
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting the interfacial dynamics and thermodynamics in polyanion cathode by carbon dots for ultrafast-charging sodium ion batteries†
Yujin
Li
a,
Yu
Mei
a,
Roya
Momen
 b,
Bai
Song
c,
Yujie
Huang
a,
Xue
Zhong
a,
Hanrui
Ding
a,
Wentao
Deng
a,
Guoqiang
Zou
a,
Hongshuai
Hou
*a and
Xiaobo
Ji
a
b,
Bai
Song
c,
Yujie
Huang
a,
Xue
Zhong
a,
Hanrui
Ding
a,
Wentao
Deng
a,
Guoqiang
Zou
a,
Hongshuai
Hou
*a and
Xiaobo
Ji
a
aState Key Laboratory of Powder Metallurgy, College of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, China. E-mail: hs-hou@csu.edu.cn
bDepartment of Chemistry and Shenzhen Grubbs Institute, Southern University of Science and Technology, Shenzhen, 518055, China
cDongying Cospowers Technology Limited Company, China
First published on 30th November 2023
Abstract
Ultrafast-charging is the focus of next-generation rechargeable batteries for widespread economic success by reducing the time cost. However, the poor ion diffusion rate, intrinsic electronic conductivity and structural stability of cathode materials seriously hinder the development of ultrafast-charging technology. To overcome these challenges, an interfacial dynamics and thermodynamics synergistic strategy is proposed to synchronously enhance the fast-charging capability and structural stability of polyanion cathode materials. As a case study, a Na3V2(PO4)3 composite (NVP/NSC) is successfully obtained by introducing an interface layer derived from N/S co-doped carbon dots. Density functional theory calculations validate that the interfacial bonding effect of V–N/S–C significantly reduces the Na+ transport energy barrier. D-band center theory analysis confirms the downward shift of the V d-band center enhances the strength of the V–O bond and considerably inhibits irreversible phase transformation. Benefitting from this interfacial synergistic strategy, NVP/NSC achieves a high capability and excellent cycling stability with a surprisingly low carbon content (2.23%) at an extremely high rate of 100C for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles (87.2 mA h g−1, 0.0028% capacity decay per cycle). Furthermore, a superior performance at 5C (115.3 mA h g−1, 92.1% capacity retention after 800 cycles) is exhibited by the NVP/NSC‖HC full cell. These findings provide timely new insights for the systematic design of ultrafast-charging cathode materials.
000 cycles (87.2 mA h g−1, 0.0028% capacity decay per cycle). Furthermore, a superior performance at 5C (115.3 mA h g−1, 92.1% capacity retention after 800 cycles) is exhibited by the NVP/NSC‖HC full cell. These findings provide timely new insights for the systematic design of ultrafast-charging cathode materials.
1 Introduction
With the increasing demand for electrical energy, the research and development of rechargeable batteries has received a great deal of attention.1,2 Lithium ion batteries (LIBs) have caught the attention of researchers with their high energy density and cycling stability.3,4 However, the uneven distribution and rising prices of lithium resources limit the sustainable development of LIBs.5 Sodium ion batteries (SIBs) are considered as a viable alternative to LIBs in some application areas due to their abundant sodium resources and low cost.6 Thus, the growing market for electric vehicles, portable electronics and large energy storage systems has attracted widespread interest in SIBs.7,8 So far, a myriad of scientists have raised a series of problems regarding the high-rate performance of SIBs at the material level and battery level, namely the so-called fast-charging.9–11 From the components and energy-storage process of SIBs, cathode materials are the hard-core elements and continuously perform reversible intercalation/deintercalation of ions during rapid charging/discharging, which shows that the energy and power densities of batteries are largely determined by cathode materials.12 However, cathode issues related to the aggressive situations of ultrafast charging, such as the deterioration of capacity, life, and safety of batteries, have not been extensively reported in a more systematic way.13To successfully achieve the ultrafast-charging performance of the cathode, it's important to holistically understand the kinetic processes taking place on/in the cathode under operating conditions. As shown in Fig. 1, the electrochemical reactions are coupled electron and Na+ ion transfer processes with respective transport pathways. The main limiting processes of the battery system towards ultrafast-charging are Na+ diffusion in the active materials, interfacial Na+ transfer at the cathode/electrolyte interface (CEI) and Na+ transport in the electrolyte.14,15 For the cathode side, realizing the ultrafast Na diffusion in the lattice architecture of cathode material particles and Na+ transfer at the CEI are key two challenges with regard to ultrafast-charging.16,17 More importantly, accelerating the transport of Na+ ions and electrons in the cathode material as much as possible is a crucial step, which highly depends on the ion diffusivity/electronic conductivity of the corresponding material.9,10,18 Meanwhile, successful realization of ultrafast-charging requires chemical and structural stabilities from the atomic towards the particle level to ensure the long cycle life since the high current density under ultrafast charging conditions would cause varying degrees of damage to the cathode materials.19 In brief, the key factors for developing ultrafast-charging cathodes lie in simultaneously improving the electronic conductivity, ionic conductivity and structural stability of the cathode.15,16 Current SIB cathodes are subjects of active interest with a high potential, in particular with respect to developing their energy density, and include layered oxides,20 polyanions,21 and Prussian blue and its analogs.22 Meanwhile, the ultrafast-charging performance improvement of these cathode materials needs to be systematically considered. For example, Na3V2(PO4)3 (NVP) is a promising polyanionic material with fast ion conductivity. However, NVP is limited by its inherent poor electronic conductivity and thus exhibits unsatisfactory rate capability and cycling stability.23 Present modification strategies focus on carbon coating,24,25 ion doping,26,27 and material nanosizing.28,29 As one of the most common and effective modification strategies, carbon coating can only improve the electronic conductivity of electrode materials.30,31 Nevertheless, the structural stability and ion transport properties are not enhanced synchronously, and thus fail to meet the ultrafast-charging operating conditions.32 Above all, systemic modification studies are urgently needed to get deeper insights into the ultrafast-charging performance of SIB cathodes so as to provide generic solutions for bridging the gap between the energy and power density.
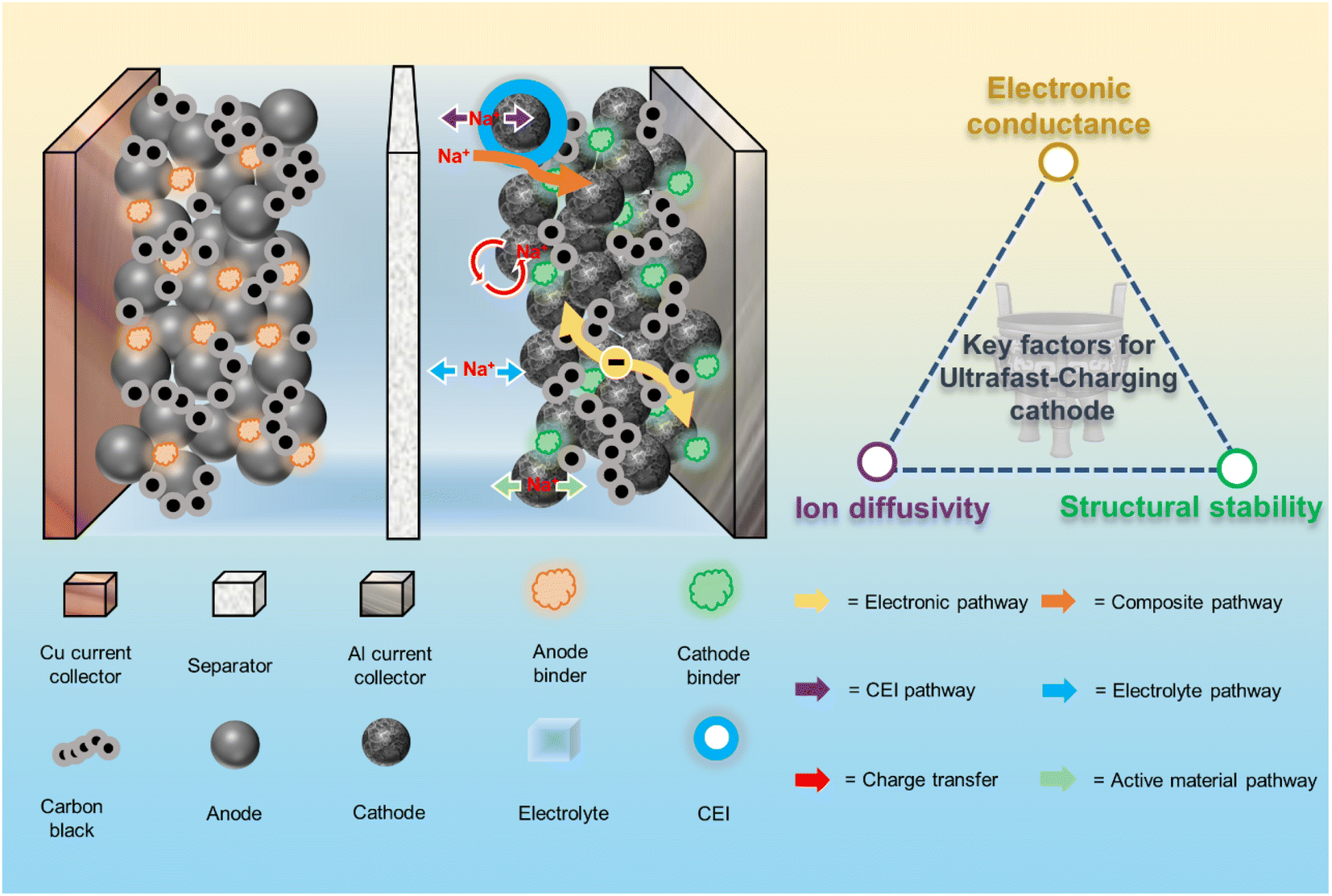 | ||
| Fig. 1 Electrochemical kinetics of the sodium-ion battery cathode and design principles for ultrafast-charging cathode materials. | ||
In this study, an interfacial dynamics and thermodynamics synergistic strategy is proposed to simultaneously improve the ionic conductivity, electronic conductivity and structural stability of SIB cathode materials by concurrently tuning interfacial bonding and energy band alignment. As a case of study, trace amounts of N/S-doped carbon dots are adopted to bond with NVP, obtaining NVP/N, S co-doped carbon composites (NVP/NSC). Density functional theory (DFT) calculations show that the bonding effects of V–N/S–C reconstruct the Na+ diffusion path in the bulk and near surface, significantly reducing the Na+ transport energy barrier. Interestingly, through d-band center theory analysis, the bonding strength enhancement of V–O and the intrinsic electronic conductivity improvement are further verified. Combining electrochemical analysis, synchrotron radiation based spectroscopic techniques and electron micrography, a detailed evaluation of the redox mechanism and structural evolution shows that high-rate Na+ diffusion is enabled and structural stability is synchronously strengthened through this synergistic strategy. Remarkably, the NVP/NSC cathode exhibits excellent rate performance (87.2 mA h g−1 at 100C) and cycling stability (72.2% capacity retention after 10000 cycles at 100C). In this case, complete charging/discharging time shrinks to only 25 s, achieving comparable ultrafast charging/discharging performance. Furthermore, through electrolyte matching engineering, the full cell assembled with hard carbon demonstrates excellent discharge capacity (115.3 mA h g−1 at 5C) and cycling stability (92.1% capacity retention after 800 cycles). This study introduces a novel perspective, highlighting the previously overlooked interfacial dynamics and thermodynamics synergistic effects that influence the ultrafast-charging performance of SIB cathodes. This underscores the importance of systematically designing strategies for high-power-density cathodes.
2 Results and discussion
2.1 Design principles of the ultrafast-charging cathode
Carbon dots (CDs), as novel zero-dimensional carbon nanomaterials (<10 nm), have received much attention in the field of electrochemical energy storage.33,34 Compared with conventional carbon materials, CDs are rich in surface functional groups and thus susceptible to bonding with a variety of electronegative elements to meet the customization of electrode material construction.35–37 Herein, CDs were sensibly screened as the carbon skeleton and N/S heteroatoms served as co-dopants for the preparation of N/S CDs to construct functionalized interfacial layers.Firstly, the Na+ migration path and diffusion kinetics of the designed NVP/NSC materials were evaluated with density functional theory (DFT) calculations to verify the role of N/S co-doped functionalized interfacial layers. Based on the optimized structural models of NVP and NVP/NSC (Fig. S1 and S2†), two diffusion paths represent the diffusion at the interface (Path I, Fig. 2a and b) and inside the bulk phase (Path II, Fig. 2f–i, S3 and S4†), respectively. The results displayed in Fig. 2c and j–m show that the diffusion energy barrier of NVP/NSC (Path I = 0.90 eV, path II = 0.12, 0.19 eV) is significantly lower than that of NVP (Path I = 1.07 eV, path II = 0.30, 0.20 eV), strongly evidencing that the bonding effects of V–N/S–C enhance the Na+ migration in both the near-surface and bulk phases of the material. In addition, the crystal structure of the material keeps consistent during cycling as fast Na+ transport facilitates homogeneous reaction; in contrast, slow Na+ transport may lead to severe phase separation in the bulk and near the surface (Fig. 2d and e).
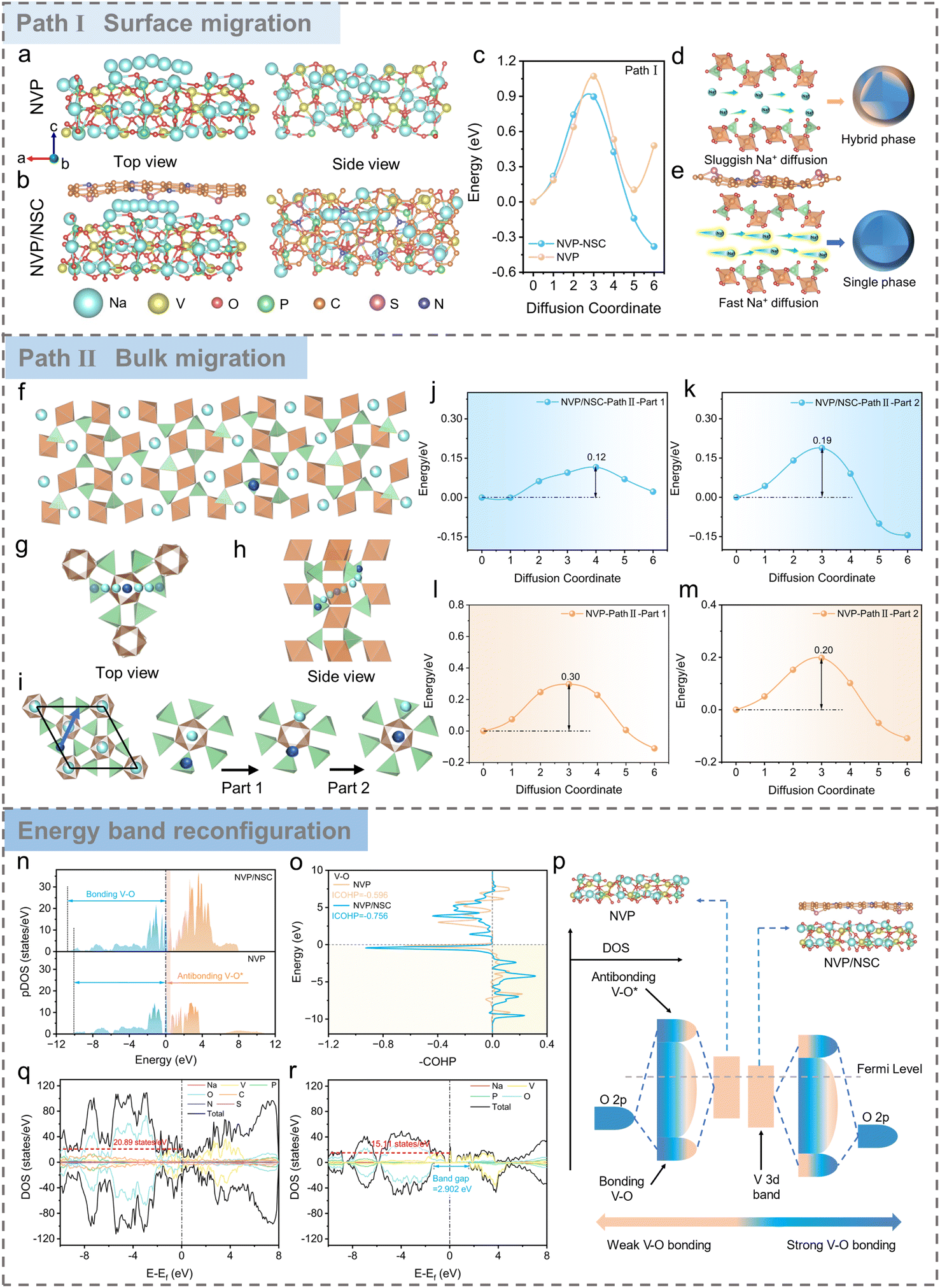 | ||
| Fig. 2 DFT calculations. (a) The diffusion paths of NVP and (b) NVP/NSC for Na+ migration along Path I (Na+ migration along the a-axis). (c) The calculated diffusion energy barrier profiles according to Path I. (d and e) Schematic of the different diffusion capabilities of NVP and NVP/NSC and their effect on the material structure during cycling. (f) Side views of NVP structure. Cyan and blue balls stand for lattice and intercalated Na atoms, respectively. (g) Top and (h) side views of Na diffusion pathways in NVP. Cyan and blue balls indicate Na ions and the most energetically favorable site for Na intercalation, respectively. (i) Top views of NVP structure and schematic of the Path II migration process. Blue arrows show a schematic of a representative pathway for Na diffusion along path II in NVP. Calculated energy paths of Na diffusion along path II in NVP/NSC (j and k) and NVP (l and m), respectively. (n) V 3d pDOS of NVP/NSC and NVP (the blue region is the bonding state and the orange region is the anti-bonding state). (o) COHP analyses of V–O bonds. (p) Schematic of the decline of the d-band center in NVP/NSC. Total density of states (tDOS) of NVP/NSC (q) and NVP (r). | ||
From the viewpoint of thermodynamics, the effect of N/S co-doped heterointerfaces on the energy band structure is further elucidated from the electronic structure point of view. The V 3d partial densities of states (pDOS) of NVP/NSC and NVP are illustrated in Fig. 2n, with the bonding orbital energy band of NVP/NSC being significantly widened due to the increase of electron delocalization, resulting in the tendency of the electrons to occupy the V–O bonding orbitals and thus effectively enhancing the V–O bonds. The stronger V–O bonds of NVP/NSC than those of NVP are confirmed by crystal orbital Hamiltonian population (COHP) analysis (Fig. 2o), which will effectively enhance the crystal structure stability of the material. D-band center theory is an effective tool to describe the transition metal bonding environment.38 The bonding state between V 3d and O 2p is a strong predictor of the cycling stability of the NVP materials. As shown in Fig. 2p, the three-dimensional spreading of bonding orbitals in NVP/NSC leads to a significant decrease in the d-band center, in which more electrons fall back to the bulk, demonstrating a more stable V–O bonding state, thus effectively suppressing the generation of hetero-phases and the destruction of the crystal structure during the cycling process. This will significantly enhance the structural stability of the material and forge the superb cycle life of NVP/NSC. In addition, the energy band gap of the spin-down state of NVP/NSC is significantly smaller than that of NVP, and the density of the spin-up state increases to 20.89 eV−1 at the Fermi energy level (Fig. 2q and r), indicating enhanced electronic conduction.
Above all, the interfacial dynamics and thermodynamics synergistic strategy consisting in tuning interfacial bonding and energy band alignment is validated through theoretical calculation. Clearly, the ionic conductivity, electronic conductivity and structural stability of SIB cathode materials would be concurrently reinforced. In this case, the fast-charging performance of the NVP cathode can be comprehensively upgraded, which will be discussed in the following sections.
2.2 Materials preparation and characterization
As outlined in the ESI Experimental section,† NVP/NSC composites were synthesized by a simple one-step solid phase ball milling method, followed by high temperature sintering. As shown in Fig. 3a, the NVP features a rhombohedral hexahedral structure and belongs to the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c space group. The [V2(PO4)3]3− unit of the NVP crystal structure consists of three [PO4] tetrahedra connected to each [VO6] octahedron. Na+ is held in separate oxygen environments in two different locations. While one Na+ ion (Na1) is in the 6b sites (six-coordinate Na–O sites, M1), the other two Na+ ions reside in the 18e sites (eight-coordinate Na–O sites, M2). Na+ ions at the M1 sites in NVP tend to remain immobilized, suggesting a direct M2-to-M2 conduction pathway.39 To clarify this crystal structure of the obtained NVP/NSC, the XRD spectra of three samples prepared with different addition content of N/S CDs (NVP/NSC-50, 75, 100) are illustrated in Fig. 3b. All diffraction peaks are indexed to the Rc space group without impurities and the high peak intensity demonstrates excellent crystallinity of all materials.40 The Rietveld refinement analysis is illustrated in Fig. 3c–e and the refinement results for samples with different amounts of N/S CDs added are shown in Table S1.† The lattice parameters of NVP/NSC-75 are a = b = 8.73285 Å, c = 21.82199 Å, V = 1441.243 Å3, which are consistent with the previously reported standard crystal structure of NVP.41 Notably, the refinement results for the samples with different amounts of carbon dots added are similar, suggesting that addition of different amounts of N/S CDs did not affect the bulk structure characteristics.
c space group. The [V2(PO4)3]3− unit of the NVP crystal structure consists of three [PO4] tetrahedra connected to each [VO6] octahedron. Na+ is held in separate oxygen environments in two different locations. While one Na+ ion (Na1) is in the 6b sites (six-coordinate Na–O sites, M1), the other two Na+ ions reside in the 18e sites (eight-coordinate Na–O sites, M2). Na+ ions at the M1 sites in NVP tend to remain immobilized, suggesting a direct M2-to-M2 conduction pathway.39 To clarify this crystal structure of the obtained NVP/NSC, the XRD spectra of three samples prepared with different addition content of N/S CDs (NVP/NSC-50, 75, 100) are illustrated in Fig. 3b. All diffraction peaks are indexed to the Rc space group without impurities and the high peak intensity demonstrates excellent crystallinity of all materials.40 The Rietveld refinement analysis is illustrated in Fig. 3c–e and the refinement results for samples with different amounts of N/S CDs added are shown in Table S1.† The lattice parameters of NVP/NSC-75 are a = b = 8.73285 Å, c = 21.82199 Å, V = 1441.243 Å3, which are consistent with the previously reported standard crystal structure of NVP.41 Notably, the refinement results for the samples with different amounts of carbon dots added are similar, suggesting that addition of different amounts of N/S CDs did not affect the bulk structure characteristics.
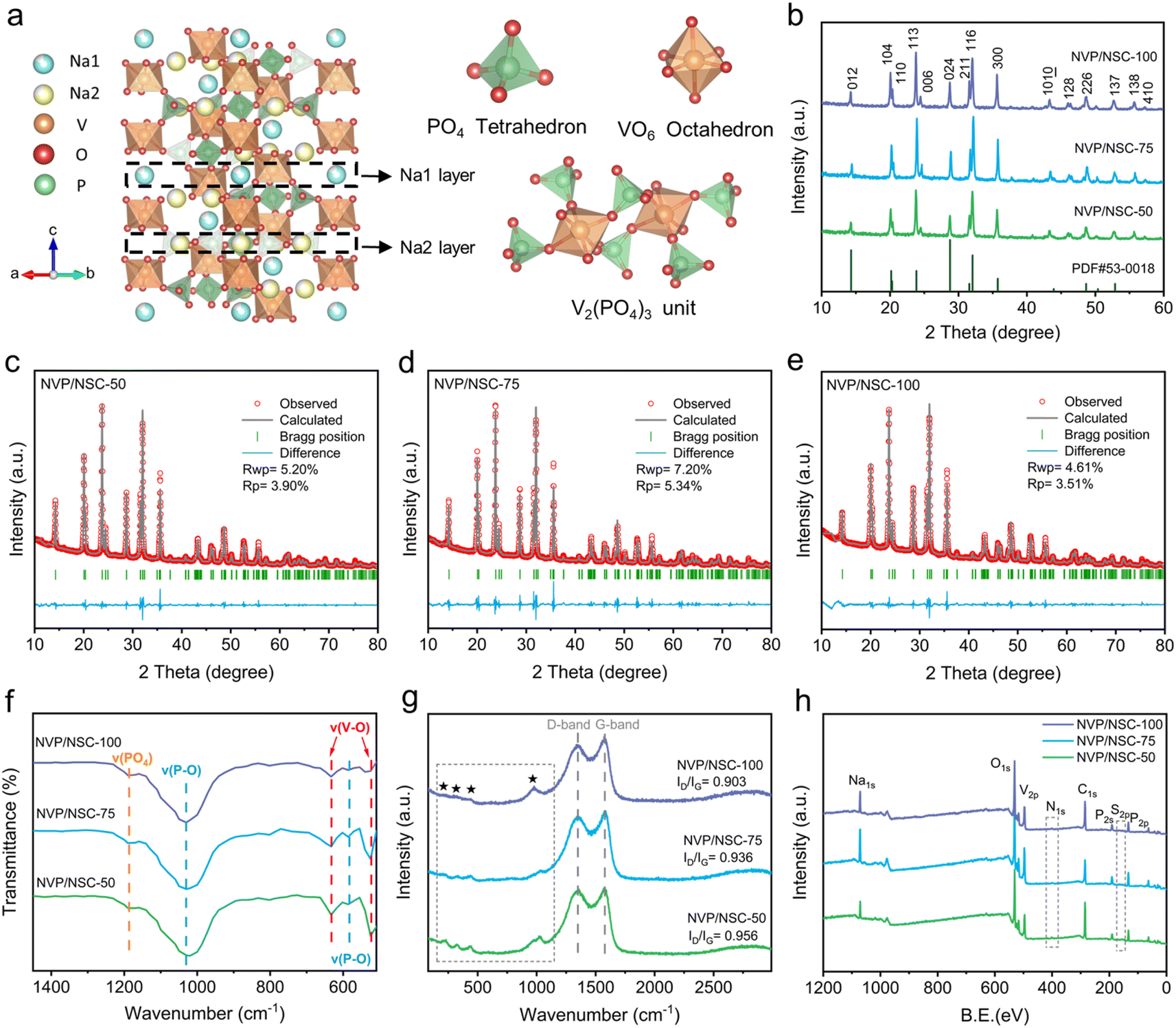 | ||
| Fig. 3 (a) Crystal structure of NVP. (b) XRD patterns of NVP/NSC samples. (c–e) Rietveld refinement of the XRD patterns of NVP/NSC samples. (f) FT-IR spectra of NVP/NSC samples. (g) Raman spectra curves of NVP/NSC samples. (h) XPS survey spectra of NVP/NSC samples. | ||
The carbon content of these NVP/NSC composites was determined by elemental analysis (EA) and the test results are shown in Table S2.† It is found that the carbon contents of NVP/NSC-50, 75 and 100 are 1.10%, 2.23% and 3.43%, respectively. The fine structure of the material was further characterized by Fourier transform infrared spectroscopy (FT-IR) and the results are shown in Fig. 3f. Vibrations of V3+–O2− bonds in the VO6 octahedra are detected at 633 cm−1 and 525 cm−1. The presence of P–O bonds in the PO4 tetrahedra is detected at 583 cm−1 and 1030 cm−1. The IR peak at 1190 cm−1 can be attributed to the stretching vibrations of the PO4 unit.42 Interestingly, the intensity of the absorption peaks of the V–O group is observed to diminish with the increase of N/S CDs incorporation, signifying that the electronegativity of the V–O group decreases due to the influence of the V–N/S–C bonds. To further investigate the graphitization of the carbon layer in all samples, Raman spectroscopic analyses were conducted (Fig. 3g). The characteristic peaks at approximately 1350.2 cm−1 and 1376.2 cm−1 are attributed to the D-band (amorphous carbon) and G-band (graphitized carbon), respectively.29 The region before 1200 cm−1 could be ascribed to the Raman fingerprint characteristics of NVP. The light peak in the range of 400–600 cm−1 points to the presence of PO4 or VO6, and the absorption peak at around 1000 cm−1 corresponds to asymmetric stretching vibrations of the PO4 unit.43 The presence of V–N/S–C bonding is reaffirmed by the gradual weakening of the absorption peaks in the fingerprint region at 400–600 nm, as this interfacial bonding significantly affects the vibrational mode of the VO6 octahedra. The relative intensity ratio of the D peak to the G peak (ID/IG) is an important indicator to characterize the degree of graphitization of the material.44 Notably, the ID/IG value is gradually decreased with the amount of N/S CDs increased, suggesting the degree of graphitization of the material progressively increases.
Subsequently, X-ray photoelectron spectroscopy (XPS) was employed to probe the elemental properties in NVP/NSC composites (Fig. 3h). Two peaks at 516.7 and 523.9 eV can be observed in the V 2p spectra of the three N/S CDs incorporated samples, which can be attributed to the spin–orbit splitting of V3+ 2p3/2 and V3+ 2p1/2 (Fig. S5a†).45 The C 1s spectra show four binding configurations (Fig. S5b†), namely C–C (284.7 eV), C–N/C–S (285.6 eV), C–O (286.7 eV) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.7 eV).46 The N 1s spectra (Fig. S5c†) show three characteristic peaks at 398.5, 400.4 and 402.0 eV, namely pyridinic N, pyrrolic N and graphitic N, respectively.47 The S 2p spectra (Fig. S5d†) display three characteristic peaks observed at 163.7, 164.6 and 168.6 eV, which can be attributed to the C–S–C (2P3/2), C–S–C (2P1/2) and C–SOX–C structures, respectively.48
O (288.7 eV).46 The N 1s spectra (Fig. S5c†) show three characteristic peaks at 398.5, 400.4 and 402.0 eV, namely pyridinic N, pyrrolic N and graphitic N, respectively.47 The S 2p spectra (Fig. S5d†) display three characteristic peaks observed at 163.7, 164.6 and 168.6 eV, which can be attributed to the C–S–C (2P3/2), C–S–C (2P1/2) and C–SOX–C structures, respectively.48
The morphology and detailed structure of the samples were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). It is found that the particle sizes of NVP/NSC-50, NVP/NSC-75 and NVP/NSC-100 decrease sequentially (Fig. 4a–c and S6†) since the crystalline growth of NVP particles is inhibited by active defects and nucleation sites on the surface of the carbon layer provided by N and S heteroatoms.49,50 Pleasingly, the sodium ion diffusion rate is expected to be improved by the decreased particle size and the shortened diffusion path. The patterns of selected area electron diffraction (SAED) spots (Fig. 4d–f) reveal that all three materials are highly ordered single-crystal structures with good crystallinity. The interplanar spacing coincides with the crystalline surface index of rhombic NVP. The structural properties of the materials were further revealed by TEM and high-resolution transmission electron microscopy (HRTEM) (Fig. S7† and 4g–i). A complete and uniform carbon layer is formed on the surface of NVP/NSC-50, NVP/NSC-75 and NVP/NSC-100, the thicknesses of which is gradually increased from 2 to 8 nm. EDS mapping was performed to further explore the elemental distribution and composition, showing a uniform distribution of Na, V, O, P, C, N and S (Fig. 4j, S8 and S9†).
 | ||
| Fig. 4 (a–c) SEM images of NVP/NSC samples. (d–f) SAED images of NVP/NSC samples. (g–i) HRTEM images of NVP/NSC samples. (j) Element mapping images for Na, V, O, P, C, N and S in NVP/NSC-75. | ||
2.3 Electrochemical performances of the sodium-ion half cell
Rate performance tests were conducted from 0.5C to 100C to investigate the ultrafast-charging performance of the NVP/NSC in Fig. 5a. Among them, NVP/NSC-50 exhibits a high specific discharge capacity at low current densities, but poor rate performance as there is a rapid drop of specific discharge capacity with the current density increased. The observed limitation in performance can be attributed to the insufficient content of the conductive carbon layer, which hinders the efficient transport of electrons, particularly at high current densities. The best rate performance is obtained by the NVP/NSC-100, but its capacity is lower than that of the NVP/NSC-75 because of the higher carbon content. Combining high specific capacity and excellent rate performance, 116.7, 114.4, 113.2, 108.6, 102.9, 98.8, 91.0, 87.1, and 83.2 mA h g−1 are delivered by NVP/NSC-75 at current densities of 0.5C, 1C, 2C, 5C, 10C, 20C, 50C, 70C and 100C, respectively. Notably, when the current density returns to 0.5C after cycling at various high current densities, the specific discharge capacity reaches an impressive value of 116.4 mA h g−1, close to the initial specific discharge capacity, implying that the extraction/insertion of Na+ in the NVP/NSC-75 electrode is still reversible even at ultra-high rates. The galvanostatic charge–discharge (GCD) curves of NVP/NSC-75, NVP/NSC-50 and NVP/NSC-100 at different current densities are illustrated in Fig. 5e–g. A noticeable discharge voltage plateau remains even at a high rate of 100C for NVP/NSC-75 and NVP/NSC-100, proving the role of synergistic interfacial dynamics and thermodynamics strategies in reducing polarization. Compared to other NVP materials modified by carbon coating (Fig. 5b and Table S3†), NVP/NSC-75 displays excellent rate capacity with a very low carbon content, indicating the unique advantages of the designed synergistic strategy of interface dynamics and thermodynamics. To further investigate the practical value of NVP/NSC, the energy density and power density of the NVP/NSC cathodes are calculated (Fig. 5c). Remarkably, NVP/NSC-75 delivers both energy and power densities (379 W h kg−1 and 33 kW kg−1), which are prominently better than those of current advanced cathodes, such as LiFePO4@C/CNT,51 rGO/C@Li3V2(PO4)3,52 NVP/C53 and NVP/rGO.54 In this case, NVP/NSC is a very promising candidate for the construction of superior electrochemical energy storage devices with both high power and high energy density.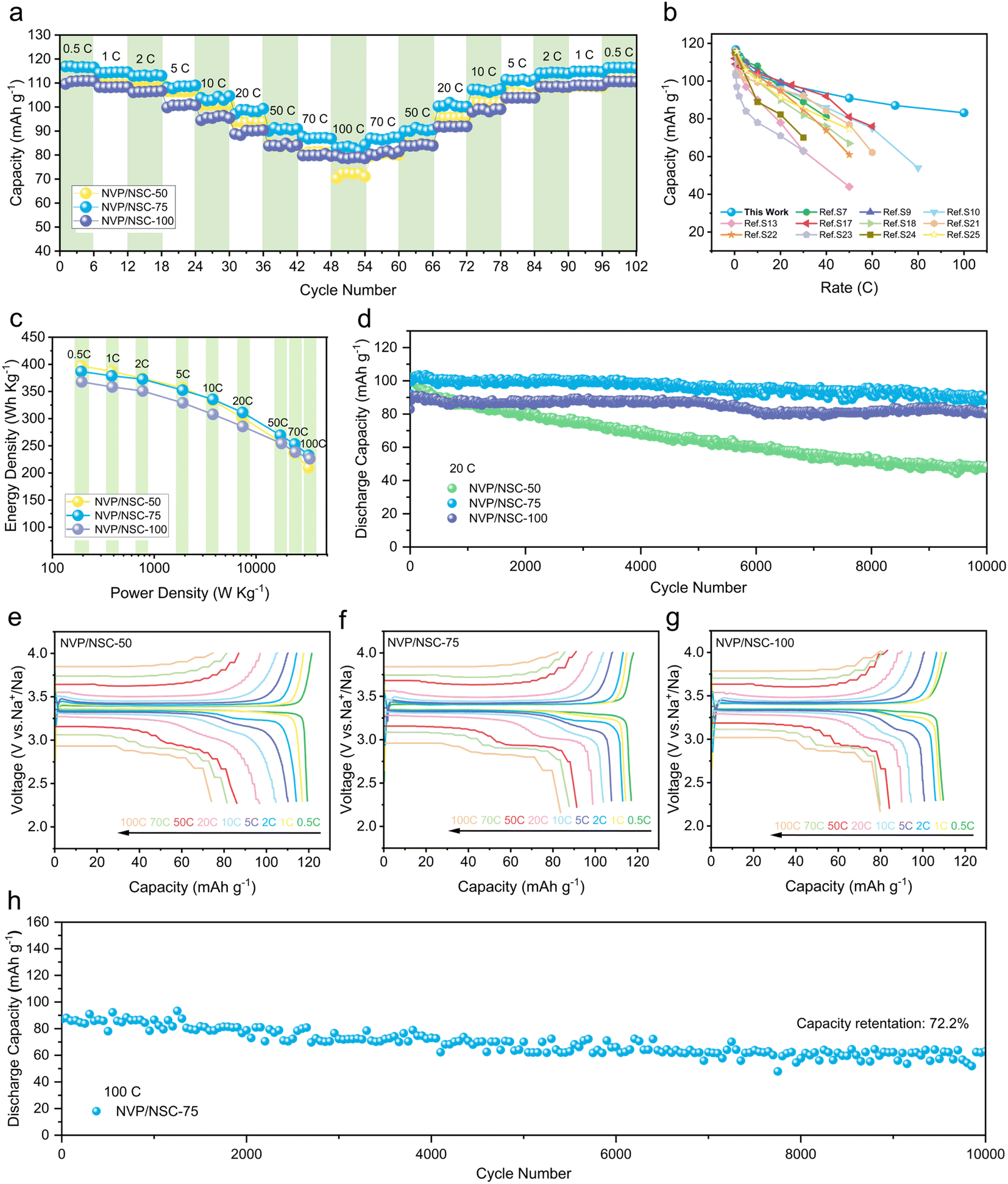 | ||
| Fig. 5 (a) Rate performances of NVP/NSC samples from 0.5 to 100C. (b) Comparison of the rate performances for NVP/NSC-75 half cells to the previous results in the literature. (c) Ragone plot at various rates of NVP/NSC samples. (d) Long-term cycling performance of NVP/NSC samples at the rate of 20C. (e–g) GCD curves of NVP/NSC samples. (h) Long-term cycling performance of NVP/NSC-75 at the rate of 100C. | ||
The cycling performance of the NVP/NSC electrodes at the rate of 1C is displayed in Fig. S10,† and the specific discharge capacities of 106.5, 115.2 and 108.9 mA h g−1 after 100 cycles are exhibited by NVP/NSC-50, NVP/NSC-75, and NVP/NSC-100, respectively. To further investigate the superior performance of NVP/NSC electrodes, the long-term cycling measurements were carried out at 10, 20 and 100C (1C = 117 mA g−1). The cycling performance of the NVP/NSC electrode at 10C for 2000 cycles is illustrated in Fig. S11.† Specific discharge capacity of 78.9, 101.6, and 96.5 mA h g−1 and capacity retention of 74.5%, 97.8%, and 99.5% are obtained by NVP/NSC-50, NVP/NSC-75 and NVP/NSC-100 respectively after 2000 cycles at 10C. This suggests that NVP/NSC-75 and NVP/NSC-100 exhibit remarkable cycling stability. Furthermore, Fig. S12† shows that the NVP/NSC-75 and NVP/NSC-100 exhibit high mid-value voltages, consistent with their excellent cycling stability. Further increasing the current density, the long-term cycling performance at a current density of 20C is given in Fig. 5d. After 10000 cycles, the specific capacity of NVP/NSC-50, NVP/NSC-75 and NVP/NSC-100 remains at 48.6, 89.3 and 81.1 mA h g−1, corresponding to the capacity retention of 47.0%, 86.7%, and 89.1%, respectively. Even more dramatic, the cycling performance of the NVP/NSC-75 at an ultra-high rate of 100C (25 s for full charge/discharge) was tested. A high initial specific capacity of 87.2 mA h g−1 is observed and remains at 63 mA h g−1 after 10000 cycles (Fig. 5h), corresponding to capacity retention of 72.2% (0.0028% capacity decay per cycle). Above all, it is clear that the cycling stability of the NVP/NSC composites can be enhanced significantly by the designed interfacial kinetic and thermodynamic strategies.
2.4 Ultrafast sodium storage mechanism
The Na+ extraction/insertion kinetics of NVP/NSC composites were investigated by CV measurements at different scan rates in the range of 0.1–1.0 mV s−1. As shown in Fig. 6a–c, a pair of redox peaks of all samples are displayed at approximately 3.4 V, representing the redox reaction between V3+ and V4+. The small reduction peak between 3.0 and 3.2 V is related to the structural reformation caused by the electrochemical behavior between Na(2) and Na(1),53 which is also observed in the GCD curves with a smaller discharge plateau. A significant increase in the redox potential difference is observed for the NVP/NSC-50 with the scan rate increasing, whereas the NVP/NSC-75 and NVP/NSC-100 maintain sharp peaks with small potential shifts, suggesting a milder electrochemical polarization. The peak current versus the square root of the scan rate is illustrated in Fig. 6d. The diffusion coefficient of Na+ (DNa+) in the cell can be obtained from the slope of the linear relationship by using the Randles–Sevcik equation:55| Ip = 2.69 × 105n3/2ACDNa+1/2ν1/2 |
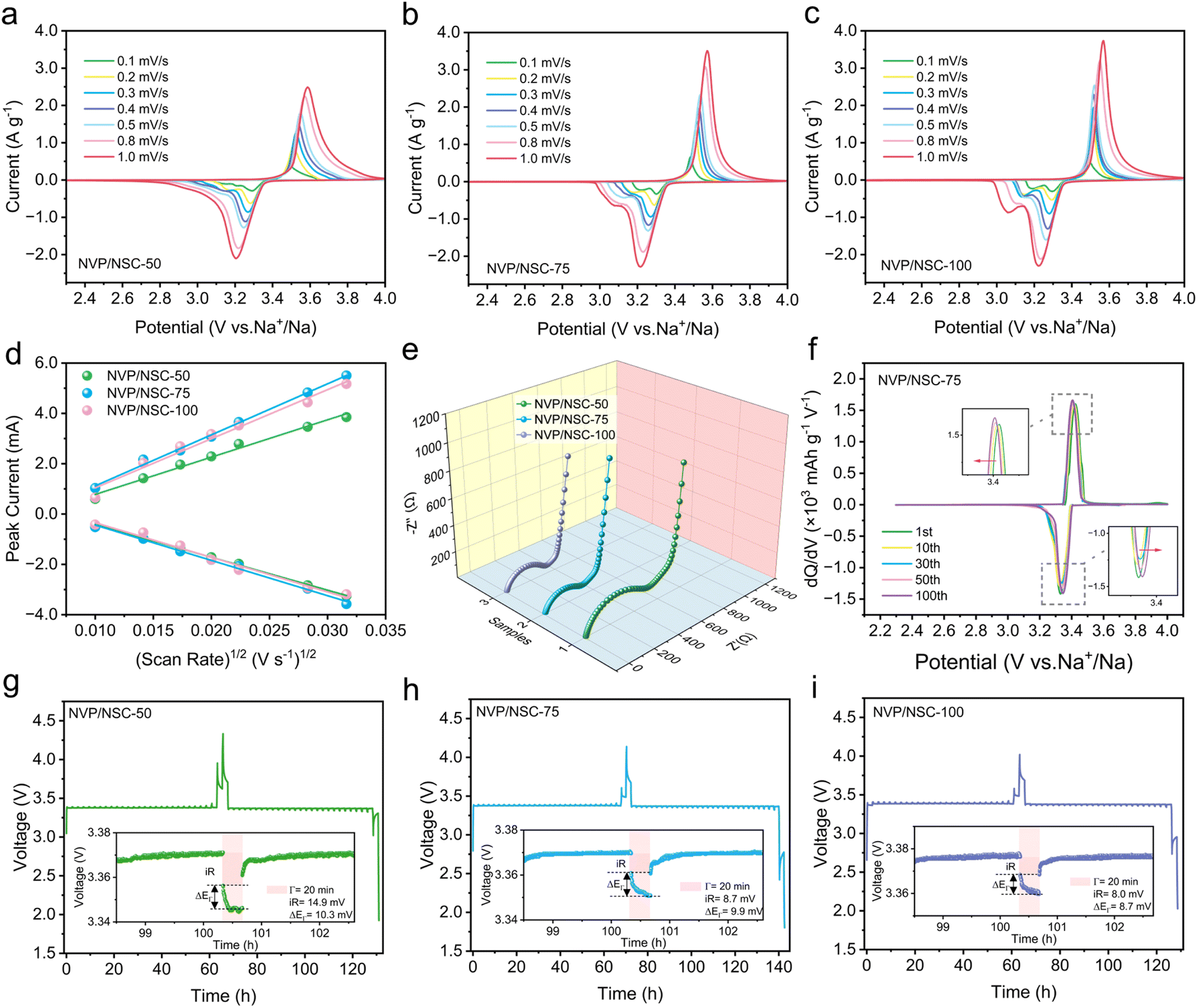 | ||
| Fig. 6 Kinetic properties of NVP/NSC-50, NVP/NSC-75 and NVP/NSC-100. CV curves at different scanning rates of (a) NVP/NSC-50, (b) NVP/NSC-75, and (c) NVP/NSC-100. (d) Linear fits for the anodic and cathodic peak currents versus scan rates of NVP/NSC samples. (e) Nyquist plots of NVP/NSC samples. (f) The dQ/dV curves of NVP/NSC-75 for the selected cycles at 1C. (g–i) Transient voltage–time profile obtained from a GITT test for NVP/NSC samples. | ||
An electrochemical impedance spectrum (EIS) test was performed to further investigate the differences in electrochemical kinetics of the electrode materials. The fitted model is displayed in Fig. S14,† where Rs denotes the ohmic impedance due to solution resistance etc., Rct corresponds to the charge transfer resistance, and CPE and Zw are associated with the double-layer capacitance and Warburg impedance during the diffusion of Na+ ions in NVP, respectively. As illustrated in Fig. 6e, the Nyquist plots of the NVP/NSC composites are all composed of a semicircle in the high-frequency region and a diagonal line in the low-frequency region. The results illustrate that the Rct of NVP/NSC-50, NVP/NSC-75 and NVP/NSC-100 are 464.6 Ω, 308.5 Ω and 292.3 Ω, indicating that the charge transfer impedance is effectively reduced by the presence of the N/S doped carbon layer and thus the Na+ diffusion kinetics can be enhanced.
The dQ/dV curves were investigated to verify the electrochemical stability of different samples during cycling (Fig. 6f, S15a and b†). All three materials show a pair of redox peaks, which is consistent with the CV and GCD test results. Severe voltage degradation, obvious capacity decay and increased polarization are displayed for NVP/NSC-50 as the number of cycles increased. In contrast, the NVP/NSC-75 and NVP/NSC-100 exhibit low polarization and good voltage plateau retention, which is responsible for the outstanding cycling performance. In addition, a gradual leftward shift of the high potential and a gradual rightward shift of the low potential plateau are demonstrated for NVP/NSC-75 as the number of charge/discharge cycles increased. This proves that there is a depolarization process in 100 cycles, which can further increase the depth of discharge and thus the capacity.
The galvanostatic intermittent titration technique (GITT) was carried out to provide comparative information on the electrode kinetics of two-phase systems by analyzing the time of voltage relaxation to equilibrium potential and the magnitude of the overpotential. As shown in Fig. 6g–i, the equilibrium potential of NVP/NSC-50 after relaxation is highest, while the equilibrium potential is lower and remains stable for NVP/NSC-75 and NVP/NSC-100, indicating the relative difficulty of Na+ extraction/insertion in NVP/NSC-50 during the electrochemical reaction. The lower overpotential and polarization of NVP/NSC-75 and NVP/NSC-100 suggest faster kinetics, resulting in enhanced electron conductivity and ion transport.
2.5 Charge compensation mechanism and failure mechanism analysis
The structural evolution mechanism of NVP/NSC-75 was revealed with the in situ X-ray diffraction analysis (XRD) in the voltage window of 2.3–4.0 V (Fig. 7a and b). During charging and discharging, a two-phase reaction occurs between Na3V2(PO4)3 and NaV2(PO4)3:| Na3V2(PO4)3 ↔ NaV2(PO4)3 + 2Na+ + 2e− |
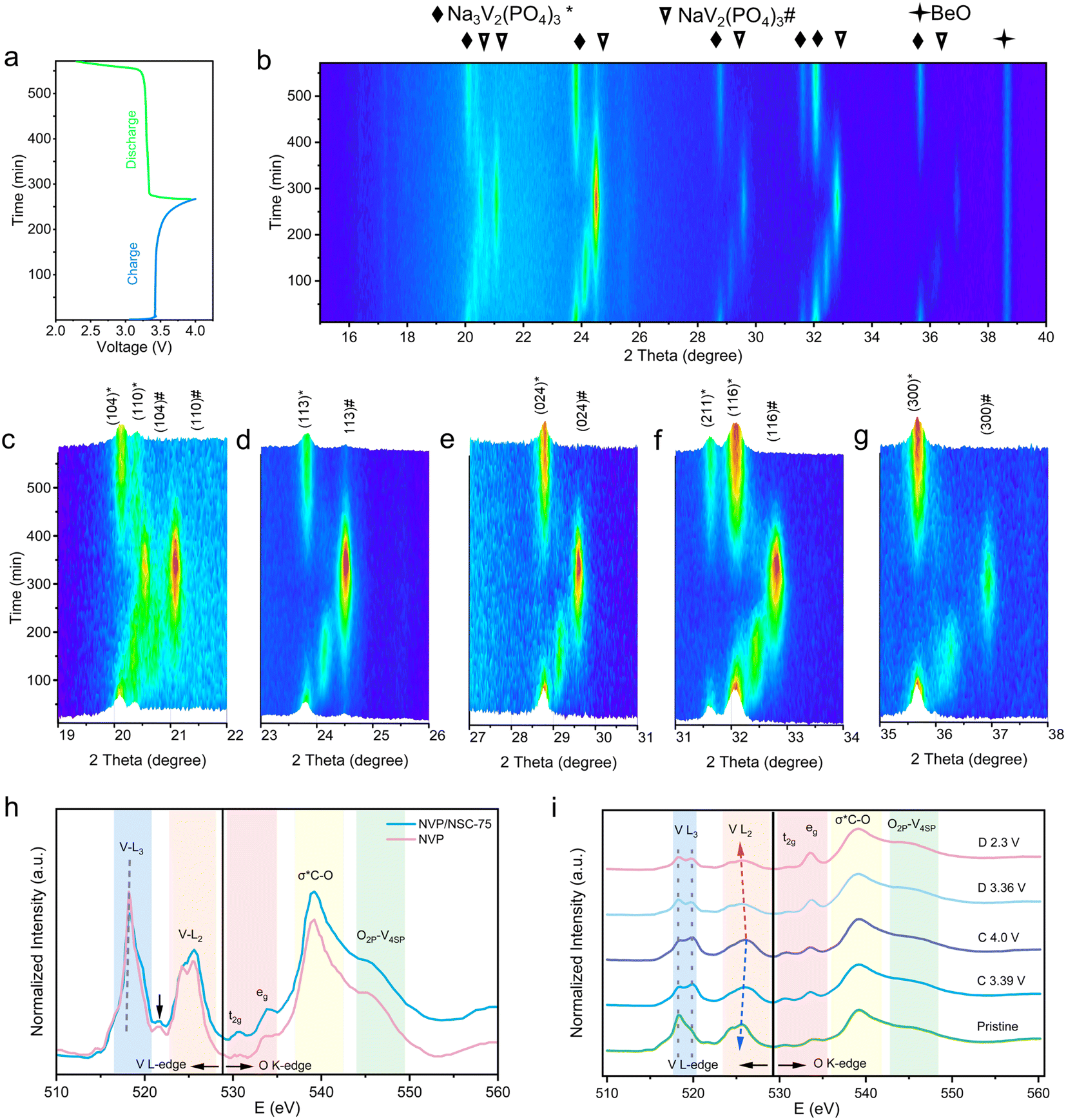 | ||
| Fig. 7 (a) GCD curves of NVP/NSC-75. (b) In situ XRD patterns and (c–g) structural evolution of the NVP/NSC-75 electrode during the extraction/intercalation of Na+. (h) Normalized sXAS spectra of the V L-edge and O K-edge for NVP and NVP/NSC-75. (i) Normalized sXAS spectra of NVP/NSC-75 at different charging/discharging states. | ||
At the initial voltage, all XRD peaks exactly correspond to the Na3V2(PO4)3 standard card. The main peaks at 20.1°, 23.8°, 28.8°, 32.1° and 35.6° are associated with the (104), (113), (024), (116) and (300) crystal planes of Na3V2(PO4)3 (Fig. 7c–g). New XRD peaks (corresponding to NaV2(PO4)3) become progressively stronger during charging accompanied by the intensity of the Na3V2(PO4)3 peaks gradually diminishing. At around 3.4 V, two phases of Na3V2(PO4)3 and NaV2(PO4)3 coexist, and the material completely transforms to the NaV2(PO4)3 phase after charging to 4.0 V. The XRD changes during the discharging process are opposite to those during the charging process: NaV2(PO4)3 gradually disappears and finally completely transforms to the Na3V2(PO4)3 phase, which demonstrates that highly reversible extraction/insertion process of sodium ions in the NVP/NSC-75 cathode.
In addition, soft X-ray absorption spectroscopy (sXAS) tests were adopted to characterize the changes during the charging and discharging process from the level of the electronic structure of the material to further investigate the mechanism of the excellent electrochemical performance of the NVP/NSC-75. The V-L edge and O-K edge of NVP/NSC-75 and NVP are shown in Fig. 7h. The V L edge includes two major peaks, the V L3 peak (514–521 eV) and the V L2 peak (521–528 eV), which originate from electronic jumps from the V 2p3/2 energy level and the 2p1/2 energy level to the V 3d unoccupied state, respectively. The fine structure of the L3 pre-edge is caused by crystal field splitting in the V-3d orbital. The energy range of the O K-edge locates at 528–550 eV. The broad peak at 539.2 eV is attributed to σ*C–O,56 and the peak at 544.8 eV is associated with the mixed state of V 4sp and O 2p.57 Further, NVP/NSC-75 was studied by an ex situ sXAS test at different states of charge (Fig. 7i). During the charging process, the bimodal intensity of V L3 changes with a gradual increase in voltage, with a gradual increase in peak intensity at 519.8 eV and a gradual decrease at 518.3 eV, demonstrating a change in electronic structure. The rightward shift of the V L2 edge indicates a gradual increase in the valence state of V (V3+ → V4+),58 corresponding to an increase in the oxidation state during the charging process. As for the discharging process, the peak intensity is gradually increased at 519.8 eV and decreased at 518.3 eV. The leftward shift of the V L2 edge back to the initial state indicates a gradual decrease in the valence state of V (V4+ → V3+). Considering all of the above, the excellent reversibility of NVP/NSC-75 is proved from the electronic structure point of view.
Furthermore, failure mechanism analysis was adopted. Firstly, an ex situ XRD analysis of NVP/NSC-75 and NVP after 100 cycles at 10C was carried out (Fig. 8a). For NVP, the diffraction peaks at 14.1°, 20.1°, etc. are prominently weaker and the peak height ratios of the two peaks at 23.7° and 32.0° are destroyed after cycling compared to the pristine NVP, suggesting that the crystal structure of NVP is damaged after high-rate cycling. In contrast, the XRD peaks of NVP/NSC-75 are intact and well maintained after cycling, and the peak height ratios do not change noticeably, signifying that the NVP/NSC-75 displays excellent crystal retention ability during high-rate cycling. The V-L edges of NVP and NVP/NSC-75 after 100 cycles at 10C are shown in Fig. 8b and c. The presence of V3+ is demonstrated by a small absorption peak pair at 521.6 eV coinciding with the V-L edge of V2O3.58 It is suggested that NVP/NSC-75 exhibits good electrochemical reversibility as the peak intensity ratio of L3/L2 for NVP/NSC-75 after 100 cycles is closer to that before cycling. However, the peak intensity ratio of L3/L2 for NVP after 100 cycles is prominently lower that before cycling, indicating that the oxidation state of V gradually increases,58 which is responsible for the poor cycling stability and reversibility of NVP (Fig. S16a and b†). The changes in interfacial impedance at different cycles were investigated by using electrochemical impedance spectroscopy (EIS). The EIS results of the samples after 100 cycles at the rate of 10C (Fig. 8d) show that the charge transfer resistance (Rct) is closely related to the charge transfer kinetics during the long-term cycling process (determined by the surface structure of the cathode material and the interface formed by electrolyte decomposition), and the Rct (278.7 Ω) of the NVP/NSC-75 electrode after cycling is obviously smaller than that of NVP (444.2 Ω), favoring a lower kinetic limitation. In addition, the relationship between the real parts of the low frequency impedance of the samples after 100 cycles is compared (Fig. 8e). It is revealed that the V–N/S–C surface bonding strategy is effective in enhancing sodium ion migration with smaller slope values.
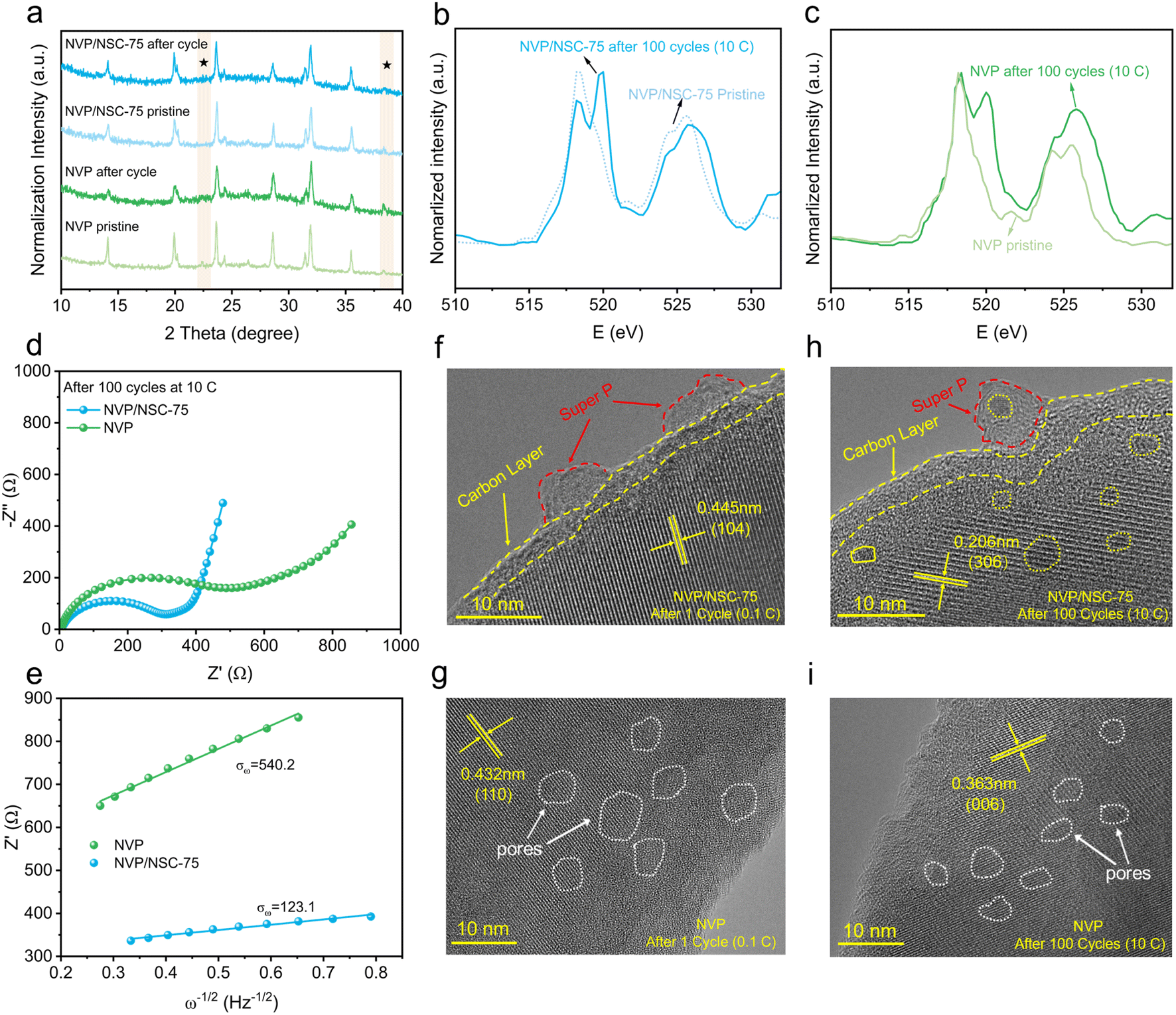 | ||
| Fig. 8 (a) Ex situ XRD patterns of NVP and NVP/NSC-75 before and after cycling. (b and c) Normalized sXAS spectra of NVP and NVP/NSC-75 after cycling. (d) Nyquist plots of samples after 100 cycles at 10C. (e) The relationship between Z′ and ω−1/2 after 100 cycles at 10C. (f–i) HRTEM images of NVP and NVP/NSC-75 samples after cycling. | ||
Finally, a TEM analysis for the cycled samples was also carried out. A large number of pores are produced in NVP after cycling (Fig. 8g, i and S17a, b†), probably due to the erosion or destruction of the material structure by the electrolyte during the cycling process. This is consistent with the poor rate performance and cycling stability of NVP at high current density. In contrast, both the carbon coating layer and bulk of NVP/NSC-75 remain intact before and after cycling (Fig. 8f, h, S17c and d†), suggesting that the interfacial bonding and energy band alignment strategy is able to effectively boost the structural stability of NVP/NSC-75 during cycling.
2.6 Full cell performances
In a further attempt to assess the potential of the NVP/NSC composites for application, the full cell was assembled by utilizing NVP/NSC-75 as the cathode and hard carbon (HC) as anode. The long cycle performance of the HC is largely influenced by the stability of the electrolyte/electrode interface, which is principally related to the composition of the electrolyte. The nature of sodium ion transport through the interface is highly dependent on the ionic conductivity of the resulting solid electrolyte interface (SEI). Compared to ester-based electrolytes, ether-based electrolytes have thinner SEI films and lower desolvation barriers at the electrolyte–electrode interface. Thus, ether electrolytes have emerged as a potential option for improving the rate performance and cycling stability of hard carbon anode materials.59,60 In this case, electrolyte matching engineering was carried out through the rate performance comparison of the HC in an ester-based electrolyte and an ether-based electrolyte (Fig. 9a). As a result, good rate performance, capacity retention and low charge/discharge plateau (∼0.1 V) are exhibited by HC half-cells in ether-based electrolytes at different current densities. Especially, a specific charge capacity of 245 mA h g−1 is achieved at a current density of 0.5 A g−1 (Fig. 9b). In contrast, the capacity of the HC half-cell decayed significantly in the ester electrolyte; the specific charge capacity at 0.5 A g−1 is only 40.5 mA h g−1 (Fig. 9c). What's worse, an insignificant charge/discharge plateau and gradual increase in the plateau with increasing current density were displayed. NVP/NSC-75 half-cells were carefully assembled with ether electrolyte in order to verify the effect of ether electrolyte on the cathode material. The test results show that NVP/NSC-75 has comparable performance in ether-based electrolytes, indicating that the electrolyte has negligible effect on the electrochemical performance of the cathode material (Fig. 9d and S18†).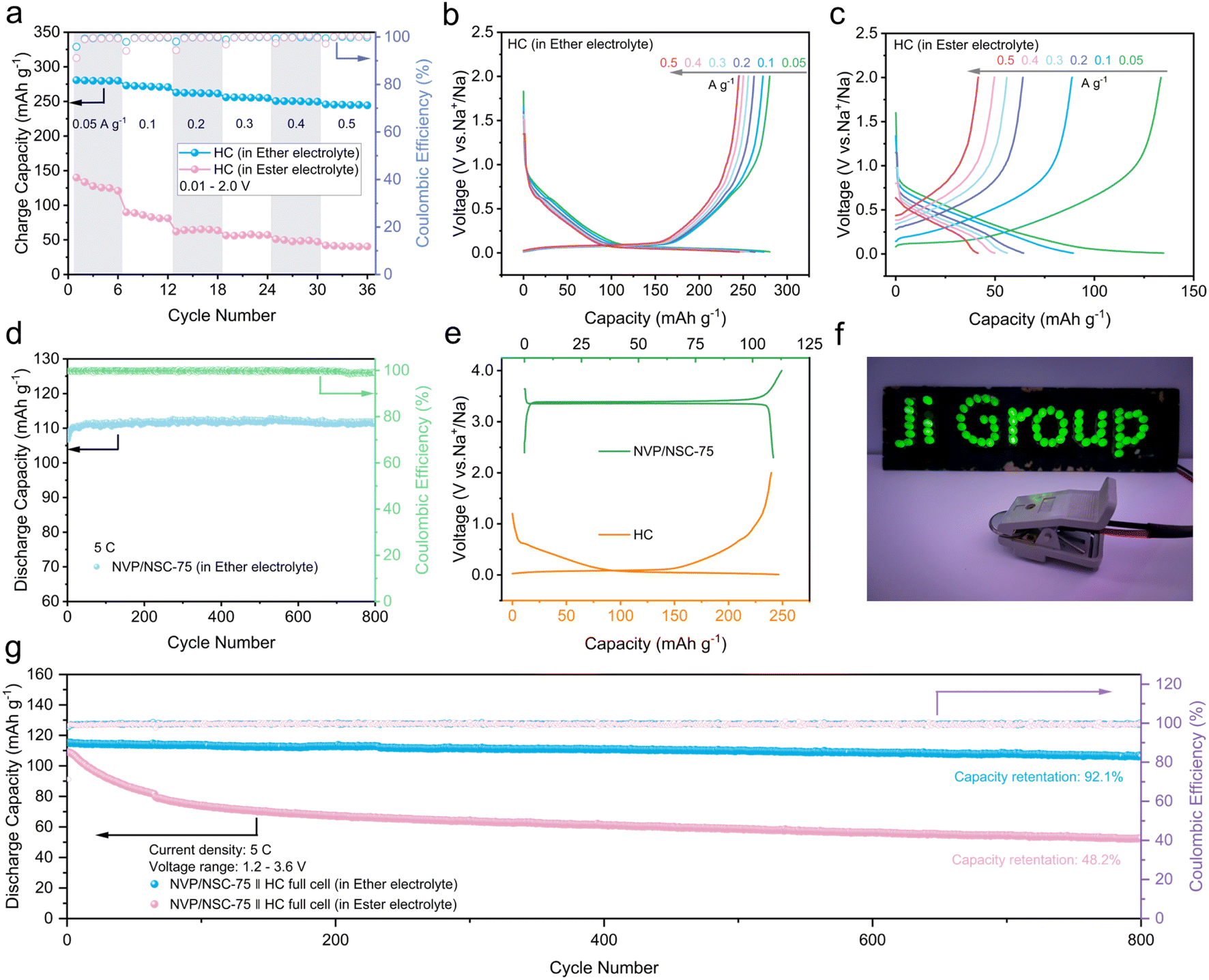 | ||
| Fig. 9 (a) Rate performance of the HC anode in ether electrolyte and ester electrolyte respectively. (b) GCD curves of the HC half-cell in ether electrolyte. (c) GCD curves of the HC half-cell in ester electrolyte. (d). Cycling performance of the NVP/NSC-75 half-cell in ether electrolyte. (e) GCD curves of NVP/NSC-75 and HC anode. (f) Photo of a lighted sign lit by the NVP/NSC-75‖HC full cell. (g) Long-term cycling performances of the NVP/NSC-75‖HC full cell at 5C for 800 cycles. | ||
The charge/discharge curves for the NVP/NSC-75 cathode and HC anode in ether electrolyte in the half-cell are shown in Fig. 9e, respectively. Prior to the full cell assembly, a pre-sodiation treatment was applied to the HC anode to offset the irreversible depletion of Na+ caused by the initial generation of SEI during cycling. The cycling performance of NVP/NSC-75‖HC full cells in ether-based and ester-based electrolytes was compared over a charge/discharge interval of 1.2–3.6 V (Fig. 9g). At a current density of 5C (1C = 117 mA g−1), the NVP/NSC-75‖HC full cell in ether-based electrolyte shows a high specific capacity of 115.3 mA h g−1 with a high initial coulombic efficiency (ICE) of 97.9% and a capacity retention rate of 92.1% after 800 cycles. In contrast, the NVP/NSC-75‖HC full cell in ester-based electrolyte has an initial specific capacity of 109.0 mA h g−1, which decayed seriously after 800 cycles, with a capacity retention rate of only 48.2%. This is consistent with the rate performance of the HC half-cell in different electrolytes, suggesting that the electrochemical performance of the full cell is mainly limited by the anode side of the HC. The NVP/NSC-75‖HC full cell after preferential electrolyte selection exhibits excellent electrochemical performance with an energy density of up to 330 W h kg−1 (Fig. S19†), which is the leading performance among NVP full cells (Table S4†).
3 Conclusions
The key factors for developing ultrafast-charging SIBs lie in simultaneously improving the electronic conductivity, ionic conductivity and structural stability of cathode materials. In this work, an interfacial dynamics and thermodynamics synergistic strategy is designed to synchronously enhance the three key factors so as to face the challenges of ultrafast-charging operations. Taking NVP as a typical case, interface bonding modifications for NVP are successfully performed by introducing a heterolayer derived from N/S co-doped carbon dots.In terms of interfacial dynamics aspects, the highly graphitized N/S co-doped carbon coating layer effectively reduced the electrical internal resistance, as confirmed by EIS. Moreover, DFT calculations demonstrated that the bonding effects of V–N/S–C could reconstruct the Na+ diffusion path in the bulk and near surface, significantly reducing the Na+ transport energy barrier. As for interfacial dynamics aspects, the d-band center of V is effectively reduced and the bonding strength of V–O would be improved, which helps to build a highly stable interface and suppress irreversible phase transitions. This view is supported by the post-cycling failure analysis and in situ structural evolution mechanisms. Tuned by this interfacial synergistic strategy, the meticulously designed NVP/NSC material exhibited excellent cycling stability and rate performance. The high specific discharge capacity of 89.3 mA h g−1 is achieved after 10000 cycles at 20C. Even at a very high current density of 100C, an initial specific discharge capacity of 87.2 mA h g−1 and a capacity retention rate of 72.2% after 10000 cycles (i.e., ultralow 0.0028% capacity decay per cycle) are exhibited by NVP/NSC-75. In addition, through electrolyte matching engineering for full cells, NVP/NSC-75‖HC full cells exhibit high specific capacity and cycling stability even at high current densities. Based on the findings above, a practical reference for the cathode material of SIBs with ultra-long life and ultrafast-charging capability is provided, which will promote the development of next-generation energy storage batteries.
Data availability
All the data supporting this article have been uploaded as part of the ESI.†Author contributions
Yujin Li: conceptualization, methodology, software, data curation, investigation, writing. Yu Mei: conceptualization, methodology, software, investigation. Roya Momen: methodology, investigation, writing – review & editing. Bai Song: resources, funding acquisition. Yujie Huang: investigation, methodology. Xue Zhong: conceptualization, methodology. Hanrui Ding: investigation. Wentao Deng: resources, methodology. Guoqiang Zou: conceptualization, methodology. Hongshuai Hou: supervision, methodology, funding acquisition, writing – review & editing. Xiaobo Ji: methodology, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52261135632, 22379165, 52074359, U21A20284) and the Science and Technology Innovation Program of Hunan Province (2021RC3014).References
- D. Larcher and J. M. Tarascon, Towards greener and more sustainable batteries for electrical energy storage, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- J. Cho, S. Jeong and Y. Kim, Commercial and research battery technologies for electrical energy storage applications, Prog. Energy Combust. Sci., 2015, 48, 84–101 CrossRef.
- J. W. Choi and D. Aurbach, Promise and reality of post-lithium-ion batteries with high energy densities, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- K. Zou, M. Jiang, Z. Zhao, S. Xie, T. Ning, L. Tan, H. Li, Y. Zhou, W. Wang, X. Wu and L. Li, Mechanistic insights into suppressing microcracks by regulating grain size of precursor for high-performance Ni-rich cathodes, Chem. Eng. J., 2023, 476, 146793 CrossRef CAS.
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. L. Notten, From Li-Ion Batteries toward Na-Ion Chemistries: Challenges and Opportunities, Adv. Energy Mater., 2020, 10, 2001310 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Research Development on Sodium-Ion Batteries, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- T. F. Liu, Y. P. Zhang, Z. G. Jiang, X. Q. Zeng, J. P. Ji, Z. H. Li, X. H. Gao, M. H. Sun, Z. Lin, M. Ling, J. C. Zheng and C. D. Liang, Exploring competitive features of stationary sodium ion batteries for electrochemical energy storage, Energy Environ. Sci., 2019, 12, 1512–1533 RSC.
- X. J. Pu, H. M. Wang, D. Zhao, H. X. Yang, X. P. Ai, S. N. Cao, Z. X. Chen and Y. L. Cao, Recent Progress in Rechargeable Sodium-Ion Batteries: toward High-Power Applications, Small, 2019, 15, 1805427 CrossRef.
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli and J. Janek, Fast Charging of Lithium-Ion Batteries: A Review of Materials Aspects, Adv. Energy Mater., 2021, 11, 2101126 CrossRef CAS.
- B. Kang and G. Ceder, Battery materials for ultrafast charging and discharging, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- D. Ansean, M. Dubarry, A. Devie, B. Y. Liaw, V. M. Garcia, J. C. Viera and M. Gonzalez, Fast charging technique for high power LiFePO4 batteries: A mechanistic analysis of aging, J. Power Sources, 2016, 321, 201–209 CrossRef CAS.
- C. Liu, Z. G. Neale and G. Cao, Understanding electrochemical potentials of cathode materials in rechargeable batteries, Mater. Today, 2016, 19, 109–123 CrossRef CAS.
- M. Yuan, H. Liu and F. Ran, Fast-charging cathode materials for lithium & sodium ion batteries, Mater. Today, 2023, 63, 360–379 CrossRef CAS.
- G. G. Eshetu, G. A. Elia, M. Armand, M. Forsyth, S. Komaba, T. Rojo and S. Passerini, Electrolytes and Interphases in Sodium-Based Rechargeable Batteries: Recent Advances and Perspectives, Adv. Energy Mater., 2020, 10, 2000093 CrossRef CAS.
- Y. Liu, Y. Zhu and Y. Cui, Challenges and opportunities towards fast-charging battery materials, Nat. Energy, 2019, 4, 540–550 CrossRef.
- W. Zhang, Y. L. Wu, Z. M. Xu, H. X. Li, M. Xu, J. W. Li, Y. H. Dai, W. Zong, R. W. Chen, L. He, Z. Zhang, D. J. L. Brett, G. J. He, Y. Q. Lai and I. P. Parkin, Rationally Designed Sodium Chromium Vanadium Phosphate Cathodes with Multi-Electron Reaction for Fast-Charging Sodium-Ion Batteries, Adv. Energy Mater., 2022, 12, 2201065 CrossRef CAS.
- C. Yan, R. Xu, Y. Xiao, J.-F. Ding, L. Xu, B.-Q. Li and J.-Q. Huang, Toward Critical Electrode/Electrolyte Interfaces in Rechargeable Batteries, Adv. Funct. Mater., 2020, 30, 1909887 CrossRef CAS.
- Z. Zhang, D. Zhao, Y. Xu, S. Liu, X. Xu, J. Zhou, F. Gao, H. Tang, Z. Wang, Y. Wu, X. Liu and Y. Zhang, A Review on Electrode Materials of Fast-Charging Lithium-Ion batteries, Chem. Rec., 2022, 22, e202200127 CrossRef CAS.
- H. Lee, E. Jo, K. Y. Chung, D. Byun, S. M. Kim and W. Chang, In-Depth TEM Investigation on Structural Inhomogeneity within a Primary LixNi0.835Co0.15Al0.015O2 Particle: Origin of Capacity Decay during High-Rate Discharge, Angew. Chem., Int. Ed., 2020, 59, 2385–2391 CrossRef CAS PubMed.
- R. Berthelot, D. Carlier and C. Delmas, Electrochemical investigation of the P2–NaxCoO2 phase diagram, Nat. Mater., 2011, 10, 74–80 CrossRef CAS.
- G. Yan, S. Mariyappan, G. Rousse, Q. Jacquet, M. Deschamps, R. David, B. Mirvaux, J. W. Freeland and J.-M. Tarascon, Higher energy and safer sodium ion batteries via an electrochemically made disordered Na3V2(PO4)(2)F3 material, Nat. Commun., 2019, 10, 585 CrossRef CAS.
- A. Simonov, T. De Baerdemaeker, H. L. B. Boström, M. L. Ríos Gómez, H. J. Gray, D. Chernyshov, A. Bosak, H.-B. Bürgi and A. L. Goodwin, Hidden diversity of vacancy networks in Prussian blue analogues, Nature, 2020, 578, 256–260 CrossRef CAS PubMed.
- S. Li, Y. Dong, L. Xu, X. Xu, L. He and L. Mai, Effect of Carbon Matrix Dimensions on the Electrochemical Properties of Na3V2(PO4)(3) Nanograins for High-Performance Symmetric Sodium-Ion Batteries, Adv. Mater., 2014, 26, 3545–3553 CrossRef CAS PubMed.
- Y. H. Jung, C. H. Lim and D. K. Kim, Graphene-supported Na3V2(PO4)(3) as a high rate cathode material for sodium-ion batteries, J. Mater. Chem. A, 2013, 1, 11350–11354 RSC.
- D. Guo, J. Qin, Z. Yin, J. Bai, Y.-K. Sun and M. Cao, Achieving high mass loading of Na3V2(PO4)(3)@carbon on carbon cloth by constructing three-dimensional network between carbon fibers for ultralong cycle-life and ultrahigh rate sodium-ion batteries, Nano Energy, 2018, 45, 136–147 CrossRef CAS.
- Y. Li, M. Chen, B. Liu, Y. Zhang, X. Liang and X. Xia, Heteroatom Doping: An Effective Way to Boost Sodium Ion Storage, Adv. Energy Mater., 2020, 10, 2000927 CrossRef CAS.
- L. Liang, X. Li, F. Zhao, J. Zhang, Y. Liu, L. Hou and C. Yuan, Construction and Operating Mechanism of High-Rate Mo-Doped Na3V2(PO4)(3)@C Nanowires toward Practicable Wide-Temperature-Tolerance Na-Ion and Hybrid Li/Na-Ion Batteries, Adv. Energy Mater., 2021, 11, 2100287 CrossRef CAS.
- J. F. Yang, D. D. Li, X. S. Wang, X. X. Zhang, J. Xu and J. T. Chen, Constructing micro-nano Na3V2(PO4)(3)/C architecture for practical high-loading electrode fabrication as superior-rate and ultralong-life sodium ion battery cathode, Energy Storage Mater., 2020, 24, 694–699 CrossRef.
- X. Cao, A. Pan, B. Yin, G. Fang, Y. Wang, X. Kong, T. Zhu, J. Zhou, G. Cao and S. Liang, Nanoflake-constructed porous Na3V2(PO4)(3)/C hierarchical microspheres as a bicontinuous cathode for sodium-ion batteries applications, Nano Energy, 2019, 60, 312–323 CrossRef CAS.
- Y. Jiang, Z. Yang, W. Li, L. Zeng, F. Pan, M. Wang, X. Wei, G. Hu, L. Gu and Y. Yu, Nanoconfined Carbon-Coated Na3V2(PO4)(3) Particles in Mesoporous Carbon Enabling Ultralong Cycle Life for Sodium-Ion Batteries, Adv. Energy Mater., 2015, 5, 1402104 CrossRef.
- Y. Fang, L. Xiao, X. Ai, Y. Cao and H. Yang, Hierarchical Carbon Framework Wrapped Na3V2(PO4)(3) as a Superior High-Rate and Extended Lifespan Cathode for Sodium-Ion Batteries, Adv. Mater., 2015, 27, 5895–5900 CrossRef CAS.
- X. Xu, F. Xiong, J. Meng, X. Wang, C. Niu, Q. An and L. Mai, Vanadium-Based Nanomaterials: A Promising Family for Emerging Metal-Ion Batteries, Adv. Funct. Mater., 2020, 30, 1904398 CrossRef CAS.
- K. Ding, Y. Ye, J. Hu, L. Zhao, W. Jin, J. Luo, S. Cai, B. Weng, G. Zou, H. Hou and X. Ji, Aerophilic Triphase Interface Tuned by Carbon Dots Driving Durable and Flexible Rechargeable Zn-Air Batteries, Nano-Micro Lett., 2023, 15, 28 CrossRef CAS.
- L. Xu, H. Tu, F. Zhu, Y. Xiang, Z. Luo, S. Fang, W. Deng, G. Zou, H. Hou and X. Ji, Carbon dots for ultrastable solid-state batteries, Smartmat, 2022, 3, 286–297 CrossRef CAS.
- C. Hu, M. Li, J. Qiu and Y.-P. Sun, Design and fabrication of carbon dots for energy conversion and storage, Chem. Soc. Rev., 2019, 48, 2315–2337 RSC.
- Y. Liu, S. Roy, S. Sarkar, J. Xu, Y. Zhao and J. Zhang, A review of carbon dots and their composite materials for electrochemical energy technologies, Carbon Energy, 2021, 3, 795–826 CrossRef CAS.
- L. Li, D. Cheng, G. Zou, H. Hou, X. Ji and L. Yang, Carbon anode from carbon dots-regulated polypyrrole for enhanced potassium storage, J. Alloys Compd., 2023, 958, 170481 CrossRef CAS.
- K. Jia, J. Ma, J. Wang, Z. Liang, G. Ji, Z. Piao, R. Gao, Y. Zhu, Z. Zhuang, G. Zhou and H.-M. Cheng, Long-Life Regenerated LiFePO4 from Spent Cathode by Elevating the d-Band Center of Fe, Adv. Mater., 2023, 35, 2208034 CrossRef CAS PubMed.
- Z. Jian, C. Yuan, W. Han, X. Lu, L. Gu, X. Xi, Y.-S. Hu, H. Li, W. Chen, D. Chen, Y. Ikuhara and L. Chen, Atomic Structure and Kinetics of NASICON NaxV2(PO4)(3) Cathode for Sodium-Ion Batteries, Adv. Funct. Mater., 2014, 24, 4265–4272 CrossRef CAS.
- P. Feng, W. Wang, K. Wang, S. Cheng and K. Jiang, Na3V2(PO4)(3)/C synthesized by a facile solid-phase method assisted with agarose as a high-performance cathode for sodium-ion batteries, J. Mater. Chem. A, 2017, 5, 10261–10268 RSC.
- W. Duan, Z. Zhu, H. Li, Z. Hu, K. Zhang, F. Cheng and J. Chen, Na3V2(PO4)(3)@C core-shell nanocomposites for rechargeable sodium-ion batteries, J. Mater. Chem. A, 2014, 2, 8668–8675 RSC.
- X. Liu, G. Feng, Z. Wu, Z. Yang, S. Yang, X. Guo, S. Zhang, X. Xu, B. Zhong and Y. Yamauchi, Enhanced sodium storage property of sodium vanadium phosphate via simultaneous carbon coating and Nb5+ doping, Chem. Eng. J., 2020, 386, 123953 CrossRef CAS.
- D. Wang, P. Cai, G.-Q. Zou, H.-S. Hou, X.-B. Ji, Y. Tian and Z. Long, Ultra-stable carbon-coated sodium vanadium phosphate as cathode material for sodium-ion battery, Rare Met., 2022, 41, 115–124 CrossRef CAS.
- Y. J. Chen, Y. L. Xu, X. F. Sun and C. Wang, Effect of Al substitution on the enhanced electrochemical performance and strong structure stability of Na3V2(PO4)(3)/C composite cathode for sodium-ion batteries, J. Power Sources, 2018, 375, 82–92 CrossRef CAS.
- Q. Ni, Y. Bai, Y. Li, L. Ling, L. Li, G. Chen, Z. Wang, H. Ren, F. Wu and C. Wu, 3D Electronic Channels Wrapped Large-Sized Na3V2(PO4)(3) as Flexible Electrode for Sodium-Ion Batteries, Small, 2018, 14, 1702864 CrossRef.
- T. Wang, Q. Xi, K. Wang, Z. Zeng, Z. Du, Z. Xu, L. Xie, W. Ai and W. Huang, Covalently binding ultrafine MoS2 particles to N, S co-doped carbon renders excellent Na storage performances, Carbon, 2021, 184, 177–185 CrossRef CAS.
- X.-R. Qi, Y. Liu, L.-L. Ma, B.-X. Hou, H.-W. Zhang, X.-H. Li, Y.-S. Wang, Y.-Q. Hui, R.-X. Wang, C.-Y. Bai, H. Liu, J.-J. Song and X.-X. Zhao, Delicate synthesis of quasi-inverse opal structural Na3V2(PO4)(3)/N-C and Na4MnV(PO4)(3)/N-C as cathode for high-rate sodium-ion batteries, Rare Met., 2022, 41, 1637–1646 CrossRef CAS.
- M. Ren, F. Li, H. Xu, W. Liu, G. Li, M. Li, C. Gao and H. Ma, CoO/CoFe2O4 bi-component nanorod core with S-doped carbon shell as excellent anode for lithium ion battery, J. Alloys Compd., 2018, 737, 442–447 CrossRef CAS.
- S. Cao, W. Shang, G.-L. Li, Z.-F. Lu, X. Wang, Y. Yan, C. Hao, S. Wang and G. Sun, Defect-rich and metal-free N, S co-doped 3D interconnected mesoporous carbon material as an advanced electrocatalyst towards oxygen reduction reaction, Carbon, 2021, 184, 127–135 CrossRef CAS.
- Z. J. Liu, F. F. Zheng, W. W. Xiong, X. G. Li, A. H. Yuan and H. Pang, Strategies to improve electrochemical performances of pristine metal-organic frameworks-based electrodes for lithium/sodium-ion batteries, Smartmat, 2021, 2, 488–518 CrossRef CAS.
- X. L. Wu, Y. G. Guo, J. Su, J. W. Xiong, Y. L. Zhang and L. J. Wan, Carbon-Nanotube-Decorated Nano-LiFePO4 @C Cathode Material with Superior High-Rate and Low-Temperature Performances for Lithium-Ion Batteries, Adv. Energy Mater., 2013, 3, 1155–1160 CrossRef CAS.
- Y. Zhang, Z. H. Zhang, Y. K. Tang, D. Z. Jia, Y. D. Huang, W. K. Pang, Z. P. Guo and Z. Zhou, LiFePO4 Particles Embedded in Fast Bifunctional Conductor rGO&C@Li3V2(PO4)(3) Nanosheets as Cathodes for High-Performance Li-Ion Hybrid Capacitors, Adv. Funct. Mater., 2019, 29, 1807895 CrossRef.
- H. Xiong, G. Sun, Z. Liu, L. Zhang, L. Li, W. Zhang, F. Du and Z.-A. Qiao, Polymer Stabilized Droplet Templating towards Tunable Hierarchical Porosity in Single Crystalline Na3V2(PO4)(3) for Enhanced Sodium-Ion Storage, Angew. Chem., Int. Ed., 2021, 60, 10334–10341 CrossRef CAS PubMed.
- Y. N. Xu, Q. L. Wei, C. Xu, Q. D. Li, Q. Y. An, P. F. Zhang, J. Z. Sheng, L. Zhou and L. Q. Mai, Layer-by-Layer Na3V2(PO4)(3) Embedded in Reduced Graphene Oxide as Superior Rate and Ultralong-Life Sodium-Ion Battery Cathode, Adv. Energy Mater., 2016, 6, 1600389 CrossRef.
- K. Kretschmer, B. Sun, J. Zhang, X. Xie, H. Liu and G. Wang, 3D Interconnected Carbon Fiber Network-Enabled Ultralong Life Na3V2(PO4)3@Carbon Paper Cathode for Sodium-Ion Batteries, Small, 2017, 13, 1603318 CrossRef PubMed.
- D. Wang, Q. Li, C. Han, Q. Lu, Z. Xing and X. Yang, Atomic and electronic modulation of self-supported nickel-vanadium layered double hydroxide to accelerate water splitting kinetics, Nat. Commun., 2019, 10, 1–12 CrossRef.
- Y. Wang, S. Wei, Z.-H. Qi, S. Chen, K. Zhu, H. Ding, Y. Cao, Q. Zhou, C. Wang, P. Zhang, X. Guo, X. Yang, X. Wu and L. Song, Intercalant-induced V t2g orbital occupation in vanadium oxide cathode toward fast-charging aqueous zinc-ion batteries, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2217208120 CrossRef CAS.
- W. Sigle, Analytical transmission electron microscopy, Annu. Rev. Mater. Res., 2005, 35, 239 CrossRef CAS.
- K. Li, J. Zhang, D. Lin, D.-W. Wang, B. Li, W. Lv, S. Sun, Y.-B. He, F. Kang, Q.-H. Yang, L. Zhou and T.-Y. Zhang, Evolution of the electrochemical interface in sodium ion batteries with ether electrolytes, Nat. Commun., 2019, 10, 725 CrossRef CAS PubMed.
- Y. Li, F. Wu, Y. Li, M. Liu, X. Feng, Y. Bai and C. Wu, Ether-based electrolytes for sodium ion batteries, Chem. Soc. Rev., 2022, 51, 4484–4536 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05593k |
| This journal is © The Royal Society of Chemistry 2024 |