
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA controlled non-radical chlorine activation pathway on hematite photoanodes for efficient oxidative chlorination reactions†
Daojian
Tang‡
ac,
Lei
Wu‡
ac,
Liubo
Li
 b,
Niankai
Fu
bc,
Chuncheng
Chen
ac,
Yuchao
Zhang
*ac and
Jincai
Zhao
ac
b,
Niankai
Fu
bc,
Chuncheng
Chen
ac,
Yuchao
Zhang
*ac and
Jincai
Zhao
ac
aKey Laboratory of Photochemistry, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: yczhang@iccas.ac.cn
bKey Laboratory of Molecular Recognition and Function, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 16th January 2024
Abstract
Photo(electro)catalytic chlorine oxidation has emerged as a useful method for chemical transformation and environmental remediation. However, the reaction selectivity usually remains low due to the high activity and non-selectivity characteristics of free chlorine radicals. In this study, we report a photoelectrochemical (PEC) strategy for achieving controlled non-radical chlorine activation on hematite (α-Fe2O3) photoanodes. High selectivity (up to 99%) and faradaic efficiency (up to 90%) are achieved for the chlorination of a wide range of aromatic compounds and alkenes by using NaCl as the chlorine source, which is distinct from conventional TiO2 photoanodes. A comprehensive PEC study verifies a non-radical “Cl+” formation pathway, which is facilitated by the accumulation of surface-trapped holes on α-Fe2O3 surfaces. The new understanding of the non-radical Cl− activation by semiconductor photoelectrochemistry is expected to provide guidance for conducting selective chlorine atom transfer reactions.
Introduction
In the past few decades, solar-driven photo(electro)chemistry has attracted enormous attention for solar fuel production1–4 and environmental remediation.5–7 The photoelectrochemical (PEC) technology combines light absorption and electrochemical processes into an integrated unit through direct semiconductor/electrolyte interfaces, offering advantages over particulate photocatalytic systems with its efficient charge separation and collection, as well as easy catalyst recycling.8,9 In recent years, PEC approaches have pointed towards a more sustainable and energy-saving alternative for redox organic transformations compared to thermal or electrochemical methods in terms of solar energy utilization.10–13Organochlorides are commonly found in many bioactive natural products or synthetic drugs and are widely employed for the construction of complex structures in organic synthesis.14–17 Conventional synthetic methods involve the use of massive amounts of explosive, corrosive, or toxic chlorine sources such as chlorine gas (Cl2), which raises concerns regarding potential environmental risks.16,18,19 The electrochemical technology for chlorine evolution has inspired an oxidative chlorination approach to generate reactive chloride species in situ, facilitating the green synthesis of high-value chemicals such as chlorohydrins, aryl chlorides, and epoxides.20–24 Alternatively, PEC-assisted oxidative chlorination is expected to be a clean and more energy-saving chemical process with reduced electrical power input.25 However, reports on the photoredox protocol for chlorine activation show challenges in PEC oxidative chlorination, where photocatalysts (e.g., TiO2,25–27 WO3,5,6etc.) with deep valence band edges are often used to drive the chloride (Cl−) oxidation (Fig. S1†). The photogenerated holes in the valence band (VB) of these photocatalysts are more positive than the 1-e− oxidation potential of Cl−, which forms chlorine radicals (Cl˙).25,27 Although Cl˙ facilitates the activation of C–H bonds,25,27,28 it can more easily trigger uncontrolled radical chain reactions, leading to polychlorination and radical coupling reactions.27,29 Thus, a compromise must be made between selectivity and conversion. As such, the development of a PEC approach to achieve more controlled activation of Cl− for C–Cl construction remains a substantial challenge.
Hematite (α-Fe2O3) is one of the most promising photoanode materials in PEC studies due to its abundance, non-toxicity, stability, and visible light absorption capabilities (Fig. S1†). Recently, α-Fe2O3 has attracted wide attention for selective oxygen transfer reactions.30,31 Under PEC conditions, long-lived surface-trapped holes have been demonstrated on α-Fe2O3,32,33 and multi-hole H2O oxidation catalysis is realized, in which the accumulation of multiple oxidizing equivalents (i.e., high-valent iron-oxo species, FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and FeVO) has been proposed as the key to efficient H2O oxidation catalysis.34–36 It is further found that α-Fe2O3 serves as an efficient oxygen atom transfer (OAT) catalyst, achieving highly selective two-electron oxygenation of substrates such as thioethers and alkenes.30 The high-valent iron-oxo characteristics of surface-trapped holes on α-Fe2O3 is reminiscent of the high-valent iron-oxo species in halogenating enzymes, which are capable of controllably oxidizing Cl− to form “Cl+” species (e.g., HClO or its equivalents) through a two-electron oxidation process.15,37,38 This enables selective electrophilic chlorination for constructing C–Cl bonds and preventing side reactions caused by single-electron radical reactions under relatively mild reaction conditions. There has been a significant advancement in the spectroscopic, physicochemical, and mechanistic investigations of homogeneous Fe catalysts, but the chlorination reaction using heterogeneous Fe or Fe oxide catalysts is rarely reported.38–40
O and FeVO) has been proposed as the key to efficient H2O oxidation catalysis.34–36 It is further found that α-Fe2O3 serves as an efficient oxygen atom transfer (OAT) catalyst, achieving highly selective two-electron oxygenation of substrates such as thioethers and alkenes.30 The high-valent iron-oxo characteristics of surface-trapped holes on α-Fe2O3 is reminiscent of the high-valent iron-oxo species in halogenating enzymes, which are capable of controllably oxidizing Cl− to form “Cl+” species (e.g., HClO or its equivalents) through a two-electron oxidation process.15,37,38 This enables selective electrophilic chlorination for constructing C–Cl bonds and preventing side reactions caused by single-electron radical reactions under relatively mild reaction conditions. There has been a significant advancement in the spectroscopic, physicochemical, and mechanistic investigations of homogeneous Fe catalysts, but the chlorination reaction using heterogeneous Fe or Fe oxide catalysts is rarely reported.38–40
Inspired by this, we envision that α-Fe2O3 could serve as an efficient PEC catalyst for controlled non-radical activation of Cl− for C–Cl construction. In this work, the Cl− oxidation process on α-Fe2O3 is investigated. We show a direct “Cl+” species formation process on α-Fe2O3 surfaces, which is facilitated by the accumulation of surface-trapped holes. As a result, α-Fe2O3 exhibits a high selectivity (up to 99%) for the synthesis of aromatic chlorides and chlorohydrins. This is in sharp contrast to the low-selective chlorination on TiO2 photoanodes, which follows the free radical chlorination pathway. This work is anticipated to offer an in-depth understanding of selective Cl− activation by semiconductor photoelectrochemistry and provide guidance for the construction of efficient chlorine atom transfer reactions.
Results and discussion
PEC Cl− oxidation behaviors on α-Fe2O3
As a proof of concept, the Cl− activation process on α-Fe2O3 was firstly investigated. α-Fe2O3 photoanodes were prepared according to our previous reports.31 Structural characterization of α-Fe2O3 is shown in Fig. S2.† The PEC tests were conducted in an H-type cell with a three-electrode configuration, where a Pt wire was used as the cathode and an Ag/AgCl electrode served as the reference electrode. The PEC performance of Cl− oxidation and H2O oxidation was investigated by using a mixed solution of H2O/CH3CN (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, pH = 6.4) with 0.1 M NaCl or NaClO4 as the electrolyte. Linear sweep voltammetry (LSV) measurements under AM 1.5G irradiation (100 mW cm−2) exhibited an onset potential of 0.6 V vs. Ag/AgCl for oxidizing H2O in a 0.1 M NaClO4 solution on the α-Fe2O3 photoanode (Fig. 1a). When the electrolyte was replaced with 0.1 M NaCl, the onset potential decreased by approximately 200 mV and the photocurrent was significantly enhanced, indicating that Cl− was preferentially oxidized on α-Fe2O3. To further explore the photocurrent behavior, LSV measurements were performed under various concentrations of Cl− solution. As shown in Fig. S3,† the presence of only 10 mM Cl− resulted in a remarkable photocurrent enhancement. Moreover, the overall photocurrent on the α-Fe2O3 photoanode increased with the concentration of Cl−, indicating that the enhancement of photocurrent was associated with Cl− oxidation.
:1, pH = 6.4) with 0.1 M NaCl or NaClO4 as the electrolyte. Linear sweep voltammetry (LSV) measurements under AM 1.5G irradiation (100 mW cm−2) exhibited an onset potential of 0.6 V vs. Ag/AgCl for oxidizing H2O in a 0.1 M NaClO4 solution on the α-Fe2O3 photoanode (Fig. 1a). When the electrolyte was replaced with 0.1 M NaCl, the onset potential decreased by approximately 200 mV and the photocurrent was significantly enhanced, indicating that Cl− was preferentially oxidized on α-Fe2O3. To further explore the photocurrent behavior, LSV measurements were performed under various concentrations of Cl− solution. As shown in Fig. S3,† the presence of only 10 mM Cl− resulted in a remarkable photocurrent enhancement. Moreover, the overall photocurrent on the α-Fe2O3 photoanode increased with the concentration of Cl−, indicating that the enhancement of photocurrent was associated with Cl− oxidation.
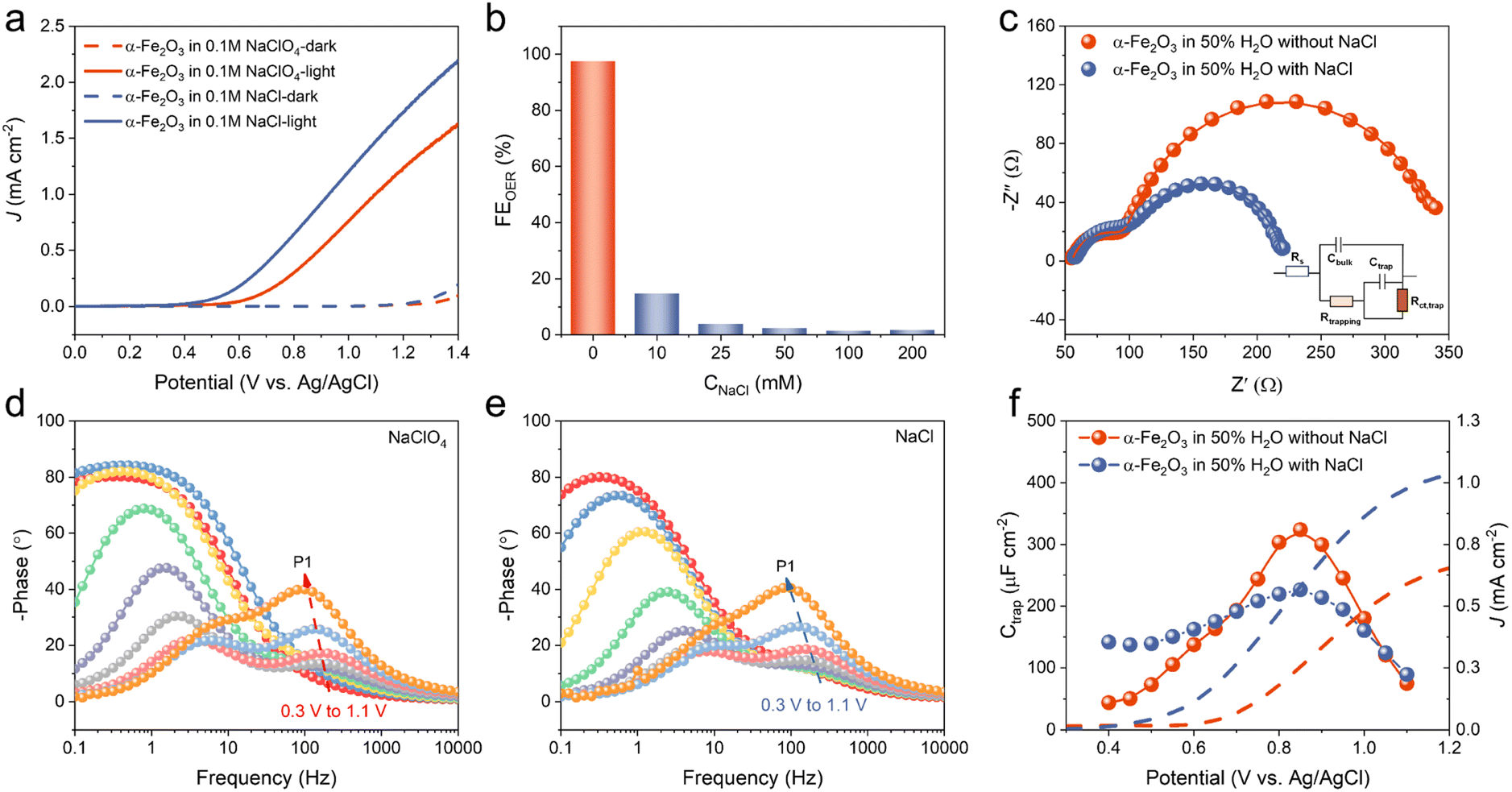 | ||
| Fig. 1 PEC Cl− oxidation activity and reaction kinetics on α-Fe2O3 photoanodes. (a) LSV curves of an α-Fe2O3 photoanode in a mixed solution of CH3CN/H2O (1:1 v/v) with 0.1 M NaCl or NaClO4 as the electrolyte under AM 1.5G irradiation. (b) The FE of the OER after 1 h of photoelectrolysis at 1.0 V vs. Ag/AgCl with different concentrations of Cl−. (c) Nyquist plots and the fitted curves of an α-Fe2O3 photoanode in 0.1 M NaClO4 solution with or without 10 mM Cl− under 470 nm irradiation at 1.0 V vs. Ag/AgCl. The inset is the equivalent circuit for EIS fitting. (d) Potential-dependent Bode plots measured in 0.1 M NaClO4 solution; (e) potential-dependent Bode plots measured in 0.1 M NaClO4 solution with 10 mM Cl−. (f) LSV curves (dashed lines) and Ctrap values (solid lines and dots) obtained on an α-Fe2O3 photoanode with 10 mM Cl− (grayish blue) and without Cl− (red) under 470 nm irradiation. | ||
Intensity modulated photocurrent spectroscopy (IMPS) measurements were performed to compare interfacial charge-transfer kinetics between the oxidations of Cl− and H2O on the α-Fe2O3 photoanode. IMPS plots showed identical semi-circles in the high frequency domain (the fourth quadrant) but a decreased radius of the low-frequency circle in the first quadrant after the addition of Cl− (Fig. S4†). Simulation of the IMPS plots showed that the presence of Cl− increased the charge-transfer rate constant (kct) from 13.4 s−1 to 21.6 s−1 and the charge-transfer efficiency (ηct) from 53.5% to 77.0%, indicating that Cl− oxidation was much faster than H2O oxidation. The inhibition of H2O oxidation was confirmed by monitoring the generation of O2 by gas chromatography (GC, Fig. S5†) after 1 h of photoelectrolysis under AM 1.5G irradiation. As shown in Fig. 1b and Table S1,† the addition of Cl− resulted in a rapid decrease in the faradaic efficiency (FE) of the oxygen evolution reaction (OER) and almost complete suppression when 25–200 mM Cl− was present. To further confirm the high selectivity of Cl− oxidation, we employed the N,N-diethyl-p-phenylenediamine (DPD) colorimetric method to quantify the active chlorine concentrations under various Cl− solution concentrations (Fig. S6†).41 The results showed that after 10 min of photoelectrolysis, the FE of active chlorine generation was more than 90% when Cl− concentrations ranged from 25 to 200 mM, which was consistent with the results obtained from OER experiments. In addition, no active chlorination generation was detected under conditions of only electricity or light, suggesting that both light and electricity were necessary in our system for Cl− oxidation (Fig. S7†). Strikingly, α-Fe2O3 exhibited a high selectivity of Cl− oxidation even at a relatively low Cl− concentration of 50 mM, showing a significant advantage compared to other reported photocatalysts for Cl− oxidation (Table S2†). We also explored the Cl− oxidation performance in an aqueous solution. As shown in Fig. S8,† the selectivity of Cl− oxidation decreased in 0.1 M Cl− aqueous solution compared to that in a H2O–MeCN mixture solution (Fig. S6†), suggesting that the presence of an organic solvent helped to inhibit the competitive H2O oxidation reaction. Moreover, a further increase of Cl− concentration could improve the selectivity of Cl− oxidation. Under 0.3 M Cl−, the FE of Cl− oxidation could reach more than 90%, which still showed good selectivity for Cl− oxidation compared to other reported photocatalysts (Table S2†). These results indicated the high reactivity of α-Fe2O3 towards Cl− oxidation.
Previous research has demonstrated that the process of H2O oxidation on α-Fe2O3 occurs through surface-trapped holes rather than direct hole transfer from the valence band.42,43 In order to uncover the underlying relationship between the Cl− oxidation and the surface-trapped holes, electrochemical impedance spectroscopy (EIS) measurements were carried out under the same conditions to investigate the interface reaction of Cl− oxidation and H2O oxidation on α-Fe2O3. As shown in Fig. 1c, the Nyquist plots for both Cl− oxidation and H2O oxidation on the α-Fe2O3 photoanode consisted of two semicircles, and its Bode plots exhibited two peaks at different frequency regions (Fig. 1d and e). This is a typical feature of surface-state mediated charge transfer rather than direct hole transfer.3,43 The high-frequency (HF) semicircle in Nyquist plots should be assigned to the surface hole-trapping process and the low-frequency (LF) semicircle should be attributed to the transfer of those trapped holes to the electrolyte. In Fig. 1c, the HF semicircles were almost unchanged upon adding 10 mM Cl−, which indicated that Cl− had little effect on the generation of surface trapped holes. To further confirm this, potential-dependent experiments were conducted. For both H2O oxidation and Cl− oxidation, the potential-dependent experiments (Fig. 1d and e) showed the phase angle of the HF region (P1, which was assigned to the surface hole-trapping process) in Bode plots increased with applied potentials, suggesting the accumulation of surface-trapped holes. Regardless of the presence of Cl−, there was no distinction in the phase angle P1, demonstrating that Cl− oxidation and H2O oxidation shared similar surface states or active sites. This was consistent with the competitive oxidation process of Cl− and H2O (Fig. 1b and S6†). The trapped holes on these surface states were previously assigned to high-valent surface iron-oxo species, i.e., FeIVO and FeVO.43,44 On the other hand, compared to H2O oxidation, the radius of the LF semicircle in Cl− oxidation was significantly reduced (Fig. 1c). This demonstrated that the oxidation of Cl− mediated by surface-trapped holes was easier than the oxidation of H2O, which correlated well with the increasing photocurrent and the IMPS results.
To quantify the two associated capacitive elements, a well-established physical model consisting of the space-charge capacitance (Cbulk) and surface-state capacitance (Ctrap) was considered (see details in Fig. S9†).3,43–45 As shown in Fig. 1f, in 0.1 M NaClO4 solution, the fitted Ctrap increased with the applied bias at low potentials and reached a maximum at approximately 0.85 V due to the accumulation of surface-trapped holes. After that, Ctrap began to decrease because of the increased consumption rate of surface-trapped holes. In the presence of Cl−, the maximum value of Ctrap was smaller than that in the 0.1 M NaClO4 solution, indicating that the consumption of surface-trapped holes by Cl− oxidation were faster than that by H2O oxidation. To further prove that Cl− oxidation was mediated by the high-valent iron-oxo species, the behavior of surface-state capacitance (Ctrap) with Cl− concentrations was investigated (Fig. S10†). The Ctrap values decreased with the concentration of Cl−, supporting that Cl− oxidation was favorably mediated by the high-valent iron-oxo species. Therefore, we anticipated an efficient PEC chlorination of α-Fe2O3 through a controlled non-radical Cl− activation pathway similar to that of halogenating enzymes.
Different oxidative chlorination behaviors on α-Fe2O3 and TiO2
Aromatic chlorides are widely applied in pharmaceuticals, agrochemicals, and organic materials for modifying their biological or physicochemical properties.14,15 They also serve as important starting materials for cross-coupling reactions.18 It is reported that aromatic chlorination can be efficiently driven via a non-radical electrophilic chlorination reaction.18,46 Therefore, aromatic chlorination of acetanilide (1a) was chosen as one model reaction to explore PEC oxidative chlorination (Fig. 2a, model reaction I). LSV measurements in Fig. S11† showed that, under AM 1.5 G irradiation, the presence of 1a did not change the onset potential and the photocurrent in the whole tested voltage range of 0.0 to 1.2 V vs. Ag/AgCl (red dash curves), which suggested that the direct oxidation of 1a should not occur on α-Fe2O3. To further confirm this, the Cl− oxidation and 1a oxidation were compared in a 100% MeCN solution with NEt4BF4 as the supporting electrolyte. As shown in Fig S12,† we observed that the direct oxidation of 1a occurred at higher overpotentials. Moreover, compared with Cl− oxidation, the onset potential of 1a oxidation anodically shifted by 0.4 V on the α-Fe2O3 photoanode, suggesting that the direct oxidation of 1a on α-Fe2O3 in the oxidative chlorination reaction system would be difficult to occur. This excluded the possibility that the chlorination reaction was conducted through activation of 1a to yield the chlorination products. | ||
| Fig. 2 Photoelectrolysis experiments of PEC oxidative chlorination. (a) The model reactions for the PEC oxidative chlorination reaction. (b) The aromatic chlorination activity compared on α-Fe2O3 and TiO2. (c) The cyclic stability test of a single α-Fe2O3 photoanode in 0.1 M NaCl with 10 mM 1a at 1.0 V vs. Ag/AgCl. (d) The UV-vis spectra of DPD colorimetric tests for the detection of active chlorine derivatives during aromatic chlorination on α-Fe2O3 and TiO2 after 2 h of photoelectrolysis at 1.0 V vs. Ag/AgCl. Inset: the digital photograph of the anolyte with DPD test solution added. (e) FE and selectivity for the chlorohydrin product on α-Fe2O3 and TiO2 after 2 h of photoelectrolysis at 1.0 V vs. Ag/AgCl in 0.1 M NaCl solution. (f) The GC-MS spectra of styrene chlorination products on α-Fe2O3 and TiO2 after 1 h of photoelectrolysis. | ||
To investigate the performance of aromatic chlorination on α-Fe2O3, chronoamperometry was conducted at 1.0 V vs. Ag/AgCl under AM 1.5G irradiation for 2 h under the same conditions. Product analysis by high-performance liquid chromatography (HPLC) showed that 1a was converted into chlorinated products 2a and 3a (Fig. S13†). The FE for chlorinated products 2a and 3a on the α-Fe2O3 photoanode was found to be 90%, with a remarkable selectivity of 98% for monochlorinated product 2a (p/o = 1.6:1) (Fig. 2b). It has been reported that the TiO2 photoanodes drive Cl− oxidation to form ˙Cl through a single-electron oxidation mechanism.25 To confirm the uniqueness of α-Fe2O3 for efficient aromatic chlorination, the oxidative chlorination of 1a on a TiO2 photoanode was investigated and compared with that on α-Fe2O3. Structural characterization of TiO2 is shown in Fig. S14.† The FE for aromatic chlorination of 1a on TiO2 was only 50%, while the selectivity for 2a was 57% (Fig. 2b). In addition, the TiO2 photoanode showed a higher 3a/2a ratio for chlorinated products compared to that on the α-Fe2O3 photoanode, suggesting a low regioselectivity for aromatic mono-chlorination. This was consistent with the polychlorination occurring during the ˙Cl-involved chlorination reaction. Potential-dependent experiments on the α-Fe2O3 photoanode revealed that a high FE (>90%) was maintained within the potential range of 0.7 to 1.0 V (Fig. S15a†). By extending the electrolysis time to 2.5 h, a yield of 95% for 2a was achieved on the α-Fe2O3 photoanode (Fig. S15b†). We evaluated the stability of the α-Fe2O3 photoanode in batch reactions. Under high conversion of the substrate (>90%), a high selectivity of 98% was maintained for the 6th run under the same reaction conditions (Fig. 2c). The X-ray diffraction (XRD), X-ray absorption fine structure spectra (XAFS) and X-ray photoelectron spectroscopy (XPS) characterizations (Fig. S16 and 17†) displayed no changes for the used α-Fe2O3, demonstrating the stability of α-Fe2O3. Moreover, the presence of obvious Cl− residues was observed after photoelectrolysis, indicating a strong absorption of Cl− on Fe2O3 surfaces (Fig. S17c and d†). More importantly, the enhanced interfacial charge transfer of Cl− oxidation resulted in a higher solar H2 production (47 μmol h−1) compared to H2O oxidation (22 μmol h−1) as the oxidative half reaction (Fig. S18†), suggesting a higher utilization efficiency of photo-induced charges and a more effective storage of solar energy through the coupled electrophilic chlorination reaction with H2 production.
Considering the high Cl− oxidation performance across a wide range of Cl− concentrations (10–200 mM, Fig. 1b and S6†), the aromatic chlorination of 1a with different Cl− concentrations was also evaluated. At the same potential (1.0 V vs. Ag/AgCl), the yield rate of 2a increased with the higher concentration of Cl−, reaching 23 μmol cm−2 h−1, while the selectivity of 2a remained up to 90% (Fig. S15c and d†). This was consistent with the linear change in Cl− oxidation photocurrent (Fig. S3b†), indicating that the rate-determining step (RDS) of aromatic chlorination was the PEC Cl− oxidation rather than the subsequent chemical process. More importantly, under low Cl− concentration (10 mM Cl−, 1 equiv. substrate concentration), the chlorination activity on the α-Fe2O3 photoanode still showed a high FE of 75%, indicating the efficient Cl− oxidation and the rapid aromatic chlorination process on the α-Fe2O3 photoanode. The effective chlorination process on the α-Fe2O3 photoanode was also supported by inhibiting the further oxidation of active chlorine species to form ClO3− (Fig. S19†). During photoelectrolysis without 1a, the analysis of Cl− oxidation products using ion chromatography (IC) showed a gradual accumulation of ClO3− over a period of 2 h (Fig. S19a†). However, when photoelectrolysis was performed in the presence of 1a, ClO3− formation was completely inhibited (Fig. S19b†) and the chlorination of 1a showed a high current efficiency (FE 98%), indicating the rapid reaction between 1a and active chlorine species. This was further supported by DPD colorimetric tests,41 in which most of the active chlorine species derived from Cl− oxidation already participated in the aromatic chlorination and only a small portion of them was residual and detected on the α-Fe2O3 photoanode (Fig. 2d). In contrast, DPD colorimetric tests showed a significant accumulation of active chlorine species on the TiO2 photoanode (Fig. 2d), suggesting that most of the active chlorine species generated from Cl− oxidation on TiO2 were unable to participate in the subsequent aromatic chlorination and were thus detected by DPD tests. ˙Cl can lead to uncontrolled hydrogen abstraction and polymerization reactions, resulting in the low aromatic chlorination activity.18 These results suggested that PEC activation of Cl− on α-Fe2O3 and TiO2 photoanodes produced distinct active chlorine species.
To further confirm this, we explored the synthesis of chlorohydrin products (Fig. 2a, model reaction II), which act as essential intermediates in industrial-scale epoxidations. ˙Cl can initiate radical chain reactions or radical coupling reactions, resulting in the formation of dimeric or polymeric products during the chlorination of alkene.47,48 After electrolysis at 1.0 V vs. Ag/AgCl for 2 h, product analysis by HPLC showed that nearly 80% conversion of 1b with up to 85% selectivity of chlorohydrin 2b was achieved on the α-Fe2O3 photoanode (Fig. 2e and S20†). The calculated FE was close to 80%. In addition to producing chlorohydrin products of 2b, a minor quantity of dichloride products of 3b was also observed (Fig. 2e). In contrast, the chlorohydrin of 1b on the TiO2 photoanode only exhibited an FE of 23% and a selectivity of 21% for 2b. It also showed a much higher production of dichlorination products 3b (Fig. 2e). The gas chromatography-mass spectrometry (GC-MS, Fig. 2f) revealed the formation of high molecular mass polymers on the TiO2 photoanode, which were not observed on the α-Fe2O3 photoanode. These results indicated that ˙Cl is not formed and the selective electrophilic chlorination process occurs on α-Fe2O3.
Scope of PEC oxidative chlorination on α-Fe2O3
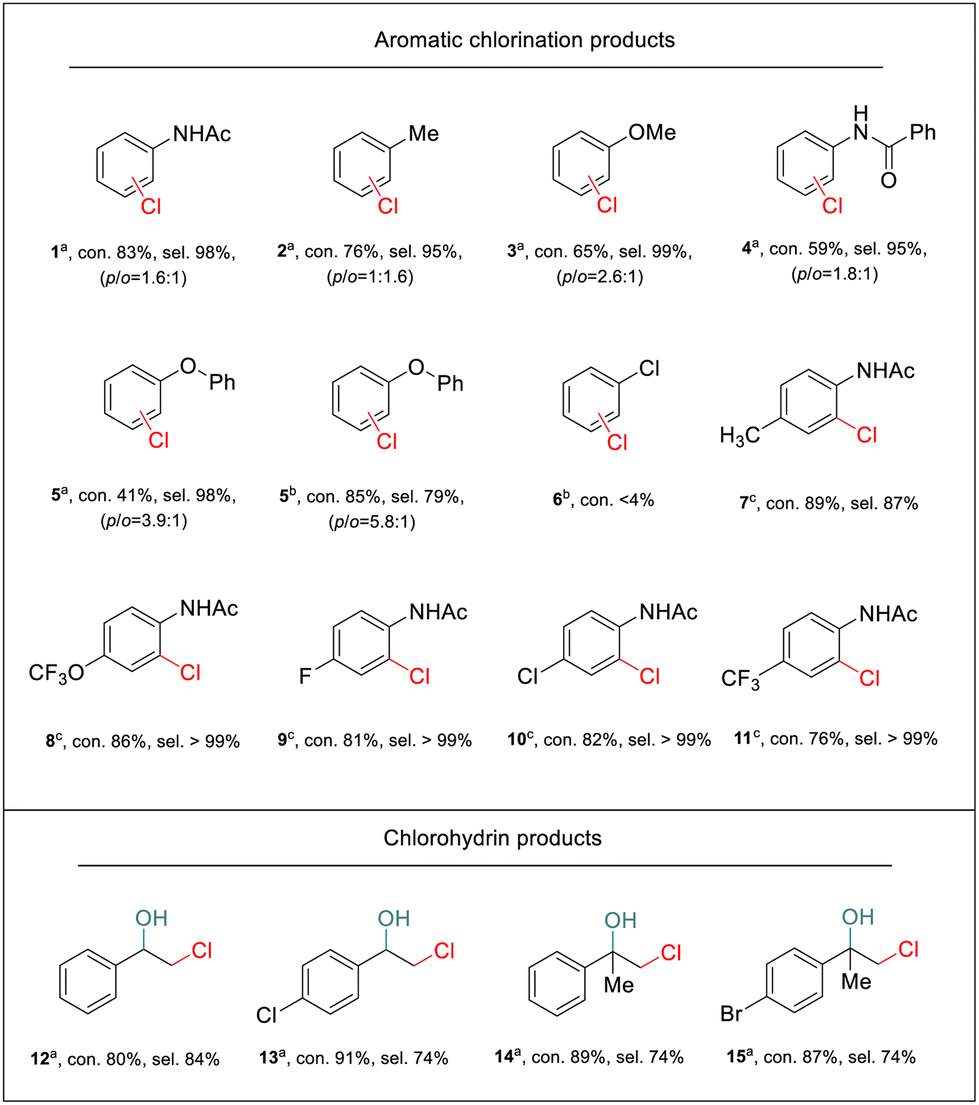
To show the general applicability of the PEC strategy for controlled oxidative chlorination on the α-Fe2O3 photoanode, a broad scope of substrates was tested. We first explored mono-substituted aromatic compounds. As shown in Table 1, aromatic compounds with strong electron-donating groups on the phenyl ring, such as methyl (2), methoxyl (3), benzamide (4), and ether (5), exhibited good regioselectivity towards monochlorination products. After 2 h of photoelectrolysis, both para- and ortho-chlorinated products were formed (Fig. S21–24†). However, due to the limited solubility of substrates 4 and 5 in the 50% H2O of MeCN solution, the conversion rate was low. When the chlorination reaction was conducted in the 16% H2O of MeCN solution, substrate 5 exhibited a high conversion with a selectivity of 79% for monochlorination products (Fig. S24†). Moreover, the substrate that only contained an electron-withdrawing group (chlorine, 6) did not undergo chlorination. This was consistent with the limitations of electrophilic chlorination, in which electrophilic “Cl+” species were the major reagent for the chlorination process. Although mono-substituted substrates were chlorinated at both the ortho and para positions due to the inherent reactivity of “Cl+” species, complete site selectivity can be achieved for disubstituted substrates. In these cases, chlorination occurred exclusively ortho to the amide group. Substrates bearing functional groups with various electron-rich and electron-deficient substituents, including methyl (7), trifluoromethoxy (8), halogen (9–10), and trifluoromethyl (11), were all chlorinated in good yields as well (Fig. S25–29†).|
a typical reaction conditions applied in this work: 0.1 M NaCl, 0.1 mmol substrate in 10 mL of CH3CN solution with 50% H2O (v/v = 1:1), PEC reactions were carried out for 2 h under AM 1.5G irradiation at 1.0 V vs. Ag/AgCl with a non-conditioned air atmosphere and pH adjustments.
b reaction in 0.1 M NEt4BF4 with 16% H2O and 50 mM NaCl for 2 h.
c reaction in 0.1 M NEt4BF4 with 16% H2O and 50 mM NaCl for 5 h.
|
|---|
|
In addition, the electrosynthesis of chlorohydrins was explored. As shown in Table 1, our PEC approach exhibited a high yield for the chlorohydrination of styrene and its derivatives (12–15) (Fig. S30–32†), thus demonstrating the universality of the PEC halogenation strategy on α-Fe2O3 photoanodes for conducting efficient chlorine atom transfer reactions.
Direct “Cl+” formation on α-Fe2O3
DPD tests showed an efficient chlorine transfer process on α-Fe2O3 (Fig. 2d). The highly selective formation of the “Cl+” species is essential for effective electrophilic chlorination.46 Many efforts have pointed out that the surface-trapped holes on α-Fe2O3 facilitate the non-radical multi-hole oxidation process.30,31 We hypothesized that α-Fe2O3 surfaces could potentially facilitate a “Cl+” species formation pathway, which would be responsible for the selective electrophilic chlorination of aromatic compounds and alkenes.Based on the kinetics of various mono-substituted benzene substrates in the aromatic chlorination reaction, the Hammett linear free energy relationship can be calculated. It showed a negative slope of −1.1 for para-chlorinated products on the α-Fe2O3 photoanode (Fig. 3a), indicating the positive charge buildup at the aromatic moiety of the substrate in the transition state. This confirmed the electrophilic chlorination pathway mediated by non-radical “Cl+” species on α-Fe2O3 photoanodes. This was further supported by toluene chlorination experiments (Fig. 3b). The “Cl+” species only triggers electrophilic aromatic substitution reactions, while highly reactive ˙Cl easily induces competitive sp3 C–H chlorination, resulting in low selectivity during the ˙Cl-mediated aromatic chlorination process (Fig. S33†). As shown in Fig. 3b, TiO2 generated few aromatic chlorination products (chlorotoluene, CT) in toluene chlorination experiments, but mainly produced the sp3 C–H chlorination product benzyl chloride (BC). In contrast, the α-Fe2O3 photoanode exhibited a high CT yield rate of 12.6 μmol cm−2 h−1 at 1.0 V vs. Ag/AgCl (Fig. 3b), suggesting that ˙Cl was the main active chlorine species on the TiO2, while the active chlorine species generated on the α-Fe2O3 were more likely to be “Cl+” species rather than ˙Cl. To further confirm this, electron paramagnetic resonance (EPR) measurements were carried out to detect possible radicals. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) was used as the spin-trapping reagent to capture the possible radicals that were generated in the photoelectrolysis. As shown in Fig. 3c, radical signals were detected only on the TiO2 photoanode. The three peaks at 3338, 3353, and 3368 G were attributed to DMPO-Cl˙, which was in situ produced by the reaction between DMPO and Cl˙ during the photoelectrolysis on TiO2 (red rhombi). The quartet peaks at 3332, 3345, 3360, and 3374 G with an intensity ratio of 1:2:2:1 corresponded to DMPO-OH˙ (yellow stars). The other six peaks indicated the formation of DMPO-OCl˙ (blue hearts).49,50 These EPR experiments supported the radical process on TiO2 and non-radical process on α-Fe2O3.
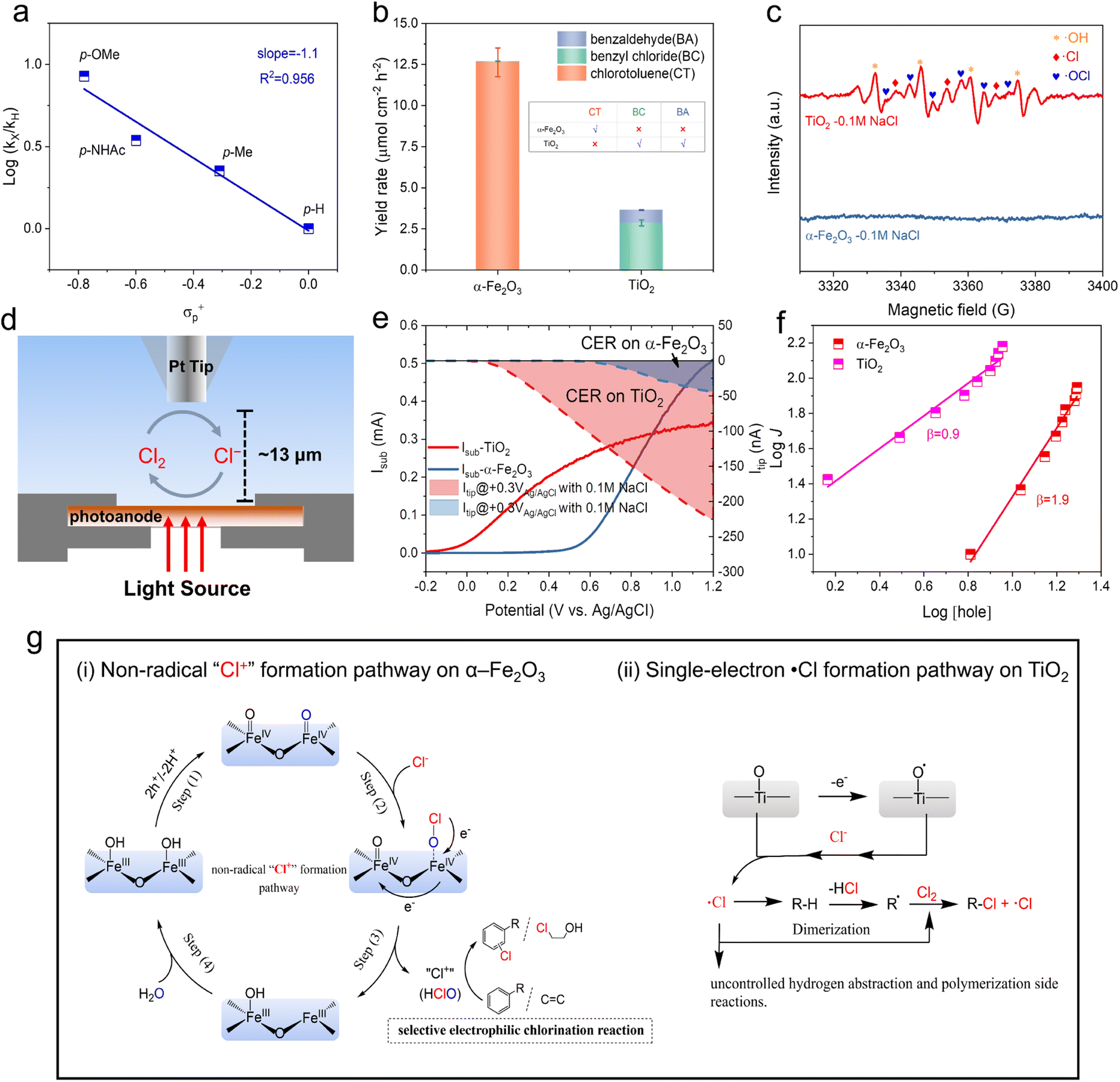 | ||
| Fig. 3 Different Cl− activation behaviors on α-Fe2O3 and TiO2. (a) Hammett plot for the para-chlorination of mono-substituted benzene on α-Fe2O3. (b) Toluene chlorination experiments for α-Fe2O3 and TiO2. (c) EPR spectra of the solution obtained from an in situ trapping method during PEC Cl− oxidation on TiO2 and α-Fe2O3, with the addition of DMPO as a radical capture reagent. (d) Schematic of SG/TC SECM experiments. (e) The substrate current and the tip current during LSV (5 mV s−1) for Cl− oxidation on α-Fe2O3 and TiO2. (f) Rate law analysis for Cl− oxidation on α-Fe2O3 and TiO2. (g) Schematic illustration for distinct PEC oxidative chlorination reaction pathways on α-Fe2O3 and TiO2. | ||
Cl2 is also one of the active chlorine species and can be formed by the self-coupling of Cl˙. To rule out the possibility that Cl2 is the main active chlorine species on α-Fe2O3, substrate generation/tip collection mode scanning electrochemical microscopy (SG/TC SECM) measurements were conducted (see more details in Fig. S35 and 36†) to directly monitor the formation of Cl2 on the photoanodes surfaces. As shown in Fig. 3d, Cl2 was used as the probe molecule to specifically detect Cl2 generated from the photoanode surface at an ultra-close distance (∼13 μm). With the increase in photocurrent (Isub) after an onset potential of −0.2 V vs. Ag/AgCl, the corresponding rise in Itip unequivocally confirmed the formation of Cl2 on TiO2 surfaces (Fig. 3e). In contrast, the production of Cl2 was significantly lower on α-Fe2O3 surfaces, especially when the applied potential exceeded 0.9 V vs. Ag/AgCl. For example, at 1.2 V vs. Ag/AgCl, the Isub for Cl− oxidation on α-Fe2O3 was significantly higher than that on TiO2. In contrast, the Itip for Cl2 production was more than 4 times lower on α-Fe2O3 compared to that on TiO2, demonstrating that Cl2 was not the main product on α-Fe2O3 surfaces. The residue amount of Cl2 on α-Fe2O3 may originate from the reaction between “Cl+” species and Cl−.
To further explore the nature of the Cl− activation process on α-Fe2O3, rate law analysis based on EIS measurements was performed to compare the hole transfer kinetics for Cl− oxidation between α-Fe2O3 and TiO2 photoanodes (see details in Fig. S9†). Cl− oxidation on α-Fe2O3 showed a second-order kinetics (Fig. 3f and S37, and Tables S4 and S5†). In contrast, Cl− oxidation on TiO2 exhibited a first-order kinetics. The second-order reaction kinetics indicated that two-hole transfer was involved in the RDS for the Cl− oxidation reaction on α-Fe2O3, indicating that “Cl+” species were directly formed from the reaction between the absorbed Cl− and the surface-trapped holes (i.e., high-valent iron-oxo species). To support this conclusion, HClO was purposely used as a model “Cl+” reagent for the chlorination of 1a. As shown in Fig. S38,† its product distribution was similar to that under the PEC reaction conditions, implying that “Cl+” species (e.g., HClO or its equivalents) were the actual active chlorine species during the PEC Cl− oxidation on α-Fe2O3 for the subsequent electrophilic chlorination. In contrast, the Cl− oxidation on TiO2 was dominated by transfer of a single surface-trapped hole, which is consistent with the free radical characteristics of TiO2.
The proposed mechanism
According to the above results, the proposed reaction mechanism was summarized in Fig. 3g. The formation of surface-trapped holes is believed to occur through hole transfers to hydroxyl groups coordinated on the surface, accompanied by a simultaneous deprotonation step (step 1).44,51 The trapping of holes could lead to the formation of the two adjacent high-valent surface iron-oxo species (see details in Fig. S39†). After the formation of surface-trapped holes, as indicated by EIS analysis (Fig. 1c and f), the nucleophilic attack of a Cl− at one of the two adjacent high-valent iron-oxo species was more favorable than that of H2O, resulting in the formation of an Fe–O–Cl intermediate complex (step 2). Next, considering the second-order characteristics of the hole kinetics (Fig. 3f), a concerted two-hole transfer process as the RDS contributed to the direct formation of “Cl+” species (e.g., HClO or its equivalents), which efficiently reacted with the substrate for the Cl transfer to form the chlorination product (step 3). Then, the chlorine transfer process left a vacant FeIII site on α-Fe2O3, which was recovered by the adsorption of a H2O molecule (step 4). In contrast, the chlorination reaction was only driven by the ˙Cl-mediated radical pathway on TiO2, which resulted in low chlorination reactivity and low utilization of Cl atoms due to the uncontrolled hydrogen abstraction and polymerization side reactions.Moreover, we also successfully constructed a self-powered PEC system for the electrophilic chlorination reaction. In this system, a solar panel (∼3 V) was used to provide a constant bias (Fig. 4a). As shown in Fig. 4b, this self-powered PEC system successfully achieved aromatic chlorination of acetanilide, showing a promising approach to harness solar energy for synthesizing valuable organic halides.
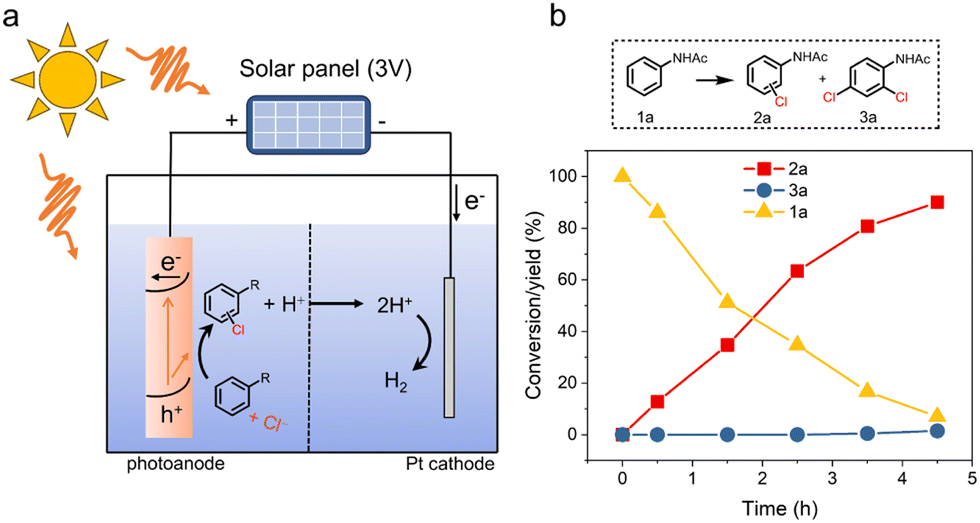 | ||
| Fig. 4 The self-powered PEC system for the aromatic chlorination reaction. (a) Schematic configuration of the self-powered PEC system. (b) Time-dependent conversions of 1a to 2a and 3a in the self-powered PEC system. | ||
Conclusion
In this study, we report a PEC strategy for selective electrophilic chlorination reactions on α-Fe2O3 photoanodes with NaCl as the chlorine source. High selectivity (up to 99%) and faradaic efficiency (up to 90%) are achieved for the chlorination of a wide range of aromatic compounds and alkenes. A systematic PEC study verifies that the direct “Cl+” formation process occurs on the α-Fe2O3 surface, which is facilitated by the accumulation of surface-trapped holes. The non-radical Cl− activation characteristic and the efficient Cl transfer process make the electrophilic chlorination reactions very efficient. Moreover, we successfully constructed a self-powered PEC system for chlorinating aromatic compounds while simultaneously maintaining exceptional catalytic activity, offering a promising approach to harness solar energy for synthesizing valuable organic halides. This work provides new insight for constructing efficient chlorine atom transfer reactions by semiconductor photoelectrochemistry.Data availability
The authors declare that all supporting data are available in the ESI† and from the corresponding author upon request.Author contributions
Y. Zhang directed the project. D. Tang conceived and carried out most of the experiments. L. Wu assisted in fabricating the photoanodes and carrying out SECM measurements. Y. Zhang, D. Tang and L. Wu wrote the manuscript, with input from others. All the authors analyzed the results and reviewed the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (No. 2022YFA1505000, 2020YFC1808401), the National Natural Science Foundation of China (No. 22072158), the Strategic Priority Research Program of Chinese Academy of Sciences (No. XDB36000000) and CAS Project for Young Scientists in Basic Research (No. YSBR-004). We thank Prof. Wanhong Ma for the helpful discussions. We thank Qiaozhen Li for helping with the analysis of the products. We also express our gratitude to Jing Xue, Jiaming Wang, Siqin Liu, Kun Dang, and Dr Mingge Wu for their support in the lab. Furthermore, we would like to acknowledge the assistance provided by the staff of the 1W1B beamline at the Beijing Synchrotron Radiation Facility (BSRF) during the XAFS measurements.Notes and references
- T. Hisatomi, J. Kubota and K. Domen, Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- C. Guo, J. Ran, A. Vasileff and S.-Z. Qiao, Rational design of electrocatalysts and photo(electro)catalysts for nitrogen reduction to ammonia (NH3) under ambient conditions, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, Electrochemical and photoelectrochemical investigation of water oxidation with hematite electrodes, Energy Environ. Sci., 2012, 5, 7626–7636 RSC.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Photocatalytic reduction of CO2 on TiO2 and other semiconductors, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- Z. Shen, Y. Zhang, C. Zhou, J. Bai, S. Chen, J. Li, J. Wang, X. Guan, M. Rahim and B. Zhou, Exhaustive denitrification via chlorine oxide radical reactions for urea based on a novel photoelectrochemical cell, Water Res., 2020, 170, 115357 CrossRef CAS PubMed.
- X. Li, M. Kan, T. Wang, Z. Qin, T. Zhang, X. Qian, Y. Kuwahara, K. Mori, H. Yamashita and Y. Zhao, The ClO· generation and chlorate suppression in photoelectrochemical reactive chlorine species systems on BiVO4 photoanodes, Appl. Catal., B, 2021, 296, 120387 CrossRef CAS.
- C.-X. Chen, S.-S. Yang, J. Ding, L. Ding, R. Wu, L.-M. Liu, J.-W. Pang, L. He, J.-Q. Jiang and N.-Q. Ren, Existence of chloride ions in high salinity wastewater accelerates the removal of micropollutants over light-driven catalysts, Appl. Catal., B, 2023, 334, 122823 CrossRef CAS.
- X.-D. Wang, Y.-H. Huang, J.-F. Liao, Z.-F. Wei, W.-G. Li, Y.-F. Xu, H.-Y. Chen and D.-B. Kuang, Surface passivated halide perovskite single-crystal for efficient photoelectrochemical synthesis of dimethoxydihydrofuran, Nat. Commun., 2021, 12, 1202 CrossRef CAS PubMed.
- S. Chu, W. Li, Y. Yan, T. Hamann, I. Shih, D. Wang and Z. Mi, Roadmap on solar water splitting: current status and future prospects, Nano Futures, 2017, 1, 022001 CrossRef.
- D. Tang, K. Dang, J. Wang, C. Chen, J. Zhao and Y. Zhang, Solar-driven green synthesis of epoxides, Sci. China: Chem., 2023, 66, 3415–3425 CrossRef CAS.
- L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Photoelectrocatalytic arene C–H amination, Nat. Catal., 2019, 2, 366–373 CrossRef CAS PubMed.
- X. Liu, Z. Chen, S. Xu, G. Liu, Y. Zhu, X. Yu, L. Sun and F. Li, Bromide-mediated photoelectrochemical epoxidation of alkenes using water as an oxygen source with conversion efficiency and selectivity up to 100%, J. Am. Chem. Soc., 2022, 144, 19770–19777 CrossRef CAS PubMed.
- T. Hardwick, A. Qurashi, B. Shirinfar and N. Ahmed, Interfacial photoelectrochemical catalysis: solar-Induced green synthesis of organic molecules, ChemSusChem, 2020, 13, 1967–1973 CrossRef CAS PubMed.
- W.-Y. Fang, L. Ravindar, K. P. Rakesh, H. M. Manukumar, C. S. Shantharam, N. S. Alharbi and H.-L. Qin, Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review, Eur. J. Med. Chem., 2019, 173, 117–153 CrossRef CAS PubMed.
- S. Engbers, R. Hage and J. E. M. N. Klein, Toward environmentally benign electrophilic chlorinations: from Chloroperoxidase to bioinspired Isoporphyrins, Inorg. Chem., 2022, 61, 8105–8111 CrossRef CAS PubMed.
- S. Han, C. Cheng, M. He, R. Li, Y. Gao, Y. Yu, B. Zhang and C. Liu, Preferential adsorption of ethylene oxide on Fe and chlorine on Ni enabled scalable electrosynthesis of ethylene chlorohydrin, Angew. Chem., Int. Ed., 2023, 62, e202216581 CrossRef CAS PubMed.
- X. Wang, Z. Chen, Q. Liu, W. Lin and X. Xiong, Amine organocatalysts for highly ortho-selective chlorination of anilines with sulfuryl chloride, Chem. Commun., 2022, 58, 13325–13328 RSC.
- L. Zhang and X. Hu, Room temperature C(sp2)–H oxidative chlorination via photoredox catalysis, Chem. Sci., 2017, 8, 7009–7013 RSC.
- F. Liu, N. Wu and X. Cheng, Chlorination reaction of aromatic compounds and unsaturated carbon–carbon bonds with chlorine on demand, Org. Lett., 2021, 23, 3015–3020 CrossRef CAS PubMed.
- L. Huang, P. Wang, Y. Jiang, K. Davey, Y. Zheng and S.-Z. Qiao, Ethylene electrooxidation to 2-chloroethanol in acidic seawater with natural chloride participation, J. Am. Chem. Soc., 2023, 145, 15565–15571 CrossRef CAS PubMed.
- M. Chung, K. Jin, J. S. Zeng and K. Manthiram, Mechanism of chlorine-mediated electrochemical ethylene oxidation in saline Water, ACS Catal., 2020, 10, 14015–14023 CrossRef CAS.
- W. R. Leow, Y. Lum, A. Ozden, Y. Wang, D.-H. Nam, B. Chen, J. Wicks, T.-T. Zhuang, F. Li, D. Sinton and E. H. Sargent, Chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density, Science, 2020, 368, 1228–1233 CrossRef CAS PubMed.
- Y. Liang, F. Lin, Y. Adeli, R. Jin and N. Jiao, Efficient electrocatalysis for the preparation of (hetero)aryl chlorides and vinyl chloride with 1,2-dichloroethane, Angew. Chem., Int. Ed., 2019, 58, 4566–4570 CrossRef CAS PubMed.
- P. Zhang, T. Wang and J. Gong, Advances in electrochemical oxidation of olefins to epoxides, CCS Chem., 2023, 5, 1028–1042 CrossRef CAS.
- Z. Li, L. Luo, M. Li, W. Chen, Y. Liu, J. Yang, S.-M. Xu, H. Zhou, L. Ma, M. Xu, X. Kong and H. Duan, Photoelectrocatalytic C–H halogenation over an oxygen vacancy-rich TiO2 photoanode, Nat. Commun., 2021, 12, 6698 CrossRef CAS PubMed.
- G. Zhang, J. Ruan and T. Du, Recent advances on photocatalytic and electrochemical oxidation for ammonia treatment from water/wastewater, ACS ES&T Engg, 2021, 1, 310–325 Search PubMed.
- J. Ye, D. Zhang, S. Salli, Y. Li, F. Han, Y. Mai, F. Rosei, Y. Li, Y. Yang, F. Besenbacher, H. Niemantsverdriet, E. Richards and R. Su, Heterogeneous photocatalytic recycling of FeX2/FeX3 for efficient halogenation of C–H bonds using NaX, Angew. Chem., Int. Ed., 2023, 62, e202302994 CrossRef CAS PubMed.
- P. Xu, P.-Y. Chen and H.-C. Xu, Scalable photoelectrochemical dehydrogenative cross-coupling of heteroarenes with aliphatic C–H Bonds, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS PubMed.
- M. Xiang, C. Zhou, X.-L. Yang, B. Chen, C.-H. Tung and L.-Z. Wu, Visible light-catalyzed benzylic C–H bond chlorination by a combination of organic Dye (Acr+-Mes) and N-chlorosuccinimide, J. Org. Chem., 2020, 85, 9080–9087 CrossRef CAS PubMed.
- Y. Zhao, C. Deng, D. Tang, L. Ding, Y. Zhang, H. Sheng, H. Ji, W. Song, W. Ma, C. Chen and J. Zhao, α-Fe2O3 as a versatile and efficient oxygen atom transfer catalyst in combination with H2O as the oxygen source, Nat. Catal., 2021, 4, 684–691 CrossRef CAS.
- L. Wu, D. Tang, J. Xue, S. Liu, J. Wang, H. Ji, C. Chen, Y. Zhang and J. Zhao, Competitive non-radical nucleophilic attack pathways for NH3 oxidation and H2O oxidation on hematite photoanodes, Angew. Chem., Int. Ed., 2022, 61, e202214580 CrossRef CAS PubMed.
- O. Zandi and T. W. Hamann, Determination of photoelectrochemical water oxidation intermediates on haematite electrode surfaces using operando infrared spectroscopy, Nat. Chem., 2016, 8, 778–783 CrossRef CAS PubMed.
- B. Klahr and T. Hamann, Water oxidation on hematite photoelectrodes: Insight into the nature of surface states through in situ spectroelectrochemistry, J. Phys. Chem. C, 2014, 118, 10393–10399 CrossRef CAS.
- F. Le Formal, E. Pastor, S. D. Tilley, C. A. Mesa, S. R. Pendlebury, M. Grätzel and J. R. Durrant, Rate law analysis of water oxidation on a Hematite surface, J. Am. Chem. Soc., 2015, 137, 6629–6637 CrossRef CAS PubMed.
- T. Takashima, K. Ishikawa and H. Irie, Detection of intermediate species in oxygen evolution on Hematite electrodes using spectroelectrochemical measurements, J. Phys. Chem. C, 2016, 120, 24827–24834 CrossRef CAS.
- C. A. Mesa, L. Francàs, K. R. Yang, P. Garrido-Barros, E. Pastor, Y. Ma, A. Kafizas, T. E. Rosser, M. T. Mayer, E. Reisner, M. Grätzel, V. S. Batista and J. R. Durrant, Multihole water oxidation catalysis on haematite photoanodes revealed by operando spectroelectrochemistry and DFT, Nat. Chem., 2020, 12, 82–89 CrossRef CAS PubMed.
- F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Nature's inventory of halogenation catalysts: oxidative strategies predominate, Chem. Rev., 2006, 106, 3364–3378 CrossRef CAS PubMed.
- H.-A. Wagenknecht and W.-D. Woggon, New active-site analogues of Chloraperoxidase—syntheses and catalytic reactions, Angew. Chem., Int. Ed., 1997, 36, 390–392 CrossRef CAS.
- Z. Cong, S. Yanagisawa, T. Kurahashi, T. Ogura, S. Nakashima and H. Fujii, Synthesis, characterization, and reactivity of hypochloritoiron(III) porphyrin complexes, J. Am. Chem. Soc., 2012, 134, 20617–20620 CrossRef CAS PubMed.
- Z. Cong, T. Kurahashi and H. Fujii, Oxidation of chloride and subsequent chlorination of organic compounds by oxoiron(IV) porphyrin π-cation radicals, Angew. Chem., Int. Ed., 2011, 50, 9935–9939 CrossRef CAS PubMed.
- Y. Yao, L. Zhao, J. Dai, J. Wang, C. Fang, G. Zhan, Q. Zheng, W. Hou and L. Zhang, Single atom Ru monolithic electrode for efficient chlorine evolution and nitrate reduction, Angew. Chem., Int. Ed., 2022, 61, e202208215 CrossRef CAS PubMed.
- C. Y. Cummings, F. Marken, L. M. Peter, K. G. Upul Wijayantha and A. A. Tahir, New insights into water splitting at mesoporous α-Fe2O3 films: a study by modulated transmittance and impedance spectroscopies, J. Am. Chem. Soc., 2012, 134, 1228–1234 CrossRef CAS PubMed.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, Water oxidation at hematite photoelectrodes: the role of surface states, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS PubMed.
- Y. Zhang, H. Zhang, A. Liu, C. Chen, W. Song and J. Zhao, Rate-limiting O–O bond formation pathways for water oxidation on Hematite photoanode, J. Am. Chem. Soc., 2018, 140, 3264–3269 CrossRef CAS PubMed.
- Y. Zhang, H. Zhang, H. Ji, W. Ma, C. Chen and J. Zhao, Pivotal role and regulation of proton transfer in water oxidation on hematite photoanodes, J. Am. Chem. Soc., 2016, 138, 2705–2711 CrossRef CAS PubMed.
- T. Hering, B. Mühldorf, R. Wolf and B. König, Halogenase-inspired oxidative chlorination using flavin photocatalysis, Angew. Chem., Int. Ed., 2016, 55, 5342–5345 CrossRef CAS PubMed.
- N. Fu, G. S. Sauer and S. Lin, Electrocatalytic radical dichlorination of alkenes with nucleophilic chlorine sources, J. Am. Chem. Soc., 2017, 139, 15548–15553 CrossRef CAS PubMed.
- N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Metal-catalyzed electrochemical diazidation of alkenes, Science, 2017, 357, 575–579 CrossRef CAS PubMed.
- Q. Wang, T. Li, C. Yang, M. Chen, A. Guan, L. Yang, S. Li, X. Lv, Y. Wang and G. Zheng, Electrocatalytic methane oxidation greatly promoted by chlorine intermediates, Angew. Chem., Int. Ed., 2021, 60, 17398–17403 CrossRef CAS PubMed.
- Z. Shen, J. Li, Y. Zhang, J. Bai, X. Tan, X. Li, L. Qiao, Q. Xu and B. Zhou, Highly efficient total nitrogen and simultaneous total organic carbon removal for urine based on the photoelectrochemical cycle reaction of chlorine and hydroxyl radicals, Electrochim. Acta, 2019, 297, 1–9 CrossRef CAS.
- C. A. Mesa, L. Francàs, K. R. Yang, P. Garrido-Barros, E. Pastor, Y. Ma, A. Kafizas, T. E. Rosser, M. T. Mayer, E. Reisner, M. Grätzel, V. S. Batista and J. R. Durrant, Multihole water oxidation catalysis on haematite photoanodes revealed by operando spectroelectrochemistry and DFT, Nat. Chem., 2020, 12, 82–89 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06337b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |