
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence109Ag NMR chemical shift as a descriptor for Brønsted acidity from molecules to materials†
Colin
Hansen‡
 ,
Scott R.
Docherty‡
,
Weicheng
Cao
,
Alexander V.
Yakimov
and
Christophe
Copéret
*
,
Scott R.
Docherty‡
,
Weicheng
Cao
,
Alexander V.
Yakimov
and
Christophe
Copéret
*
ETH Zurich, Department of Chemistry and Applied Biosciences, Vladimir Prelog Weg 1-5, CH-8093 Zurich, Switzerland. E-mail: ccoperet@inorg.chem.ethz.ch
First published on 17th January 2024
Abstract
Molecular-level understanding of the acid/base properties of heterogeneous catalysts requires the development of selective spectroscopic probes to establish structure–activity relationships. In this work we show that substituting the surface protons in oxide supports by isolobal N-heterocyclic carbene (NHC) Ag cations and measuring their 109Ag nuclear magnetic resonance (NMR) signatures enables to probe the speciation and to evaluate the corresponding Brønsted acidity of the substituted OH surface sites. Specifically, a series of silver N-heterocyclic carbene (NHC) Ag(I) complexes of general formula [(NHC)AgX] are synthesized and characterized, showing that the 109Ag NMR chemical shift of the series correlates with the Brønsted acidity of the conjugate acid of X− (i.e., HX), thus establishing an acidity scale based on 109Ag NMR chemical shift. The methodology is then used to evaluate the Brønsted acidity of the OH sites of representative oxide materials using Dynamic Nuclear Polarization (DNP-)enhanced solid-state NMR spectroscopy.
Introduction
Heterogeneous catalysis largely relies on the use of high surface area oxide materials as catalysts or supports. Notably, these oxides contain surface OH groups that often determine the reactivity of heterogeneous catalysts. Therefore, evaluating the strength and the speciation of surface hydroxyl groups and their Brønsted acidity, is critical to establish detailed structure–activity relationships and it remains an intense field of research with many challenges.1–3 In many instances, the Brønsted acidity of specific (–OH) sites in materials is difficult to assess directly, due to the absence of spectroscopic signatures directly associated with acidity, dynamic averaging and the typical presence of a distribution of OH sites.Approaches involving probe molecules, in conjunction with either calorimetry/temperature-programmed desorption (TPD), or spectroscopy (IR/NMR) are often conducted to understand the strength or the nature of acid sites in materials respectively. However, interpretation of these data is often complicated by the presence of various OH sites with different Brønsted acidities, along with Lewis acid sites (LAS), whose presence can interfere with the analysis, due to overlapping signals or dynamics.4,5 In many instances, studies directed at determining the Brønsted acidity of materials are limited to the measurement of their acid strength, i.e. their ability to (reversibly) protonate a given probe molecule, such as pyridine or ammonia.6–14 However, evaluating both the types and the strength of acid sites remains a challenge. Thus, developing bespoke probe molecules, that are able to evaluate the specific Brønsted acidity of various surface OH sites of a support is of importance; the probe would ideally react specifically with OH groups, respond to each of them with a specific (spectroscopic) signature and not be susceptible to dynamic exchange between sites (i.e. bound covalently) to avoid averaging information.
Towards this goal, we explore the use of a ‘chemical surrogate’ for protons (H+) as a means of obtaining insights into the Brønsted acidity of a given BAS. We reason that the substitution of protons by an isolobal equivalent with a distinct spectroscopic signature related to NMR chemical shift could be a possible approach.15 [(NHC)AgR] with a reactive and readily protonolysed R group is chosen for this purpose, because Ag+ is formally isolobal to H+, due to the closed-shell nature of the d10 electron configuration (Scheme 1a) and because Ag has favourable NMR properties with two I = ½ isotopes, namely 107Ag and 109Ag.16,17
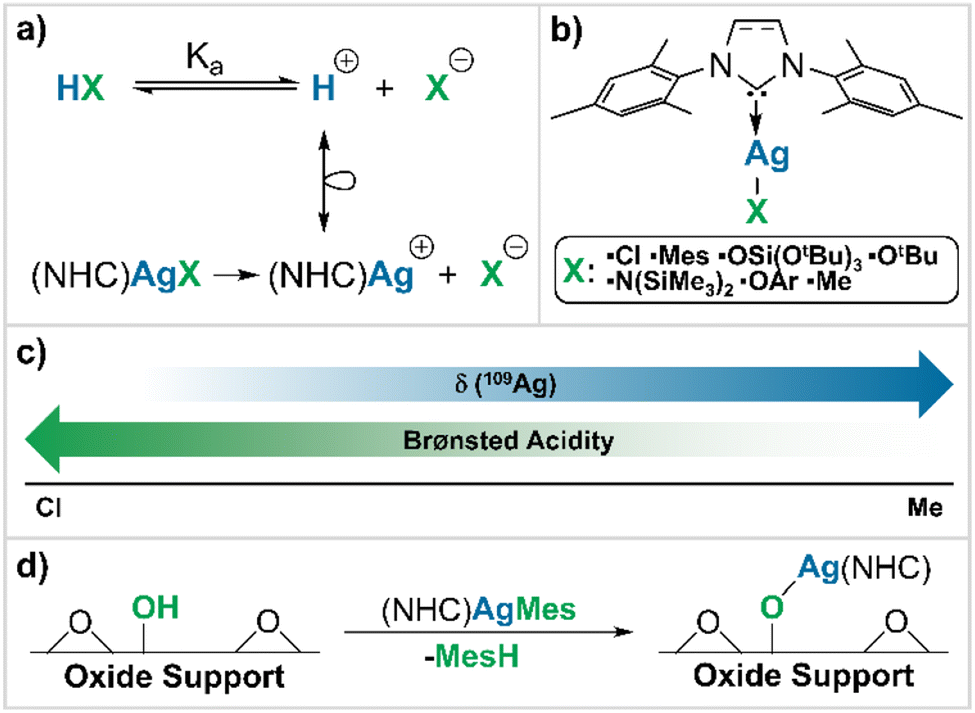 | ||
| Scheme 1 General approach to the determination of a correlation between δ109Ag and Brønsted acidity of the conjugate acid of the studied X-type ligands. (a) Isolobal analogy between Brønsted acid HX and the corresponding (NHC)AgX complex; (b) series of (NHC)AgX complexes explored in this work; (c) general trends between Brønsted acidity and δ109Ag; (d) grafting reaction of the molecular probe onto a generic oxide support. | ||
Since NMR chemical shifts of metals are, to a first approximation, directly related to the σ-donating ability of their ligands,15 we reasoned that a correlation between 109Ag NMR chemical shift (δ109Ag) and the nature of an anionic ligand (X−) should exist, making δ109Ag a potential descriptor for the acidity of the corresponding Brønsted acid, HX. To establish the possible correlation between 109Ag chemical shift and Brønsted acidity, a library of monomeric Ag(I) N-heterocyclic carbene (NHC) complexes of general structure (NHC)AgX are synthesized (Scheme 1b). Here we show a linear relationship between δ109Ag and the Brønsted acidity of the conjugate acid of X− (HX) (Scheme 1c),18 which we use to evaluate the Brønsted acidity of surface hydroxyl groups on a series of representative oxide supports commonly employed in heterogeneous catalysis, namely silica, alumina and silica–alumina (Scheme 1d).
Results and discussion
We first synthesize a series of monomeric, isostructural, molecular Ag complexes, (SIMes)AgX (A–X). To cover a broad range of Brønsted acidity, ligands are chosen from chloride (HCl, high Brønsted acidity, pKa = −5.9 (ref. 19)) to methyl (MeH, very low Brønsted acidity, pKa = 48 (ref. 20)). Salt metathesis is used to prepare (SIMes)AgMes (A–Mes) [Mes = 2,4,6-trimethylphenyl, SIMes = 1,3-bis-(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene], (SIMes)AgOtBu (A–OtBu), (SIMes)AgOSi(OtBu)3 (A–TBOS) [TBOS = tris-tert-butoxysiloxy-], (SIMes)AgN(SiMe3)2 (A–HMDS) and (SIMes)AgMe (A–Me) by starting from (SIMes)AgCl (A–Cl) (see Scheme 2a).21–24 (SIMes)AgOAr [Ar = 2,6-(tBu)-4-(Me)-C6H2] (A–BHT) is synthesized via protonolysis, by reaction of (A–Mes) with 1 equivalent of 2,6-di-tert-butyl-4-methylphenol, giving the product in good yield (Scheme 2b) (see ESI S2† for experimental details).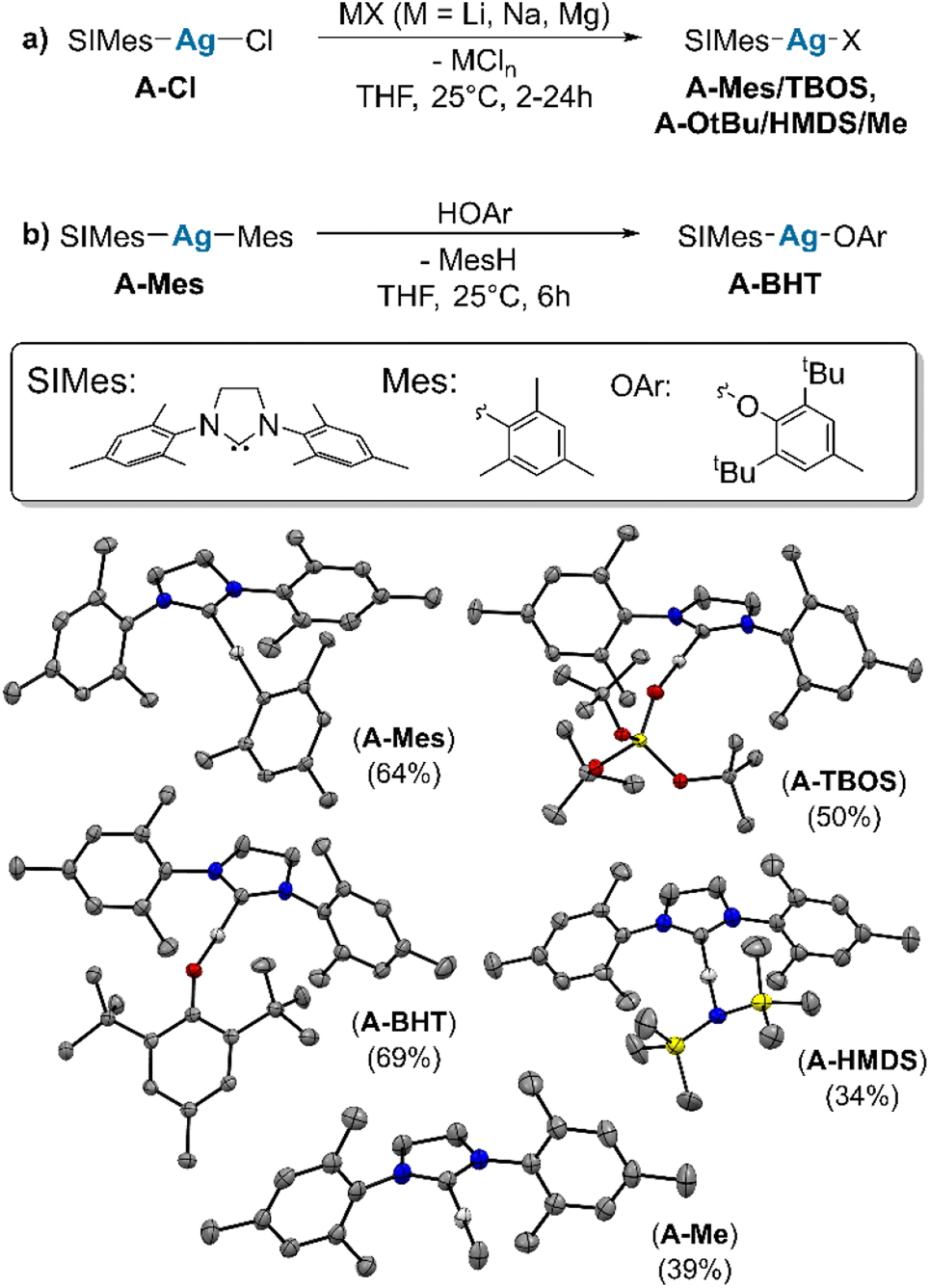 | ||
| Scheme 2 Synthesis of molecular compounds of general structure (SIMes)AgX via salt metathesis (a) and protonolysis (b). (Ar = 2,6-(tBu)-4-(Me)-C6H2). Crystal structures of all crystallized complexes and corresponding yields. (TBOS = –OSi(OtBu)3, HMDS = –N(SiMe3)2) (crystal structures depicted at 50% probability with the exception of A–HMDS which has ellipsoids at 25% probability for better visibility). | ||
Single crystals suitable for X-ray diffraction (XRD), were obtained for all compounds (see Scheme 2), except for A–OtBu. Issues with the crystallization of (NHC)M(OtBu) were also reported for related (SIDipp)AgOtBu complexes [SIDipp = 1,3-bis(2,6-diisopropyl-phenyl)-imidazolidine-2-ylidene] and other (NHC)M(OtBu) complexes of coinage metals.25–29 XRD data confirms the identity of the compounds in the solid state, and is fully consistent with data obtained by 1H and 13C NMR spectroscopy (all spectra can be found in the ESI S2†). The structural features are similar across the series, in that the NHC–Ag–X angle lies very close to 180° in all complexes (see ESI S5†).
The 13C NMR spectra of all complexes show the signal corresponding to the carbenic carbon directly bound to Ag, where the coupling between the carbon and Ag appears as two doublets, associated with the coupling with the two silver isotopes (107Ag and 109Ag, both I = ½; isotope ratio 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :48) that have distinguishable J-couplings (1J13C–107Ag ≈ 113–220 Hz, 1J13C–109Ag ≈ 130–253 Hz). It is also notable that for (SIMes)AgX complexes, the chemical shift of the carbenic carbon falls within a narrow range (Δδiso < 6 ppm across the series) illustrating that δ13C provides limited information regarding the nature of X− in this series.
:48) that have distinguishable J-couplings (1J13C–107Ag ≈ 113–220 Hz, 1J13C–109Ag ≈ 130–253 Hz). It is also notable that for (SIMes)AgX complexes, the chemical shift of the carbenic carbon falls within a narrow range (Δδiso < 6 ppm across the series) illustrating that δ13C provides limited information regarding the nature of X− in this series.
Subsequently, values for δ109Ag were obtained from two-dimensional 1H–109Ag HMQC experiments.18,30 In all complexes cross-peaks between 109Ag and 1H are observed in the HMQC spectra (see ESI S3† for 2D spectra). The observed δ109Ag ranged from 539 ppm in (A–Cl) to 810 ppm in (A–Me), following what is expected from the σ-donation of the X-type ligand related to its electronegativity (see Fig. 1). To assess the influence of the choice of NHC, (IMes)AgCl (B–Cl) [IMes = 1,3-Bis-(2,4,6-trimethylphenyl)-imidazol-2-yliedene], (IMes)AgMes (B–Mes), and (IMes)AgOSi(OtBu)3 (B–TBOS) are also synthesized and their δ109Ag measured. Comparison between the SIMes complexes and the IMes complexes reveals a systematic increase in δ109Ag (ca. 5–10 ppm), illustrating the importance of maintaining the same NHC when comparing series.
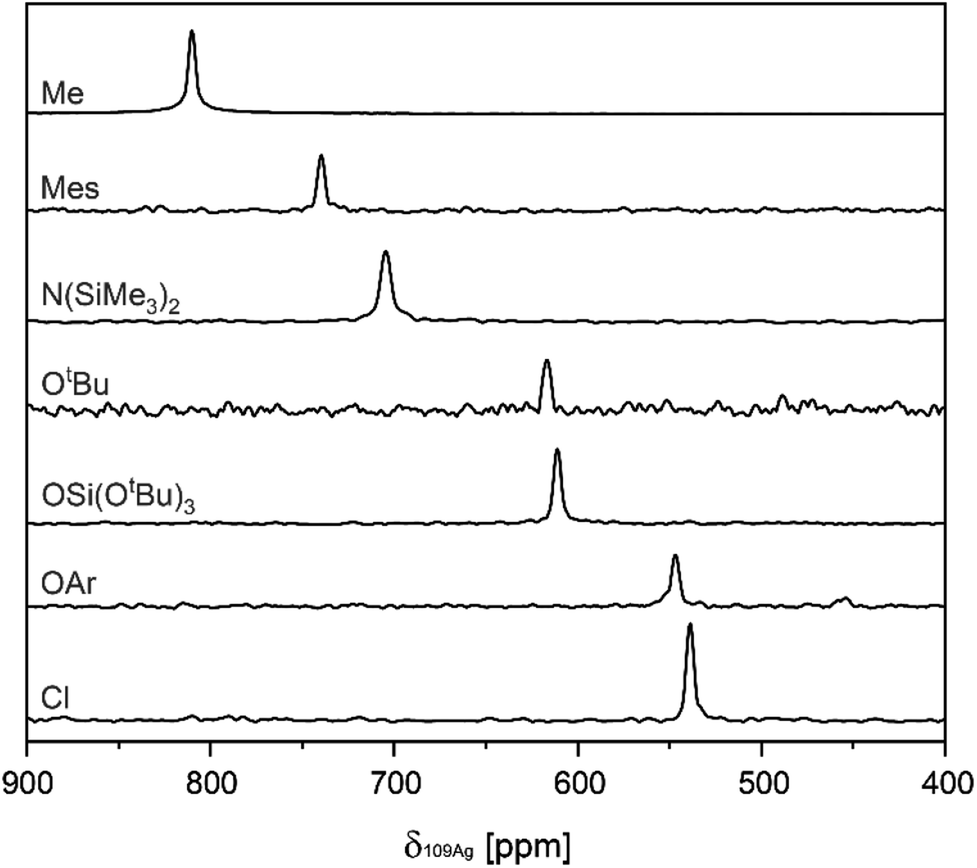 | ||
| Fig. 1 Overview of projections extracted from 1H–109Ag HMQC spectra, containing values for δ109Ag in the A series. | ||
Next, we explored the relationship between δ109Ag and the Brønsted acidity of the conjugate acid (HX). As pKa values are strongly dependent on solvation effects,31 we chose first to evaluate gas phase acidity (deprotonation enthalpy, DPE) using density functional theory (DFT) (i.e. the enthalpy of the release of a proton and formation of the anion in gas phase, ΔHacid (ref. 32)). The obtained values are in good agreement with those obtained in literature (R2 = 0.955) (see ESI S6† for further details), enabling comparison of relative acidities for all systems.33 Notably, a linear relationship is obtained between δ109Ag and ΔHacid, with two significant outliers (A–OtBu and A–HMDS) (Fig. 2 [top], R2 = 0.812), which can be explained by different behaviour of the molecular compound in solution vs. gas phase. Similarly, δ109Ag correlates linearly (Fig. 2 [bottom], R2 = 0.906) with the experimental pKa values in water, found in literature or estimated from data for closely related structures (see ESI S6† for tabulated data).19,20,34 Overall, the presented data shows that there is a strong linear correlation between δ109Ag and X-type ligand acidity/basicity, showing that δ109Ag can be used as a quantitative descriptor for Brønsted acidity of X–H groups.
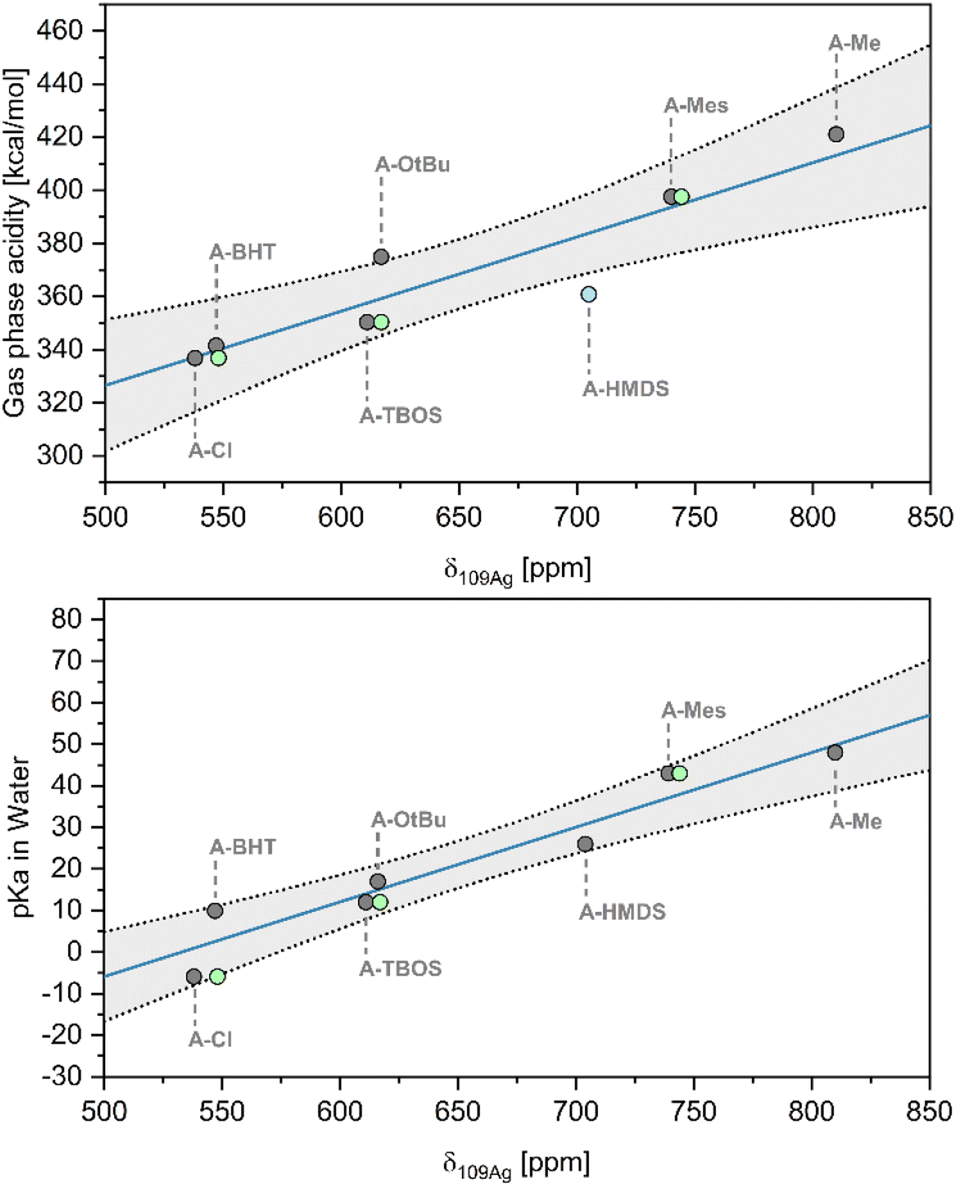 | ||
| Fig. 2 Linear correlations between δ109Ag and calculated gas phase acidity (DPE) ([top] (R2 = 0.812)) and literature pKa values in H2O ([bottom] (R2 = 0.906)). Grey circles show the saturated complexes (A), and green circles represent unsaturated NHC complexes (B). | ||
Having established that δ109Ag correlates with Brønsted acidity, we extended our efforts to the study of oxide supports relevant to heterogeneous catalysis, choosing to interrogate the acidity of widely-used oxide supports that are known to contain Brønsted acid sites (BAS) of different strength – silica, alumina, and silicated alumina (SiO2, γ-Al2O3, Si/γ-Al2O3). Towards this goal, the partially dehydroxylated oxide supports are contacted with a solution of A–Mes, as such molecular compounds react selectively via deprotonation of surface –OH groups to release MesH (see Scheme 1d). This reaction is evidenced by IR spectroscopy through the disappearance of –OH bands at >3600 cm−1, as well as the emergence of C–H stretching bands (3000–2700 cm−1) (see ESI S2† for further experimental details). Elemental analysis and 13C NMR confirms grafting and the formation of (NHC)Ag–O![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) surface sites and the replacement of protons by the isolobal [Ag(NHC)]+ surface fragment (see ESI S4†).
surface sites and the replacement of protons by the isolobal [Ag(NHC)]+ surface fragment (see ESI S4†).
Subsequently, 109Ag solid-state NMR spectroscopy is used to interrogate the nature of surface sites present. To overcome the low inherent sensitivity of 109Ag NMR, we turn to dynamic nuclear polarization surface enhanced NMR spectroscopy (DNP SENS), which is used to reduce measurement times in NMR by orders of magnitude.35–37 Using this approach, it is possible to extract the isotropic 109Ag NMR chemical shift (δiso(109Ag)) and spinning sideband manifold for the 109Ag NMR signal of (NHC)Ag–O grafted on each of the studied oxides. Notably, judicious choice of the DNP formulation is required, since the materials are not compatible with either aqueous or halide-containing formulations, that are often used for DNP, due to the inherent reactivity of the NHC–M fragments.38 We thus opt for the use of a toluene-based DNP formulation to avoid possible side reactions of the DNP formulation with surface sites (proton enhancements (εH) = 9–16, see ESI S4†). Fig. 3a and b show the 109Ag solid-state NMR spectra for A–Mes grafted on γ-Al2O3 and SiO2 (Ag@γ-Al2O3 and Ag@SiO2) recorded with magic angle spinning (MAS) at 8 kHz. The δiso(109Ag) for (SIMes)Ag@γ-Al2O3 is 598 ppm (FWHM = 59 ppm), while δiso(109Ag) for (SIMes)Ag@SiO2 is centered at 569 ppm (FWHM = 60 ppm) (see ESI S4† for an exhaustive analysis of solid-state NMR of grafted complexes and their simulated spectra with the full spinning sideband manifold). These signals are assigned to [(SIMes)Ag] grafted on Al–OH and Si–OH, respectively (Fig. 3a and b). By interpolation, gas phase acidities of approximately 354 kcal mol−1 and 346 kcal mol−1 are obtained for the surface hydroxyl groups of γ-Al2O3 and SiO2. Analogously pKa values of approx. 12 and 7 were estimated, respectively. The difference in pKa (ΔpKa ≈ 5), is consistent with previously reported Brønsted acidity trends.39
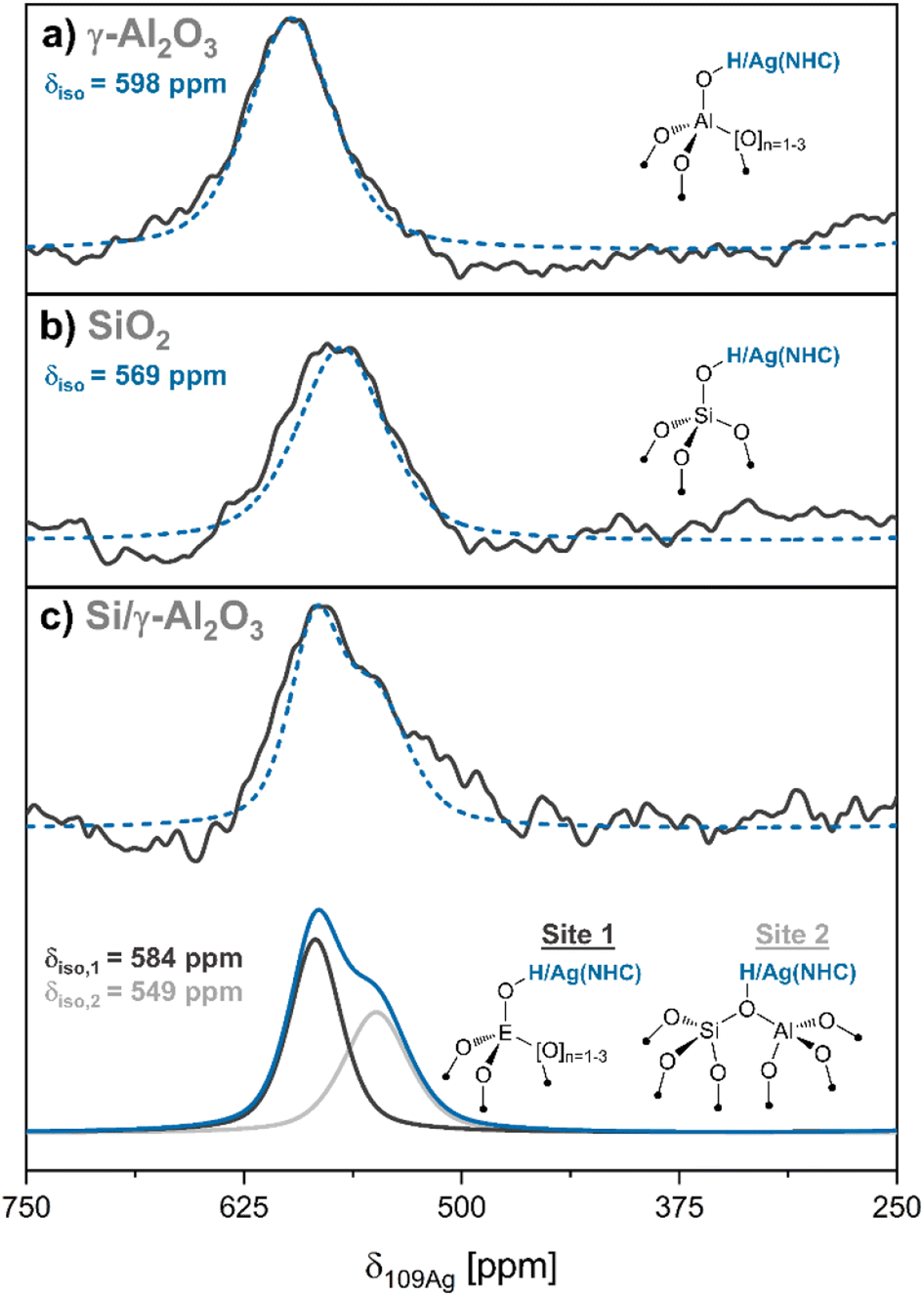 | ||
| Fig. 3 Isotropic signals in DNP-enhanced 109Ag NMR spectra of oxide-supported (SIMes)Ag@MOx (recorded with MAS at 8 kHz, see ESI S4† for the full spectra including spinning sideband manifolds) (fitted spectra in blue and experimental spectra in black) (a) (SIMes)Ag@γ-Al2O3 (δiso = 598 ppm). (b) (SIMes)Ag@SiO2 (δiso = 569 ppm). (c) [top] Experimental spectrum of Ag@Si/γ-Al2O3. [bottom] Fitted spectrum of Ag@Si/γ-Al2O3 showing isotropic chemical shifts of 584 ppm and 549 ppm (decomposition shown in green and light blue). The sites associated with the signals are shown next to the respective signals (E = Si/Al). | ||
We next look at evaluating the Brønsted acidity of a silicated alumina material (Si/γ-Al2O3), prepared by dispersion of Si on alumina (experimental description and characterization by 29Si NMR and 15N pyridine experiments can be found in ESI S4†).40–44 Pyridine adsorption experiments and 15N NMR show the formation of pyridinium, which is not observed in either SiO2 or γ-Al2O3, indicating an increased Brønsted acidity of this Si/γ-Al2O3.40,45,46 Following reaction with A–Mes to form Ag@Si/γ-Al2O3 (Fig. 3c), 109Ag DNP SENS shows the presence of two main species, with peaks centered at 584 ppm (FWHM = 35 ppm) and 549 ppm (FWHM = 45 ppm), consistent with the presence of two families of sites. The site associated with a δiso(109Ag) of 549 ppm, clearly indicates the presence of stronger Brønsted acid sites in Si/γ-Al2O3 (DPE = 340 kcal mol−1, pKa ≈ 3) when compared to SiO2 or γ-Al2O3, which is consistent with the observation that pyridinium forms on Si/γ-Al2O3. This site is ascribed to the presence of Brønsted acidic pseudo-bridging silanol sites, where a silanol interacts with a neighboring, coordinatively-unsaturated Al site (Fig. 3c).47,48 The second type of site, centered at 584 ppm (associated with a DPE of ca. 350 kcal mol−1 and a pKa of approx. 9) is closer to the shift found for alumina itself, indicating the presence of residual Al–OH.
Conclusions
A set of linear two-coordinate Ag complexes, NHC–Ag–X, with anionic ligands spanning a broad range of σ-donating ability (from –Cl to –Me) are successfully synthesized and fully characterized. This series displays a linear relationship between δiso(109Ag) and the Brønsted acidity of the conjugate acid of the anionic ligand. The grafting of NHC–Ag–Mes on oxide supports (silica, alumina, and silica–alumina) generates the corresponding Osurface–Ag surface sites, whose 109Ag NMR chemical shift (δiso(109Ag)) can be measured by DNP SENS, thereby enabling to determine the presence of different surface sites and to evaluate the Brønsted acidity of the corresponding surface hydroxyl groups. This approach is likely general and can be extended to a broad range of supports, provided NHC–AgMes reacts with protic groups at the surface. We are currently expanding the types of probe molecules and exploring the determination of acidity on other supports.Experimental
Experimental procedures, purification procedures for commercial chemicals, instrument specifications, and characterization data are covered in greater detail in the ESI.†The ESI† is provided as a separate document.
Author contributions
Synthesis and characterization were performed by C. H. and S. R. D. Single crystal XRD was performed by C. H. NMR experiments were performed by C. H., S. R. D., and W. C. Calculations at the DFT-level were performed by C. H. The project was conceptualized by S. R. D. and C. C. All authors participated in formal analysis, as well as writing and editing of the work.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
C. C. and S. R. D. acknowledge the Swiss National Science Foundation (grants 200021_169134, and 200020B_1920) and the NCCR Catalysis. C. C. and A. V. Y. gratefully acknowledge ETH+ Project SynthMatLab for the financial support. We gratefully acknowledge SwissCat+ for the usage of the DNP 400 MHz spectrometer. Prof. Peter Chen, Christian Ehinger and Christoph Kaul are thanked for informative discussions. We also thank Dr Darryl Nater, Dr Michael Wörle, Christian Ehinger and Yuya Kakiuchi for help with single crystal XRD.Notes and references
- A. Bhan and E. Iglesia, Acc. Chem. Res., 2008, 41, 559–567 CrossRef CAS PubMed.
- G. Busca, Chem. Rev., 2007, 107, 5366–5410 CrossRef CAS PubMed.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- C. Bornes, M. Fischer, J. A. Amelse, C. F. G. C. Geraldes, J. Rocha and L. Mafra, J. Am. Chem. Soc., 2021, 143, 13616–13623 CrossRef CAS PubMed.
- C. Bornes, C. F. G. C. Geraldes, J. Rocha and L. Mafra, Microporous Mesoporous Mater., 2023, 360, 112666 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- N. Cardona-Martínez and J. A. Dumesic, J. Catal., 1990, 125, 427–444 CrossRef.
- C. Morterra and G. Cerrato, Langmuir, 1990, 6, 1810–1812 CrossRef CAS.
- E. P. Parry, J. Catal., 1963, 2, 371–379 CrossRef CAS.
- V. Zholobenko, C. Freitas, M. Jendrlin, P. Bazin, A. Travert and F. Thibault-Starzyk, J. Catal., 2020, 385, 52–60 CrossRef CAS.
- M. E. Z. Velthoen, S. Nab and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2018, 20, 21647–21659 RSC.
- C. V. Hidalgo, H. Itoh, T. Hattori, M. Niwa and Y. Murakami, J. Catal., 1984, 85, 362–369 CrossRef CAS.
- J. R. Anderson, K. Foger, T. Mole, R. A. Rajadhyaksha and J. V. Sanders, J. Catal., 1979, 58, 114–130 CrossRef CAS.
- J. O. Ehresmann, W. Wang, B. Herreros, D.-P. Luigi, T. N. Venkatraman, W. Song, J. B. Nicholas and J. F. Haw, J. Am. Chem. Soc., 2002, 124, 10868–10874 CrossRef CAS PubMed.
- C. P. Gordon, L. Lätsch and C. Copéret, J. Phys. Chem. Lett., 2021, 12, 2072–2085 CrossRef CAS PubMed.
- R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 711–724 CrossRef.
- D. G. Evans and D. M. P. Mingos, J. Organomet. Chem., 1982, 232, 171–191 CrossRef CAS.
- G. H. Penner and W. Li, Inorg. Chem., 2004, 43, 5588–5597 CrossRef CAS PubMed.
- A. Trummal, L. Lipping, I. Kaljurand, I. A. Koppel and I. Leito, J. Phys. Chem. A, 2016, 120, 3663–3669 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- P. de Frémont, N. M. Scott, E. D. Stevens, T. Ramnial, O. C. Lightbody, C. L. B. Macdonald, J. A. C. Clyburne, C. D. Abernethy and S. P. Nolan, Organometallics, 2005, 24, 6301–6309 CrossRef.
- M. Paas, B. Wibbeling, R. Fröhlich and F. E. Hahn, Eur. J. Inorg. Chem., 2006, 2006, 158–162 CrossRef.
- T. Ramnial, C. D. Abernethy, M. D. Spicer, I. D. McKenzie, I. D. Gay and J. A. C. Clyburne, Inorg. Chem., 2003, 42, 1391–1393 CrossRef CAS PubMed.
- C. A. Citadelle, E. L. Nouy, F. Bisaro, A. M. Z. Slawin and C. S. J. Cazin, Dalton Trans., 2010, 39, 4489–4491 RSC.
- B. K. Tate, C. M. Wyss, J. Bacsa, K. Kluge, L. Gelbaum and J. P. Sadighi, Chem. Sci., 2013, 4, 3068–3074 RSC.
- D. S. Laitar, P. Müller, T. G. Gray and J. P. Sadighi, Organometallics, 2005, 24, 4503–4505 CrossRef CAS.
- E. Y. Tsui, P. Müller and J. P. Sadighi, Angew. Chem., Int. Ed., 2008, 47, 8937–8940 CrossRef CAS PubMed.
- N. P. Mankad, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 3369–3371 CrossRef CAS.
- P. L. Arnold, Heteroat. Chem., 2002, 13, 534–539 CrossRef CAS.
- K. Zangger and L. M. Armitage, Met.-Based Drugs, 1999, 6, 239–245 CrossRef CAS PubMed.
- K. K. Samudrala and M. P. Conley, Chem. Commun., 2023, 59, 4115–4127 RSC.
- S. Rayne and K. Forest, Nature Precedings, 2010 DOI:10.1038/npre.2010.5061.1.
- G. N. Merrill and S. R. Kass, J. Phys. Chem., 1996, 100, 17465–17471 CrossRef CAS.
- D. J. Belton, O. Deschaume and C. C. Perry, FEBS J., 2012, 279, 1710–1720 CrossRef CAS PubMed.
- A. Yakimov, J. Xu, K. Searles, W.-C. Liao, G. Antinucci, N. Friederichs, V. Busico and C. Copéret, J. Phys. Chem. C, 2021, 125, 15994–16003 CrossRef CAS.
- A. V. Yakimov, D. Mance, K. Searles and C. Copéret, J. Phys. Chem. Lett., 2020, 11, 3401–3407 CrossRef CAS PubMed.
- L. Lätsch, E. Lam and C. Copéret, Chem. Sci., 2020, 11, 6724–6735 RSC.
- N. Kaeffer, D. Mance and C. Copéret, Angew. Chem., Int. Ed., 2020, 59, 19999–20007 CrossRef CAS PubMed.
- J. A. Schwarz, C. T. Driscoll and A. K. Bhanot, J. Colloid Interface Sci., 1984, 97, 55–61 CrossRef CAS.
- I. B. Moroz, K. Larmier, W.-C. Liao and C. Copéret, J. Phys. Chem. C, 2018, 122, 10871–10882 CrossRef CAS.
- S. Greiser, G. J. G. Gluth, P. Sturm and C. Jäger, RSC Adv., 2018, 8, 40164–40171 RSC.
- A. Dessombz, G. Coulibaly, B. Kirakoya, R. W. Ouedraogo, A. Lengani, S. Rouziere, R. Weil, L. Picaut, C. Bonhomme, F. Babonneau, D. Bazin and M. Daudon, C. R. Chim., 2016, 19, 1573–1579 CrossRef CAS.
- A. G. M. Rankin, P. B. Webb, D. M. Dawson, J. Viger-Gravel, B. J. Walder, L. Emsley and S. E. Ashbrook, J. Phys. Chem. C, 2017, 121, 22977–22984 CrossRef CAS PubMed.
- W. R. Gunther, V. K. Michaelis, R. G. Griffin and Y. Román-Leshkov, J. Phys. Chem. C, 2016, 120, 28533–28544 CrossRef CAS PubMed.
- I. B. Moroz, A. Lund, M. Kaushik, L. Severy, D. Gajan, A. Fedorov, A. Lesage and C. Copéret, ACS Catal., 2019, 9, 7476–7485 CrossRef CAS.
- M. Kaushik, C. Leroy, Z. Chen, D. Gajan, E. Willinger, C. R. Müller, F. Fayon, D. Massiot, A. Fedorov, C. Copéret, A. Lesage and P. Florian, Chem. Mater., 2021, 33, 3335–3348 CrossRef CAS.
- K. Larmier, C. Chizallet, S. Maury, N. Cadran, J. Abboud, A.-F. Lamic-Humblot, E. Marceau and H. Lauron-Pernot, Angew. Chem., Int. Ed., 2017, 56, 230–234 CrossRef CAS PubMed.
- M. Valla, A. J. Rossini, M. Caillot, C. Chizallet, P. Raybaud, M. Digne, A. Chaumonnot, A. Lesage, L. Emsley, J. A. van Bokhoven and C. Copéret, J. Am. Chem. Soc., 2015, 137, 10710–10719 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2257386–2257392. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04067d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |