
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNIR-II photothermal conversion and imaging based on a cocrystal containing twisted components†
Tao
Li
ab,
Jia-Chuan
Liu
a,
En-Ping
Liu
c,
Bai-Tong
Liu
 d,
Jing-Yu
Wang
a,
Pei-Yu
Liao
a,
Jian-Hua
Jia
a,
Yuanning
Feng
*b and
Ming-Liang
Tong
*a
d,
Jing-Yu
Wang
a,
Pei-Yu
Liao
a,
Jian-Hua
Jia
a,
Yuanning
Feng
*b and
Ming-Liang
Tong
*a
aKey Laboratory of Bioinorganic and Synthetic Chemistry of Ministry of Education, School of Chemistry, Institute of Green Chemistry and Molecular Engineering, Sun Yat-Sen University, Guangzhou, Guangdong 510006, China. E-mail: tongml@mail.sysu.edu.cn
bDepartment of Chemistry and Biochemistry, The University of Oklahoma, 101 Stephenson Parkway, Norman, Oklahoma 73019, USA. E-mail: yf@ou.edu
cSchool of Materials Science and Engineering, Tianjin University, Tianjin 300072, China
dDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA
First published on 28th November 2023
Abstract
On account of the scarcity of molecules with a satisfactory second near-infrared (NIR-II) response, the design of high-performance organic NIR photothermal materials has been limited. Herein, we investigate a cocrystal incorporating tetrathiafulvalene (TTF) and tetrachloroperylene dianhydride (TCPDA) components. A stable radical was generated through charge transfer from TTF to TCPDA, which exhibits strong and wide-ranging NIR-II absorption. The metal-free TTF–TCPDA cocrystal in this research shows high photothermal conversion capability under 1064 nm laser irradiation and clear photothermal imaging. The remarkable conversion ability—which is a result of twisted components in the cocrystal—has been demonstrated by analyses of single crystal X-ray diffraction, photoluminescence and femtosecond transient absorption spectroscopy as well as theoretical calculations. We have discovered that space charge separation and the ordered lattice in the TTF–TCPDA cocrystal suppress the radiative decay, while simultaneously strong intermolecular charge transfer enhances the non-radiative decay. The twisted TCPDA component induces rapid charge recombination, while the distorted configuration in TTF–TCPDA favors an internal non-radiative pathway. This research has provided a comprehensive understanding of the photothermal conversion mechanism and opened a new way for the design of advanced organic NIR-II photothermal materials.
Introduction
Near-infrared (NIR) photothermal materials can convert1–3 absorbed NIR light into thermal energy through attenuating radiative relaxation. Various NIR-absorbing photothermal materials including inorganic materials,4–7 conjugated polymers,8–10 inorganic–organic hybrid materials11–15 and small organic molecules16–19 have been explored to promote efficient thermal production. Organic photothermal materials have been discovered as some of the most promising photothermal agents with economical cost, improved biocompatibility,20 and potential biodegradability.21 In order to synthesize high-yielding photothermal conversion materials, great efforts have been made in two aspects. One is to improve22 NIR absorption capacity by expanding the molecular π-conjugated area or connecting electron donor–acceptor (D–A) fragments covalently. The other one is to attenuate23–25 the radiative transition process by increasing the concentration of radicals or enhancing the quenching effect. It is challenging to achieve NIR-II absorption in organic photothermal materials following the traditional strategy26,27 since the narrow band gap is difficult to overcome. Compared with those focusing on visible and NIR-I light, the literature25,28 focusing on organic photothermal agents with a competent response to NIR-II light is lacking. Although high-intensity light can cause damage to skin and tissues, most photothermal materials require29 input energy far above the maximum permissible exposure (MPE, i.e., 1064 nm, 1 W cm−2). In order to solve this problem, an important goal is to investigate new building blocks combined with unique design strategies which will lead to a decrease in the required irradiation intensity so as to achieve safe phototherapy in vivo.In recent years, researchers have been interested in30–33 the design and synthesis of distorted aromatic compounds. In contrast to planar, nonplanar aromatic molecules in general are able to reduce supramolecular aggregation and increase solubility. These distorted π-conjugated molecular scaffolds enable new possibilities to direct a desired packing arrangement which entails34–38 the alteration of the physical properties, such as energy levels of the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs), charge transfer (CT) efficiency as well as electron distribution and mobility. One of them is the twisted molecule, which has received39,40 extensive attention in the field of photo-responsive organic compounds. The existence of wiggle motion in the twisted molecule leads to flexible molecular conformation which promotes stronger non-radiative transitions.
Cocrystals are obtained41–43 typically by the self-assembly of two alternative molecular building blocks, i.e., an electron-rich donor (D) and an electron-poor acceptor (A). Attributed to the adjustable energy gaps, solvent processability, and easy recyclability44 as a type of crystalline materials, cocrystals present bright prospects in the fields of field-effect transistors,45 ferroelectricity materials,46 dynamic photomechanics,47–49 and nonlinear optics.50 Because the energy absorption and transfer can be adjusted through changing their constituting components, cocrystals have been applied in photothermal conversion. Hu et al. reported23 in 2018 a cocrystal containing two molecular components based on D–A interactions to prepare NIR-I photothermal materials. There is a lack of literature which report25 the development of cocrystals with satisfactory NIR-II-absorbing performance. To develop cocrystals that are capable of fulfilling the NIR-II photothermal conversion requires overcoming the current design bottleneck.
Herein, a D–A type molecule, namely TTF–TCPDA, is designed based on a tetrathiafulvalene (TTF) donor and a 1,6,7,8-tetrachloro-3,4,9,10-perylenetetracarboxylic dianhydride (TCPDA) acceptor as the components. Substituted Cl atoms on each side of the perylene backbone repel each other, inducing a twisted conformation in the cocrystal. Single-crystal X-ray diffraction (SC-XRD) and theoretical calculation reveal strong intermolecular interactions and unique spin states in TTF–TCPDA, respectively. The TTF component tends to transfer one electron intermolecularly to its adjacent TCPDA unit which contains a twisted π-conjugated backbone, forming a stable radical cation. The stable radical is generated via CT in this D–A system, resulting in strong and wide-ranging NIR-II absorption. According to the characterization of ultrafast transient absorption (TA), photoluminescence spectroscopy and theoretical calculations, the photothermal performance depends on presumably the twisted molecular backbone that enables efficient space charge separation and inhibits radiative recombination. The twisted conformation in the TTF–TCPDA cocrystal is conducive to the realization of internal non-radiative pathways.
Results and discussion
(Super)structural determination
The solid-state superstructure of the TTF–TCPDA cocrystal was characterized (Fig. 1 and Table S1†) by SC-XRD. The TCPDA backbone is twisted (Scheme S4†) through its long axis with a torsional angle of 34° between two dichloronaphthalic anhydride units. Each TCPDA interacts (Fig. 1a) with four neighbouring TTF components through four [C–H⋯O] intermolecular hydrogen bonds (2.61 Å), extending into a two-dimensional (2D) layer. The alternative arrangement of the interlayer superstructure is observed (Fig. 1b) in the TTF–TCPDA cocrystal. The centroid-to-centroid distance between two donors is determined to be 7.35 Å, whereas it is 8.02 Å between two dichloronaphthalic anhydride acceptors. The highly ordered TTF–TCPDA cocrystal consists of TCPDA and TTF in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio. Each TCPDA faces two TTFs, which are axis-symmetrically located in parallel at both sides of TCPDA. The 2D layers are stacked (Fig. 1c) in an ABAB packing mode which contains [π⋯π] interactions between TCPDA and TTF components. The face-to-face distance between TTF and dichloronaphthalic anhydride units is ∼3.6 Å (Fig. 1d), while the dihedral angle is less than 3°. These parameters are favourable for D–A interactions. The phase purity of products is confirmed (Fig. S2†) by powder X-ray diffraction (PXRD), in which the positions of diffraction peaks differ (Fig. S3†) significantly from those in either TTF or TCPDA alone.
:2 ratio. Each TCPDA faces two TTFs, which are axis-symmetrically located in parallel at both sides of TCPDA. The 2D layers are stacked (Fig. 1c) in an ABAB packing mode which contains [π⋯π] interactions between TCPDA and TTF components. The face-to-face distance between TTF and dichloronaphthalic anhydride units is ∼3.6 Å (Fig. 1d), while the dihedral angle is less than 3°. These parameters are favourable for D–A interactions. The phase purity of products is confirmed (Fig. S2†) by powder X-ray diffraction (PXRD), in which the positions of diffraction peaks differ (Fig. S3†) significantly from those in either TTF or TCPDA alone.
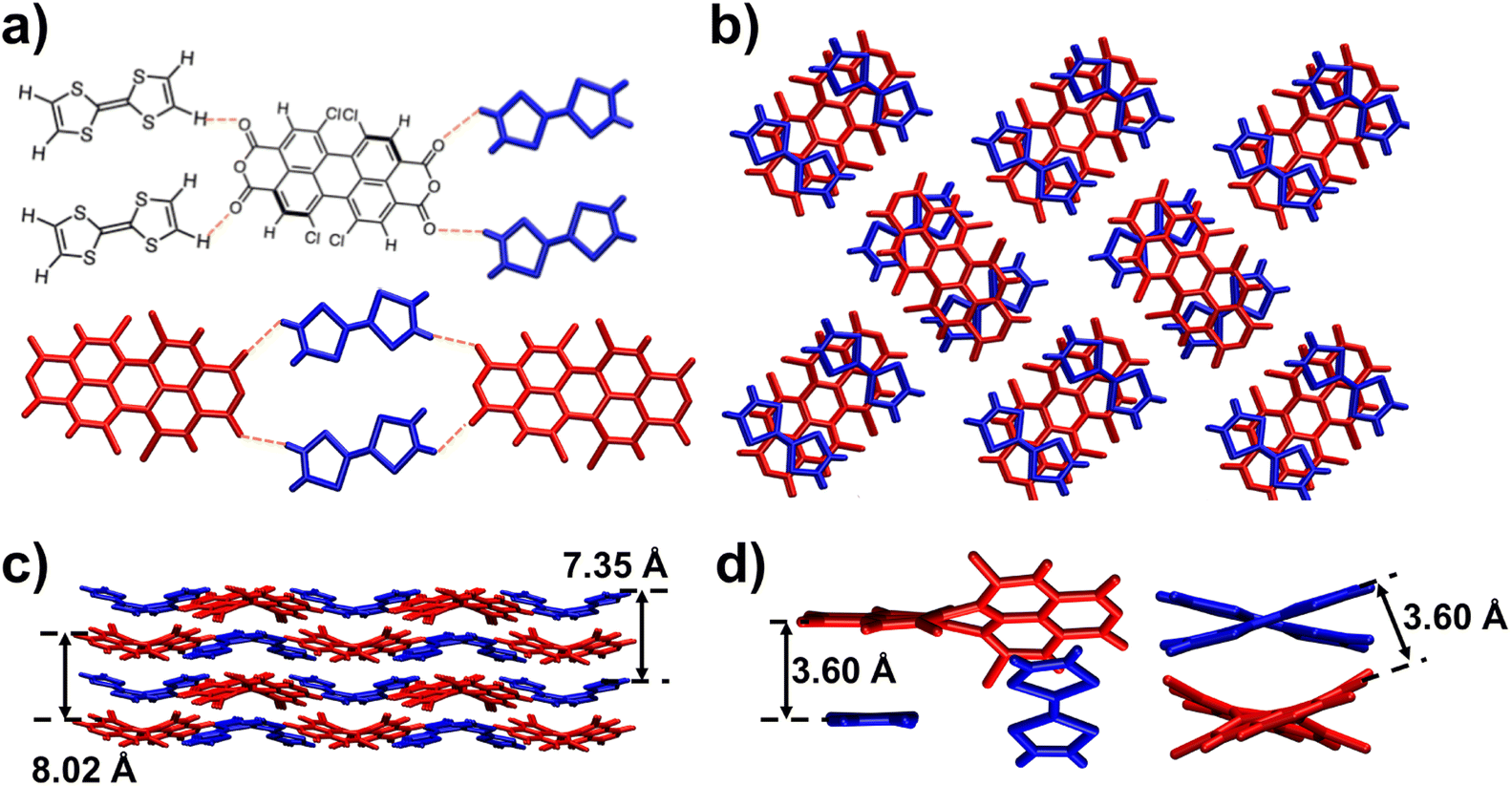 | ||
| Fig. 1 XRD superstructure of the TTF–TCPDA cocrystal. (a) Chemical structures and tubular representations of TTF and TCPDA components, while showing the intermolecular hydrogen bonds in the cocrystal. (b) A bilayer stick representation of the TTF and TCPDA components in the cocrystal indicating the face-to-face overlaying between TTF and TCPDA components. (c) ABAB packing mode viewed along the b-axis. (d) Stick representation of 2TTF–TCPDA groups indicating the twisted backbone of the TCPDA and the [π⋯π] interaction. | ||
Detailed analyses of the geometry and bond parameters resulting from SC-XRD crystallography give us detailed information on the intermolecular interaction. Instead of (Scheme S1†) a planar structure, the TTF unit exhibits (Scheme S2†) a torsional angle of 2.7° between two thiophenes in the TTF–TCPDA cocrystal. The central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and average C–S bond distances are 1.32 and 1.72 Å, respectively, whereas the bond length distortion for both left and right pentagons rings is distributed asymmetrically. These bond parameters are inconsistent with reported51–54 values of neutral TTF, suggesting the occurrence of the radical cation state. Bond lengths and angles in the TCPDA component are changed in the cocrystal—i.e., the distortion of the central hexagonal ring of TCPDA is reduced, and the torsional angle is reduced from 41.9 (Scheme S3†) to 34.3° (Scheme S4†), revealing the strong [π⋯π] stacking between D and A fragments in TTF–TCPDA. This angle decrease indicates that the hexagonal ring in the core of TCPDA changes55 from helical to planar aromaticity. We hypothesize that the CT process occurs during the assembly process of TTF–TCPDA cocrystals, which induces the deformation of TTF through partial CT and generates TTF˙+ radical cations.
C and average C–S bond distances are 1.32 and 1.72 Å, respectively, whereas the bond length distortion for both left and right pentagons rings is distributed asymmetrically. These bond parameters are inconsistent with reported51–54 values of neutral TTF, suggesting the occurrence of the radical cation state. Bond lengths and angles in the TCPDA component are changed in the cocrystal—i.e., the distortion of the central hexagonal ring of TCPDA is reduced, and the torsional angle is reduced from 41.9 (Scheme S3†) to 34.3° (Scheme S4†), revealing the strong [π⋯π] stacking between D and A fragments in TTF–TCPDA. This angle decrease indicates that the hexagonal ring in the core of TCPDA changes55 from helical to planar aromaticity. We hypothesize that the CT process occurs during the assembly process of TTF–TCPDA cocrystals, which induces the deformation of TTF through partial CT and generates TTF˙+ radical cations.
Physical characterization
TTF–TCPDA crystalline samples show (Fig. S4†) a color darker than the amorphous TCPDA solid. Solid-state UV-Vis-NIR absorption analyses of TTF, TCPDA and TTF–TCPDA were carried out (Fig. 2a) to investigate this color difference. The TTF–TCPDA cocrystal exhibits (Fig. S5†) a broad absorption band in the NIR-II region, which results presumably from the occurrence of radical cationic (TTF˙+) and CT from TTF to TCPDA. To confirm the formation of radicals, electron paramagnetic resonance (EPR) and electron spin resonance (ESR) spectroscopies were performed. The EPR signal of TTF–TCPDA (Fig. 2b) with a factor of 2.0033 shows good stability in air (Fig. S6†) and under light (Fig. S7†), whereas no significant signal is observed in the ESR spectrum of individual TTF or TCPDA. In order to further understand the proposed radical generation mechanism, cyclic voltammetry (CV) was carried out (Fig. 2c) for TTF, TCPDA, and TTF–TCPDA. The CV plot of TTF–TCPDA exhibits three redox potential signals located at 0.17, 0.46, and −0.69 V (vs. Ag/Ag+), which can be attributed to TTF/TTF˙+, TTF˙+/TTF2+, and TCPDA/TCPDA˙− redox couples, respectively. Compared with the redox state of TTF and TCPDA alone (Fig. S8†), the potentials of TCPDA in TTF–TCPDA cocrystals (Fig. S9†) do not shift significantly, whereas the oxidative signal of TTF (TTF/TTF˙+) upshifts 0.19 V. This phenomenon is further validated by X-ray photoelectron spectroscopy (XPS) analysis. The S 2p core-level XPS spectrum of TTF–TCPDA can be deconvoluted (Fig. 2d) into two bands that are assigned51,56 to TTF0 (164.0 eV) and TTF˙+ (165.1 eV), respectively. These analyses indicate that TTF undergoes a CT process and forms radical cationic TTF˙+ during the assembly.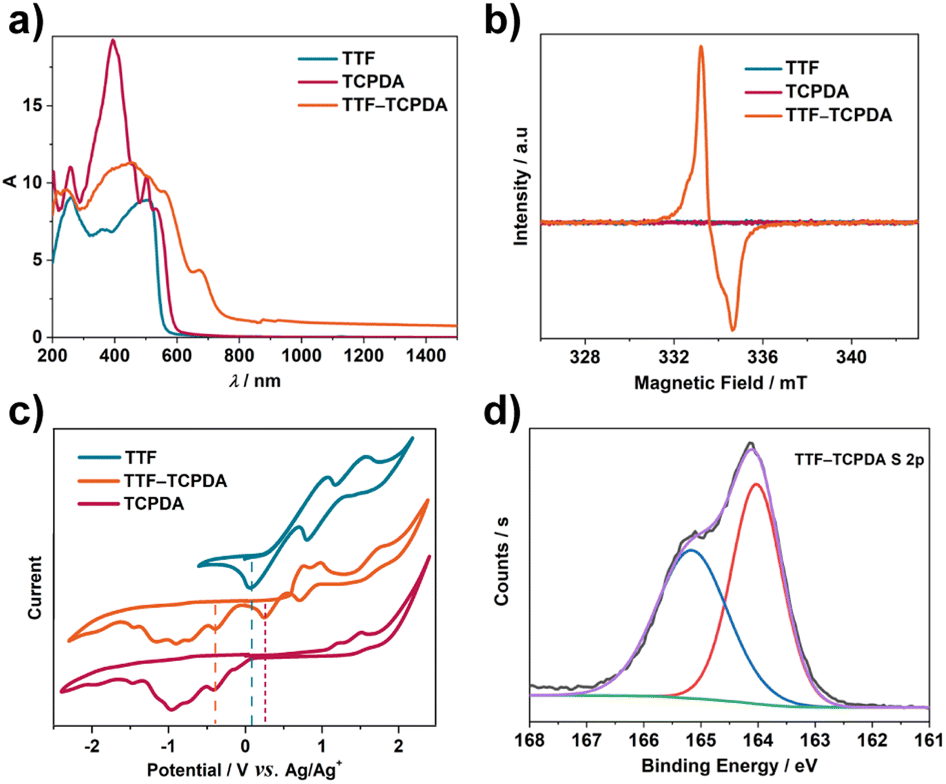 | ||
| Fig. 2 (a) UV-Vis-NIR diffuse reflectance spectra, (b) overlaid solid-state EPR spectra, (c) cyclic voltammetry graph at the same scan rates (0.03 mV s−1), and (d) fitting of XPS spectra of S 2p orbitals of TTF–TCPDA cocrystals. | ||
Theoretical calculation
To attain a deeper comprehension of the geometric and electronic properties of the TTF–TCPDA cocrystal and its components, density functional theory (DFT) calculations were conducted (Fig. 3) at the B3LYP/6-311+G** D3(BJ) level. Visualized diagrams demonstrate that the HOMOs of TTF–TCPDA are distributed mainly on the electron donating TTF component with an energy level of −5.20 eV, whereas the LUMOs of TTF–TCPDA are localized mostly on the electron accepting TCPDA component with an energy level of −4.27 eV. The calculated HOMO–LUMO energy gap (ΔEgap) of TTF–TCPDA is 0.94 eV, which is smaller than that of individual components. During the formation of cocrystals of TTF and TCPDA, their bond length and angle undergo distortion, which matched with calculated results. The result shows that (Fig. S10†) that the HOMO level of the distorted TTF increased by 0.16 eV, whereas the LUMO energy level of the distorted TCPDA decreased by 0.04 eV. The change in energy on the orbital indicates the occurrence of intermolecular CT between D and A, which further demonstrates that TTF donates one electron to form radical cationic TTF˙+. In addition, the interaction energy (ΔEint) is calculated to be −19.22 kcal mol−1 for the TTF–TCPDA D–A system (Table S2†), confirming the key role of π⋯π stacking in structural stability. According to the calculated electrostatic potential distribution (Fig. S11†) on the molecular surface, TCPDA is relatively easy to attract electrons, whereas TTF is prone to repel electrons. The excited state calculation using time-dependent density functional theory (TD-DFT) demonstrates (Fig. S12 and Table S3†) that the sharp band in the visible region and the broad band in the NIR region involve the transition from the donor's to acceptor's orbital, confirming intermolecular CT in the cocrystal. The calculated absorption spectrum of the TTF–TCPDA matches (Fig. S13†) with the experimental absorption spectrum. The consistent calculation provides theoretical evidence for the occurrence of CT.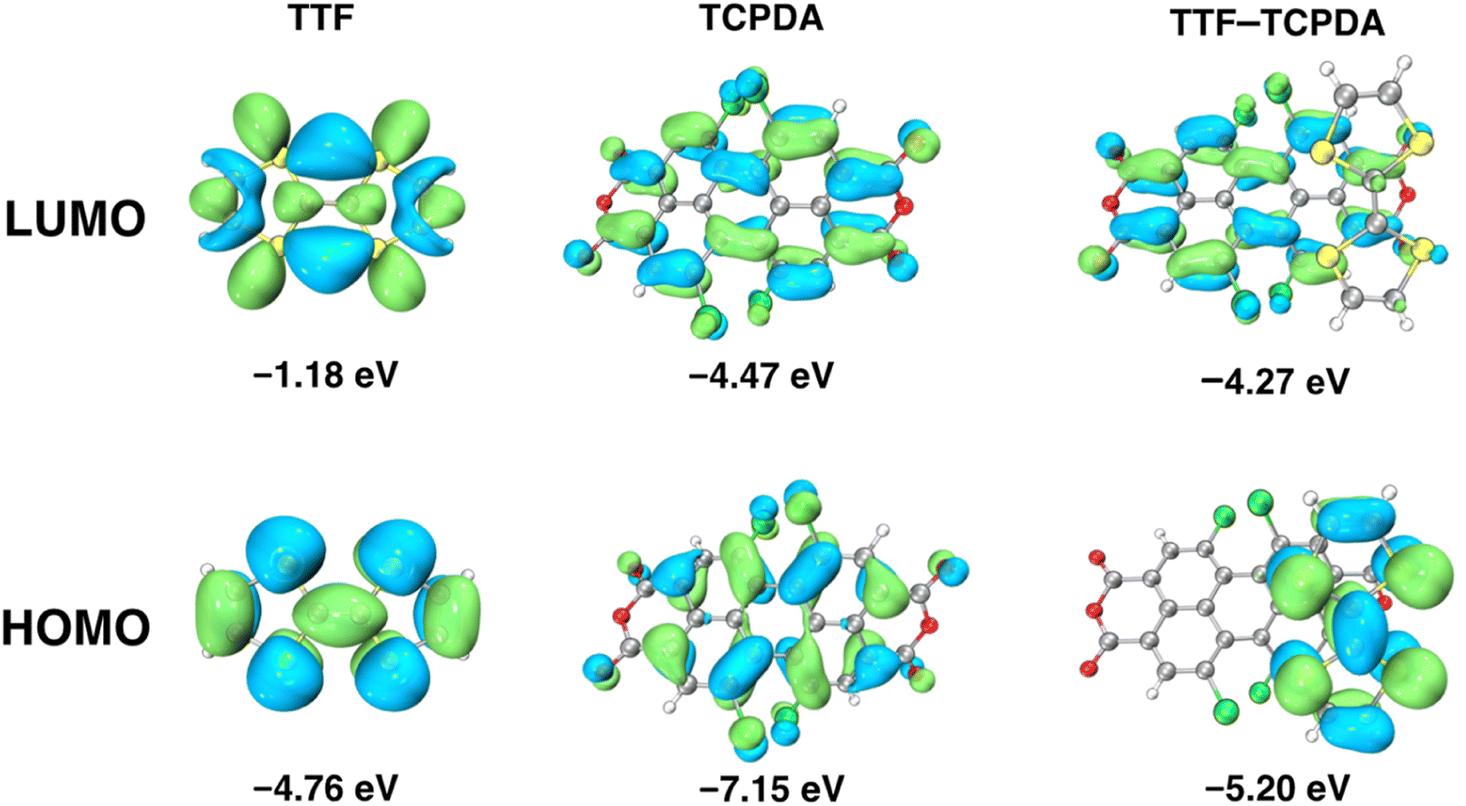 | ||
| Fig. 3 Frontier molecular orbitals of TTF, TCPDA and TTF–TCPDA along with their corresponding energy gaps, i.e., 5.79, 2.68 and 0.93 eV, respectively. | ||
NIR-II photothermal conversion measurement
Inspired by the wide absorption spectrum (Fig. S5†) and excellent thermal stability (Fig. S14†), we explored the photothermal conversion performance of the TTF–TCPDA cocrystal by recording (Fig. 4a) the temperature changes under an IR camera over time. Under the irradiation of a 1064 nm laser (0.1 W cm−2) for 20 s, the temperature of TTF–TCPDA powder increases (ΔT = 35 °C) dramatically, from 22 to 57 °C. When the laser intensity increases to 0.5 W cm−2, the ΔT boosts to 145 °C. In comparison, the temperature rises of either TTF or TCPDA as well as their simple mixture are negligible (Fig. S15†) under the same conditions.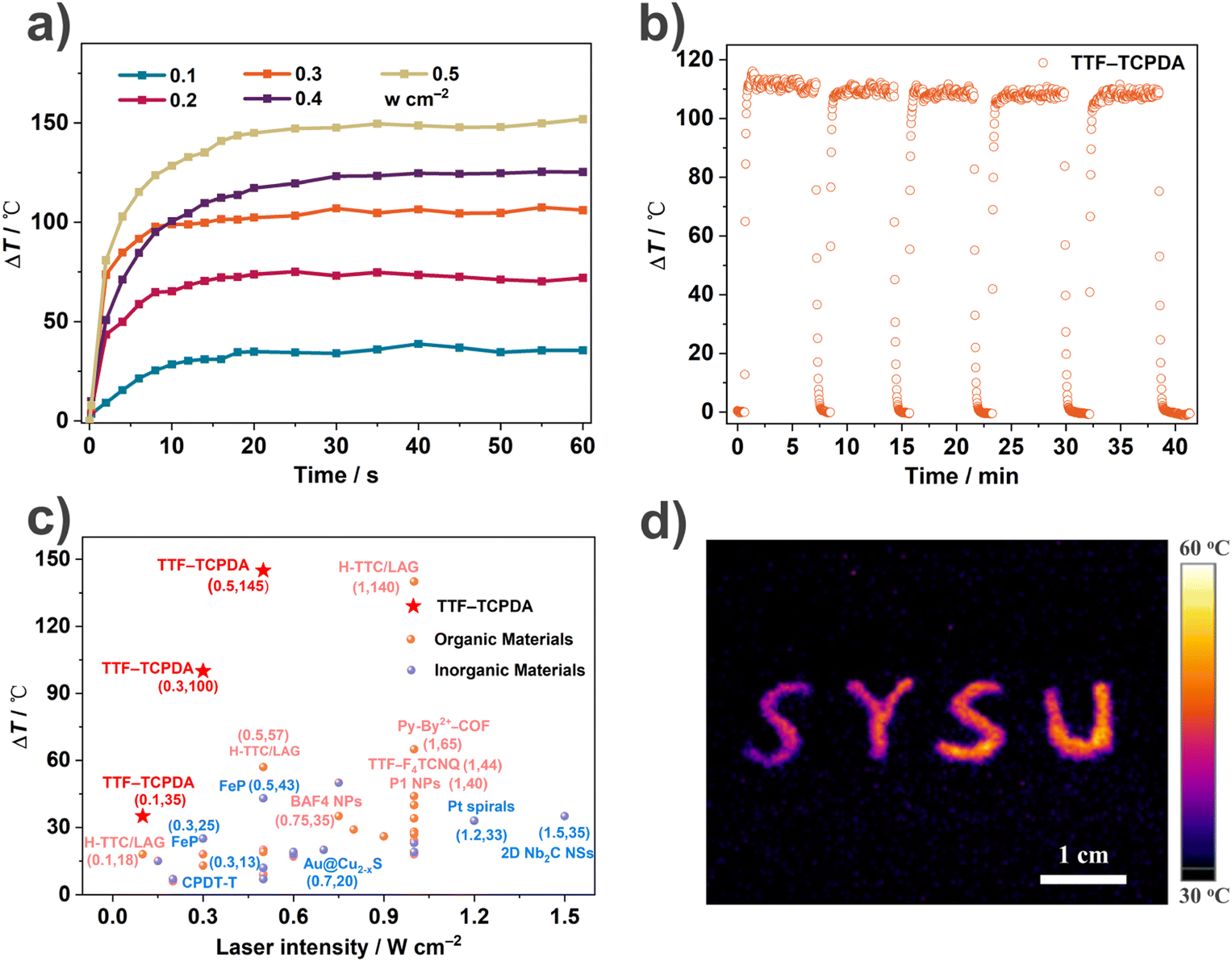 | ||
| Fig. 4 (a) Photothermal conversion behaviour of TTF–TCPDA powder under the irradiation of a 1064 nm laser with different intensities (0.1 to 0.5 W cm−2). (b) Anti-photobleaching properties of TTF–TCPDA powder during five cycles of heating–cooling processes under 1064 nm laser (0.5 W cm−2) irradiation. (c) Temperature rises under 1064 nm laser irradiation of various materials in previous literature are summarized and compared with our TTF–TCPDA cocrystal which exhibits superior photothermal conversion properties. (d) Thermographic images of TTF–TCPDA powder. | ||
The photothermal effect depends on NIR laser intensity linearly (Fig. 4a) from 0.1 to 0.5 W cm−2, revealing the controllability of photothermal behaviour. After five irradiation cycles, imperceptible degradation of the photothermal performance of TTF–TCPDA powder is observed (Fig. 4b), indicating its satisfactory thermal and photostability. The photothermal conversion of TTF–TCPDA powder can be recorded using thermographic (Fig. 4d) cameras under the irradiation of a 1064 nm laser (0.1 W cm−2). Compared with (Fig. 4c and Table S4†) other reported NIR-II photothermal materials, the TTF–TCPDA cocrystal has remarkable performance in terms of temperature rises under 1064 nm laser irradiation with different intensities. The conversion performance is still competitive when the irradiation wavelength is blue-shifted (Fig. S16†) to 660, 808 or 980 nm.
Mechanism analysis
The outstanding photothermal behaviour of TTF–TCPDA including intense absorption in the NIR-II region and high photothermal conversion efficiency encouraged deeper investigation of the mechanism. In order to demonstrate the radical formation resulting from intermolecular CT during the assembly, the spin density distribution of TTF–TCPDA is calculated (Fig. S17†), illustrating that it delocalized mainly on central C and S atoms in TTF. The atom sustaining the spin is located in the core of the molecule, confirming that the spin state of solid-state TTF–TCPDA depends on intermolecular D–A interactions. The calculated results are consistent with the CV and XPS data (Fig. 2c and d). The band gap between the lowest singly unoccupied molecular orbital (SUMO) and the singly occupied molecular orbital (SOMO) of the organic radical is constantly smaller than that between the LUMO and HOMO of the closed-shell molecule,21,57,58 which is further reduced through the radical formation driven by intermolecular CT. This phenomenon indicates presumably the strong π⋯π interaction between TTF and TCPDA components. The energy gap and radical intermediate in TTF–TCPDA play a synergistic role in promoting the NIR-II bio-window absorption.Femtosecond transient absorption (fs-TA) characterization was carried out (Fig. 5a) to investigate the excited state dynamics of the radical TTF–TCPDA, especially for charge separation and recombination. Three broad absorption bands are observed and assigned (Fig. 5b) to the excited state absorption. There is an excited state absorption band in the range of 800–1200 nm originating from the absorption of the TTF˙+ radical cation and belonging to23,59 the CT1 excited state. TA kinetics have no essential change (Fig. S18 and S19†) at different detection wavelengths in the range of 800–1200 nm. Kinetic analysis shows (Fig. 5c) that the TA spectrum decays with three components with time constants of 2.8 ± 0.4, 91 ± 1, and 922 ± 30 ps, and the evolution-associated spectra show no change in shape during these processes. This kinetic analysis suggests that the CT exciton that is formed immediately upon excitation can migrate through the cocrystal while simultaneously experiencing CT exciton annihilation and charge recombination. The longest-lived population still undergoes sub-nanosecond charge recombination, which enables the molecule to complete the energy dissipation cycle of the excited state rapidly following excitation without trapping the energy in metastable states such as localized triplet excitons, which is favorable for the photothermal conversion.
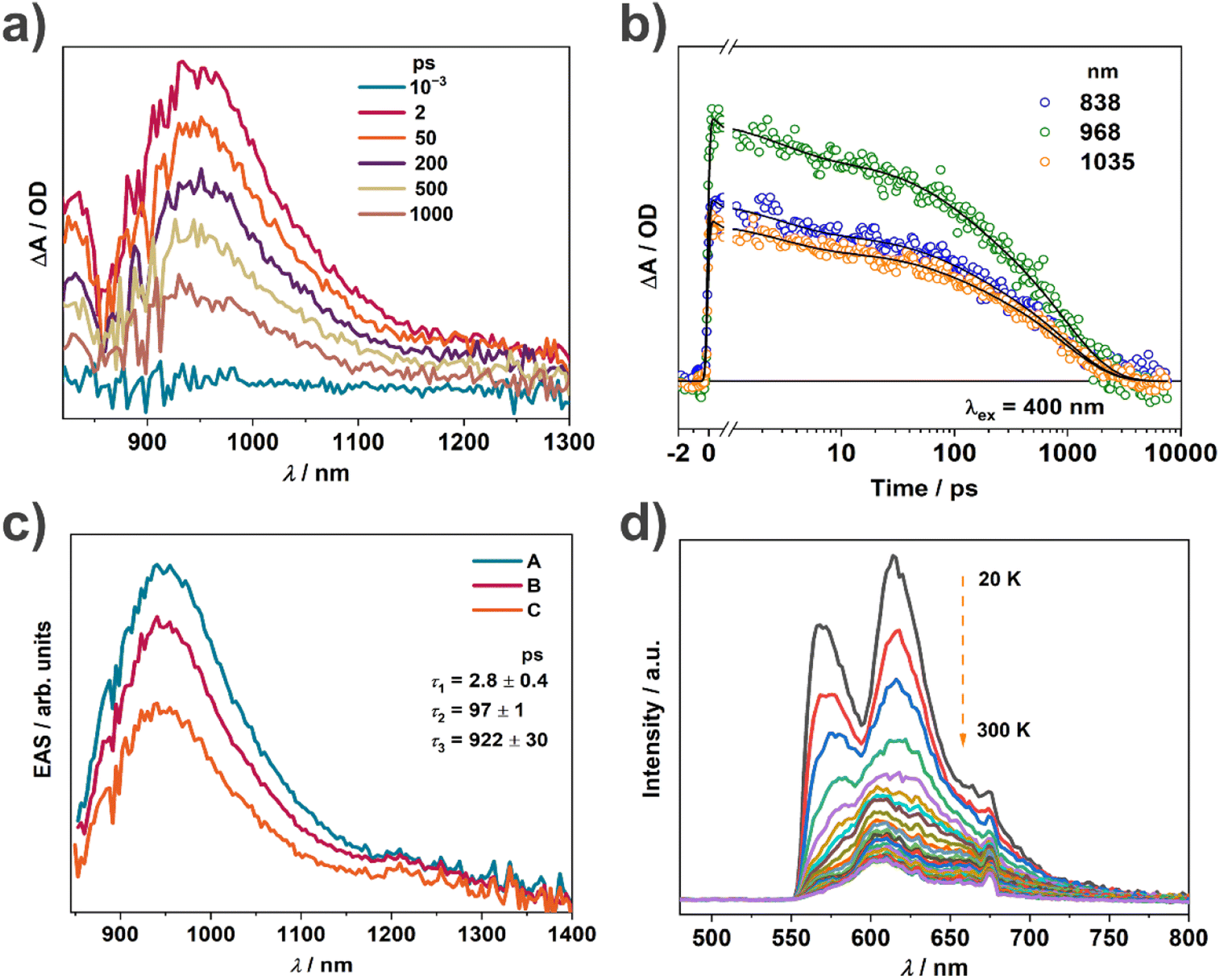 | ||
| Fig. 5 (a) Overlaid femtosecond transient absorption (fs-TA) spectra of TTF–TCPDA after laser irradiation for 10−3, 2, 50, 200, 500 and 1000 ps, respectively. (b) Fs-TA kinetic fitting results of TTF–TCPDA at 839, 968 and 1035 nm, respectively. (c) Evolution-associated spectra of the fs-TA data following kinetic analysis. (d) Variable temperature fluorescence spectra (λex = 365 nm) of TTF–TCPDA from 20 to 300 K. | ||
According to the energy gap law, a small energy gap favors the non-radiative decay process, which weakens the fluorescence intensity. The TTF–TCPDA cocrystal exhibits weak fluorescence with low quantum efficiency, which is quenched (Fig. S20†) upon aggregation almost completely. This results from the synergistic effect of TTF and TCPDA components in TTF–TCPDA cocrystals as a result of the face-to-face packing mode. The dense stacking superstructure enhances60,61 (i) the intramolecular and intermolecular interactions, (ii) the π-electron delocalization and (iii) radical stability, resulting in diminishing of the photon emission process. Variable temperature fluorescence experiments indicate (Fig. 5d) that the fluorescence intensity increases when temperature decreases. The vibration and rotation of molecules are restricted significantly, leading to the dominance of radiative decay at lower temperatures. These results validate that intramolecular motions play a crucial role in the non-radiative decay of TTF–TCPDA cocrystals.
We further employed DFT calculations to decipher the different conformations of TCPDA to reveal energy changes between different twisted conformations. We conjectured that the main molecular motion of TCPDA in the free state was dynamic twisting motion because of the conformational flexibility in the backbone. TCPDA can undergo multistep conformational transformations and is conducive to the dissipation of excited energy through non-radiative relaxation. To confirm this hypothesis, we used the intrinsic reaction coordinate (IRC) to search (Fig. 6a) for the energy landscape of the conformational changing process of the isolated optimized TCPDA geometry. Changes in the torsional angle between two dichloronaphthalic anhydride units result in different energy levels. Transient state geometry ③ was found (Fig. 6b) to be 55 and 102 kJ mol−1 higher than geometry ① and ②, respectively. The energy landscape demonstrated that the TCPDA is prone to undergo successive conformational distortion processes. The crystal structure of free TCPDA appeared to have no stationary points on the IRC path. We hypothesize that the twisted TCPDA in the cocrystal is revealed partially by the torsional angle of 2.7° between two thiophene rings in TTF. The continuous energy dissipation resulting from multiple twisted conformations in the cocrystal enhances the internal non-radiative pathway, which increases presumably the photothermal conversion efficiency.
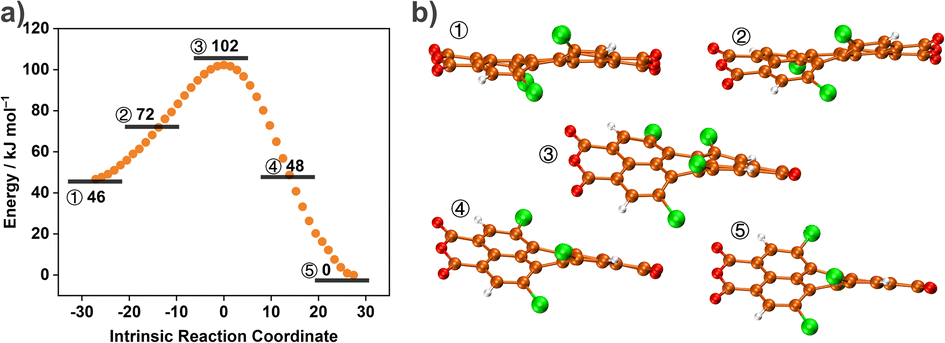 | ||
| Fig. 6 (a) Energy landscape for the conformational transformation of the TCPDA at the B3LYP/def2-SVP D3(BJ) level. (b) Stick-and-ball representations of calculated TCPDA conformations at five representative intrinsic reaction coordinates (IRC). | ||
Conclusions
We propose a supramolecular strategy to incorporate TTF and TCPDA, obtaining a TTF–TCPDA cocrystal. Stable TTF radicals are generated in TTF–TCPDA through charge transfer from the TTF donor to TCPDA acceptor, exhibiting paramagnetic signals and wide-range NIR-II absorption. Under 1064 nm laser irradiation (0.1 W cm−2), the temperature of TTF–TCPDA powder increases significantly and quickly—i.e., ΔT = 35 °C in 20 s. Compared with other photothermal materials, TTF–TCPDA exhibits superior conversion efficiency under low-intensity irradiation, capability for NIR-II imaging and a linearly controllable photothermal behaviour. No photothermal conversion occurs in either components or their mixture under the same conditions. Single crystal X-ray diffraction analysis, theoretical calculation, ultrafast transient absorption, and photo-luminescence spectroscopy demonstrated that the effective photothermal conversion is due to the twisted components. Introduction of twisted structures elevates space charge separation and inhibits radiative recombination. The twisted conformation and intermolecular interaction in TTF–TCPDA cocrystals promote non-radiative relaxation by accelerating charge recombination and facilitating intermolecular charge transfer. To the best of our knowledge, this is the first example of using multiple twisted conformation-induced components in a cocrystal to increase the efficiency of photothermal conversion. The facile preparation of the TTF–TCPDA material and deep analysis of the photothermal properties provide a promising platform and new ideas for rational design of radical-based photothermal materials and a comprehensive understanding of the photothermal conversion mechanism.Data availability
The data that support the findings of this study are available in the main text and the ESI.†Author contributions
Prof. M.-L. T. guided the project. Dr Y. F. provided constructive suggestions. T. L. designed and carried out the synthesis and characterization in this work. J.-C. L. conducted the DFT calculations. Dr E.-P. L. conducted the photothermal measurement. Dr B.-T. L. conducted the experiments on structural determination. J.-Y. W. conducted electrochemical measurements. Y. F. assisted in the fluorescence titration. P.-Y. L. conducted the variable temperature fluorescence measurements. T. L. drafted the manuscript. J.-C. L., Dr B.-T. L., Prof. J.-H. J. Dr Y. F. and Prof. M.-L. T. commented on the manuscript. T. L. and Dr Y. F. responded to the reviewers and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Ryan M. Young at Northwestern University for his enormous contribution to the data analysis on transient absorption spectra for the revised manuscript. The authors thank Mr Zihao Wen for his assistance with the calculated IRC data. We gratefully acknowledge the financial support from the National Natural Science Foundation of China (grant no. 22071274, 22131011, and 21821003) and the Pearl River Talent Plan of Guangdong (2017BT01C161). Y. F. would like to thank the University of Oklahoma for generous financial support as startup funds.Notes and references
- Y. Yang, Y. Yang, S. Chen, Q. Lu, L. Song, Y. Wei and X. Wang, Nat. Commun., 2017, 8, 1559 CrossRef PubMed.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. S. Casalongue, V. Daniel and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS PubMed.
- Y. Lu, D. Liu, Y.-J. Lin and G.-X. Jin, Chem. Sci., 2020, 11, 11509–11513 RSC.
- Z. Yu, W. K. Chan, Y. Zhang and T. T. Y. Tan, Biomaterials, 2021, 269, 120459 CrossRef CAS PubMed.
- J. Chen, Z. Ye, F. Yang and Y. Yin, Small Sci., 2021, 1, 2000055 CrossRef CAS.
- Y. Lyu, J. Li and K. Pu, Small Methods, 2019, 3, 1900553 CrossRef CAS.
- Y. Liu, P. Bhattarai, Z. Dai and X. Chen, Chem. Soc. Rev., 2019, 48, 2053–2108 RSC.
- Z. Huang, Y.-H. Luo, W.-Y. Geng, Y. Wan, S. Li and C.-S. Lee, Small Methods, 2021, 5, 2100036 CrossRef CAS PubMed.
- Z. Mi, P. Yang, R. Wang, J. Unruangsri, W. Yang, C. Wang and J. Guo, J. Am. Chem. Soc., 2019, 141, 14433–14442 CrossRef CAS PubMed.
- X. Tang, Z. Chen, Q. Xu, Y. Su, H. Xu, S. Horike, H. Zhang, Y. Li and C. Gu, CCS Chem., 2022, 4, 2842–2853 CrossRef CAS.
- X. Ye, L. H. Chung, K. Li, S. Zheng, Y. L. Wong, Z. Feng, Y. He, D. Chu, Z. Xu, L. Yu and J. He, Nat. Commun., 2022, 13, 6116 CrossRef CAS PubMed.
- Y. Lu, D. Liu, Y.-J. Lin and G.-X. Jin, Chem. Sci., 2020, 11, 11509–11513 RSC.
- Q. Ma, P. Yin, M. Zhao, Z. Luo, Y. Huang, Q. He, Y. Yu, Z. Liu, Z. Hu, B. Chen and H. Zhang, Adv. Mater., 2019, 31, 1808249 CrossRef PubMed.
- B. Lu, Y. Chen, P. Li, B. Wang, K. Mullen and M. Yin, Nat. Commun., 2019, 10, 767 CrossRef CAS PubMed.
- L.-L. Dang, T.-T. Li, T.-T. Zhang, Y. Zhao, T. Chen, X. Gao, L.-F. Ma and G.-X. Jin, Chem. Sci., 2022, 13, 5130–5140 RSC.
- C. Xu, R. Ye, H. Shen, J. W. Y. Lam, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202204604 CrossRef CAS PubMed.
- G. Chen, J. Sun, Q. Peng, Q. Sun, G. Wang, Y. Cai, X. Gu, Z. Shuai and B. Z. Tang, Adv. Mater., 2020, 32, e1908537 CrossRef PubMed.
- J.-S. Ni, X. Zhang, G. Yang, T. Kang, X. Lin, M. Zha, Y. Li, L. Wang and K. Li, Angew. Chem., Int. Ed., 2020, 59, 11298–11302 CrossRef CAS PubMed.
- S. Gao, G. Wei, S. Zhang, B. Zheng, J. Xu, G. Chen, M. Li, S. Song, W. Fu, Z. Xiao and W. Lu, Nat. Commun., 2019, 10, 2206 CrossRef PubMed.
- D. Xi, M. Xiao, J. Cao, L. Zhao, N. Xu, S. Long, J. Fan, K. Shao, W. Sun, X. Yan and X. Peng, Adv. Mater., 2020, 32, 1907855 CrossRef CAS PubMed.
- L. Zhao, Y. Liu, R. Xing and X. Yan, Angew. Chem., Int. Ed., 2020, 59, 3793–3801 CrossRef CAS PubMed.
- G. Wu, F. Li, B. Tang and X. Zhang, J. Am. Chem. Soc., 2022, 144, 14962–14975 CrossRef CAS PubMed.
- Y. Wang, W. Zhu, W. Du, X. Liu, X. Zhang, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2018, 57, 3963–3967 CrossRef CAS PubMed.
- T. Yan, Y.-Y. Li, J. Su, H.-Y. Wang and J.-L. Zuo, Chem.–Eur. J., 2021, 27, 11050–11055 CrossRef CAS PubMed.
- S. Tian, H. Bai, S. Li, Y. Xiao, X. Cui, X. Li, J. Tan, Z. Huang, D. Shen, W. Liu, P. Wang, B. Z. Tang and C. S. Lee, Angew. Chem., Int. Ed., 2021, 60, 11758–11762 CrossRef CAS PubMed.
- L. Wu, F. Li, Y. Rao, B. Wen, L. Xu, M. Zhou, T. Tanaka, A. Osuka and J. Song, Angew. Chem., Int. Ed., 2019, 58, 8124–8128 CrossRef CAS PubMed.
- Z. Chen, Y. Liu, W. Wagner, V. Stepanenko, X. Ren, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 5729–5733 CrossRef CAS PubMed.
- S. Zhang, W. Guo, J. Wei, C. Li, X.-J. Liang and M. Yin, ACS Nano, 2017, 11, 3797–3805 CrossRef CAS PubMed.
- C. Xu and K. Pu, Chem. Soc. Rev., 2021, 50, 1111–1137 RSC.
- M. Juríček, N. L. Strutt, J. C. Barnes, A. M. Butterfield, E. J. Dale, K. K. Baldridge, J. F. Stoddart and J. S. Siegel, Nat. Chem., 2014, 6, 222–228 CrossRef PubMed.
- T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2016, 138, 3587–3595 CrossRef CAS PubMed.
- Y.-Z. Tan, B. Yang, K. Parvez, A. Narita, S. Osella, D. Beljonne, X. Feng and K. Müllen, Nat. Commun., 2013, 4, 2646 CrossRef PubMed.
- M. Gsänger, J. H. Oh, M. Könemann, H. W. Höffken, A.-M. Krause, Z. Bao and F. Würthner, Angew. Chem., Int. Ed., 2010, 49, 740–743 CrossRef PubMed.
- Z. Sun, W. Fan, Y. Han, W. Yuan, Y. Ni, J. Wang, H. Wei, Y. Zhao, Z. Sun and J. Wu, Chem. Sci., 2023, 14, 7922–7927 RSC.
- A. Artigas, D. Hagebaum-Reignier, Y. Carissan and Y. Coquerel, Chem. Sci., 2021, 12, 13092–13100 RSC.
- M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267–276 CrossRef CAS.
- T. Liu, J. Yang, F. Geyer, F. S. Conrad-Burton, R. Hernández Sánchez, H. Li, X. Zhu, C. P. Nuckolls, M. L. Steigerwald and S. Xiao, Angew. Chem., Int. Ed., 2020, 59, 14303–14307 CrossRef CAS PubMed.
- Z. Jin, Q. Cheng, S. T. Bao, R. Zhang, A. M. Evans, F. Ng, Y. Xu, M. L. Steigerwald, A. E. McDermott, Y. Yang and C. Nuckolls, J. Am. Chem. Soc., 2022, 144, 13973–13980 CrossRef CAS PubMed.
- Y. Li, A. Yagi and K. Itami, J. Am. Chem. Soc., 2020, 142, 3246–3253 CrossRef CAS PubMed.
- C. Schaack, A. M. Evans, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2022, 144, 42–51 CrossRef CAS PubMed.
- L. Sun, W. Zhu, X. Zhang, L. Li, H. Dong and W. Hu, J. Am. Chem. Soc., 2021, 143, 19243–19256 CrossRef CAS PubMed.
- J. Zhang, W. Xu, P. Sheng, G. Zhao and D. Zhu, Acc. Chem. Res., 2017, 50, 1654–1662 CrossRef CAS PubMed.
- D. Yan and D. G. Evans, Mater. Horiz., 2014, 1, 46–57 RSC.
- D. Yan, Chem.–Eur. J., 2015, 21, 4880–4896 CrossRef CAS PubMed.
- P. Yu, Y. Zhen, H. Dong and W. Hu, Chem, 2019, 5, 2814–2853 CAS.
- A. S. Tayi, A. K. Shveyd, A. C.-H. Sue, J. M. Szarko, B. S. Rolczynski, D. Cao, T. J. Kennedy, A. A. Sarjeant, C. L. Stern, W. F. Paxton, W. Wu, S. K. Dey, A. C. Fahrenbach, J. R. Guest, H. Mohseni, L.-X. Chen, K.-L. Wang, J. F. Stoddart and S. I. Stupp, Nature, 2012, 488, 485–489 CrossRef CAS PubMed.
- S. Li, B. Lu, X. Fang and D. Yan, Angew. Chem., Int. Ed., 2020, 59, 22623–22630 CrossRef CAS PubMed.
- X.-G. Yang, Z.-M. Zhai, X.-M. Lu, L.-F. Ma and D. Yan, ACS Cent. Sci., 2020, 6, 1169–1178 CrossRef CAS PubMed.
- S. Li and D. Yan, ACS Appl. Mater. Interfaces, 2018, 10, 22703–22710 CrossRef CAS PubMed.
- Y. Wang, H. Wu, P. Li, S. Chen, L. O. Jones, M. A. Mosquera, L. Zhang, K. Cai, H. Chen, X.-Y. Chen, C. L. Stern, M. R. Wasielewski, M. A. Ratner, G. C. Schatz and J. F. Stoddart, Nat. Commun., 2020, 11, 4633 CrossRef CAS PubMed.
- J. Su, N. Xu, R. Murase, Z.-M. Yang, D. M. D'Alessandro, J.-L. Zuo and J. Zhu, Angew. Chem., Int. Ed., 2021, 60, 4789–4795 CrossRef CAS PubMed.
- J. Castells-Gil, S. Mañas-Valero, I. J. Vitórica-Yrezábal, D. Ananias, J. Rocha, R. Santiago, S. T. Bromley, J. J. Baldoví, E. Coronado, M. Souto and G. Mínguez Espallargas, Chem.–Eur. J., 2019, 25, 12636–12643 CrossRef CAS PubMed.
- M. Souto, J. Romero, J. Calbo, I. J. Vitórica-Yrezábal, J. L. Zafra, J. Casado, E. Ortí, A. Walsh and G. M. Espallargas, J. Am. Chem. Soc., 2018, 140, 10562–10569 CrossRef CAS PubMed.
- L.-S. Xie, E. V. Alexandrov, G. Skorupskii, D. M. Proserpio and M. Dinca, Chem. Sci., 2019, 10, 8558–8565 RSC.
- B.-T. Liu, X.-H. Pan, D.-Y. Zhang, R. Wang, J.-Y. Chen, H.-R. Fang and T.-F. Liu, Angew. Chem., Int. Ed., 2021, 60, 25701–25707 CrossRef CAS PubMed.
- H.-Y. Wang, J.-Y. Ge, C. Hua, C.-Q. Jiao, Y. Wu, C. F. Leong, D. M. D'Alessandro, T. Liu and J.-L. Zuo, Angew. Chem., Int. Ed., 2017, 56, 5465–5470 CrossRef CAS PubMed.
- D. Schmidt, M. Son, J. M. Lim, M.-J. Lin, I. Krummenacher, H. Braunschweig, D. Kim and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 13980–13984 CrossRef CAS PubMed.
- B. Tang, W.-L. Li, Y. Chang, B. Yuan, Y. Wu, M.-T. Zhang, J.-F. Xu, J. Li and X. Zhang, Angew. Chem., Int. Ed., 2019, 58, 15526–15531 CrossRef CAS PubMed.
- J. Guo, H. Ohkita, H. Benten and S. Ito, J. Am. Chem. Soc., 2009, 131, 16869–16880 CrossRef CAS PubMed.
- J. Liu, Y. Cui, Y. Pan, Z. Chen, T. Jia, C. Li and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202117087 CrossRef CAS PubMed.
- J. Sun, E. Zhao, J. Liang, H. Li, S. Zhao, G. Wang, X. Gu and B. Z. Tang, Adv. Mater., 2022, 34, 2108048 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2219621 and 2219622. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03532h |
| This journal is © The Royal Society of Chemistry 2024 |