
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ studies of reversible solid–gas reactions of ethylene responsive silver pyrazolates†
H. V. Rasika
Dias
 *a,
Devaborniny
Parasar
a,
Andrey A.
Yakovenko
*b,
Peter W.
Stephens
*c,
Álvaro
Muñoz-Castro
*d,
Mukundam
Vanga
a,
Pavel
Mykhailiuk
ef and
Evgeniy
Slobodyanyuk
e
*a,
Devaborniny
Parasar
a,
Andrey A.
Yakovenko
*b,
Peter W.
Stephens
*c,
Álvaro
Muñoz-Castro
*d,
Mukundam
Vanga
a,
Pavel
Mykhailiuk
ef and
Evgeniy
Slobodyanyuk
e
aDepartment of Chemistry and Biochemistry, The University of Texas at Arlington, Arlington, Texas 76019, USA. E-mail: dias@uta.edu
bX-Ray Science Division, Advanced Photon Source, Argonne National Laboratory, Argonne, Illinois 60439, USA. E-mail: ayakovenko@anl.gov
cDepartment of Physics and Astronomy, Stony Brook University, Stony Brook, NY 11794-3800, USA. E-mail: peter.stephens@stonybrook.edu
dFacultad de Ingeniería, Arquitectura y Diseño, Universidad San Sebastián, Bellavista 7, Santiago, 8420524, Chile. E-mail: alvaro.munozc@uss.cl
eEnamine Ltd., Winston Churchill Street 78, 02094 Kyiv, Ukraine
fTaras Shevchenko National University of Kyiv, Faculty of Chemistry, Volodymyrska 60, 01601 Kyiv, Ukraine
First published on 29th November 2023
Abstract
Solid–gas reactions and in situ powder X-ray diffraction investigations of trinuclear silver complexes {[3,4,5-(CF3)3Pz]Ag}3 and {[4-Br-3,5-(CF3)2Pz]Ag}3 supported by highly fluorinated pyrazolates reveal that they undergo intricate ethylene-triggered structural transformations in the solid-state producing dinuclear silver–ethylene adducts. Despite the complexity, the chemistry is reversible producing precursor trimers with the loss of ethylene. Less reactive {[3,5-(CF3)2Pz]Ag}3 under ethylene pressure and low-temperature conditions stops at an unusual silver–ethylene complex in the trinuclear state, which could serve as a model for intermediates likely present in more common trimer–dimer reorganizations described above. Complete structural data of three novel silver–ethylene complexes are presented together with a thorough computational analysis of the mechanism.
Introduction
Trinuclear silver(I) complexes of fluorinated pyrazolates have attracted significant interest because many of them show interesting π-acid properties, luminescence, argentophilic contacts, and useful applications.1–8 For example, {[3,5-(CF3)2Pz]Ag}3 (Fig. 1, [Ag–H]3) reported by Dias et al.,9 is a strong π-acid and displays rich π-acid/π-base chemistry with unsaturated hydrocarbons leading to sandwich complexes of various types.10 It also serves as a sensor for arenes such as benzene and toluene.11,12 With o-terphenyl,13 it produces a white light emitting material while the treatment of [Ag–H]3 with phenylacetylene produces a Ag13 cluster with the breakup of the Ag3N6 core.14 The silver complex has also been utilized in the desulfurization of fossil fuels.15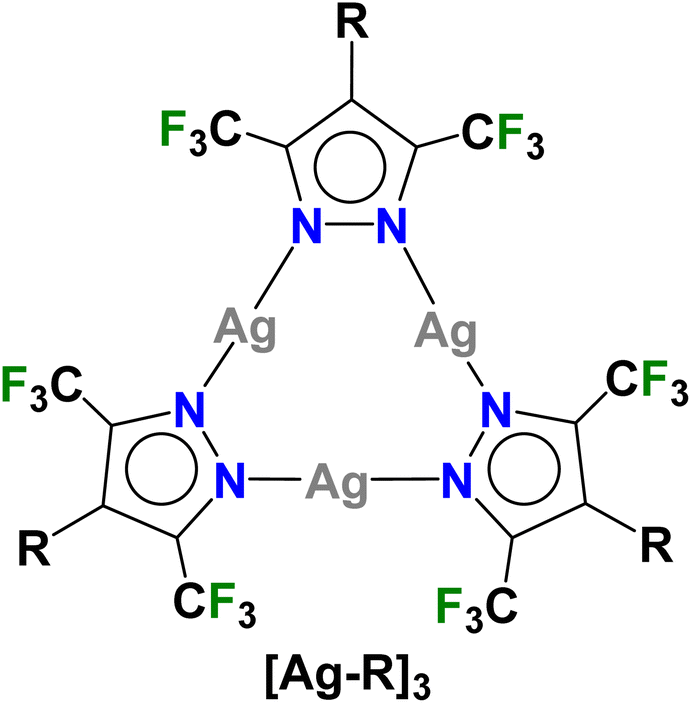 | ||
| Fig. 1 Trinuclear silver(I)-pyrazolates utilized in this work, {[4-R-3,5-(CF3)2Pz]Ag}3 ([Ag–R]3, R = H, Br, CF3). | ||
In contrast to the aromatic hydrocarbons, the chemistry of industrially relevant gaseous hydrocarbons such as ethylene with silver pyrazolates has not been explored. Silver–ethylene complexes are of particular interest since silver is the metal of choice for partial oxidation of ethylene, which is a major industrial process.16,17 They are challenging to stabilize and quite labile due to the relatively weak silver(I)–ethylene interactions.18–24 Reversible binding of ethylene to silver, however, is valuable in applications such as the separation of ethylene from ethylene–ethane mixtures using silver complexes and silver-doped materials.25–27 The copper(I) analogs of [Ag–H]3 such as {[4-R-3,5-(CF3)2Pz]Cu}3 ([Cu–R]3, R = H, CF3) are effective in the selective separation of ethylene from ethane containing mixtures.28,29
Motivated by the fundamental interest and novelty, we embarked on an in-depth study of ethylene chemistry of silver(I) pyrazolates {[4-R-3,5-(CF3)2Pz]Ag}3 ([Ag–R]3, R = H, Br, CF3) with different pyrazolyl ring substituents that also utilizes solid–gas30–33 synthesis and in situ powder X-ray diffraction (PXRD) measurements at 17-BM beamline at the Argonne National Laboratory (ANL) advanced photon source. As evident from the following account, this undertaking was successful and led to the stabilization of an unusual trinuclear silver–ethylene complex in a crystalline state. We also uncovered two unprecedented dinuclear silver–ethylene complexes with bridging pyrazolates, of which, only one could be obtained via a traditional solution method.
Results and discussion
Traditional solution chemistry
The per-fluorinated silver(I) complex {[3,4,5-(CF3)3Pz]Ag}3 ([Ag–CF3]3) was utilized first for this purpose because it possesses powerful Lewis acidic silver sites and is expected to be more reactive towards ethylene compared to the less-fluorinated analogs. The [Ag–CF3]3 was obtained very conveniently via a reaction between the corresponding pyrazole [3,4,5-(CF3)3Pz]H34 and silver(I) oxide. It is a colorless, air-stable solid and has been characterized by several techniques including NMR spectroscopy, and single crystal and powder X-ray crystallography. It crystallizes with a molecule of dichloromethane in the asymmetric unit (Fig. 2, see ESI for additional details and Fig. S7–S8†) and displays short intermolecular Ag⋯Cl and Ag⋯F contacts. There are no argentophilic interactions as observed in [Ag–H]3 or electron-rich systems like {[3,5-(Ph)2Pz]Ag}3 and {[3,5-(i-Pr)2Pz]Ag}3.35–37 | ||
| Fig. 2 Molecular structure of [Ag–CF3]3·CH2Cl2 (top) and [Ag–CF3·(C2H4)]2 (bottom) obtained from solution process and single crystal X-ray diffraction studies. | ||
More importantly, [Ag–CF3]3 reacts with ethylene in CH2Cl2 at low temperatures and produces a product which can be crystallized from the same mixture at −25 °C under an ethylene blanket (Scheme 1). The variable temperature 19F NMR spectroscopic data show that this transformation takes place below −10 °C in CDCl3 (Fig. S4†). The analysis of crystalline solid using single crystal X-ray diffraction reveals that it is a dinuclear species [Ag–CF3·(C2H4)]2 (Fig. 2), and a rare isolable silver–ethylene complex.20,22,23,38–54 Solid samples, however, lose ethylene rapidly upon removal from the ethylene atmosphere at room temperature and return to the ethylene-free trimer form [Ag–CF3]3 (Scheme 1).
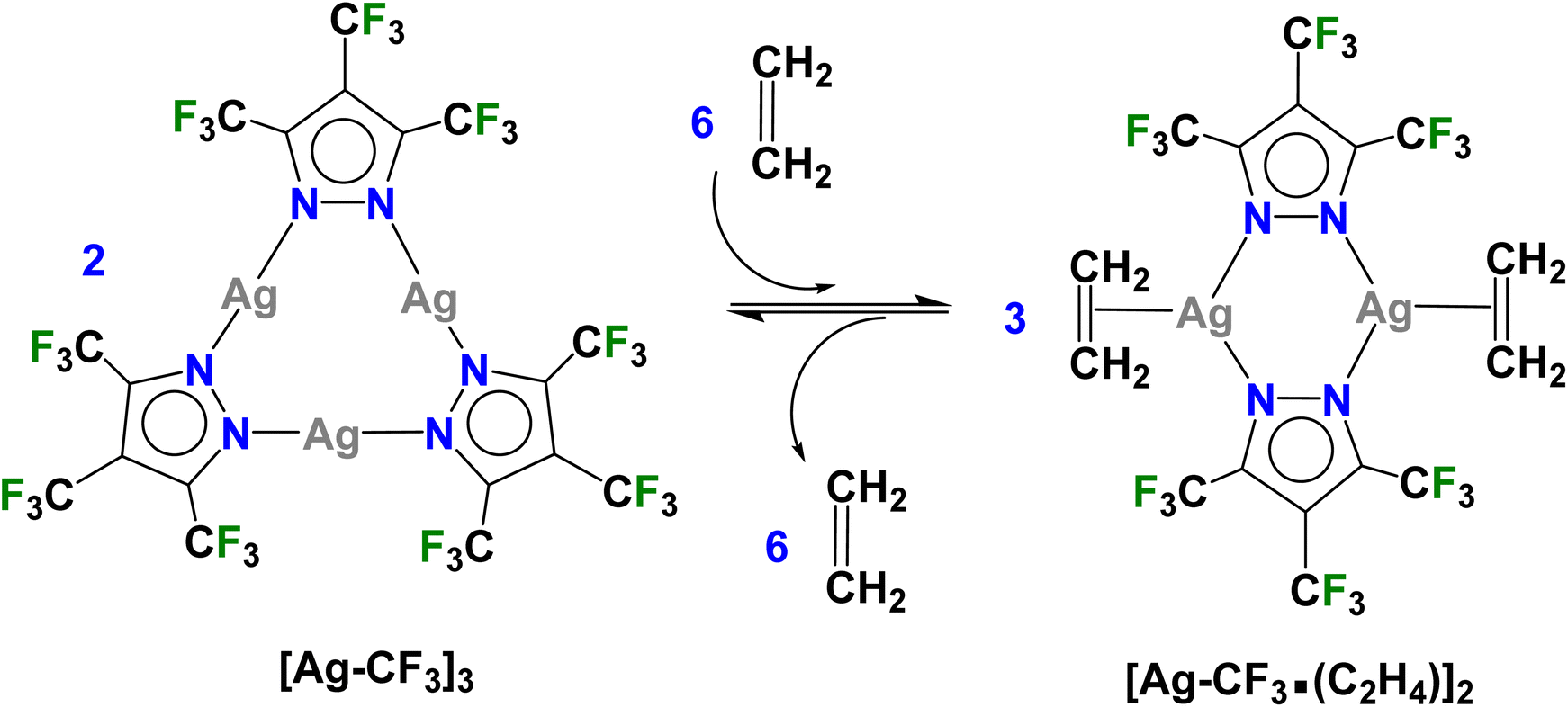 | ||
| Scheme 1 Ethylene responsive trinuclear silver(I)-pyrazolate [Ag–CF3]3 that undergoes structural changes upon addition of ethylene to form [Ag–CF3·(C2H4)]2 and reverts to [Ag–CF3]3 upon removal of ethylene. | ||
There are two chemically similar but crystallographically different molecules of [Ag–CF3·(C2H4)]2 in the asymmetric unit. The silver sites are trigonal planar and Ag2N4 cores adopt a boat shape. Although there are no analogous dinuclear silver–ethylene complexes for a direct comparison, a few silver–ethylene complexes such as [PhB(3-(CF3)Pz)3]Ag(C2H4)41 and {[H2C(3,5-(CF3)2Pz)2]Ag(C2H4)}[SbF6]20 with a three coordinate silver sites supported by N-donor ligands are known. The average Ag–N (2.231 Å) and Ag–C (2.282 Å) distances of [Ag–CF3·(C2H4)]2 are similar to those observed in [PhB(3-(CF3)Pz)3]Ag(C2H4) (av. Ag–N and Ag–C are 2.261 and 2.264 Å, respectively).
Next, we focussed on the related {[3,5-(CF3)2Pz]Ag}3 ([Ag–H]3),9 which is a molecule based on less fluorinated pyrazolate possessing relatively less electrophilic silver sites. Our attempts to observe the silver–ethylene complex from a reaction between [Ag–H]3 and ethylene in CH2Cl2 solution were unsuccessful even at −50 °C. It is understandable since ethylene–silver bonds in general are quite weak while the Ag–N bonds in [Ag–H]3 are relatively strong considering that it features a better electron-donating pyrazolate55 than the one present in [Ag–CF3]3.
In situ solid–gas chemistry
We then decided to investigate these processes, using solid materials and study the progress of the reaction “live” using in situ PXRD at ANL synchrotron beamline. Recent developments show that in situ, in crystallo, and solid–gas chemistry are valuable techniques that enable synthesis and characterization of organometallic species that are difficult or impossible to observe under solution-phase conditions.30–33 Remarkably, crystals of [Ag–CF3]3 upon exposure to ethylene (3–5 bar at 295 K, Fig. S10†), converted smoothly to the same dinuclear silver–ethylene complex [Ag–CF3·(C2H4)]2 (Fig. S13†), mimicking process that occurs in solution. The PXRD based molecular structure of the solid–gas generated [Ag–CF3·(C2H4)]2 is very similar to that obtained from traditional solution chemistry (and single crystal X-ray crystallography, Fig. 2). It is a reversible process (as in the solution) and affords ethylene free precursor [Ag–CF3]3 (Fig. 3) upon purging crystalline [Ag–CF3·(C2H4)]2 with helium at 295 K (Fig. S11 and S14†). Furthermore, these solid–gas reactions, despite the complexity and break-up and formation of several bonds and rearrangement of molecular fragments, are quite fast as evident from the PXRD patterns. Although the progress of both the forward and reverse reaction involving [Ag–CF3]3 can be followed using in situ PXRD, the trimer–dimer transition under the conditions noted above generates the products directly with no evidence of crystalline phases attributable to intermediates. | ||
| Fig. 3 Molecular structure of [Ag–CF3]3 obtained by in situ powder X-ray diffraction studies of the materials from solid–gas chemistry. | ||
To see if we can detect transient species, we proceeded with in situ studies of the less reactive [Ag–H]3 with ethylene. In contrast to [Ag–CF3]3, the reaction of solid [Ag–H]3 with ethylene did not proceed at 295 K even under high ethylene pressure up to 60 bar (ESI Fig. S16†), nor when cooled to 173 K under ∼1 bar of ethylene flow. However, to our delight, the solid–gas reaction proceeded as we lowered the temperature of polycrystalline [Ag–H]3 while subjecting the sample to higher ethylene pressure. Specifically, the transformation was evident from the in situ PXRD experiment as the PXRD lines of [Ag–H]3 started to disappear around 223 K at 10 bar (or 206 K at 5 bar) of ethylene with the generation of a new crystalline phase (Fig. S17 and S19†). This new phase does not change even upon further cooling to 173 K under ethylene. The process of ethylene uptake by [Ag–H]3 is reversible, and the product converts back to ethylene free [Ag–H]3 upon warming to about 262 K even under 10 bar of ethylene (Fig. S18†). The PXRD data analysis revealed the structure of the product (illustrated in Scheme 2), which turned out to be not the dinuclear species encountered with [Ag–CF3]3, but an unusual silver–ethylene complex {[3,5-(CF3)2Pz]Ag(C2H4)}3 ([Ag–H·(C2H4)]3) that retains the trinuclear form.
 | ||
| Scheme 2 Reaction of [Ag–H]3 with 10 bar ethylene below temperature of 223 K (or 5 bar of ethylene below 206 K) leading the trinuclear silver–ethylene complex {[3,5-(CF3)2Pz]Ag(C2H4)}3 ([Ag–H·(C2H4)]3) with a nine-membered Ag3N6 core. | ||
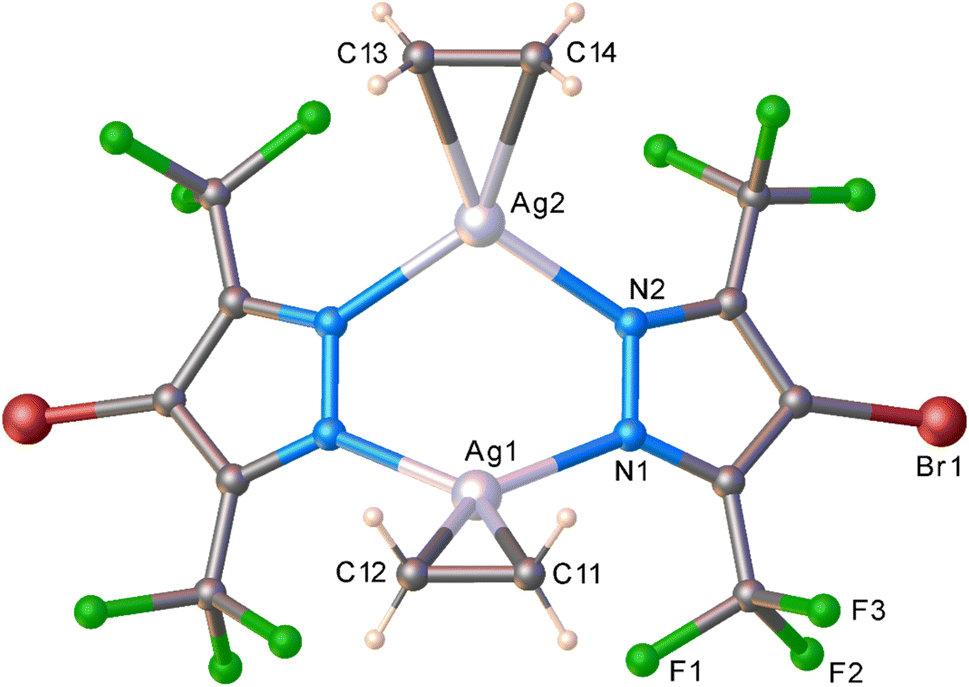
The molecular structure of this unprecedented species [Ag–H·(C2H4)]3 is illustrated in Fig. 4 (and S23†). It is a trinuclear silver complex featuring a nine-membered Ag3N6 metallacycle, and three trigonal-planar silver–ethylene sites. The Ag3N6 core of [Ag–H·(C2H4)]3 displays significant puckering compared to the planar configuration found in [Ag–H]3 (and the related [Ag–CF3]3, see Fig. 3).9 This large deviation from planarity is a result of the interaction of ethylene with silver sites from opposite faces, but the interactions are perhaps not strong enough to break the Ag–N bonds at low-temperature conditions. The compound [Ag–H·(C2H4)]3 may possibly be a model for a likely intermediate present in more facile reaction of [Ag–CF3]3 with ethylene, just prior to the breakup of trimers to produce the corresponding dinuclear metal–ethylene complexes.
 | ||
| Fig. 4 In situ PXRD based molecular structure of the silver–ethylene [Ag–H·(C2H4)]3 complex generated by in situ solid–gas chemistry (top). Selected atoms showing only the Ag3N6(C2H4)3 moiety and the distorted Ag3N6 core (bottom). | ||
Postulating that this ethylene loaded trimer phase [Ag–H·(C2H4)]3 might be a transition state between unloaded trimer and loaded dimer phases observed for other metal pyrazolates, experiments were carried out at even higher pressures and lower temperatures to see if a further transition to a loaded dimer “[Ag–H·(C2H4)]2” could be observed. First, the in situ PXRD data were collected at 45 bar of C2H4 from room temperature down to 110 K (just above the freezing point of C2H4). The pressure was then increased to 70 bar of ethylene and the sample warmed to room temperature (which led to [Ag–H]3 formation). We did not observe any evidence of new crystalline phase under both these conditions (see Fig. S25†).
Encouraged by the success with [Ag–H]3 that led to the characterization of a rare species in the ethylene bound yet pre trimer → dimer transformation stage, we also probed the chemistry of [Ag–Br]3 with ethylene. Note that these planar, trinuclear metal adducts display interesting and different extended structures and therefore, the outcome of solid-state chemistry with ethylene is not necessarily predictable through extrapolation. For example, in contrast to [Ag–H]3 which crystallizes forming zig-zag columns with argentophilic interactions,9,56[Ag–Br]3 trimers form extended structures with inter-trimer Ag⋯Br contacts57 (while [Ag–CF3]3 reported here shows inter-trimer Ag⋯F interactions between trimers).
Traditional solution chemistry with ∼1 bar ethylene thus far did not yield an isolable silver–ethylene complex from [Ag–Br]3 in CH2Cl2. The in situ PXRD data of the solid–gas reaction of polycrystalline [Ag–Br]3 also do not show any phase changes even at 173 K under flow of ethylene (∼1 bar). However, at 10 bar of ethylene, a notable change was observed at 220 K (Fig. S26†). Data analysis indicated that it directly progressed to the dimer stage producing {[4-Br-3,5-(CF3)2Pz]Ag(C2H4)}2 ([Ag–Br·(C2H4)]2) (Fig. 5 and S30†), which is in contrast to the [Ag–H]3 chemistry but similar to that observed with [Ag–CF3]3 and ethylene. Upon warming, [Ag–Br·(C2H4)]2 loses ethylene and returns to the precursor trimer at 295 K, even under 10 bar of ethylene (Fig. S27 and S28†). The dinuclear silver(I)–ethylene complex [Ag–Br·(C2H4)]2 adopts a slightly deeper a boat configuration with a closer Ag⋯Ag separation (3.35(2) Å) within the six-membered Ag2N4 core relative to that observed with [Ag–CF3·(C2H4)]2 (which has Ag⋯Ag separations at 3.49(2) Å). Ethylene ligands are η2-bonded to silver sites, as expected. Overall, trinuclear [Ag–Br]3 and [Ag–CF3]3 show unprecedented ethylene triggered solid–gas chemistry leading to dinuclear silver–ethylene complexes featuring Ag2N4 cores while [Ag–H]3 enabled the observation of an ethylene bound silver trimer that retains the metalacyclic Ag3N6 core.
 | ||
| Fig. 5 Molecular structure of in situ generated [Ag–Br·(C2H4)]2 based on powder X-ray diffraction data. | ||
Computational study
In order to further understand ethylene driven molecular reorganization processes described above, we undertook a detailed computational study of ethylene reactions of [Ag–CF3]3, [Ag–Br]3 and [Ag–H]3 (Fig. 6). The Gibbs free energy profiles at 298 K were computed to uncover reaction paths at room temperature in the molecular calculations at the TZ2P/BP86-D3 level of theory. For this purpose, thermodynamic quantities from vibrational frequencies accounting for enthalpy and entropy changes for the proposed reaction mechanism were obtained (see ESI† for additional details). As the first step (Fig. 6, 1), the formation of an adduct between the trinuclear silver pyrazolate and three molecules of C2H4 was predicted, prior to the deformation of the Ag3N6 core as a transition state (TS1), which is further relaxed to the intermediate 2 (such as [Ag–H·(C2H4)]3). The formation of 2 involves a computed Gibbs free energy (298.15 K) of −16.8, −15.9 and −13.7 kcal mol−1, respectively (Table S10†), in comparison to the initial reactants, for [Ag–CF3]3, [Ag–Br]3 and [Ag–H]3. The observed deformation of the Ag3N6 core from precursors to the intermediates is not favored in the absence of ethylene, by about 50 kcal mol−1 (Table S8†) for all the species, showing that such processes is driven exclusively by the initial coordination of C2H4 to the bare Ag3N6 core (step 1). The process of forming [Ag–R·(C2H4)]3 from [Ag–R]3 and gaseous ethylene (R = H, Br, CF3) is not favorable entropically and can be influenced significantly by lower temperatures. Thus, intermediate 2 is more likely to be characterized, especially at lower temperature, as experimentally realized in this work in the reaction involving [Ag–H]3. | ||
| Fig. 6 Gibbs free energy diagram for the proposed mechanism for dimer formation, involving [Ag–H]3, [Ag–Br]3, and [Ag–CF3]3 at 298 K. Values given per [Ag–R]3 unit in kcal mol−1 (R = H, Br, or CF3). | ||
Intermediate 2 is a key step prior to the trimer → dimer transformation. After the formation and relaxation of this intermediate, the next step is to release one [Ag–R·(C2H4)] unit (i.e., ethylene bound metal-pyrazolate) given as the second transition state (TS2), which is the rate-determinant step leading to the dimer. Calculations of the bonding energy of Ag2N4–AgN2 (Table S9†) for –H, –Br and –CF3, indicate that it is easier to break-up [Ag–CF3]3 and [Ag–Br]3 species (−64.3 and −65.2 kcal mol−1, respectively), in comparison to [Ag–H]3 counterpart (−83.8 kcal mol−1). From the Gibbs free energy profiles (Table S10 and Fig. S31†), the activation barriers related to the 1 → TS1 process can be evaluated, which amount to 5.0, 4.7, and 5.6 kcal mol−1 for –CF3, –Br, and –H at 298 K, respectively. For the 2 → TS2 process, the related values are 10.8, 13.4, and 14.9 kcal mol−1, denoting a slightly larger activation barrier for the [Ag–H]3 complex.
In the final step, the loss of a [Ag–R·(C2H4)] unit from [Ag–R·(C2H4)]3, leads to the formation of one dimer species [Ag–R·(C2H4)]2 (TS2), where the released unit further aggregates with another [Ag–R·(C2H4)] fragment from a parallel reaction, resulting in the formation of a second dimer species (3). Calculated Gibbs free energy for step 3 amounts to −30.1, −25.5, and −24.1 kcal mol−1 for [Ag–CF3]3, [Ag–Br]3 and [Ag–H]3, respectively. Overall, [Ag–H·(C2H4)]2 formation is slightly less energetically favorable process, while [Ag–CF3·(C2H4)]2 formation is the most facile, which is consistent with the experimental observations, and denoted by the slightly less stabilized transition states and activation barriers, in addition to the bonding energy of Ag2N4–AgN2 fragments prior formation of TS2. The formation of trinuclear-tris-ethylene intermediate 2 is favored at lower temperatures but the experimental conditions must be just right to trap this species before it breaks-up to even more energetically favorable dimers 3. The silver(I) and [3,5-(CF3)2Pz]− ligand combination provides the ideal ingradients to trap the elusive species [Ag–H·(C2H4)]3.
Conclusion
In summary, after a careful investigation that involved strategic variations of pyrazolyl ring substituents and solid–gas synthesis under different temperature–pressure combinations, and synchrotron based, in situ PXRD, we successfully trapped and structurally characterized a remarkable, trinuclear silver–ethylene complex {[3,5-(CF3)2Pz]Ag(C2H4)}3 ([Ag–H·(C2H4)]3) with a severely distorted, yet intact Ag3N6 core, that can be viewed as a model for fleeting intermediates likely exist in ethylene driven, trimer–dimer transformations observed in related [Ag–CF3]3 and [Ag–Br]3 systems. Furthermore, this study reveals for the first time, ethylene triggered structural transformations of trinuclear silver(I) pyrazolates in the solid-state leading to dinuclear species {[3,4,5-(CF3)3Pz]Ag(C2H4)}2 ([Ag–CF3·(C2H4)]2) and {[4-Br-3,5-(CF3)2Pz]Ag(C2H4)}2 ([Ag–Br·(C2H4)]2), and molecular structures of two rare dinuclear, silver–ethylene complexes. This work also demonstrates the power of in situ synthesis and ab initio PXRD structure determination over traditional solution chemistry for the isolation and study of labile species. Computational studies indicated that the silver(I) and [3,5-(CF3)2Pz]− ligand combination provides the ideal ingradients to stabilize [Ag–H·(C2H4)]3. Further in situ, solid–gas studies guided by computational work in search of rare species from other metal complexes are currently underway.Data availability
All data associated with this article can be found in the ESI.†Author contributions
Conceived the original idea and project administration (H. V. R. D.), methodology and experimental design (H. V. R. D., A. A. Y., P. W. S., P. M., A. M.-C., E. S.), chemical syntheses, spectroscopy, and data analysis (D. P., M. V., E. S.), DFT calculations and data analysis (A. M.-C.), single and powder XRD and solid–gas synthesis (H. V. R. D., A. A. Y., P. W. S.), writing and proof-reading process (all authors).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under grant number (CHE-1954456, H. V. R. D.). A. M.-C. thanks the financial support from FONDECYT 1221676. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CH11357.References
- J. Zheng, Z. Lu, K. Wu, G.-H. Ning and D. Li, Chem. Rev., 2020, 120, 9675–9742 CrossRef CAS PubMed.
- J. Luo, X. Luo, M. Xie, H.-Z. Li, H. Duan, H.-G. Zhou, R.-J. Wei, G.-H. Ning and D. Li, Nat. Commun., 2022, 13, 7771 CrossRef CAS.
- R. Galassi, M. A. Rawashdeh-Omary, H. V. R. Dias and M. A. Omary, Comments Inorg. Chem., 2019, 39, 287–348 CrossRef CAS.
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS.
- J. Zheng, H. Yang, M. Xie and D. Li, Chem. Commun., 2019, 55, 7134–7146 RSC.
- M. A. Omary, M. A. Rawashdeh-Omary, M. W. A. Gonser, O. Elbjeirami, T. Grimes, T. R. Cundari, H. V. K. Diyabalanage, C. S. P. Gamage and H. V. R. Dias, Inorg. Chem., 2005, 44, 8200–8210 CrossRef CAS PubMed.
- A. A. Titov, O. A. Filippov, L. M. Epstein, N. V. Belkova and E. S. Shubina, Inorg. Chim. Acta, 2018, 470, 22–35 CrossRef CAS.
- R. Hahn, F. Bohle, W. Fang, A. Walther, S. Grimme and B. Esser, J. Am. Chem. Soc., 2018, 140, 17932–17944 CrossRef CAS PubMed.
- H. V. R. Dias, S. A. Polach and Z. Wang, J. Fluorine Chem., 2000, 103, 163–169 CrossRef CAS.
- H. V. R. Dias and C. S. P. Gamage, Angew. Chem., Int. Ed., 2007, 46, 2192–2194 CrossRef CAS PubMed.
- M. A. Rawashdeh-Omary, M. D. Rashdan, S. Dharanipathi, O. Elbjeirami, P. Ramesh and H. V. R. Dias, Chem. Commun., 2011, 47, 1160–1162 RSC.
- M. A. Omary, O. Elbjeirami, C. S. P. Gamage, K. M. Sherman and H. V. R. Dias, Inorg. Chem., 2009, 48, 1784–1786 CrossRef CAS PubMed.
- S.-Z. Zhan, F. Ding, X.-W. Liu, G.-H. Zhang, J. Zheng and D. Li, Inorg. Chem., 2019, 58, 12516–12520 CrossRef CAS.
- S.-K. Peng, Z. Lu, M. Xie, Y.-L. Huang, D. Luo, J.-N. Wang, X.-W. Zhu, X. Li, X.-P. Zhou and D. Li, Chem. Commun., 2020, 56, 4789–4792 RSC.
- R. Liu, W. Zhang, D. Wei, J.-H. Chen, S. W. Ng and G. Yang, Dalton Trans., 2019, 48, 16162–16166 RSC.
- H. Kestenbaum, A. L. de Oliveira, W. Schmidt, F. Schüth, W. Ehrfeld, K. Gebauer, H. Löwe, T. Richter, D. Lebiedz, I. Untiedt and H. Züchner, Ind. Eng. Chem. Res., 2002, 41, 710–719 CrossRef CAS.
- J.-X. Liu, S. Lu, S.-B. Ann and S. Linic, ACS Catal., 2023, 13, 8955–8962 CrossRef CAS.
- R. H. Hertwig, W. Koch, D. Schröder, H. Schwarz, J. Hrušák and P. Schwerdtfeger, J. Phys. Chem., 1996, 100, 12253–12260 CrossRef CAS.
- H. V. R. Dias and C. J. Lovely, Chem. Rev., 2008, 108, 3223–3238 CrossRef CAS PubMed.
- J. Mehara, B. T. Watson, A. Noonikara-Poyil, A. O. Zacharias, J. Roithová and H. V. R. Dias, Chem.–Eur. J., 2022, 28, e202103984 CrossRef CAS PubMed.
- M. S. Nechaev, V. M. Rayón and G. Frenking, J. Phys. Chem. A, 2004, 108, 3134–3142 CrossRef CAS.
- K. Klimovica, K. Kirschbaum and O. Daugulis, Organometallics, 2016, 35, 2938–2943 CrossRef CAS PubMed.
- I. Krossing and A. Reisinger, Angew. Chem., Int. Ed., 2003, 42, 5725–5728 CrossRef CAS.
- H. V. R. Dias and J. Wu, Eur. J. Inorg. Chem., 2008, 509–522 CrossRef CAS.
- Y. Yin, Z. Zhang, C. Xu, H. Wu, L. Shi, S. Wang, X. Xu, A. Yuan, S. Wang and H. Sun, ACS Sustainable Chem. Eng., 2020, 8, 823–830 CrossRef CAS.
- R. B. Eldridge, Ind. Eng. Chem. Res., 1993, 32, 2208–2212 CrossRef.
- M. G. Cowan, W. M. McDanel, H. H. Funke, Y. Kohno, D. L. Gin and R. D. Noble, Angew. Chem., Int. Ed., 2015, 54, 5740–5743 CrossRef CAS PubMed.
- D. Parasar, A. H. Elashkar, A. A. Yakovenko, N. B. Jayaratna, B. L. Edwards, S. G. Telfer, H. V. R. Dias and M. G. Cowan, Angew. Chem., Int. Ed., 2020, 59, 21001–21006 CrossRef CAS PubMed.
- N. B. Jayaratna, M. G. Cowan, D. Parasar, H. H. Funke, J. Reibenspies, P. K. Mykhailiuk, O. Artamonov, R. D. Noble and H. V. R. Dias, Angew. Chem., Int. Ed., 2018, 57, 16442–16446 CrossRef CAS PubMed.
- K. A. Reid and D. C. Powers, Chem. Commun., 2021, 57, 4993–5003 RSC.
- F. M. Chadwick, T. Krämer, T. Gutmann, N. H. Rees, A. L. Thompson, A. J. Edwards, G. Buntkowsky, S. A. Macgregor and A. S. Weller, J. Am. Chem. Soc., 2016, 138, 13369–13378 CrossRef CAS PubMed.
- F. M. Chadwick, A. I. McKay, A. J. Martinez-Martinez, N. H. Rees, T. Krämer, S. A. Macgregor and A. S. Weller, Chem. Sci., 2017, 8, 6014–6029 RSC.
- J. C. Goodall, M. A. Sajjad, E. A. Thompson, S. J. Page, A. M. Kerrigan, H. T. Jenkins, J. M. Lynam, S. A. Macgregor and A. S. Weller, Chem. Commun., 2023, 59, 10749–10752 RSC.
- E. Y. Slobodyanyuk, O. S. Artamonov, O. V. Shishkin and P. K. Mykhailiuk, Eur. J. Org Chem., 2014, 2487–2495 CrossRef CAS.
- A. A. Mohamed, L. M. Pérez and J. P. Fackler, Inorg. Chim. Acta, 2005, 358, 1657–1662 CrossRef CAS.
- H. V. R. Dias and H. V. K. Diyabalanage, Polyhedron, 2006, 25, 1655–1661 CrossRef CAS.
- Y. Morishima, D. J. Young and K. Fujisawa, Dalton Trans., 2014, 43, 15915–15928 RSC.
- H. V. R. Dias, Z. Wang and W. Jin, Inorg. Chem., 1997, 36, 6205–6215 CrossRef CAS.
- H. V. R. Dias and X. Wang, Dalton Trans., 2005, 2985–2987 RSC.
- H. A. Chiong and O. Daugulis, Organometallics, 2006, 25, 4054–4057 CrossRef CAS.
- H. V. R. Dias, J. Wu, X. Wang and K. Rangan, Inorg. Chem., 2007, 46, 1960–1962 CrossRef CAS.
- A. Reisinger, N. Trapp and I. Krossing, Organometallics, 2007, 26, 2096–2105 CrossRef CAS.
- S. Uchida, R. Kawamoto, H. Tagami, Y. Nakagawa and N. Mizuno, J. Am. Chem. Soc., 2008, 130, 12370–12376 CrossRef CAS PubMed.
- X. Kou and H. V. R. Dias, Dalton Trans., 2009, 7529–7536 RSC.
- A. Reisinger, N. Trapp, C. Knapp, D. Himmel, F. Breher, H. Rüegger and I. Krossing, Chem.–Eur. J., 2009, 15, 9505–9520 CrossRef CAS PubMed.
- H. V. R. Dias and J. Wu, Organometallics, 2012, 31, 1511–1517 CrossRef CAS.
- N. B. Jayaratna, I. I. Gerus, R. V. Mironets, P. K. Mykhailiuk, M. Yousufuddin and H. V. R. Dias, Inorg. Chem., 2013, 52, 1691–1693 CrossRef CAS PubMed.
- M. Stricker, B. Oelkers, C. P. Rosenau and J. Sundermeyer, Chem.–Eur. J., 2013, 19, 1042–1057 CrossRef CAS PubMed.
- S. G. Ridlen, J. Wu, N. V. Kulkarni and H. V. R. Dias, Eur. J. Inorg. Chem., 2016, 2573–2580 CrossRef CAS.
- M. Navarro, J. Miranda-Pizarro, J. J. Moreno, C. Navarro-Gilabert, I. Fernández and J. Campos, Chem. Commun., 2021, 57, 9280–9283 RSC.
- M. Vanga, A. Muñoz-Castro and H. V. R. Dias, Dalton Trans., 2022, 51, 1308–1312 RSC.
- B. T. Watson, M. Vanga, A. Noonikara-Poyil, A. Muñoz-Castro and H. V. R. Dias, Inorg. Chem., 2023, 62, 1636–1648 CrossRef CAS PubMed.
- M. Fianchini, C. F. Campana, B. Chilukuri, T. R. Cundari, V. Petricek and H. V. R. Dias, Organometallics, 2013, 32, 3034–3041 CrossRef CAS.
- H. V. R. Dias and M. Fianchini, Angew. Chem., Int. Ed., 2007, 46, 2188–2191 CrossRef CAS PubMed.
- I. I. Gerus, R. X. Mironetz, I. S. Kondratov, A. V. Bezdudny, Y. V. Dmytriv, O. V. Shishkin, V. S. Starova, O. A. Zaporozhets, A. A. Tolmachev and P. K. Mykhailiuk, J. Org. Chem., 2012, 77, 47–56 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- C. V. Hettiarachchi, M. A. Rawashdeh-Omary, D. Korir, J. Kohistani, M. Yousufuddin and H. V. R. Dias, Inorg. Chem., 2013, 52, 13576–13583 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthesis of [Ag–CF3]3 and [Ag–CF3·(C2H4)]2via solution methods, and the in situ solid phase synthesis of [Ag–CF3·(C2H4)]2, [Ag–H·(C2H4)]3 and [Ag–Br·(C2H4)]2, and the reverse processes, and molecular structure determinations using crystal X-ray crystallography and PXRD. Computational analysis of the ethylene uptake by silver pyrazolates, reaction pathways, additional figures, and references. CCDC 2256899, 2256900, 2266816–2266818 and 2267047. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04182d |
| This journal is © The Royal Society of Chemistry 2024 |