
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic ramoplanin analogues are accessible by effective incorporation of arylglycines in solid-phase peptide synthesis†
Edward
Marschall
 abc,
Rachel W.
Cass
abc,
Komal M.
Prasad
abc,
James D.
Swarbrick
d,
Alasdair I.
McKay
e,
Jennifer A. E.
Payne
abc,
Max J.
Cryle
*abc and
Julien
Tailhades
*abc
abc,
Rachel W.
Cass
abc,
Komal M.
Prasad
abc,
James D.
Swarbrick
d,
Alasdair I.
McKay
e,
Jennifer A. E.
Payne
abc,
Max J.
Cryle
*abc and
Julien
Tailhades
*abc
aDepartment of Biochemistry and Molecular Biology, The Monash Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia. E-mail: max.cryle@monash.edu; julien.tailhades@monash.edu
bEMBL Australia, Monash University, Clayton, VIC 3800, Australia
cARC Centre of Excellence for Innovations in Peptide and Protein Science, Clayton, VIC 3800, Australia
dDepartment of Microbiology, Monash University, Clayton, VIC 3800, Australia
eDepartment of Chemistry, Monash University, Clayton, VIC 3800, Australia
First published on 29th September 2023
Abstract
The threat of antimicrobial resistance to antibiotics requires a continual effort to develop alternative treatments. Arylglycines (or phenylglycines) are one of the signature amino acids found in many natural peptide antibiotics, but their propensity for epimerization in solid-phase peptide synthesis (SPPS) has prevented their use in long peptide sequences. We have now identified an optimized protocol that allows the synthesis of challenging non-ribosomal peptides including precursors of the glycopeptide antibiotics and an analogue of feglymycin (1 analogue, 20%). We have exploited this protocol to synthesize analogues of the peptide antibiotic ramoplanin using native chemical ligation/desulfurization (1 analogue, 6.5%) and head-to-tail macrocyclization in excellent yield (6 analogues, 3–9%), with these compounds extensively characterized by NMR (U-shaped structure) and antimicrobial activity assays (two clinical isolates). This method significantly reduces synthesis time (6–9 days) when compared with total syntheses (2–3 months) and enables drug discovery programs to include arylglycines in structure–activity relationship studies and drug development.
Introduction
Peptide therapeutics have progressed rapidly in recent years, fuelled by advances in both biological and synthetic sciences.1 Such compounds are known for high activity and selective modes of action as well as the ability to target larger protein–protein interactions, which are typically hard to access with small molecules. However, peptides can suffer from poor pharmacokinetics (PK), with in vivo stability often a major challenge for peptides.2 Increased interest in bioactive peptides has seen the replacement of proteogenic amino acids (AAs) with non-proteogenic analogues become a common strategy to improve the PK properties or to undertake structure–activity relationship (SAR) studies of candidate peptides.3 In this regard, non-ribosomal peptides (NRPs) are a major source of inspiration in terms of non-proteogenic amino acids, structural diversity, and post-synthesis modifications.4 The arylglycines5,6 are one of the signature amino acids found in many medically important NRPs (Fig. 1) including clinical antibiotics such as β-lactams7 (ampicillin, cephalexin) or glycopeptide antibiotics (GPAs – vancomycin, teicoplanin).8,9 Solution-based synthesis approaches are acceptable for small molecules and short peptides containing arylglycines (Fig. 1) such as arylomycin (6 AAs)10,11 or vancomycin (7 AAs)9 but become inefficient for longer sequences such as feglymycin (13 AAs),12,13 longer type V GPAs14,15 or the lipoglycodepsipeptide family16,17 that includes ramoplanin,18 enduracidin19 and chersinamycin (17 AAs).20 In recent years arylglycines have also become a regular inclusion in small molecule SAR campaigns, showing that these residues hold great promise in medicinal chemistry.21–24 Such efforts have been supported by the development of chemical methodologies to access enantiomerically pure arylglycines, which can be further modified for use in solution or on solid-phase.6,25,26 Despite all of this, they are rarely included in solid-phase peptide synthesis (SPPS)-driven SAR studies despite their potential as proteolytically stable and rigid alternatives to phenylalanine, tyrosine or hydrophobic residues. SAR studies, which typically require milligram scales, can benefit greatly from SPPS, as this can deliver rapid access to small quantities of peptide in a largely automated way as well as the possibility to work at a larger scale.27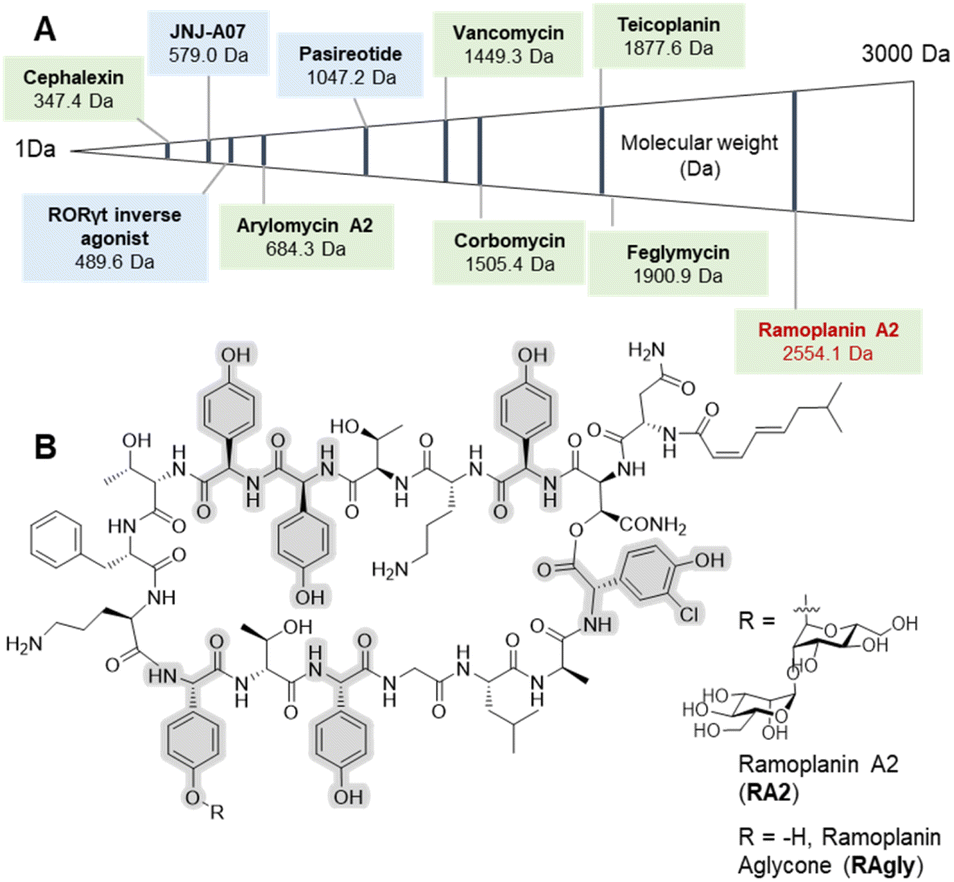 | ||
| Fig. 1 Selected arylglycine containing bioactive compounds up to 3000 Da including both small molecules and peptides. (A) Molecules boxed in green (cephalexin,7 vancomycin,8,9 arylomycin A2,10,11 teicoplanin,8,9 corbomycin,15 feglymycin12,13 and ramoplanin A2 and aglycone14,15) are either natural products or derived from natural products (semi-synthetics); molecules boxed in blue (JNJ-A07,20 the Related Orphan Receptor-γt (RORγt) inverse agonist21 and pasireotide22) are synthetic compounds. (B) Chemical structure of ramoplanin A2 (RA2) and aglycone (RAgly – without the two D-mannoses). | ||
The common Fmoc/tBu SPPS methodology uses elongation under repeated basic treatments, which can lead to side reactions including aspartimide formation and amino acid epimerization;28 this is particularly troublesome for arylglycines.29,30 In solution, the majority of the work has adopted the carbodiimide activation (EDC) with various additives (Oxyma, HOBt or HOAt)6,31 or DEPBT with an inorganic base (typically NaHCO3).12,32,33 These strategies limit the epimerization of the sensitive arylglycine α-proton by using a sterically hindered base or a heterogeneous mixture. The application of carbodiimide/additive coupling is also well-established on solid-phase31,34 leaving Fmoc removal as the main issue for SPPS. One way to limit the epimerization of arylglycine on solid-phase is to use a short exposure to a sterically hindered base (DBU).35 We optimized a methodology for arylglycine installation in Fmoc/tBu SPPS utilising these approaches, which we have validated through the effective synthesis of active and structured ramoplanin analogues.
Ramoplanin is one of the largest known NRPs and is active against all Gram-positive pathogens including Vancomycin-Resistant Enterococci (VRE) and Clostridioides difficile, where it has successfully completed non-inferiority (phase II) trials.36,37 Ramoplanin A2 is a lipodepsipeptide antibiotic38 comprising a 16-amino acid macrocycle including an ester functionality, two D-mannose moieties at position 11, and a fatty acid at the N-terminus (Fig. 1). NMR analysis has contributed to the determination of the structure39 and the mechanism of action of ramoplanin.40,41 The U-shaped structure forms a tight complex with both Lipid I and Lipid II in Gram-positive bacteria. In doing so, it prevents transglycosylases from crosslinking Lipid II on the extra-cellular surface42 as well as inducing membrane depolarization43 as is seen for lipo-GPAs (telavancin and oritavancin).8 Total synthesis of the ramoplanin aglycone was first achieved by the Boger group in 2003.33 In 2007, the β-hydroxyasparagine residue (L-β-OH-Asn2) was successfully replaced with diaminopropionic acid (L-Dap2), leading to a further series of ramoplanin aglycone analogues.32 Overall, studies investigating the modification of the ramoplanin aglycone (alanine scanning – ornithine capping) enabled the identification of some residues crucial for antimicrobial activity (D-Orn10).31 Although ramoplanin SAR is largely under investigated; except for the replacement of the lipid moiety responsible for toxicity.44,45 Recent reviews indicate ramoplanin as being in development,36,37 but the majority of this research was conducted almost 15 years ago. This development delay is at least in part due to the difficulty and time needed to modify these complex peptides either through chemical synthesis32,33 or synthetic biology.46,47 A direct route to the synthesis of ramoplanin aglycone analogues would therefore revitalize interest in this antimicrobial class, identify many target molecules for non-ribosomal peptide synthetase redesign and revigorate in vivo studies to define the role of ramoplanin in the antimicrobial resistance context.48
Results and discussion
Optimization of solid-phase peptide synthesis protocol
We began our investigation using our reported SPPS methodology employed for peptides such as GPA precursors used in in vitro cytochrome P450 cyclization(s).49,50 These include the heptapeptide cores of vancomycin51 and teicoplanin,52 which contain 3 and 5 arylglycines (4-hydroxyphenylglycine (Hpg) and 3,5-dihydroxyphenylalanine (Dpg) residues) respectively. Our efforts to synthesize such peptides identified a SPPS protocol using a sterically hindered base (DBU) with multiple shorter deprotection cycles for Fmoc removal, combined with COMU/lutidine for coupling side chain unprotected Fmoc-(L/D)Hpg-OH and Fmoc-(L)Dpg-OH residues.35,53 While this is sufficient for GPA precursors, the disparity in results between the linear heptapeptides of vancomycin (1, 3/7 arylglycines) and teicoplanin (2a, 5/7 arylglycines) indicated that this protocol was not as general as desired (see ESI, Fig. S1 and Table S1†) and would likely be inefficient for the synthesis of analogues of ramoplanin or longer GPA sequences such as corbomycin.15 Importantly, the high conversion of linear GPA peptides into GPA aglycones using an in vitro cytochrome P450 cyclization was a positive read-out of the enantiopurity of these peptides.49–52 Consequently, we adopted the use of DIC/Oxyma instead of COMU/lutidine for peptide coupling since both coupling reagents form an activated Oxyma-ester.34,53Impressively, the use of DIC/Oxyma was sufficient to increase the linear teicoplanin peptide yield (from 32% for 2a to 43% for 2b) and improve peptide purity almost 4-fold (from 12% for 2a to 47% for 2b at 214 nm) (see ESI, Fig. S1 and Table S1†). Encouraged by these results, we sought to investigate temperature-assisted coupling via conventional and microwave heating. Temperature-assisted coupling is highly effective in reducing synthesis time and accessing longer peptide sequences.54 We based our approach on reported conditions for the coupling of epimerization-prone cysteine and histidine residues.55 The improved coupling reaction performed at 50 °C for 10 min led to substantial improvements in peptide recovery without compromising peptide purity with both microwave (2c) and conventional heating (2d) (Fig. 2A – 2d, see ESI, Fig. S1 and Table S1†). Applying this methodology to the longer corbomycin GPA sequence (9 AAs, 6/9 arylglycines) now afforded high yield and purity using an automatic microwave-assisted peptide synthesizer (Fig. 2A – 3, see ESI, Fig. S1 and Table S1†).
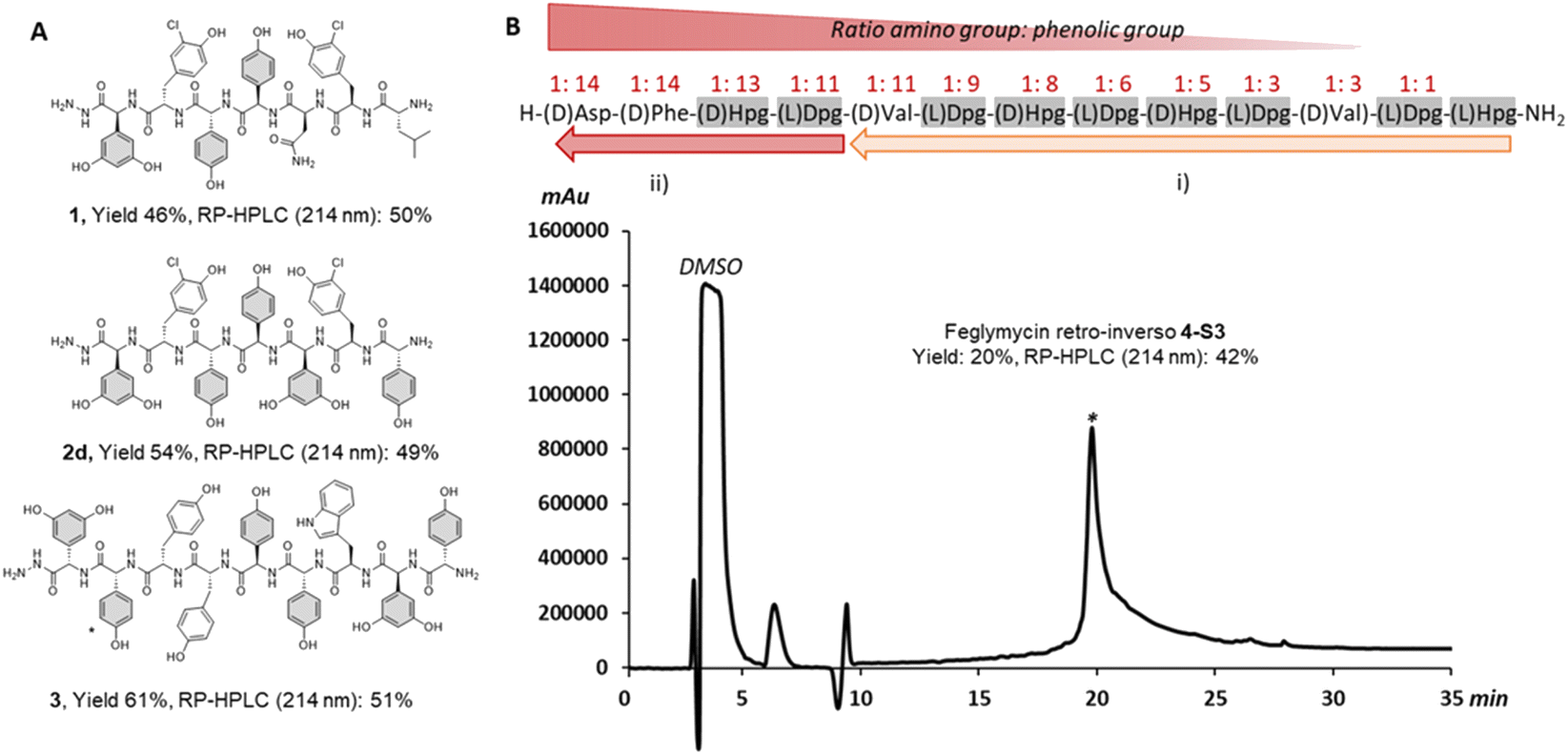 | ||
| Fig. 2 Summary of arylglycine-rich peptides benefiting from DIC/Oxyma temperature assisted coupling. (A) Linear vancomycin peptide (1 obtained with COMU/Lutidine at RT),45,47 teicoplanin peptide (2d obtained by conventional heating) and corbomycin peptide precursors (3 obtained by microwave heating). (B) Above, the sequence of a retro-inverso feglymycin variant (4) and the SPPS strategy: (i) single coupling; (ii) double coupling. (B) Below, RP-HPLC chromatogram at 214 nm of the crude peptide (4-S3) obtained with SpheriTide™ Rink amide resin (0.19 mmol g−1). *(D)-Dpg is the natural residue at this position in corbomycin | ||
As a further proof of concept, we also targeted a highly challenging feglymycin analogue (13 AAs, 8/13 arylglycines),12,13 a retro-inverso peptide sequence (4).56 Monitoring the peptide elongation (using conventional heating) highlighted high purity until the final 4 amino acids. The synthesis failure (4-S1) was attributed to a mixture of on-resin aggregation (polystyrene matrix) and the unwanted phenolic reactivity consuming the activated Fmoc-amino acids.
Next, we used a double coupling strategy for the final 4 amino acids on Rink amide resin using either a polystyrene or a SpheriTide matrix. After TFA cleavage and RP-HPLC, the final retro-inverso feglymycin peptides were isolated with 3% (4-S2) and 20% (4-S3) yield in only 3 days (Fig. 2B – 4-S3, see ESI, Fig. S2†). Importantly, a high number of unprotected phenolic groups is either tolerated (GPA 1-3) or can be overcome by classical SPPS strategies (4-S2-3).
Control of arylglycine epimerization in ramoplanin sequence
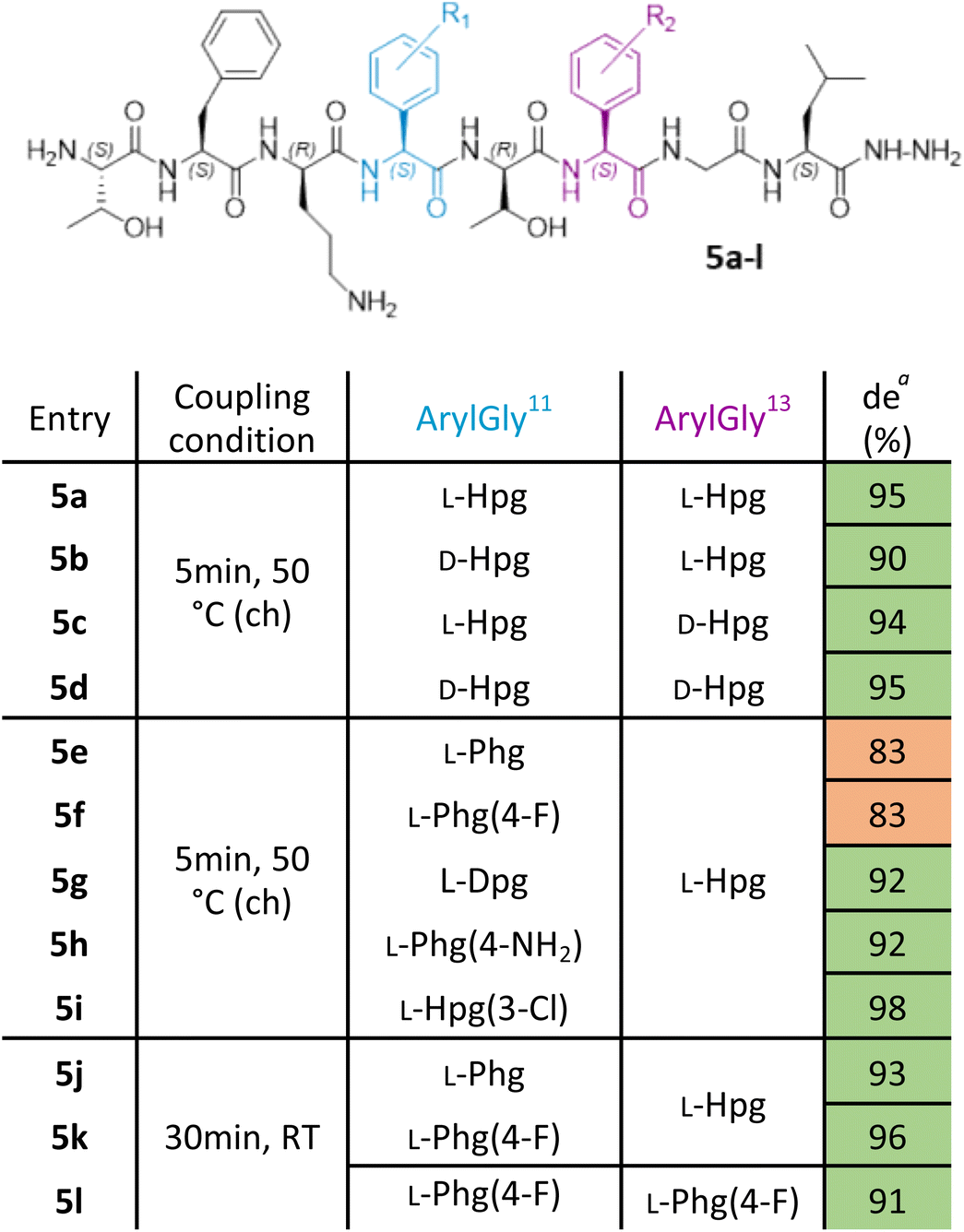
With this improved protocol in hand, we next sought to ensure that this could be implemented for SAR studies as well as allowing the synthesis of larger peptides. To this end, our investigation now turned to the synthesis of analogues of ramoplanin aglycone. For our initial target, we used the Thr8 to Leu15 octapeptide fragment (Table 1) from ramoplanin A2 containing two arylglycines and explored elongation conditions with Fmoc-arylglycines prepared from commercially available amino acids. This octapeptide sequence was selected to determine the diastereomeric excess (de) by chiral RP-HPLC as it was the starting sequence of our first SPPS-inspired total synthesis strategy (Scheme 1A). As controls, the 4 diastereomers of the octapeptides were synthesized in parallel using DIC/Oxyma protocol with conventional heating (Table 1. Entries 5a–d) to ensure the feasilbility of the diastereoisomeric separation by chiral RP-HPLC. This demonstrated excellent diastereomeric purity in these sequences (>90%) using 50 °C DIC/Oxyma coupling. We then broadened the scope of our syntheses to include L-Dpg, L-Hpg(3-Cl), L-Phg, L-Phg(4-F) and L-Phg(4-NH2) for future implementation in SAR (Table 1. Entries 5e–i). Here, we observed that the peptides containing substituted arylglycines showing higher diastereomeric purity (>90%) (Table 1. Entries 5g–i) than those without (80–85%) (Table 1. Entries 5e and 5f).| a Calculated by chiral RP-HPLC UV spectra at 254 nm. ch: conventional heating. |
|---|
|
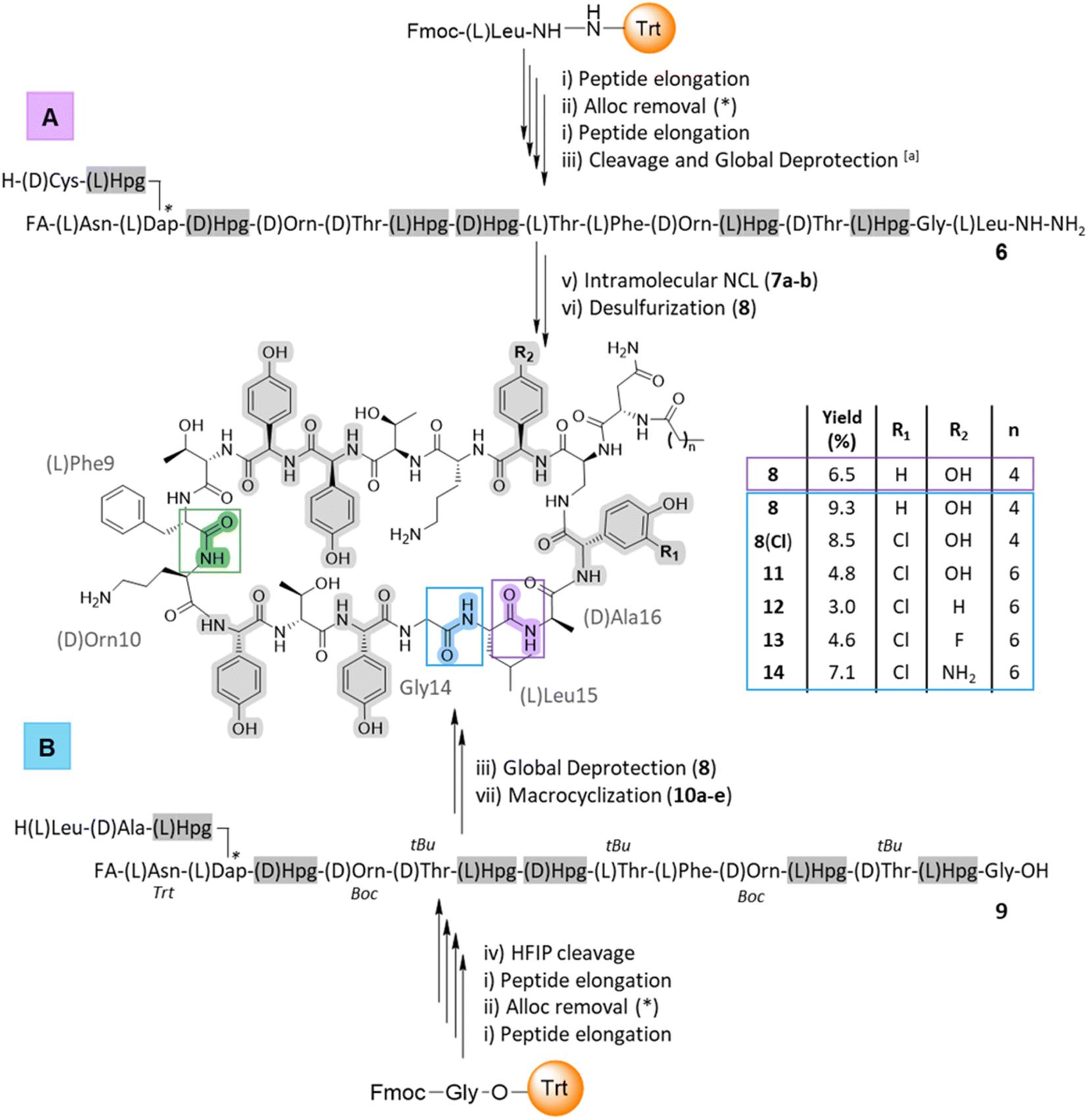 | ||
Scheme 1 SPPS and macrocyclization for the synthesis of ramoplanin aglycone analogues. (A) Native chemical ligation and desulfurization (site highlighted in purple – R = –H); (B) macrolactamization (site highlighted in blue – R = –H or –Cl). Protocol: (i) (a) DBU/DMF (v/v′; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :99), 3 × 30 s, RT; (b) Fmoc-AA-OH (3 eq.), DIC (3 eq.), Oxyma (6 eq.), DMF, 10 min, 50 °C or for Fmoc-arylglycine 3 (compounds 11–14) (b)′ Fmoc-AA-OH (3 eq.), DIC (3 eq.), Oxyma (6 eq.), DMF, 1 h, RT; (ii) Pd(PPh3)4 (0.1 eq.), phenylsilane (5 eq.), TEA (0.2 eq.), DCM, 2 × 45 min, RT; (iii) cocktail K: TFA/TIPS/water (v/v′/v′′; 95:2.5:2.5), a TFA/DODT/TIPS/H2O (v/v′/v′′/v′′′; 95:2: 2:1), 1 h, RT; (iv) HFIP/DCM (v/v′; 30:70), 3 × 30 min, RT; (v) (a) NaNO2 (5 eq.), GnHCl pH 3, 15 min, −15 °C; (b) (4-amino)thiophenol (40 eq.), GnHCl pH 7, 15 min, −15 °C; then NaOH (1 M) until pH 7, 1 h, −15 °C to RT; (d) TCEP pH 7, 15 h, RT; (vi) VA-044, tBuSH, TCEP, GnHCl pH 7, 4 h, 37 °C; (vii) PyBOP, TEA, DCM/DMF (50:50), 2 h, RT. The site of macrocyclization used in total synthesis is highlighted in green. *Represents Alloc removal position and timing. Hpg residues are highlighted in grey and orange in the structure and sequence respectively. :99), 3 × 30 s, RT; (b) Fmoc-AA-OH (3 eq.), DIC (3 eq.), Oxyma (6 eq.), DMF, 10 min, 50 °C or for Fmoc-arylglycine 3 (compounds 11–14) (b)′ Fmoc-AA-OH (3 eq.), DIC (3 eq.), Oxyma (6 eq.), DMF, 1 h, RT; (ii) Pd(PPh3)4 (0.1 eq.), phenylsilane (5 eq.), TEA (0.2 eq.), DCM, 2 × 45 min, RT; (iii) cocktail K: TFA/TIPS/water (v/v′/v′′; 95:2.5:2.5), a TFA/DODT/TIPS/H2O (v/v′/v′′/v′′′; 95:2: 2:1), 1 h, RT; (iv) HFIP/DCM (v/v′; 30:70), 3 × 30 min, RT; (v) (a) NaNO2 (5 eq.), GnHCl pH 3, 15 min, −15 °C; (b) (4-amino)thiophenol (40 eq.), GnHCl pH 7, 15 min, −15 °C; then NaOH (1 M) until pH 7, 1 h, −15 °C to RT; (d) TCEP pH 7, 15 h, RT; (vi) VA-044, tBuSH, TCEP, GnHCl pH 7, 4 h, 37 °C; (vii) PyBOP, TEA, DCM/DMF (50:50), 2 h, RT. The site of macrocyclization used in total synthesis is highlighted in green. *Represents Alloc removal position and timing. Hpg residues are highlighted in grey and orange in the structure and sequence respectively. | ||
Gratifyingly, reducing coupling temperature to room temperature for the residues without protic side chains was sufficient to improve diastereomeric purity to >90% (Table 1. Entries 5j–l). With our protocol benchmarked on these truncated ramoplanin peptides, we next turned to the synthesis of the entire ramoplanin aglycone analogue via SPPS followed by intramolecular macrocyclization.
Total synthesis of [Dap2] ramoplanin analogues
Cyclic peptides have numerous assembly routes, which makes the choice of the site of macrocyclization one of the most important decisions in their synthesis.57 Intramolecular cyclization is often performed by a peptide coupling reaction on a fully protected peptide or by ligation on an unprotected peptide. In the total synthesis of ramoplanin aglycone, the macrocyclization was achieved between Phe9 and D-Orn10 (Scheme 1, green junction).32,33 Looking at ramoplanin from an SPPS perspective, there are several possible positions to perform macrocyclization: the Gly14/Leu15 junction by conventional macrocyclization on a fully or partially protected peptide, the Hpg13/Gly14 junction by auxiliary-mediated ligation and auxiliary removal58 or the Leu15/D-Ala16 junction by native chemical ligation (NCL) and desulfurization.59Threonine ligation and oxazolidine cleavage is a strategy that requires the formation of an arylglycine ester for the D-Hpg7/aThr8 and Hpg11/D-aThr12 junctions (risk of epimerization) or is not directly compatible with the adjacent amino acid for the D-Orn4/D-aThr5 junction.60 Taking advantage of the rate at which peptides can be produced with SPPS it was possible to rapidly synthesize several sequences (Scheme 1) and thus study different macrocyclization strategies from linear precursors to identify the optimal route to produce [Dap2] ramoplanin analogues.
Intramolecular native chemical ligation (NCL) followed by desulfurization at the Leu15/D-Ala16 junction was chosen based on our experience preparing peptide thioesters from hydrazides61 and the possibility to use commercially available D-cysteine, which would be converted into D-alanine after desulfurization. Using our optimized SPPS protocol (50 μmol scale), peptide elongation on a Liberty Blue (CEM) synthesizer was achieved from Leu15 to the fatty acid. As a proof-of-concept, we have chosen the hexanoic acid (C6) since the N-terminal of ramoplanin tolerates a variety of groups.44,45,62 This synthesis utilised unprotected phenolic groups on Hpg residues, L- and D-threonine residues and L-Dap(Alloc) in position 2 (Scheme 1A). After Alloc removal and completion of the synthesis, linear precursor 6 was isolated with 44% RP-HPLC purity (at 214 nm) and 87% peptide recovery (110 mg, 44 μmol). After purification, intramolecular NCL was achieved in two steps according to Li's protocol,63 albeit with the substitution of MPAA (NCL yield for 7a: 28%) for (4-amino)thiophenol to facilitate purification (MPAA and the product were found to partially co-elute in RP-HPLC; NCL yield for 7b: 40.5%, 16.4 mg) (See ESI, Fig. S2†). Subsequent desulfurization64 using TCEP and VA-044 was achieved in 5 h and afforded 8 in an overall yield of 6.5% (calculated from the starting resin – 0.05 mmol) in an 8 days procedure (See ESI, Fig. S3†). This strategy also allowed the introduction of a thiol moiety, which is a versatile handle for a wide range of further modifications.65
The second strategy – macrocyclization of a partially protected peptide at the Gly14/Leu15 junction (Scheme 1B) – was chosen due to improved organic solvent solubility. Using organic solvents instead of an aqueous buffer would help overcome aggregation, one of the major limitations of the NCL and subsequent desulfurization strategy. Aggregation of the linear ramoplanin peptide has been widely reported in aqueous conditions during the synthesis/purification process32,33 as well as with ramoplanin A2.40 As with the previous strategy, peptide elongation was performed from Gly14 to the fatty acid (hexanoic acid) including the coupling of five Hpg residues with unprotected side chains and the L-Dap(Alloc) in position 2. After manual Alloc removal and completion of the synthesis, the linear precursor 9 was isolated with 51% purity (at 214 nm) and a 77% recovery (105 mg) after HFIP/DCM cleavage. Macrocyclization on a 1 mg pilot scale was investigated using different coupling reagents31 in a mixture of DMF/DCM (1:1) at a concentration of 0.5 mM (See ESI, Table S2 and Fig. S4†). The optimized reaction protocol utilised Py-BOP/TEA (19% peptide purity for 10d) and was completed in the shortest time (1 h). The macrocyclization was then scaled to 100 mg and the solvent was removed after 2 h. Global deprotection of the crude peptide using cocktail K (Scheme 1B) led to ramoplanin analogue 8 in an overall yield of 9.3% (calculated from the starting resin – 0.05 mmol) in a 6 days protocol. LCMS co-injection using material from each synthetic route confirmed that these were the same molecule (See ESI, Fig. S5†).
After the characterization of 8 by structural NMR (Fig. 3) and careful analysis of the alanine scanning results,32 we applied the macrocyclization strategy to incorporate alternate aryglycine residues (vide infra, D-Hpg, D-Phg, D-Phg(4-F) and D-Phg(4-NH2), Table 1). The largest reduction in antimicrobial activity after the D-Orn10 replacement by D-Ala10 was observed with the replacement of D-Hpg3 with D-Ala3.32 As such we synthesised compounds 8(Cl) and 11–14 to provide insights into the role of D-Hpg3 and L-Hpg(3-Cl)17 located inside the U-shaped structure.34 For the library generated at position 3, the Fmoc-aryglycines D-Hpg3 (11), D-Phg (12), D-Phg(4-F) (13) and D-Phg(4-NH2) (14) were incorporated at room temperature to maintain the amino acid chirality using a parallel SPPS approach. Also, an extended C8 fatty acid was introduced as an N-terminal aliphatic group to enhance the antimicrobial properties as suggested previously.44,45 The increased hydrophobicity resulted in lower overall yields of 3–7% for compounds 11–14 while the yield of 8(Cl) remained unchanged (Scheme 1B).
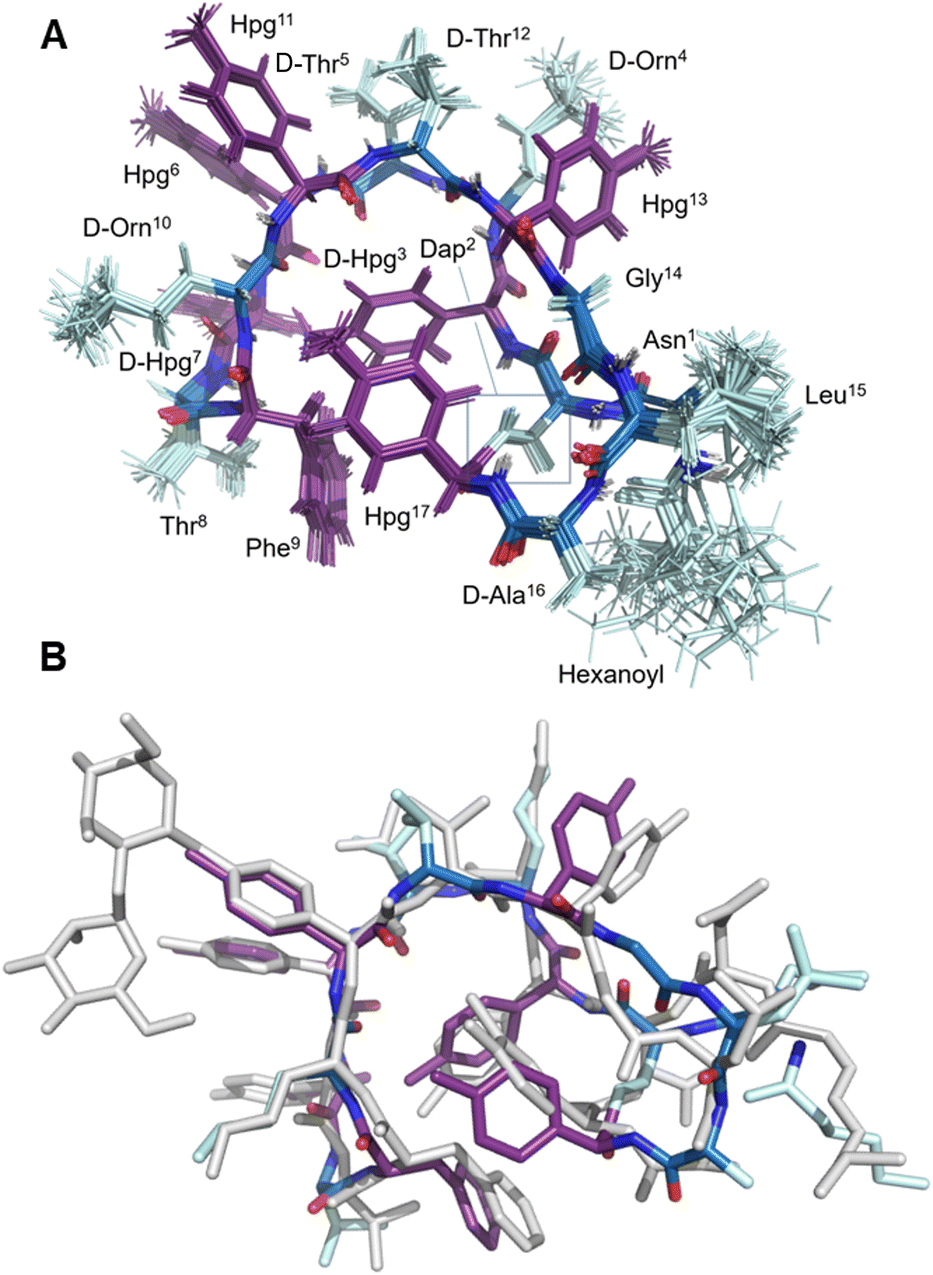 | ||
| Fig. 3 Structural characterization of 8. (A) Labelled view of the final ensemble of 20 structures of 8; (B) comparative superposition of the ramoplanin A2 NMR structure (1DSR) and the NMR structure of ramoplanin aglycone 8 | ||
Confirmation of a U-shaped structure and antimicrobial activity
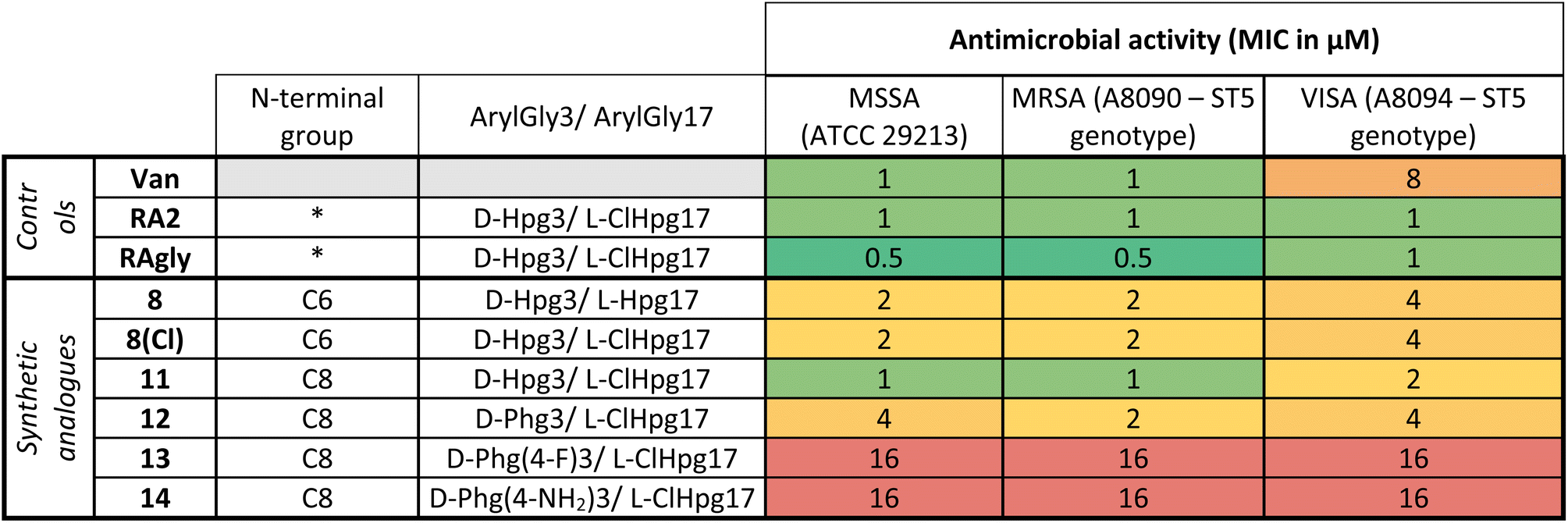
Once the syntheses were complete, we validated the structure and activity of 8. The high yield obtained enabled investigation of the structure of 8 using NMR, commonly absent from synthetic efforts thus far. Ramoplanin A2 is a highly structured molecule in solution with a characteristic ‘U-shaped’ β-turn motif (PDB: 1DSR)39 that was also found for our synthetic analogue (Fig. 3A). The 1H and 1H-attached 13C and 15N resonances of 8 were assigned using 2D homonuclear and heteronuclear NMR.66 Backbone assignments were made using the standard sequential NOE-based assignment strategy using 2D-TOCSY and 2D-NOESY data and sidechain assignments were further aided using the 2D 13C–1H H2BC experiment (see ESI, Table S3 and Fig. S6†). The final structure ensemble was calculated in CYANA67 and is based on 34230 distance restraints (Table S3†) which included 104 long-range NOEs and had good precision (backbone RMSD = 0.17 Å, heavy atom RMSD = 0.49 Å) and residual restraint violations (Fig. S6†). The structure of 8 is highly similar to the deposited NMR or X-ray structure of ramoplanin A2 and important side chains involved in Lipid II interaction (Hpg6, D-Hpg7, L-Thr8 and D-Orn10) superpose well (Fig. 3B).39,68 Nine long-range NOEs were observed within the D-Hpg3, L-Phe9, L-Hpg17 triad, including a broadened side chain OH of D-Hpg3 (Fig. S7†) that revealed an aromatic core giving the characteristic U-shaped antiparallel structure of ramoplanin A2. The hexanoyl lipid appears to be largely unstructured in 8 owing to the paucity of identified NOEs therein, but it is nevertheless suitably oriented to engage upon membrane binding. Unexpectedly, L-Hpg17 is more face-on into the core compared to the equivalent Hpg(3-Cl)17 in ramoplanin A2, possibly due to the different cyclisation (amide/ester) or by the favourable burial of the hydrophobic chlorine atom in ramoplanin A2 (Fig. 3B). D-Hpg3 is also observed facing the backbone of L-Thr8 in RA2 while slightly shifted in 8, where it now faces the backbone of D-Hpg7.As controls for the antimicrobials assays, we used vancomycin (Van), ramoplanin A2 (RA2) and ramoplanin aglycone (RAgly); the latter was obtained by hydrolysis of the sugars from ramoplanin A2 as previously reported (38% yield).17 We tested these 3 controls and 6 synthetic analogues against clinical isolates of Gram-positive bacteria including; methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant S. aureus (MRSA69), and vancomycin-intermediate S. aureus (VISA70) (Table 2 and ESI Fig. 8–10†). Whilst we observed a 2-4-fold loss of activity for 8 and 8(Cl) compared to RA2 and RAgly (See ESI, Table S4 and Fig. S8†), these results were encouraging given that the activity for 8 and 8(Cl) is 2-4-fold higher than the activity reported for a hexanoyl-ramoplanin depsipeptide obtained by modifying ramoplanin A2 (4 μM on MSSA, 4 μM on MRSA and 16 μM on VISA).44,45 The 3-Cl substituent of Hpg17 did not have a significant impact on the MIC despite its interesting structural properties.32,71 In agreement with Ciabatti,44,45 switching from a C6 to a C8 aliphatic chain (11) resulted in improved activity, which is 2-fold higher compared to 8 and 8(Cl). The nature of the amino acid in position 3 also appears to be critical since replacing D-Hpg3 lead to moderate (12) or significant loss (13–14) of activity. Without a clear ramoplanin/Lipid II interaction such as in vancomycin with binding to the D-Ala–D-Ala region,8,9 it remains unclear whether the subtle change impacts only the structure and/or binding to Lipid II. This position had been noted as important in the alanine scan performed by Boger and co-workers;32 although our synthetic approach combined with insights from NMR structures improves our understanding of the important role of the phenolic group in this position. Importantly our data on VISA shows that as the best-in-class antibiotic vancomycin loses activity, the activity of ramoplanin analogues remain largely constant. Beyond S. aureus, ramoplanin A2 has been considered a possible agent for the treatment of vancomycin resistant Enterococci (VRE)72,73 and C. difficile gut infections.74,75 More recently, ramoplanin A2 has also showed in vitro synergistic activity against nontuberculous Mycobacterium.76 Taken together, this highlights the value of continued investigation into this class of antibiotics as important agents that can decrease the burden of bacterial antimicrobial resistance.77
|
Conclusions
We have developed a robust methodology for the synthesis of arylglycine-containing peptides including ramoplanin, feglymycin and GPA precursors. The incorporation of multiple arylglycine residues showed the importance of controlling the temperature during coupling (critical for enantiopurity) and peptide synthesis parameters such as the resin matrix (critical when unprotected phenolic groups are present). The synthesis of 6 ramoplanin analogues was achieved in good yields (3–9%) and more rapidly than is possible via total synthesis. The structural characterization of the [Dap2] ramoplanin analogue (8) represents the first structure of synthetic aglycone of the ramoplanin family, and guided an arylglycine-focused SAR study as well as providing insights into the modifications generated in the seminal work by the Ciabatti18,44,45 and Boger17,32,33 groups. Antibacterial studies also indicate significant potential for synthetic ramoplanin analogues to tackle antimicrobial resistance as potent antimicrobial activity is maintained towards methicillin- and vancomycin-resistant strains. We anticipate that this method will both revitalize interest in ramoplanin-type antibiotics and encourage the inclusion of arylglycine residues in future SPPS-driven SAR campaigns, taking advantage of these important non-proteinogenic residues that have previously been excluded.Data availability
The chemical shifts have been deposited at the BMRB (code 52188) and the coordinates at the PDB (code 8V4B, PDB DOI: https://doi.org/10.2210/pdb8v4b/pdb).Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript. The research was designed by JT and MJC; EM and JT performed all synthesis, liquid chromatography and mass spectrometry. JDS and AIK performed the NMR experiments and structure calculations (JDS). RWC, KMP and JAEP performed antimicrobial testing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Health and Medical Research Council for funding (APP2003325). This research was conducted by the Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012) and funded by the Australian Government. The authors thank Dr Nitin Patil (Monash) for helpful discussions and Prof. Anton Peleg (Monash) for access to bacterial strains.Notes and references
- B. G. de la Torre and F. Albericio, Molecules, 2020, 25, 2293 CrossRef CAS PubMed.
- L. Diao and B. Meibohm, Clin. Pharmacokinet., 2013, 52, 855–868 CrossRef CAS PubMed.
- Y. Ding, J. P. Ting, J. Liu, S. Al-Azzam, P. Pandya and S. Afshar, Amino Acids, 2020, 52, 1207–1226 CrossRef CAS PubMed.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- R. S. Al Toma, C. Brieke, M. J. Cryle and R. D. Süssmuth, Nat. Prod. Rep., 2015, 32, 1207–1235 RSC.
- J. Tailhades, Int. J. Pept. Res. Ther., 2021, 28, 10 CrossRef.
- E. Wick Warren, Appl. Microbiol., 1967, 15, 765–769 CrossRef PubMed.
- E. Marschall, M. J. Cryle and J. Tailhades, J. Biol. Chem., 2019, 294, 18769–18783 CrossRef CAS PubMed.
- A. Okano, N. A. Isley and D. L. Boger, Chem. Rev., 2017, 117, 11952–11993 CrossRef CAS PubMed.
- D. S. Peters, F. E. Romesberg and P. S. Baran, J. Am. Chem. Soc., 2018, 140, 2072–2075 CrossRef CAS PubMed.
- T. C. Roberts, P. A. Smith and F. E. Romesberg, J. Nat. Prod., 2011, 74, 956–961 CrossRef CAS PubMed.
- F. Dettner, A. Hänchen, D. Schols, L. Toti, A. Nußer and R. D. Süssmuth, Angew. Chem., Int. Ed., 2009, 48, 1856–1861 CrossRef CAS PubMed.
- S. Fuse, Y. Mifune, H. Nakamura and H. Tanaka, Nat. Commun., 2016, 7, 13491 CrossRef CAS PubMed.
- M. H. Hansen, E. Stegmann and M. J. Cryle, Curr. Opin. Biotechnol., 2022, 77, 102767 CrossRef CAS PubMed.
- E. J. Culp, N. Waglechner, W. Wang, A. A. Fiebig-Comyn, Y.-P. Hsu, K. Koteva, D. Sychantha, B. K. Coombes, M. S. Van Nieuwenhze, Y. V. Brun and G. D. Wright, Nature, 2020, 578, 582–587 CrossRef CAS PubMed.
- D. G. McCafferty, P. Cudic, B. A. Frankel, S. Barkallah, R. G. Kruger and W. Li, Biopolymers, 2002, 66, 261–284 CrossRef CAS PubMed.
- S. Walker, L. Chen, Y. Hu, Y. Rew, D. Shin and D. L. Boger, Chem. Rev., 2005, 105, 449–476 CrossRef CAS PubMed.
- R. Ciabatti, J. K. Kettenring, G. Winters, G. Tuan, L. Zerilli and B. Cavalleri, J. Antibiot., 1989, 42, 254–267 CrossRef CAS PubMed.
- E. Higashide, K. Hatano, M. Shibata and K. Nakazawa, J. Antibiot., 1968, 21, 129–137 CrossRef.
- K. T. Morgan, J. Zheng and D. G. McCafferty, ChemBioChem, 2021, 22, 176–185 CrossRef CAS PubMed.
- L. F. Weigel, C. Nitsche, D. Graf, R. Bartenschlager and C. D. Klein, J. Med. Chem., 2015, 58, 7719–7733 CrossRef CAS PubMed.
- S. J. F. Kaptein, O. Goethals, D. Kiemel, A. Marchand, B. Kesteleyn, J.-F. Bonfanti, D. Bardiot, B. Stoops, T. H. M. Jonckers, K. Dallmeier, P. Geluykens, K. Thys, M. Crabbe, L. Chatel-Chaix, M. Münster, G. Querat, F. Touret, X. de Lamballerie, P. Raboisson, K. Simmen, P. Chaltin, R. Bartenschlager, M. Van Loock and J. Neyts, Nature, 2021, 598, 504–509 CrossRef CAS PubMed.
- S. von Berg, Y. Xue, M. Collins, A. Llinas, R. I. Olsson, T. Halvarsson, M. Lindskog, J. Malmberg, J. Jirholt, N. Krutrök, M. Ramnegård, M. Brännström, A. Lundqvist, M. Lepistö, A. Aagaard, J. McPheat, E. L. Hansson, R. Chen, Y. Xiong, T. G. Hansson and F. Narjes, ACS Med. Chem. Lett., 2019, 10, 972–977 CrossRef CAS PubMed.
- H. A. Schmid, Mol. Cell. Endocrinol., 2008, 286, 69–74 CrossRef CAS PubMed.
- K. L. Reddy and K. B. Sharpless, J. Am. Chem. Soc., 1998, 120, 1207–1217 CrossRef CAS.
- R. M. Williams and J. A. Hendrix, Chem. Rev., 1992, 92, 889–917 CrossRef CAS.
- T. Bruckdorfer, O. Marder and F. Albericio, Curr. Pharm. Biotechnol., 2004, 5, 29–43 CAS.
- R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27 CrossRef CAS PubMed.
- M. A. Elsawy, C. Hewage and B. Walker, J. Pept. Sci., 2012, 18, 302–311 CrossRef CAS PubMed.
- C. Liang, M. A. M. Behnam, T. R. Sundermann and C. D. Klein, Tetrahedron Lett., 2017, 58, 2325–2329 CrossRef CAS.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- J. Nam, D. Shin, Y. Rew and D. L. Boger, J. Am. Chem. Soc., 2007, 129, 8747–8755 CrossRef CAS PubMed.
- W. Jiang, J. Wanner, R. J. Lee, P.-Y. Bounaud and D. L. Boger, J. Am. Chem. Soc., 2003, 125, 1877–1887 CrossRef CAS PubMed.
- R. Subirós-Funosas, R. Prohens, R. Barbas, A. El-Faham and F. Albericio, Chem.–Eur. J., 2009, 15, 9394–9403 CrossRef PubMed.
- C. Brieke and M. J. Cryle, Org. Lett., 2014, 16, 2454–2457 CrossRef CAS PubMed.
- P. Fulco and R. P. Wenzel, Expert Rev. Anti-Infect. Ther., 2006, 4, 939–945 CrossRef CAS PubMed.
- D. K. Farver, D. D. Hedge and S. C. Lee, Ann. Pharmacother., 2005, 39, 863–868 CrossRef CAS PubMed.
- S. Zhang, Y. Chen, J. Zhu, Q. Lu, M. J. Cryle, Y. Zhang and F. Yan, Nat. Prod. Rep., 2023, 40, 557–594 RSC.
- M. Kurz and W. Guba, Biochemistry, 1996, 35, 12570–12575 CrossRef CAS PubMed.
- P. Cudic, J. K. Kranz, D. C. Behenna, R. G. Kruger, H. Tadesse, A. J. Wand, Y. I. Veklich, J. W. Weisel and D. G. McCafferty, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 7384–7389 CrossRef CAS PubMed.
- M.-C. Lo, H. Men, A. Branstrom, J. Helm, N. Yao, R. Goldman and S. Walker, J. Am. Chem. Soc., 2000, 122, 3540–3541 CrossRef CAS.
- Y. Hu, J. S. Helm, L. Chen, X.-Y. Ye and S. Walker, J. Am. Chem. Soc., 2003, 125, 8736–8737 CrossRef CAS PubMed.
- M. Cheng, J. X. Huang, S. Ramu, M. S. Butler and M. A. Cooper, Antimicrob. Agents Chemother., 2014, 58, 6819–6827 CrossRef PubMed.
- R. Ciabatti, S. I. Maffioli, G. Panzone, A. Canavesi, E. Michelucci, P. S. Tiseni, E. Marzorati, A. Checchia, M. Giannone, D. Jabes, G. Romanò, C. Brunati, G. Candiani and F. Castiglione, J. Med. Chem., 2007, 50, 3077–3085 CrossRef CAS PubMed.
- R. Ciabatti, S. Maffioli, A. Checchia, G. Romano, G. Candiani and G. Panzone, WO2003076460A1, 2003.
- W. L. Thong, Y. Zhang, Y. Zhuo, K. J. Robins, J. K. Fyans, A. J. Herbert, B. J. C. Law and J. Micklefield, Nat. Commun., 2021, 12, 6872 CrossRef CAS PubMed.
- A. J. Hoertz, J. B. Hamburger, D. M. Gooden, M. M. Bednar and D. G. McCafferty, Bioorg. Med. Chem., 2012, 20, 859–865 CrossRef CAS PubMed.
- D. M. P. D. Oliveira, B. M. Forde, T. J. Kidd, P. N. A. Harris, M. A. Schembri, S. A. Beatson, D. L. Paterson and M. J. Walker, Clin. Microbiol. Rev., 2020, 33, e00181–e00119 CrossRef PubMed.
- J. Tailhades, Y. Zhao, Y. T. C. Ho, A. Greule, I. Ahmed, M. Schoppet, K. Kulkarni, R. J. A. Goode, R. B. Schittenhelm, J. J. De Voss and M. J. Cryle, Angew. Chem., Int. Ed., 2020, 59, 10899–10903 CrossRef CAS PubMed.
- Y. Zhao, Y. T. C. Ho, J. Tailhades and M. Cryle, ChemBioChem, 2021, 22, 43–51 CrossRef CAS PubMed.
- J. Tailhades, Y. Zhao, M. Schoppet, A. Greule, R. J. A. Goode, R. B. Schittenhelm, J. J. De Voss and M. J. Cryle, Org. Lett., 2019, 21, 8635–8640 CrossRef CAS PubMed.
- Y. Zhao, R. J. A. Goode, R. B. Schittenhelm, J. Tailhades and M. J. Cryle, J. Org. Chem., 2020, 85, 1537–1547 CrossRef CAS PubMed.
- A. El-Faham, R. S. Funosas, R. Prohens and F. Albericio, Chem.–Eur. J., 2009, 15, 9404–9416 CrossRef CAS PubMed.
- S. L. Pedersen, A. P. Tofteng, L. Malik and K. J. Jensen, Chem. Soc. Rev., 2012, 41, 1826–1844 RSC.
- M. Ieronymaki, M. E. Androutsou, A. Pantelia, I. Friligou, M. Crisp, K. High, K. Penkman, D. Gatos and T. Tselios, Biopolymers, 2015, 104, 506–514 CrossRef CAS PubMed.
- M. Chorev and M. Goodman, Acc. Chem. Res., 1993, 26, 266–273 CrossRef CAS.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- J. Offer, Pept. Sci., 2010, 94, 530–541 CrossRef CAS PubMed.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed.
- H. Liu and X. Li, Acc. Chem. Res., 2018, 51, 1643–1655 CrossRef CAS PubMed.
- J. Tailhades, M. Schoppet, A. Greule, M. Peschke, C. Brieke and M. J. Cryle, Chem. Commun., 2018, 54, 2146–2149 RSC.
- Y. Rew, D. Shin, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2004, 126, 1041–1043 CrossRef CAS PubMed.
- J.-S. Zheng, S. Tang, Y. Guo, H.-N. Chang and L. Liu, ChemBioChem, 2012, 13, 542–546 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem.–Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- J. D. Swarbrick, J. A. Karas, J. Li and T. Velkov, Chem. Commun., 2020, 56, 2897–2900 RSC.
- P. Güntert, in Protein NMR Techniques, ed. A. K. Downing, Humana Press, Totowa, NJ, 2004, pp. 353–378, DOI:10.1385/1-59259-809-9:353.
- J. B. Hamburger, A. J. Hoertz, A. Lee, R. J. Senturia, D. G. McCafferty and P. J. Loll, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 13759–13764 CrossRef CAS PubMed.
- A. Y. Peleg, D. Monga, S. Pillai, E. Mylonakis, R. C. Moellering Jr and G. M. Eliopoulos, Int. J. Infect. Dis., 2009, 199, 532–536 CrossRef PubMed.
- M. M. Mwangi, S. W. Wu, Y. Zhou, K. Sieradzki, H. de Lencastre, P. Richardson, D. Bruce, E. Rubin, E. Myers, E. D. Siggia and A. Tomasz, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 9451–9456 CrossRef CAS PubMed.
- J. Han, J. Chen, L. Shao, J. Zhang, X. Dong, P. Liu and D. Chen, Plos One, 2016, 11, e0154121 CrossRef PubMed.
- U. Stiefel, J. Pultz Nicole, S. Helfand Marion and J. Donskey Curtis, Antimicrob. Agents Chemother., 2004, 48, 2144–2148 CrossRef CAS PubMed.
- M. T. Wong, C. A. Kauffman, H. C. Standiford, P. Linden, G. Fort, H. J. Fuchs, S. B. Porter, R. P. Wenzel and V. R. E. C. S. G. Ramoplanin, Clin. Infect. Dis., 2001, 33, 1476–1482 CrossRef CAS PubMed.
- J. Freeman, S. D. Baines, D. Jabes and M. H. Wilcox, J. Antimicrob. Chemother., 2005, 56, 717–725 CrossRef CAS PubMed.
- W. K. Smits, D. Lyras, D. B. Lacy, M. H. Wilcox and E. J. Kuijper, Nat. Rev. Dis. Primers, 2016, 2, 16020 CrossRef PubMed.
- D. B. Aziz, J. W. P. Teo, V. Dartois and T. Dick, Front. Microbiol., 2018, 9, 932 CrossRef PubMed.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. Kashef Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01944f |
| This journal is © The Royal Society of Chemistry 2024 |