
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen spillover enhances alkaline hydrogen electrocatalysis on interface-rich metallic Pt-supported MoO3†
Rajib
Samanta‡
ab,
Biplab Kumar
Manna‡
ab,
Ravi
Trivedi
cd,
Brahmananda
Chakraborty
 be and
Sudip
Barman
*ab
be and
Sudip
Barman
*ab
aSchool of Chemical Sciences, National Institute of Science Education and Research (NISER), HBNI, Bhubaneswar, Orissa 752050, India. E-mail: sbarman@niser.ac.in; Tel: +91 6742494183
bHomi Bhabha National Institute, Training School Complex, Anushakti Nagar, Mumbai 400094, India
cDepartment of Physics, Karpagam Academy of Higher Education, Coimbatore 641021, India
dCentre for High Energy Physics, Karpagam Academy of Higher Education, Coimbatore 641021, India
eHigh Pressure & Synchroton Radiation Physics Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India
First published on 8th December 2023
Abstract
Efficient and cost-effective electrocatalysts for the hydrogen oxidation/evolution reaction (HOR/HER) are essential for commercializing alkaline fuel cells and electrolyzers. The sluggish HER/HOR reaction kinetics in base is the key issue that requires resolution so that commercialization may proceed. It is also quite challenging to decrease the noble metal loading without sacrificing performance. Herein, we report improved HER/HOR activity as a result of hydrogen spillover on platinum-supported MoO3 (Pt/MoO3-CNx-400) with a Pt loading of 20%. The catalyst exhibited a decreased over-potential of 66.8 mV to reach 10 mA cm−2 current density with a Tafel slope of 41.2 mV dec−1 for the HER in base. The Pt/MoO3-CNx-400 also exhibited satisfactory HOR activity in base. The mass-specific exchange current density of Pt/MoO3-CNx-400 and commercial Pt/C are 505.7 and 245 mA mgPt−1, respectively. The experimental results suggest that the hydrogen binding energy (HBE) is the key descriptor for the HER/HOR. We also demonstrated that the enhanced HER/HOR performance was due to the hydrogen spillover from Pt to MoO3 sites that enhanced the Volmer/Heyrovsky process, which led to high HER/HOR activity and was supported by the experimental and theoretical investigations. The work function value of Pt [Φ = 5.39 eV) is less than that of β-MoO3 (011) [Φ = 7.09 eV], which revealed the charge transfer from Pt to the β-MoO3 (011) surface. This suggested the feasibility of hydrogen spillover, and was further confirmed by the relative hydrogen adsorption energy [ΔGH] at different sites. Based on these findings, we propose that the H2O or H2 dissociation takes place on Pt and interfaces to form Pt–Had or (Pt/MoO3)–Had, and some of the Had shifted to MoO3 sites through hydrogen spillover. Then, Had at the Pt and interface, and MoO3 sites reacted with H2O and HO− to form H2 or H2O molecules, thereby boosting the HER/HOR activity. This work may provide valuable information for the development of hydrogen-spillover-based electrocatalysts for use in various renewable energy devices.
Introduction
The world's environmental problems and energy consumption are gradually increasing with the growing population.1 Due to increasing fossil fuel consumption and expanding environmental concerns, the switch to the use of hydrogen fuel is urgently required.2,3 Hydrogen is a potential alternative to fossil fuels because of its high gravimetric energy density as well as its carbon-free H2O emissions.4,5 Wind, tidal, and solar energy are several renewable alternatives to fossil fuels, but these are regional and seasonal.6,7 Therefore, researchers have investigated several methods to store renewable energies and use them during energy crises.8,9In this context, there has recently been great interest in electrolyzers and fuel cells. There are mainly two commercially available techniques for water electrolyzers and fuel cells – (i) anion exchange membranes, or AEMs, and (ii) proton exchange membranes, or PEMs.10,11 PEM-based devices are comparatively more durable with a high power density as compared to AEM-based devices.12 However, the high cost of the components is the fundamental disadvantage of PEM-based systems.13,14 The AEM is less expensive, and comparatively less expensive materials are used for the oxygen evolution reaction (OER) and the oxygen reduction reaction (ORR) in alkaline medium.12,15 Therefore, AEM technology is favored, and commercial Pt/C has been extensively used as a state-of-the-art catalyst for alkaline electrolyzers and fuel cells.16 Pt/C in basic media exhibits nearly 2–3 orders of magnitude lower activity compared to acid media activity.17 Hence, a greater amount of Pt/C is needed in basic medium to achieve the same activity as that found in acid media.18 Moreover, the durability of this catalyst is insufficient for industrial application.18 Therefore, the development of a novel approach that boosts the activity and longevity as well as lowering the costs of the components in base medium is extremely desirable.
To design a new catalyst for the alkaline HER/HOR, it is essential to understand the mechanisms of the reactions. The alkaline HER is associated with two main steps: the dissociation of water is the first step (Volmer step), followed by the formation of Had and HOad. In the second step, two Had combine together to form hydrogen (H2) (Tafel step), or Had interacts with H2O to form H2, followed by the desorption of HOad (Heyrovsky step).19,20 For the alkaline HOR, dissociation of H2 occurs on the surface of the metal to form the M–Had intermediate. Then, M–Had interacts with HOad (bifunctional mechanism) or HO− of the electrolyte (hydrogen binding energy (HBE) theory) to produce H2O.21,22
The two most acceptable mechanisms for the alkaline HER/HOR are (a) the HBE theory23,24 and (b) the oxophilicity effect.17,25 Sheng et al.23 correlated the HER activity of different metals with HBE and obtained a volcano-type plot, which suggests the existence of an optimal HBE (Sabatier's principle). The reason for the decreased HER/HOR activity as well as the rate-controlled process in alkaline medium remains unclear. Most probably, the very strong M–H adsorption decreases the HOR activity in basic solution as compared to acid solution, which was suggested by Durst et al.26
Currently, the hydrogen spillover phenomena have recently created a new opportunity for improving the HER/HOR performance of metal/support electrocatalysts using hydrogen-deficient components (support) and a hydrogen-rich Pt substrate.27,28 Therefore, the designs of such catalysts can improve the catalytic activity in base. For the hydrogen-spillover-based catalysts (HSBCs), there are three main steps involved in the alkaline HER/HOR: (1) the dissociation of H2O on metal to form M–Had and M–OHad, followed by the desorption of OHad or dissociation of H2 to form M–Had; (2) the diffusion of hydrogen and then hydrogen spillover from metal to support; and (3) finally, desorption of H2 and H2O on supports (ΔGH-support > 0).29,30
Several studies and observations regarding hydrogen spillover have recently been published. For example, Ma and co-workers demonstrated how large differences in work functions (the least amount of energy required for an electron to escape from the surface when released from the interior, depending on surface roughness, crystal orientation, chemical composition, and surface termination) between metals and supports cause strongly charged molecules to accumulate at the Schottky junction interface, which prevents hydrogen spillover across metal and support interfaces by sequentially increasing interfacial proton adsorption.29 They synthesized several catalysts (Pt-alloy-CoP) with different ΔΦ (ΔΦ = work function difference between Pt-alloy and CoP) by alloying different metals (such as Ir, Pd, Au, Rh, and Ag). It was observed that the activity of Pt-alloy-CoP increased with decreasing ΔΦ value.
They also calculated the free energy for each step of hydrogen spillover for Pt/CoP (larger ΔΦ) and Pt2Ir1/CoP (smaller ΔΦ), and observed strong hydrogen adsorption at the Pt/CoP interface (ΔGH = −0.36 eV). This delayed the hydrogen spillover, whereas a moderate ΔGH at the Pt2Ir1/CoP interface increased the hydrogen spillover and enhanced the HER performance accordingly.29 Thus, by combining metal and support, this strategy can kinetically promote hydrogen adsorption and desorption.31 Therefore, this design concept reduces novel metal consumption and cost of the catalysts while providing competitive catalytic performance.32
The use of a small amount of Pt on a suitable support can modify the electronic structure by synergistic interaction, which improves the hydrogen spillover as well as the catalytic activity.33 This strategy was adequately exemplified by the Pt/WO3,34 Pt/CoP,27,29 Pt/CoOx,35 Pt/SiO2,36 Pt/RuCeOx,32 and Pt/hypo-d-metal oxide systems.37 Molybdenum oxides (MoO3−x, 0 ≤ x ≤ 1) with a variety of stoichiometries and mixed oxidation states are among the nonprecious inorganic electrocatalysts that offer highly controllable chemical and physical properties with moderate electrocatalytic activity.38 Therefore, MoO3−x can be used as a support material with Pt nanoparticles to produce a high-performance electrocatalyst.
In this work, a heterointerface-rich Pt/MoO3-CNx-400 composite was prepared for the HER/HOR in alkaline medium. The catalyst possesses approximately 4.5 and 2 times higher HER and HOR activity as compared to commercial Pt/C with high durability, respectively. The experimental results suggest that HBE is the prime descriptor for the alkaline HER/HOR of Pt/MoO3-CNx-400. We demonstrate that the high HER/HOR activity of Pt/MoO3-CNx-400 is due to the hydrogen spillover from Pt to the MoO3 surface, as suggested by experimental and theoretical findings. Based on our findings, we propose a hydrogen-spillover-based mechanism to explain the enhanced HER/HOR activity of the catalyst in base.
Results and discussion
Characterization
The preparation of the Pt/MoO3-CNx-400 nanocomposite was carried out by three facile steps. First, formamide was treated with microwave irradiation for three hours at 180 °C to prepare nitrogen-doped carbon (CNx). Then, PtMo/CNx was prepared by the sodium borohydride reduction of MoCl5 and H2PtCl6·6H2O in the presence of the as-prepared CNx, followed by ultrasonication. Finally, calcination of PtMo/CNx was carried out at 400 °C for 120 minutes to prepare Pt/MoO3-CNx-400 (see the ESI† for details). For comparison purposes, we synthesized samples of Mo/CNx-400, Pt/CNx-400, Pt/MoO3-CNx-400 (with different Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo ratios), and Pt/MoO3-CNx (with different synthesis temperatures) using the same method.
:Mo ratios), and Pt/MoO3-CNx (with different synthesis temperatures) using the same method.
During calcination, growth of Pt nanoparticles on the MoO3 sheets occurs, which leads to the formation of heterointerfaces. Additionally, heating causes a significant synergistic interaction, which results in the catalyst's high catalytic activity. Transmission electron microscopy (TEM) and field emission scanning electron microscopy (FESEM) were used to analyze the morphology, shape, and structure of all the catalysts. Fig. 1a and S1a† display FESEM images of Pt/MoO3-CNx-400 and PtMo/CNx, respectively, which show small particles spread over the nanosheet. The particles over the nanosheet were also further confirmed by the TEM images of Pt/MoO3-CNx-400 (Fig. 1b and c). The average size of the particles for Pt/MoO3-CNx-400 was found to be approximately 13.3 nm (inset in Fig. 1b). TEM images of pre-heated PtMo/CNx are shown in Fig. S1b–d,† which also reveals that the particles are spread over the sheet. The average particle size of PtMo/CNx is approximately 2.32 nm (inset in Fig. 1d). Because of the small size of the particles, the electrochemical surface area is greater, which can increase the catalytic activity of the catalysts.
 | ||
| Fig. 1 (a) FE-SEM, (b and c) TEM, (d) HRTEM, and (e) SAED images of Pt/MoO3-CNx-400. (f–k) STEM and elemental mapping images of Pt/MoO3-CNx-400. (l) Elemental mapping overlay of Pt, Mo, and O. | ||
HRTEM analysis of the catalysts was also carried out to examine the phases of the catalysts. Fig. 1d, S1a and b† present HRTEM images of Pt/MoO3-CNx-400. The lattice fringes were calculated from the HRTEM images, and were found to be 2.28, 3.79, and 3.27 Å, which corresponded to the presence of the Pt(111), MoO3(−101), and MoO3 (011) planes, respectively. The presence of heterointerfaces was also found after examination of the HRTEM images. During the calcination of PtMo/CNx, growth of Pt nanoparticles occurred on the MoO3 nanosheets, which led to the formation of Pt/MoO3 boundaries. These heterointerfaces can alter the electronic structure of the elements in the junction, which can enhance the catalytic activity. The selected area electron diffraction (SAED) spectrum of Pt/MoO3-CNx-400 suggests that the compound is polycrystalline in nature (Fig. 1e). The interlayer spacing (d-spacing) was calculated from the SAED images of the catalysts. The interlayer spacings of 2.28, 1.98, and 1.40 Å present the (111), (200), and (220) planes of Pt, whereas the values of 3.79, 3.27, and 1.57 Å present the (−101), (011), and (014) planes of MoO3, respectively. Fig. S2c and d† show HRTEM and SAED images of PtMo/CNx, respectively.
All these data suggest the presence of metallic Pt and MoO3 after the calcination of PtMo/CNx. The elemental mappings of Pt/MoO3-CNx-400 and PtMo/CNx are shown in Fig. 1f–l and S3a–g,† respectively. This suggests the presence of C, N, O, Mo, and Pt in both catalysts. Fig. 1l and S3g† show a non-uniform distribution of Mo and Pt over the entire compound, suggesting the formation of a hetero-composite, not an alloy. A line scan profile of Mo and Pt was carried out on the Pt/MoO3-CNx-400 catalyst to determine the distribution of the elements (Fig. S3h and i†). Discontinuous Mo and Pt spectra occurred throughout the line drawn on the Pt/MoO3-CNx-400 compound, suggesting the presence of boundaries between the Pt and Mo composites. Powder X-ray diffraction (p-XRD) was also carried out to confirm the crystalline phases of the sample.
The p-XRD patterns of Pt/MoO3-CNx-400 and PtMo/CNx are shown in Fig. 2a. The standard powder diffraction files (PDF) of metallic Pt (PDF# 00-004-0802) and MoO3 (PDF# 01-085-2405) were used to confirm the phases of the materials. The peaks at 39.7, 46.1, and 67.3 arose due to the (111), (200), and (220) planes of metallic Pt, while the peaks at 23.7, 27.4, 29.2, 33.7, 49.4, 55.5, and 59.1 correspond to the (−101), (011), (101), (110), (020), (−121), and (014) planes of MoO3, respectively. Therefore, Pt and MoO3 are formed in Pt/MoO3-CNx-400 upon calcination of PtMo/CNx.
 | ||
| Fig. 2 (a) p-XRD spectra of Pt/MoO3-CNx-400 and PtMo/CNx. (b) XPS survey scan spectrum of Pt/MoO3-CNx-400. (c–g) High-resolution XPS spectra of Mo 3d, Pt 4f, C 1s, O 1s, and N 1s for Pt/MoO3-CNx-400, respectively. (h and i) N2 physisorption isotherm and pore size distribution plots for Pt/MoO3-CNx-400, respectively. | ||
To confirm the existence of metallic Pt and MoO3 in the compound, the X-ray photoelectron spectroscopy (XPS) analysis of Pt/MoO3-CNx-400 was carried out. Fig. 2b presents the XPS survey scan spectrum of Pt/MoO3-CNx-400, suggesting the existence of Mo, Pt, N, C, and O in the composite. The deconvoluted XPS spectrum of Mo 3d is shown in Fig. 2c. The small peaks at 231.44 and 234.43 eV arose due to the presence of 3d5/2 and 3d3/2 of Mo5+, whereas the peaks at 232.64 and 235.82 eV correspond to Mo6+ 3d5/2 and Mo6+ 3d3/2, respectively.39 Six peaks were used to fit the deconvoluted Pt 4f XPS spectrum. The peaks at 71.12 and 74.44 eV were attributed to the existence of Pt0 4f7/2 and Pt0 4f5/2, whereas the peaks at 71.75 and 75.1 eV corresponded to the presence of Pt2+ 4f7/2 and Pt2+ 4f5/2, respectively (Fig. 2d).40 Satellite peaks were found at 73.0 and 76.44 eV. This again confirms the presence of Pt and MoO3 in the compound.
Five peaks were used to fit the deconvoluted XPS spectrum of C 1s (Fig. 2e). The peak at 283.9 eV arose due to the presence of a metal carbon bond, whereas the peaks at 284.7, 285.3, 286.4, and 288.7 eV were due to the presence of sp2 C, sp3 C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C–N, and OC–O, respectively.41,42 The O 1s spectrum was also fitted with three peaks (Fig. 2f). The peaks at 530.3 and 531.2 eV correspond to the Mo–O bond and C–O-containing functional groups, whereas the peak at 532.6 eV suggests the presence of the adsorbed hydroxyl group.43Fig. 2g represents the deconvoluted N 1s XPS spectra of Pt/MoO3-CNx-400. The peaks at 398.2, 399.2, and 401.1 eV correspond to the presence of sp2 N/CN, sp3 N, and C–N–H/N–O, respectively.44 Fig. S4a† shows the comparison of the Pt 4f XPS spectra for Pt/MoO3-CNx-400 and Pt/CNx-400, while Fig. S4b† shows the comparison of the Mo 3d XPS spectra for Pt/MoO3-CNx-400 and Mo/CNx-400. These data show that the Pt 4f peak of Pt/MoO3-CNx-400 was positively shifted compared to the Pt/CNx-400, while the Mo 3d peak of Pt/MoO3-CNx-400 was negatively shifted compared to Mo/CNx-400, suggesting electron transfer from Pt to MoO3.
O/C–N, and OC–O, respectively.41,42 The O 1s spectrum was also fitted with three peaks (Fig. 2f). The peaks at 530.3 and 531.2 eV correspond to the Mo–O bond and C–O-containing functional groups, whereas the peak at 532.6 eV suggests the presence of the adsorbed hydroxyl group.43Fig. 2g represents the deconvoluted N 1s XPS spectra of Pt/MoO3-CNx-400. The peaks at 398.2, 399.2, and 401.1 eV correspond to the presence of sp2 N/CN, sp3 N, and C–N–H/N–O, respectively.44 Fig. S4a† shows the comparison of the Pt 4f XPS spectra for Pt/MoO3-CNx-400 and Pt/CNx-400, while Fig. S4b† shows the comparison of the Mo 3d XPS spectra for Pt/MoO3-CNx-400 and Mo/CNx-400. These data show that the Pt 4f peak of Pt/MoO3-CNx-400 was positively shifted compared to the Pt/CNx-400, while the Mo 3d peak of Pt/MoO3-CNx-400 was negatively shifted compared to Mo/CNx-400, suggesting electron transfer from Pt to MoO3.
Brunauer–Emmett–Teller (BET) analysis was carried out to determine the surface area, porosity, and pore size of the compounds. Fig. 2h displays the nitrogen adsorption/desorption isotherm of Pt/MoO3-CNx-400. This indicates that the isotherm increases with increasing relative pressure, and hysteresis loops arise at 0.1–0.95 in the relative pressure region. The surface area of the compound was also calculated from the isotherm, which shows a surface area of 14.2 m2 g−1 for Pt/MoO3-CNx-400. Fig. 2i shows the pore size distribution data for Pt/MoO3-CNx-400, as calculated by density functional theory (DFT), and the average pore size of the composite was 1.2–3.35 nm in diameter. Therefore, there is a microporous as well as mesoporous structure in the compound, which could offer accessible channels for electrolytes and ions that would increase the electrocatalytic efficiency of the process.
The electrochemical surface area (ECSA) of the catalysts was calculated from CO-stripping experiments. It was found that the ECSA value of Pt/MoO3-CNx-400 was 42.2 m2 g−1 in base, whereas the ECSA values of PtMo/CNx, Pt/CNx-400, and commercial Pt/C were 20, 41.3, and 36.1 m2 g−1, respectively (Fig. S4c–f†). Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis was performed to determine the exact loading of metals. The total metal loading was 46%, and the weight ratio of Pt and Mo was 1:1.3 (Table S1†).
HER activity of Pt/MoO3-CNx-400 in alkaline medium
A three-electrode system associated with a potentiostat (Autolab, Metrohm) was used to measure the electrochemical HER performance of all catalysts, with a rotation speed of 1600 rpm and scan speed of 10 mV s−1. A catalyst-loaded glassy carbon electrode was used as the working electrode, while Pt and Ag/AgCl electrodes were used as counter and reference electrodes, respectively. The HER activity of Pt/MoO3-CNx-400 was compared with that of PtMo/CNx, Pt/MoO3-400, Pt/CNx-400, Mo/CNx-400, and commercial Pt/C in base medium. Fig. 3a and S5a† show the iR-corrected and non-iR-corrected HER curves of all these catalysts in the base. Pt/MoO3-CNx-400 required less overpotential to reach any potential as compared to other catalysts. The HER LSV curves of Pt/MoO3-CNx-400 with different metal loadings, different Pt and Mo ratios, and different calcination temperatures are shown in Fig. S5b–d,† respectively. | ||
| Fig. 3 (a) iR-corrected HER curves of Pt/MoO3-CNx-400, PtMo/CNx, Pt/CNx-400, Mo/CNx-400, and commercial Pt/C in 1.0 M KOH. (b) Potential at 10 mA cm−2 current density. (c and d) Tafel slopes and Nyquist plots of the catalysts in base, respectively. (e–g) Current density, mass activity, and surface activity plots of these catalysts at −0.1 V (RHE), respectively. (h) Chronopotentiometric stability of Pt/MoO3-CNx-400 and commercial Pt/C at −10 mA cm−2 current density in base. (i) H2 faradaic efficiency of Pt/MoO3-CNx-400 in 1.0 M KOH at 10 mA cm−2 current density. | ||
These indicate that Pt/MoO3-CNx-400, with a calcination temperature 400 °C and metallic ratio 1:1.3, is more favourable for the HER performance. Pt/MoO3-CNx-400 requires only 66.8 mV of overpotential to achieve a current density of 10 mA cm−2, while PtMo/CNx, Pt/MoO3-400, Pt/CNx-400, and commercial Pt/C require 88, 76.7, 80.4, and 93.4 mV overpotentials, respectively (Fig. 3b). This indicates high HER activity of the catalyst. The Tafel slope of all catalysts was calculated using the Tafel equation (see the ESI†) to determine their reaction kinetics. The Tafel slopes of Pt/MoO3-CNx-400, commercial Pt/C, Pt/MoO3-400, PtMo/CNx, and Pt/CNx-400 are 41.2, 82.6, 64.3, 63.2, and 52.7 mV dec−1, respectively (Fig. 3c).
This shows that there is a lower Tafel slope value for Pt/MoO3-CNx-400, which indicates faster HER kinetics as compared to other catalysts. In addition, the impedance spectra proved that there are faster reaction kinetics for Pt/MoO3-CNx-400. The Nyquist plots for Pt/MoO3-CNx-400, commercial Pt/C, Pt/MoO3-400, PtMo/CNx, and Pt/CNx-400 at −0.02 V (RHE) are shown in Fig. 3d. All Nyquist plots were fitted using an equivalent circuit to calculate the charge transfer resistance (Rct) of the compounds (Fig. S5e†). Pt/MoO3-CNx-400 possesses a lower Rct value of 9.99 Ω, which is much less as compared to the other catalysts, suggesting the rapid charge transfer of the catalyst.
Fundamental parameters such as current density, mass activity, and surface-specific activity of the catalysts were evaluated from the HER LSV plots at −0.1 V (RHE). The Pt/MoO3-CNx-400 shows a 50.2 mA cm−2 current density at −0.1 V (RHE) potential, which is much higher as compared to the other catalysts (Fig. 3e). The mass activities of Pt/MoO3-CNx-400, commercial Pt/C, Pt/MoO3-400, PtMo/CNx, and Pt/CNx-400 were 3514, 784, 1580.2, 926.8, and 1344 mA mgmetal−1, respectively (Fig. 3f). The surface-specific activity of Pt/MoO3-CNx-400 was 8.327 mA cmmetal−2, whereas those for commercial Pt/C, Pt/MoO3-400, PtMo/CNx, and Pt/CNx-400 were 2.17, 2.21, 4.63, and 3.25 mA cmmetal−2 (Fig. 3g).
Table S2† presents a comparison of all the activity parameters of these compounds. The HER activity parameters for Pt/MoO3-CNx-400 were also compared to the recently reported catalysts in Table S3.† All these results suggest that Pt/MoO3-CNx-400 is an excellent HER catalyst in base media. The enhanced activity of this catalyst may be due to hydrogen spillover, which is discussed later. The long-term stability of a catalyst is also highly important for its industrialization. Chronopotentiometry was performed at −10 mA cm−2 current density for 24 hours to determine the stability of Pt/MoO3-CNx-400 and commercial Pt/C in the presence of Nafion binder (Fig. 3h). After a 24 hour stability test, the commercial Pt/C required additional overpotential of approximately 35 mV to reach 10 mA cm−2 current density, while Pt/MoO3-CNx-400 required approximately an additional 9 mV of overpotential, suggesting the satisfactory stability of Pt/MoO3-CNx-400. The chronoamperometry tests of these catalysts were also carried out using Nafion binder (Fig. S5f†), which suggested very satisfactory stability of Pt/MoO3-CNx-400 as compared to commercial Pt/C.
After the stability test, Pt/MoO3-CNx-400 was characterized using p-XRD, XPS, and TEM analyses (Fig. S6†). The p-XRD data clearly show the metallic Pt peaks as found in the parent compound, while a low-intensity MoO3 peak was found after the stability test (Fig. S6a†). XPS characterization of the sample was also carried out after the stability test to confirm the presence of metallic Pt and MoO3 in the compound (Fig. S6b and c†). Similar Pt 4f and Mo 3d XPS spectra were revealed after and before the stability test of Pt/MoO3-CNx-400. Fig. S6d–g† presents the TEM analysis of Pt/MoO3-CNx-400 after the stability test, suggesting a morphology that is similar to that of the catalyst. This indicates that the compound remained nearly the same, even after the stability test, and suggests that the catalyst is stable throughout electrocatalysis. The evolved hydrogen (H2) was also quantified by gas chromatography to calculate the faradaic efficiency. The evolved H2 plotted against time based on theoretical and experimental calculations is shown in Fig. 3i. It shows that there was approximately 93% faradaic efficiency after 120 minutes of reaction at −10 mA cm−2 current density. All these results suggest that Pt/MoO3-CNx-400 is one of the most optimal catalysts for the alkaline HER.
HOR performance of Pt/MoO3-CNx-400 in alkaline medium
The HOR performances of Pt/MoO3-CNx-400, commercial Pt/C, Pt/MoO3-400, PtMo/CNx, Mo/CNx-400, and Pt/CNx-400 were observed in H2-saturated 0.1 M KOH solution. The iR-corrected and non-iR-corrected HOR polarisation curves of all these catalysts are shown in Fig. 4a and S7a,† respectively. They suggest that the highest HOR activity was exhibited by Pt/MoO3-CNx-400, as compared to the other catalysts. The HOR conducted with different Pt-loaded Pt/MoO3-CNx-400 catalysts is shown in Fig. S7b,† suggesting that catalytic activity increases with increasing Pt loading up to 20%, after which there is less of an increase. The HOR polarization curves for Pt/MoO3-CNx-400 with different rotation speeds in base are presented in Fig. 4b. They indicate that the limiting current density (JL) increases with increasing rotation speed. The JL is the summation of the kinetic and diffusion currents JK and JD, respectively. The Koutecký–Levich (K–L) equation (eqn (1)) was used to calculate JK as follows:| 1/JL = 1/JK + 1/JD = 1/JK + 1/BC0ω1/2 | (1) |
| JD = 0.62nFD2/3υ−1/6C0ω1/2 = BC0ω1/2 | (2) |
 | ||
| Fig. 4 (a) HOR LSV plots of Pt/MoO3-CNx-400, PtMo/CNx, Pt/MoO3-400, commercial Pt/C, Mo/CNx-400, and Pt/CNx-400 in base. (b) HOR curves of Pt/MoO3-CNx-400 at different rotation speeds in base. (c) Corresponding K–L plot of the catalyst. (d) Butler–Volmer fitting plots of these catalysts in base. (e–h) i0, i0,m, i0,s, and MA (at 0.4 V RHE) plots of all catalysts in base, respectively. (i) Chronoamperometric HOR stability of Pt/MoO3-CNx-400 and commercial Pt/C in base. | ||
The K–L plot was obtained from Fig. 4b at 0.35 V (RHE), which displays a straight line passing through the origin, indicating that H2 mass diffusion controls the HOR kinetics (Fig. 4c). The BC0 value for Pt/MoO3-CNx-400 was found to be 0.0687 mA (cm2 rpm)−1/2 in 0.1 M KOH.
The Nernstian diffusion model was used to explain HOR kinetics. Eqn (3) was used to correct the HOR diffusion overpotential.
| ηdiffusion = −(RT/F)ln(1 − J/JL) | (3) |
Eqn (4) was used to express diffusion current (JD) in the pure diffusion region:
| JD = J(1 − exp(−2ηF/RT)) | (4) |
The Butler–Volmer equation was used to determine the catalyst's exchange current density (eqn (5))
| JK = i0(e(αFη/RT) − e−((α−1)Fη/RT)) | (5) |
The Butler–Volmer fitting curves of the compounds were estimated from Fig. 4a using the Butler–Volmer equation, as shown in Fig. 4d. The exchange current densities (i0) were also calculated from the Butler–Volmer fitting. The i0 of Pt/MoO3-CNx-400 was found to be 2.58 mA cm−2 in base, which is higher than that for other catalysts (Fig. 4e). The intrinsic fundamental properties such as mass-specific and surface-specific exchange current densities were calculated for the catalysts. The mass-specific exchange current density (i0,m) for Pt/MoO3-CNx-400 was 505.7 mA mgmetal−1, while that for commercial Pt/C, Pt/MoO3-400, PtMo/CNx, and Pt/CNx-400 were 245, 427.3, 52, and 301.8 mA mgmetal−1, respectively (Fig. 4f). The catalyst showed a surface-specific exchange current density (i0,s) of 1.198 mA cmmetal−2, which is superior to those of other catalysts (Fig. 4g). The mass activity of Pt/MoO3-CNx-400 at −0.2 V (RHE) was 490 mA mgmetal−1, which was 1.31, 1.096, 1.445, and 1.163 times higher than that of commercial Pt/C, Pt/MoO3-400, PtMo/CNx, and Pt/CNx-400, respectively (Fig. 4h).
Table S4† presents a comparison of all activity parameters for these catalysts. Additionally, the activity parameters for Pt/MoO3-CNx-400 were compared to those of recently reported HOR catalysts (Table S5†). The long-term stability of the catalyst is essential for its industrial application. A comparison of HOR stability for Pt/MoO3-CNx-400 and commercial Pt/C is shown in Fig. 4i and S7c† in the absence and presence of Nafion binder. There was increased stability for Pt/MoO3-CNx-400 as compared to commercial Pt/C. Therefore, Pt/MoO3-CNx-400 is one of the most optimal catalysts for the HER and HOR in base.
Mechanistic insights
To gain mechanistic insights, several experimental and theoretical studies were performed. There are two mechanisms that are widely used to explain the sluggish kinetics of HER/HOR in alkaline medium: (i) the HBE theory23,24 and (ii) the OHBE theory.25 Gasteiger and co-workers reported that the HBE is the key descriptor for HER/HOR in alkaline media.26 Strmcnik et al.25 reported that hydroxyl adsorption improves the HOR activity (oxophilicity). Therefore, to determine whether the reaction is occurring through the HBE theory or OHBE theory, we performed HOR and CV experiments in different pH (buffer) solutions. Fig. S7d† and 5a show HOR and normalised HOR plots for Pt/MoO3-CNx-400 in different pH solutions (pH range 1–13), and the HOR performance gradually decreased with increasing pH. The half-wave potential is defined as the overpotential at half of the normalised limiting current density (i = 0.5ilim). The half-wave potentials slowly transition to positive potentials with increasing pH.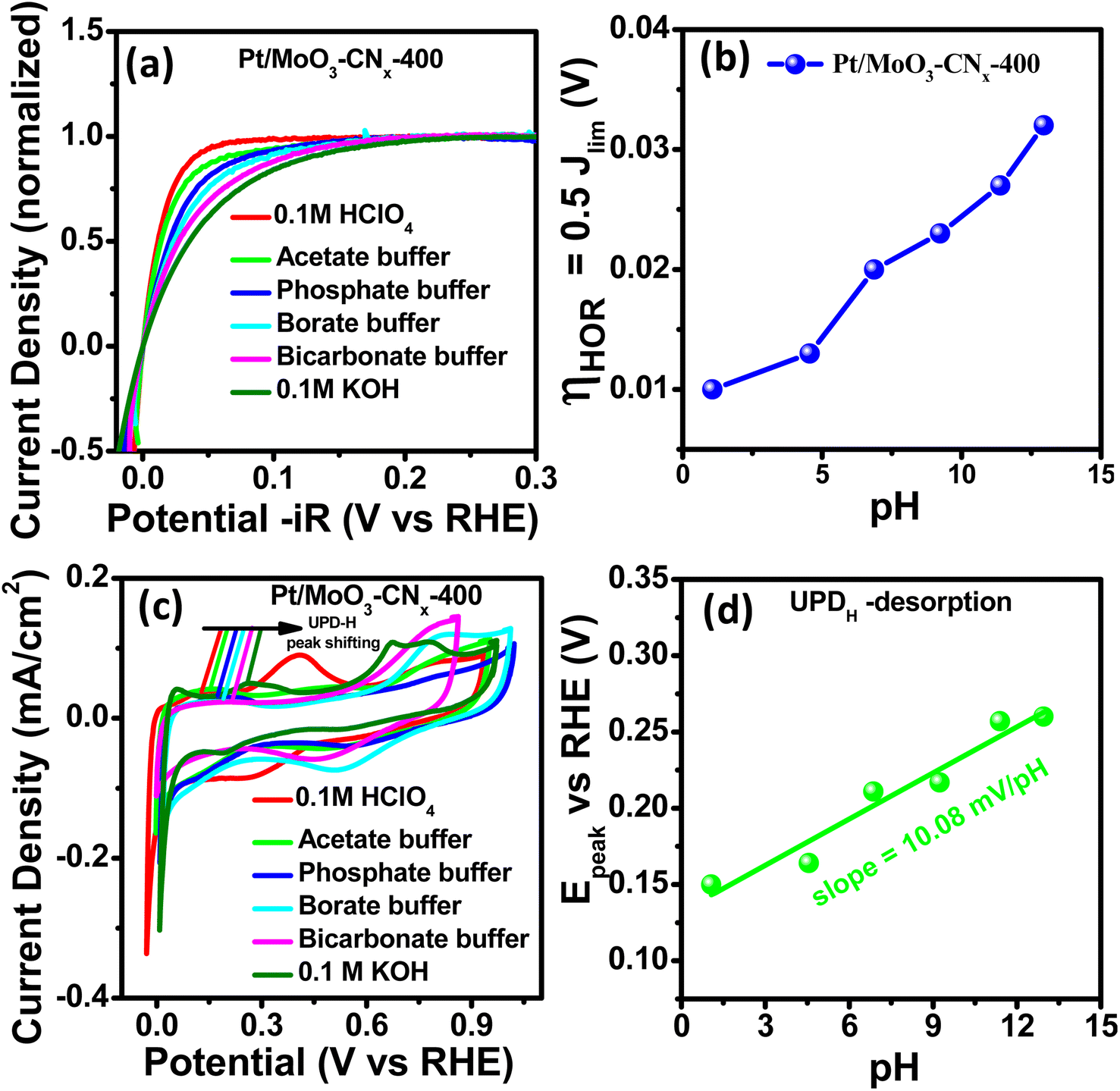 | ||
| Fig. 5 (a) Normalized HOR plots for Pt/MoO3-CNx-400 in different pH solutions. (b) Overpotential at 0.5 × Jlimvs. pH plot of the catalyst. (c) CV curves for Pt/MoO3-CNx-400 in different pH solutions. (d) Epeak of UPD–H vs. pH plot of Pt/MoO3-CNx-400. | ||
Fig. 5b represents the change in half-wave potential with solution pH. Auinger et al.45 proposed that upon changing the pH of the solution, the HOR/HER performance in non-buffer solution remained unaltered due to the local pH gradient. In contrast, Koper and colleagues showed that the HOR/HER undergo changes throughout a broad pH range on polycrystalline Pt surfaces in phosphate buffer.46 The HER/HOR reaction kinetics are fully affected by the pH in buffer solutions, unlike non-buffer solution, where the pH does not affect kinetics. We studied the surface properties, especially the hydrogen binding energy (HBE) value of Pt/MoO3-CNx-400, to understand the mechanism behind the pH-dependent HER/HOR properties of the catalyst. It was reported by several groups that the UPD-H desorption peak was not affected by pre-adsorbed species, and its location in the CV can be directly linked to the HBE of the relevant active site.47 The relationship between the UPD–Hdes peak potential in CV and HBE of the associated active site may be easily determined using the Langmuir adsorption assumption according to the following equation:
| ΔGM–H = −F·Epeak | (6) |
485 C mol−1) and hydrogen binding energy, and Epeak is estimated from the CV data.
We investigated the CV data for Pt/MoO3-CNx-400 in various buffer solutions under the same electrochemical circumstances to compute the HBE of the catalyst. The corresponding CV profile diagram for the Pt/MoO3-CNx-400 composite in these buffers is displayed in Fig. 5c. It shows two peaks for each CV, which correspond to H-desorption and under potential hydrogen desorption (UPD–H). Fig. 5d indicates the peak positions moving towards positive potentials with increasing pH of the electrolyte. Therefore, HBE also increases with increasing electrolyte pH. The Epeak is linearly dependent on the pH, with a slope of 10.08 mV per pH. Additionally, we investigated the effect of anion on HBE in base. Fig. S7e† shows that the UPD–H peak position is almost unchanged upon addition of 0.1 M KNO3, 0.1 M KClO3, and 0.1 M KCl separately in 0.1 M KOH solution. This result indicates that anion adsorption did not affect the HBE of Pt/MoO3-CNx-400 surfaces.
Stimming and co-workers reported that the Pt electrode reaction kinetics were controlled by its intermediate, Had, and metal bond length (M–H).48 Durst et al.26 and Yan et al.47 also proposed that the HBE controls HOR/HER performances on noble metal surfaces (Pt, Ir) in various pH solutions. From the HOR and CV profiles, it can be concluded that the HER/HOR activity of Pt/MoO3-CNx-400 in different electrolytes is fully controlled by electrolyte pH. Therefore, HBE is the sole descriptor for HER/HOR on Pt/MoO3-CNx-400. The HER/HOR activity of Pt/MoO3-CNx-400 in base is lower than its acid media activity, but its HER and HOR activity is approximately 4.5 and 2 times higher than that of commercial Pt/C, respectively. This enhanced activity may be due to the hydrogen spillover from Pt to MoO3 sites and other factors.
Experimental evidence for hydrogen spillover in Pt/MoO3-CNx-400
Hydrogen spillover has been previously recognized as a highly significant factor in heterogeneous catalyst support properties. Therefore, one would expect that hydrogen adsorption and desorption in the Pt/MoO3-CNx-400 catalyst will significantly differ if hydrogen spillover is indeed present. Hydrogen spillover was previously confirmed by observing a color change between the catalyst and a WO3 mixture for the HER.29,30Fig. S8† shows similar color change images of Pt/MoO3-CNx-400 + WO3, MoO3 + WO3, and WO3. MoO3 + WO3, and WO3 did not show any color change, but the Pt/MoO3-CNx-400 + WO3 mixture exhibited a dark blue color after the HER test. The observed color change is due to the migrated hydrogen spillover, which reacts with WO3 to form the dark blue HxWO3 complex. Moreover, previous research suggests that hydrogen spillover can be supported by in situ kinetics of hydrogen adsorption and desorption. The hydrogen desorption kinetics of the catalysts are determined using in situ operando CV experiments. The change in hydrogen desorption peaks was also observed in the double layer region during CV scanning. Pt black/WO3 and Pt black were used for comparison as hydrogen spillover and non-hydrogen spillover catalysts, respectively. The CV curves of Pt black, Pt black/WO3, and Pt/MoO3-CNx-400 show the shifting of the proton desorption peak with changes in the scan rate (Fig. 6a–c).
 | ||
| Fig. 6 (a–c) CV plots for Pt/MoO3-CNx-400, Pt black + WO3, and Pt black with different scan rates in 0.1 M KOH, respectively. (d) Hydrogen desorption peak position vs. scan rate plots of these catalysts. (e–g) Nyquist plots of Pt/MoO3-CNx-400, Pt black + WO3, and Pt black at different HER overpotentials in 0.1 M KOH, respectively (inset: equivalent circuit). (h) Corresponding electrochemical impedance spectroscopy (EIS)-derived Tafel plots obtained from the hydrogen adsorption resistance R2. (i–k) Nyquist plots of Pt/MoO3-CNx-400, Pt black + WO3, and Pt black at different HOR overpotentials in 0.1 M KOH, respectively (inset: equivalent circuit). (l) Corresponding EIS-derived Tafel plots of the catalysts. | ||
A plot of hydrogen desorption peak positions vs. scan rates was employed to compare their corresponding fitted slopes because that is the rational method used to quantify hydrogen desorption kinetics. Fig. 6d shows the decrease of slopes in the order of Pt black (8.4 × 10−5) > Pt black/WO3 (6.3 × 10−5) > Pt/MoO3-CNx-400 (1.4 × 10−5). The lower slope value of Pt/MoO3-CNx-400 indicates its faster hydrogen desorption kinetics. The hydrogen spillover effects were found to promote hydrogen desorption kinetics in metal–support electrocatalysts, as reported by several groups.29,30 Therefore, hydrogen spillover can contribute to the rapid hydrogen desorption kinetics of Pt/MoO3-CNx-400. Similarly, hydrogen adsorption kinetics also support the possible hydrogen spillover process.
EEIS was performed at different overpotentials to investigate the Pt black, Pt black/WO3, and Pt/MoO3-CNx-400 catalysts in the HER region (Fig. 6e–g). The recorded Nyquist plots were fitted with a double-parallel equivalent circuit model (inset: Fig. 6e). It has been previously recognized that the second parallel component R2 (representing the hydrogen adsorption resistance) reflects the behaviour of hydrogen adsorption on catalyst surfaces.29 The catalysts possess different R2 values at different potentials. The hydrogen adsorption kinetics of the catalysts can be quantified by plotting logR2vs. overpotential (Fig. 6h).
The Tafel slopes were calculated from the plots by virtue of Ohm's law. A lower Tafel slope was exhibited by Pt/MoO3-CNx-400, suggesting the flourishing hydrogen adsorption rate, which may be due to the enhanced hydrogen spillover. Similarly, Nyquist plots at different overpotentials for the catalysts at the HOR region suggest that the increased hydrogen adsorption rate might be due to the enhanced hydrogen spillover (Fig. 6i–l). The above experiments suggest the possible hydrogen spillover on Pt/MoO3-CNx-400. It is difficult to directly observe the electrocatalytic hydrogen spillover phenomenon. However, some theoretical concepts have been recently reported to prove the hydrogen spillover phenomenon.
Theoretical modeling of the hydrogen spillover mechanism
To explore additional theoretical insight enabling efficient hydrogen spillover for the HER, a work-function-dependent HER activity was performed to determine the kinetics of the interfacial spillover process on a Pt-supported β-MoO3 (011) surface.Inspired by the hydrogen spillover mechanism on Ru-WO3−x,49 work function calculations for the Pt4 nanocluster and β-MoO3 (011) were carried out to obtain a theoretical concept regarding how hydrogen spillover can occur. The computational details and structural evolution are provided in the ESI and Fig. S9,† respectively. Smaller work functions of nanoparticles (NPs) rather than the surface revealed that charge transfer from NPs to the surface facilitated the hydrogen transfer process.49,50 The work function of the Pt nanocluster was obtained to be Φ1 = 5.39 eV smaller than that of the β-MoO3 (011) surface work function (Φ2 = 7.09 eV), revealing charge transfer from Pt nanoclusters to the β-MoO3 surface, which can also be verified by a charge density difference plot, as shown in Fig. 7. The red colours show a charge loss region at the Pt cluster, whereas the blue colour on the β-MoO3 (011) surface indicates a charge gain region, i.e., electrons are transferring from the Pt to the β-MoO3 (011) surface.
 | ||
| Fig. 7 (a) Schematic illustration of the hydrogen spillover mechanism over a Pt4@MoO3 (011) surface. (b) Charge density difference plot across the Pt4@MoO3 (011) surface. | ||
To further confirm the hydrogen spillover mechanism, the change in the Gibbs free energy was computed at each step of hydrogen transfer from the Pt4 cluster to the surface of β-MoO3 (011). The energy profile was obtained by sampling hydrogen at different sites on Pt4@MoO3 (011), and is shown in Fig. 8. The Gibbs free energy of adsorption of the hydrogen atom was calculated by the following equation:49
| ΔGH = E[surface + H*] − E[surface] − 1/2E[H2] + ΔEZPE − TΔSH | (7) |
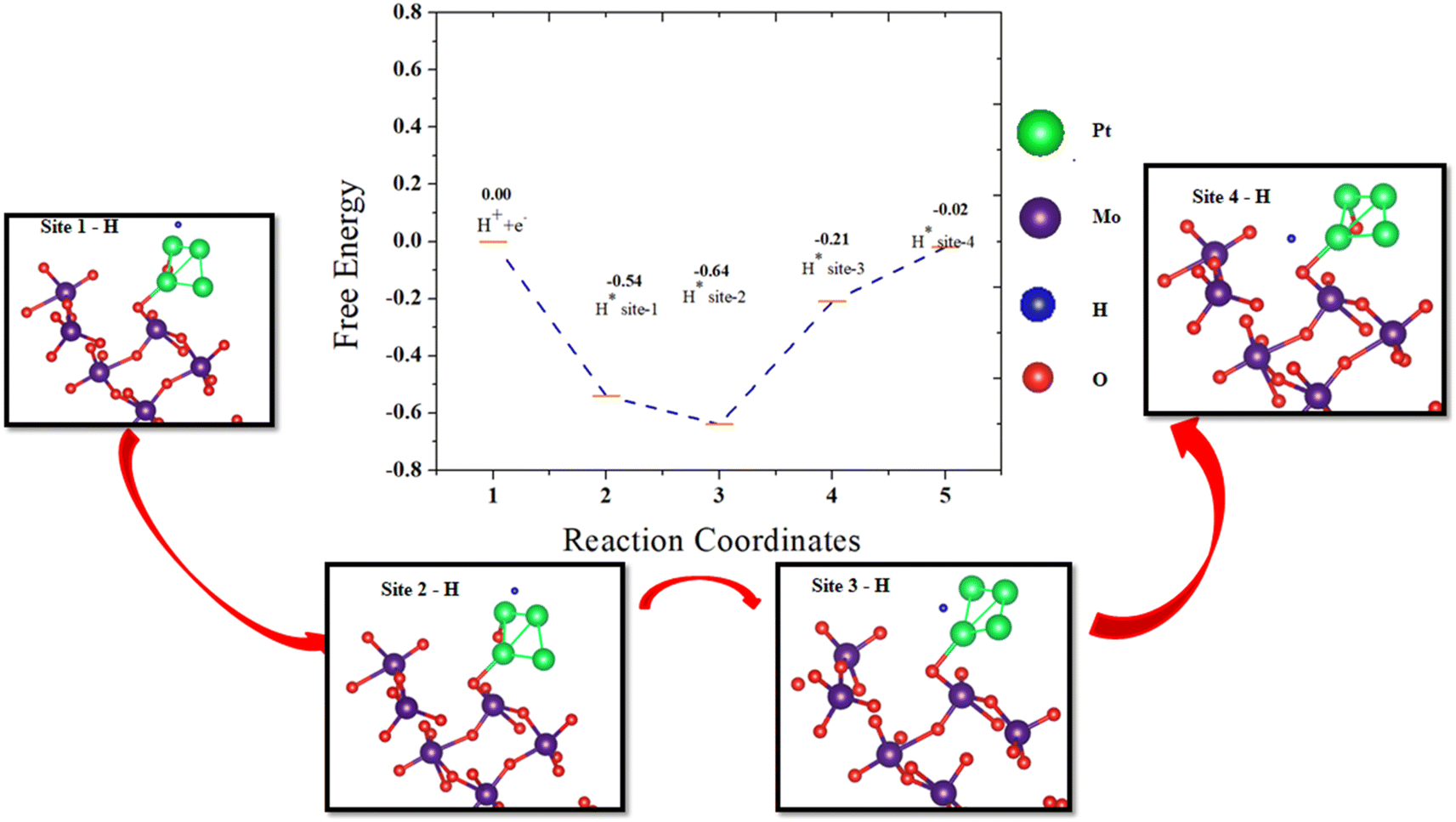 | ||
| Fig. 8 Schematic presentation of a free energy diagram for the HER on the surface of Pt@MoO3 (011). Optimized H* adsorption structures from site 1 to site 4 at different locations on Pt@MoO3 (011) are shown in the black boxes according to the red arrows. | ||
The total energy of the hydrogen attached to the Pt4@β-MoO3 (011) surface, total energy of the Pt4@β-MoO3 (011) surface, total energy of the H2 molecule in the gas phase, zero point energy (0.05 eV), and contribution from entropy (0.20 eV) at 298 K are noted by E[surface + H*], E[surface], E[H2], ΔEZPE, and TΔSH, respectively. The Gibbs free energy (ΔGH) values at the Pt4 cluster over β-MoO3 (011) are −0.54 eV, −0.64 eV, −0.21 eV, and −0.02 eV at site-1 to site-4, respectively. The kinetic energy barrier of hydrogen transfer from site-1 to site-2 is 0.1 eV, site-2 to site-3 is 0.43 eV, and site-3 to site-4 is 0.19 eV, which indicates greatly increased hydrogen adsorption and leads to highly efficient HER activity and rapid release of active sites for the spillover process on the β-MoO3 (011) surface. Similar energy barrier values were considered as favorable for H transfer on Ru-WO3−x.49
Under alkaline conditions using KOH solution, the morphology and structure of the Pt4@β-MoO3 (011) were stable. The difference in the free energy of Pt4@β-MoO3 (011) under vacuum and alkaline media was 0.14 eV, which was less and exhibits the feasibility of Pt4@β-MoO3 (011) in alkaline media. If we compare our results with recent work performed on the Pt/CoP49 or Ru-WO3−x surface,29 the significant changes in ΔGH at the interface of the Pt4/MoO3 (011) system indicate great hydrogen adsorption, which reduces the kinetic barrier and leads to highly efficient HER activity and rapid release of active sites.
The gradual increase in the UPD–H peak potential and corresponding decrease in HOR activity with increasing pH indicates that the reaction goes through the hydrogen binding energy (HBE) mechanism. The Tafel plot suggested that the reactions on Pt/MoO3-CNx-400 go through the Volmer–Heyrovsky pathway. The activity of the catalyst decreases with increasing pH, and the HER/HOR activity of the catalyst in base remains approximately 4.5 and 2 times higher than that of commercial Pt/C, respectively. This increase in the catalytic activity is mainly due to the hydrogen spillover from Pt to MoO3, as suggested by the experimental and theoretical findings.
Based on these results, we proposed a hydrogen spillover-induced mechanism to explain the enhanced HER/HOR activity of Pt/MoO3-CNx-400 in base. During the HER, H2O dissociates on metallic Pt and interfaces to form Pt–Had or (Pt/MoO3)–Had and OH−. Some of the adsorbed hydrogen migrates to the MoO3 sites due to hydrogen spillover, which thereby boosts the Volmer step. Finally, Had at Pt and the interface, and MoO3 sites react with H2O to form H2 molecules, enhancing the Heyrovsky step, which leads to the high HER activity of Pt/MoO3-CNx-400. Similarly, for the HOR, H2 dissociates on Pt and interfaces to form Pt–Had and (Pt/MoO3)–Had. Then, some adsorbed hydrogen may transfer to the MoO3 site to form MoO3–Had by the hydrogen spillover process. Had subsequently interacts with the OH− of the electrolyte to form H2O, which leads to the high HOR performance of the catalyst. A schematic representation of the hydrogen spillover-induced alkaline HER/HOR mechanism is shown in Scheme 1.
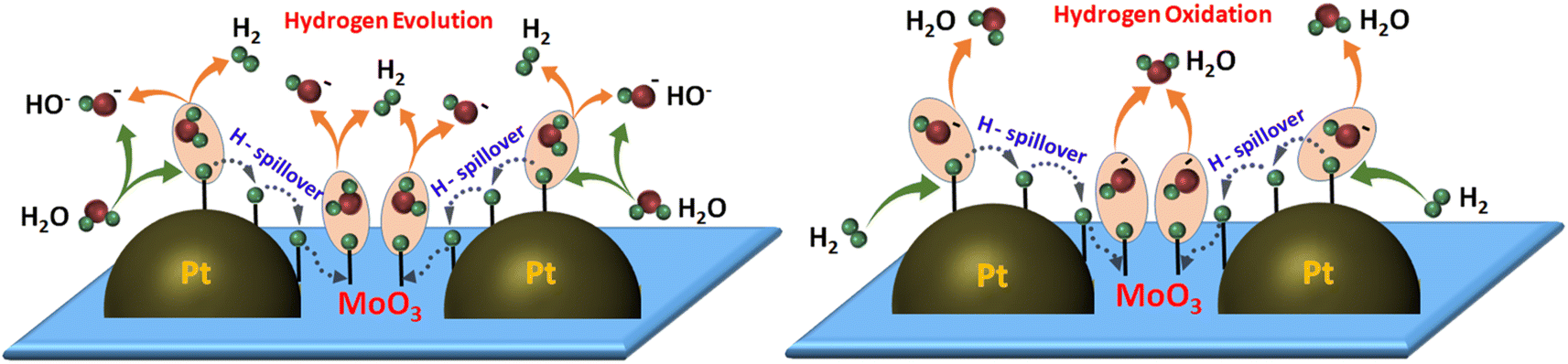 | ||
| Scheme 1 Schematic representation of the hydrogen spillover-induced HER/HOR mechanism in alkaline medium. | ||
Factors contributing to the high catalytic performance of Pt/MoO3-CNx-400
Several factors are responsible for the excellent HOR/HER activity of Pt/MoO3-CNx-400:(i) The hydrogen spillover is the most important factor for the high catalytic activity of Pt/MoO3-CNx-400. It was reported by Wang and co-workers that the smaller work function of nanoparticles compared to the surface revealed charge transfer from nanoparticles to the surface, which facilitates the hydrogen transfer process and subsequently boosts the HER.49 We also found that the work function of the Pt nanocluster is 5.39 eV, which is smaller than the β-MoO3 (011) surface work function (Φ2 = 7.09 eV), revealing charge transfer from Pt nanoclusters to the β-MoO3 surface (Fig. 7a). The charge density difference plot also suggests the same (Fig. 7b).
The energy profile diagram suggests that hydrogen spillover occurs from the Pt to the MoO3 sites, as shown in Fig. 8, which illustrates that there is little higher adsorption energy at the Pt and interface sites, while MoO3 possesses weak adsorption energy. Therefore, hydrogen can easily adsorb at the Pt and interface sites and desorb from the MoO3 site. With less of an energy barrier, hydrogen can move from Pt or the interface to the MoO3 site instead of direct desorption occurring from Pt or interfaces, which thereby increases the HER/HOR activity.
(ii) The synergistic interaction among Pt, MoO3, and CNx components could play an important role in high catalytic performance. For example, Xi et al.51 reported that the enhanced HER activity of the (Mo3S13)2− cluster co-catalyst and WSe2 photocathode was due to the synergistic interaction between the components. The presence of synergistic interaction and electronic modulation was confirmed by the Pt 4f and Mo 3d XPS peak shifting of Pt/MoO3-CNx-400 compared to Pt/CNx-400 and Mo/CNx-400, respectively (Fig. S4a and b†). The figure shows a positive shift of Pt 4f of Pt/MoO3-CNx-400 compared to Pt/CNx-400 and a negative shift of Mo 3d of Pt/MoO3-CNx-400 compared to Mo/CNx-400, suggesting electron transfer from Pt to the MoO3 site. This indicates electronic modulation of Pt and MoO3 in the Pt/MoO3-CNx-400 composite.
EIS and HER measurements were used to study the synergistic interaction. The HER planarization curve and Nyquist plot of Pt/MoO3-CNx-400 were compared with other single components and physical mixtures (Fig. S10†). The higher HER activity of Pt/MoO3-CNx-400 compared to other components and physical mixture suggests that synergistic interaction occurs between the components. The Nyquist plots were also fitted to determine the Rct values, which were found to be 1410, 17.6, 75.9, and 9.99 Ω for Mo/CNx-400, Pt/CNx-400, the physical mixture, and Pt/MoO3-CNx-400, respectively. This indicates that there is a lower Rct value for Pt/MoO3-CNx-400 as compared to other components and the physical mixture, which suggests that strong synergistic interactions occur among its components.
(iii) The existence of interfaces as active catalytic sites has been reported by a number of groups. For example, Sun and co-workers reported that the interface between nickel and nickel nitrides boosts the HER/HOR activity of the catalyst.52 The presence of interfaces can be easily seen in Fig. 1d, S2a and b†). The line scan change of Pt to Mo or vice versa also suggests the presence of boundaries between Pt and Mo (Fig. S3h and i†). Thus, the presence of interfaces in Pt/MoO3-CNx-400 is another driving factor for this superior electrochemical activity.
(iv) Another important factor for the high activity of the catalyst is the ECSA, which is generally proportional to the electrochemical activity of a catalyst. CO-stripping experiments were performed to calculate the ECSAs for the catalysts (Fig. S4c–f†), and they showed that the highest ECSA value of 42.2 m2 g−1 was obtained for Pt/MoO3-CNx-400, as compared to other catalysts. The greater HER/HOR performance of Pt/MoO3-CNx-400 may be due to the high ECSA of the catalyst.
Conclusions
The interface-rich compound Pt/MoO3-CNx-400 was prepared by a simple borohydride reduction and calcination process for the alkaline HER/HOR. p-XRD, TEM, and XPS analyses were performed to characterize the presence of interfaces and the phases of the components. The catalyst exhibited high HER/HOR activity as well as stability compared to commercial Pt/C, showing 10 mA cm−2 current density at 66.8 mV overpotential. In addition, the HOR activity of the catalyst was 2 times higher than that of commercial Pt/C. The HBE is the main descriptor for the alkaline HER/HOR of the Pt/MoO3-CNx-400 composite. The hydrogen spillover from Pt to MoO3 enhanced the Heyrovsky step, thereby enhancing the HER activity. Theoretically, it was unveiled that the Pt4/MoO3 (011) catalyst promoted the free energy change in hydrogen transfer over Pt4/MoO3 (011), which was favorable for HER activity. The charge difference and low work function of Pt nanoclusters also revealed that the electron transfer from Pt to the MoO3 (011) surface enhanced the HER activity by reducing the charge accumulation at the interface.Based on our findings, we propose a hydrogen-spillover-based mechanism to explain the high alkaline HER/HOR activity. We demonstrated that the high HER/HOR activity of Pt/MoO3-CNx-400 is due to the hydrogen spillover, synergistic interactions between the components, presence of interfaces, and high ECSA of the catalyst. This work may provide mechanistic insight that can be utilized to design a catalyst for the alkaline HER/HOR for development of several renewable technologies such as electrolyzers and fuel cells.
Data availability
All experimental and characterization data and detailed experimental procedures are available in the published article and ESI.†Author contributions
S. B. conceived the research and supervised the work. R. S. and B. K. M. performed the synthesis, characterization, and electrochemical performance tests of the catalyst. B. K. M. and R. S. equally contributed to this work. R. K. T. performed the DFT calculations, and B. C. supervised the DFT calculations. S. B. and R. S. wrote the paper. R. K. T. and B. C. wrote the theoretical parts of this manuscript.Conflicts of interest
There are no competing financial interests to declare.Acknowledgements
We are thankful to the Department of Atomic Energy (DAE) and NISER for financial support (RIN4002). We are also thankful to the Centre for Interdisciplinary Science (CIS) and NISER for providing TEM and FE-SEM measurements. R. K. T. and B. C. would like to thank Dr Nandini Garg, Dr T. Sakuntala, Dr S. M. Yusuf, and Dr A. K. Mohanty for support and encouragement. B. C. would also like to thank the staff at the BARC computer division for use of the supercomputing facility.References
- T. Ahmad and D. Zhang, A critical review of comparative global historical energy consumption and future demand: The story told so far, Energy Rep., 2020, 6, 1973–1991 CrossRef.
- P. J. Megía, A. J. Vizcaíno, J. A. Calles and A. Carrero, Hydrogen Production Technologies: From Fossil Fuels toward Renewable Sources. A Mini Review, Energy Fuels, 2021, 35, 16403–16415 CrossRef.
- Y. Yang, L. Zhang, Z. Hu, Y. Zheng, C. Tang, P. Chen, R. Wang, K. Qiu, J. Mao, T. Ling and S.-Z. Qiao, The Crucial Role of Charge Accumulation and Spin Polarization in Activating Carbon-Based Catalysts for Electrocatalytic Nitrogen Reduction, Angew. Chem., Int. Ed., 2020, 59, 4525–4531 CrossRef CAS.
- N. K. Oh, J. Seo, S. Lee, H.-J. Kim, U. Kim, J. Lee, Y.-K. Han and H. Park, Highly efficient and robust noble-metal free bifunctional water electrolysis catalyst achieved via complementary charge transfer, Nat. Commun., 2021, 12, 4606 CrossRef CAS.
- R. Samanta, R. Mishra, B. K. Manna and S. Barman, Two-Dimensional Amorphous Cobalt Oxide Nanosheets/N-Doped Carbon Composites for Efficient Water Splitting in Alkaline Medium, ACS Appl. Nano Mater., 2022, 5, 17022–17032 CrossRef CAS.
- A. Shekhawat, R. Samanta and S. Barman, MOF-Derived Porous Fe3O4/RuO2-C Composite for Efficient Alkaline Overall Water Splitting, ACS Appl. Energy Mater., 2022, 5, 6059–6069 CrossRef CAS.
- G. E. Halkos and E.-C. Gkampoura, Reviewing Usage, Potentials, and Limitations of Renewable Energy Sources, Energies, 2020, 13, 2906 CrossRef.
- J. A. Turner, Sustainable Hydrogen Production, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- A. Kovač, D. Marciuš and L. Budin, Solar hydrogen production via alkaline water electrolysis, Int. J. Hydrogen Energy, 2019, 44, 9841–9848 CrossRef.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Green hydrogen from anion exchange membrane water electrolysis: a review of recent developments in critical materials and operating conditions, Sustainable Energy Fuels, 2020, 4, 2114–2133 RSC.
- R. Samanta, R. Mishra, B. K. Manna and S. Barman, IrO2 modified Crystalline-PdO nanowires based bi-functional electro-catalyst for HOR/HER in acid and base, Renewable Energy, 2022, 191, 151–160 CrossRef CAS.
- D. Li, A. R. Motz, C. Bae, C. Fujimoto, G. Yang, F.-Y. Zhang, K. E. Ayers and Y. S. Kim, Durability of anion exchange membrane water electrolyzers, Energy Environ. Sci., 2021, 14, 3393–3419 RSC.
- A. Kampker, H. Heimes, M. Kehrer, S. Hagedorn, P. Reims and O. Kaul, Fuel cell system production cost modeling and analysis, Energy Rep., 2023, 9, 248–255 CrossRef.
- S. Krishnan, V. Koning, M. Theodorus de Groot, A. de Groot, P. G. Mendoza, M. Junginger and G. J. Kramer, Present and future cost of alkaline and PEM electrolyser stacks, Int. J. Hydrogen Energy, 2023, 48(83), 32313–32330 CrossRef CAS.
- D. R. Dekel, Review of cell performance in anion exchange membrane fuel cells, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: Insights into materials design, Science, 2017, 355, eaad4998 CrossRef.
- M. T. M. Koper, A basic solution, Nat. Chem., 2013, 5, 255–256 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Noble metal-free hydrogen evolution catalysts for water splitting, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. Zheng, W. Sheng, Z. Zhuang, B. Xu and Y. Yan, Universal dependence of hydrogen oxidation and evolution reaction activity of platinum-group metals on pH and hydrogen binding energy, Sci. Adv., 2016, 2, e1501602 CrossRef.
- T. Tang, L. Ding, Z.-C. Yao, H.-R. Pan, J.-S. Hu and L.-J. Wan, Synergistic Electrocatalysts for Alkaline Hydrogen Oxidation and Evolution Reactions, Adv. Funct. Mater., 2022, 32, 2107479 CrossRef CAS.
- Y. Cong, B. Yi and Y. Song, Hydrogen oxidation reaction in alkaline media: From mechanism to recent electrocatalysts, Nano Energy, 2018, 44, 288–303 CrossRef CAS.
- X. Mu, S. Liu, L. Chen and S. Mu, Classification, Activity Regulation and Challenges, Small Struct., 2023, 4, 2200281 CrossRef CAS.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Correlating the hydrogen evolution reaction activity in alkaline electrolytes with the hydrogen binding energy on monometallic surfaces, Energy Environ. Sci., 2013, 6, 1509–1512 RSC.
- W. Sheng, Z. Zhuang, M. Gao, J. Zheng, J. G. Chen and Y. Yan, Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy, Nat. Commun., 2015, 6, 5848 CrossRef CAS PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption, Nat. Chem., 2013, 5, 300–306 CrossRef CAS.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- J. Li, H.-X. Liu, W. Gou, M. Zhang, Z. Xia, S. Zhang, C.-R. Chang, Y. Ma and Y. Qu, Ethylene-glycol ligand environment facilitates highly efficient hydrogen evolution of Pt/CoP through proton concentration and hydrogen spillover, Energy Environ. Sci., 2019, 12, 2298–2304 RSC.
- J. Park, S. Lee, H.-E. Kim, A. Cho, S. Kim, Y. Ye, J. W. Han, H. Lee, J. H. Jang and J. Lee, Investigation of the Support Effect in Atomically Dispersed Pt on WO3−x for Utilization of Pt in the Hydrogen Evolution Reaction, Angew. Chem., Int. Ed., 2019, 58, 16038–16042 CrossRef CAS.
- J. Li, J. Hu, M. Zhang, W. Gou, S. Zhang, Z. Chen, Y. Qu and Y. Ma, A fundamental viewpoint on the hydrogen spillover phenomenon of electrocatalytic hydrogen evolution, Nat. Commun., 2021, 12, 3502 CrossRef CAS.
- J. Dai, Y. Zhu, Y. Chen, X. Wen, M. Long, X. Wu, Z. Hu, D. Guan, X. Wang, C. Zhou, Q. Lin, Y. Sun, S.-C. Weng, H. Wang, W. Zhou and Z. Shao, Hydrogen spillover in complex oxide multifunctional sites improves acidic hydrogen evolution electrocatalysis, Nat. Commun., 2022, 13, 1189 CrossRef CAS.
- H. Shen, H. Li, Z. Yang and C. Li, Magic of hydrogen spillover: Understanding and application, Green Energy Environ., 2022, 7, 1161–1198 CrossRef CAS.
- T. Liu, W. Gao, Q. Wang, M. Dou, Z. Zhang and F. Wang, Selective Loading of Atomic Platinum on a RuCeOx Support Enables Stable Hydrogen Evolution at High Current Densities, Angew. Chem., Int. Ed., 2020, 59, 20423–20427 CrossRef CAS.
- K. Chen, S. Deng, Y. Lu, M. Gong, Y. Hu, T. Zhao, T. Shen and D. Wang, Molybdenum-doped titanium dioxide supported low-Pt electrocatalyst for highly efficient and stable hydrogen evolution reaction, Chin. Chem. Lett., 2021, 32, 765–769 CrossRef CAS.
- H. Tian, X. Cui, L. Zeng, L. Su, Y. Song and J. Shi, Oxygen vacancy-assisted hydrogen evolution reaction of the Pt/WO3 electrocatalyst, J. Mater. Chem. A, 2019, 7, 6285–6293 RSC.
- Y.-N. Zhou, X. Liu, C.-J. Yu, B. Dong, G.-Q. Han, H.-J. Liu, R.-Q. Lv, B. Liu and Y.-M. Chai, Boosting hydrogen evolution through hydrogen spillover promoted by Co-based support effect, J. Mater. Chem. A, 2023, 11, 6945–6951 RSC.
- D. V. Esposito, I. Levin, T. P. Moffat and A. A. Talin, H2 evolution at Si-based metal–insulator–semiconductor photoelectrodes enhanced by inversion channel charge collection and H spillover, Nat. Mater., 2013, 12, 562–568 CrossRef CAS PubMed.
- J. M. Jaksic, D. Labou, G. D. Papakonstantinou, A. Siokou and M. M. Jaksic, Novel Spillover Interrelating Reversible Electrocatalysts for Oxygen and Hydrogen Electrode Reactions, J. Phys. Chem. C, 2010, 114, 18298–18312 CrossRef CAS.
- R. S. Datta, F. Haque, M. Mohiuddin, B. J. Carey, N. Syed, A. Zavabeti, B. Zhang, H. Khan, K. J. Berean, J. Z. Ou, N. Mahmood, T. Daeneke and K. Kalantar-zadeh, Highly active two dimensional α-MoO3−x for the electrocatalytic hydrogen evolution reaction, J. Mater. Chem. A, 2017, 5, 24223–24231 RSC.
- H. N. Tran, S. Park, F. T. A. Wibowo, N. V. Krishna, J. H. Kang, J. H. Seo, H. Nguyen-Phu, S.-Y. Jang and S. Cho, 17% Non-Fullerene Organic Solar Cells with Annealing-Free Aqueous MoOx, Adv. Sci., 2020, 7, 2002395 CrossRef CAS PubMed.
- B. P. Vinayan and S. Ramaprabhu, Platinum–TM (TM = Fe, Co) alloy nanoparticles dispersed nitrogen doped (reduced graphene oxide-multiwalled carbon nanotube) hybrid structure cathode electrocatalysts for high performance PEMFC applications, Nanoscale, 2013, 5, 5109–5118 RSC.
- J. Senthilnathan, Y.-F. Liu, K. S. Rao and M. Yoshimura, Submerged Liquid Plasma for the Synchronized Reduction and Functionalization of Graphene Oxide, Sci. Rep., 2014, 4, 4395 CrossRef PubMed.
- C. L. Chiang and J. M. Yang, 11 - Flame retardance and thermal stability of polymer/graphene nanosheet oxide composites, Novel Fire Retardant Polymers and Composite Material, 2017, pp. 295–312, DOI:10.1016/B978-0-08-100136-3.00011-X.
- Y. Li, Y. Wang, J. Lin, Y. Shi, K. Zhu, Y. Xing, X. Li, Y. Jia and X. Zhang, Solution-plasma-induced oxygen vacancy enhances MoOx/Pt electrocatalytic counter electrode for bifacial dye-sensitized solar cells, Front. Energy Res., 2022, 10 DOI:10.3389/fenrg.2022.924515.
- C. Varodi, F. Pogăcean, A. Cioriţă, O. Pană, C. Leoştean, B. Cozar, T. Radu, M. Coroş, R. I. Ştefan-van Staden and S.-M. Pruneanu, Nitrogen and Sulfur Co-Doped Graphene as Efficient Electrode Material for L-Cysteine Detection, Chemosensors, 2021, 9, 146 CrossRef CAS.
- M. Auinger, I. Katsounaros, J. C. Meier, S. O. Klemm, P. U. Biedermann, A. A. Topalov, M. Rohwerder and K. J. J. Mayrhofer, Near-surface ion distribution and buffer effects during electrochemical reactions, Phys. Chem. Chem. Phys., 2011, 13, 16384–16394 RSC.
- R. Gisbert, G. García and M. T. M. Koper, Adsorption of phosphate species on poly-oriented Pt and Pt(111) electrodes over a wide range of pH, Electrochim. Acta, 2010, 55, 7961–7968 CrossRef CAS.
- J. Zheng, Z. Zhuang, B. Xu and Y. Yan, Correlating Hydrogen Oxidation/Evolution Reaction Activity with the Minority Weak Hydrogen-Binding Sites on Ir/C Catalysts, ACS Catal., 2015, 5, 4449–4455 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- Y.-H. Kwon, X. Yang, Z. Wu, Z. Fan, W. Xu and C.-M. Jon, Anodic Process of Nano-Ni Hydroxides for the Urea Oxidation Reaction and Its Electrochemical Removal with a Lower Energy Input, J. Phys. Chem. C, 2022, 126, 12492–12499 CrossRef CAS.
- H. Q. Fu, M. Zhou, P. F. Liu, P. Liu, H. Yin, K. Z. Sun, H. G. Yang, M. Al-Mamun, P. Hu, H.-F. Wang and H. Zhao, Hydrogen Spillover-Bridged Volmer/Tafel Processes Enabling Ampere-Level Current Density Alkaline Hydrogen Evolution Reaction under Low Overpotential, J. Am. Chem. Soc., 2022, 144, 6028–6039 CrossRef CAS PubMed.
- F. Xi, F. Bozheyev, X. Han, M. Rusu, J. Rappich, F. F. Abdi, P. Bogdanoff, N. Kaltsoyannis and S. Fiechter, Enhancing Hydrogen Evolution Reaction via Synergistic Interaction between the [Mo3S13]2– Cluster Co-Catalyst and WSe2 Photocathode, ACS Appl. Mater. Interfaces, 2022, 14, 52815–52824 CrossRef CAS.
- F. Song, W. Li, J. Yang, G. Han, P. Liao and Y. Sun, Interfacing nickel nitride and nickel boosts both electrocatalytic hydrogen evolution and oxidation reactions, Nat. Commun., 2018, 9, 4531 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials details, details of the instruments, electrochemical experiments, ECSA calculation, calculation of Tafel slope, mass activity, other electrochemical data, post stability characterization data, and HER/HOR comparison tables. See DOI: https://doi.org/10.1039/d3sc04126c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |