
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImplementing vanadium peroxides as direct air carbon capture materials†
Eduard Garrido
Ribó‡
a,
Zhiwei
Mao‡
 a,
Jacob S.
Hirschi
a,
Taylor
Linsday
a,
Karlie
Bach
a,
Eric D.
Walter
b,
Casey R.
Simons
c,
Tim J.
Zuehlsdorff
a and
May
Nyman
*a
a,
Jacob S.
Hirschi
a,
Taylor
Linsday
a,
Karlie
Bach
a,
Eric D.
Walter
b,
Casey R.
Simons
c,
Tim J.
Zuehlsdorff
a and
May
Nyman
*a
aDepartment of Chemistry, Oregon State University, Corvallis, OR 97331, USA. E-mail: may.nyman@oregonstate.edu
bPacific Northwest National Laboratory, Environmental Molecular Sciences Laboratory, Richland, WA 99352, USA
cCAMCOR, University of Oregon, Eugene, OR 97331, USA
First published on 21st December 2023
Abstract
Direct air capture (DAC) removal of anthropogenic CO2 from the atmosphere is imperative to slow the catastrophic effects of global climate change. Numerous materials are being investigated, including various alkaline inorganic metal oxides that form carbonates via DAC. Here we explore metastable early d0 transition metal peroxide molecules that undergo stabilization via multiple routes, including DAC. Specifically here, we describe via experiment and computation the mechanistic conversion of A3V(O2)4 (tetraperoxovanadate, A = K, Rb, Cs) to first a monocarbonate VO(O2)2(CO3)3−, and ultimately HKCO3 plus KVO4. Single crystal X-ray structures of rubidium and cesium tetraperoxovanadate are reported here for the first time, likely prior-challenged by instability. Infrared spectroscopy (FTIR), powder X-ray diffraction (PXRD), 51V solid state NMR (nuclear magnetic resonance), tandem thermogravimetry-mass spectrometry (TGA-MS) along with calculations (DFT, density functional theory) all converge on mechanisms of CO2 capture and release that involve the vanadium centre, despite the end product of a 300 days study being bicarbonate and metavanadate. Electron Paramagnetic Resonance (EPR) Spectroscopy along with a wet chemical assay and computational studies evidence the presense of ∼5% adventitous superoxide, likely formed by peroxide reduction of vanadium, which also stabilizes via the reaction with CO2. The alkalis have a profound effect on the stability of the peroxovanadate compounds, stability trending K > Rb > Cs. While this translates to more rapid CO2 capture with heavier alkalis, it does not necessarily lead to capture of more CO2. All compounds capture approximately two equivalents CO2 per vanadium centre. We cannot yet explain the reactivity trend of the alkali peroxovanadates, because any change in speciation of the alkalis from reactions to product is not quantifiable. This study sets the stage for understanding and implementing transition metal peroxide species, including peroxide-functionalized metal oxides, for DAC.
Introduction
Burning fossil fuels produces high levels of greenhouse gases including CO2, which has led to alarming changes in the Earth's climate, ecosystems and geography.1,2 To mitigate the negative impacts of climate change, it is essential to transition to low emissions energy technologies, and to also decrease the current high levels of CO2 in the atmosphere. A number of technologies have been developed and deployed to decrease legacy and future CO2 levels, respectively by direct air capture (DAC), and at-the-source capture.3 Experts agree, however, that no single technology can meet all the demands of rapid and high capacity capture, energy-efficient regeneration and cyclability, and conversion of CO2 to useful products. Moreover, global, local and seasonal atmospheric differences including temperature, temperature swings, relative humidity, atmospheric pressure, and presence of competing gas phase molecules can either promote or inhibit carbon capture chemistries. Therefore, expanding our chemistry toolbox for carbon dioxide removal can supplement, enhance and diversify DAC technologies.4–7 Perhaps one of the most intriguing feature of solid phase materials for DAC is their potential to be integrated into existing infrastructure, buildings or vehicles, becoming a more decentralized and flexible solution for carbon capture.8The simplest and most common carbon capture chemistries are amine solutions and basic oxides (both synthetic and earth materials). Aqueous amines is one of the most mature chemical technologies, implemented at the source.9 Amines are inexpensive and effective, but they undergo degradation with reuse, can be corrosive, and require high temperatures and pressures to regenerate via CO2 release.10 Basic oxides, i.e. MgO and CaO are inexpensive and lightweight, and they present similar strengths and limitations as amine solutions, with surface passivation as an additional challenge.11–14 Other effective solid-phase materials are most often basic oxides or basic hybrid materials, with or without porosity, including zeolites,15–17 hydrotalcites (synthetic or mine tailings),18,19 amine-functionalized silica,20,21 and metal organic frameworks (MOFs).22–24
The implementation of peroxide or superoxide ligation of metals (i.e. alkalis, alkaline earths) is an interesting approach to solid DAC materials. For example, potassium superoxide has been used since the dawn of space travel to remove CO2 and replenish O2 within enclosed space vehicles.25 These compounds are simple, lightweight, inexpensive and very reactive. However, their reactivity with CO2 and the resultant product is very sensitive to lattice water and atmospheric water. For example, either carbonate or bicarbonate can form, where the former has a decomposition temperature greater than 600 °C, and the latter has a decomposition temperature less than 150 °C; a critical difference for regeneration.
Peroxometalates of the early d0 transition metals include titanium,26 molybdenum,27 tungsten,28 niobium,29 tantalum,30 chromium31 and vanadium.32 These have been known for decades as either homoleptic anions, or more often mixed organic-inorganic hybrid complexes, and heteroligation is one strategy to tune stability/reactivity. Due to their high oxidative reactivity (especially MoVI and VV), they have been studied for biological and catalytic applications.33,34 Notable to the periodic table relationship between early d0 and f0 metals, UVI as the linear uranyl species (UO22+) also forms stable peroxide complexes. Transition metal peroxide complexes are typically pseudo-tetrahedral with four bonded peroxides (M(O2)4x−), while uranyl binds three peroxides, equatorial to the linear yl-dioxo complex (Fig. 1). Though rarely mentioned in the literature because it is deemed a ‘failed experiment’, uranyl triperoxides, as both solids and solutions, will often form uranyl carbonates, via reaction with atmospheric CO2. However, it is uncertain if this is related to the basicity of the solution, the peroxide reactivity, or both. Forbes et al.35,36 described synthesis of uranyl diperoxide-monosuperoxide, where superoxide is formed from atmospheric O2via benzyl alcohol oxidation; and this reactive species converts to the uranyl tricarbonate. While the focus of this prior paper is on the superoxide, the end product is uranyl tricarbonate via direct air capture, meaning both the peroxide and superoxide react with atmospheric CO2, when bonded to uranyl. However, the mechanism of conversion of uranyl peroxide/uranyl peroxide-superoxide to uranyl carbonate via DAC is not well-understood, since it happens spontaneously without intent (with both peroxide and superoxide ligands), under a variety of conditions (in solution and as solids). The reactivity of the related transition metal peroxometalates has been largely unexplored. This possibility was mentioned in a review paper37 concerning the reactivity of K3V(O2)4 with atmospheric CO2, which in turn cites a reference that reports the reactivity of dimeric [V2O3(O2)4]4− with CO2.38
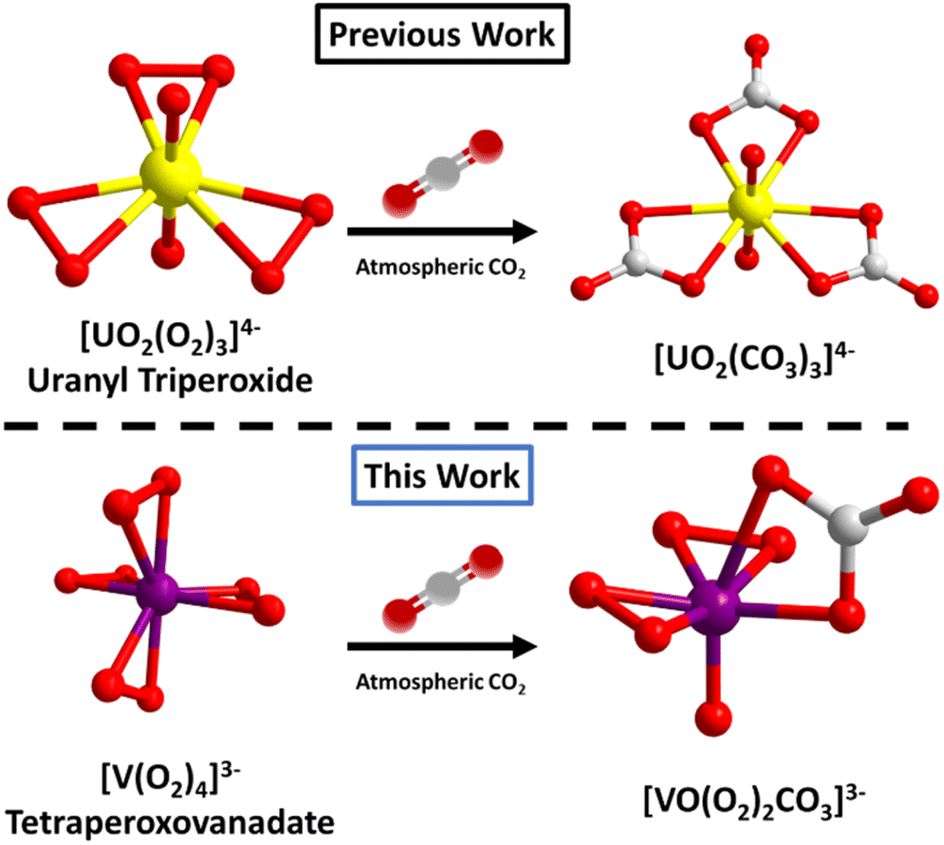 | ||
| Fig. 1 Comparing reaction of f0-actinide peroxide and d0 transition metal peroxide (superoxide) with atmospheric CO2. Under some conditions, uranyl triperoxide (and similar compounds) converts to a uranyl tricarbonate (top).35 In this work we report a related reaction pathway with tetraperoxovanadates (bottom). | ||
Considering transition metal peroxometalates as candidates for DAC, it is hypothetically desirable to have the maximum number of peroxide ligands bound to the metal centre in order to maximize efficiency, so these M(O2)4x− species are intriguing candidates. Mixed ligand peroxocarbonato metalates have been synthesized and studied, i.e. for second harmonic generation materials.39,40 However, the transition from peroxo complexes to carbonates by reaction with atmospheric CO2 has not been extensively investigated. In a related study, Cummins demonstrated the affinity of Mo for carbonate ligands via a reaction between orthomolybdate (MoO42−) and two equivalents of CO2 in organic solvent.41 Moreover, reduction of CO32− to formate was achieved, demonstrating the potential of early d0 transition metals for not only capture, but conversion.
Here we present a detailed study of the reaction of tetraperoxovanadate with atmospheric CO2 in ambient lab conditions, using K3V(O2)4 (ref. 42) (compound 1) and synthesized and structurally described here for the first time, Rb3V(O2)4·2H2O2·H2O (compound 2) and Cs3V(O2)4·3H2O (compound 3). A combination of thermogravimetric analysis – mass spectrometry (TGA-MS), 51V MAS NMR (magic angle spinning nuclear magnetic resonance spectrometry), quantitative carbon analysis, infrared spectroscopy (FTIR) and powder X-ray diffraction (PXRD) benchmarks the conversion of K3V(O2)4 immediately to K3VO(O2)2(CO3), and ultimately a mixture containing mostly KVO3 plus KHCO3 over several months. Parallel studies of the Rb and Cs analogues show the same reaction pathway, higher reactivity rates, but also a possible competing reaction of autodegradation, or water-promoted degradation. The vanadyl-bound CO2 is thermally removed below 300 °C for all three alkali compounds simultaneously with the bound peroxide, indicating a concerted mechanism which is supported by computational studies. While the alkali affects reactivity of the peroxovanadate complex, the vanadium plays a clear role in both CO2 capture, and its subsequent release. Finally, we also consider the role of superoxides both experimentally and computationally. These studies suggest that superoxide is present in small concentrations, and are likewise reactive towards DAC.
Results and discussion
Synthesis
Warning: These compounds can be shock sensitive in the dry form, especially with heavier alkali counterions. K, Rb and Cs salts of V(O2)43− were synthesized adapting reported procedures.42 Details for each compound are included in the ESI.† Summarily, V2O5 was suspended in a basic solution of AOH (A = K, Rb, Cs) and introduced into an ice bath. Then cold 30% H2O2 was added, causing a colour change from brown to green and ultimately purple. The intermediate colour could be due to transient reduction of vanadium, but the final obtained product is pentavalent, based on alkalis for charge-balance as well as experimental precedence (discussed below). After a short time, the solid A3V(O2)4 is obtained by precipitation with cold ethanol. Crystals suitable for single crystal X-ray diffraction (SCXRD) were obtained by ethanol vapour diffusion at low temperature. The ratios of metal to alkali to peroxide in reaction solutions were commonly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:10 but, a number of other combinations were also attempted. For instance, when the amount of base is increased, the isolation of the material became challenging due to the solid being very effervescent and decomposing quickly. Similarly, when the peroxide amount was lowered to stoichiometric ratios, the isolated crystalline material underwent rapid colour change, from deep purple to yellow. In this case, the small amount of peroxide present is not enough to coordinate to the vanadium and form the tetraperoxovanadate. Despite these challenges, the syntheses of these compounds are reproducible with high yields. Considering all three compounds are obtained from the same procedure, the prior paucity of structures of the Rb and Cs analogues was somewhat surprising, perhaps owed to their high reactivity, similar to the alkali uranyl triperoxides.43 Single crystals of the Cs-analogue were only obtained when the samples were flushed with argon gas. This was not a necessary for the Rb-analogue, but it helped obtain better quality crystals. This reactivity hints at a clear trend of the alkalis influencing reactivity of the peroxovanadates.
:5:10 but, a number of other combinations were also attempted. For instance, when the amount of base is increased, the isolation of the material became challenging due to the solid being very effervescent and decomposing quickly. Similarly, when the peroxide amount was lowered to stoichiometric ratios, the isolated crystalline material underwent rapid colour change, from deep purple to yellow. In this case, the small amount of peroxide present is not enough to coordinate to the vanadium and form the tetraperoxovanadate. Despite these challenges, the syntheses of these compounds are reproducible with high yields. Considering all three compounds are obtained from the same procedure, the prior paucity of structures of the Rb and Cs analogues was somewhat surprising, perhaps owed to their high reactivity, similar to the alkali uranyl triperoxides.43 Single crystals of the Cs-analogue were only obtained when the samples were flushed with argon gas. This was not a necessary for the Rb-analogue, but it helped obtain better quality crystals. This reactivity hints at a clear trend of the alkalis influencing reactivity of the peroxovanadates.
Structure description
K3V(O2)4 (1) was described prior,42 but the Rb and Cs analogues are described here for the first time, below (full details on lattice packing and models used for DFT calculations shown in Fig. S1–S4†). All crystallographic information is compiled in Table S1.† Here we briefly note that the simulated PXRD from the single-crystal structures for 2 and 3 do not match that of the experimental (Fig. S5 and S6).† We attribute this to the ease of partial to complete removal of the free peroxide molecules in the lattice upon isolating the dry powder for PXRD. While compounds 2 and 3 contain extra-framework species according to the single crystal X-ray data (2 peroxide molecules and one water molecule per formula unit for 2; 3 lattice waters per formula unit for 3), the bulk characterization suggests otherwise. Thermogravimetry (Table S2†) of the pristine materials showed weight loss consistent with A3V(O2)4 for all three compounds. Only the Rb analogue has ∼2% excess weight loss, the equivalent of 8.9 mass units, or approximately half a water molecule or a quarter peroxide molecule. Experiments discussed later suggest a small amount of residual reactive oxygen species (peroxide or superoxide) remains in the lattice in the bulk material. The PXRD for K3V(O2)4 also does not match the simulated PXRD from single crystal-data, despite not having extra-framework molecules. In this case (and certainly also relevant for the Rb and Cs analogues); we attribute this lattice mismatch to the observed evolution of oxygen from the solid, disrupting long-range order. Nonetheless, bulk characterization shows the peroxoanions charge-balanced by the appropriate number of alkalis with a starting composition of A3V(O2)4 comprises the bulk solids. In fact, a starting bulk composition of A3V(O2)4 for all three compounds means their reactivity is more readily compared, with the alkali being the main difference.Despite the lattices characterized by SCXRD being different that the studied material, they are worth describing here because; (1) they are new structures, and (2) this provides opportunity to compare the influence of the different alkalis on O–O and V–O bond distance. Finally, for the sake of accuracy in naming, we refer to the bulk material as K3V(O2)4, Rb3V(O2)4, and Cs3V(O2)4; or K-analogue, Rb-analogue and Cs-analogue.
All three compounds feature the V(O2)43− anion (Fig. 2a). The peroxide ligands are in a pseudo-tetrahedral arrangement around VV with Op–Op (Op = peroxide oxygen) bond lengths ranging from 1.38–1.52 Å, and V–Op bond lengths ranging from 1.86–1.98 Å. Notably, the bond lengths found in all the crystal structure are consistent with peroxide; superoxide O–O bond lengths are shorter (∼1.2–1.3 Å).44 In addition, superoxide-peroxide disorder in the electron density maps was not detected. Therefore, the ∼5% adventitious superoxide discussed later is not stoichiometric. While the V–Op bond lengths increase slightly with increasing alkali size (Table 1), the Op–Op bond lengths are notably shorter for 1 than for 2 and 3. Both the total number of bonded alkalis per peroxovanadate and the A–O bond lengths are nearly identical for 2 and 3, suggesting it is merely packing effects. We also note there is considerably more A–Op bonding for 1 than for 2 and 3; either because (1) K+ is smaller and more can pack around the anion, or (2) while 1 crystallizes with no additional lattice molecules, 2 contains free peroxide and lattice water, and 3 contains lattice water, meaning there are more ligands besides the vanadium-bound peroxide to which the alkalis can bond. Bond valence sum (BVS) calculations are not feasible since parameters for a V–O1− bond are not available in standard databases, but number of charge-balancing cations confirms pentavalent vanadium, despite the deep purple colour of these compounds (Fig. S7a†).
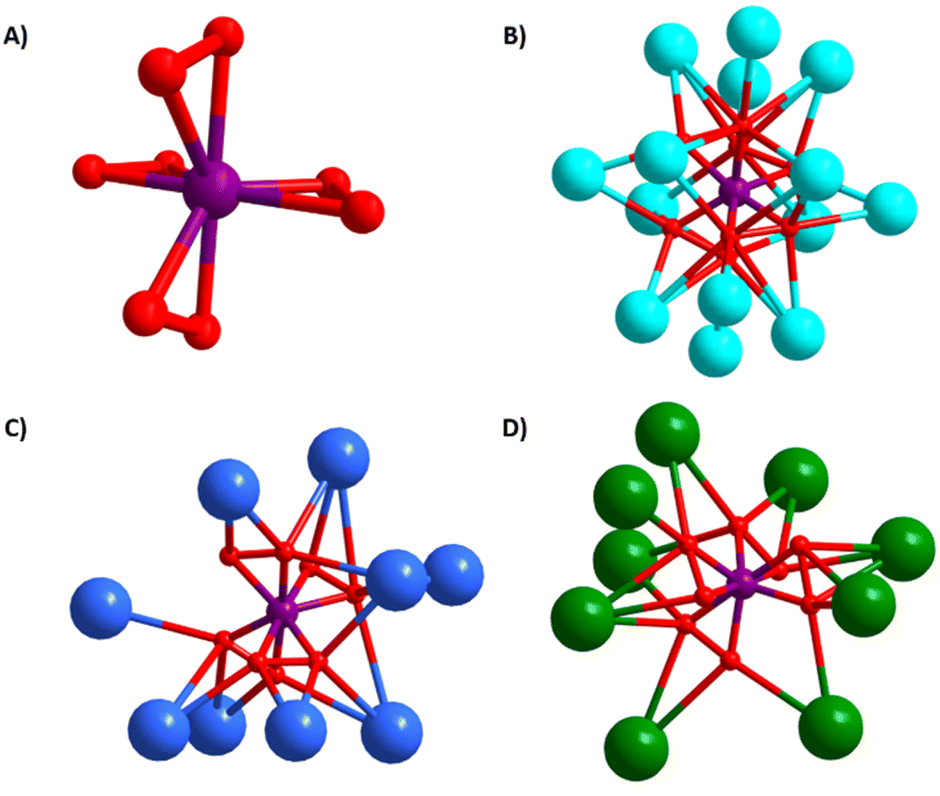 | ||
| Fig. 2 (A) Ball-and-stick representation of the tetraperoxovanadate core common to all synthetized structures and illustrating the alkali--[V(O2)4]3− bonding for: (B) the K-analogue (1), (C) the Rb-analogue (2), and (D) the Cs-analogue (3). Vanadium is purple, potassium is light blue, rubidium is dark blue, caesium is green and oxygen is red. | ||
| Alkali cation | V–Op, avg | Op–Op, avg | A+–Op, avg | # A+ bonded to each [V(O2)4]· | # A+–Op bonds per [V(O2)4]· |
|---|---|---|---|---|---|
| K (1) | 1.9181 | 1.5508 | 2.8822 | 14 | 31 |
| Rb (2) | 1.9198 | 1.4654 | 3.0982 | 9 | 21 |
| Cs (3) | 1.9276 | 1.4762 | 3.0985 | 9 | 20 |
The Rb-analogue (2) crystallizes in the monoclinic system with the space group P121/n1 (V = 1153.65(6) Å3) and is fully formulated Rb3[V(O2)4]·2H2O2·H2O. The V-Op bond lengths range between 1.876(6)−1.985(5) Å, and Op–Op distances are 1.461 (7)–1.470 (9) Å. The Op–V–Op angle is ∼45° (±2°) for all four peroxides, but the V–Op–Op angles vary between 60 and 70°, consistent with the wide range of V–Op bond distances.
There are two additional hydrogen peroxide molecules in the lattice. The hydrogen atoms were not located in the electron density map, but they are necessary for charge-balance, and when included in the solid-state calculations, result in a stable structure with an H-bonding network (Fig. S1 and S2†). Protonation is expected, since deprotonation of peroxide is the first step to decomposition, and we would expect a free peroxide anion to be unstable. In addition, the free peroxide oxygen atoms are located 2.7 Å from the vanadium-bound peroxide ligands, consistent with hydrogen-bonding. A prior structure reported for the Na-analogue of V(O2)43− also contained free peroxide molecules.32 The hydrogen peroxide molecules are bonded terminally, bridging and side-on to Rb with bond lengths ranging from 2.98 to 3.43 Å (Fig. S1a†). Finally, a water molecule coordinates between Rb2 and Rb3, a lattice arrangement reported in prior structures.28,32 The [V(O2)4]3− anions are aligned along the a-axis in the lattice. The Rb1 and Rb3 each form four-atom parallel rows that are broken by a zigzag column of Rb2 cations (Fig. S1b†). This bears resemblance to the prior-reported sodium analogue structure, where the cations interconnect with one another through bound water in octahedral configuration.32
Compound 3 crystallizes in the monoclinic space group P121/n1 (V = 1162.57 (8) Å3) and is fully formulated Cs3[V(O2)4]·3H2O. The [V(O2)4]3− of compound 3 is more symmetric than that of compound 2; with deviation between the bonds significantly lower in compound 3; almost three times lower for V–O bonds and over five times lower for O–O bonds. The same is true for the O–V–O and V–O–O angles, which also have an average of 45° and 67° respectively, but with a smaller range in both cases. This suggests the distortions in 2 arise from H-bonding to the free peroxide.
Compound 3 has three lattice water molecules per formula unit, each water molecule sits between four Cs+ and one tetraperoxovanadate. The lack of free hydrogen peroxide molecules in the lattice is a notable difference between compounds 2 and 3, but is also not unprecedented for this family of compounds and the DFT calculated unit cell of Cs3[V(O2)4]·3H2O with waters explicitly included converged to a stable geometry (Fig. S3 and S4†). The most-studied Na3V(O2)4 compound has been crystallized with a range of H2O2 and H2O in its lattice,32 and it is likely that other ordered lattices for K, Rb and Cs analogues could be isolated, with or without the inclusion of free H2O and H2O2.
The lattice of 3 features alternating layers of alkalis and tetraperoxovanadates, stacked in the b-direction (Fig. S3c†). Along the a-axis (Fig. S3d†), parallel layers of Cs2 and Cs3 alternate with double layers of Cs1. Viewed along the c-direction, the Cs+ cations appear as triads of all three independent Cs atoms (Fig. S3b†), bearing resemblance to the Na3[V(O2)4] lattice.32 The main difference, as discussed above, is that the Na-triads are interconnected through lattice water for the Na-analogue.
Reaction with atmospheric CO2
All analogues of A3V(O2)4 react immediately upon air exposure, if not stored cold and preferably (esp. Rb and Cs analogues) under an inert atmosphere, best depicted by a colour change, from purple to yellow (Fig. S7†). This colour change is commensurate with reaction of the unstable oxygen ligands (peroxide and adventitious superoxide), either via auto-decomposition or capture of atmospheric CO2, discussed below. The macroscopic colour change occurs more rapidly for Rb3V(O2)4 and Cs3V(O2)4; no purple is visible after just 5 days, while K3V(O2)4 retains a purple hue two weeks to a month. This is in agreement with previous reports that describe larger alkali tetraperoxovanadates (and also Mo and W counterparts) as more reactive, or even explosive.45 That said, this faster reactivity does not necessarily translate to higher carbon capture, as revealed by characterization of the degradation products via Fourier transform infrared (FTIR), thermogravimetric analysis/mass spectrometry (TGA-MS), powder X-ray diffraction (PXRD), 51V solid state MAS NMR (magic angle spinning nuclear magnetic resonance) and elemental analysis, described below.To investigate the changes of A3V(O2)4, the K-analogue was left on a lab bench under ambient conditions (temperature ∼21 °C). FT-IR spectra were recorded over time as a first indication of moieties present in the decomposition products (Fig. 3, S8 and S9 for K, Rb and Cs analogues, respectively). Here we describe the spectral bands citing K3V(O2)4, and bands for Rb3V(O2)4 and Cs3V(O2)4 are summarized in Table S3.† It is apparent that these compounds react with atmospheric CO2 to form carbonates due to the presence of characteristic bidentate carbonate peaks at 1570 cm−1, 1340 cm−1, and 1050 cm−1.46 The increasing absorbance at 935 cm−1 and 735 cm−1 were assigned to V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching and V–O vibration respectively, both indicating a change in the vanadium-oxygen bonding mode. This was further confirmed by the peak growth at 740 cm−1, which matches the V–O–C vibrational mode and implies that the carbonate is bonded to the vanadium centre. The absence of these peaks in the fresh K3V(O2)4 evidences DAC is the source. Other peaks were assigned to O–O stretching (peroxide, 850 cm−1) and V–(O2) vibrations (625 cm−1, 590 cm−1, and 560 cm−1). Overall, the new spectrum resembles that of synthesized K3[VO(O2)2CO3].39 We can describe the reaction as:
O stretching and V–O vibration respectively, both indicating a change in the vanadium-oxygen bonding mode. This was further confirmed by the peak growth at 740 cm−1, which matches the V–O–C vibrational mode and implies that the carbonate is bonded to the vanadium centre. The absence of these peaks in the fresh K3V(O2)4 evidences DAC is the source. Other peaks were assigned to O–O stretching (peroxide, 850 cm−1) and V–(O2) vibrations (625 cm−1, 590 cm−1, and 560 cm−1). Overall, the new spectrum resembles that of synthesized K3[VO(O2)2CO3].39 We can describe the reaction as:
| K3V(O2)4 + CO2 → K3[VO(O2)2CO3] + O2 | (1) |
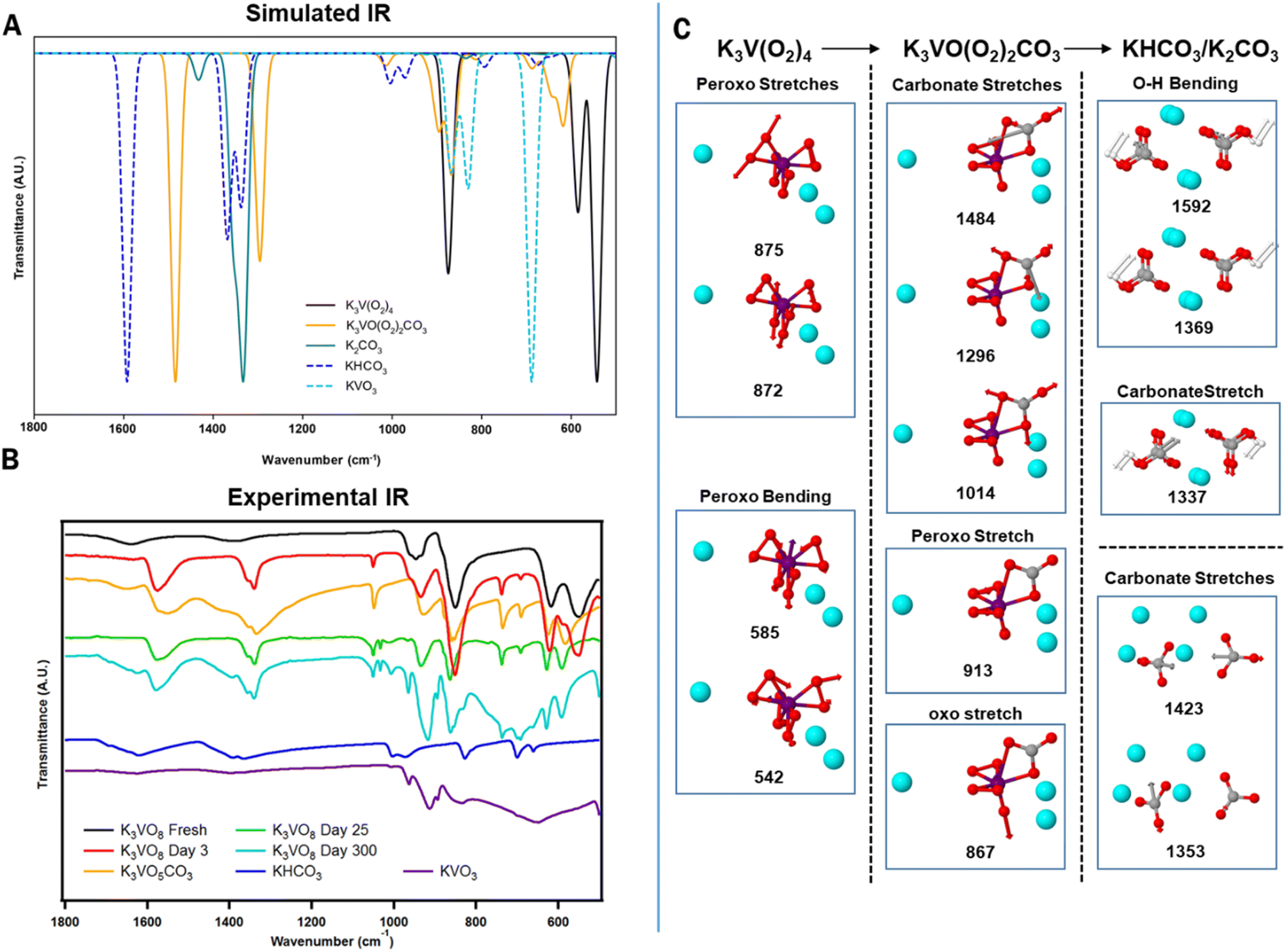 | ||
| Fig. 3 Comparison of the calculated IR spectrum of K3V(O2)4 and its possible decomposition products (A) with the experimental data (B). Representative carbonate peaks for K3[VO(O2)2(CO3)] (1570 cm−1, 1340 cm−1, and 1050 cm−1) are observed in Day 3, Day 25, and Day 300 samples. The spectrum of Day 300 also contains IR peaks representative of KHCO3 (1630 cm−1, 1390 cm−1, and 1010 cm−1) and KVO3 (965 cm−1, and 890 cm−1). The modes of vibration to which they are assigned are shown in (C). | ||
Solid-state density-functional theory (DFT) calculations were carried out in CASTEP47 (see ESI† for details) to assist in the interpretation of the experimental IR spectra. Phonon calculations of the starting material, K3V(O2)4, the monocarbonate (K3VO(O2)2CO3), and other species thought to be involved in decomposition/side reactions were simulated in this manner, namely KOH, K2O, K3VO4, and K2CO3. Comparison between calculated and observed peaks in solid-state IR is complicated by the experimental inevitably of there being a superposition of reactant, product, and other species. Other discrepancies can be attributed to inaccuracies in the unit cells, lack of surface terminations in the calculations and the method's own limitations. Nonetheless, a few supported comments can be made here. If only considering motions above 500 cm−1, K3V(O2)4 has effectively four signals: V–O–O bending motions at 542 and 585 cm−1, and O–O stretching motions at 871 and 876 cm−1, as would be expected for a homoleptic metal-peroxide compound.48 K3VO(O2)2CO3 also has signals in this region including a VO stretch at 867 cm−1, an asymmetric peroxo stretch at 895 cm−1, and a symmetric peroxo stretch at 914 cm−1. Distinct from K3V(O2)4, however, K3VO(O2)2CO3 is predicted to have three intense peaks above 1000 cm−1 attributed to the carbonate group. These include symmetric [CO3]2− stretch at 1014 cm−1, and asymmetric [CO3]2− stretches at 1296 cm−1 and 1484 cm−1. This collection of peaks are found experimentally only after the A3V(O2)4 sample has been exposed to the air. Further confirmation of this transition comes from PXRD patterns. Fig. 4 displays a structural transition from K3V(O2)4 to K3[VO(O2)2CO3] by ingrowth of peaks that are consistent with the latter compound. Longer ambient exposure times bring a new set of peaks at 2theta (°) = 17.2, 24.3, 24.7, 34.2, 34.6, and 36.9; indicating a mixture of KHCO3 and KVO3. Given the order of peak appearance, we deduce that these simple salts form via capture of additional CO2, thus marking a possible end-product of the reaction, which can be formulated as:
| K3[VO(O2)2CO3] + H2O + CO2 → KVO3 + 2KHCO3 + O2 | (2) |
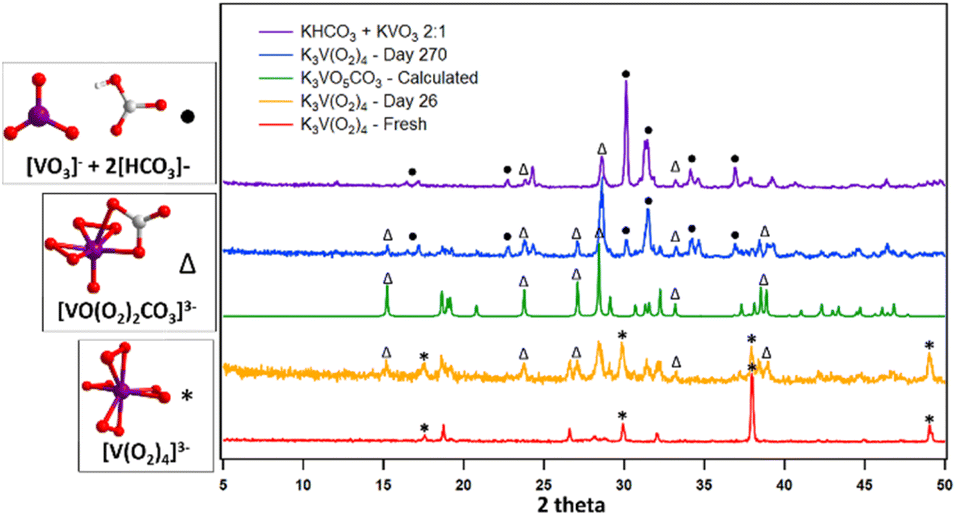 | ||
| Fig. 4 PXRD patterns of K3V(O2)4 at different time stamps compared to the synthesized K3VO(O2)2CO3 as an intermediate and KVO3 and KHCO3 as end products. Peaks corresponding to compounds containing the key molecular species are shown on the left are marked accordingly. | ||
It is worth noting, however, that peaks for both K3[VO(O2)2CO3] and K3[V(O2)4] persist, despite the ingrowth of peaks for the secondary decomposition products, which is further verified by TGA-MS experiments, discussed later.
We synthesized prior-reported K3[VO(O2)2CO3] to use as a standard for various spectroscopic characterizations (see ESI† for synthesis details).42 The 51V MAS NMR of synthesized K3[VO(O2)2CO3] and 1 aged for nine months is shown in Fig. 5. K3[VO(O2)2CO3] has a single 51V peak at 789 ppm. The central peak was identified as the non-shifting peak upon changing spin speeds (Fig. S10†), and the remainder are spinning side bands. The single 13C peak for this compound is located at 161 ppm. Aging K3V(O2)4 for 9 months yields a 51V MAS NMR spectrum with two peak envelopes (Fig. 5b). The minor envelope (spinning sidebands marked by red stars) is K3[VO(O2)2CO3], and the major envelope (spinning sidebands marked by black stars) is KVO3,49 consistent with both IR and PXRD. Notably, we were not able to collect data on any fresh V(O2)43−. A single attempt led to ‘explosion’ of the sample rotor, because the excessive heat generated by spinning rapidly decomposed peroxide ligands.
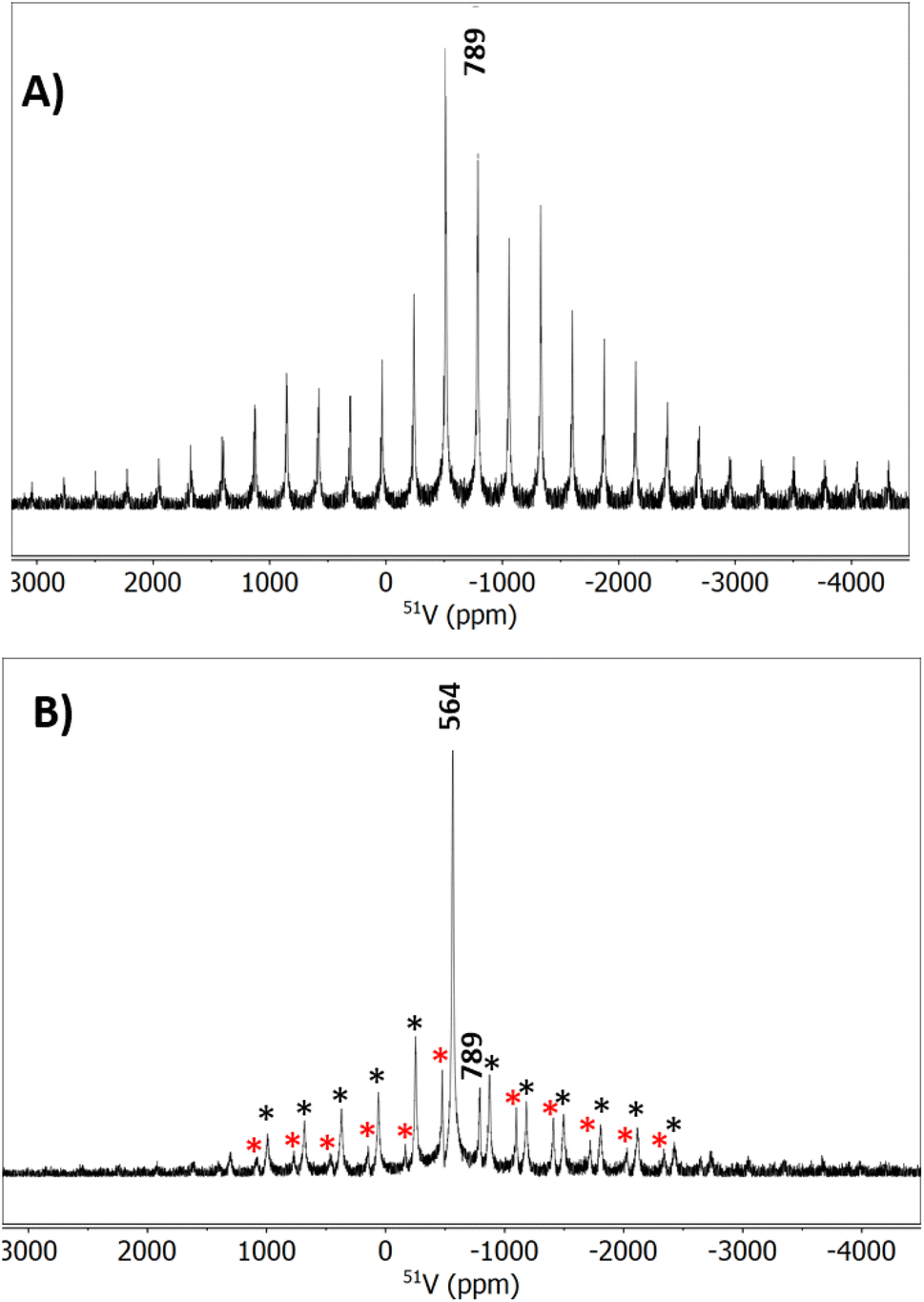 | ||
| Fig. 5 51V MAS NMR for (A) K3VO(O2)2CO3 and (B) K3V(O2)4 aged 9 months. The red stars indicate spinning sidebands for the peak at 789 ppm (K3VO(O2)2CO3) and the black stars indicate spinning sidebands for KVO3 (564 ppm). | ||
To gain insight into the possible mechanistic steps associated with the conversion of gaseous CO2 into complexed [CO3]2−, cluster-model (Fig. 6A) DFT calculations in Gaussian50 were performed on one unit of the tetraperoxovanadate complex with three potassium ions explicitly included (see ESI† for details). The resulting proposed mechanism (summarized in Fig. 6B; see Fig. S11† for complete diagram) consists of five steps for the CO2 addition: (1) barrierless addition of CO2 to K3V(O2)4, forming K3V(O2)3(κ1-CO4); (2) isomerization of K3V(O2)3(κ1-CO4) to K3V(O2)3(κ2-CO4), (3) transfer of an oxygen atom from the peroxycarbonate to one of the peroxo ligands to yield '(η2-O3)(κ1-CO3), (4) detachment of one of the coordinating O atoms in η2-O3, meaning η2-O3 becomes η1-O3; (5) cleavage of the dangling O2 from η1-O3, producing K3VO(O2)2(κ1-CO3). The formation of a peroxycarbonate ligand is the natural starting point from chemical intuition and is consistent with previous computational and experimental studies that demonstrate the ability of peroxo-containing metal species to form stable peroxycarbonate complexes.51,52 Additionally, the transfer of an oxygen atom to form an ozone intermediate in step 3 (see Fig. S11†) has some literature precedent in zirconium structures featuring η2-O3 involved in the epoxidation of propene.53 Although the cluster-model product geometry, K3VO(O2)2(κ1-CO3), does feature the carbonate ligand bound in a κ1-fashion that is different from both the experimental and solid-state computational structures, this binding mode can be rationalized on the basis of the lack of crystal packing in the cluster (vacuum) calculation and the small atomic radius of V5+. We performed analogous simulations with the heavier group congeners Nb and Ta, and found that only Ta exhibits bidentate bonding of the carbonate to the metal centre (see Fig. S12 and S13†). As shown in Fig. 6B, the overall net change in Gibbs free energy for the CO2 addition is −64.8 kcal mol−1 with a rate limiting barrier of 24.3 kcal mol−1. Given the predictions of highly exergonic free energy changes, surmountable kinetic barriers for a room-temperature reaction, and qualitatively matching IR simulations, we believe the reaction described here is a plausible pathway for the initial carbon capture process observed, invoking conversion of K3V(O2)4 to K3VO(O2)2CO3.
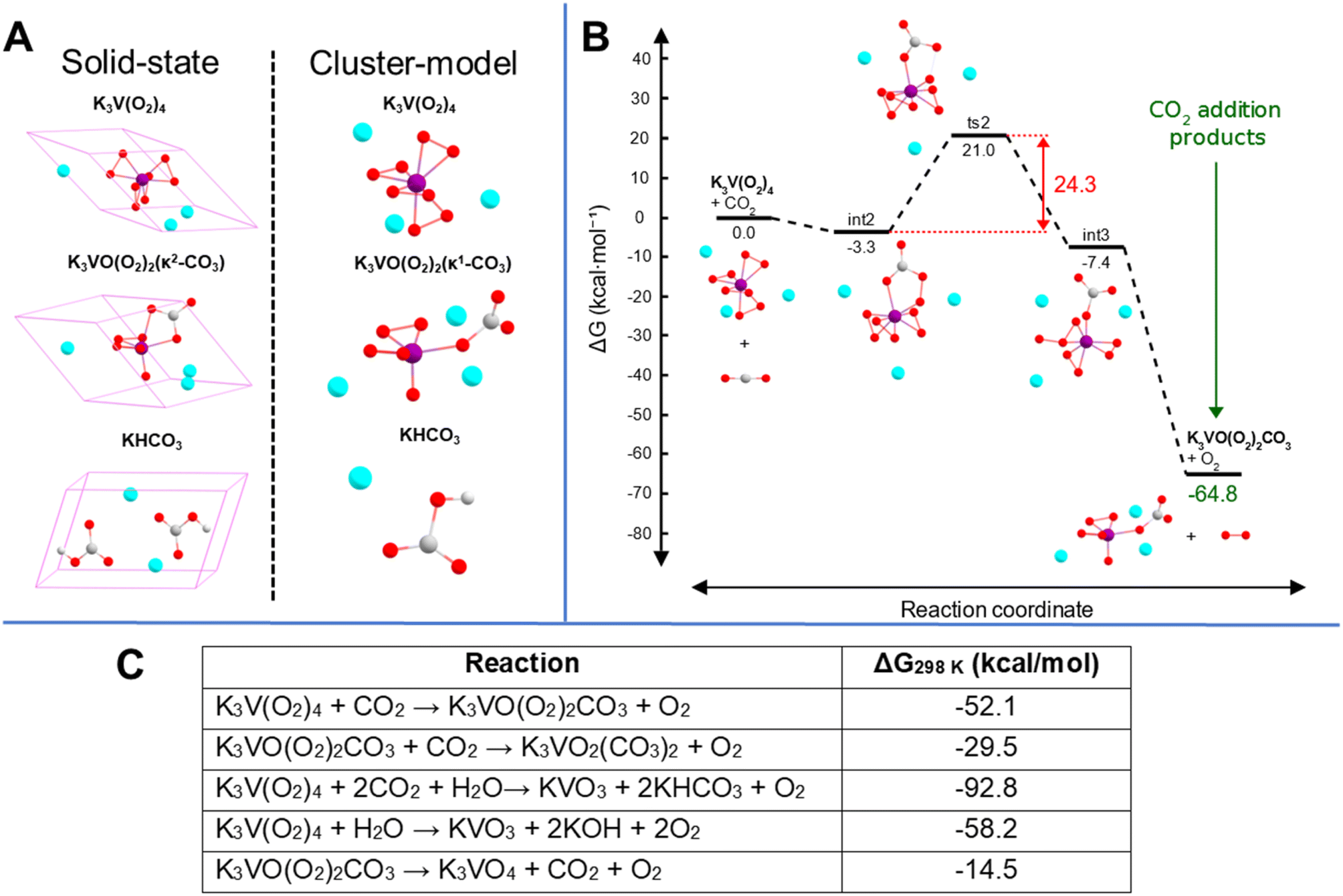 | ||
| Fig. 6 (A) Two computational modelling approaches used: solid-state DFT with periodic boundary conditions to simulate IR spectra and gas-phase cluster-model DFT to describe CO2 capture by K3V(O2)4. (B) Simplified reaction mechanism, displaying only the thermodynamics and rate-limiting steps associated with the addition of CO2 to K3V(O2)4. (C) Room-temperature solid-state thermodynamics of some selected reactions. | ||
We attempted to explain the relative reactivity dependence of V(O2)43− on the alkalis (Cs > Rb > K) using computation. If an idealized K3V(O2)4 structure is assumed for the Rb and Cs analogues, the calculated solid-state relative electronic energy of Rb3V(O2)4 is destabilized by 25.0 kcal mol−1 relative to K3V(O2)4, while Cs3V(O2)4 is destabilized by 48.9 kcal mol−1 compared to the K-analogue. Interestingly, despite the notable difference in stability between all three compounds, the ΔG of the decomposition reactions (described in eqn (1) and (2)) are rather similar. We had expected this, due to the higher intrinsic reactivity of the Rb and Cs analogues. However, the presence of a larger alkali destabilizes both the starting materials and the products of the reaction, meaning that the net effect is comparable between all three reactions, so the experimentally observed difference in reactivity cannot be attributed to simple electrostatic differences in the counterions. Our computational method is limited by the potential role of reagents that cannot be accounted for in a balanced chemical reaction, e.g. water, free hydrogen peroxide, hydroxide, and even the chemical role of the alkalis.
The PXRD patterns of Rb3V(O2)4 and Cs3V(O2)4, while displaying similar behaviour to those of K3V(O2)4 initially, show no evidence for the A3[VO(O2)2CO3] intermediate beyond the 90 days mark (Fig. S5 and S6†). This suggests that after a 3 months period, all the CO2 captured is transformed to [HCO3]− and the vanadium complex has fully decomposed to its simplest metavanadate form, with no remaining peroxide. While this information can be extracted from the PXRD data; as the material decomposes over time it also loses crystallinity, making peak assignment somewhat more challenging. Therefore, in order to confirm this observation as well as determine temperature and mechanism of CO2 release and monitor the thermal degradation pathway, thermogravimetric analysis-mass spectrometry (TGA-MS) was employed.
Fig. 7 displays the simultaneous thermogravimetric analysis and mass spectrometry (TGA-MS) analysis of K3V(O2)4 with aging (up to 9 months) in the ambient lab atmosphere, and the data discussed below is summarized in Table S4.† Mass spectrometry identifies volatile species evolved from K3V(O2)4, and observed changes with time are consistent with PXRD, IR and 51V NMR analyses. The fresh K3V(O2)4 served as a reference and a starting point of the carbon capture reaction (Fig. 7A). The loss of molecular oxygen originated from the four peroxide ligands bound to the metal centre occurs at 120 °C. The absence of any CO2 signals in the mass spectrum indicates the fresh product is pure, prior to initiating carbon capture.
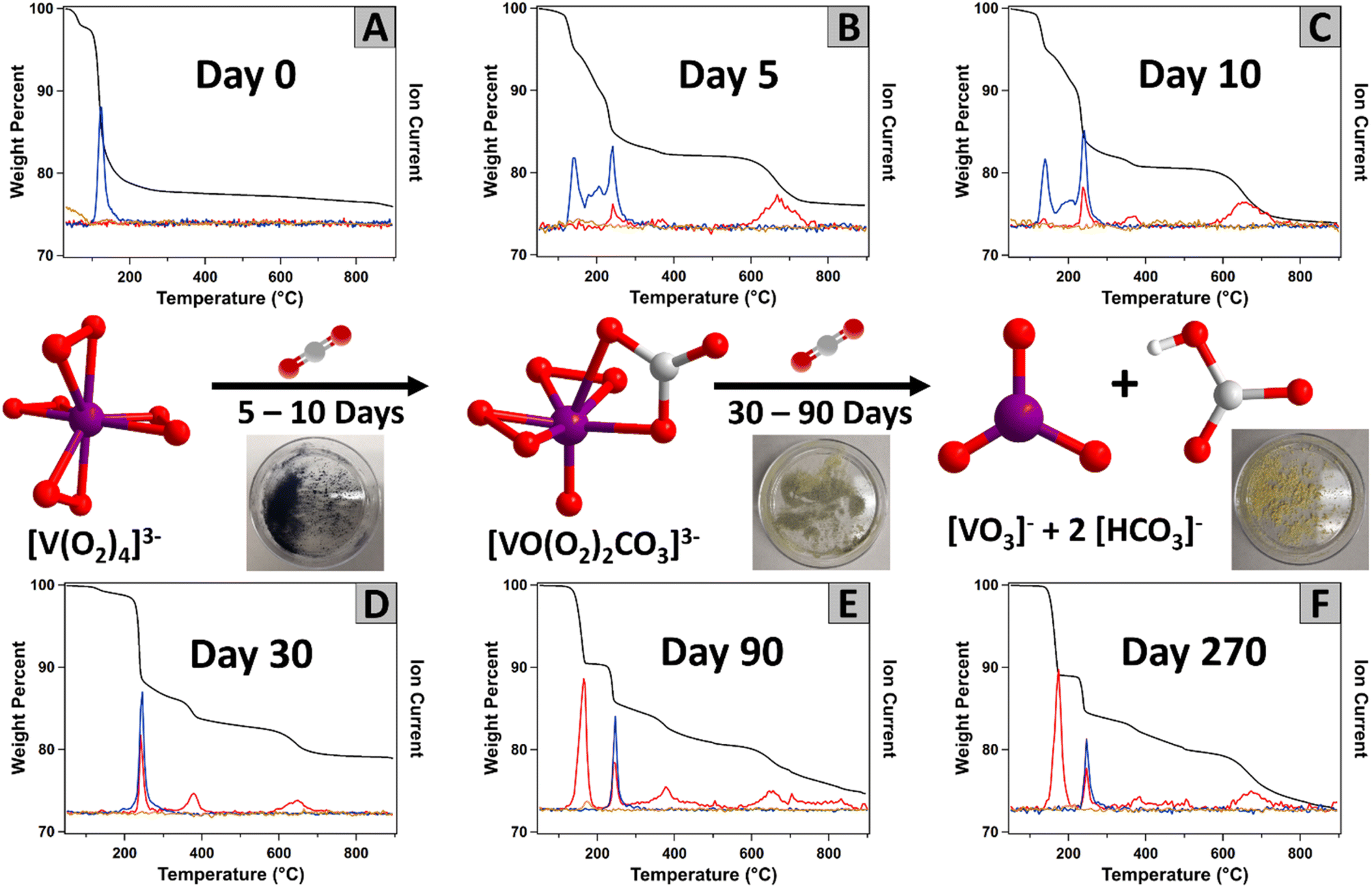 | ||
| Fig. 7 Evolution of TGA-MS of K3V(O2)4 following exposure to ambient air with time (increasing time from (A) to (F)). The thermogravimetric signal is black. The red, blue, and light orange traces are the signals of CO2, O2 and H2O by tandem mass spectrometry, respectively. Photographs show the colour evolution of the solid materials with aging in ambient conditions. | ||
After 5 days on the lab benchtop, the oxygen peak from unreacted tetraperoxovanadates remains, but a new simultaneous release of oxygen and carbon dioxide appears at 240 °C (Fig. 7B). This simultaneous release is assigned to peroxides and carbonates bonded to vanadium, namely the newly formed K3VO(O2)2CO3, also observed by PXRD (Fig. S15†). This was confirmed by performing the TGA-MS experiment on the standard (synthesized) K3VO(O2)2CO3, which displayed a comparable simultaneous weight loss of O2 and CO2 (Fig. S16†). We used computation to understand the mechanism of simultaneous release. These studies revealed favourable thermodynamics for the two-step sequential removal of CO2 and O2 from K3VO(O2)2CO3 to form K3VO4 at room and elevated temperatures (Fig. S14†). A new peak at 370 °C that broadens in later spectra also appears after 10 days of ambient air exposure. This CO2 release appears to originate from decomposition of the formed alkali carbonate, catalysed by the vanadium present in the mixture (discussed below). In addition, higher temperature CO2 loss (670 °C) originating from carbonates bonded to alkali metals is observed. The CO2 has a weight percent of 8.29% and the O2 is 13.41%, where the water only accounts for 2.26%. Similar signals can be found after 10 days (Fig. 7C), with an increase in the carbon dioxide signal (weight percent of CO2, O2, and H2O were 10.37%, 13.54%, and 2.16%, respectively).
With continued ambient exposure, the oxygen signal at 140 °C is completely absent after 30 days (Fig. 7D), indicating that any remaining K3V(O2)4 decomposed to other materials. At this point, the multiple CO2 signals have increased (total weight loss 12.36%), and less O2 (weight loss 8.48%) was released. The CO2 peak at 165 °C, dominates after 90 days of exposure (Fig. 7E). This peak is assigned to the decomposition of bicarbonate and suggests that this new carbon environment is the most prevalent form in the material. We confirmed this by preparing a mixture of potassium bicarbonate and KVO3 (as observed by PXRD) and performing the TGA-MS experiment on this mixture (Fig. S17†). In this reference spectrum, the peak appearing at 300–500 °C can also be observed, confirming it belongs to a vanadium catalysed release of CO2. Between day 90 and day 270, the TGA-MS data exhibited little change. The weight loss is dominated by CO2 (weight loss 21.16%) and some remaining O2 from the [VO(O2)2CO3]3− intermediate (weight loss 2.88%). A series of IR spectra were run of this sample after being heated at different temperatures. As expected, there is a decrease in the intensity of the carbonate bands after the sample is heated to temperatures higher than 300 °C in favour of bands assigned to VO stretches (Fig. S18†). In summary, the TGA-MS and PXRD data both show an evolution of V(O2)43− to VO(CO3)(O2)23− to VO3− plus bicarbonate, with all anions charge-balanced by potassium. At each step of air exposure, VO(CO3)(O2)23− is always present.
The thermal behaviour of the Rb and Cs analogues following ambient air exposure was investigated in the same way as K3V(O2)4. Details for these results for Rb3V(O2)4 are in Fig. S19–S24, summarized in Table S5, and in Fig. S25–S30, summarized in Table S6,† for Cs3V(O2)4. There are distinct differences between the three analogues. While the oxygen release occurs in a single step at 120 °C for 1, the oxygen loss for both Rb3V(O2)4 and Cs3V(O2)4 are represented by two temperature maxima, 130 °C and 180 °C. The shifts observed in the CO2 peak positions at 300–400 °C and 550–750 °C can be explained by the varying thermal stabilities of the alkali carbonates, to which these peaks are assigned (Fig. S31†). In turn, the minimal shift for the CO2 released at 250 °C confirms this peak to correspond to vanadium-bonded carbonate. However, the biggest difference between all three compounds appears after 90 days when the simultaneous release of CO2 and O2 at 250 °C, characteristic of the [VO(O2)2CO3]3− complex, disappears for Rb3V(O2)4 and Cs3V(O2)4. This is confirmed by the PXRD data that shows after 90 days, the [VO(O2)2CO3]3− complex is fully decomposed to bicarbonate and metavanadate (Fig. S5 and S6†). This key difference between the K-analogue and Rb3V(O2)4 and Cs3V(O2)4 can, once again, be explained based on the relative stability of the different compounds.
To quantify CO2 capture per tetraperoxovanadate molecule, we used CHN microanalysis at similar time stamps as the above-described analyses. In addition to this, the weight loss and the integration of the peak areas from the TGA-MS spectra was also used to validate the results. Fig. 8 shows the capture of CO2 occurs most rapidly the first few days of atmospheric exposure, at which time the total carbon captured changes notably on a daily basis (full details of CHN analysis are shown in Tables S7–S9 and Fig. S32–S35†). This is consistent with the energetically favourable conversion of V(O2)43− to [VO(O2)2CO3]3− (ΔG = −51.6 kcal mol−1, −53.2 kcal mol−1 and −51.4 kcal mol−1 for K, Rb and Cs analogues, respectively, in the solid-state). As discussed above, this intermediate continues to degrade into even more stable products, namely metavanadate and bicarbonate (ΔG = −41.2 kcal mol−1, −41.5 kcal mol−1 and −41.1 kcal mol−1 for K, Rb and Cs analogues, respectively). The possibility of binding the second CO2 molecule to the peroxovanadate complex was hypothesized briefly. Computational calculations show a favoured process, with a net free energy of −50.4 kcal mol−1 as a cluster-model (−29.5 kcal mol−1 in the solid-state) with a rate limiting barrier of 24.5 kcal mol−1. However, this product was never observed. After the initial transformation, the capture rate seems to slow down and plateau at 1.5–2.0 CO2 per molecule. The fact that all three compounds reach a similar endpoint is expected since the reaction path is the same for all three, and they have similar overall change in free energy (ΔG). Therefore, even though their thermodynamic behaviour and reaction pathway is similar, Cs3V(O2)4 reacts faster. Furthermore, the prospect of binding a third CO2 molecule through a reaction with the remaining peroxo ligand in hypothetical K3VO2(CO3)2 to form K3VO(κ1-CO3)3 was considered, but was found to be uphill in relative free energy by 3.2 kcal mol−1, and the fourth addition product, K3V(κ1-CO3)4, did not result in a stable complex with cluster calculations.
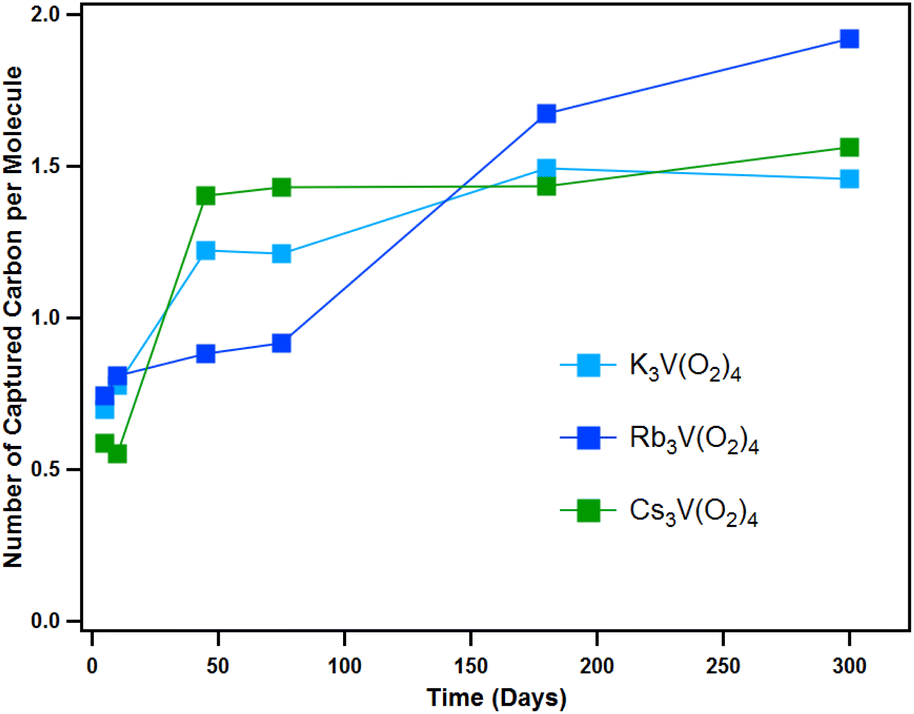 | ||
| Fig. 8 Carbon capture performance for the three A3V(O2)4 analogues under ambient conditions evaluated by CHN analysis. | ||
The alkali countercations play a role in the reactivity of V(O2)43−, with a clear trend in increasing reactivity with increasing alkali size. However, the exact mechanism, and therefore the role of the alkalis is still poorly understood, despite the observed consequences. On the other hand, the number of CO2 molecules per metal centre (1.5 to 2) is similar for K3V(O2)4, Rb3V(O2)4, and Cs3V(O2)4; suggesting the effect of the alkalis is largely kinetic. At the end of the experiment time (300 days), the K-analogue exhibits carbonate directly bound to the vanadium as well as carbonate bound to the alkalis, based on release temperature in TGA-MS and PXRD. On the other hand, the Rb and Cs analogues only exhibit carbonate bound to the alkalis after 300 days of ambient exposure. Competing decomposition pathways of the peroxometalates that complicate mechanistic understanding is auto-degradation (eqn (3)) or water-promoted degradation (eqn (4)):
| V(O2)43− → VO43− + 2O2 | (3) |
| H2O + V(O2)43− → VO31− + 2OH− | (4) |
The role of the alkalis is also not readily probed by computation since we have no evidence for its change in speciation. Finally, the role of water, whose presence is coupled to alkali charge-density, cannot be dismissed in any of the solid-state reactions; DAC, auto-degradation, or water-promoted degradation.
The vanadium-bound carbonate for K3V(O2)4 (observed for up to 300 days of monitoring), coupled with the computational studies indicating monodentate-bound carbonate is more stable than chelating carbonate, leads us to some hypotheses about the progression of carbon capture for K3V(O2)4. It is possible that the vanadium is the steady-state point of CO2 capture and conversion to CO32−via reactivity of the peroxides, and V–OCO2 binding is followed by migration of CO32− to the alkalis, relieving steric hindrance and strain at the vanadium centre (Fig. S12†).
A final hypothesis considered for this DAC process was the presence of superoxide species in the material. Superoxide, O2−, is a potential decomposition product of peroxide, especially in the presence of a redox active metal such as vanadium. As discussed in the introduction; (1) a uranyl peroxide-superoxide anion was shown to evolve to a uranyl tricarbonate via direct air capture,35 and (2) potassium superoxide has been used for air purification for space travel, given its reaction with CO2 to release O2. To identify and quantify superoxides, two approaches were employed: an experimental approach using a superoxide scavenger and Electron Paramagnetic Resonance (EPR) spectroscopy to detect unpaired electrons. First, following a reported procedure,35 K3V(O2)4, Rb3V(O2)4, and Cs3V(O2)4 were suspended in DMSO and mixed with nitro blue tetrazolium chloride (NBT). A colour change was observed immediately in the solution, suggesting the presence of superoxide bound to the vanadium or in the lattice. While there was no appreciable difference in the rate at which the reagent solutions containing the three compounds changed colour, we saw potential to finally establish a reactivity trend for the alkalis-analogues (discussed later). It is known that larger alkalis stabilize superoxide better due to poorer polarization of O–O bonds. Hence if the formation of superoxides is necessary for the CO2 capture to proceed, it could explain the increased reactivity with alkali size. To supplement these results, we also analysed both hydroxide and hydroxide-peroxide solutions for the presence of superoxide, where the latter has been reported.54 The mixture of hydroxides and NBT in DMSO without peroxide also gave a colour change indicating superoxide, suggesting this technique must be used with caution. However, water did not give a false positive, so further tests were performed in water (see ESI† for details). In the presence of superoxide, NBT is reduced to form formazan, which produces a colour change that can be monitored via UV-vis.55 The aqueous NBT solutions with added KOH-peroxide, RbOH-peroxide, and CsOH-peroxide, all showed linear increase in the formazan absorption peak, and therefore increase in formation of superoxides with time (Fig. S36†). The concentration of superoxide trended CsOH > RbOH > KOH, suggesting increasing basicity increases peroxide decomposition in water, with superoxide as one of the decomposition products. We also measured TMAOH-peroxide and NH4OH-peroxide (TMA = tetramethylammonium) because (1) NH4OH is a weak base, and (2) TMAOH is generally considered a strong base, and (3) TMA exhibits minimal bonding/association interactions that we expect from the alkalis and provides competing stabilization/destabilization effects. Indeed, TMAOH exhibited similar superoxide presence as KOH according to the NBT test, while minimal superoxide was detected for the NH4OH solution (Fig. S36†). UV-vis of K3V(O2)4, Rb3V(O2)4, and Cs3V(O2)4 measured in aqueous NBT solutions also showed an increase in superoxide concentration, which plateaued over time (Fig. S37–S39† and 9A). Rb3V(O2)4 showed the maximum absorbance, perhaps due to residual free peroxide (Table S2†). This apparent increase in superoxide concentration with time may also be an artifact of the NBT test, and the results should be considered with caution. Additionally, dissolution of V(O2)43− in water leads to dissociation of the peroxide ligands and formation of orthovanadate. Nonetheless, similar results were obtained from EPR and the aqueous NBT test, discussed below. Similar tests performed on samples exposed to ambient air for three days showed little to no absorbance for Rb3V(O2)4, and Cs3V(O2)4, and absorbance similar to that of the fresh sample of K3V(O2)4 (Fig. S40†). Unlike Rb3V(O2)4, and Cs3V(O2)4, K3V(O2)4 had retained much of its purple colour, characteristic of less decomposition. This indicates that the peroxide (and superoxide) present in the fresh Rb3V(O2)4, and Cs3V(O2)4 materials soon decomposes upon atmospheric exposure, by reaction with CO2 and/or water. Infrared analysis supports this data (Fig. S8 and S9†). The peaks at 2500–3200 cm−1 present in fresh Rb3V(O2)4 and Cs3V(O2)4, consistent with either water, peroxide, or other reactive oxygen species such as superoxide,56 disappear after one day. We used aqueous solutions of KO2-NBT to create a calibration curve and quantify superoxide in the peroxovanadates (Fig. S41†). Concentration of superoxide vs. time in all three compounds best fits a power trend line (details in the ESI, Table S10†). Measuring the beginning of the reaction proved impossible since upon dissolution the three compounds retained their natural purple colour before the peroxide dissociates from the vanadium-centre, which interfered with the absorbance caused by the reaction with NBT. Extrapolation to the initiation of the reaction, it appears that the solids contained an initial 4–5% of superoxide (Fig. 9C). Because the NBT test are done a liquid medium (DMSO or water) that seems to promote superoxide formation (from peroxide in water and from an unknown source in DMSO), and the CO2 capture studies are performed in the solid-state, additional evidence and quantification of superoxide in the solid-state was necessary.
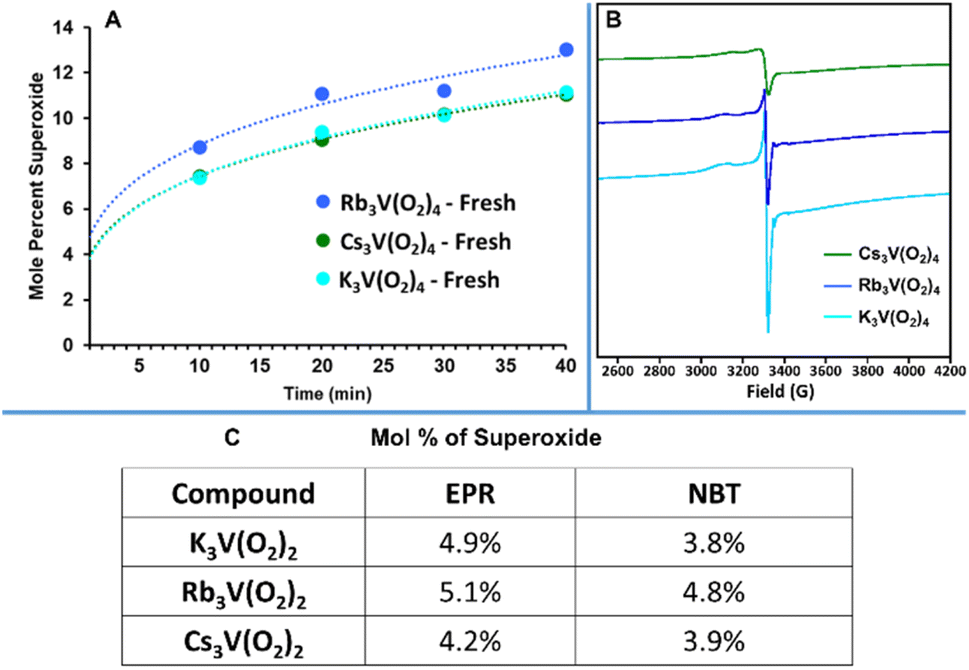 | ||
| Fig. 9 Quantification of superoxide present in K3V(O2)4, Rb3V(O2)4, and Cs3V(O2)4 by: (A) UV-vis data from NBT tests, based on a KO2 standard (Fig. S41†). The trendline of the data points was extrapolated to time ≈ 0 to estimate the starting concentration of superoxide in the solid material (error on data points ∼5%, based on triplicate measurements). (B) EPR spectra of pristine K3V(O2)4, Rb3V(O2)4, and Cs3V(O2)4 recorded at 100 K, using a copper imidazolate standard. (C) Comparison of superoxide quantification present in pristine K3V(O2)4, Rb3V(O2)4, and Cs3V(O2)4 from the EPR tests and NBT tests. | ||
Additional evidence was provided by EPR of K3V(O2)4, Rb3V(O2)4, and Cs3V(O2)4 recorded at 100 K (Fig. 9B). These spectra all show an axial lineshape associated with V5+/superoxide over a broad background peak,57,58 with the g-values tabulated in Table S11.† The g-values show a clear correlation with the cation. Variable temperature spectra were collected from 100 K up to 260 K for K3V(O2)4, Rb3V(O2)4, and Cs3V(O2)4 (Fig. S42†). With increased temperature, the spectra coalesced into a symmetric peak, likely due to increased motion. If the superoxide is bonded to vanadium in the solid-state, we expect hyperfine splitting of the O2− signal into eight peaks from the quadrupolar vanadium nucleus (51V, 99.75% natural abundance, spin 7/2).57 However, not observing this in the solid-state could be due to coupling of proximal spins, typical solid-state broadening, or evidence that the superoxide is not bound to the vanadium-centres. Dilute solution measurements to determine if V-superoxide bonds are present were not possible because the compounds are only soluble in water, and the peroxide ligands dissociate in water. Attempted dissolution in DMSO did not evidence any purple coloration of the solution. However, when spun down to remove the solids, the resulting supernatant exhibited a frozen solution spectrum similar to “free” superoxide in DMSO (KO2 in DMSO is shown for comparison, Fig. S43†). This indicates that the superoxide detected was released from the solid lattice. We determined number of spins (100 K) in the peroxovanadates by double integration of the spectra and comparison with a spin standard (see ESI and Table S10† for details). This revealed that less than 5% (mole percent) superoxide is present per vanadium (Fig. 9C). In summary, both NBT and EPR measurements confirmed that peroxide, not superoxide is the most prevalent reactive oxygen species present in the bulk material The ∼5% superoxide detected may be produced at steady-state as a decomposition product of peroxide, and then further reacts with water or with CO2, also at steady-state.
To complement the experimental determination of the role of superoxide in DAC, we computationally explored the reaction of hypothetical triperoxosuperoxovanadate, [V(O22−)3(O2−)]2/3− with CO2. The salient points are summarized here, while the details are presented in the ESI.† First, [VIV(O22−)3(O2−)]3− instead of [VV(O22−)3(O2−)]2− was chosen as the superoxide model compound because; (1) it is only 1–3 kcal mol−1 less stable than the parent [VV(O22−)4]3− (Table S12†) and therefore accessible, (2) the formation of [VIV(O22−)3(O2−)]3− is a likely mechanism of superoxide formation from peroxide, and (3) the total charge on the anion is constant between [VIV(O22−)3(O2−)]3− and [VV(O22−)4]3−, allowing for easier direct comparison. Notably, the CO2 favourably binds the peroxide ligand, not the superoxide ligand (likely due to the longer O–O bond with more polarizable nature for peroxide), and the vanadium reverts to VV in the end-product (Fig. S44†). The superoxide lowers the barrier of the rate-determining step by ∼3 kcal mol−1, and the energy difference of the complete reaction of K3V(O2)4 + CO2 → K3VO(CO3)(O2)2 for the complex with superoxide vs. without superoxide is likewise <3 kcal mol−1. In summary, computation suggests that vanadium-bound peroxide and superoxide could both be important in the observed CO2 DAC capture reaction.
Practical Aspects of V(O2)43− DAC molecules. Compiled in Table S13† is performance of a variety of leading materials for DAC (including the peroxovanadates reported here) or carbon capture in CO2-rich environments, selected and summarized from two recent reviews.59,60 The range is 0.4 to 17 mmol g−1; and K3V(O2)4 has similar CO2 capture capacity (∼4.9 mmol g−1) as carbon-based adsorbents.
To further put the peroxovanadate performance into context, the DAC behaviour of sodium peroxide (Na2O2) was demonstrated. Total DAC capacity reached a maximum of 9.6 carbon weight% after 3 days (Fig. S45†). Both rate and capacity are greater than that of A3V(O2)4. On the other hand, TGA-MS analyses shows no CO2 release up to 900 °C (Fig. S46 and S47†). This is due to the formation of sodium carbonate, which has a high decomposition temperature. Both Na2O2 and its decomposition products are hygroscopic, evidenced by the increased hydrogen weight% and higher weight loss at lower temperatures, due to water present in the solid (Fig. S47†).
Finally, recovering the starting Na2O2 is not trivial since its commercial synthesis is based on the oxygenation of sodium oxide,61 which would be hard to obtain from sodium carbonate due its aforementioned high thermal stability. Concerning reusability of the peroxovanadate materials for DAC; the peroxovanadate salts are easily dissolved in water at any stage of the process from (CO3)VO(O2)23− to the end product of VO3-plus HCO3−. Dissolution in KOH plus H2O2 followed by precipitation via addition of alcohol, per the synthesis regenerates the A3V(O2)4 materials. We have performed this reaction to demonstrate its feasibility. Since the synthesis is robust and single step; the source of vanadium and the CO32− present in the reprocessing reaction does not alter the product. The challenge to implementing this in an industrial setting is the cooling required for maximum yield of product.
Conclusions
In this study, we have experimentally and computationally demonstrated that reactive alkali vanadium peroxide compounds (containing up to 5% adventitious superoxide) undergo stabilization via direct air capture of CO2, forming VO(CO3)(O2)23−. The re-release of vanadium-bound CO2 by heating (i.e. regeneration step) is simultaneous with vanadium-bound peroxide, with a low release temperature of 250 °C. Alkali carbonate and alkali bicarbonate are also products of carbon capture, especially with long aging times. However, the K-analogue shows evidence that the carbon capture is always via the vanadium-centre, followed by migration of the carbonate to the alkali. In addition, we provide evidence that vanadium catalyses lower temperature release of alkali-bound carbonate (520 °C vs. 850 °C). These studies all show that the alkalis and vanadium are synergistic in both capture and release of atmospheric CO2. The reactivity of vanadium peroxides increases with alkali countercation size (Cs > Rb > K, as shown here); yet the underlying role of the alkalis is still poorly understood. Ongoing experiments and computation include detailed analysis of the Li and Na analogues of vanadium tetraperoxide to better understand the complex role of the alkalis. We are also investigating the DAC performance of the d0 transition metal peroxide neighbours including Nb, Ta, Mo, W and Ti by computation and experiment. Emerging trends in reactivity will inform and enable the use of a range of peroxide-functionalized inorganic oxides in DAC.Data availability
Crystallographic data for compounds 2 and 3 have been deposited at the CCDC under 2264013 and 2264012, respectively, and can be obtained from https://www.ccdc.cam.ac.uk/structures/. Computational input files and structures can be accessed through the public GitHub repository (https://github.com/tjz21/DAC_peroxovanadates) or through Zenodo (https://doi.org/10.5281/zenodo.10266510).Author contributions
EGR methodology, investigation, visualization and writing; ZM methodology and investigation, JSH investigation, visualization and writing, TL investigation, KB investigation, EDW investigation and writing, CRS investigation, TJZ writing and supervision, MN conceptualization, supervision, writing and visualization, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Department of Energy under the grant DE-SC0022278. The authors would also like to thank Kylie Welch and Miguel Goni for their help with carbon microanalysis. The EPR data collection at PNNL (EDW) was supported by project award 10.46936/staf.proj.2023.61003/60008901 from the Environmental Molecular Sciences Laboratory, a DOE Office of Science User Facility sponsored by the Biological and Environmental Research program under Contract No. DE-AC05-76RL01830.Notes and references
- K. O. Yoro and M. O. Daramola, in Advances in Carbon Capture, ed. M. R. Rahimpour, M. Farsi and M. A. Makarem, Woodhead Publishing, 2020, pp. 3–28, DOI:10.1016/B978-0-12-819657-1.00001-3.
- D. Shindell and C. J. Smith, Climate and air-quality benefits of a realistic phase-out of fossil fuels, Nature, 2019, 573, 408–411 CrossRef CAS.
- T. Wilberforce, A. Olabi, E. T. Sayed, K. Elsaid and M. A. Abdelkareem, Progress in carbon capture technologies, Sci. Total Environ., 2021, 761, 143203 CrossRef CAS.
- A. Gambhir and M. Tavoni, Direct Air Carbon Capture and Sequestration: How It Works and How It Could Contribute to Climate-Change Mitigation, One Earth, 2019, 1, 405–409 CrossRef.
- S. Deutz and A. Bardow, Life-cycle assessment of an industrial direct air capture process based on temperature–vacuum swing adsorption, Nat. Energy, 2021, 6, 203–213 CrossRef CAS.
- P. Gabrielli, M. Gazzani and M. Mazzotti, The role of carbon capture and utilization, carbon capture and storage, and biomass to enable a net-zero-CO2 emissions chemical industry, Ind. Eng. Chem., 2020, 59, 7033–7045 CrossRef CAS.
- X. Shi, Y. Lin and X. Chen, Development of sorbent materials for direct air capture of CO2, MRS Bull., 2022, 47, 405–415 CrossRef CAS.
- R. W. J. Edwards and M. A. Celia, Infrastructure to enable deployment of carbon capture, utilization, and storage in the United States, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, E8815–E8824 CAS.
- N. McCann, D. Phan, X. Wang, W. Conway, R. Burns, M. Attalla, G. Puxty and M. Maeder, Kinetics and Mechanism of Carbamate Formation from CO2(aq), Carbonate Species, and Monoethanolamine in Aqueous Solution, J. Phys. Chem. A, 2009, 113, 5022–5029 CrossRef CAS PubMed.
- F. O. Ochedi, J. Yu, H. Yu, Y. Liu and A. Hussain, Carbon dioxide capture using liquid absorption methods: a review, Environ. Chem. Lett., 2021, 19, 77–109 CrossRef CAS.
- Y. Hu, Y. Guo, J. Sun, H. Li and W. Liu, Progress in MgO sorbents for cyclic CO 2 capture: a comprehensive review, J. Mater. Chem. A, 2019, 7, 20103–20120 RSC.
- P. E. Sánchez Jiménez, A. Perejón, M. Benítez Guerrero, J. M. Valverde, C. Ortiz and L. A. Pérez Maqueda, High-performance and low-cost macroporous calcium oxide based materials for thermochemical energy storage in concentrated solar power plants, Appl. Energy, 2019, 235, 543–552 CrossRef.
- N. McQueen, P. Kelemen, G. Dipple, P. Renforth and J. Wilcox, Ambient weathering of magnesium oxide for CO2 removal from air, Nat. Commun., 2020, 11, 3299 CrossRef CAS PubMed.
- M. Samari, F. Ridha, V. Manovic, A. Macchi and E. J. Anthony, Direct capture of carbon dioxide from air via lime-based sorbents, Mitig. Adapt. Strateg. Glob. Chang., 2020, 25, 25–41 CrossRef.
- D. Fu, Y. Park and M. E. Davis, Zinc Containing Small-Pore Zeolites for Capture of Low Concentration Carbon Dioxide, Angew. Chem., Int. Ed., 2022, 61, e202112916 CrossRef CAS.
- D. Fu, Y. Park and M. E. Davis, Confinement effects facilitate low-concentration carbon dioxide capture with zeolites, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2211544119 CrossRef CAS.
- R. Gonzalez-Olmos, A. Gutierrez-Ortega, J. Sempere and R. Nomen, Zeolite versus carbon adsorbents in carbon capture: A comparison from an operational and life cycle perspective, J. CO2 Util., 2022, 55, 101791 CrossRef CAS.
- C. C. Turvey, S. A. Wilson, J. L. Hamilton, A. W. Tait, J. McCutcheon, A. Beinlich, S. J. Fallon, G. M. Dipple and G. Southam, Hydrotalcites and hydrated Mg-carbonates as carbon sinks in serpentinite mineral wastes from the Woodsreef chrysotile mine, New South Wales, Australia: Controls on carbonate mineralogy and efficiency of CO2 air capture in mine tailings, Int. J. Greenhouse Gas Control, 2018, 79, 38–60 CrossRef CAS.
- D. Suescum-Morales, J. R. Jiménez and J. M. Fernández-Rodríguez, Review of the Application of Hydrotalcite as CO2 Sinks for Climate Change Mitigation, Chem. Eng., 2022, 6, 50 CAS.
- S. A. Didas, S. Choi, W. Chaikittisilp and C. W. Jones, Amine–Oxide Hybrid Materials for CO2 Capture from Ambient Air, Acc. Chem. Res., 2015, 48, 2680–2687 CrossRef CAS.
- A. Cherevotan, J. Raj and S. C. Peter, An overview of porous silica immobilized amines for direct air CO2 capture, J. Mater. Chem. A, 2021, 9, 27271–27303 RSC.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Carbon capture and conversion using metal–organic frameworks and MOF-based materials, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- C. G. Piscopo and S. Loebbecke, Strategies to Enhance Carbon Dioxide Capture in, Met.-Org. Frameworks, 2020, 85, 538–547 CAS.
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Sorbents for the Direct Capture of CO2 from Ambient Air, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS.
- H. Jakob, S. Leininger, T. Lehmann, S. Jacobi and S. Gutewort, Peroxo Compounds, Inorganic, in Ullmann's Encyclopedia of Industrial Chemistry, 2007, DOI:10.1002/14356007.a19_177.pub2.
- M. Dakanali, E. T. Kefalas, C. P. Raptopoulou, A. Terzis, G. Voyiatzis, I. Kyrikou, T. Mavromoustakos and A. Salifoglou, A New Dinuclear Ti(IV)−Peroxo−Citrate Complex from Aqueous Solutions. Synthetic, Structural, and Spectroscopic Studies in Relevance to Aqueous Titanium(IV)−Peroxo−Citrate Speciation, Inorg. Chem., 2003, 42, 4632–4639 CrossRef CAS PubMed.
- M. Grzywa, M. Różycka and W. Łasocha, Crystal structure of potassium tetraperoxomolybdate (VI) K2[Mo(O2)4], Powder Diffr., 2005, 20, 203–206 CrossRef CAS.
- M. Grzywa, W. Nitek and W. Łasocha, Synthesis, characterization and crystal structures of lithium tetraperoxomolybdate (VI) tetrahydrate Li2[Mo(O2)4]·4H2O and lithium tetraperoxotungstate (VI) tetrahydrate Li2[W(O2)4]·4H2O, J. Mol. Struct., 2007, 828, 111–115 CrossRef CAS.
- H. Haeuseler, M. Wagener and H. Müller, Preparation, Crystal Structure, and Vibrational Spectra of Rubidiumperoxoniobate (Rb3NbO8), Z. Naturforsch. B, 1997, 52, 1082–1086 CrossRef CAS.
- G. Wehrum and R. Hoppe, On the constitution of peroxotantalates (V) with alkali metals: on the structure of K3Ta(O2)4, Z. Anorg. Allg. Chem., 1993, 619 Search PubMed.
- R. Stomberg and C. Brosset, The Crystal Structure of Potassium Perchromate, Acta Chem. Scand., 1960, 14, 2 CrossRef.
- T.-J. Won, C. L. Barnes, E. O. Schlemper and R. C. Thompson, Two crystal structures featuring the tetraperoxovanadate (V) anion and a brief reinvestigation of peroxovanadate equilibria in neutral and basic solutions, Inorg. Chem., 1995, 34, 4499–4503 CrossRef CAS.
- Z. W. Yu, P. A. Jansson, B. I. Posner, U. Smith and J. W. Eriksson, Peroxovanadate and insulin action in adipocytes from NIDDM patients. Evidence against a primary defect in tyrosine phosphorylation, Diabetologia, 1997, 40, 1197–1203 CrossRef CAS PubMed.
- V. Conte and B. Floris, Vanadium and molybdenum peroxides: synthesis and catalytic activity in oxidation reactions, Dalton Trans., 2011, 40, 1419–1436 RSC.
- D. V. Kravchuk, N. N. Dahlen, S. J. Kruse, C. D. Malliakas, P. M. Shand and T. Z. Forbes, Isolation and reactivity of uranyl superoxide, Angew. Chem., Int. Ed., 2021, 60, 15041–15048 CrossRef CAS PubMed.
- D. V. Kravchuk and T. Z. Forbes, Thermodynamics and Chemical Behavior of Uranyl Superoxide at Elevated Temperatures, ACS Mater. Au, 2022, 2, 33–44 CrossRef CAS.
- P. Schwendt, J. Tatiersky, L. Krivosudský and M. Šimuneková, Peroxido complexes of vanadium, Coord. Chem. Rev., 2016, 318, 135–157 CrossRef CAS.
- P. Schwendt and D. Uskert, Thermal-Decomposition of Potassium Mu-Oxo- and Mu-Hydroxo-Bis(Oxo-Diperoxovanadate), Chem. Zvesti, 1981, 35, 229–233 CAS.
- G. Arrambide, J. Rivadeneira, S. Etcheverry, B. Parajón-Costa, D. Gambino and E. Baran, Spectroscopic Behavior and Biological Activity of K3[VO(O2)2CO3]·H2O, Biol. Trace Elem. Res., 2009, 132, 176–183 CrossRef CAS PubMed.
- G. Zou, Z. Lin, H. Zeng, H. Jo, S.-J. Lim, T.-S. You and K. M. Ok, Cs3VO(O2)2CO3: an exceptionally thermostable carbonatoperoxovanadate with an extremely large second-harmonic generation response, Chem. Sci., 2018, 9, 8957–8961 RSC.
- I. Knopf, T. Ono, M. Temprado, D. Tofan and C. C. Cummins, Uptake of one and two molecules of CO2 by the molybdate dianion: a soluble, molecular oxide model system for carbon dioxide fixation, Chem. Sci., 2014, 5, 1772–1776 RSC.
- M. Grzywa and W. Łasocha, Crystal structure of potassium and ammonium tetraperoxovanadates (V), Z. Kristallogr. - Cryst. Mater., 2007, 222, 95–98 CrossRef CAS.
- A. Arteaga, L. Zhang, S. Hickam, M. Dembowski, P. C. Burns and M. Nyman, Uranyl–Peroxide Capsule Self-Assembly in Slow Motion, Eurasian J. Chem., 2019, 25, 6087–6091 CrossRef CAS PubMed.
- D. V. Kravchuk, N. N. Dahlen, S. J. Kruse, C. D. Malliakas, P. M. Shand and T. Z. Forbes, Isolation and reactivity of uranyl superoxide, Angew. Chem., Int. Ed., 2021, 60, 15041–15048 CrossRef CAS PubMed.
- M. Grzywa, W. Łasocha and D. Rutkowska-Żbik, Structural investigation of tetraperoxo complexes of Mo (VI) and W (VI): X-ray and theoretical studies, J. Solid State Chem., 2009, 182, 973–982 CrossRef CAS.
- K. Coenen, F. Gallucci, B. Mezari, E. Hensen and M. van Sint Annaland, An in-situ IR study on the adsorption of CO2 and H2O on hydrotalcites, J. CO2 Util., 2018, 24, 228–239 CrossRef CAS.
- S. Clark, M. Segall, C. Pickard, P. Hasnip, M. Probert, K. Refson and M. C. Payne, First Principles Methods Using CASTEP, Z. Kristallogr. - Cryst. Mater., 2005, 220, 567 CrossRef CAS.
- D. Bayot, M. Devillers and D. Peeters, Vibrational spectra of eight-coordinate niobium and tantalum complexes with peroxo ligands: A theoretical simulation, Eur. J. Inorg. Chem., 2005, 2005, 4118–4123 CrossRef.
- E. Mutlu, T. Cristy, S. W. Graves, M. J. Hooth and S. Waidyanatha, Characterization of aqueous formulations of tetra-and pentavalent forms of vanadium in support of test article selection in toxicology studies, Environ. Sci. Pollut. Res., 2017, 24, 405–416 CrossRef CAS.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision A. 03, Gaussian, Inc., Wallingford CT, 2016, 3 Search PubMed.
- W. Ma, Y. Qiao, N. Theyssen, Q. Zhou, D. Li, B. Ding, D. Wang and Z. Hou, A mononuclear tantalum catalyst with a peroxocarbonate ligand for olefin epoxidation in compressed CO2, Catal. Sci. Technol., 2019, 9, 1621–1630 RSC.
- M. Yamashita, K. Goto and T. Kawashima, Fixation of both O2 and CO2 from air by a crystalline palladium complex bearing N-heterocyclic carbene ligands, J. Phys. Chem. B, 2005, 127, 7294–7295 CAS.
- T. A. Manz and B. Yang, Selective oxidation passing through η3-ozone intermediates: applications to direct propene epoxidation using molecular oxygen oxidant, RSC Adv., 2014, 4, 27755–27774 RSC.
- D. Wang, J. Zou, H. Cai, Y. Huang, F. Li and Q. Cheng, Effective degradation of Orange G and Rhodamine B by alkali-activated hydrogen peroxide: roles of HO2− and O2, Environ. Sci. Pollut. Res., 2019, 26, 1445–1454 CrossRef CAS.
- B. H. Bielski, G. G. Shiue and S. Bajuk, Reduction of nitro blue tetrazolium by CO2-and O2-radicals, J. Phys. Chem., 1980, 84, 830–833 CrossRef CAS.
- P. A. Giguere and K. B. Harvey, An Infrared Study of Hydrogen Bonding in Solid H2O2 and H2O-H2O2 Mixtures, J. Mol. Spectrosc., 1959, 3, 36–45 CrossRef CAS.
- V. N. Shetti, M. J. Rani, D. Srinivas and P. Ratnasamy, Chemoselective Alkane Oxidation by Superoxo−Vanadium(V) in Vanadosilicate Molecular Sieves, J. Am. Chem. Soc., 2006, 110, 677–679 CAS.
- K. Sobańska, A. Krasowska, T. Mazur, K. Podolska-Serafin, P. Pietrzyk and Z. Sojka, Diagnostic Features of EPR Spectra of Superoxide Intermediates on Catalytic Surfaces and Molecular Interpretation of Their g and A Tensors, Top. Catal., 2015, 58, 796–810 CrossRef.
- D. Panda, V. Kulkarni and S. K. Singh, Evaluation of amine-based solid adsorbents for direct air capture: a critical review, React. Chem. Eng., 2023, 8, 10–40 RSC.
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Industrial carbon dioxide capture and utilization: state of the art and future challenges, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- E. Dönges, Lithium and Sodium Peroxides, Academic Press, NY, 1963 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: For supplementary figures and tables and text describing synthesis, crystallography, spectroscopic information, and computational studies. CCDC 2264012 and 2264013. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05381d |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |