
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkaline-earth ion stabilized sub-nano-platinum tin clusters for propane dehydrogenation†
Zhenpu
Lu
 ab,
Ran
Luo
ac,
Sai
Chen
ab,
Donglong
Fu
ab,
Guodong
Sun
abc,
Zhi-Jian
Zhao
abd,
Chunlei
Pei
*ab and
Jinlong
Gong
*abcd
ab,
Ran
Luo
ac,
Sai
Chen
ab,
Donglong
Fu
ab,
Guodong
Sun
abc,
Zhi-Jian
Zhao
abd,
Chunlei
Pei
*ab and
Jinlong
Gong
*abcd
aKey Laboratory for Green Chemical Technology of Ministry of Education, School of Chemical Engineering and Technology, Tianjin University, China. E-mail: jlgong@tju.edu.cn; chunlei.pei@tju.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin 300072, China
cJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China
dHaihe Laboratory of Sustainable Chemical Transformations, Tianjin 300192, China
First published on 15th December 2023
Abstract
The strong promotion effects of alkali/alkaline earth metals are frequently reported for heterogeneous catalytic processes such as propane dehydrogenation (PDH), but their functioning principles remain elusive. This paper describes the effect of the addition of calcium (Ca) on reducing the deactivation rate of platinum–tin (Pt–Sn) catalyzed PDH from 0.04 h−1 to 0.0098 h−1 at 873 K under a WHSV of 16.5 h−1 of propane. The Pt–Sn–Ca catalyst shows a high propylene selectivity of >96% with a propylene production rate of 41 molC3H6 (gPt h)−1 and ∼1% activity loss after regeneration. The combination of characterization and DFT simulations reveals that Ca acts as a structural promoter favoring the transition of Snn+ in the parent catalyst to Sn0 during reduction, and the latter is an electron donor that increases the electron density of Pt. This greatly suppresses coke formation from deep dehydrogenation. Moreover, it was found that Ca promotes the formation of a highly reactive and sintering-resistant sub-nano Pt–Sn alloy with a diameter of approximately 0.8 nm. These lead to high activity and selectivity for the Pt–Sn–Ca catalyst for PDH.
Introduction
Propylene, a significant chemical feedstock, has encountered mounting conflict between supply and demand in recent years.1 The propane dehydrogenation (PDH) process, which can directly yield propylene, has attracted great attention due to its economic viability2,3 and continues to increase in the share of global propylene production.4,5 Pt-based catalysts are widely utilized in the chemical industry for PDH.6–8 The key to their success is the high stability realized by the addition of alkali metal K.9 A similar stabilization effect has also been observed with alkaline-earth metals such as Ca for PDH.9,10 In fact, both alkali and alkaline-earth metals have been recognized as “universal” additives in a variety of other processes, such as Fischer–Tropsch synthesis,11–14 ammonia synthesis,15,16etc.Generally, these promoters are broadly classified into electronic or structural additives.17,18 Alkali/alkaline-earth metals were found to be as electron donors that modify the electron density of active sites and they consequently alter the adsorption energy of reaction intermediates.19–21 However, alkali/alkaline-earth metals are known to appear in the ionic state rather than the metallic counterpart due to their strong metallic nature.22 An alternate perspective postulates that alkali/alkaline-earth ions interact with the active metal center profile by binding with the surrounding oxygen atoms, thereby stabilizing the active metal and preventing its sintering at high temperatures.23–26 Nevertheless, the exact working mechanisms of their promoting effect as either electronic or structural additives in catalysis continue to be the subject of ongoing debate.
