-based catalysts
Marie-Nour
Kaydouh
 a,
Nissrine
El Hassan
*a,
Ahmed I.
Osman
*b,
Hamid
Ahmed
c,
Naif
Alarifi
*d,
Anis H.
Fakeeha
c,
Abdulrahman
Bin Jumah
c and
Ahmed S.
Al-Fatesh
*c
a,
Nissrine
El Hassan
*a,
Ahmed I.
Osman
*b,
Hamid
Ahmed
c,
Naif
Alarifi
*d,
Anis H.
Fakeeha
c,
Abdulrahman
Bin Jumah
c and
Ahmed S.
Al-Fatesh
*c
aPetroleum Engineering Program, School of Engineering, Lebanese American University, P.O. Box 36, Byblos, Lebanon. E-mail: nissrine.elhassan@lau.edu.lb
bSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast BT9 5AG, Northern Ireland, UK. E-mail: aosmanahmed01@qub.ac.uk
cChemical Engineering Department, College of Engineering, King Saud University, P.O. Box 800, Riyadh 11421, Saudi Arabia. E-mail: aalfatesh@ksu.edu.sa
dInstitute of Refining and Petrochemicals Technologies, King Abdulaziz City for Science and Technology (KACST), P.O. Box 6086, Riyadh 11442, Kingdom of Saudi Arabia. E-mail: naalarifi@kacst.gov.sa
First published on 24th April 2024
Abstract
CO2 methanation presents an intriguing avenue for utilizing carbon dioxide and generating methane as synthetic natural gas. This study delves into the innovative synthesis of MgO–Al2O3 mixed oxide support, a Co-active phase, and either Sr or Ce promoters to enhance the performance of Ni-based catalysts in CO2 methanation. The primary objective is to identify the optimal ratio of Mg to Al for supporting 5 wt% Ni, followed by assessing the synergistic utilization of Co and Ni, along with different promoters, on the most promising support. Despite exhibiting comparable textural and basic characteristics, the increase of Mg content in Al2O3 introduces a delay in NiO reduction by promoting the formation of a NiO–MgO solid solution. The Ni5/Mg63Al37 catalyst presents the highest CO2 conversion of 92% and CH4 yield of 82% at 400 °C. This catalytic activity surpasses that of Co5/Mg63Al37 and Ni2.5Co2.5/Mg63Al37, mainly due to easier reduction of the monometallic Ni-based sample. Examining the impact of promoters on Ni2.5Co2.5/Mg63Al37 catalyst reveals the advantageous influence of Ce in terms of facilitative reduction and improved basicity. However, the promoting effect of Sr remains less discernible, potentially due to the already increased basicity resulting from the utilization of the MgO–Al2O3 support.
Introduction
Carbon dioxide undergoes hydrogenation in a methanation reaction to produce methane, as depicted in eqn (1). This process is pivotal in mitigating the greenhouse effect and combating global warming. Indeed, the reduction of CO2 emissions is nowadays a common challenge worldwide.1 Furthermore, it offers a transition from the carbon capture and storage (CCS) strategy to a more pragmatic and favorable carbon capture and utilization (CCU) approach.2 Depending on the specific operational conditions, this reaction may be accompanied by concurrent side reactions, as illustrated in eqn (2) to (4). These side reactions can potentially yield undesired byproducts like carbon monoxide (CO) or consume methane, consequently leading to diminished product yield and selectivity.3 Alternatively, side reactions (eqn (5) to (9)) can also occur, resulting in solid carbon deposits that may accumulate on the catalyst surface, ultimately triggering catalyst deactivation.| CO2(g) + 4H2(g) = CH4(g) + 2H2O(g) | (1) |
| CO2(g) + H2(g) = CO(g) + H2O(g) | (2) |
| CO2(g) + CH4(g) = 2CO(g) + 2H2(g) | (3) |
| CO(g) + 3H2(g) = CH4(g) + H2O(g) | (4) |
| CO2(g) + 2H2(g) = C(s) + 2H2O(g) | (5) |
| CO2(g) + CH4(g) = 2C(s) + 2H2O(g) | (6) |
| 2CO(g) = C(s) + CO2(g) | (7) |
| CH4(g) = C(s) + 2H2(g) | (8) |
| CO(g) + H2(g) = C(s) + H2O(g) | (9) |
The utilization of mixed oxides as catalytic supports is gaining increasing attention due to their ability to exhibit tunable properties and modified metal–support interactions,13 in addition to the synergetic effect from the individual oxide components.14 The use of MgO, for instance, is an attractive approach due to its strong basicity that can enhance the adsorption of CO2 and improve the reaction yield.15 Enhancing the basicity of the catalysts creates additional adsorption sites for CO2 and favors the formation of more reactive reaction intermediates.16 The advantageous promotional effect of introducing Mg to Al2O3 on Ni-based17–19 and Co-based20 catalytic systems has been established to improve catalyst stability in dry reforming17,18,20 and combined steam and dry reforming of methane.19 In CO2 methanation, research indicates that adding Mg to the Pd/SiO2 catalyst increased the CO2 conversion from 40.8 to 59.2% and the CH4 selectivity from 10.4 to 95.3% at 450 °C.21 Similarly, co-impregnating Ni and Mg on SiO2 enabled the catalyst to maintain stability at 67% CO2 conversion and over 98% CH4 selectivity for a minimum of 50 hours.22 Impregnating up to 2.5% of Mg on Ni-based USY zeolites results in 15% and 20% enhancements in CO2 conversion and CH4 selectivity, respectively.23 Likewise, proper Mg loading on Ni-5 Mg/SBA-15-AE sample can efficiently catalyze CO2 activation,24 to reach 50% conversion at 295 °C, compared to 315 °C on unpromoted Ni/SBA-15-AE sample. These studies23,24 suggest that the amount of Mg-loading greatly impacts the extent of CO2 methanation. In line with these findings, Li et al.25 established an optimal Ni-to-Mg mass ratio of 0.26 for achieving high CO2 conversion rates. While these studies primarily used Mg as a promoter to enhance the methanation activity, its importance also resides in its use as a catalytic support. In this regard, Julkapli and Bagheri26 reviewed the use of MgO as a heterogeneous catalyst support and highlighted its capability to modify the acid–base properties of the catalyst, resulting in higher selectivity and lower formation of by-products. However, as a single oxide, MgO shows low thermal stability, particularly in humid and aqueous environments.26 As a component in mixed oxide supports, Mg plays a vital role in enhancing the basicity of the samples and increasing the metal–support interactions, which reflect in improved catalytic activity, as validated in several applications such as dry reforming of methane,27,28 hydrotreating,29 water gas shift,30 and ethanol coupling reactions.31 In CO2 methanation, the presence of Mg2+ strong basic sites in Ni/La2O3–MgO–CeO2 enhances the Ni interaction with the support and this emerges as a key factor to achieve high catalytic activity.32 In a similar context, the improved methanation activity of an MgO-modified ZrO2 support for Ru-based catalysts is attributed to better active phase dispersion and fast reaction intermediates decomposition and hydrogenation to methane.33 Likewise, the addition of basic MgO to Al2O3 alters the acidity of the catalyst, resulting, for example, in improved reactant conversion to syngas with an absence of carbon deposition during DRM,27 enhanced sulfidation of CoMo active metals for water gas shift reaction,30 and boosted ethanol coupling selectivity to butanol.31 However, most of these studies28,29,31 use a fixed MgO–Al2O3 support with a constant MgO content and evaluate the effects of metal nature, active metal loading, promoter, or calcination temperature. The effect of increasing MgO content in Ni/Al2O3 catalysts have been recently evaluated for the dry reforming of methane reaction.34–36 Results show that the MgO content affects the Ni particles stability and interaction with the support, thus altering the hydrogen yield and carbon deposition. Yet, to the best of our knowledge, the effect of variation of Mg content in MgO–Al2O3 supports on CO2 methanation has not been studied in literature before.
Concerning the active phase, several studies have explored the utilization of cobalt as an alternative for CO2 methanation,37–45 primarily due to its activity in Fischer–Tropsch synthesis, with a tendency to generate more methane than heavier hydrocarbons.46,47 As a result, cobalt has been examined across various oxides and mixed oxide supports, including TiO2, ZrO2, Al2O3, SiO2, CeO2,37–40 among others. The outcomes of these investigations have demonstrated diversity, dependent upon the specific support nature38,39 and the preparation method employed.37 Le et al.39 identified CeO2 as a suitable support for Co, while Li et al.38 highlighted ZrO2 as the optimal choice, attributing the observed deactivation on Al2O3 to the presence of acid sites on this support. Nonetheless, studies have consistently shown the enhanced catalytic activity of cobalt compared to other metals. Notably, Co/Al2O3 exhibited significantly greater activity in CO2 methanation compared to Ni/Al2O3 at GHSV = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1, even at lower metal loading, with the discrepancy attributed to divergent reaction intermediates during the process.41 The distinct reaction intermediates were also linked to the influence of additives used in conjunction with Co/Al2O3 (such as Na, K, Mg, or Ca), leading to varying CH4 yields in each case.40 Indeed, while single metal oxide catalysts are subject to poisoning and low thermal stability, the use of multiple oxides can help overcome these problems and achieve enhanced properties, such as better metal dispersion, resulting in improved catalytic activity.48 In this regard, Co exhibited superior methanation activity when incorporated alongside Ni on Al2O3 support compared to Fe and Mo.42 This points toward the potential of bimetallic catalysts to enhance catalytic properties through modifications in Ni dispersion, the fundamental characteristics of the support, and the Ni–support interaction.13,49 For instance, the utilization of Ni–Co supported on CaAl2O4/Al2O3 leads to an enhanced coking resistance during partial methane oxidation.50 In CO2 methanation, the bimetallic Ni–Co catalyst supported on ordered mesoporous alumina showed excellent stability at 500 °C for 60 hours, while the Ni-based catalyst exhibited a 14.8% decrease in CO2 conversion.51 Nevertheless, the favorable impact of bimetallic Ni–Co catalysts, as reported in certain studies,51,52 did not manifest evidently in other investigations53,54 since large metal particles were formed using Co/Ni/Al2O3 and resulted in lower CH4 yield, in addition to CO formation.53 Hasrack et al.54 showed comparable stability at 250 °C for 5 hours, with a slightly higher performance of 1Co15Ni/CeO2 catalyst.
000 h−1, even at lower metal loading, with the discrepancy attributed to divergent reaction intermediates during the process.41 The distinct reaction intermediates were also linked to the influence of additives used in conjunction with Co/Al2O3 (such as Na, K, Mg, or Ca), leading to varying CH4 yields in each case.40 Indeed, while single metal oxide catalysts are subject to poisoning and low thermal stability, the use of multiple oxides can help overcome these problems and achieve enhanced properties, such as better metal dispersion, resulting in improved catalytic activity.48 In this regard, Co exhibited superior methanation activity when incorporated alongside Ni on Al2O3 support compared to Fe and Mo.42 This points toward the potential of bimetallic catalysts to enhance catalytic properties through modifications in Ni dispersion, the fundamental characteristics of the support, and the Ni–support interaction.13,49 For instance, the utilization of Ni–Co supported on CaAl2O4/Al2O3 leads to an enhanced coking resistance during partial methane oxidation.50 In CO2 methanation, the bimetallic Ni–Co catalyst supported on ordered mesoporous alumina showed excellent stability at 500 °C for 60 hours, while the Ni-based catalyst exhibited a 14.8% decrease in CO2 conversion.51 Nevertheless, the favorable impact of bimetallic Ni–Co catalysts, as reported in certain studies,51,52 did not manifest evidently in other investigations53,54 since large metal particles were formed using Co/Ni/Al2O3 and resulted in lower CH4 yield, in addition to CO formation.53 Hasrack et al.54 showed comparable stability at 250 °C for 5 hours, with a slightly higher performance of 1Co15Ni/CeO2 catalyst.
Alternatively, an increasing focus has been directed towards incorporating promoters into Ni-based catalysts, aiming to enhance specific properties while benefitting from the accessibility and cost-effectiveness associated with these additives.55 For example, the addition of strontium (Sr) to Ni/CeZrOx increased the basicity of the catalyst and resulted in an enhancement of CO2 adsorption.56 It also improved the metal–support interaction when used as a promoter on Ni-based catalysts supported on Al2O3,57–59 thus boosting the catalytic performance during CO2 methanation. The addition of Sr to Ni/SiO2 catalysts increased CO2 conversion from 64.7 to 70.5% and CH4 selectivity from 97.5 to 98.9% at 350 °C and 15000 ml h−1 g−1 while preventing Ni metal sintering.60 Similarly, the utilization of CeO2 is advantageous owing to its inherent oxygen storage capacity, which can help address the issue of carbon deposition on the catalyst's surface.61 Cerium introduces oxygen vacancies that can lead to higher conversion of CO to CO2 during propane steam reforming, for example, preventing carbon deposition and resulting in higher hydrogen production.62 It has been reported that oxygen defects on the surface of the catalyst play an important role in enhancing CO2 adsorption, activation, and conversion by hydrogenation into methane.63 Liu et al.64 revealed an improvement in CO2 conversion from 45 to 71% upon the addition of 2 wt% CeO2 to Ni/Al2O3 catalyst during CO2 methanation at 300 °C and 15000 ml h−1 g−1. A higher CeO2 loading can result in active site coverage and consequent activity loss. Likewise, a 2.5 wt% Ce addition to 10 wt% Ni supported on Al2O3 boosted CO2 conversion from 58 up to 71% and CH4 selectivity from 94 to 96%, while reducing CO selectivity.65
Therefore, it is intriguing to leverage the benefits of each catalyst constituent to achieve superior performance in CO2 methanation. To the best of our knowledge, only a limited number of studies have explored such intricate combinations. Varbar et al.66 evaluated the effect of Co addition to 20 wt% Ni/MgAl2O4 in methane oxidation and found that the addition of 3 wt% Co is optimum to limit side reactions. Zhang et al.67 varied the amount of CeO2 added to Ni/MgO–Al2O3 in CO methanation and determined an optimum content of 10 wt% CeO2 for high performance. The use of cobalt-promoted Ni–Mg–Al hydrotalcite-derived catalysts68 or Co–Ni–Mg–Al mixed-oxides69 can be promising in CO2 methanation, yet these studies do not focus on the effect of each component of the catalyst.
Therefore, the innovation intrinsic to our present work resides in the comprehensive assessment of the impact of each catalyst constituent. While limited literature evaluates the composition of mixed-oxide support,34–36 we assess in this work the increase of Mg-content in MgO–Al2O3 support and its effect on catalyst basicity and resulting activity in CO2 methanation. Furthermore, the preceding literature shows that cobalt can be a potential active phase for CO2 methanation and methane production, yet its performance depends on the nature of support and the type of promoters along which it is tested. This is due to different surface properties such as the interaction of the metal with its support, its dispersion, reducibility, and basicity. It is thus suggested in this work to test this candidate along with nickel on an optimized MgO–Al2O3 support. While the effect of using Ni- and/or Co-based catalysts vary from one study to another, the effect of mono- and bi-metallic catalysts and the possible synergetic effect between the two metals will be explored on the best MgO–Al2O3 support. Also, on these samples, we additionally explore possible benefits from incorporating suitable promoters for CO2 methanation: Sr will be added as a promoter to enhance catalyst basicity, while Ce will be tested in this composition to improve metal dispersion and metal oxides reducibility and overcome carbon deposition problems. These enhancements are expected to improve the catalytic performance during CO2 methanation. Such promising combinations have not been tested for CO2 methanation before.
Experimental
Catalyst preparation
The supports used in this work were obtained as a gift from SASOL Anckelmannsplatz 1, 20537, Hamburg, Germany. As specialized providers of alumina-based catalyst carriers, they offered these supports to be used and tested in CO2 methanation, among other catalytic applications. They were composed of different alumina and magnesium oxide ratios, specifically including 20, 30, 63, and 70 wt% MgO balanced with Al2O3. The catalysts were prepared via the impregnation method. For the first series of samples, 20 mL of distilled water and the appropriate amount of nickel nitrate hexahydrate [Ni(NO3)2·6H2O, 98%, Alfa Aesar] to get 5.0 wt% loading of nickel oxide and the required amount of support (mixture of alumina and magnesium oxide) were mixed and stirred at room temperature in a crucible. After 30 minutes, the solution was dried while being stirred at 80 °C. The catalysts were then calcined at 600 °C for 3 hours. The resulting samples are denoted Ni5/MgxAl100−x (x = 20, 30, 63, or 70 wt%).For the second series of samples, the corresponding amount of Ni(NO3)2·6H2O and/or Co(NO3)2·6H2O to get 5.0 wt% Co or 2.5% Ni + 2.5% Co was dissolved in 30 ml of distilled water with stirring and heating. Afterwards, 63% MgO and 37% Al2O3 support were added, and the stirring was continued for 30 minutes. The obtained catalysts were dried at 120 °C for 20 hours and then calcined for 5 hours at 600 °C. The promoted catalysts were prepared by incorporating 3 wt% of either Ce or Sr along the active metals before the support addition stage, using their corresponding nitrates (Ce(NO3)3·6H2O; Sr(NO3)2). The resulting samples are referred to as Co5/Mg63Al37 (Ni-free sample), Ni2.5Co2.5/Mg63Al37, Ni2.5Co2.5Ce3/Mg63Al37 or Ni2.5Co2.5Sr3/Mg63Al37.
Catalyst characterization
The textural properties of the samples were evaluated using a Micromeritics Tristar II 3020 surface area and porosity analyzer after degassing at 200 °C for 3 h using N2. The XRD data were recorded on a Rigaku (Miniflex) diffractometer equipped with Cu Kα radiation and operated at 40 kV and 40 mA. The diffractograms were collected in a 2θ range between 5 and 90° at a step 0.02°. The Diffrac. EVA v4.2.1 software was used to identify the crystallite species present and estimate their size. The temperature-programmed desorption (TPD) data were acquired using Micromeritics Auto Chem II 2920, after running 30 ml min−1 of a 10% CO2/He mixture over the sample for 30 minutes at 50 °C. Then, the temperature increased to 900 °C, and the amount of CO2 desorbed was found at various temperatures using a TCD detector. The total amount of CO2 desorbed was thus calculated by integration of the resulting peaks. The FTIR data were taken using a Perkin Elmer GX spectrophotometer. The spectra were registered at the range 400–4000 cm−1 employing a KBr pellet. The TEM images were obtained using a transmission electron microscope JEOL JEM-2100F, USA, operated at 120 kV. A laser Raman (NMR-4500) spectrometer (JASCO, Japan) was employed to collect the Raman spectra of the spent catalysts using an objective lens of 100× magnification. The excitation beam was fixed at 532 nm wavelength, and the laser intensity was set at 1.6 mW for a 10-second exposure time at 3 accumulations. A Micromeritics Auto-Chem II 2920 device was used to perform H2 temperature-programmed reduction (H2-TPR). The catalyst sample was heated for one hour at 200 °C in an argon environment prior to the measurements, and it was then cooled to room temperature. Using a gas mixture of H2/Ar (v/v, 10/90) at a flow rate of 40 mL min−1, 0.07 g of the sample was heated at a rate of 10 °C min−1 to 1000 °C. The H2 consumption was captured using a TCD detector.Catalyst testing
The catalytic activity was evaluated in a fixed-bed stainless-steel continuous-flow reactor (PID Eng. & Tech), having an internal diameter of 0.94 cm and a length of 30 cm. The reaction temperature was controlled and monitored by using a thermocouple placed in the center of the catalytic bed. A schematic of the reaction setup is shown in Fig. 1. At the beginning of the test, 0.12 g of each sample was first reduced in situ under a hydrogen flow of 30 mL min−1 at 800 °C for 2 hours at atmospheric pressure. Then, the reactor was cooled, and a total reactive flow of 30 mL min−1, composed of 16 mL min−1 H2, 4 mL min−1 CO2 and 10 mL min−1 N2, was introduced at an equivalent gas hourly space velocity (GHSV) of 15000 mL h−1 gcat−1. The mixture was carried by argon gas in a gas chromatograph (GC). The methanation reaction was performed at 400 °C for 300 minutes at atmospheric pressure. The reaction temperature, pressure, and other variables were monitored through the reactor panel. A SHIMADZU GC-2014 equipped with Porapak Q and Molecular Sieve 5A columns and a thermal conductivity detector analyzed the reaction products and feed using a combination of series and bypass connections. To quantify the results from catalytic experiments, the peak areas of the components separated by the GC were analyzed. These peak areas are proportional to the amount of each component present. While a basic comparison can be done using peak areas alone, a calibration process with standard gases is usually employed for more accurate quantification. This involves running known concentrations of reactants and products (external standards) through the GC to create a calibration curve. The peak areas of the samples are then compared to this curve to determine their actual concentrations. Additionally, an internal standard gas (such as N2) can be added to both samples and calibration standards. By comparing the peak area ratio of the target component to the internal standard, variations in injection volume and detector response are accounted for, leading to more reliable results. The catalytic tests were repeated twice for all the catalysts for reproducibility.
The CO2 conversion and CH4 yield were evaluated using eqn (10) and (11):
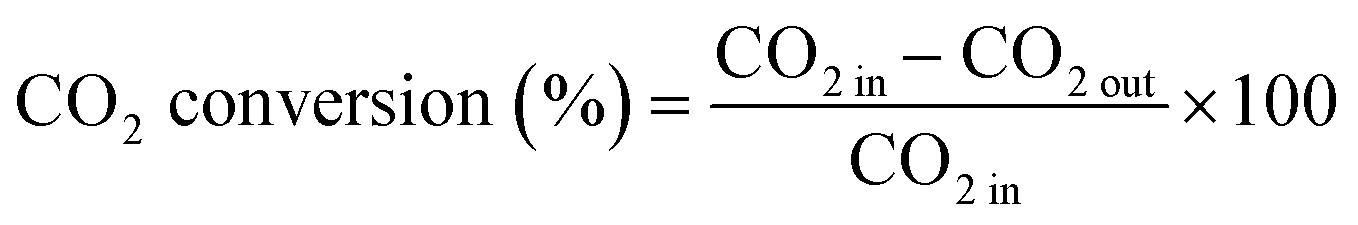 | (10) |
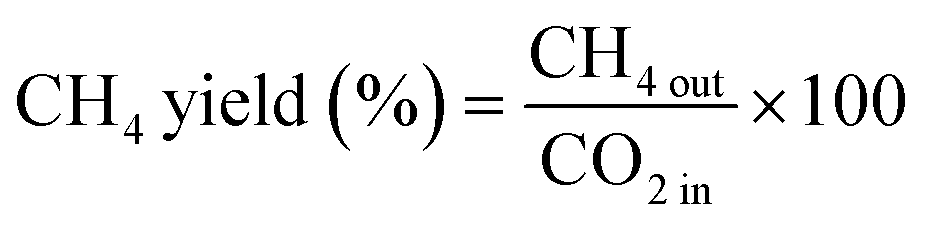 | (11) |
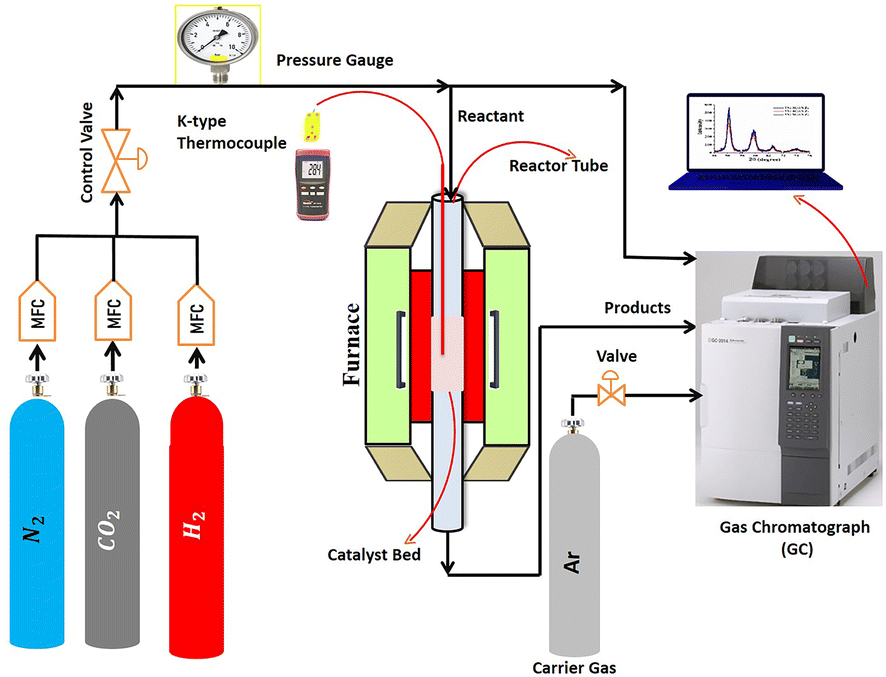 | ||
| Fig. 1 Reaction setup for the CO2 methanation testing. | ||
Results and discussion
During CO2 methanation, the presence of metallic Ni0 sites is highly essential since they constitute the active sites that promote the adsorption and dissociation of H2.6,11,70 Consequently, a reduction step is systematically applied, prior to the methanation reaction to convert nickel oxide into metallic nickel.57 For these reasons, the samples are examined in this work in their reduced states to understand their properties. Hence, only the textural properties of the calcined samples are shown, while the remaining characterization results of the BET, XRD, CO2-TPD and FTIR experiments are presented for the samples in their reduced states.Effect of support composition on CO2 methanation performance
All the calcined34,35 and reduced samples exhibited type IV N2 sorption isotherms (Fig. 2), characteristic of mesoporous materials, comparable to those reported for similar MgO–Al2O3 mixed oxide supports.31 The H4 hysteresis loop observed for all samples is the consequence of MgO presence in the samples.28 The analysis of textural properties (Table 1) indicated a gradual decline in surface area and pore volume with the increase in the percentage of MgO. Specifically, the BET surface area and pore volume decreased from 184 m2 g−1 and 0.53 cm3 g−1 for the reduced Ni5/Mg20Al80 to around 197 m2 g−1 and 0.29 cm3 g−1 for reduced Ni5/Mg63Al37 and 161.2 m2 g−1 and 0.27 cm3 g−1 for reduced Ni5/Mg70Al30, respectively. This can be explained by possible pore blockage as the MgO content increases in the support.35 Furthermore, a similar trend was observed for the pore size, which dropped from 8.4 nm for reduced Ni5/Mg20Al80 to approximately 5.5 nm for reduced Ni5/Mg63Al37. This is attributed to the presence of more Mg species in the pores, as also observed in Mg-containing Ni/SBA-15 catalytic systems.24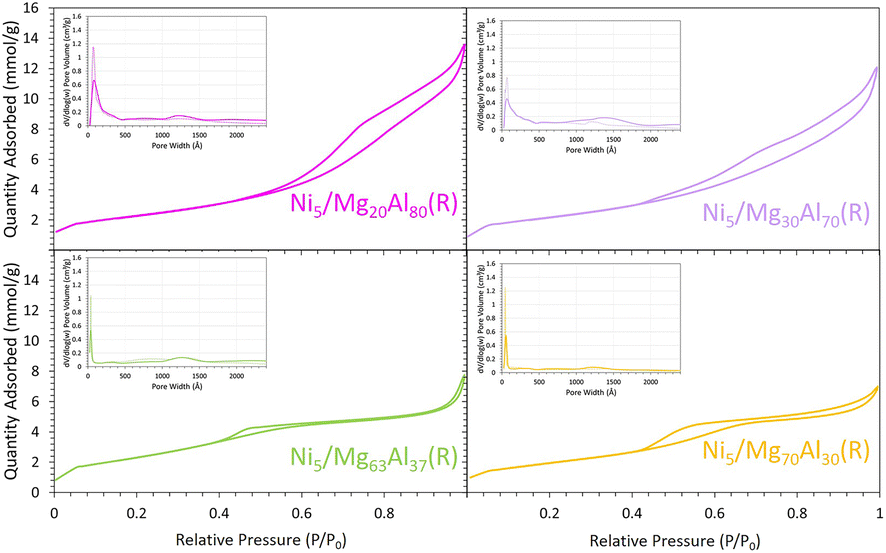 | ||
| Fig. 2 N2 sorption isotherms and pore size distribution (inset figures, solid line: adsorption, dashed line: desorption) for the reduced Ni5/MgxAl100−x (where x = 20, 30, 63 or 70 wt%) samples. | ||
| Sample | BET surface area (m2 g−1) | BJH adsorption cumulative pore volume (cm3 g−1) | BJH desorption cumulative pore volume (cm3 g−1) | BJH adsorption average pore width (Å) | BJH desorption average pore width (Å) |
|---|---|---|---|---|---|
| Ni5/Mg20Al80(R) | 183.7 | 0.49 | 0.53 | 94 | 84 |
| Ni5/Mg30Al70(R) | 176.5 | 0.44 | 0.47 | 91 | 80 |
| Ni5/Mg63Al37(R) | 197.3 | 0.29 | 0.29 | 53 | 55 |
| Ni5/Mg70Al30(R) | 161.2 | 0.25 | 0.27 | 58 | 55 |
| Co5/Mg63Al37(R) | 185.3 | 0.28 | 0.29 | 58 | 57 |
| Ni2.5Co2.5/Mg63Al37(R) | 212.3 | 0.30 | 0.31 | 52 | 55 |
| Ni2.5Co2.5Sr3/Mg63Al37(R) | 182.3 | 0.29 | 0.29 | 57 | 59 |
| Ni2.5Co2.5Ce3/Mg63Al37(R) | 175.7 | 0.27 | 0.29 | 56 | 60 |
The H2-TPR profiles, reported in previous studies,34,35 exhibit a prominent reduction peak ranging from 700 to 1000 °C. This indicates two important points: firstly, the absence of free NiO species that are typically reduced between 300 and 400 °C;15 and secondly, the positioning of this peak at 830, 840, 860, and 925 °C for an Mg content of 20, 30, 63 and 70%, respectively. This implies that an increase in the Mg content of the support delays the reduction process and makes the reducibility of the active phase more difficult. This phenomenon is commonly attributed to the formation of either a stable NiO–MgO solid solution,15,23 or MgNiO2 (ref. 24) upon the incorporation of Mg, suggesting a strong interaction between NiO and MgO in both cases. In this regard, Kumar et al.29 reported stronger metal–support interaction of Mo/MgO–Al2O3 compared to single oxide supports. Similarly, Lian et al.30 reported a higher metal–support interaction upon the increase in Mg content in Co–Mo/MgO–Al2O3 samples. Furthermore, during the reduction, hydrogen consumption rises proportionally with the increment in Mg content. Specifically, it increases from 0.96 mmol g−1 for Ni5/Mg20Al80 and Ni5/Mg30Al70 samples to about 1.22 mmol g−1 for Ni5/Mg63Al37 and Ni5/Mg70Al30 samples.
Following reduction, the X-ray diffractograms of the reduced samples (Fig. 3) exhibit prominent peaks at about 37, 43, and 63°, which can be primarily attributed to magnesium nickel oxide Mg0.7Ni0.3O (ref. 04-023-4643) or MgO (ref. 00-003-0998), both having a Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m cubic lattice, corresponding to characteristic interplanar spacings (111), (200) and (220), respectively. These peaks can also be assigned to spinel MgAl2O4 (ref. 00-005-0672) having a Fdm structure, indexed as (311), (400) and (440), respectively. A more detailed examination of the significant peak unveils a shift towards spinel MgAl2O4 (∼44.8°) at low Mg content and a shift towards MgO (∼43.1°) at high Mg content.15,23 This observation supports the earlier discussion on the formation of a mixed NiO–MgO phase at elevated Mg content, as evident from the TPR results. Moreover, it is in full agreement with published data that show a reduction of the spinel formation upon the increment of Mg content due to the promotion of the mixed solid phase.15,23,27,28 On these profiles, the presence of very small peaks at 51 and 75° confirms the presence of metallic Ni0 species.
m cubic lattice, corresponding to characteristic interplanar spacings (111), (200) and (220), respectively. These peaks can also be assigned to spinel MgAl2O4 (ref. 00-005-0672) having a Fdm structure, indexed as (311), (400) and (440), respectively. A more detailed examination of the significant peak unveils a shift towards spinel MgAl2O4 (∼44.8°) at low Mg content and a shift towards MgO (∼43.1°) at high Mg content.15,23 This observation supports the earlier discussion on the formation of a mixed NiO–MgO phase at elevated Mg content, as evident from the TPR results. Moreover, it is in full agreement with published data that show a reduction of the spinel formation upon the increment of Mg content due to the promotion of the mixed solid phase.15,23,27,28 On these profiles, the presence of very small peaks at 51 and 75° confirms the presence of metallic Ni0 species.
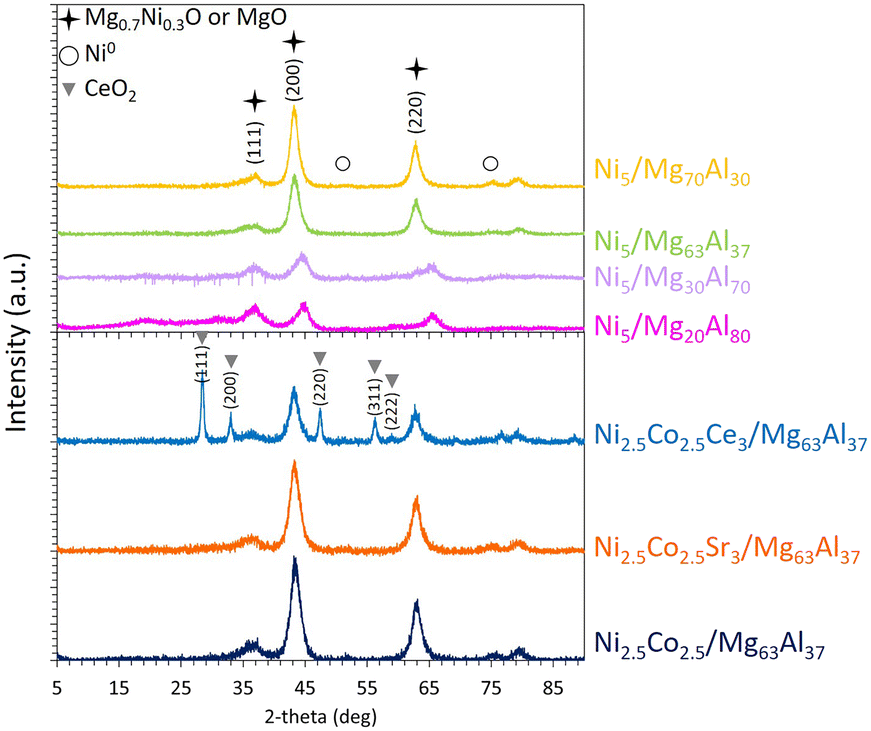 | ||
| Fig. 3 XRD patterns of the reduced Ni5/MgxAl100−x (where x = 20, 30, 63, or 70 wt%) catalysts and Co-containing catalysts supported on Mg63Al37, along with the characteristic (hkl) interplanar planes of the identified species. | ||
Furthermore, the assessment of the crystalline domains using the major peak at 43° indicates an increase in their sizes from 4.9 and 4.6 nm for Ni5/Mg20Al80 and Ni5/Mg30Al70 reduced samples, respectively, to 5.8 nm for Ni5/Mg63Al37 and 6.9 nm for Ni5/Mg70Al30 reduced samples. This suggests that the increase in Mg content slightly reduces the dispersion of the active phase, as similarly observed in other studies.18,25 For instance, Karam et al.18 reported an increase in the NiO crystalline domain size from 4.6 nm for Ni6–Mg5–Al2O3 to 5.4 and 7.7 nm for Ni6–Mg15–Al2O3 and Ni6–Mg26–Al2O3, respectively. Conversely, in Mg-modified Ni/SiO2 catalysts, an increase in Mg-loading led to improved metal dispersion.22 This discrepancy can be attributed to different preparation methods employed in each study.
The evaluation of the basicity of the reduced samples is conducted using CO2-TPD (Fig. 4). The resulting peaks can be conventionally decomposed into three major regions: the 50 to 200 °C range for low basicity, 200 to 400 °C for moderate basicity, and above 400 °C for strong basicity.57 Irrespective of the Mg content, all peaks are predominantly centered around 250 °C, indicating the presence of mainly moderate basic sites on all samples. Notably, the Ni5/Mg63Al37 sample displays the highest amount of desorbed CO2 of 0.29 mmol g−1, in contrast to the values of 0.11, 0.16, and 0.22 mmol g−1 for Ni5/Mg20Al80, Ni5/Mg30Al70 and Ni5/Mg70Al30, respectively. Comparably, the total basicity of catalysts increased with increasing Mg content from 0.32 μmol m−2 for Ni–3Mg/SBA-15-WI to 0.39 and 0.41 μmol m−2 for Ni–5Mg/SBA-15-WI and Ni–7Mg/SBA-15-WI, respectivel.24 Similarly, the total amount of CO2 desorbed increased from 1.84 mmol g−1 to 5.19 mmol g−1 as the Mg/Al ratio of the support increased from 0.3 to 2.27 This implies that the magnitude of Mg loading substantially influences the extent of CO2 adsorption on the catalyst, consequently impacting the catalytic performance, a topic that will be elaborated on in the subsequent section.
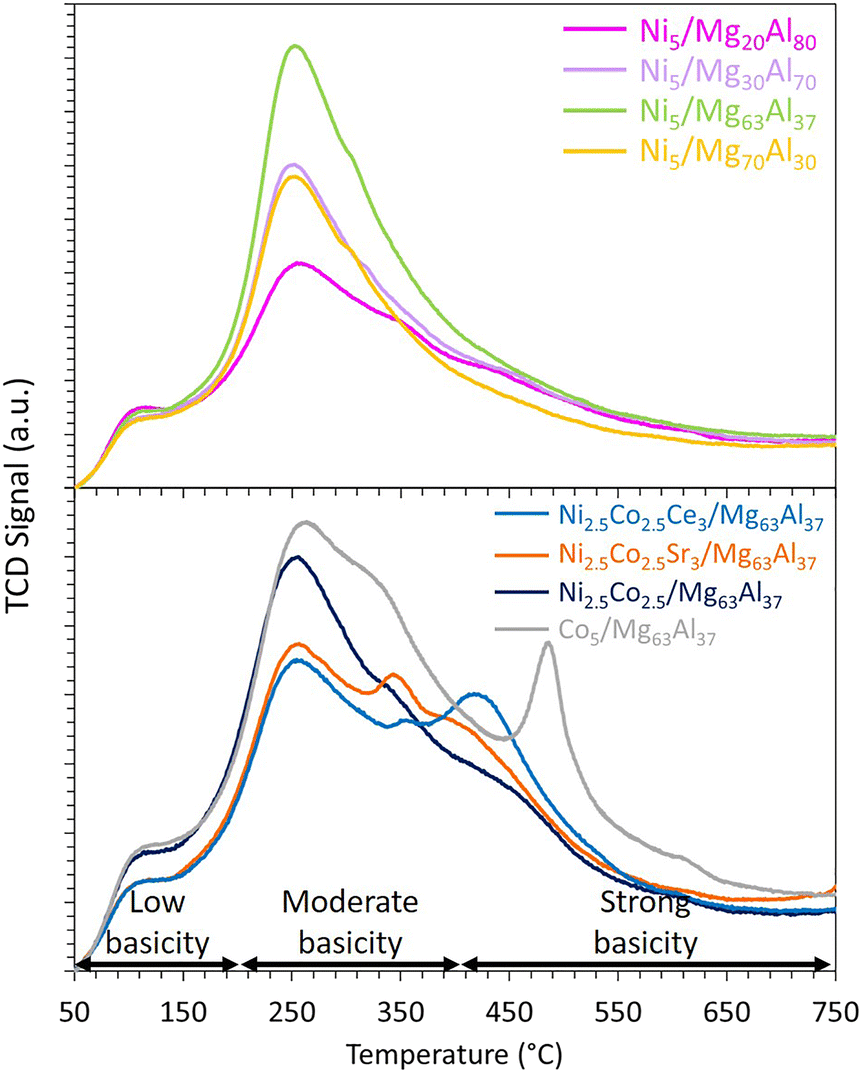 | ||
| Fig. 4 CO2-TPD results of reduced Ni5/MgxAl100−x (where x = 20, 30, 63 or 70 wt%) catalysts and Co-containing catalysts supported on Mg63Al37. | ||
The FTIR results (Fig. 5) reveal transmission bands at 3430 and 1630 cm−1 that can be assigned to stretching and bending vibrations of –OH hydroxyl groups or adsorbed water on the surface of the samples, respectively.70,71 The transmission bands at 2890 and 2350 cm−1 are attributed to –C–H stretch,58 and to CO2 adsorption on the samples,72 respectively. The bands between 400 and 500 cm−1 correspond to Ni–O stretching73 and Ni–O–Ni bond.74
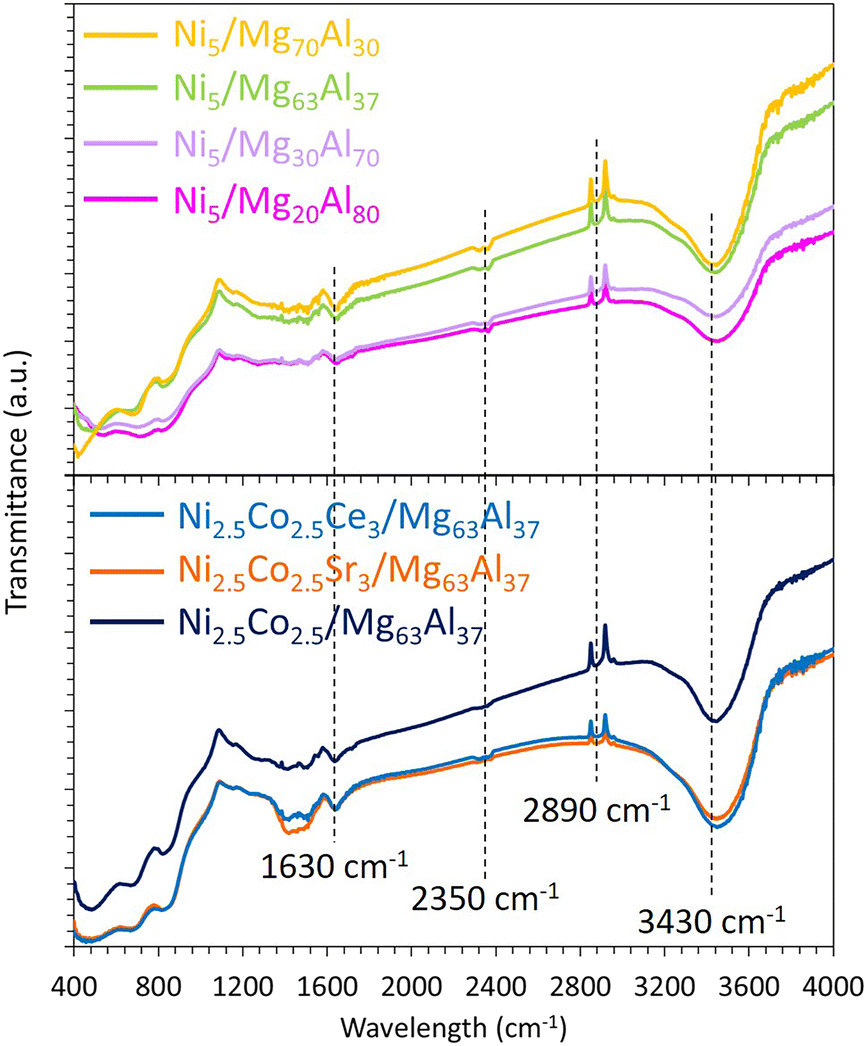 | ||
| Fig. 5 FTIR spectra of the reduced Ni5/MgxAl100−x (where x = 20, 30, 63 or 70 wt%) catalysts and Co-containing catalysts supported on Mg63Al37. | ||
The CO2 methanation performance of the reduced samples is evaluated at 400 °C (Fig. 6). Overall, all samples exhibit stability and maintain a constant level of CO2 conversion and CH4 yield for about 300 min. This finding aligns with previous research,75 which suggests that the presence of medium basic sites facilitates the hydrogenation step and enhances CO2 methanation.
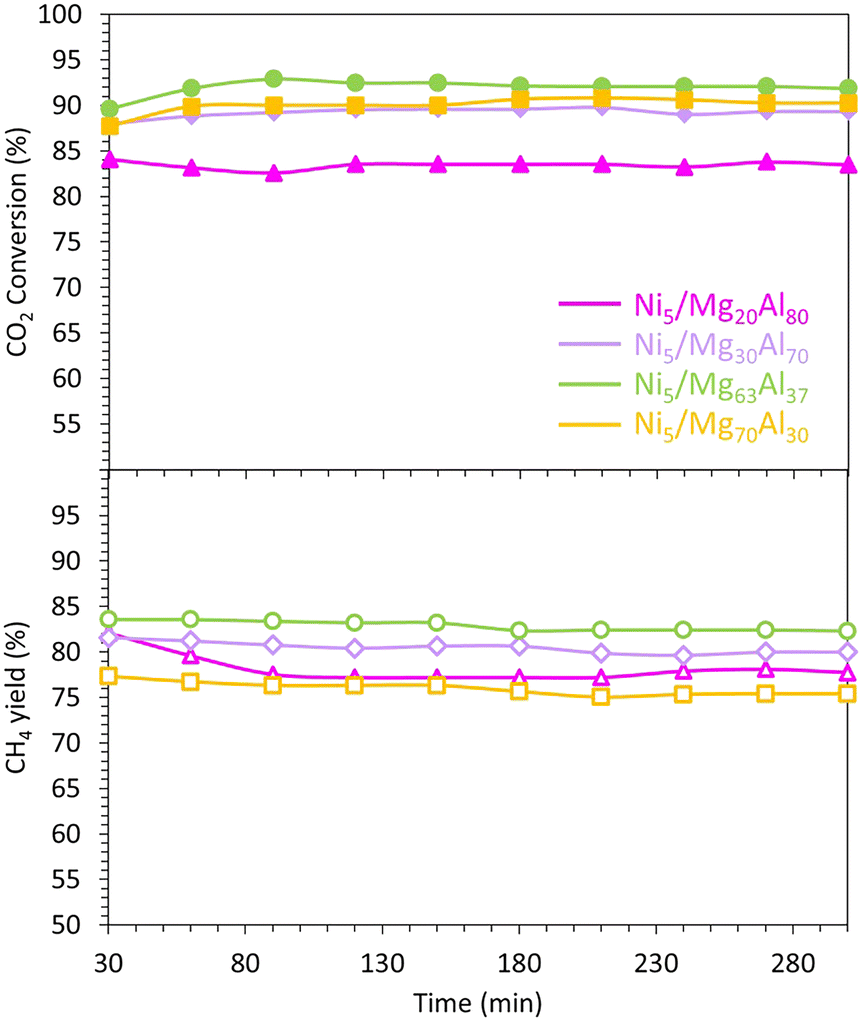 | ||
| Fig. 6 Catalytic test results of the Ni5/MgxAl100−x (where x = 20, 30, 63 or 70 wt%) catalysts. The methanation reaction was performed at 400 °C for 300 minutes, at an equivalent gas hourly space velocity of 15000 mL g−1 h−1. | ||
In more specific terms, the CO2 conversions are approximately 83% for Ni5/Mg20Al80, 89% for Ni5/Mg30Al70, 92% for Ni5/Mg63Al37 and 90% for Ni5/Mg70Al30. Notably, the Ni5/Mg30Al70 catalyst demonstrates the highest CO2 conversion, while the Ni5/Mg20Al80 catalyst exhibits the lowest activity. The trend in CO2 conversion seems to align with the amount of CO2 desorbed during the CO2-TPD experiment (Fig. 8). This suggests that the catalyst with the strongest affinity for CO2 displays the highest activity during the reaction.
However, despite the Ni5/Mg70Al30 sample showing relatively high CO2 conversion, it is the least selective and produces the lowest CH4 yield. Indeed, the CH4 yield changes as follows: Ni5/Mg70Al30 (75%) < Ni5/Mg20Al80 (78%) < Ni5/Mg30Al70 (80%) < Ni5/Mg63Al37 (82%). These findings imply that increased Mg content enhances the catalytic activity by improving both CO2 conversion and CH4 yield. Nevertheless, an optimal Mg content of 63% seems to strike a balance, as further increase negatively impacts catalytic performance, leading to a significant drop in both CO2 conversion and CH4 yield. Similar findings were also obtained on Ni–Mgx/coconut shell carbon (CSC) catalysts,25 where the high performance of the optimum catalyst (x = 0.26) was assigned to its strong basicity and strong adsorption affinity for CO2.
Interestingly, the XRD profiles of the Ni5/Mg63Al37 catalyst before and after testing (Fig. 7a and a′) show preservation of the peaks and their relative intensities, as observed on the reduced sample earlier. This indicates the absence of metal sintering and the preservation of a good active phase dispersion throughout the test. On the Raman spectra (Fig. 7b), the absence of the G-band at around 1580 cm−1 and the D-band near 1350 cm−1, typical of carbon-containing materials, reveals the absence of carbon deposition on the used Ni5/Mg63Al37 catalyst. These observations are also validated on the TEM images of the Ni5/Mg63Al37 catalyst before and after testing (Fig. 7c and c′) that confirm the absence of carbon deposits and show good maintenance of active phase dispersion without apparent metal agglomeration, suggesting high thermal stability of the sample under the employed conditions.
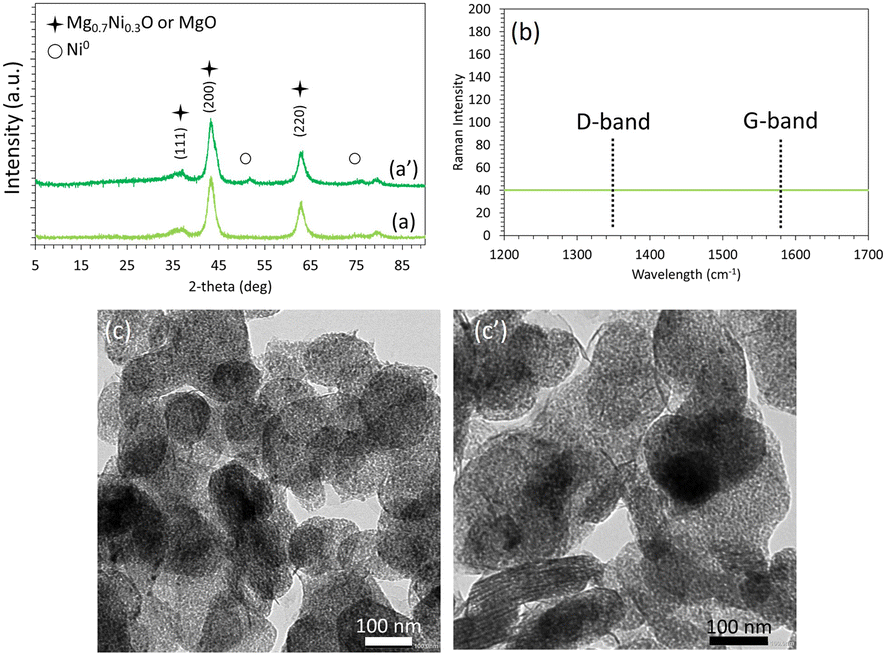 | ||
| Fig. 7 XRD profiles (a and a′), Raman spectra (b) and TEM images (c and c′) of the reduced (a and c) and spent (a′, b and c′) Ni5/Mg63Al37 catalysts. | ||
To better clarify the effect of Mg loading on the catalytic performance, the catalytic test results are coupled with the amount of CO2 desorbed during CO2-TPD and the maximum reduction temperature observed during TPR (Fig. 8). The results illustrate that the extent of CO2 conversion (shown in red in Fig. 8) follows the same pattern as the amount of CO2 desorbed during CO2-TPD (depicted as a histogram in Fig. 8). CO2 conversion increases with the increase in Mg content up to 63 wt% and then declines beyond this threshold, aligning with the trend observed in the amount of CO2 desorbed during CO2-TPD. This suggests that the most active catalyst, namely Ni5/Mg63Al37 sample, exhibits the highest CO2 desorption, signifying the highest CO2 adsorption affinity. Moreover, the NiO reduction occurs at a reasonable temperature (black curve in Fig. 8). Consequently, the Mg63Al37 support is chosen for further investigation in the subsequent phases of the study.
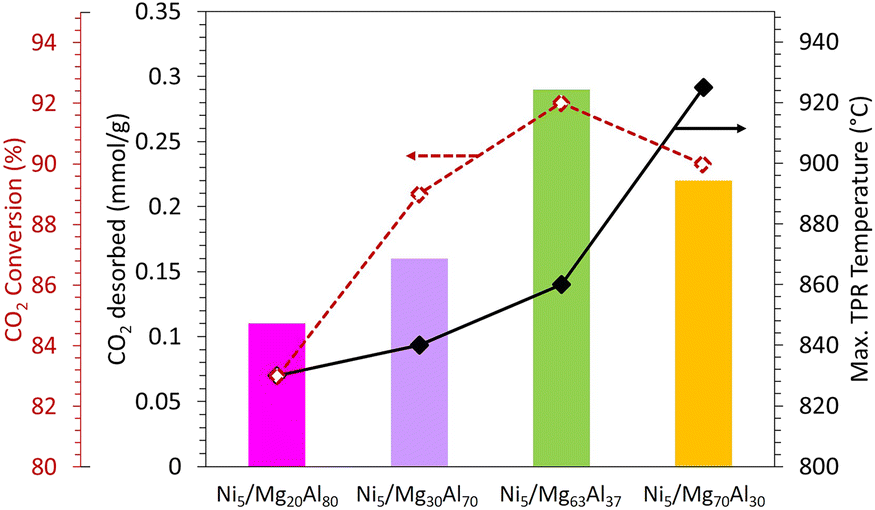 | ||
| Fig. 8 Amount of CO2 desorbed by CO2-TPD (mmol g−1), CO2 conversion (%) and maximum TPR temperature (°C) obtained on Ni5/MgxAl100−x (where x = 20, 30, 63 or 70 wt%) catalysts. | ||
The absence of similar catalysts tested in literature for CO2 methanation makes the comparison of performance challenging. While the 5Ni–7Mg–Al2O3 and 5Ni–26Mg–Al2O3 show compositions close to the samples used in this work,15 they present lower conversions than those of the current study. This can be primarily attributed to the much higher gas hourly space velocity of 86100 mL g−1 h−1 used during the test15 compared to 15000 mL g−1 h−1 applied in the present work. Thus, differences in the operating conditions can make the catalytic comparison impractical. Yet, the optimum properties discussed above and the superior catalytic performance of the Ni5/Mg63Al37 catalyst reflect in higher performance compared to some catalytic systems reported in the literature (Table 2). Indeed, at the same reaction temperature of 400 °C, the CO2 conversion of the mentioned catalyst is higher than that of 30Ni/Al2O3·0.5SiO2 (ref. 14) and other Ni–Mgx samples supported on coconut shell carbon (CSC),25 even when the latter are tested at lower GHSV. The samples presented in this work are tested under more drastic conditions, i.e. higher GHSV of 15000 mL g−1 h−1, yet they still show higher performance than those tested under milder conditions using a GHSV of 9000 and 10000 mL g−1 h−1. Such differences can be associated with variations in catalyst composition or differences in the preparation methods adopted in each study. In this regard, Burger et al.76 showed that employing different preparation methods for Ni–Al catalysts can result in completely different chemical interactions that translate into diverse behaviors in CO2 methanation upon Fe-doping. In more detail, the large difference between the CO2 conversion obtained in this work and that of 30Ni/Al2O3·0.5SiO2 can be assigned to the absence of MgO, and consequently of basic sites, on the sample, limiting thus CO2 adsorption and conversion. More globally, an evaluation of the TPR profiles of the various samples compared in Table 2 reveals that most of these samples are reducible at temperatures near 700 °C, which are much lower than that of Ni5/Mg63Al37 (860 °C). This implies that higher metal–support interactions are achieved on Ni5/Mg63Al37, reflecting in higher catalytic performance. This comparison highlights the beneficial effect of using MgO–Al2O3 in optimized proportions as a catalytic support for Ni-based samples in CO2 methanation.
| Sample | Catalyst weight (mg) | Reaction temperature (°C) | H2:CO2 |
GHSV (mL g−1 h−1) | CO2 conversion (%) | Ref. |
|---|---|---|---|---|---|---|
| Ni5/Mg63Al37 | 120 | 400 | 4 | 15000 |
92 | This study |
| 5Ni–7Mg–Al2O3 | 200 | 400 | 4 | 89400 |
60 | 15 |
| 5Ni–26Mg–Al2O3 | 65 | |||||
| 30Ni/Al2O3·0.5SiO2 | 200 | 400 | 3.5 | 9000 | 72 | 14 |
| Ni–Mg0.13/CSC | 300 | 400 | 4 | 10000 |
80 | 25 |
| Ni–Mg0.26/CSC | 88 | |||||
| Ni–Mg0.39/CSC | 85 |
Effect of the bimetallic catalyst
On the Mg63Al37 support, the use of monometallic Co and bimetallic Ni–Co active phases is investigated. The properties of the reduced samples are first evaluated. While the monometallic and bimetallic samples demonstrate comparable textural properties (Fig. 9 and Table 1) and exhibit identical particle size in the range of 5.7–5.8 nm based on the XRD data (Fig. 3), the TPR (Fig. 10) and CO2-TPD (Fig. 4) results present distinct differences.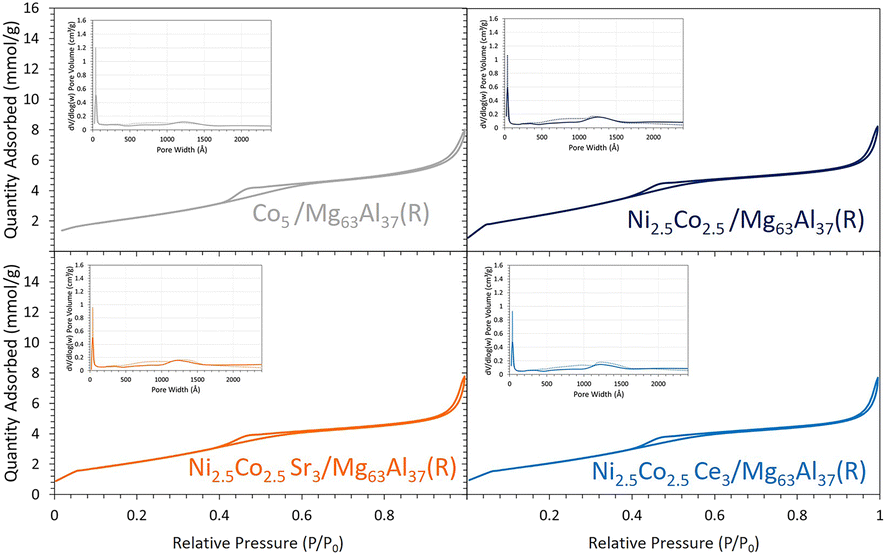 | ||
| Fig. 9 N2 sorption isotherms and pore size distribution (inset figures, solid line: adsorption, dashed line: desorption) for the reduced Co-containing catalysts supported on Mg63Al37. | ||
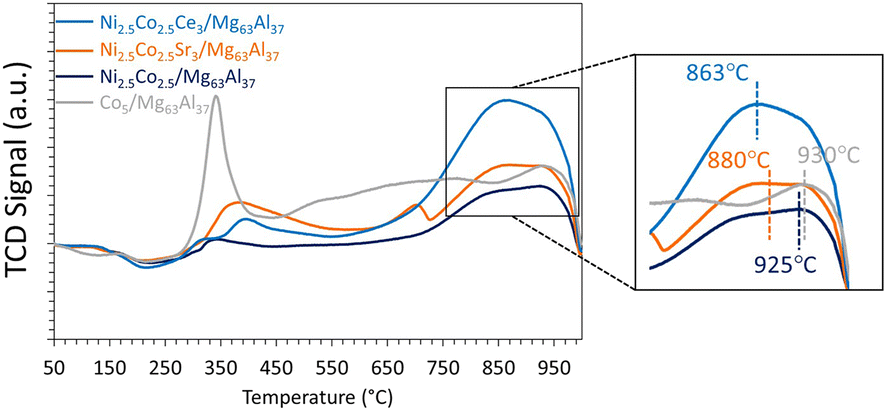 | ||
| Fig. 10 H2-TPR profiles of Co-containing catalysts supported on Mg63Al37. | ||
To elaborate, the reduction of the Co5/Mg63Al37 sample shows two major reduction peaks at around 340 and 930 °C. These peaks correspond to the reduction of Co3O4 to CoO and subsequently to metallic cobalt.44 Similarly, the reduction of the bimetallic Ni2.5Co2.5/Mg63Al37 sample requires elevated temperatures for complete reduction, with the primary reduction peak centered at 925 °C, as also reported elsewhere.36 This indicates that the introduction of Co leads to a stronger metal–support interaction, a phenomenon observed in previous studies.51 It is worth noting that the monometallic Ni-based sample demonstrates the easiest reduction, with a peak observed at 860 °C, as discussed earlier.
The XRD patterns of the Co-containing samples is comparable to those of the Co-free samples (Fig. 3), with an absence of clear peaks characteristic of either Co3O4 or metallic Co.62 This can be attributed to the low Co-content used in these samples or to a high dispersion of small Co particles that cannot be detected by XRD.
The CO2-TPD results (Fig. 4) indicate that the addition of cobalt contributes to the enhancement of basicity. This is evident as the Co5/Mg63Al37 sample exhibits a distinct peak at approximately 490 °C, indicating the presence of strong basic sites within this sample. Indeed, Alabi et al.28 reported the formation of a stronger basic site on Co catalysts supported on Mg–Al compared to Ni catalysts and assigned this to stronger metal–support interaction. On the bi-metallic Ni2.5Co2.5/Mg63Al37 sample, an additional peak is observed at around 450 °C, suggesting a boost of the basicity compared to the mono-metallic Ni-based sample. However, when interpreting these observations in relation to the catalytic test results, it becomes evident that the basicity of the catalyst is not the sole factor influencing the catalytic performance during CO2 methanation. The nature of the active phase, as well as the ease of reduction, are also crucial parameters contributing to the attainment of a catalyst that is both highly active and stable.
On the Mg63Al37 support, the use of Co and Ni–Co active phases is investigated during the methanation reaction at 400 °C and 15000 mL g−1 h−1 (Fig. 11). The initial CO2 conversion recorded is as follows: Ni5/Mg63Al37 (90%) > Ni2.5Co2.5/Mg63Al37 (85%) > Co5/Mg63Al37 (63%). The CH4 yield follows the same trend: Ni5/Mg63Al37 (84%) > Ni2.5Co2.5/Mg63Al37 (72%) > Co5/Mg63Al37 (63%). Consequently, the catalytic activity seems to be affected by the type of active phase employed since the Co-based catalyst shows lower catalytic performance compared to the Ni-based one. This result does not agree with the findings of Liang et al.,41 where the Co-based catalyst supported on Al2O3 showed higher activity and better stability than the Ni-based one under comparable operating conditions at 400 °C. In this case, the discrepancy can be essentially related to the different nature of the support (MgO–Al2O3 mixed oxide) employed in this study. After 150 min on stream, the Co5/Mg63Al37 sample reveals a rise in CO2 conversion from 63% up to 67%, accompanied by a decrease in CH4 yield from 63% to 61%. This increase in CO2 conversion can be due to the occurrence of side reactions such as RWGS (eqn (2)), DRM (eqn (3)), or reactions that generate solid carbon (eqn (5) and (6)). At the reaction temperature of 400 °C, the change in Gibbs free energy ΔG is 13.965 kJ for eqn (2), 68.319 kJ for eqn (3), −25.127 kJ for eqn (5), and −9.864 kJ for eqn (6). Consequently, the first two reactions (eqn (2) and (3)) are not favorable under the reaction conditions (ΔG > 0 at 400 °C), then it is more likely that the increase in CO2 conversion and decrease in CH4 yield is due to the simultaneous occurrence of eqn (5) and (6) that consume CO2 and result in solid carbon on the surface of the catalyst. This effect is alleviated upon the use of bimetallic catalysts since the Ni2.5Co2.5/Mg63Al37 sample maintains a constant level of activity throughout the test. Notably, the bimetallic Ni–Co catalyst prepared with the optimal support demonstrates superior performance compared to analogous catalytic systems tested in the literature on different supports, even with higher active phase loadings. For instance, while Ni2.5Co2.5/Mg63Al37 achieved about 85% CO2 conversion at 400 °C and 15000 mL g−1 h−1, a 30Ni–5Co/Al2O3 sample reaches no more than 65% CO2 conversion under the same test conditions, despite a lower GHSV of 9000 mL g−1 h−1.55 Likewise, the utilization of 15Ni–12.5Co on Al2O3 support attains a maximum of 45% CO2 conversion at 300 °C and 9000 mL g−1 h−1.52 These findings underscore the significance of optimizing the catalytic support to achieve high performance in CO2 methanation.
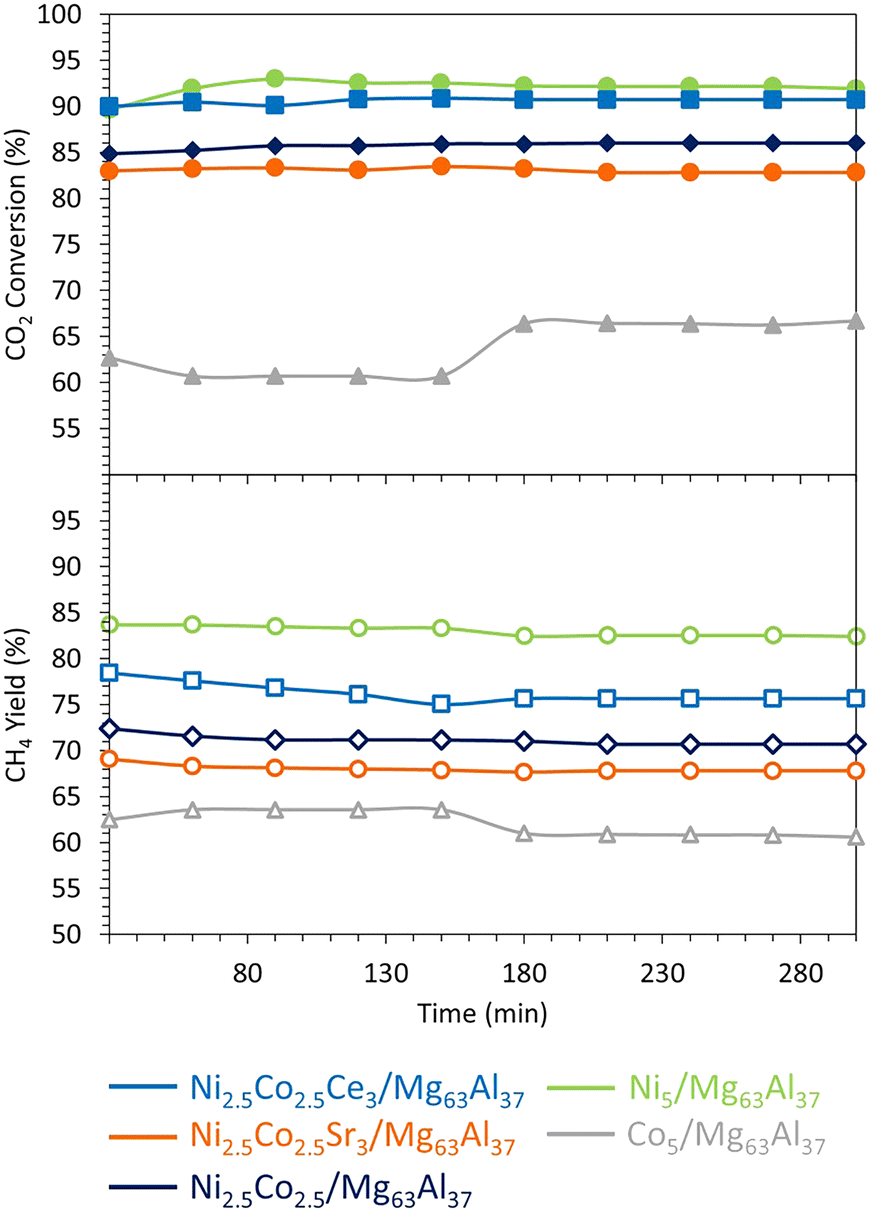 | ||
| Fig. 11 Catalytic test results of the Co-containing catalysts supported on Mg63Al37. The methanation reaction was performed at 400 °C for 300 minutes, at an equivalent gas hourly space velocity of 15000 mL g−1 h−1. | ||
Effect of Ce and Sr promoters
Subsequently, to improve the catalytic performance of the bimetallic Ni2.5Co2.5/Mg63Al37 sample, the latter is promoted with either Sr or Ce. The promoted samples present a type IV isotherm (Fig. 9), characteristic of mesoporous materials. The BET surface area for all reduced samples falls within the range of 176 to 212 m2 g−1 with a pore volume of about 0.3 cm3 g−1 and a pore size in the range of 5.5–6.0 nm (Table 1). These textural properties are similar to those of the bimetallic Ni2.5Co2.5/Mg63Al37 sample.The TPR profiles of the promoted samples (Fig. 10) show a minor reduction peak at around 400 °C, attributed to cobalt oxide reduction, and a second more pronounced peak, above 800 °C, attributable to the reduction of nickel oxide. The maximum of this peak shifts from 925 °C for Ni2.5Co2.5/Mg63Al37 to 880 °C for Ni2.5Co2.5Sr3/Mg63Al37 and to 863 °C for Ni2.5Co2.5Ce3/Mg63Al37. Hence, it becomes evident that the incorporation of Ce reduces the reduction temperature, thereby enhancing the reducibility of nickel oxide.
Post-reduction, the XRD results (Fig. 3) indicate consistent major peaks at 37, 43, and 63° previously assigned to Fmm cubic magnesium nickel oxide Mg0.7Ni0.3O (ref. 04-023-4643). In the Ni2.5Co2.5Ce3/Mg63Al37 sample, additional peaks at 28.5, 33, 47.5, 56.5 and 59° correspond to Fmm cerium oxide structure CeO2 (ref. 04-016-6171). The assessment of the particle size using the most intense peak at about 44° demonstrates a particle size of 5.2 nm for Ni2.5Co2.5Sr3/Mg63Al37 and 6.3 nm for Ni2.5Co2.5Ce3/Mg63Al37.
As previously mentioned, the addition of cobalt improved the Ni-based sample's basicity. The introduction of Ce as a promoter further enhances the basicity, evident from the emergence of a peak at around 425 °C (Fig. 4), indicative of the presence of strong basic sites on Ni2.5Co2.5Ce3/Mg63Al37 sample. Surprisingly, the incorporation of Sr results in moderate basicity, unlike previously reported data,56 where the addition of 4% Sr to Ni/CeZr samples contributes to the formation of very strong basic sites on the catalyst's surface. This can be due to the nature of support and its interaction with the active metals since the addition of MgO alters the basicity of alumina support, and the effect of Sr addition on the basicity of the sample might be hindered.
The catalytic stability test results at 400 °C (Fig. 11) show an enhancement of the catalytic activity of the Ni2.5Co2.5/Mg63Al37 sample upon the addition of Ce. Indeed, the Ni2.5Co2.5Ce3/Mg63Al37 catalyst presents a CO2 conversion of 90% and a CH4 yield of 78% that remains constant throughout the test duration. These values are comparable to those of Ni5/Mg63Al37 but higher than those of Ni2.5Co2.5/Mg63Al37 catalyst (only 85% CO2 conversion and 72% CH4 yield), highlighting the beneficial impact of Ce addition to the Ni–Co bimetallic catalyst.
The promotional effect of Ce is further examined on the TGA curves of the spent catalysts (Fig. 12), since Ni2.5Co2.5Ce3/Mg63Al37 catalyst shows only 6% weight loss compared to about 11% for Ni5/Mg63Al37 and 10% for Ni2.5Co2.5/Mg63Al37. On the TGA profiles, the major weight loss before 200 °C corresponds to the removal of water and other chemisorbed species.35 Additional weight loss between 200 and 500 °C is attributed to the combustion of amorphous carbon and that above this range (>500 °C) results from the oxidation of graphitic carbon.35 On all the samples, the very limited weight loss up to 500 °C (about 2%) suggests the removal of reactive amorphous carbon.35 This is also confirmed by the absence of any filamentous carbon nanotubes on the TEM images of the spent Ni2.5Co2.5Ce3/Mg63Al37 (Fig. 13). Furthermore, the highly active and stable Ni2.5Co2.5Ce3/Mg63Al37 and Ni5/Mg63Al37 catalysts maintain good dispersion of the active phase, without apparent metal agglomeration or carbon deposition on their surfaces (Fig. 7 and 13). This consistency in active metal dispersion is evident in TEM images before and after the catalytic tests, as indicated by red arrows on Fig. 13, implying the absence of particle sintering and the robust thermal stability of these samples under the employed conditions. The Ni2.5Co2.5Sr3/Mg63Al37 sample, however, exhibits lower catalytic performance compared to the unpromoted sample (83% CO2 conversion and 69% CH4 yield). This may arise from the fact that the MgO–Al2O3 support itself already provides basic sites for CO2 adsorption, and the Sr loading might not be sufficiently high to significantly enhance basicity beyond what the support itself offers.
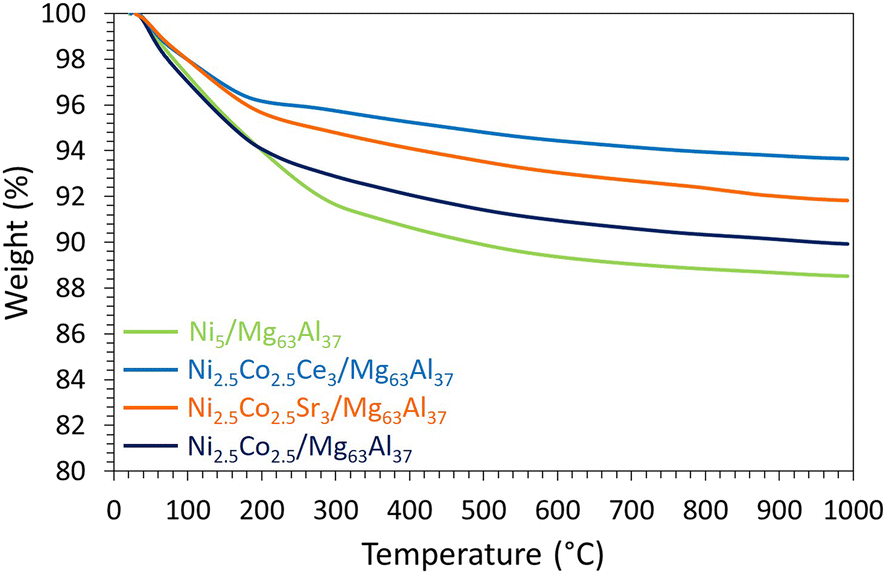 | ||
| Fig. 12 TGA curves of the spent catalysts supported on Mg63Al37. | ||
 | ||
| Fig. 13 TEM images of the (a) reduced and (a′) spent Ni2.5Co2.5Ce3/Mg63Al37 catalyst. | ||
The highly performing Ni2.5Co2.5Ce3/Mg63Al37 sample is further examined under the same operating conditions but for longer duration (Fig. 14). It is shown that, after more than 25 hours on stream, this sample maintains high and constant CO2 conversion of 90% with a stable CH4 yield at about 75%. After this period, the evaluation of the spent catalyst by TGA shows a limited weight loss that does not exceed 11%. As detailed earlier, the major weight loss of about 7% is assigned to the removal of chemisorbed species and the remaining 4% are primarily assigned to the elimination of amorphous carbon. The XRD profiles present the same characteristic peaks observed on the reduced sample and reveal a preservation of the active sites dispersion with an absence of significant particles sintering. These results confirm, once again, the beneficial effect of Ce addition to the bimetallic Ni–Co catalyst that leads to superior catalytic activity through the enhancement of NiO reducibility and the improvement of basicity. The promotional effect of ceria is comparable to that observed on Ni/MgO–Al2O3–CeO2 catalysts for CO methanation in terms of improved metal oxide reducibility, Ni dispersion and resistance to carbon deposition.67 Nevertheless, the catalytic activity observed in this work is higher than that observed on similar samples containing higher metal loadings. In more detail, the CO2 conversion reached a maximum of 70 and 75% using 15% Ni/Al2O3 promoted with up to 15% Co (ref. 52) (9000 mL g−1 h−1) and 15% Ni/Al2O3 promoted with up to 20% Ce (30000 h−1),63 respectively at 400 °C and H2:CO2 ratio of 4. This comparison highlights the importance of properly selecting the support and promoter of Ni-based catalysts to achieve beneficial improvement of the catalytic performance in CO2 methanation.
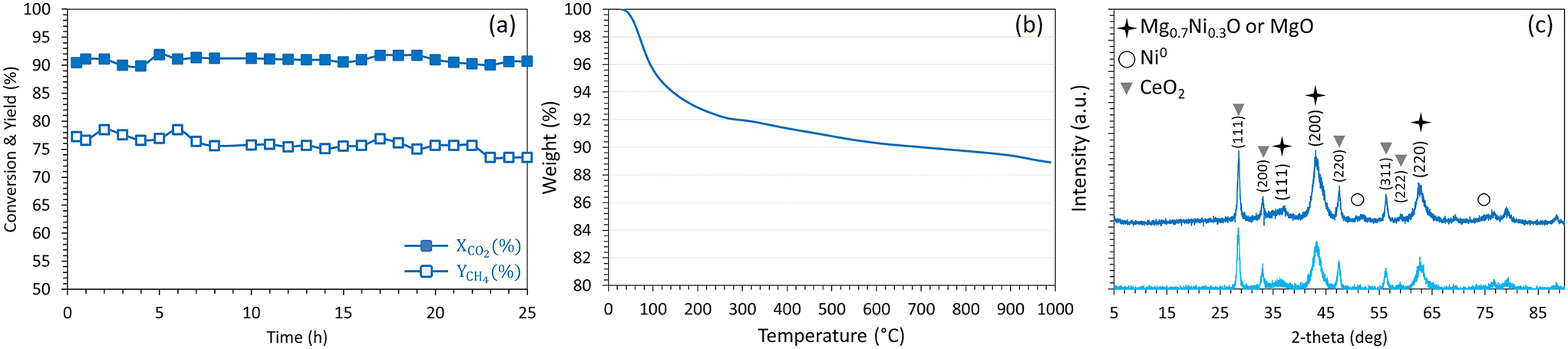 | ||
| Fig. 14 (a) CO2 conversion and CH4 yield, (b) TGA results and (c) XRD profiles (light blue: reduced, dark blue: spent) of Ni2.5Co2.5Ce3/Mg63Al37 catalyst. | ||
A comparison of the data obtained in this study (Table 3) can provide valuable insights into the pivotal factors influencing the catalytic performance in CO2 methanation. Notably, it becomes evident that the most high-performing catalysts, namely Ni5/Mg63Al37 and Ni2.5Co2.5Ce3/Mg63Al37, exhibit a distinctive characteristic of facile reduction, even in the presence of medium basic sites on their surfaces. This affinity for easy reduction emerges as a paramount factor for attaining enhanced catalytic activity. Indeed, the presence of highly dispersed particles with strong basic sites, like in the case of Ni2.5Co2.5Sr3/Mg63Al37 sample, is not sufficient to ensure good catalytic activity because the reduction of this sample required higher temperatures than the highly performing catalysts. Consequently, these findings suggest that incorporating a secondary active phase and/or introducing a promoter holds tangible benefits only when it synergistically facilitates easier reduction.
| Sample | TPR peak max. (°C) | TPD basicity | TPD CO2 desorbed (mmol g−1) | Particle size XRD (nm) | CO2 conv. (%) |
|---|---|---|---|---|---|
| Ni5/Mg63Al37 | 860 | Medium | 0.29 | 5.8 | 92 |
| Ni2.5Co2.5/Mg63Al37 | 925 | Medium strong (450 °C) | 0.10 | 5.7 | 85 |
| Ni2.5Co2.5Ce3/Mg63Al37 | 863 | Medium strong (425 °C) | 0.08 | 6.3 | 90 |
| Ni2.5Co2.5Sr3/Mg63Al37 | 880 | Medium strong (350 °C) | 0.05 | 5.2 | 83 |
Conclusions
Given the urgent need for global warming mitigation, CO2 methanation is a pivotal pathway for the reduction of CO2 emissions. This study explores the incorporation of MgO to Al2O3 as support for Ni and/or Co active phases for the hydrogenation of CO2 into methane. As the amount of MgO in Al2O3 support increases, the NiO reduction shifts towards higher temperatures, resulting in stronger metal–support interaction due to the formation of NiO–MgO solid solution. The highest catalytic activity of 92% CO2 conversion and 82% CH4 yield at 400 °C is achieved using a Ni5/Mg63Al37 sample that shows the highest amount of CO2 desorbed by CO2-TPD. The catalytic activity of this sample is higher than those of Co5/Mg63Al37 and Ni2.5Co2.5/Mg63Al37, owing to easier reduction on Ni5/Mg63Al37, suggesting that Ni is better than Co under the conditions employed in this work. On the bimetallic sample, the addition of Ce as a promoter is favorable since it boosts the catalytic activity by facilitating the reduction and improving the basicity. This improvement reflects in high catalytic stability for more than 25 hours on steam, with limited carbon deposition and high resistance to sintering. The effect of Sr addition remains limited, probably because the use of an optimized MgO–Al2O3 support already enhances the basicity of the catalyst. It is interesting next to evaluate the feasibility of the promising catalysts presented in this study for large-scale applications. This can be achieved by testing them under more industrially attractive conditions, at lower temperatures and higher pressures.Disclaimer
The views and opinions expressed in this paper do not necessarily reflect those of the European Commission or the Special EU Programmes Body (SEUPB).Author contributions
Investigation, data acquisition, conceptualization, writing – original draft: Marie-Nour Kaydouh, Nissrine El Hassan, Ahmed I. Osman, Hamid Ahmed, Ahmed S. Al-Fatesh. Methodology, writing, review & editing: Naif Alarifi, Anis H. Fakeeha, Abdulrahman bin Jumah, Ahmed S. Al-Fatesh. Writing – original draft, writing – review & editing; all authors review and edit the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was funded by Researchers Supporting Project number (RSP2024R368), King Saud University, Riyadh, Saudi Arabia. The authors would like to extend their sincere appreciation to the Researchers Supporting Project number (RSP2024R368), King Saud University, Riyadh, Saudi Arabia. Dr. Ahmed I. Osman wishes to acknowledge the support of The Bryden Centre project (Project ID VA5048), which was awarded by The European Union's INTERREG VA Programme, managed by the Special EU Programmes Body (SEUPB), with match funding provided by the Department for the Economy in Northern Ireland and the Department of Business, Enterprise and Innovation in the Republic of Ireland. This work was also supported by the Lebanese American University President's Intramural Research Fund PIRF I0046.Notes and references
- R. Ramezani, L. Di Felice and F. Gallucci, JPhys Energy, 2023, 5, 024010 CrossRef.
- S. Hafeez, E. Harkou, S. M. Al-Salem, M. A. Goula, N. Dimitratos, N. D. Charisiou, A. Villa, A. Bansode, G. Leeke, G. Manos and A. Constantinou, React. Chem. Eng., 2022, 7, 795 RSC.
- J. Ren, H. Lou, N. Xu, F. Zeng, G. Pei and Z. Wang, J. Energy Chem., 2023, 80, 182 CrossRef CAS.
- T. Burger, P. Donaubauer and O. Hinrichsen, Appl. Catal., B, 2021, 282, 119408 CrossRef CAS.
- M. Song, L. Shi, X. Xu, X. Du, Y. Chen, W. Zhuang, X. Tao, L. Sun and Y. Xu, J. CO2 Util., 2022, 64, 102150 CrossRef CAS.
- L. Karam, M. C. Bacariza, J. M. Lopes, C. Henriques, J. Reboul, N. El Hassan and P. Massiani, J. CO2 Util., 2021, 51, 101651 CrossRef CAS.
- S. Danaci, L. Protasova, J. Lefevere, L. Bedel, R. Guilet and P. Marty, Catal. Today, 2016, 273, 234 CrossRef CAS.
- J. Guilera, J. del Valle, A. Alarcón, J. A. Díaz and T. Andreu, J. CO2 Util., 2019, 30, 11 CrossRef CAS.
- H. Chen, Z. Guo, S. Xiang, H. Jianga and Y. Teng, React. Chem. Eng., 2023, 8, 2632 RSC.
- S. M. Jogdand, P. R. Bedadur, A. Torris, U. K. Kharul, V. Satyam Naidu and R. Nandini Devi, React. Chem. Eng., 2021, 6, 1655 RSC.
- J. Ashok, S. Pati, P. Hongmanorom, Z. Tianxi, C. Junmei and S. Kawi, Catal. Today, 2020, 356, 471 CrossRef CAS.
- M. Paviotti, B. Faroldi and L. Cornaglia, J. Environ. Chem. Eng., 2021, 9, 105173 CrossRef CAS.
- L. Li, W. Zeng, M. Song, X. Wu, G. Li and C. Hu, Catalysts, 2022, 12, 244 CrossRef CAS.
- S. V. Moghaddam, M. Rezaei, F. Meshkani and R. Daroughegi, Int. J. Hydrogen Energy, 2018, 43, 19038 CrossRef CAS.
- L. Karam, C. M. Bacariza, J. M. Lopes, C. Henriques, P. Massiani and N. El Hassan, Int. J. Hydrogen Energy, 2020, 45, 28626 CrossRef CAS.
- M. Yusuf Shahul Hamid, A. Abdul Jalil, A. Farhana Abdul Rahman and T. Amran Tuan Abdullah, React. Chem. Eng., 2019, 4, 1126 RSC.
- T. Margossian, K. Larmier, S. M. Kim, F. Krumeich, A. Fedorov, P. Chen, C. R. Müller and C. Copéret, J. Am. Chem. Soc., 2017, 139, 6919 CrossRef CAS PubMed.
- L. Karam, M. Armandi, S. Casale, V. El Khoury, B. Bonelli, P. Massiani and N. El Hassan, Energy Convers. Manage., 2020, 225, 113470 CrossRef CAS.
- K. Jabbour, P. Massiani, A. Davidson, S. Casale and N. El Hassan, Appl. Catal., B, 2017, 201, 527 CrossRef CAS.
- Y. Guo, J. Lu, Q. Liu, X. Bai, L. Gao, W. Tu and Z.-j. Wang, Catal. Commun., 2018, 116, 81 CrossRef CAS.
- J.-N. Park and E. W. McFarland, J. Catal., 2009, 266, 92 CrossRef CAS.
- M. Guo and G. Lu, Catal. Commun., 2014, 54, 55 CrossRef CAS.
- M. C. Bacariza, I. Graça, S. Bebiano, J. M. Lopes and C. Henriques, Energy Fuels, 2017, 31, 9776 CrossRef CAS.
- P. Hongmanorom, J. Ashok, G. Zhang, Z. Bian, M. H. Wai, Y. Zeng, S. Xi, A. Borgna and S. Kawi, Appl. Catal., B, 2021, 282, 119564 CrossRef CAS.
- X. Li, Y. Wang, G. Zhang, W. Sun, Y. Bai, L. Zheng, X. Han and L. Wu, ChemistrySelect, 2019, 4, 838 CrossRef CAS.
- N. M. Julkapli and S. Bagheri, Rev. Inorg. Chem., 2016, 36, 1 CAS.
- W. O. Alabi, B. M. Adesanmi, H. Wang and C. Patzig, Int. J. Hydrogen Energy, 2024, 51, 1087 CrossRef CAS.
- W. O. Alabi, K. O. Sulaiman, H. Wang, Y. Hu and C. Patzig, J. CO2 Util., 2020, 37, 180 CrossRef CAS.
- M. Kumar, F. Aberuagba, J. Gupta, K. Rawat, L. Sharma and G. M. Dhar, J. Mol. Catal. A: Chem., 2004, 213, 217 CrossRef CAS.
- Y. Lian, H. Wang, Q. Zheng, W. Fang and Y. Yang, J. Nat. Gas Chem., 2009, 18, 161 CrossRef CAS.
- A. Vikár, F. Lónyi, A. Makoye, T. Nagy, G. Novodárszki, R. Barthos, B. Szabó, J. Valyon, M. R. Mihályi, D. Deka and H. E. Solt, Molecules, 2023, 28, 3788 CrossRef PubMed.
- G. I. Siakavelas, N. D. Charisiou, A. AlKhoori, S. AlKhoori, V. Sebastian, S. J. Hinder, M. A. Baker, I. V. Yentekakis, K. Polychronopoulou and M. A. Goula, J. CO2 Util., 2021, 51, 101618 CrossRef CAS.
- S. Cisneros, L. Santa-Taborda, L. M. Quintana, A. I. M. Rabee, H. Abed, N. Rockstroh, S. Bartling, M. Romero-Sáez, H. Atia, A. B. Dongil, A. Brückner and J. Rabeah, Chem. Eng. J., 2023, 474, 145646 CrossRef CAS.
- A. A. M. Abahussain, A. S. Al-Fatesh, N. Patel, S. B. Alreshaidan, N. A. Bamatraf, A. A. Ibrahim, A. Y. Elnour, J. K. Abu-Dahrieh, A. E. Abasaeed, A. H. Fakeeha and R. Kumar, Nanomater., 2023, 13, 2984 CrossRef CAS PubMed.
- Y. A. Al Baqmaa, A. S. Al Fatesh, A. A. Ibrahim, A. A. Bagabas, F. S. Almubadde, A. I. Alromaeh, J. K. Abu Dahrieh, A. E. Abasaeed and A. H. Fakeeha, Res. Chem. Intermed., 2023, 49, 5015 CrossRef CAS.
- A. A. Ibrahim, A. H. Fakeeha, A. E. Abasaeed, I. Wazeer, A. Bentalib, N. S. Kumar, J. K. Abu-Dahrieh and A. S. Al-Fatesh, Catalysts, 2023, 13, 1420 CrossRef CAS.
- W. Li, Y. Liu, M. Mu, F. Ding, Z. Liu, X. Guo and C. Song, Appl. Catal., B, 2019, 254, 531 CrossRef CAS.
- W. Li, X. Nie, X. Jiang, A. Zhang, F. Ding, M. Liu, Z. Liu, X. Guo and C. Song, Appl. Catal., B, 2018, 220, 397 CrossRef CAS.
- T. A. Le, M. S. Kim, S. H. Lee and E. D. Park, Top. Catal., 2017, 60, 714 CrossRef CAS.
- Z. Zhang, X. Zhang, L. Zhang, J. Gao, Y. Shao, D. Dong, S. Zhang, Q. Liu, L. Xu and X. Hu, J. Energy Inst., 2020, 93, 1581 CrossRef.
- C. Liang, H. Tian, G. Gao, S. Zhang, Q. Liu, D. Dong and X. Hu, Int. J. Hydrogen Energy, 2020, 45, 531 CrossRef CAS.
- R. Razzaq, H. Zhu, L. Jiang, U. Muhammad, C. Li and S. Zhang, Ind. Eng. Chem. Res., 2013, 52, 2247 CrossRef CAS.
- J. J. C. Struijs, V. Muravev, M. A. Verheijen, E. J. M. Hensen and N. Kosinov, Angew. Chem., 2023, 62, e202214864 CrossRef CAS PubMed.
- M. Gäßler, J. Stahl, M. Schowalter, S. Pokhrel, A. Rosenauer, L. Mädler and R. Güttel, ChemCatChem, 2022, 14, e202200286 CrossRef.
- J. Tu, H. Wu, Q. Qian, S. Han, M. Chu, S. Jia, R. Feng, J. Zhai, M. Hea and B. Han, Chem. Sci., 2021, 12, 3937 RSC.
- W. Li, G. Zhang, X. Jiang, Y. Liu, J. Zhu, F. Ding, Z. Liu, X. Guo and C. Song, ACS Catal., 2019, 9, 2739 CrossRef CAS.
- B. A. Oni, O. S. Tomomewo, S. E. Sanni and V. O. Ojo, Mater. Today Commun., 2023, 36, 106802 CrossRef CAS.
- K. Frikha, L. Limousy, J. Bouaziz, K. Chaari and S. Bennici, Materials, 2020, 13, 4607 CrossRef CAS PubMed.
- M. J. Moradi, G. Moradi, A. Heydarinasab and A. Rashidi, Mater. Today Commun., 2023, 34, 105226 CrossRef CAS.
- A. C. Koh, L. Chen, W. K. Leong, B. F. Johnson, T. Khimyak and J. Lin, Int. J. Hydrogen Energy, 2007, 32, 725 CrossRef CAS.
- Q. Liu, B. Bian, J. Fan and J. Yang, Int. J. Hydrogen Energy, 2018, 43, 4893 CrossRef CAS.
- P. Shafiee, S. M. Alavi and M. Rezaei, Int. J. Hydrogen Energy, 2021, 46, 3933 CrossRef CAS.
- N. Fatah, A. Jalil, N. Salleh, M. Hamid, Z. Hassan and M. Nawawi, Int. J. Hydrogen Energy, 2020, 45, 18562 CrossRef CAS.
- G. Hasrack, M. C. Bacariza, C. Henriques and P. Da Costa, Catalysts, 2022, 12, 36 CrossRef CAS.
- S. V. Moghaddam, M. Rezaei, F. Meshkani and R. Daroughegi, Int. J. Hydrogen Energy, 2018, 43, 16522 CrossRef.
- M. Mikhail, P. Da Costa, J. Amouroux, S. Cavadias, M. Tatoulian, M. E. Galvez and S. Ognier, Appl. Catal., B, 2021, 294, 120233 CrossRef CAS.
- E. H. Cho, Y.-K. Park, K. Y. Park, D. Song, K. Y. Koo, U. Jung, W. R. Yoon and C. H. Ko, Chem. Eng. J., 2022, 428, 131393 CrossRef CAS.
- F. Namvar, M. Hajizadeh-Oghaz, M. A. Mahdi, S. H. Ganduh, F. Meshkani and M. Salavati-Niasari, Int. J. Hydrogen Energy, 2023, 48, 3862 CrossRef CAS.
- C. Liang, X. Hu, T. Wei, P. Jia, Z. Zhang, D. Dong, S. Zhang, Q. Liu and G. Hu, Int. J. Hydrogen Energy, 2019, 44, 8197 CrossRef CAS.
- M. Guo and G. Lu, RSC Adv., 2014, 4, 58171 RSC.
- A. Alarcón, J. Guilera, J. A. Díaz and T. Andreu, Fuel Process. Technol., 2019, 193, 114 CrossRef.
- J. Y. Do, R. K. Chava, N. Son, J. Kim, N.-K. Park, D. Lee, M. W. Seo, H.-J. Ryu, J. H. Chi and M. Kang, Catalysts, 2018, 8, 413 CrossRef.
- M.-J. Kim, J.-R. Youn, H. J. Kim, M. W. Seo, D. Lee, K. S. Go, K. B. Lee and S. G. Jeon, Int. J. Hydrogen Energy, 2020, 45, 24595 CrossRef CAS.
- H. Liu, X. Zou, X. Wang, X. Lu and W. Ding, J. Nat. Gas Chem., 2012, 21, 703 CrossRef CAS.
- L. Zhou, Q. Wang, L. Ma, J. Chen, J. Ma and Z. Zi, Catal. Lett., 2015, 145, 612 CrossRef CAS.
- M. Varbar, S. M. Alavi, M. Rezaei and E. Akbari, Res. Chem. Intermed., 2022, 48, 1129 CrossRef CAS.
- L. Zhang, D.-X. Gu, L. Yi and Y. Zhang, Catal. Lett., 2017, 147, 1172 CrossRef CAS.
- P. Summa, K. Ś. Da Costa, Y. Wang, B. Samojeden, M. Rønning, C. Hu, M. Motak and P. Da Costa, Appl. Mater. Today, 2021, 25, 101211 CrossRef.
- P. Summa, K. Ś. Da Costa, J. Gopakumar, B. Samojeden, M. Motak, M. Rønning, W. Van Beek and P. Da Costa, Appl. Mater. Today, 2023, 32, 101795 CrossRef.
- R. A. El-Salamony, A. S. Al-Fatesh, K. Acharya, A. A. M. Abahussain, A. Bagabas, N. S. Kumar, A. A. Ibrahim, W. U. Khan and R. Kumar, Catalysts, 2023, 13, 113 CrossRef CAS.
- S. Ramkumar and G. Rajarajan, Appl. Phys. A: Mater. Sci. Process., 2017, 123, 401 CrossRef.
- M. Kaur and K. Pal, J. Mater. Sci.: Mater. Electron., 2020, 31, 10903 CrossRef CAS.
- Y. Budipramana, E. Taslim and F. Kurniawan, ARPN J. Eng. Appl. Sci., 2014, 9, 2074 Search PubMed.
- M. Parvas, M. Haghighi and S. Allahyari, Arabian J. Chem., 2019, 12, 1298 CrossRef CAS.
- Q. Pan, J. Peng, T. Sun, S. Wang and S. Wang, Catal. Commun., 2014, 45, 74 CrossRef CAS.
- T. Burger, H. M. S. Augenstein, F. Hnyk, M. Döblinger, K. Köhler and O. Hinrichsen, ChemCatChem, 2020, 12, 649 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |