
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photosensitizer-singlet oxygen sensor conjugated silica nanoparticles for photodynamic therapy and bioimaging†
Jeladhara
Sobhanan
 ab,
Kenji
Ono
c,
Takuya
Okamoto
ad,
Makoto
Sawada
c,
Paul S.
Weiss
e and
Vasudevanpillai
Biju
*ad
ab,
Kenji
Ono
c,
Takuya
Okamoto
ad,
Makoto
Sawada
c,
Paul S.
Weiss
e and
Vasudevanpillai
Biju
*ad
aGraduate School of Environmental Science, Hokkaido University, Sapporo, Hokkaido 060-0810, Japan. E-mail: biju@es.hokudai.ac.jp
bDepartment of Chemistry, Rice University, Houston, Texas 77005, USA
cResearch Institute of Environmental Medicine, Nagoya University, Nagoya 464-8601, Japan
dResearch Institute for Electronic Science, Hokkaido University, Sapporo, Hokkaido 001-0020, Japan
eCalifornia NanoSystems Institute and the Departments of Chemistry and Biochemistry, Bioengineering, and Materials Science and Engineering, University of California Los Angeles, CA 90095-1487, USA
First published on 14th December 2023
Abstract
Intracellular singlet oxygen (1O2) generation and detection help optimize the outcome of photodynamic therapy (PDT). Theranostics programmed for on-demand phototriggered 1O2 release and bioimaging have great potential to transform PDT. We demonstrate an ultrasensitive fluorescence turn-on sensor-sensitizer-RGD peptide-silica nanoarchitecture and its 1O2 generation–releasing–storing–sensing properties at the single-particle level or in living cells. The sensor and sensitizer in the nanoarchitecture are an aminomethyl anthracene (AMA)-coumarin dyad and a porphyrin or CdSe/ZnS quantum dots (QDs), respectively. The AMA in the dyad quantitatively quenches the fluorescence of coumarin by intramolecular electron transfer, the porphyrin or QD moiety generates 1O2, and the RGD peptide facilitates intracellular delivery. The small size, below 200 nm, as verified by scanning electron microscopy and differential light scattering measurements, of the architecture within the 1O2 diffusion length enables fast and efficient intracellular fluorescence switching by the tandem ultraviolet (UV)-visible or visible-near-infrared (NIR) photo-triggering. While the red emission and 1O2 generation by the porphyrin are continually turned on, the blue emission of coumarin is uncaged into 230-fold intensity enhancement by on-demand photo-triggering. The 1O2 production and release by the nanoarchitecture enable spectro-temporally controlled cell imaging and apoptotic cell death; the latter is verified from cytotoxic data under dark and phototriggering conditions. Furthermore, the bioimaging potential of the TCPP-based nanoarchitecture is examined in vivo in B6 mice.
Introduction
Reactive oxygen species (ROS)-mediated photodynamic therapy (PDT) is a promising tool in cancer management. The unique oxidizing power of metastable singlet oxygen (1O2) received considerable attention in synthetic chemistry, environmental remediation, and therapeutics.1–3 Notably, 1O2 released by photosensitizer (PS) drugs triggers the disruption of the tumor microvasculature, oxidative stress-induced cellular damage, and impaired membrane transport functions.4–6 The cytotoxic 1O2 produced by PS drugs precisely delivered in a tumor milieu avoids or minimizes undesired toxicity to normal cells and tissues due to systemic overdose or nonspecific drug localization. Furthermore, the nonspecific toxicity of PS drugs is minimized by local phototriggering. Therefore, the detection and quantification of 1O2 produced by a PS drug at the desired location are of utmost significance in PDT. An array of fluorescence (FL) probes built on polycyclic and heterocyclic aromatics has been developed to detect or sense 1O2.7–12 However, the performances of such probes are limited due to inadequate FL turn-on features, low sensitivity, and complex syntheses. Nevertheless, anthracene derivatives dominate as safe 1O2 sensors and carriers, and substituents at the 9,10-positions of anthracene modulate the 1O2 sensitivity.13–17 In addition, the self-limiting factor of 1O2 invokes the hypoxic microenvironment in tumors and thus proliferates the cancer metastasis or builds up a resistance towards PDT.18–22 Therefore, localized generation, controlled supply, and accurate detection of 1O2 using probes with high spectroscopic/microscopic sensitivity are critical for advancing PDT. The co-existence of 1O2 sensing and controlled releasing using a sensor-sensitizer system would help overcome the challenges associated with photooxygenation techniques.23–26Nanomaterials offer myriad advantages over traditional drug delivery systems (DDS). Various photoresponsive, biocompatible nanomaterials have been recently developed for this purpose.27–33 Mesoporous silica nanoparticles (MSN) are among the most promising theranostic platforms for cancer imaging and therapy.34–44 Caged drugs/contrast agents in MSN by chemical functionalization protect their intrinsic properties, promote aqueous solubility, and increase tumoral accumulation.34–43 Also, the mesoporous structure and high surface-to-volume ratios of MSNs increase the drug/contrast agent loading and delivery efficiencies.42,43 Recently, Meng and Nel developed lipid bilayer-coated MSN (silicasomes) for intratumoral delivery of chemotherapeutics such as oxaliplatin, gemcitabine, or paclitaxel to the Kras-derived pancreatic cancer in mice.38,39 Similarly, they studied the antitumor effects of irinotecan-loaded silicasomes in pancreatic and colorectal cancers, utilizing the ability of irinotecan to neutralize the lysosomal acidity.40,41 Likewise, MSN-coupled 1O2 FL probes are widely applied in PDT. For example, Jiao et al. prepared a silica nanocarrier (FSNC) functionalized with the PS protoporphyrin IX, a 2-pyridone derivative, as the 1O2 storage/release unit, and a cyanine derivative as the 1O2 self-monitoring unit for fractional PDT.45 Under light-induced 1O2 generation, the FSNC forms endoperoxide and releases the stored 1O2 under the dark by a cycloreversion mechanism, monitored continuously by the FL bleaching of the cyanine dye. Also, they demonstrated the biocompatibility and bioimaging potentials of NIR-dyes covalently incorporated in silica nanoparticles.46 In another report, they constructed hydrophobic domains in nanosilica using a modified silane coupling agent and preserved the FL of NIR dyes by preventing aggregation.47 A polymeric nanocarrier for enhanced phototherapy is another example.48 Here, dual NIR laser-regulated 1O2 trapping by an anthracene-BODIPY conjugate enabled the sustained 1O2 release. Also, Stang and coworkers developed an organoplatinum(II) supramolecular metallacycle by coordination-driven self-assembly of dipyridyl anthracene donor – Pt(II) acceptor for reversible 1O2 capture and release, which showed high photooxygenation and thermolysis rates.49 Nonetheless, programmed nanomaterials integrating multiple fluorescent PS drugs and fluorogenic sensors for 1O2 storing, sensing, and controlled releasing from a single framework are highly sought after for next-generation PDT.
Here, we describe and demonstrate a sensor-sensitizer-based multifunctional mesoporous silica nanoarchitecture and its on-demand ability to produce, to store, to release, and to sense 1O2 continuously at the single-particle level and in living cells. The nanoscale (200 nm diameter) silica and multiple sensor–sensitizer conjugates within the diffusion length of 1O2 in cells enable efficient 1O2 storing–sensing–releasing even at the single-cell and single-particle levels. Here, a porphyrin [tetrakis(4-carboxyphenyl)porphyrin (TCPP)] and an uncaged coumarin provide the spectrally and visibly distinct bimodal FL to the nanoarchitecture. Indeed, the blue FL is enormously increased (230-fold) with time under photosensitization of the PS. The FL of the coumarin in the nanoarchitecture is quantitatively quenched by lock-in photoinduced intramolecular electron transfer (PIET), which is nearly 100% efficient from an aminomethyl anthracene (AMA). This efficient PIET enables the highest 1O2 sensitivity to the sensor molecule or the nanoarchitecture. The FL intensity enhancement by the one- (UV-vis) or two- (NIR) photon-triggered uncaging (releasing coumarin FL by oxidation of the AMA part) represents 1O2 sensing. Solution-based, single-particle and single-cell experiments demonstrate outstanding 1O2-induced FL turn-on efficiency at a controlled rate for sensor 4 compared to previously reported sensors such as Si-DMA (ca. 10-fold),50 or SOSG (ca. 50-fold).51 Furthermore, an arginine-rich peptide conjugate facilitates cellular uptake of the nanoarchitecture. Along with the intracellular production and release of 1O2 by the nanoarchitecture, the colocalized red FL of the PS and the intense blue FL of the uncaged sensor offer spectrally-resolved imaging and PDT modalities. The cytotoxicity of the nanoarchitecture is evaluated by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay in MCF7 cells under dark and photo-triggering conditions, which showed significant phototoxicity. We selected TCPP considering its carboxylic groups for functionalization and its photophysical properties, such as broad absorption in the UV-vis region, deep red FL (>650 nm), and 1O2 production (10% in water and 4.3% in DMF).52 We prepared a nanoarchitecture analogous to the TCPP-based one using near-infrared (NIR) emitting CdSe/ZnS quantum dots (QD) instead of TCPP, considering the wealth of information about QDs for bioimaging.53–58 Also, we considered the ability of the QDs to generate 1O2.56–58 Further, we examined the in vivo FL imaging potential of the SS-MSNP-RGD nanoarchitecture by applying it to B6 mice intravenously or subcutaneously.
Experimental section
Materials and methods
All chemicals used in this research were analytical grade and used as received unless otherwise stated. Silica nanoparticles and (+) biotin-N-hydroxysuccinimide ester (biotin-NHS) were obtained from Sigma Aldrich. Potassium carbonate (K2CO3) and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) were obtained from FUJIFILM Wako Pure Chemical Corporation, Japan. 7-Amino-4-methyl coumarin, N,N-dioctylamine, 9,10-bis(chloromethyl)anthracene, and TCPP were obtained from Tokyo Chemical Industry (TCI), Japan. Qdot™ 655 (QD655)-streptavidin conjugate, fetal bovine serum (FBS), penicillin/streptomycin (P/S), and trypsin were obtained from ThermoFisher Scientific, USA. RGD-SH (CDCRGDCFC) was purchased from GenScript, Japan. The MTT assay kit was obtained from Rosche (Sigma-Aldrich) and used as described by the manufacturer. Optical densities of MTT samples were measured using a microplate reader (Multiskan Sky TCD, ThermoFisher, USA). All solvents were reagent grade from FUJIFILM Wako Pure Chemical Corporation, Japan.Absorption spectra were recorded using a UV-visible spectrophotometer (Evolution 220, ThermoFisher Scientific), and FL spectra were recorded using a Hitachi F4500 Spectrofluorometer. Nuclear magnetic resonance (NMR) measurements were performed in a JEOL 400 MHz NMR spectrometer. A diode-pumped solid-state (DPSS) 532 nm green laser (Spectraphysics or Shanghai Dream Laser Technology) or a mercury vapor lamp (365 nm) was used for the photoirradiation experiment at the single-particle or ensemble levels. The scanning electron microscope (SEM) images (Fig. 1d) were recorded in a Hitachi HD-2000 microscope operated at 200 kV. Microtoming of cell samples was carried out using an ultramicrotome EM UC7i (Leica-microsystems), and TEM images of the microtomed samples were recorded at 25 kV on a Hitachi field emission (FE) SEM (SU8230) equipped with a STEM detecter. Zeta potentials of the samples were measured in an ELS-Z 2 system (Otsuka Electronics Co., Ltd.). Dynamic light scattering (DLS) experiments were conducted on an FDLS-3000 system (Otsuka Electronics Co., Ltd.) FL images of the B6 mice were obtained using a Maestro small animal imaging system (PerkinElmer).
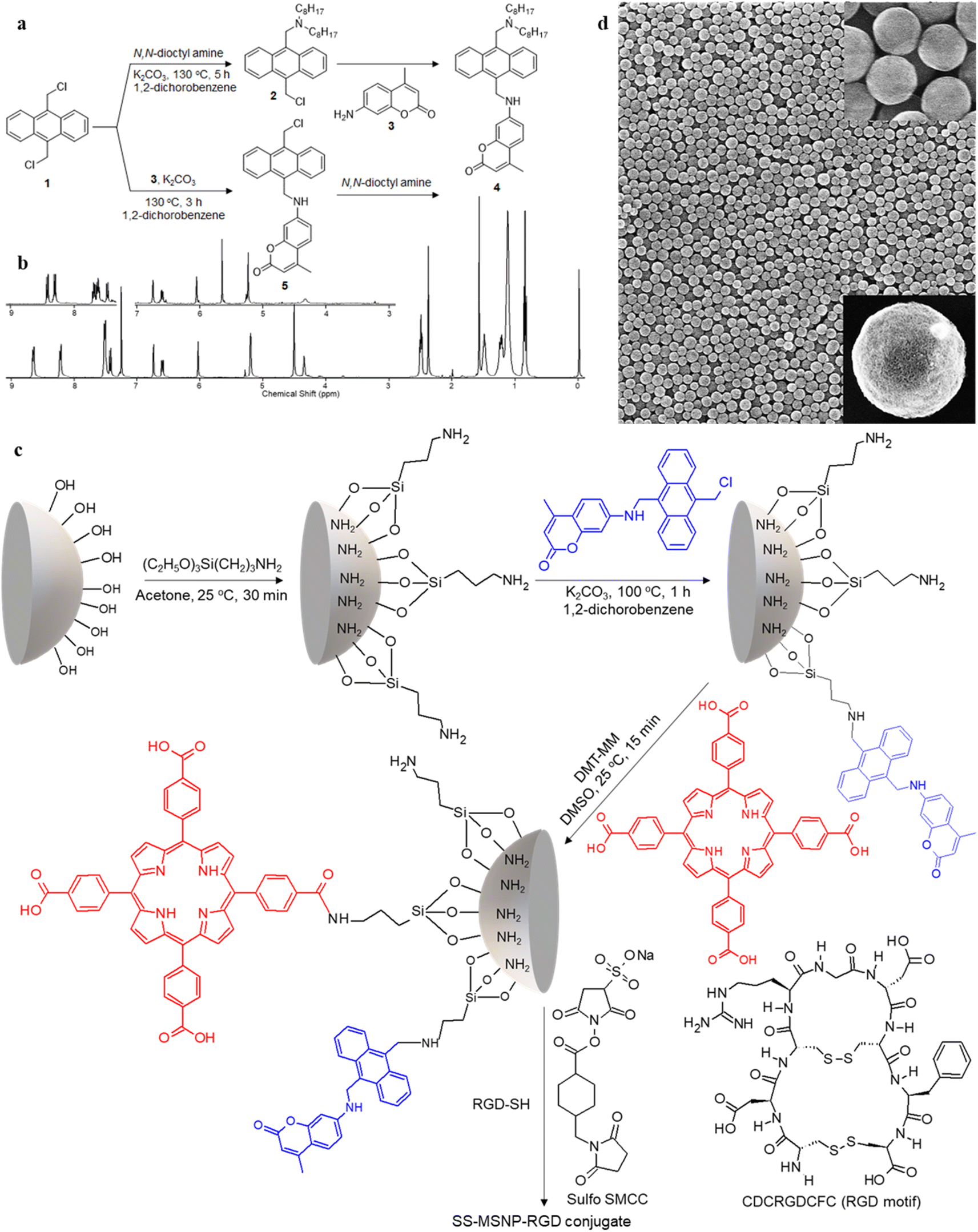 | ||
| Fig. 1 Synthesis of the sensor and sensor-sensitizer-silica nanoarchitectures (SS-MSNPs). A scheme showing the synthesis of (a) sensor 4 and the intermediate 5, and (c) S-MSNP and SS-MSNP conjugates. The structures of the amine-to-thiol cross-linker (Sulfo SMCC), and RGD-SH and its sequence are also shown in ‘c’. (b) 1H NMR spectra of sensor 4, and the intermediate 5 (inset), and (d) scanning electron microscopy images of MSNPs before (low magnification) and after surface modification (inset). | ||
Synthesis and characterization
1H NMR (400 MHz, CDCl3, Fig. 1b): δ = 8.64–8.67 (d, 2H, Ar–H), 8.21–8.23 (d, 2H, Ar–H), 7.51–7.53 (m, 4H, Ar–H), 7.42–7.45 (d, 1H, Ar–H), 6.75 (s, 1H, Ar–H), 6.60–6.62 (d, 1H, Ar–H), 6.04 (s, 1H, allylic), 5.21 (d, 2H, CH2–NH), 4.52 (s, 2H, Ar–CH2–N), 4.36 (t, 1H, NH), 2.51–2.53 (t, 4H, N–CH2), 2.39 (s, 3H, CH3), 1.4–1.55 (q, 4H, N–CH2–C), 1–1.35 (m, 2H, C–CH2–C), 0.5–0.8 (t, 6H, C–CH3). 13C NMR (100 MHz, THF-D8) 14.63, 18.53, 23.73, 28.09, 28.61, 30.49, 33.00, 41.37, 52.39, 54.49, 98.30, 109.99, 110.78, 125.76, 125.94, 127.72, 127.04, 130.38, 131.51, 132.47, 133.36, 153.14, 153.55, 157.70, 161.05. HR-ESI-MS m/z for C42H54O2N2 – [M+] – calcd: 618.8971 found: 619.4229.
1H NMR (400 MHz, CDCl3, Fig. 1b): δ = 8.41–8.43 (d, 2H, Ar–H), 8.29–8.43 (d, 2H, Ar–H), 7.62–7.67 (m, 4H, Ar–H), 7.44–7.46 (d, 1H, Ar–H), 6.76 (s, 1H, Ar–H), 6.61–6.63 (d, 1H, Ar–H), 6.06 (s, 1H, allylic), 5.66 (s, 2H, CH2–Cl), 5.24 (s, 2H, CH2–NH), 4.33 (s, 1H, NH). 13C NMR (400 MHz, CDCl3): δ = 18.72, 31.03, 38.97, 40.72, 98.18, 110.02, 110.46, 111.18, 124.42, 124.76, 125.80, 126.79, 126.89, 128.32, 129.13, 129.67, 129.84, 130.38, 130.48, 151.21, 153.01, 156.20, 161.97. 13C NMR NMR (100 MHz, DMSO-D6) 17.89, 96.41, 107.46, 108.88, 110.24, 124.34, 125.04, 125.90, 126.13, 126.42, 129.08, 129.78, 131.32, 133.60, 152.18, 153.58, 155.53, 160.60.
Results and discussion
1O2 generation and sensing in solutions
The 1O2 generation, storage, sensing, and delivery efficiencies of the nanoarchitecture samples are based on highly efficient PIET from the bis-AMA to coumarin in sensor 4 (Fig. 1a). Sensor 4 was synthesized in one pot by a sequential base-catalyzed nucleophilic substitution reaction between 9,10-bis(chloromethyl)anthracene (1), 7-amino-4-methylcoumarin (3) and N,N-dioctylamine. The bis AMA group is critical in the quantitative electron transfer-induced FL quenching and 230-fold FL intensity enhancement during the sensing. Conversely, the initial coumarin FL was not efficiently quenched in the corresponding intermediate (5). Therefore, it does not show the 1O2 sensitivity like 4 because of poor PIET from the chloromethyl anthracene to coumarine parts. The nanoarchitecture integrated with a 1O2 sensor and a suitable PS, such as TCPP or QDs, overcomes various limitations in conventional PDT.34–37 The red FL from TCPP or QD and the enhanced blue emission from the sensor offer 1O2 sensing and two-color imaging modalities during PDT. To develop SS-MSNP, the intermediate 5 and TCPP or 5 and QD were attached covalently to the amino-functionalized MSNPs (Fig. 1a and c). The covalent attachment prevents the undesired sensor/sensitizer release or leaching, thereby maintaining photoactivity.59–65 We selected MSNPs considering the advantages of mesopores for efficiently loading drugs and contrast agents. Nevertheless, directly incorporating sensors or sensitizers in nonporous silica during NP synthesis45–47 does not limit the current work. Fig. 1d shows the SEM images of the functionalized MSNPs. The particles showed a uniformly distributed spherical morphology with a 200 nm average diameter. The S-MSNP, SS-MSNP, and SS-MSNP-RGD samples were further characterized by zeta potentials (+16.91 mV for S-MNSP, +15.52 mV for SS-MSNP, and −15.02 for SS-MSNP-RGD), and DLS measurements (the size distributions are shown in Fig. S1†).The pivotal role of 4 in S-MSNP and SS-MSNP as an excellent 1O2 sensor and donor was investigated by measuring its photophysical properties at the ensemble and single-particle levels. Fig. 2a shows the photosensitization and 1O2 sensing processes on an SS-MSNP similar photochemical processes take place in a solution of 4 and TCPP. First, we examined the FL spectra of 4 under UV light irradiation without and with 1O2 scavengers (NaN3 or CuCl2) to distinguish between the roles of UV-induced anthracene dimerization66 and 1O2-mediated anthracene oxidation in blocking PIET from AMA to coumarin. The UV-induced FL intensity enhancement of 4, without any 1O2 scavenger, exceeded 230-fold (Fig. 2b and d). Conversely, the FL intensity enhancement was <60-fold under UV irradiation (Fig. S2†) in the presence of NaN3 or CuCl2. Therefore, we assume 4 generates 1O2 under UV irradiation, and AMA oxidation by 1O2 blocks PIET and enhances the FL intensity by >170-fold. The >170-fold enhancement factor includes the FL of the 1O2-4 intermediate and coumarin released after UV-triggered oxidation of AMA. Nevertheless, unlike visible light absorbing 1O2 sensors such as Si-DMA50 or SOSG,514 is highly stable under room light.
 | ||
| Fig. 2 1O2 generation, sensing, and releasing. (a) The SS-MSNP structure and its 1O2 generation and sensing processes. (b and c) FL spectra (λex = 320 nm) of (b) 4 (10 μM in CH3CN, without TCPP) before and after irradiation at 365 nm for 95 min at 2 min intervals, (c) 4 (10 μM in CH3CN) in a TCPP solution (5 μM) before and after photoactivation at 532 nm (50 mW cm−2) for 30 min (blue bar) followed by UV illumination (365 nm, 5 mW cm−2) for 82 min (red bar) showing the sensitization-sensing process followed by UV-induced 1O2 releasing/AMA oxidation. (d) Time-trace of the peak FL intensities at different photoirradiation conditions in (b) and (c). (e–g) Photographs of (left) a mixture of 4 and TCPP and (right) 4 (e) under visible light before photoactivation, (f) <1 min under UV light after 30 min 532 nm laser irradiation (50 mW cm−2) for 30 min, and (g) ca. 5 min under UV light after 30 min 532 nm laser irradiation (50 mW cm−2). (h) A scheme of 1O2 reaction on the sensor. | ||
The selectivity of 4 to 1O2 was determined by irradiating a solution of 4 (10 μM) and TCPP (5 μM) in acetonitrile with 532 nm laser (50 mW cm−2). Here, TCPP generates 1O2, and the AMA moiety in the sensor scavenges the 1O2, forming a mixture of highly fluorescent 9,10-endoperoxide67,68 and a less-fluorescent intermediate that further transforms into the endoperoxide on-demand by UV or NIR triggering. In this two-step process, the initial FL intensity enhancement (under 532 nm photosensitization of TCPP) was determined at 50-fold (Fig. 2c, d and S3†), with an overall enhancement of 230-fold under UV illumination, showing a 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22 ratio between the intermediate and the endoperoxide. We estimated this ratio from the maximum intensity enhancement factor (∼230-fold), the FL quantum yield of the parent coumarin (0.63), and the inflation of the FL intensity by changing the photoirradiation from 532 to 365 nm. The endoperoxide formation (Fig. 2h) follows the thermal cleavage of the O–O and C–C bonds common to alkyl-substituted anthracenes.69 The decay of the anthracene vibronic bands at ca. 356, 376, and 396 nm and the corresponding increase in the intensity at ca. 325 nm in the absorption spectra (Fig. S4†) suggest 1O2-mediated photo-oxidation of anthracene and the formation of anthraquinone. The FL turn-on ability (230-fold) of the sensor is the highest compared to 1O2 probes reported to date, such as SiDMA50 and SOSG,51 which is due to the efficient PIET and FL quenching in 4 favored by the bis aminomethyl moiety introduced in anthracene.
:22 ratio between the intermediate and the endoperoxide. We estimated this ratio from the maximum intensity enhancement factor (∼230-fold), the FL quantum yield of the parent coumarin (0.63), and the inflation of the FL intensity by changing the photoirradiation from 532 to 365 nm. The endoperoxide formation (Fig. 2h) follows the thermal cleavage of the O–O and C–C bonds common to alkyl-substituted anthracenes.69 The decay of the anthracene vibronic bands at ca. 356, 376, and 396 nm and the corresponding increase in the intensity at ca. 325 nm in the absorption spectra (Fig. S4†) suggest 1O2-mediated photo-oxidation of anthracene and the formation of anthraquinone. The FL turn-on ability (230-fold) of the sensor is the highest compared to 1O2 probes reported to date, such as SiDMA50 and SOSG,51 which is due to the efficient PIET and FL quenching in 4 favored by the bis aminomethyl moiety introduced in anthracene.
Two-step vis-UV-induced FL intensity enhancement is further confirmed from the UV-triggered, time-dependent FL images of a solution containing 4 with or without TCPP. A solution of 4 with TCPP produced 1O2, whereas 4 without TCPP was the negative control (without 1O2 generation). These samples help validate the efficiency of 4 in trapping the 1O2 produced, releasing it into a solution or undergoing oxidation. The 1O2 generated by TCPP is captured and released by the sensor as indicated by the brilliant magenta emission (1O2-positive) – a combination of the TCPP red emission and the uncaged sensor's brilliant blue emission. Conversely, no color change was observed for the negative control sample under 532 nm photoirradiation. However, intense blue FL was observed from the negative control sample under prolonged UV-only irradiation (Fig. 2e–g). Like 4, the S-MSNP nanoarchitecture showed excellent 1O2 caging and releasing- or oxidation-induced FL intensity enhancement in the heterogeneous solution phase with free TCPP in the solution. Similarly, enormous FL intensity enhancement was detected for the SS-MSNP with the sensor and sensitizer covalently on the particles (Fig. S5†). The time-traced FL intensity values reveal that the intraparticle 1O2 photogeneration-capturing-releasing by SS-MSNP is more efficient than S-MSNP dispersed in a TCPP solution.
1O2 generation and sensing at the single particle level
Single-particle FL studies help understand the photostability of S-MSNP and SS-MSNP and the microscopic-level kinetics of 1O2 generation and sensing for cell biological applications. Therefore, we examined the 1O2 generation, storing, sensing, and releasing efficiencies of the S-MSNP and SS-MSNP by recording single-particle FL images and intensity trajectories. FL images of SS-MSNP particles dispersed on a cover glass and resolved into the sensor's and sensitizer's FL are shown in Fig. 3a–c. The as-prepared SS-MSNPs exhibited intense red emission of TCPP, detected through a 620 nm LP filter by exciting at 532 nm (Fig. 3b). Conversely, the initial blue FL at ca. 440 nm from SS-MSNP or S-MSNP, corresponding to the coumarin moiety, was below the detection limit without the electron multiplication mode of an EMCCD camera. This is due to the FL quenching by PIET in 4. However, the FL intensity of S-MSNP and SS-MSNP was constantly increased with continuous 365 nm excitation, consistent with the FL uncaging of the intermediate.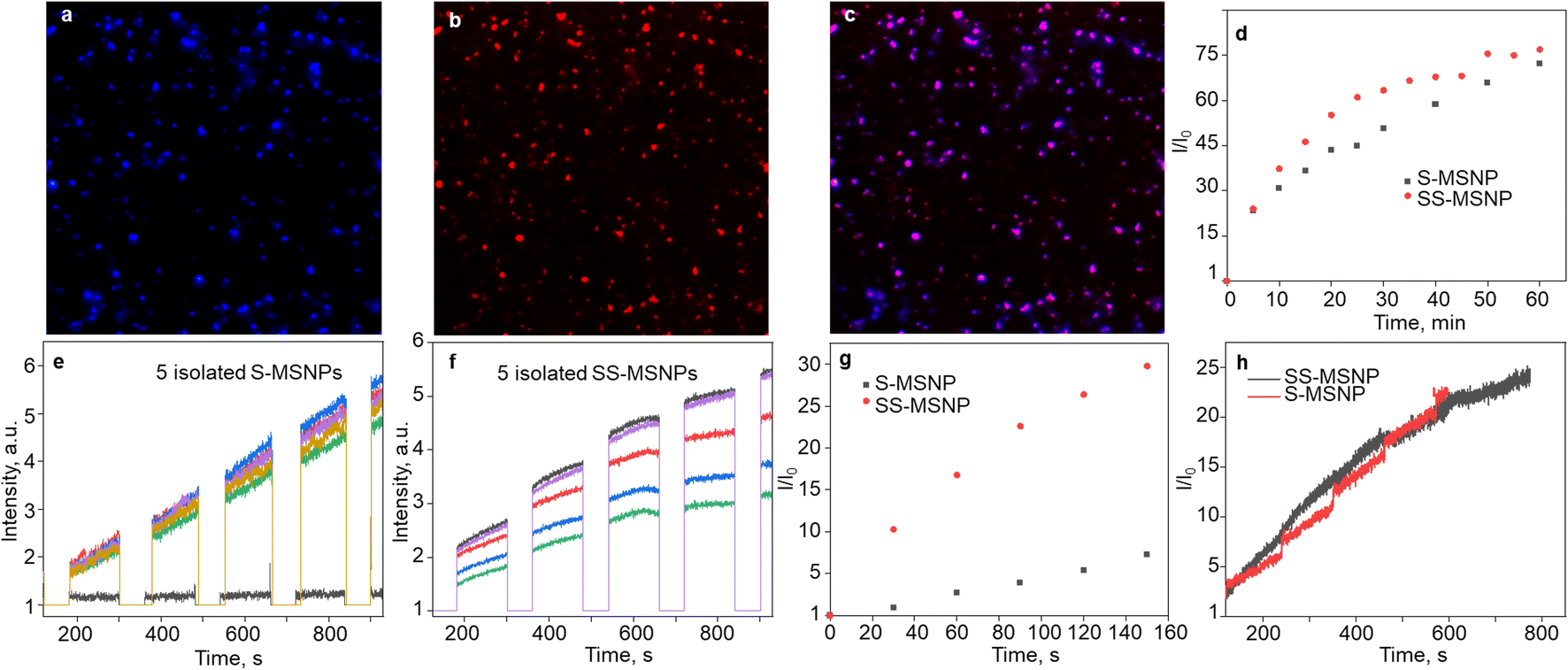 | ||
| Fig. 3 Single-particle 1O2 generation-storing-sensing-releasing properties of S-MSNPs and SS-MSNPs. (a and b) Single particle FL images (100 μm × 100 μm) of SS-MSNPs collected using (a) 420–480 nm band-pass filter, (b) 580 nm LP filter, and (c) the overlay of (a) and (b). Excitation wavelengths: (a) 365 nm (5 mW cm−2), (b) 532 nm (30 mW cm−2). (d) Time-trace of the peak (440 nm) FL intensities of the S-MSNPs in a TCPP solution (5 μM) and SS-MSNPs in water before and after photoactivation at 532 nm (5 W cm−2) for 60 min. (e and f) Single-particle FL intensity trajectories showing 1O2 production detected for (e) S-MSNPs in a TCPP solution and (f) SS-MSNPs in water under photoactivation of TCPP with 532 nm laser irradiation for 30 s (photosensitization) followed by UV illumination (0.5 W cm−2; FL releasing and detection for 120 s intervals). (g and h) Difference FL intensity profiles of S-MSNP single particles in a TCPP solution and SS-MSNP single particles in water separated into the FL intensity traces during (g) photosensitization-induced 1O2 generation and detection and (h) UV-induced 1O2 releasing and detection. A 60 s shutter temporally separated the sensitization and 1O2 release. | ||
The 1O2 capturing-releasing efficiency of the sensors in S-MSNP dispersed in a TCPP solution (Fig. 3d and e) or SS-MSNP (Fig. 3d and f) in water was examined by FL enhancement kinetic measurements of single particles. Single particle photosensitization (1O2 generation and storing) and UV-induced 1O2 release were carried out in 30–120 s sequences, as shown in Fig. 3e and f. Following the photosensitization of TCPP in an S-MSNP-TCPP solution or on the SS-MSNP surface by 532 nm laser excitation, the UV-light triggered time-dependent 1O2 releasing was detected by exciting the samples with 365 nm light (Fig. 3d). The excitation powers for photosensitization (5 W cm−2) and UV-induced FL uncaging (0.5 W cm−2) were set at higher levels in single particle experiments than ensemble solution samples to achieve appreciable signal-to-noise ratios. As a result, the single-particle photosensitization time and UV irradiation time were set at 30 s and 120 s. The FL intensity trajectories from more than 100 single particles were collected and analyzed. The single-particle FL data were deconvoluted to quantify the 1O2 capturing-storing and releasing abilities of the nanoarchitecture. The FL intensity of SS-MSNP shows a remarkable time-dependent increase compared to S-MSNP (Fig. 3g). The higher FL intensity of SS-MSNP than S-MSNP at any time after 532 nm photosensitization suggests that the 1O2 storing and sensing efficiency is greater when the sensor and PS are in close proximity. Conversely, the 1O2 produced by TCPP molecules in the solution is less efficiently captured and sensed by S-MSNP, presumably due to the random diffusion or the large degree of diffusion freedom for 1O2 produced in the solution. In other words, the sensors on SS-MSNP efficiently cage and sense 1O2 produced by TCPP on the same nanoarchitecture. Fig. 3h shows the UV-induced 1O2-releasing abilities of S-MSNP and SS-MSNP single particles. Both systems show temporally controlled 1O2 release. Also, we examined the photostability of S-MSNPs and SS-MSNPs under UV or 532 nm excitation (Fig. S6†). SS-MSNPs excited with the 532 nm laser in the presence of NaN3 showed steady FL at 440 or 650 nm, indicating both the sensor and TCPP remain stable. Also, S-MSNP or SS-MSNP excited with 365 nm light showed steady FL, suggesting low photodimerization efficiency for the sensors covalently conjugated to the Si NPs. Therefore, the SS-MSNP acts as a nanoreactor that promotes the 1O2 storing and light-triggered sustained 1O2 release. These results demonstrate the potential of SS-MSNP nanoarchitecture as an efficient 1O2 sensor and reservoir for PDT, particularly for fractional PDT.
1O2 generation and sensing in cells
We investigated the bioimaging imaging and PDT potentials of the sensor- and sensitizer-conjugated MSNPs. First, we treated the conjugates with the breast cancer cell MCF7. Fig. 4a–i shows FL images of the cells incubated with S-MSNP, TCPP-MSNP, SS-MSNP, or SS-MSNP-RGD for 1 h under ambient conditions. Also, the cells were stained with the nucleus staining dye Syto 16 (green FL in Fig. 4a, d, and f). The blue FL of S-MSNPs distributed in the cytoplasm indicates nonspecific endocytosis of the NPs. The appreciably intense intracellular blue FL suggests blockage of PIET in the sensor by endogenous 1O2, pointing towards the localization of S-MSNPs to 1O2-rich domains, such as the mitochondria. Fig. 4d–f shows FL images of the cells labeled with Syto 16 and the TCPP-MSNP conjugate. The intracellular red FL shows the effective uptake of TCPP-MSNP by MCF7 cells. Similarly, cells labeled with SS-MSNPs showed intracellular blue FL (Fig. 4g) of the oxidized sensor and red FL (Fig. 4h) of TCPP. Although we do not rule out the role of multiple COOH groups in TCPP on the endocytosis of TCPP-MSNP or SS-MSNP, the primary amino groups introduced during the silanization step (Fig. 1c) are common to S-MSNP, TCPP-MSNP, and SS-MSNP, which should have a role in the cellular uptake of these NPs. | ||
| Fig. 4 Intracellular imaging and 1O2 detection. Fluorescence (FL) images of cells labeled with 50 μg mL−1 of (a–c) S-MSNP, (d–f) TCPP-MSNP, (g–i) SS-MSNP and (j–l) SS-MSNP-RGD conjugates showing FL from the (b, g, and j) sensor, (a and d), Syto 16, (e, h, and k) TCPP, (c, f, i, and l) overlaid images. (m and n) MTT cell viability histograms for MCF cells treated with different concentrations of SS-MSNP-RGD assembly under (m) 500–650 nm band-pass filtered light irradiation (10 mW cm−2) for 30 min, and (n) dark. (o) An intracellular single-particle FL intensity trajectory for MCF7 cells incubated with SS-MSNP-RGD conjugate and photosensitized with a 532 nm laser. The FL intensity was recorded every 30 s after the photosensitization. The images are (a–c, g–l) 150 × 150 μm2 and (d–f) 200 × 200 μm2. | ||
Despite the nonspecific intracellular delivery of SS-MSNP, we applied the RGD-conjugated SS-MSNPs (SS-MSNP-RGD) to MCF7 cells to improve the cellular uptake and to understand the role of αvβ3 integrin-mediated endocytosis of the conjugate.70Fig. 4j–l shows the FL images of cells incubated with the SS-MSNP-RGD conjugate. The intense intracellular blue and red FL suggests efficient cellular uptake of the conjugate. Nevertheless, cells preincubated with the (arginine)8 (R8) peptide that blocks αvβ3 integrin and treated with SS-MSNP-RGD showed appreciable intracellular blue and red FL, suggesting αvβ3 integrin-mediated endocytosis is not the only intracellular delivery mechanism. Therefore, like the conjugates without RGD (Si-TCPP, S-MSNP, SS-MSNP, and SS-MSNP; Fig. 4a–g), macropinocytosis is involved in the intracellular pathway of SS-MSNP-RGD, whereas RGD-free conjugates are delivered by macropinocytosis alone. Although multiple intracellular delivery routes are involved for SS-MSNP and SS-MSNP-RGD, a uniform and efficient intracellular NP distribution for the RGD conjugate denotes passive and active targeting for nanoparticle-guided therapy.36 Despite the confocal FL images showing effective accumulation of the RGD conjugate inside the cells, microtoming and cross-section TEM imaging helped us locate the NPs inside. Here, the cells cultured on 1 cm × 1 cm polypropylene plates deposited in a cell culture plate were washed with PBS, labeled with SS-MSNP-RGD for 30 min, gently washed with PBS (3 times), and dried under a vacuum. The presence of the cells on the polypropylene plates was identified by optical imaging, followed by microtoming and TEM imaging. Fig. S8† shows the cross-section TEM image of a cell treated with SS-MSNP-RGD. The dark contrast corresponds to the NPs in the subcellular compartment.
Following the cellular uptake of SS-MSNP-RGD nanoarchitecture, we examined the intracellular 1O2 generation and release by single-cell FL measurements and time-lapsed FL imaging. The cells incubated with the nanoarchitecture were photo-irradiated continuously using a 532 nm laser, and the FL trajectories were recorded for each step of the 532 nm laser and UV illuminations. The time-correlated FL intensity trajectory (Fig. 4o) suggests intracellular 1O2 generation, storing, and release, like the single particle results in Fig. 3. Further, the cell morphology changes were observed by time-lapse imaging of cells under continuous light irradiation (Fig. S9†). With time under irradiation, we detected apoptosis with corpuscles and cell shrinkage.44 These results confirm that 1O2 generated and released continuously in the cells accelerates cell death mainly by the apoptotic pathway.
Low dark cytotoxicity and high phototoxicity are important factors in PDT. We examined the cytotoxicity of S-MSNP and SS-MSNP-RGD to MCF7 cells. MTT cytotoxicity histograms are shown in Fig. 4m and n. We found the lethal dose (LD) for 50% cell viability (LD50) under the dark at ≫100 μg mL−1 SS-MSNP-RGD. Also, >80% cell viability was evident for the cells treated with 10 or 1 μg mL−1 SS-MSNP-RGD solutions in the dark (Fig. 4n). Comparable dark cell viability values were obtained for S-MSNP (Fig. S10†). Phototoxicity of SS-MSNP-RGD to MCF7 cells was evaluated under the above labeling conditions. The labeled cells were irradiated with 500–650 nm band-pass filtered light (10 mW cm−2) for 30 min before the MTT treatment. Phototoxicity of SS-MSNP-RGD for 1, 10, and 50 μg mL−1 sample solutions are shown in Fig. 4m, showing appreciable phototoxicity for 50 μg mL−1 (52%) and 10 μg mL−1 (42%) samples. The phototoxicity is attributed to 1O2 generation by the sensitizer in the NPs, which is consistent with the ensemble and single-particle spectroscopic and microscopic data.
Apart from SS-MSNP-RGD, we used NIR-emitting CdSe/ZnS QD for testing 1O2 production and sensing in cells. QDs have been widely employed in PDT and cell/tissue imaging experiments in cell/animal models due to deep tissue penetration, improved water solubility, and biocompatibility.53–55 Recently, Vetrone and coworkers demonstrated a multifunctional MSNP theranostic integrated with QDs, Fe3O4, and DOX for combined bimodal (NIR-fluorescence and magnetic resonance) imaging, drug delivery, hyperthermia, and phototherapy.55 Also, the ability of QDs to produce ROS makes them promising for PDT.56 Therefore, we prepared MSNPs conjugated with the sensor, CdSe/ZnS QDs (Fig. 5a), and evaluated the 1O2 production of the S-QD-MSNP nanoarchitecture by photosensitizing at 532 nm with different laser power densities. In Fig. 5b, the FL intensity trajectories recorded by 60 s 532 nm irradiation, followed by 120 s UV illumination, showed appreciable FL (440 nm, coumarin) intensity increases at high excitation power (≥40 mW cm−2). The high excitation intensity requirement is due to the low 1O2 quantum yield of QDs.57,58 The 1O2 generation and trapping behavior of the S-QD-MSNP at the ensemble level also showed a similar trend (Fig. S7†), in agreement with the single-particle studies. Therefore, the sensor is promising to prepare sensor-sensitizer systems using different nanoparticle and molecular photosensitizers.
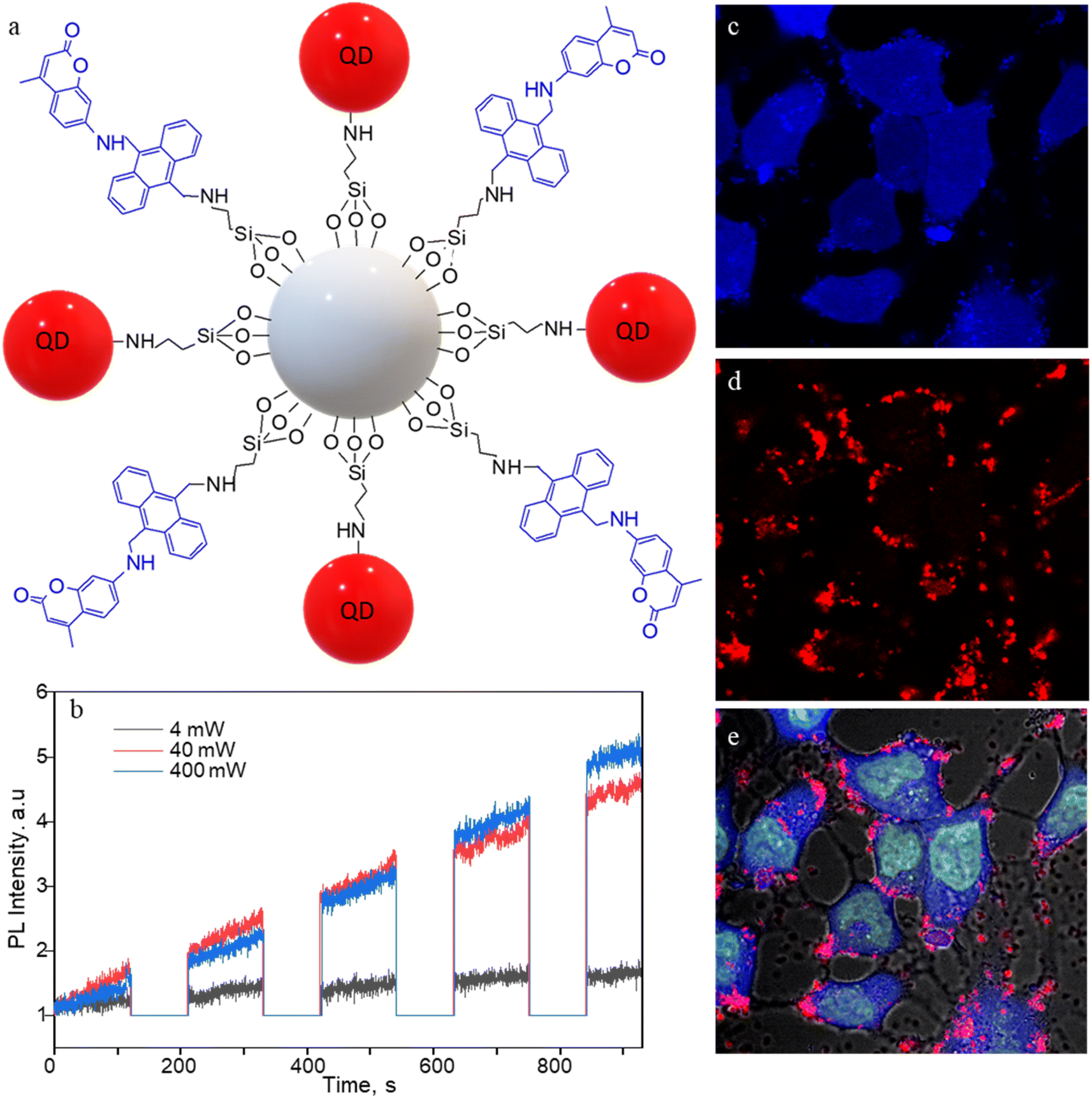 | ||
| Fig. 5 CdSe/ZnS QD- and sensor-conjugated MSNPs (S-QD-MSNP) for 1O2 generation and intracellular imaging. (a) The structure of S-QD-MSNP nanoarchitecture. (b) FL intensity trajectories (440 nm) showing 1O2 production by QDs under 532 nm laser irradiation (60 s) and UV illumination (120 s) sequence at different photosensitization powers. (c–e) FL images of MCF7 cells labeled with SYTO-16 (5 μM solution) and the S-QD-MSNP nanoarchitecture (50 μg mL−1) showing the emission from the (c) sensor, (d) QD655, and (e) the overlaid image. The photosensitization and 1O2 release are temporally separated by 30 s shutter. The images are 150 × 150 μm2. | ||
In vivo experiments
We examined the in vivo bioimaging potential of SS-MSNP-RGD in B6 mice subcutaneously and intravenously. First, we examined and optimized the fluorescence of the samples using a small-animal imaging system by applying different band-pass filters and excitation light sources. Fig. S11† shows the bright-field and FL images of S-MSNP, SS-MSNP, and SS-MSNP-RGD samples. S-MSNP showed blue-green fluorescence, of which the long wavelength part of the coumarin dye was collected due to the small Stoke's shift (λex = 445–490 nm band-pass filtered light, and the FL was collected through a 515 nm LP filter). Fig. S11c and d† shows the emission from the sensitizer in TCPP-MSNP and SS-MSNP-RGD samples. Subsequently, we examined the images of the B6 mice subcutaneously injected with the TCPP-MSNP or SS-MSNP-RGD samples. The characteristic FL (>670 nm) of the sensitizer (Fig. S11e and f†) was detected, which efficiently penetrated the skin tissue. Conversely, the FL from the sensor was not detected in mice subcutaneously or intravenously injected with S-MSNP or SS-MSNP samples, which is due to the poor penetration of the sensor FL (ca. 440 nm) through the skin/tissue and the overlapping sensor FL with the mice autofluorescence.We found higher FL intensities in the mice liver 1 to 24 h post intravenous injection of the S-MSNP or SS-MSNP-RGD conjugates (Fig. S11i–l†). The FL signals correspond to 670 nm or longer wavelengths solely from the sensitizer parts. Conversely, the sensor FL was not detected even when the SS-MSNP-RGD sample-injected mice were exposed to 532 nm light or an SS-MSNP-RGD sample was photosensitized at 532 nm for 30 min before the injection. This absence of the blue signals is attributed to its overlapping with the tissue autofluorescence or its reabsorption by tissues. The 670 nm FL intensity in the liver of the mice injected with the TCPP-MSNP was increased within 24 h post-injection (Fig. S11k†), showing a gradual accumulation. Conversely, for a mouse injected with SS-MSNP-RGD, the FL from the liver was decreased within 24 h (Fig. S11l†) with an increase in the FL intensity in the bladder. These observations suggest that the RGD conjugation helps to circulate and excrete the NPs. Further imaging, toxicity, and pharmacokinetic investigations are necessary to assess the bioimaging or phototherapeutic potentials of the above sensor, sensitizer, or nanoparticles. Also, NIR-emitting sensors and sensitizers are needed for in vivo applications.
Summary
We constructed a fluorescence turn-on photosensitizer-sensor-peptide nanoarchitecture for intracellular 1O2 generation, storing, controlled release, and sensing. The on-demand 1O2 sensing-releasing-assisted fluorescence intensity enhancement and fluorescence color change of this architecture were uncovered by fluorescence measurements of solutions, living cells, or single particles. The small size of the nanoarchitecture, within the 1O2 diffusion length, promoted tandem photo-triggered fluorescence switching and enabled bimodal fluorescence cell imaging activated by on-demand intracellular sensor oxidation and 1O2 release. Under continuous laser irradiation, 1O2-induced cell death was also observed. The MTT assay results revealed low dark toxicity and high phototoxicity of the sensor-sensitizer-silica-peptide conjugates to MCF7 cells. Also, preliminary in vivo studies on B6 mice intravenously or subcutaneously injected with the conjugates help elucidate the in vivo application of the conjugate. Nevertheless, the blue emission from the sensor is masked by reabsorption and the tissue autofluorescence, suggesting the significance of developing sensors and sensitizers absorbing and emitting in the NIR biological window.Data availability
Data supporting the findings of this study are available within the article ESI.†Author contributions
V. B. and P. W. conceived the idea. V. B. led the project. J. S. conducted the chemical synthesis, bioconjugate reactions, fluorescence detection studies, and cell imaging. K. O. and M. S. conducted the in vivo experiment. T. O. performed STEM and microtoming experiments. J. S. wrote the manuscript draft. All authors contributed to the copy-editing of the manuscript. J. S., P. W. and V. B. finalized the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by the JSPS Grant-in-Aid for Scientific Research B (23H01781). We acknowledge the Hokkaido University Photoexcitonix program, a DX doctoral fellowship, the Nanotechnology PLATFORM, the NIKON imaging facility, and the Crossover Alliance to Create the Future with People, Intelligence, and Materials.References
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
- W. Yang, F. Zhang, H. Deng, L. Lin, S. Wang, F. Kang, G. Yu, J. Lau, R. Tian, M. Zhang, Z. Wang, L. He, Y. Ma, G. Niu, S. Hu and X. Chen, ACS Nano, 2020, 14, 620–631 CrossRef CAS.
- Y. F. Xiao, W. C. Chen, J. X. Chen, G. Lu, S. Tian, X. Cui, Z. Zhang, H. Chen, Y. Wan, S. Li and C. S. Lee, ACS Appl. Mater. Interfaces, 2022, 14, 5112–5121 CrossRef CAS.
- K. Liu, X. Liu, Q. Zeng, Y. Zhang, L. Tu, T. Liu, X. Kong, Y. Wang, F. Cao, S. A. G. Lambrechts, M. C. G. Aalders and H. Zhang, ACS Nano, 2012, 6, 4054–4062 CrossRef CAS.
- N. Zhao, B. Ding, Y. Zhang, J. L. Klockow, K. Lau, F. T. Chin, Z. Cheng and H. Liu, J. Control. Release, 2020, 324, 330–340 CrossRef CAS.
- Z. Cao, D. Li, J. Wang and X. Yang, Acta Biomater., 2021, 130, 17–31 CrossRef CAS PubMed.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Cy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS.
- P. De Bonfils, E. Verron, P. Nun and V. Coeffard, ChemPhotoChem, 2021, 5, 847–856 CrossRef CAS.
- B. Chen, Y. Yang, Y. Wang, Y. Yan, Z. Wang, Q. Yin, Q. Zhang and Y. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 18533–18544 CrossRef CAS.
- T. Chen, P. Hou, Y. Zhang, R. Ao, L. Su, Y. Jiang, Y. Zhang, H. Cai, J. Wang, Q. Chen, J. Song, L. Lin, H. Yang and X. Chen, Angew. Chem., Int. Ed., 2021, 60, 15006–15012 CrossRef CAS.
- T. L. Lam, K. C. Tong, C. Yang, W. L. Kwong, X. Guan, M. De Li, V. Kar-Yan Lo, S. Lai-Fung Chan, D. Lee Phillips, C. N. Lok and C. M. Che, Chem. Sci., 2019, 10, 293–309 RSC.
- D. Liang, Y. Zhang, Z. Wu, Y. J. Chen, X. Yang, M. Sun, R. Ni, J. Bian and D. Huang, Sensors Actuators, B, 2018, 266, 645–654 CrossRef CAS.
- Z. Yuan, S. Yu, F. Cao, Z. Mao, C. Gao and J. Ling, Polym. Chem., 2018, 9, 2124–2133 RSC.
- R. Bresolí-Obach, J. Nos, M. Mora, M. L. Sagristà, R. Ruiz-González and S. Nonell, Methods, 2016, 109, 64–72 CrossRef.
- M. Sun, S. Krishnakumar, Y. Zhang, D. Liang, X. Yang, M. W. Wong, S. Wang and D. Huang, Sensors Actuators, B, 2018, 271, 346–352 CrossRef CAS.
- X. Liu, P. Dai, T. Gu, Q. Wu, H. Wei, S. Liu, K. Y. Zhang and Q. Zhao, J. Inorg. Biochem., 2020, 209, 1111096 CrossRef.
- J. Zhu, J. Zou, J. Zhang, Y. Sun, X. Dong and Q. Zhang, J. Mater. Chem. B, 2019, 7, 3303–3309 RSC.
- J. Deng, F. Liu, L. Wang, Y. An, M. Gao, Z. Wang and Y. Zhao, Biomater. Sci., 2019, 7, 429–441 RSC.
- J. Chen, D. Li, B. Huo, F. Zhang, X. Zhao, G. Yuan, D. Chen, M. Song and J. Xue, Mol. Pharm., 2019, 16, 3703–3710 CrossRef CAS.
- D. Wang, B. Xue, T. Y. Ohulchanskyy, Y. Liu, A. Yakovliev, R. Ziniuk, M. Xu, J. Song, J. Qu and Z. Yuan, Biomaterials, 2020, 251, 120088 CrossRef CAS PubMed.
- T. Chen, P. Hou, Y. Zhang, R. Ao, L. Su, Y. Jiang, Y. Zhang, H. Cai, J. Wang, Q. Chen, J. Song, L. Lin, H. Yang and X. Chen, Angew. Chem., Int. Ed., 2021, 60, 15006–15012 CrossRef CAS PubMed.
- W. Lv, M. Cao, J. Liu, Y. Hei and J. Bai, Acta Biomater., 2021, 135, 617–627 CrossRef CAS.
- J. Fan, M. Yang, J. Zhang, D. Shabat and X. Peng, ACS Sensors, 2020, 5, 3158–3164 CrossRef.
- D. C. Yang, S. Wang, X. L. Weng, H. X. Zhang, J. Y. Liu and Z. Lin, ACS Appl. Mater. Interfaces, 2021, 13, 33905–33914 CrossRef CAS PubMed.
- K. Deng, B. Wu, C. X. Wang, Q. Wang, H. Yu, J. M. Li, K. H. Li, H. Y. Zhao and S. W. Huang, Adv. Healthcare Mater., 2020, 9, 2000533 CrossRef CAS.
- H. Lai, J. Yan, S. Liu, Q. Yang, F. Xing and P. Xiao, Angew. Chem., Int. Ed., 2020, 59, 10431–10435 CrossRef CAS.
- Q. Liu, J. Tian, Y. Tian, Q. Sun, D. Sun, F. Wang, H. Xu, G. Ying, J. Wang, A. K. Yetisen and N. Jiang, ACS Nano, 2021, 15, 515–525 CrossRef CAS PubMed.
- D. Wang, H. Wu, S. Z. F. Phua, G. Yang, W. Qi Lim, L. Gu, C. Qian, H. Wang, Z. Guo, H. Chen and Y. Zhao, Nat. Commun., 2020, 11, 357 CrossRef CAS PubMed.
- R. Kv, T. I. Liu, I. L. Lu, C. C. Liu, H. H. Chen, T. Y. Lu, W. H. Chiang and H. C. Chiu, J. Control. Release, 2020, 328, 87–99 CrossRef CAS.
- X. Ke, D. Wang, C. Chen, A. Yang, Y. Han, L. Ren, D. Li and H. Wang, Nanoscale Res. Lett., 2014, 9, 1–8 CrossRef CAS.
- M. Gangopadhyay, A. Jana, Y. Rajesh, M. Bera, S. Biswas, N. Chowdhury, Y. Zhao, M. Mandal and N. D. P. Singh, ChemistrySelect, 2016, 1, 6523–6531 CrossRef.
- Y. Wan, G. Lu, W. C. Wei, Y. H. Huang, S. Li, J. X. Chen, X. Cui, Y. F. Xiao, X. Li, Y. Liu, X. M. Meng, P. Wang, H. Y. Xie, J. Zhang, K. T. Wong and C. S. Lee, ACS Nano, 2020, 14, 9917–9928 CrossRef CAS.
- S. Farooq and R. E. de Araujo, High Performance Gold Nanoshells for Singlet Oxygen Generation Enhancement, Photodiagn. Photodyn. Ther., 2021, 35, 102466 CrossRef CAS.
- C. Mendoza, N. Emmanuel, C. A. Páez, L. Dreesen, J. C. M. Monbaliu and B. Heinrichs, ChemPhotoChem, 2018, 2, 890–897 CrossRef CAS.
- F. F. E. Kohle, S. Li, M. Z. Turker and U. B. Wiesner, ACS Biomater. Sci. Eng., 2020, 6, 256–264 CrossRef CAS.
- R. Liu, Y. Gao, N. Liu and Y. Suo, Photodiagn. Photodyn. Ther., 2021, 33, 102156 CrossRef CAS.
- J. Kim, H. Lee, J. Y. Lee, K. H. Park, W. Kim, J. H. Lee, H. J. Kang, S. W. Hong, H. J. Park, S. Lee, J. H. Lee, H. D. Park, J. Y. Kim, Y. W. Jeong and J. Lee, Appl. Catal., B, 2020, 270, 118862 CrossRef CAS.
- X. Liu, J. Jiang, C. H. Chang, Y. P. Liao, J. J. Lodico, I. Tang, E. Zheng, W. Qiu, M. Lin, X. Wang, Y. Ji, K. C. Mei, A. E. Nel and H. Meng, Small, 2021, 17, 1–13 CAS.
- H. Meng, M. Wang, H. Liu, X. Liu, A. Situ, B. Wu, Z. Ji, C. H. Chang and A. E. Nel, ACS Nano, 2015, 9, 3540–3557 CrossRef CAS PubMed.
- X. Liu, J. Jiang, Y. P. Liao, I. Tang, E. Zheng, W. Qiu, M. Lin, X. Wang, Y. Ji, K. C. Mei, Q. Liu, C. H. Chang, Z. A. Wainberg, A. E. Nel and H. Meng, Adv. Sci., 2021, 8, 1–17 Search PubMed.
- X. Liu, J. Jiang, R. Chan, Y. Ji, J. Lu, Y. P. Liao, M. Okene, J. Lin, P. Lin, C. H. Chang, X. Wang, I. Tang, E. Zheng, W. Qiu, Z. A. Wainberg, A. E. Nel and H. Meng, ACS Nano, 2019, 13, 38–53 CrossRef CAS PubMed.
- M. Das, A. Solanki, A. Ganesh and S. Thakore, Photodiagn. Photodyn. Ther., 2021, 34, 102259 CrossRef CAS PubMed.
- D. Tarn, C. E. Ashley, M. I. N. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- P. Dogra, N. L. Adolphi, Z. Wang, Y.-S. Lin, K. S. Butler, P. N. Durfee, J. G. Croissant, A. Noureddine, E. N. Coker, E. L. Bearer, V. Cristini and C. J. Brinker, Nat. Commun., 2018, 9, 4551 CrossRef PubMed.
- L. Jiao, X. Zhang, J. Cui, X. Peng and F. Song, ACS Appl. Mater. Interfaces, 2019, 11, 25750–25757 CrossRef CAS PubMed.
- L. Jiao, F. Song, B. Zhang, H. Ning, J. Cui and X. Peng, J. Mater. Chem. B, 2017, 5, 5278–5283 RSC.
- L. Jiao, Y. Liu, X. Zhang, G. Hong, J. Zheng, J. Cui, X. Peng and F. Song, ACS Cent. Sci., 2020, 6, 747–759 CrossRef CAS PubMed.
- C. Yang, M. Su, P. Luo, Y. Liu, F. Yang and C. A. Li, Small, 2021, 17, 2101180 CrossRef CAS.
- Y. Q. He, W. Fudickar, J. H. Tang, H. Wang, X. Li, J. Han, Z. Wang, M. Liu, Y. W. Zhong, T. Linker and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 2601–2608 CrossRef CAS PubMed.
- S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 11707–11715 CrossRef CAS PubMed.
- A. Gollmer, J. Arnbjerg, F. H. Blaikie, B. W. Pedersen, T. Breitenbach, K. Daasbjerg, M. Glasius and P. R. Ogilby, Photochem. Photobiol., 2011, 87, 671–679 CrossRef CAS PubMed.
- S. Liu, Y. Hu, C. Hu, Y. Xiong and M. Duan, J. Porphyrins Phthalocyanines, 2019, 23, 1–8 CrossRef.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- E. S. Shibu, K. Ono, S. Sugino, A. Nishioka, A. Yasuda, Y. Shigeri, S. I. Wakida, M. Sawada and V. Biju, ACS Nano, 2013, 7, 9851–9859 CrossRef CAS.
- F. Yang, A. Skripka, M. S. Tabatabaei, S. H. Hong, F. Ren, Y. Huang, J. K. Oh, S. Martel, X. Liu, F. Vetrone and D. Ma, Chem. Mater., 2019, 31, 3201–3210 CrossRef CAS.
- E. Yaghini, K. F. Pirker, C. W. Kay, A. M. Seifalian and A. J. MacRobert, Small, 2014, 10, 5106–5115 CrossRef CAS PubMed.
- F. Pinaud, D. King, H. P. Moore and S. Weiss, J. Am. Chem. Soc., 2004, 126, 6115–6123 CrossRef CAS PubMed.
- S. Yamashita, M. Hamada, S. Nakanishi, H. Saito, Y. Nosaka, S. Wakida and V. Biju, Angew. Chem., Int. Ed., 2015, 54, 3892–3896 CrossRef CAS.
- P. Vallecorsa, G. Di Venosa, M. B. Ballatore, D. Ferreyra, L. Mamone, D. Sáenz, G. Calvo, E. Durantini and A. Casas, BMC Cancer, 2021, 21, 547 CrossRef CAS PubMed.
- Z. Dai, X. Liang, M. Chen, P. Bhattarai and S. Hameed, ACS Nano, 2020, 14, 13569–13583 CrossRef.
- L. Yang, J. Zhou, Z. Wang, H. Li, K. Wang, H. Liu and F. Wu, Dye. Pigment., 2020, 182, 108664 CrossRef CAS.
- B. R. Xie, Y. Yu, X. H. Liu, J. Y. Zeng, M. Z. Zou, C. X. Li, X. Zeng and X. Z. A. Zhang, Biomaterials, 2021, 272, 120782 CrossRef CAS PubMed.
- L. B. Silva, K. A. D. F. Castro, C. E. A. Botteon, C. L. P. Oliveira, R. S. da Silva and P. D. Marcato, Front. Bioeng. Biotechnol., 2021, 9, 679128 CrossRef.
- H. Montaseri, C. A. Kruger and H. Abrahamse, Int. J. Mol. Sci., 2020, 21, 3358 CrossRef CAS.
- Y. Gao, D. Gao, J. Shen and Q. Wang, Front. Chem., 2020, 8, 598722 CrossRef PubMed.
- R. Kohara, K. Yuyama, Y. Shigeri and V. Biju, ChemPhotoChem, 2017, 1, 299–303 CrossRef CAS.
- H. Fidder, A. Lauer, W. Freyer, B. Koeppe and K. Heyne, J. Phys. Chem. A, 2009, 113, 6289–6296 CrossRef CAS PubMed.
- M. Nathusius, D. Sleeman, J. Pan, F. Rominger, J. Freudenberg, U. H. F. Bunz and K. Müllen, Chem.–Eur. J., 2021, 27, 16606–16610 CrossRef CAS.
- W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071–15082 CrossRef CAS.
- D. Manzanares and V. Ceña, Pharmaceutics, 2020, 12, 371 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S11. See DOI: https://doi.org/10.1039/d3sc03877g |
| This journal is © The Royal Society of Chemistry 2024 |