
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpin polarized electron dynamics enhance water splitting efficiency by yttrium iron garnet photoanodes: a new platform for spin selective photocatalysis†
Harshad
Gajapathy
 a,
Savini
Bandaranayake
a,
Emily
Hruska
a,
Aravind
Vadakkayil
b,
Brian P.
Bloom
b,
Stephen
Londo
a,
Jackson
McClellan
a,
Jason
Guo
c,
Daniel
Russell
c,
Frank M. F.
de Groot
d,
Fengyuan
Yang
c,
David H.
Waldeck
b,
Martin
Schultze
e and
L. Robert
Baker
*a
a,
Savini
Bandaranayake
a,
Emily
Hruska
a,
Aravind
Vadakkayil
b,
Brian P.
Bloom
b,
Stephen
Londo
a,
Jackson
McClellan
a,
Jason
Guo
c,
Daniel
Russell
c,
Frank M. F.
de Groot
d,
Fengyuan
Yang
c,
David H.
Waldeck
b,
Martin
Schultze
e and
L. Robert
Baker
*a
aDepartment of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: baker.2364@osu.edu
bDepartment of Chemistry, University of Pittsburgh, 15260 Pittsburgh, Pennsylvania, USA
cDepartment of Physics, The Ohio State University, Columbus, Ohio 43210, USA
dInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, 3584CG Utrecht, The Netherlands
eInstitute of Experimental Physics, Graz University of Technology, Petersgasse 16, Graz 8010, Austria
First published on 24th January 2024
Abstract
This work presents a spectroscopic and photocatalytic comparison of water splitting using yttrium iron garnet (Y3Fe5O12, YIG) and hematite (α-Fe2O3) photoanodes. Despite similar electronic structures, YIG significantly outperforms widely studied hematite, displaying more than an order of magnitude increase in photocurrent density. Probing the charge and spin dynamics by ultrafast, surface-sensitive XUV spectroscopy reveals that the enhanced performance arises from (1) reduced polaron formation in YIG compared to hematite and (2) an intrinsic spin polarization of catalytic photocurrents in YIG. Ultrafast XUV measurements show a reduction in the formation of surface electron polarons compared to hematite due to site-dependent electron–phonon coupling. This leads to spin polarized photocurrents in YIG where efficient charge separation occurs on the Td sub-lattice compared to fast trapping and electron/hole pair recombination on the Oh sub-lattice. These lattice-dependent dynamics result in a long-lived spin aligned hole population at the YIG surface, which is directly observed using XUV magnetic circular dichroism. Comparison of the Fe M2,3 and O L1-edges show that spin aligned holes are hybridized between O 2p and Fe 3d valence band states, and these holes are responsible for highly efficient, spin selective water oxidation by YIG. Together, these results point to YIG as a new platform for highly efficient, spin selective photocatalysis.
1. Introduction
Motivated by the need to identify stable, earth-abundant materials with optical and electronic properties that can be tailored for solar to chemical energy conversion, this work examines the carrier dynamics of two stable oxide semiconductors, yttrium iron garnet, and hematite, and correlates these properties with their water splitting efficiency. Photocatalytic water splitting on semiconductor electrodes offers a promising route to clean H2 production.1–4 This has the potential to facilitate a H2 fuel economy5,6 as well as reduce emissions from chemical processes requiring large quantities of H2, such as the Haber–Bosch process7,8 and Fischer–Tropsch9,10 synthesis. While much effort has been devoted to understanding and controlling the redox potential and transport properties in candidate semiconductor photocatalysts, the role of electron spin is often overlooked when designing artificial photosynthetic systems.In Nature, photosystem II performs water oxidation with remarkable efficiency using sunlight as the only energy source.11,12 While a number of artificial devices have been demonstrated with photosynthetic efficiencies comparable to natural systems, most of these are ultimately limited by the stability, selectivity, or cost of the catalyst responsible for the water oxidation half-reaction. Notably, biological photosynthesis operates in chiral environments, and it has been proposed that this feature can promote efficiency because of the inherent spin selectivity of chiral matter.13–16 This supposition is supported by theoretical studies which suggest that the CaMn4O5 cofactor of photosystem II acts as a spin control for enhancing water oxidation17 and by studies that extend the principal of spin polarized enhanced water oxidation to artificial systems. For instance, chiral metal oxides have been shown to act as spin filters and result in spin selective photocatalysis, which reduces the reaction overpotential and increases the faradaic efficiency of the reaction.18–25 Because O2 forms on a triplet potential energy surface, spin selective photocatalysis can guide the reaction towards a triplet product and increase the kinetics of O2 generation.26–28
Despite the application of chiral oxides for photocatalytic water splitting, the concept of spin polarized photocatalysis has yet to be evaluated for achiral magnetic semiconductors in which spin polarization results from magnetic exchange interactions rather than structural chirality. For electrocatalytic water oxidation in the dark, a number of studies have demonstrated that certain magnetic oxides display enhanced efficiency, presumably because of spin polarized transport within aligned domains of a magnetized anode.29–33 Extending this approach to photocatalysis requires a combined understanding of the ground and excited state magnetic structure as well as the underlying photophysics that controls the spin states of transient, photoexcited redox carriers. To date, most spectroscopic studies have focused on studying spin polarized photoemission through a chiral layer34–39 rather than spin accumulation at a catalyst surface, even though the latter is primarily responsible for driving spin selective catalysis. Owing to the challenge of spin selective spectroscopy with chemical state specificity and surface or interface sensitivity, the photoinduced spin polarization of a catalyst surface has not been directly observed for either a chiral or magnetic system.40
Although YIG is widely employed as a magnetic insulator in spintronic devices, it is not often used as a photocatalyst. One exception is a recent combinatorial synthesis of approximately 30 complex oxides that identified YIG as a promising photoanode for water oxidation.41 On the other hand, hematite has been extensively explored as a photoanode for water solar splitting because of its ideal band gap, strong solar absorption, high chemical stability, and well-aligned band edge potential relative to the water oxidation half-reaction. In addition, Fe is one of the most abundant elements, making hematite almost universally accessible at a negligible cost. Despite these advantages, the photocatalytic efficiency of hematite remains low because of its poor electron carrier mobility, which is related to the efficient formation of small electron polarons that act as deep trap states.42–46 Significantly improving electron transport in Fe oxide-based semiconductors could have a transformative effect on the field of solar fuels.
Using femtosecond time-resolved surface-sensitive Extreme Ultraviolet (XUV) spectroscopy, we directly compare the polaron formation efficiency in YIG to that in hematite and find that polaron formation in YIG is significantly reduced compared to hematite. A comparison of water oxidation by these two materials in the dark and under illumination confirms that YIG displays an 8-fold enhancement in photocurrent over hematite at a fixed potential of 2.0 V vs. RHE. This dramatic enhancement of photocatalytic activity also correlates with a significantly improved O2 yield as quantified by rotating ring-disk measurements at pH 8. Finally, we confirm that the enhanced activity by YIG is a direct result of spin polarized photocurrents. To demonstrate this, we employ surface-sensitive XUV magnetic circular dichroism to directly observe the accumulation of spin aligned charge carriers at the photocatalyst surface under illumination. These measurements can directly probe the magnetic states of Fe in the thin film using the difference in M2,3-edge absorption of Fe due to the polarization of light.47–50 These XUV dichroism measurements at the Fe M2,3-edge and the O L1-edge reveal that spin aligned holes accumulate in the hybridized O 2p/Fe 3d valence band giving rise to spin polarized Fe4+ redox-active states in the Oh sublattice of YIG. Together, these results provide a detailed understanding of dramatically enhanced water splitting efficiency using a spin selective metal oxide photocatalyst and open the door for probing the effects of spin polarized currents in photosynthetic systems based on earth-abundant chiral and magnetic oxides.
2. Results and discussion
Fig. 1(a) shows the ground state static absorption spectrum of YIG and hematite. They both show an absorption feature with the peak centered at 55 eV. This feature corresponds to the 3p to 3d electronic excitation in Fe3+ atoms by the XUV probe. Because YIG and hematite contain Fe3+, they share the same M2,3-edge absorption peak. Despite subtle differences in lineshape between hematite, which contains only Oh Fe3+ centers, and YIG, which contains both Oh and Td centers, it is not possible to cleanly resolve the Oh and Td Fe3+ contributions due to strong overlap in the linear XUV spectrum. As we show below, however, these sites can be resolved using XUV magnetic circular dichroism owing to the opposite spin alignment of the Oh and Td sub-lattices in YIG. | ||
| Fig. 1 Results of XUV-RA experiments on YIG (black) and hematite (red). (a) Ground state static absorption spectrum. (b) Transient absorption spectrum at 100 fs time delay between pump and probe. (c) Transient absorption spectrum at 1 ps time delay between pump and probe. | ||
Although the ground state Fe M2,3-edge of YIG and hematite are similar, they display very different responses in the ultrafast transient XUV measurements. Pumping with a 400 nm (3.1 eV) light pulse induces above bandgap excitation in both YIG (2.6–2.8 eV bandgap)51,52 and hematite (2.0–2.2 eV bandgap).53,54 The upper valence band of these materials comprises primarily O 2p orbitals and the conduction band comprises primarily Fe 3p orbitals54,55 so that an above bandgap excitation constitutes an electron transfer from O to Fe atoms.56 This charge transfer reduces Fe3+ atoms to Fe2+ and causes decrease in the M2,3-edge absorption of Fe3+ which manifests as a bleach feature at 55 eV. Fig. 1(b) compares the transient XUV spectrum of YIG and hematite at 100 fs following photoexcitation, but before the relaxation of the photoexcited electron has occurred. As shown, both spectra display the characteristic bleach feature associated with a free conduction band electron in Fe 3d states.44,45
Following photoexcitation, the excited electrons in the Fe atoms of hematite couple with optical phonons to form a small electron polaron.42,44–46 It has been shown previously that this trapped electron–polaron has an XUV spectral signature at the Fe M2,3-edge around 52 eV.57Fig. 1(c) compares the transient spectra for YIG and hematite at 1 ps after photoexcitation following polaron formation. These data show that the polaronic yield is significantly reduced in the YIG as compared to hematite; i.e., the absorbance at 52 eV is weaker for a similar amount of bleach signal at 55 eV. Because hematite contains Fe atoms exclusively in Oh lattice sites whereas YIG contains Fe atoms in both Td and Oh lattice sites (see Fig. 2(a)), one possible explanation for reduced polaron efficiency in YIG is that the electrons excited to the Oh Fe3+ centers strongly couple to optical phonons, while the electrons in the Td Fe3+ are less susceptible to polaron formation. Because the excited electron in the Oh sub-lattice is localized in the low energy t2g orbital compared to an e orbital for the Td sub-lattice, this hypothesis is consistent with theoretical predictions that t2g electrons have a stronger electron–phonon coupling and higher efficiency of small polaron formation than the e electrons.58
 | ||
| Fig. 2 (a) Crystal structure of YIG showing the Fe3+ atoms in Td (blue) and Oh (red) lattice sites with opposite spins. (b) Illustrated band structure for YIG depicting how the interface potential leads to the drift of electrons to bulk and holes to the surface. Selective polaron formation in the Oh sub-lattice leads to spin selective trapping, which is expected to spin polarize the photoinduced current. | ||
Note that YIG is a ferrimagnet with Td and Oh electrons having opposite spins due to an antiferromagnetic coupling. In both lattice sites, Fe3+ atoms are in a high spin state with a 3d5 electronic configuration. By Pauli exclusion, the electrons excited by the pump pulse from the O 2p valence band into the Fe 3d conduction band must have a spin opposite to the already occupied Fe 3d electrons. Consequently, the conduction band electrons photoexcited to the Td and Oh lattice sites must have opposite spins. From the transient XUV measurement, we hypothesize that the small polaron formation occurs only to the electrons in the Oh lattice sites in YIG. These trapped electrons have opposite spin relative to the electrons in the Td lattice sites which do not form a polaron. Therefore, the polaronic electron trapping in the case of YIG is spin dependent. This indicates that electron transport in YIG is likely spin polarized because spin aligned electrons in the Td lattice are significantly more mobile compared to electrons having the opposite spin, which form self-trapped, small electron polarons in the Oh lattice. As YIG is an n-type semiconductor, the surface bands have higher energy compared to the bulk. Accordingly, the non-trapped conduction electrons of one particular spin in the Td sub-lattice are free to move from surface to bulk,59 while the other spin electrons remain trapped as small polarons in the Oh sub-lattice (see Fig. 2(b) for a summary of the spin dependent electron transport). We note that the XUV spectra showing reduced polaron formation in YIG are consistent with reported conductivity measurements,60–64 which measure the mobility of electrons in YIG to be within the range of 0.1 to 0.5 cm2 V−1 s−1 whereas the mobility of electrons in hematite is reported to be on the order of 10−2 cm2 V−1 s−1. The one-order of magnitude higher electron mobility in YIG compared to hematite can be attributed to reduced electron trapping in YIG due to the resistance of Td electrons to form small polarons. These measurements are consistent with previous reports of electron transport in YIG based on a large polaron hopping model;60,65,66 therefore, it is likely that the electrons excited to the Td atoms form a large polaron.
To demonstrate the influence of ultrafast polaron formation on photocatalytic water splitting efficiency, Pastor et al. used photocurrent measured during in situ water oxidation as a direct readout for a pump–push experiment, where a visible pump pulse excites the hematite bandgap, a sub-picosecond delay allows time for polaron formation, followed by an IR push pulse to de-trap the electron.42 This experiment reported a photocurrent enhancement by the IR pump pulse at the exact time constant measured for surface polaron formation by XUV reflection–absorption.44,46 These results confirm that the formation of polarons in hematite actively limits water oxidation efficiency by hindering the removal of majority carriers from the depletion region resulting in eventual charge carrier recombination and loss of photoactive holes.67–70 Consistent with the XUV measurements, we find that YIG displays nearly an order of magnitude higher photocurrent density for water oxidation relative to hematite. This can be observed in Fig. 3, which shows the current density of hematite (panel a) and YIG (panel b) with and without illumination. While dark currents are similar between these two materials, the light-driven photocatalytic response is significantly higher in YIG. Fig. 3(c) plots the difference between the total current in light and dark. At 2.15 V vs. RHE, YIG produces a 15-fold photocurrent enhancement relative to hematite increasing from 0.4 to 6.3 mA cm−2 at an illumination intensity of 100 mW cm−2.
 | ||
| Fig. 3 Linear sweep voltammograms for hematite (a) and YIG (b) catalyst-loaded electrodes in the dark and upon irradiation. (c) Difference between dark and light current densities. | ||
While the enhanced activity of YIG over hematite can be attributed in part to reduced polaron formation and greater electron mobility, below we show that the spin polarization of the photocurrent is also important. Photoexcitation of conduction band electrons leaves behind holes in the valence band. The electrons that trap as small polarons eventually recombine with their corresponding holes at a long time, circa 350–500 ps.45,71 Before recombination, these holes can act as an electron acceptor at the anode during water oxidation. Assuming the conducting electrons in YIG are spin polarized, the holes left behind must be spin polarized as well. Because molecular O2 has a triplet ground state, compared to singlet water, spin is not a conserved quantity during the O2 evolution reaction (OER) when considering only the reactants. However, considering the spin of the redox active holes, which drive water oxidation, spin polarized currents show enhanced activity toward O2 production since the transfer of two spin aligned holes balances the angular momentum of the triplet product. Consequently, spin polarized photocurrents may also play an important role in improving the efficiency of water oxidation.
To illustrate the significantly enhanced efficiency for O2 evolution along the triplet reaction pathway by YIG compared to hematite, Fig. 4(a) shows the partial current density, the part of the total current density responsible for O2 generation as measured using a rotating ring disk electrode. The O2 partial current from the bare magnetic substrate is low indicating that negligible amounts of O2 are produced in the absence of either the hematite or YIG catalyst. Hematite produces more O2 than the bare magnetic electrode, but YIG has the highest performance among the three electrodes. O2 partial current is only reported up to 1.85 V vs. RHE because bubble formation interferes with O2 detection by the ring electrode at higher current densities. This significant enhancement in O2 generation efficiency by YIG cannot be explained simply due to the difference in active area between hematite and YIG. To illustrate this, Fig. 4(b) shows a similar electrochemically active surface area for both YIG and hematite. Fig. 4(c) (inset) also shows that despite having lower absorbance and a slightly higher band gap than hematite, YIG still displays significantly enhanced photocurrent. Note that the data in Fig. 3 and 4 were obtained under a constant magnetic field, with a field strength high enough to saturate the out-of-plane magnetization of YIG and to ensure spin alignment of the respective sub-lattices across domains in the polycrystalline catalyst. Fig. S6 (see ESI†) shows the photocurrent without magnetization using a glassy carbon electrode. By comparison, the measurements using glassy carbon electrodes do not show such enhancement in YIG relative to hematite. Thus, the enhanced activity of YIG towards O2 evolution under magnetization indicates that spin polarization plays a crucial role in the high performance of YIG.
 | ||
| Fig. 4 (a) O2 partial current density for bare magnetic electrode, hematite and YIG. (b) Double layer capacitance for YIG and hematite catalysts. (c) Tauc plots showing UV-Vis absorption by YIG and hematite catalysts confirming band gaps of 2.5 eV and 2.2 eV, respectively. | ||
While spin polarized currents have been predicted to explain enhanced efficiency for a variety of chiral and magnetic catalysts,13,20,34,72–75 it has remained a significant challenge to visualize the actual spin accumulation at the catalyst surface. In this study, we show that surface-sensitive XUV Magnetic Circular Dichroism (MCD) is able to directly detect the spin polarized holes that drive spin selective OER. Here, the MCD signal is defined by the difference in absorption of circularly polarized light under the different sample magnetizations as described below. Fig. 5(a) depicts the excitation of the different spin states by the circularly polarized XUV light. As illustrated by the diagram, the magnetic field selectively stabilizes one sub-lattice in the sample while destabilizing the other, and electrons in the stabilized sub-lattice absorb at a different photon energy compared to the destabilized spins. Fig. 5(b) shows the MCD spectrum of YIG taken at the Fe M2,3-edge. Ligand field multiplet simulations of the MCD spectrum were performed using the software package CTM4XAS (see ESI Section 3†).76–79 Here the black line represents the experimental MCD spectrum, the grey line represents the simulated spectrum, and blue and red dashed lines represent individual contributions from the Fe3+ Td and Fe3+ Oh sub-lattices, respectively. As shown, combined simulations of the Td and Oh lattice sites in YIG closely reproduce the measured experimental spectrum.77 Because these sub-lattices are aligned by the external magnetic field with opposite spins, the line shapes of the Td and Oh contributions to the total MCD spectrum are opposite. Fig. 5(b) shows the ground state MCD spectrum and defines the initial state of YIG.
 | ||
| Fig. 5 (a) Illustrated band structure of YIG and associated XUV-MCD absorption at the Fe M2,3-edge. (b) and (c) Comparison of experimental and simulated XUV-MCD spectra measured in the dark (b) and during illumination with a 1 kHz, 400 nm excitation beam (c). | ||
When illuminated with visible light the spectral features change because of the light-induced charge transfer from O 2p valence band states to Fe 3d conduction band states. To observe this effect on interfacial spin polarization, we employed a 400 nm pump pulse and broadband, circularly polarized XUV probe pulse each at a 1 kHz frequency. This experimental design means that if the relative pump–probe time delay is such that the probe pulse arrives at the sample before the pump pulse by a few ps, then the time delay between a pump pulse and the subsequent probe is approximately 1 ms defined by the laser repetition rate. As the probe–pump–probe–pump train arrives at the sample with a 1 kHz repetition rate, if the sample doesn't return to its ground state in 1 ms, we can probe the corresponding long-lived excited state spectrum using the probe. Measuring the XUV-MCD spectrum under these conditions enables observation of long-lived spin polarized charge accumulation at the YIG surface. Any pump-induced changes to the MCD spectrum under these conditions represent spin polarized carriers having a sufficient lifetime to influence photocatalytic processes such as water oxidation.
The MCD spectrum of YIG under 400 nm illumination using this 1 ms time delay between pump and probe is shown in Fig. 5(c). There are two light-induced changes to the YIG spectrum resulting from 400 nm illumination. First, we observe a sharp dip at 40 eV. This feature is well below the Fe M2,3-edge and matches previous reports of XUV absorption at the O L1-edge corresponding to an O 2s to 2p transition.56,78,79 Second, we observe a change in the Fe M2,3-edge line shape with the loss of intensity at 58 eV and the appearance of a new feature at 60 eV. Considering first the O L1-edge, no MCD signal is discernible in the ground state O L1-edge spectrum. This observation is consistent with a full 2p6 electron configuration in Fe2O3, where Fe metal centers are in a 3+ oxidation state and O ligands are in a 2- oxidation state. The completely paired 2p6 electron configuration precludes any O contribution to ground-state magnetization. However, surface band bending in n-type YIG can drive holes to accumulate at the surface following photoexcitation. These holes are expected to have significant O 2p character, giving rise to unpaired electrons in the O 2p valence band. Accumulation of surface holes will only be detected in an MCD measurement to the extent that they display a net spin polarization. Consequently, the appearance of this spectral signature at the O L1-edge upon illumination represents the direct detection of spin aligned holes in O 2p valence band states. The reason that the MCD signal at the O L1-edge appears as a single negative dip rather than a peak and a dip similar to the various contributions to the Fe M2,3-edge is consistent with a spin aligned hole in an O 2p5 electron configuration as described in the ESI Section 4.†
Because the valence band of YIG also includes contributions from Fe 3d orbitals, hybridization of the O 2p ligand hole with Fe 3d states is expected.80 Previously, we showed that the position of the O L1-edge absorption reflects the extent of O 2p and metal 3d valence band hybridization, with a redshift corresponding to a more strongly hybridized valence band.56 Here the O L1-edge feature appears at 40 eV, which is several eV red-shifted compared to previous reports of photoexcited holes in Fe2O3, Co3O4, and NiO, suggesting that YIG shows greater metal–oxygen bond covalency compared to hematite and that the photoexcited holes should have both O 2p and Fe 3d character. This is consistent with previous reports that identify a combination of O 2p ligand holes81–95 and high valent 4+ Fe96–101 as the primary redox active states for water splitting in a variety of metal oxide catalysts.
The mixing of the ligand hole with Fe 3d states is evident in Fe M2,3-edge MCD spectrum above 60 eV. These changes to the Fe M2,3-edge MCD spectrum can be reproduced by including a contribution from Fe4+ Oh centers in the ligand field multiplet simulation. This new contribution is shown as the green dashed line in Fig. 5(c). The incorporation of a Fe4+ hole on the Td sub-lattice is unable to reproduce the measured, excited state MCD signal (see ESI Section 3†). Consequently, we conclude that the spin polarized hole primarily hybridizes between O 2p and Oh Fe 3d states. This is equivalent to stating that the unpaired electrons generated by the creation of a valence band hole are ferromagnetically coupled to the Fe Td sub-lattice, which is consistent with this sub-lattice controlling the net magnetic moment in YIG. This unpaired electron in O atoms is stabilized due to ferromagnetic exchange interaction between the O and the Fe Td atoms, so it has the least energy when it is spin aligned with the Td lattice. This explains the long-lived spin polarization in the material which can persist even on the ms time scale. Together, these two new features in the Fe M2,3 and O L1-edges correspond to the accumulation of spin polarized holes in the surface of YIG, which are shared between hybridized O 2p and Oh Fe 3d valence band orbitals.
Based on these findings Fig. 6 summarizes the excited state spin polarized electron dynamics in YIG that give rise to spin selective photocatalytic water oxidation. Initial photoexcitation with visible light simultaneously excites both the Oh and Td sub-lattices. Photoexcited electrons in the Oh sub-lattice rapidly trap as small electron polarons as evidenced by Fig. 1, while electrons in the Td sub-lattice have much higher mobility and are free to exit the surface depletion region via drift into bulk. Because of spin selective electron trapping in the Oh sub-lattice, subsequent electron–hole pair recombination gives rise to a residual spin polarized hole population at the YIG surface. These holes reside in the hybridized valence band with shared O 2p and Oh Fe 3d character and live for greater than 1 ms allowing them to drive spin selective water oxidation at the YIG surface as shown in Fig. 6. It is this spin polarized ligand hole population that is responsible for guiding water oxidation along the triplet pathway leading to improved kinetics for O2 production.
 | ||
| Fig. 6 Illustration of selective polaron formation and subsequent electron–hole pair recombination in Oh sites, leading to spin polarized hole accumulation at the YIG surface. These spin polarized holes represent the redox equivalents that drive spin selective water oxidation along the triplet pathway favoring the generation of O2. | ||
We note that experimental considerations necessitate the use of different samples for XUV and PEC measurements. To avoid non-resonant surface scattering, XUV-RA measurements require samples that are smooth on the scale of the XUV wavelengths, which is realized here by the epitaxial growth of YIG on GGG. However, the insulating nature of the GGG substrate precludes PEC measurements of these samples. Instead, PEC measurements employ nanoparticles of YIG and hematite drop cast on a conductive magnetic rotating disk electrode. This provides the added benefit of increasing the electrochemically active surface area of the photocatalytic samples. Despite differences in the exact sample morphology, the comparison between YIG and hematite in terms of both XUV-RA and PEC measurements, which both consist of well-characterized materials matching the specified phase and composition, form the basis for reliable conclusions regarding the origin of enhanced performance by YIG for photocatalytic water splitting.
3. Conclusion
In summary, this work provides a comparison of YIG and hematite as photoanodes for water oxidation. YIG shows nearly an order of magnitude enhancement in photocurrent density relative to hematite and a significantly enhanced yield for the production of O2. Direct observation of surface electron dynamics using surface-sensitive XUV spectroscopy reveals that this improved performance results from reduced polaron formation efficiency in YIG relative to hematite, which gives rise to spin selective electron–hole pair recombination and spin polarized hole accumulation at the YIG surface. Surface-sensitive XUV-MCD measurements provide the first direct observation of spin polarized charge accumulation, showing that these holes are long-lived (>1 ms) and are hybridized between the Oh Fe 3d and O 2p valence band states. These spin polarized charged carriers represent the redox equivalents responsible for driving efficient water oxidation along the triplet pathway leading to O2 generation. Together these findings provide a detailed picture of the spin selective electron dynamics responsible for significantly enhanced photocatalytic activity in YIG relative to widely studied hematite and highlight YIG as a new platform for spin selective photocatalysis. All indications suggest that these findings will be general to any ferrimagnetic semiconductor, where electron mobility and recombination rates differ between the various sub-lattices, opening the door to studies of spin polarized electron dynamics and photocatalysis in a range of magnetically ordered semiconductors.4. Experiments and methods
4.1. Sample preparation
For the XUV-RA and XUV-MCD measurements 20 nm thick Y3Fe5O12 (YIG) epitaxial thin films were grown on (111) oriented Gd3Ga5O12 (GGG) substrates using the off-axis sputtering technique.102 These samples were characterized using XRD, XRR, AFM, and magnetometric measurements (see ESI Section 1†). The Fe2O3 (hematite) samples were prepared by sputtering Fe metal onto SiO2 substrate and thermally annealed in air at 520 °C for 30 min. The characterization of these samples was previously reported by Husek et al.444.2. XUV reflection–absorption
Extreme Ultraviolet Reflection–Absorption (XUV-RA)44,103 spectroscopy was used to perform ground state and time-resolved measurements of small polaron formation in hematite and YIG. The samples were pumped using a 400 nm beam with an area of 1.85 mm2 and a fluence of 7.5 mJ pulse−1 cm−2 incident at the sample at 70° relative to surface normal. To produce the XUV probe beam, an 800 nm driving field mixed with a 400 nm symmetry breaking field was used for high harmonic generation in helium gas. The driving field was filtered using a thin aluminum filter, which gives a high energy cutoff of 72 eV for the XUV probe beam. The XUV probe was focused using a toroid mirror and was incident on the sample at an angle of 82° relative to the surface normal. Experiments are performed at a 1 kHz repetition rate for both pump and probe beams. The specular reflectance of the XUV probe from the sample was spectrally dispersed and imaged onto a CCD detector using a concave variable line-spaced grating. The ground state XUV-RA spectrum was calculated for all energies based on the intensity of the XUV beam reflected from the YIG or hematite sample, Isample, compared to the intensity reflected from a single crystal silicon reference, Ireference, using the formula,
For time-resolved measurements, the time delay between the pump pulse and the probe pulse was controlled using a linear stage in the pump beamline, and the absolute time delay was calibrated using the results from the hematite sample, which shows an instrument response function of approximately 80 fs. The transient signal was determined from the intensity of XUV light with the pump-on Ipump-on and pump-off Ipump-off conditions for all the time delays using the formula,
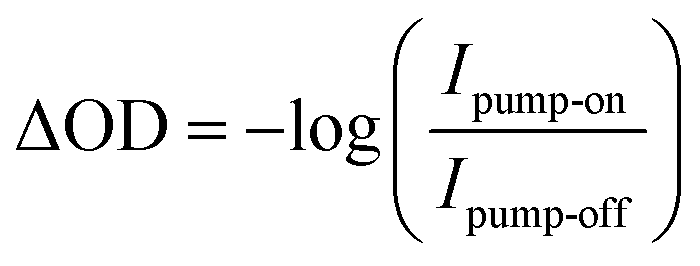
4.3. XUV magnetic circular dichroism
The XUV Magnetic Circular Dichroism (MCD) measurements were performed on the same beamline described above with the following modifications. The sample was pumped with the same 400 nm beam but with an area of 6.42 mm2 and a fluence of 7.5 mJ pulse−1 cm−2 incident at the sample at 82° relative to the surface normal. The XUV beam was right circularly polarized using a four Mo/B4C multilayer mirror reflector.104–107 The sample was magnetized by a switchable magnet with an out-of-plane magnetic field of ±300 mT. The MCD signal is given by the intensity of the right circular polarized light from the north magnetized sample Inorth and the intensity of the right circular polarized light from the south magnetized sample Isouth using the formula,
XUV-MCD measurements were performed for both the ground (400 nm pump beam off) and excited state (400 nm pump beam on) of YIG. Because XUV measurements at near-grazing angle reflection have a probe depth less than 3 nm,46,103,108,109 both XUV-RA and XUV-MCD spectra are surface specific, showing surface polaron formation and state-resolved surface spin accumulation, respectively.
4.4. Electrochemical catalysis
Hematite (Thermo Fisher) and YIG (Sigma Aldrich) catalyst solutions were prepared by mixing 5 mg of nanoparticle powder with 125 μL of Nafion perfluorinated resin solution (Aldrich) and 2.5 mL of a water/isopropyl alcohol (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) mixture. The catalyst solutions were sonicated for 20 min to ensure a homogeneous suspension and then immediately a 2 μL aliquot was drop-cast onto a permanent magnet (K&J Magnetics) which was used as a disk electrode. Note that the magnet disk electrode has a geometric area = 0.12 cm2 and was polished to a mirror finish using 0.05-micron-sized alumina slurry prior to drop-casting the catalyst ink. The electrode was then dried in an oven at 70 °C for 30 min to evaporate the solvents upon which the electrode was ready for electrochemical measurements. Faradaic efficiency measurements for the oxygen evolution reaction were performed in the dark, and upon irradiation, using a rotating ring-disk electrode (RRDE-31, ALS) apparatus with a CH Instruments 750c bipotentiostat. Note, the ring electrode was platinum. To irradiate the catalyst, a CM1-FO1 UV-enhanced aluminum mirror was placed under the electrochemical cell, and a xenon arc lamp, with a power of 100 mW, was focused on the electrode surface. Electrochemical measurements were made in a 0.02 M pH 8 phosphate buffer solution, purged with Ar for 30 minutes, with the catalyst-coated permanent magnet disk and a platinum ring (ALS) as the two working electrodes. The platinum ring was electrochemically cleaned by cycling the potential between −0.03 V and 1.37 V vs. RHE at a scan rate of 500 mV s−1 for 50 cycles while the catalyst-loaded permanent magnet disk electrode was kept at an open circuit. To measure the faradaic efficiency, a linear sweep voltammogram of the catalyst-loaded permanent magnet electrode was recorded at a 10 mV s−1 scan rate and 1600 rpm rotation rate. The ring electrode was held at a constant potential of 0.06 V vs. RHE such that the oxygen produced at the disk was reduced at the ring. The faradaic efficiency, FE was calculated using the equation,
:1 v/v) mixture. The catalyst solutions were sonicated for 20 min to ensure a homogeneous suspension and then immediately a 2 μL aliquot was drop-cast onto a permanent magnet (K&J Magnetics) which was used as a disk electrode. Note that the magnet disk electrode has a geometric area = 0.12 cm2 and was polished to a mirror finish using 0.05-micron-sized alumina slurry prior to drop-casting the catalyst ink. The electrode was then dried in an oven at 70 °C for 30 min to evaporate the solvents upon which the electrode was ready for electrochemical measurements. Faradaic efficiency measurements for the oxygen evolution reaction were performed in the dark, and upon irradiation, using a rotating ring-disk electrode (RRDE-31, ALS) apparatus with a CH Instruments 750c bipotentiostat. Note, the ring electrode was platinum. To irradiate the catalyst, a CM1-FO1 UV-enhanced aluminum mirror was placed under the electrochemical cell, and a xenon arc lamp, with a power of 100 mW, was focused on the electrode surface. Electrochemical measurements were made in a 0.02 M pH 8 phosphate buffer solution, purged with Ar for 30 minutes, with the catalyst-coated permanent magnet disk and a platinum ring (ALS) as the two working electrodes. The platinum ring was electrochemically cleaned by cycling the potential between −0.03 V and 1.37 V vs. RHE at a scan rate of 500 mV s−1 for 50 cycles while the catalyst-loaded permanent magnet disk electrode was kept at an open circuit. To measure the faradaic efficiency, a linear sweep voltammogram of the catalyst-loaded permanent magnet electrode was recorded at a 10 mV s−1 scan rate and 1600 rpm rotation rate. The ring electrode was held at a constant potential of 0.06 V vs. RHE such that the oxygen produced at the disk was reduced at the ring. The faradaic efficiency, FE was calculated using the equation,where iring and idisk are the current densities of the ring and disk electrodes, respectively, and N is the collection efficiency. Note, N is a constant whose value depends only on the geometrical aspects of the ring disk electrode and was determined to be, 0.44, from calibration experiments using a ferri/ferrocyanide redox couple. The faradaic efficiency for each electrode was measured in the dark and under irradiation, three times to ensure that photodegradation was not occurring. Moreover, each measurement was replicated for at least three independently prepared catalyst electrodes to mitigate deviations in electrode preparation. A control experiment using the ring electrode in the dark and under illumination without any catalyst deposition confirm that photoexcitation of the ring electrode did not influence the reported faradaic efficiency measurements (see ESI Section 2†).
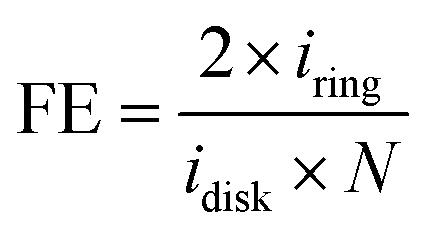
Data availability
All relevant data has been included in the manuscript and ESI.†Author contributions
L. R. B. and H. G. conceived the idea and planned the experiments. J. G. and D. R., with guidance from F. Y. prepared and characterized YIG samples. M. S. developed the XUV phase retarder to enable XUV-MCD measurements. H. G. and S. L. with guidance from M. S. and L. R. B. designed and assembled the XUV-MCD spectrometer. H. G., S. B., E. H., and S. L. with guidance from L. R. B. performed XUV-RA and XUV-MCD measurements. A. V. and B. P. B. with guidance from D. H. W. performed photo-electrocatalysis measurements. H. G. and J. M. with guidance from F. M. M. G. and L. R. B. simulated XUV-MCD spectra using charge transfer multiplet theory. A. V., B. P. B., and D. H. W. analyzed photo-electrocatalysis data. H. G. and L. R. B. analyzed the XUV-RA and XUV-MCD data. H. G. and L. R. B. wrote the paper. All authors reviewed and edited the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Design, assembly, and demonstration of XUV-MCD capabilities on the existing XUV beamline, as well as growth and characterization of epitaxial YIG samples for XUV measurements, were supported by the Center for Emergent Materials, an NSF MRSEC, under award number DMR-2011876. Measurements of relative polaron formation efficiencies in YIG and hematite were supported by the United States Air Force Office of Scientific Research under AFOSR Award No. FA95550-19-1-0184. Ligand multiplet simulations of XUV-MCD spectra were supported by a Camille Dreyfus Teacher-Scholar Award to L. R. B. Electrochemical and photoelectrochemical activity and selectivity measurements were supported by United States National Science Foundation under NSF Award No. CHE-2140249 to D. H. W.References
- Z. Xing, X. Zong, J. Pan and L. Wang, Chem. Eng. Sci., 2013, 104, 125–146 CrossRef CAS.
- C.-H. Liao, C.-W. Huang and J. C. Wu, Catalysts, 2012, 2, 490–516 CrossRef CAS.
- C. Acar, I. Dincer and G. F. Naterer, Int. J. Energy Res., 2016, 40, 1449–1473 CrossRef CAS.
- R. D. Tentu and S. Basu, Curr. Opin. Electrochem., 2017, 5, 56–62 CrossRef CAS.
- S. van Renssen, Nat. Clim. Change, 2020, 10, 799–801 CrossRef.
- DOE, US Department of Energy, Washington, DC, 2022.
- M. Capdevila-Cortada, Nat. Catal., 2019, 2, 1055 CrossRef.
- V. Kyriakou, I. Garagounis, A. Vourros, E. Vasileiou and M. Stoukides, Joule, 2020, 4, 142–158 CrossRef CAS.
- G. Zang, P. Sun, A. Elgowainy, A. Bafana and M. Wang, Environ. Sci. Technol., 2021, 55, 3888–3897 CrossRef CAS PubMed.
- A. Rahbari, A. Shirazi, M. B. Venkataraman and J. Pye, AIP Conf. Proc., 2020, 170011 CrossRef CAS.
- W. Lubitz, M. Chrysina and N. Cox, Photosynth. Res., 2019, 142, 105–125 CrossRef CAS PubMed.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Acc. Chem. Res., 2020, 53, 2659–2667 CrossRef CAS PubMed.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Annu. Rev. Biophys., 2022, 51, 99–114 CrossRef CAS PubMed.
- S. F. Ozturk and D. D. Sasselov, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2204765119 CrossRef CAS PubMed.
- B. P. Bloom, A. R. Waldeck and D. H. Waldeck, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2210505119 CrossRef CAS PubMed.
- Y. Jiao, R. Sharpe, T. Lim, J. H. Niemantsverdriet and J. Gracia, J. Am. Chem. Soc., 2017, 139, 16604–16608 CrossRef CAS PubMed.
- D. Waldeck, R. Naaman and Y. Paltiel, APL Mater., 2021, 9, 040902 CrossRef CAS.
- K. Ghosh, W. Zhang, F. Tassinari, Y. Mastai, O. Lidor-Shalev, R. Naaman, P. Möllers, D. Nürenberg, H. Zacharias and J. Wei, et al. , J. Phys. Chem. C, 2019, 123, 3024–3031 CrossRef CAS.
- S. Ghosh, B. P. Bloom, Y. Lu, D. Lamont and D. H. Waldeck, J. Phys. Chem. C, 2020, 124, 22610–22618 CrossRef CAS.
- T. Bai, J. Ai, L. Liao, J. Luo, C. Song, Y. Duan, L. Han and S. Che, Angew. Chem., Int. Ed., 2021, 60, 9421–9426 CrossRef CAS PubMed.
- Y. Li, X. Wang, J. Miao, J. Li, X. Zhu, R. Chen, Z. Tang, R. Pan, T. He and J. Cheng, Adv. Mater., 2020, 32, 1905585 CrossRef CAS PubMed.
- W. Zhang, K. Banerjee-Ghosh, F. Tassinari and R. Naaman, ACS Energy Lett., 2018, 3, 2308–2313 CrossRef CAS.
- A. Vadakkayil, C. Clever, K. N. Kunzler, S. Tan, B. P. Bloom and D. H. Waldeck, Nat. Commun., 2023, 14, 1067 CrossRef CAS PubMed.
- H. Im, S. Ma, H. Lee, J. Park, Y. S. Park, J. Yun, J. Lee, S. Moon and J. Moon, Energy Environ. Sci., 2023, 16, 1187–1199 RSC.
- W. Mtangi, F. Tassinari, K. Vankayala, A. Vargas Jentzsch, B. Adelizzi, A. R. Palmans, C. Fontanesi, E. Meijer and R. Naaman, J. Am. Chem. Soc., 2017, 139, 2794–2798 CrossRef CAS PubMed.
- J. Li, J. Ma, Z. Ma, E. Zhao, K. Du, J. Guo and T. Ling, Adv. Energy Sustainability Res., 2021, 2, 2100034 CrossRef CAS.
- T. Wu and Z. J. Xu, Curr. Opin. Electrochem., 2021, 30, 100804 CrossRef CAS.
- F. A. Garcés-Pineda, M. Blasco-Ahicart, D. Nieto-Castro, N. López and J. R. Galán-Mascarós, Nat. Energy, 2019, 4, 519–525 CrossRef.
- X. Ren, T. Wu, Y. Sun, Y. Li, G. Xian, X. Liu, C. Shen, J. Gracia, H.-J. Gao and H. Yang, et al. , Nat. Commun., 2021, 12, 2608 CrossRef CAS PubMed.
- T. Wu, X. Ren, Y. Sun, S. Sun, G. Xian, G. G. Scherer, A. C. Fisher, D. Mandler, J. W. Ager and A. Grimaud, et al. , Nat. Commun., 2021, 12, 3634 CrossRef CAS PubMed.
- R. R. Chen, Y. Sun, S. J. H. Ong, S. Xi, Y. Du, C. Liu, O. Lev and Z. J. Xu, Adv. Mater., 2020, 32, 1907976 CrossRef CAS PubMed.
- T. Lim, J. Niemantsverdriet and J. Gracia, ChemCatChem, 2016, 8, 2968–2974 CrossRef CAS.
- P. V. Möllers, J. Wei, S. Salamon, M. Bartsch, H. Wende, D. H. Waldeck and H. Zacharias, ACS Nano, 2022, 16, 12145–12155 CrossRef PubMed.
- B. Göhler, V. Hamelbeck, T. Markus, M. Kettner, G. Hanne, Z. Vager, R. Naaman and H. Zacharias, Science, 2011, 331, 894–897 CrossRef PubMed.
- M. Kettner, D. K. Bhowmick, M. Bartsch, B. Göhler and H. Zacharias, Adv. Mater. Interfaces, 2016, 3, 1600595 CrossRef.
- P. V. Möllers, S. Ulku, D. Jayarathna, F. Tassinari, D. Nürenberg, R. Naaman, C. Achim and H. Zacharias, Chirality, 2021, 33, 93–102 CrossRef PubMed.
- M. Kettner, B. Gohler, H. Zacharias, D. Mishra, V. Kiran, R. Naaman, C. Fontanesi, D. H. Waldeck, S. S. ȩk and J. Pawłowski, et al. , J. Phys. Chem. C, 2015, 119, 14542–14547 CrossRef CAS.
- M. Kettner, V. V. Maslyuk, D. Nürenberg, J. Seibel, R. Gutierrez, G. Cuniberti, K.-H. Ernst and H. Zacharias, J. Phys. Chem. Lett., 2018, 9, 2025–2030 CrossRef CAS PubMed.
- J. Fransson, Isr. J. Chem., 2022, e202200046 CrossRef CAS.
- L. Zhou, A. Shinde, D. Guevarra, M. H. Richter, H. S. Stein, Y. Wang, P. F. Newhouse, K. A. Persson and J. M. Gregoire, J. Mater. Chem. A, 2020, 8, 4239–4243 RSC.
- E. Pastor, J.-S. Park, L. Steier, S. Kim, M. Grätzel, J. R. Durrant, A. Walsh and A. A. Bakulin, Nat. Commun., 2019, 10, 3962 CrossRef PubMed.
- C. Lohaus, A. Klein and W. Jaegermann, Nat. Commun., 2018, 9, 4309 CrossRef PubMed.
- J. Husek, A. Cirri, S. Biswas and L. R. Baker, Chem. Sci., 2017, 8, 8170–8178 RSC.
- L. M. Carneiro, S. K. Cushing, C. Liu, Y. Su, P. Yang, A. P. Alivisatos and S. R. Leone, Nat. Mater., 2017, 16, 819–825 CrossRef CAS PubMed.
- S. Biswas, S. Wallentine, S. Bandaranayake and L. R. Baker, J. Chem. Phys., 2019, 151, 104701 CrossRef PubMed.
- H. Höchst, D. Zhao and D. L. Huber, Surf. Sci., 1996, 352, 998–1002 CrossRef.
- T. Koide, T. Shidara, K. Ymaguchi, A. Fujimori, H. Fukutani, N. Kimizuka and S. Kimura, Proceedings of the 11th International Conference on Vacuum Ultraviolet Radiation Physics, 1996, pp. 275–278 Search PubMed.
- M. Pretorius, J. Friedrich, A. Ranck, M. Schroeder, J. Voss, V. Wedemeier, D. Spanke, D. Knabben, I. Rozhko and H. Ohldag, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 14133 CrossRef.
- N. Telling, A. Haznar, G. Van der Laan, M. Roper, F. Schedin and G. Thornton, Phys. B, 2004, 345, 157–160 CrossRef.
- R. Metselaar and P. Larsen, Solid State Commun., 1974, 15, 291–294 CrossRef.
- A. Kumar, R. Kumar, N. Verma, A. Anupama, H. K. Choudhary, R. Philip and B. Sahoo, Opt. Mater., 2020, 108, 110163 CrossRef.
- S. Mochizuki, Phys. Status Solidi A, 1977, 41, 591–594 CrossRef.
- C. Xia, Y. Jia, M. Tao and Q. Zhang, Phys. Lett. A, 2013, 377, 1943–1947 CrossRef CAS.
- W.-K. Li and G.-Y. Guo, Phys. Rev. B, 2021, 103, 014439 CrossRef CAS.
- S. Biswas, J. Husek, S. Londo and L. R. Baker, Nano Lett., 2018, 18, 1228–1233 CrossRef CAS PubMed.
- I. M. Klein, H. Liu, D. Nimlos, A. Krotz and S. K. Cushing, J. Am. Chem. Soc., 2022, 144, 12834–12841 CrossRef CAS PubMed.
- Y. Takada and M. Masaki, J. Supercond. Novel Magn., 2007, 20, 629–633 CrossRef CAS.
- M. Battiato, K. Carva and P. M. Oppeneer, Phys. Rev. Lett., 2010, 105, 027203 CrossRef CAS.
- P. Larsen and R. Metselaar, Phys. Rev. B: Solid State, 1976, 14, 2520 CrossRef CAS.
- J. Costantini, F. Brisard, A. Meftah, F. Studer and M. Toulemonde, Nucl. Instrum. Methods Phys. Res., Sect. B, 1994, 91, 288–293 CrossRef CAS.
- N. Thiery, V. V. Naletov, L. Vila, A. Marty, A. Brenac, J.-F. Jacquot, G. de Loubens, M. Viret, A. Anane and V. Cros, et al. , Phys. Rev. B, 2018, 97, 064422 CrossRef CAS.
- B. Warnes, F. Aplan and G. Simkovich, Solid State Ionics, 1984, 12, 271–276 CrossRef CAS.
- P. Liao and E. A. Carter, J. Appl. Phys., 2012, 112, 013701 CrossRef.
- B. Deka, S. Ravi and D. Pamu, et al. , Ceram. Int., 2017, 43, 10468–10477 CrossRef.
- H. Li, Ceram. Int., 2020, 46, 15408–15416 CrossRef CAS.
- O. Zandi and T. W. Hamann, Phys. Chem. Chem. Phys., 2015, 17, 22485–22503 RSC.
- C. Li, Z. Luo, T. Wang and J. Gong, Adv. Mater., 2018, 30, 1707502 CrossRef PubMed.
- I. S. Cho, H. S. Han, M. Logar, J. Park and X. Zheng, Adv. Energy Mater., 2016, 6, 1501840 CrossRef.
- D. K. Bora, A. Braun and E. C. Constable, Energy Environ. Sci., 2013, 6, 407–425 RSC.
- Y. Fan, Y. Lin, K. Wang, K. H. Zhang and Y. Yang, Phys. Rev. B, 2021, 103, 085206 CrossRef CAS.
- V. Gatard, J. Deseure and M. Chatenet, Curr. Opin. Electrochem., 2020, 23, 96–105 CrossRef CAS.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem, 2019, 3, 250–260 CrossRef.
- Z. Bian, K. Kato, T. Ogoshi, Z. Cui, B. Sa, Y. Tsutsui, S. Seki and M. Suda, Adv. Sci., 2022, 9, 2201063 CrossRef PubMed.
- Y. Zhang, C. Liang, J. Wu, H. Liu, B. Zhang, Z. Jiang, S. Li and P. Xu, ACS Appl. Energy Mater., 2020, 3, 10303–10316 CrossRef.
- E. Stavitski and F. M. De Groot, Micron, 2010, 41, 687–694 CrossRef PubMed.
- H. B. Vasili, B. Casals, R. Cichelero, F. Macià, J. Geshev, P. Gargiani, M. Valvidares, J. Herrero-Martin, E. Pellegrin and J. Fontcuberta, et al. , Phys. Rev. B, 2017, 96, 014433 CrossRef.
- S. Londo, S. Biswas, I. V. Pinchuk, A. Boyadzhiev, R. K. Kawakami and L. R. Baker, J. Phys. Chem. C, 2022, 126, 2669–2678 CrossRef.
- S. Londo, S. Biswas, J. Husek, I. V. Pinchuk, M. J. Newburger, A. Boyadzhiev, A. H. Trout, D. W. McComb, R. Kawakami and L. R. Baker, J. Phys. Chem. C, 2020, 124, 11368–11375 CrossRef.
- U. Bergmann, C. Horne, T. Collins, J. Workman and S. Cramer, Chem. Phys. Lett., 1999, 302, 119–124 CrossRef.
- G. Chen, C. Wei, Y. Zhu and H. Huang, EcoMat, 2023, 5, e12294 CrossRef.
- H. Sun, X. Xu, Y. Song, W. Zhou and Z. Shao, Adv. Funct. Mater., 2021, 31, 2009779 CrossRef.
- P. Strasser, Acc. Chem. Res., 2016, 49, 2658–2668 CrossRef PubMed.
- C. Yang and A. Grimaud, Catalysts, 2017, 7, 149 CrossRef.
- Y. Wang, Y. Jiang, Y. Zhao, X. Ge, Q. Lu, T. Zhang, D. Xie, M. Li and Y. Bu, Chem. Eng. J., 2023, 451, 138710 CrossRef.
- M. Liu, L. Y. S. Lee and K.-Y. Wong, Handbook of Nanomaterials and Nanocomposites for Energy and Environmental Applications, 2021, pp. 1335–1367 Search PubMed.
- D. A. Kuznetsov, B. Han, Y. Yu, R. R. Rao, J. Hwang, Y. Román-Leshkov and Y. Shao-Horn, Joule, 2018, 2, 225–244 CrossRef CAS.
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647–4671 RSC.
- S. R. Ede and Z. Luo, J. Mater. Chem. A, 2021, 9, 20131–20163 RSC.
- H. Jung, S. Choung and J. W. Han, Nanoscale Adv., 2021, 3, 6797–6826 RSC.
- G. Assat and J.-M. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef.
- W. Li, J. Shi, K. H. Zhang and J. L. MacManus-Driscoll, Mater. Horiz., 2020, 7, 2832–2859 RSC.
- W. Li, H. Tian, L. Ma, Y. Wang, X. Liu and X. Gao, Mater. Adv., 2022, 3, 5598–5644 RSC.
- R. L. Bouwmeester and A. Brinkman, Rev. Phys., 2021, 6, 100056 CrossRef.
- C. K. Chan, H. Tüysüz, A. Braun, C. Ranjan, F. La Mantia, B. K. Miller, L. Zhang, P. A. Crozier, J. A. Haber, J. M. Gregoire, et al., Solar Energy for Fuels, 2015, pp. 253–324 Search PubMed.
- J. Y. Chen, L. Dang, H. Liang, W. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090–15093 CrossRef PubMed.
- H. Xiao, H. Shin and W. A. Goddard III, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5872–5877 CrossRef PubMed.
- H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2016, 138, 313–318 CrossRef PubMed.
- F.-T. Tsai, Y.-T. Deng, C.-W. Pao, J.-L. Chen, J.-F. Lee, K.-T. Lai and W.-F. Liaw, J. Mater. Chem. A, 2020, 8, 9939–9950 RSC.
- X. Wang, J. Dai, C. Zhou, D. Guan, X. Wu, W. Zhou and Z. Shao, ACS Mater. Lett., 2021, 3, 1258–1265 CrossRef.
- G. Fu, W. Li, J.-Y. Zhang, M. Li, C. Li, N. Li, Q. He, S. Xi, D. Qi and J. L. MacManus-Driscoll, et al. , Small, 2021, 17, 2006930 CrossRef PubMed.
- F. Yang and P. C. Hammel, J. Phys. D: Appl. Phys., 2018, 51, 253001 CrossRef.
- A. Cirri, J. Husek, S. Biswas and L. R. Baker, J. Phys. Chem. C, 2017, 121, 15861–15869 CrossRef CAS.
- H. Höchst, R. Patel and F. Middleton, Nucl. Instrum. Methods Phys. Res., Sect. A, 1994, 347, 107–114 CrossRef.
- F. Willems, C. Smeenk, N. Zhavoronkov, O. Kornilov, I. Radu, M. Schmidbauer, M. Hanke, C. von Korff Schmising, M. Vrakking and S. Eisebitt, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 220405 CrossRef.
- C. von Korff Schmising, D. Weder, T. Noll, B. Pfau, M. Hennecke, C. Strüber, I. Radu, M. Schneider, S. Staeck and C. M. Günther, et al. , Rev. Sci. Instrum., 2017, 88, 053903 CrossRef PubMed.
- F. Siegrist, J. A. Gessner, M. Ossiander, C. Denker, Y.-P. Chang, M. C. Schröder, A. Guggenmos, Y. Cui, J. Walowski and U. Martens, et al. , Nature, 2019, 571, 240–244 CrossRef CAS PubMed.
- S. Bandaranayake, E. Hruska, S. Londo, S. Biswas and L. R. Baker, J. Phys. Chem. C, 2020, 124, 22853–22870 CrossRef CAS.
- S. Biswas and L. R. Baker, Acc. Chem. Res., 2022, 55, 893–903 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03016d |
| This journal is © The Royal Society of Chemistry 2024 |