Herein, we demonstrate that alkaline-earth metals play multiple roles in the catalytic dehydrogenation on platinum–tin catalysts. Calcium can not only stabilize sub-nano Pt–Sn clusters by binding to the AlV sites on the Al2O3 surface to inhibit the sintering of clusters, but also promote the transition from tin oxide species to Sn0 species during reduction, which leads to the formation of a highly reactive sub-nano platinum–tin alloy with a diameter of approximately 0.8 nm. The effects of the PtSn3Ca5/Al2O3 catalyst are evidenced by its highly active and stable performance in the propane dehydrogenation reaction. Specifically, the catalyst displays an extremely low deactivation rate of 0.0098 h−1 and propylene selectivity exceeding 96% at 600 °C under a propane weight hourly space velocity (WHSV) of 16.5 h−1, with a corresponding propylene production rate of 41 molC3H6 (gPt h)−1, and shows only ∼1% activity loss after regeneration under an air atmosphere. Such remarkable attributes hold great promise for advancing the PDH process.
Results and discussion
PtSn3/Al2O3 and PtSn3Ca5/Al2O3 catalysts were prepared first via the incipient wetness impregnation method. The PtSn3Ca5/Al2O3 catalyst (Fig. 1a) exhibited high catalytic stability in the PDH process with 14% C3H8/14% H2/N2 at 873 K. No apparent loss in activity was observed after 1% O2 regeneration under 773 K on the PtSn3Ca5/Al2O3 catalyst (Fig. 1b), which exhibited better stability for regeneration compared with the catalyst without the addition of calcium (Fig. S1†). Performance testing (Fig. 1c and d) under industrial relevant conditions27 (873 K, WHSV of C3H8 = 16.5 h−1, and C3H8/H2 = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) showed a high C3H6 production rate of 41 molC3H6 (gPt h)−1 with a markedly lower deactivation rate of 0.0098 h−1 for PtSn3Ca5/Al2O3 compared to that of the commercial Pt–Sn–K/Al2O3 catalyst (0.04 h−1) and reported Al2O3 supported catalysts (Table S1†). To show the potential of the PtSn3Ca5/Al2O3 catalyst activated under the optimized conditions, we tested the catalytic performance of the catalyst under a WHSV of ∼100 h−1 (Fig. S2†). The results show a low deactivation rate of 0.007 h−1 in a course of 45 hours on stream with a stable propylene selectivity of >98%, among the best performing catalyst in the literature (Fig. S3 and Table S1†). Besides, the introduction of other alkaline-earth ions (Mg and Sr) also shows enhanced PDH performance (Fig. S4†). These results altogether suggest that the addition of alkaline-earth ions especially Ca greatly promotes the stability and activity of the Pt–Sn catalyst for PDH.
:1) showed a high C3H6 production rate of 41 molC3H6 (gPt h)−1 with a markedly lower deactivation rate of 0.0098 h−1 for PtSn3Ca5/Al2O3 compared to that of the commercial Pt–Sn–K/Al2O3 catalyst (0.04 h−1) and reported Al2O3 supported catalysts (Table S1†). To show the potential of the PtSn3Ca5/Al2O3 catalyst activated under the optimized conditions, we tested the catalytic performance of the catalyst under a WHSV of ∼100 h−1 (Fig. S2†). The results show a low deactivation rate of 0.007 h−1 in a course of 45 hours on stream with a stable propylene selectivity of >98%, among the best performing catalyst in the literature (Fig. S3 and Table S1†). Besides, the introduction of other alkaline-earth ions (Mg and Sr) also shows enhanced PDH performance (Fig. S4†). These results altogether suggest that the addition of alkaline-earth ions especially Ca greatly promotes the stability and activity of the Pt–Sn catalyst for PDH.
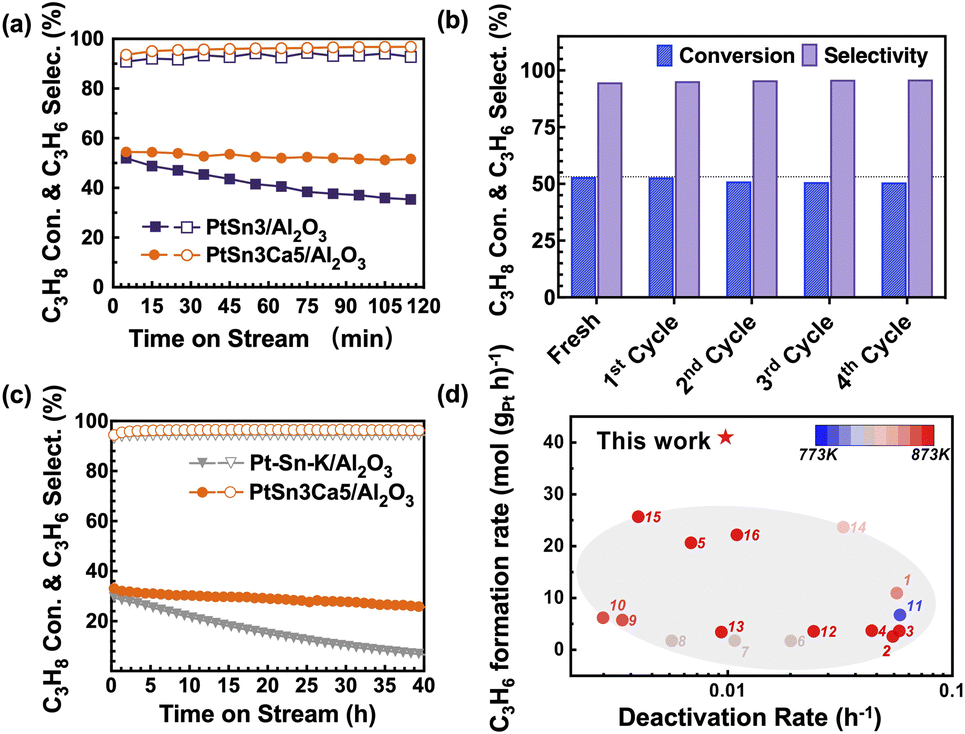 | ||
| Fig. 1 Catalytic performance and stability test for propane dehydrogenation. (a) Propane conversion and propylene selectivity as a function of reaction time over the prepared PtSn3/Al2O3 and PtSn3Ca5/Al2O3 at 873 K. Catalytic conditions: atmospheric pressure, C3H8/H2 = 1/1, with balance N2 for a total flow rate of 50 mL min−1, WHSV of propane = 8.25 h−1, and 100 mg of sample. (b) Initial catalytic performances on the PtSn3Ca5/Al2O3 catalyst for each of the five dehydrogenation–regeneration cycles. Catalytic conditions: atmospheric pressure, C3H8/H2 = 1/1, with balance N2 for a total flow rate of 50 mL min−1, WHSV of propane = 8.25 h−1 and 100 mg of sample. (c) Stability test as a function of reaction time over the prepared Pt–Sn–K/Al2O3 and PtSn3Ca5/Al2O3 at 873 K. Catalytic conditions: 0.13 MPa, C3H8/H2 = 2/1, total flow rate 21 mL min−1, no N2 dilution, WHSV of propane = 16.5 h−1 and 100 mg of sample. (d) Productivity of C3H6versus deactivation rate for the Al2O3 supported catalysts described in this work and the literature (see Table S1†). | ||
The role of alkaline-earth ions was further investigated with calcium as a representative. High-resolution 27Al solid state MAS NMR spectra28 (see details in Fig. S5†) indicate that Ca atoms bind to the Alpenta3+ sites at the Al2O3 surface via oxygen bridges. A previous study reported that Pt atoms also coordinate to the same sites on Al2O3.29,30 Thus, it can be inferred that the Ca atoms are closely adjacent to the Pt atoms in PtSn3Ca5/Al2O3, consistent with the results from EDS mapping (Fig. S6†). The dark-field electron microscopy images (Fig. 2a–d) of the fresh catalysts show higher dispersion of Pt (∼0.8 ± 0.3 nm) in the PtSn3Ca5/Al2O3 catalyst compared to the Ca-free analog (∼1.0 ± 0.6 nm). H2-chemisorption experiments of the fresh catalysts suggest that Ca enhanced the exposure of platinum, as evidenced by 1.3 times greater hydrogen adsorption on PtSn3Ca5/Al2O3 than that on PtSn3/Al2O3 (Table S2†). The study of the used catalysts after three dehydrogenation cycles shows that the dispersion of Pt–Sn NPs in PtSn3Ca5/Al2O3 remained intact with a negligible increase to 0.9 ± 0.3 nm, while that for the Ca-free material increased by 30% to ∼1.3 ± 0.5 nm. This implies that the introduction of Ca into the catalyst resulted in significant inhibition of catalyst sintering, and thus the size of the sub-nanometer particle remains intact even after undergoing three cycling regeneration reactions. DFT calculations (Fig. S7†) indicate that the migration of CaOx has a higher activation barrier than SnOx (2.14 eV for CaOx and 1.83 eV for SnOx) on the alumina surface. The aggregation of SnOx and CaOx could inhibit the migration compared with the aggregation of two SnOx clusters. The high migration barrier on CaSnOx is preserved after loading of Pt. Further experiments indicate that the catalyst's coke resistance is augmented with the inclusion of calcium. As is shown in Fig. 2e and f, the carbon generation on both catalysts increased rapidly at the beginning and gradually stabilized at different temperatures. Both the final coke content and the coke formation rate in the linear and burst zones increased with temperature, which is similar to the literature.31 The apparent activation energy (Fig. S8†) calculated through the monolayer–multilayer coke growth model (MMCGM)31,32 for both monolayer coking (70.85 kJ mol−1) and multilayer coking (178.94 kJ mol−1) on PtSn3Ca5/Al2O3 is higher than that on PtSn3/Al2O3 (32.70 kJ mol−1 and 119.60 kJ mol−1, respectively), suggesting that the coke formation on the former catalyst was inhibited by the introduction of calcium. Altogether, these results demonstrate that the addition of Ca prevented the sintering of Pt–Sn nano-clusters as well as coking deposits during dehydrogenation cycles under PDH conditions.
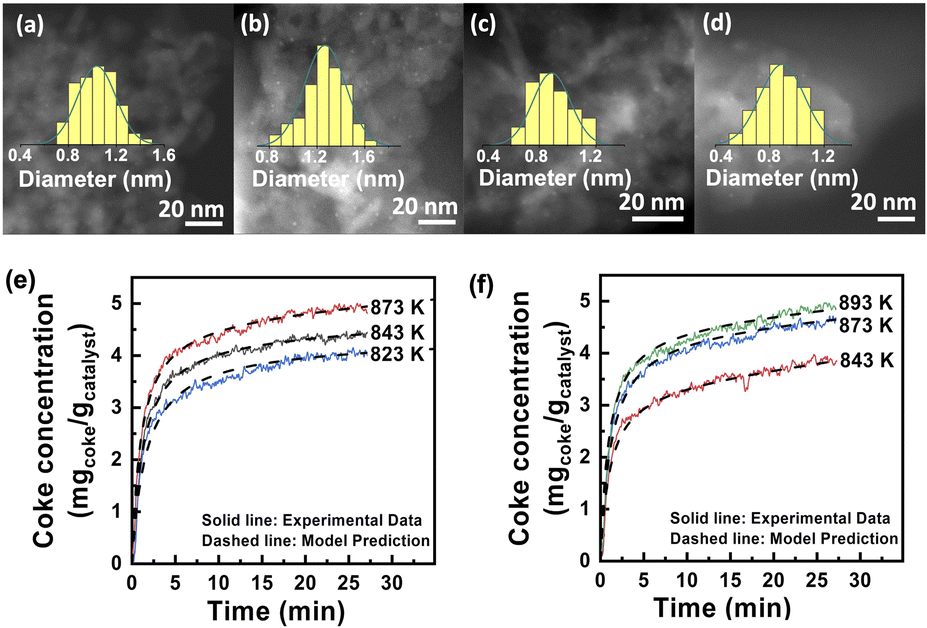 | ||
| Fig. 2 (a) HAADF-STEM images of the PtSn3/Al2O3 catalyst after reduction in a flow of 18 vol% H2/N2 at 873 K for 1 hour; (b) HAADF-STEM images of the PtSn3/Al2O3 catalyst after three dehydrogenation–regeneration cycles; catalytic conditions: atmospheric pressure, C3H8/H2 = 1/1, with balance N2 for a total flow rate of 50 mL min−1, WHSV of propane = 8.25 h−1 and 100 mg of sample; (c) HAADF-STEM images of the PtSn3Ca5/Al2O3 catalyst after reduction in a flow of 18 vol% H2/N2 at 873 K for 1 hour; (d) HAADF-STEM images of the PtSn3Ca5/Al2O3 catalyst after three dehydrogenation cycles; catalytic conditions: atmospheric pressure, C3H8/H2 = 1/1, with balance N2 for a total flow rate of 50 mL min−1, WHSV of propane = 8.25 h−1 and 100 mg of sample. Coke concentration in the catalyst as a function of time for different temperatures on (e) the PtSn3/Al2O3 catalyst and (f) PtSn3Ca5/Al2O3; catalytic conditions: 10 mg of catalysts, total flow rate = 110 mL min−1, and C3H8/N2 ratio = 1/10. | ||
Next, the working principles of the aforementioned sintering and coking resistance effects from Ca were comprehensively investigated. The introduction of Ca into the catalyst shifted the vibrational frequency of the linearly bonded CO band to 2076 cm−1 (Fig. 3a), indicating the perturbation of the electronic structure of surface Pt atoms.33,34 The vibrational frequency of the linearly bonded CO band can be attributed primarily to the size of the Pt particles35 and the electronic state of Pt.4,36,37 Given that PtSn3Ca5/Al2O3 and PtSn3/Al2O3 possess similar particle sizes (as shown in Fig. 2a and c), the difference in the CO linear adsorption peak position primarily arises from fluctuations in the electron density on Pt. The redshift of the vibrational frequency of the linearly bonded CO band indicates a strong backdonation of electrons from Pt atoms, suggesting an increase in the electron density on the Pt atoms by the addition of Ca on PtSn3Ca5/Al2O3.
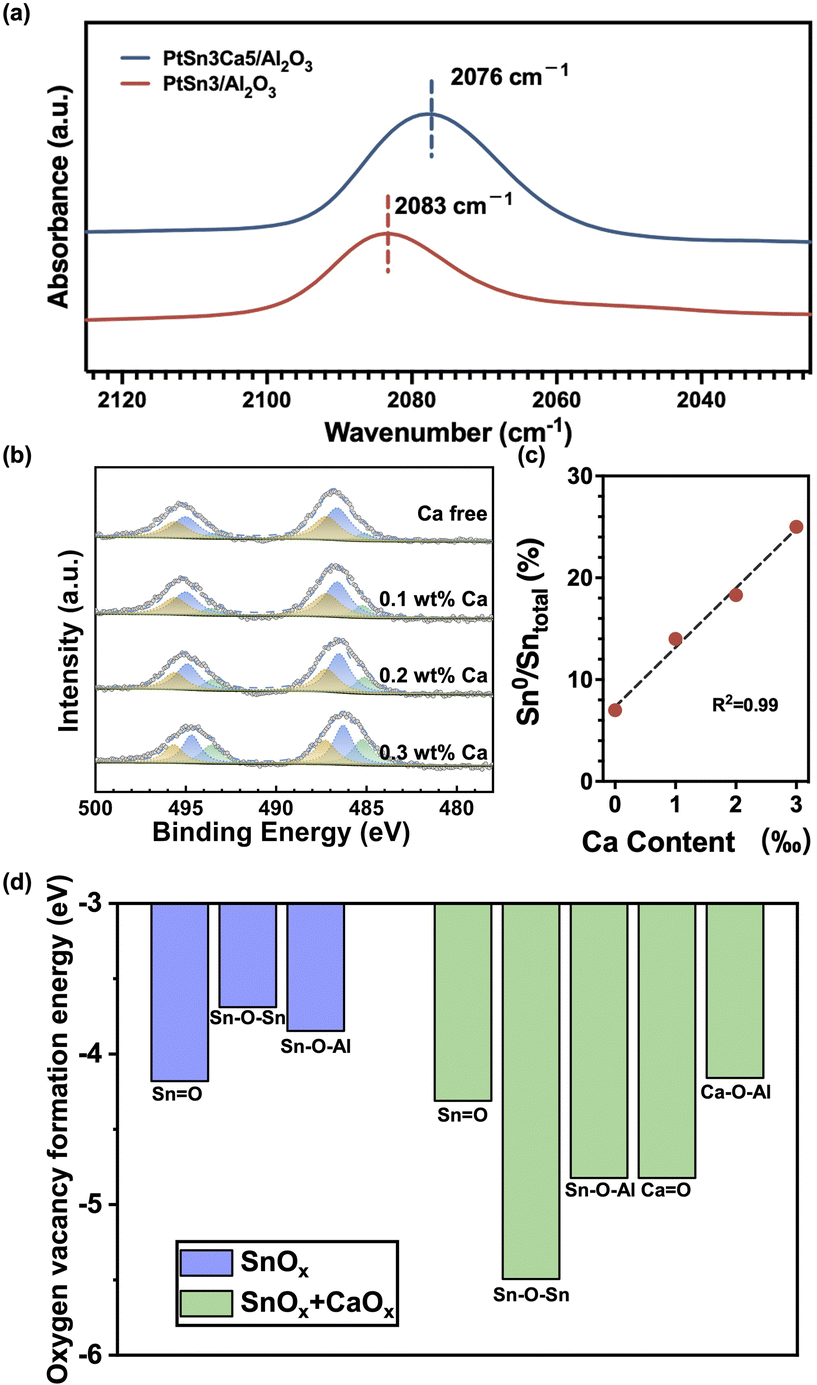 | ||
| Fig. 3 (a) CO-DRIFTS of PtSn3/Al2O3 and PtSn3Ca5/Al2O3; (b) Quasi in situ XPS spectra of Sn 3d in a series of PtSn3/Al2O3 catalysts with 0 wt% Ca, 0.1 wt% Ca, 0.2 wt% Ca or 0.3 wt% Ca (from top to bottom); (c) relationship between the Ca content and the ratio of Sn0 and Sntotal. (d) Oxygen vacancy formation energies of different oxygen species in SnOx/Al2O3 and SnOx/Al2O3 with CaOx. | ||
However, the Ca 2p XPS spectra of reduced PtCa5/Al2O3 and CaO/Al2O3 (Fig. S9†) show no shift of the Ca 2p peak, which means that there is no significant electronic interaction between this closely contacted platinum and calcium.19,20Quasi in situ XPS results obtained from reduced PtSn3/Al2O3 with different Ca contents (Ca-free, 0.1 wt%, 0.2 wt%, and 0.3 wt% Ca) in Fig. 3b show that the proportion of metallic Sn increases linearly with the calcium content (Fig. 3c), consistent with previous work.38 Further DFT calculations indicate that the oxygen vacancy formation energies of Sn–Ov–Sn and Sn–Ov–Al decrease obviously after the addition of CaOx (Fig. 3d and S10†). This corroborates the results from the XPS that the introduction of calcium facilitates SnOx reduction. Additionally, the DFT results (Fig. S10†) show direct interaction between Sn and Ca as the Sn–O–Ca species was observed in the Sn and Ca containing catalyst in the reduction process. These results together with CO-DRIFTS experiments demonstrate that the addition of Ca benefited the reduction of Snn+ to Sn0, which indirectly increased the electron density of Pt, thus promoting the desorption of the product propylene and inhibiting coke deposition.39,40
Further study was performed to analyze the impact of structural changes on the reaction performance. C3H8-D2 temperature-programmed surface reaction (C3H8-D2-TPSR) analysis, which is widely used to determine the intrinsic ability to activate the C–H bonds of propane,35,41 is carried on reduced PtSn3/Al2O3 and PtSn3Ca5/Al2O3 (Fig. S11†). The rise in C–H activation temperature with increasing calcium content suggests that the dehydrogenation barrier for PtSn3Ca5/Al2O3 was potentially greater than that for PtSn3/Al2O3.35 Detailed kinetic investigations were carried out on these catalysts at relatively low partial pressures of propane and hydrogen, in order to gain deeper insights into the impact of Ca incorporation on the C–H activation process. The reaction orders in propane and hydrogen on different catalysts were calculated from the slopes of ln(TOF) versus ln(p) plots, as shown in Fig. 4a and b. The reaction order for propane remained almost constant (∼1.0), regardless of the content of Ca, while that for hydrogen differed greatly, changing from −0.21 to −0.01 with the Ca content increasing from 0 to 0.3 wt%. The increase of the reaction order for hydrogen with the Ca content potentially indicated a shift in the rate-determining step (RDS), suggesting a modification in the active species present on the catalyst surface. Note that the determination of the RDS is related to the reaction order for propane and hydrogen.41 For PtSn3/Al2O3 with a Ca content between 0 and 0.2 wt%, no obvious RDS can be identified, and it seems that the first and second C–H bond activations would jointly dominate the overall reaction rate. As the reaction order for hydrogen was close to zero on PtSn3Ca5/Al2O3, the first C–H bond activation was most likely the RDS on this catalyst, which indicates that the active site on the PtSn3Ca5/Al2O3 surface is relatively homogeneous. Besides, isotopic experiments42 (details in the ESI†) showed that a kinetic isotope effect (KIE) of 1.65 has been found on PtSn3Ca5/Al2O3, while the KIE was 2.39 for PtSn3/Al2O3 (Fig. S12†). Thus, the change in the KIE confirmed the result obtained from the DFT calculation and kinetic analysis that the structure of the catalysts changed while Ca was added to the PtSn3/Al2O3 catalysts.
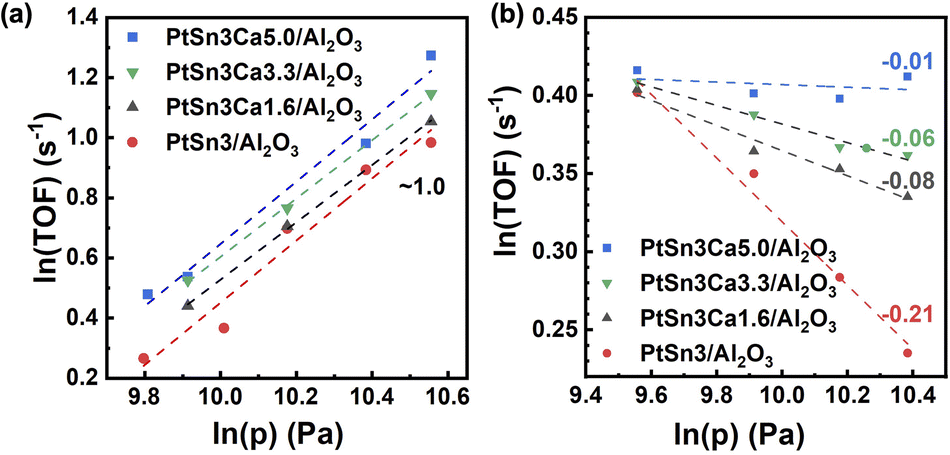 | ||
| Fig. 4 TOF of propane as a function of (a) propane partial pressure and (b) hydrogen partial pressure on PtSn3/Al2O3 catalysts with different Ca contents; catalytic conditions: atmospheric pressure, 873 K, and 5 mg of catalysts. | ||
Conclusions
In summary, we fabricated well-defined Pt–Sn/Al2O3 catalysts doped with Ca ions with a markedly improved propylene production rate of 41 molC3H6 (gPt h)−1 as well as a slow deactivation rate of 0.0098 h−1, compared to the Ca free analog. Characterization and kinetic analysis were employed to unravel the underlying mechanism of the promotion effect for propane dehydrogenation. It was found that Ca doping promoted the reduction of Snn+ to Sn0 species. The reduced tin species further led to a pronounced electronic modification of platinum. Subsequent kinetic analysis of the carbon accumulation clarified that this electron-rich Pt surface greatly inhibited the carbon accumulation process. Further kinetic analysis of the reaction process revealed that the introduction of calcium affected the surface hydrogen desorption process, and that the first C–H bond activation is the rate-determining step of the propane dehydrogenation process over the calcium-containing catalyst. The catalyst design concept advanced in this study opens a new horizon in the exploration of alkaline-earth metals in catalysis.Data availability
The data that supports the findings of this study is available from the corresponding author upon reasonable request.Author contributions
Zhenpu Lu: investigation; visualization; formal analysis; validation; writing – original draft. Ran Luo: investigation; formal analysis; validation; visualization. Sai Chen: investigation; formal analysis. Guodong Sun: investigation; formal analysis. Zhi-Jian Zhao: investigation; formal analysis; writing – review & editing. Donglong Fu: investigation; formal analysis; writing – review & editing. Chunlei Pei: investigation; formal analysis and funding acquisition. Jinlong Gong: methodology; supervision; formal analysis; writing – review & editing; resources and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2021YFA1501302), the National Natural Science Foundation of China (No. 22121004 and 22122808), the Haihe Laboratory of Sustainable Chemical Transformations, the Program of Introducing Talents of Discipline to Universities (BP0618007) and the XPLORER PRIZE. The authors also acknowledge generous computing resources at the High Performance Computing Center of Tianjin University.Notes and references
- Y. Zhou, F. Wei, H. Qi, Y. Chai, L. Cao, J. Lin, Q. Wan, X. Liu, Y. Xing, S. Lin, A. Wang, X. Wang and T. Zhang, Nat. Catal., 2022, 5, 1145–1156 CrossRef CAS.
- C. Li and G. Wang, Chem. Soc. Rev., 2021, 50, 4359–4381 RSC.
- Y. Xing, G. Bi, X. Pan, Q. Jiang, Y. Tan, Y. Su, L. Kang, B. Li, L. Li, A. Wang, J. Ma, X. Yang, X. Yan Liu and T. Zhang, J. Energy Chem., 2023, 83, 304–312 CrossRef CAS.
- A. H. Motagamwala, R. Almallahi, J. Wortman, V. O. Igenegbai and S. Linic, Science, 2021, 373, 217–222 CrossRef CAS PubMed.
- D. Li, J. Bi, Z. Xie, L. Kong, B. Liu, X. Fan, X. Xiao, Y. Miao and Z. Zhao, Sci. China: Chem., 2023, 66, 2389–2399 CrossRef CAS.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- C. L. Ye, M. Peng, Y. Li, D. S. Wang, C. Chen and Y. D. Li, Sci. China Mater., 2023, 66, 1071–1078 CrossRef CAS.
- M. Monai, M. Gambino, S. Wannakao and B. M. Weckhuysen, Chem. Soc. Rev., 2021, 50, 11503–11529 RSC.
- X.-Q. Gao, Z.-H. Yao, W.-C. Li, G.-M. Deng, L. He, R. Si, J.-G. Wang and A.-H. Lu, ChemCatChem, 2023, 15, e202201691 CrossRef CAS.
- A. J. Urquhart, J. M. Keel, F. J. Williams and R. M. Lambert, J. Phys. Chem. B, 2003, 107, 10591–10597 CrossRef CAS.
- A. N. Pour, S. M. K. Shahri, H. R. Bozorgzadeh, Y. Zamani, A. Tavasoli and M. A. Marvast, Appl. Catal., A, 2008, 348, 201–208 CrossRef.
- D. Gao, W. Li, H. Wang, G. Wang and R. Cai, Trans. Tianjin Univ., 2022, 28, 245–264 CrossRef CAS.
- Z.-H. Zhang, Z. Sun and T.-Q. Yuan, Trans. Tianjin Univ., 2022, 28, 89–111 CrossRef.
- A. Nielsen, Catal. Rev., 1971, 4, 1–26 CrossRef.
- G. Ertl, D. Prigge, R. Schloegl and M. Weiss, J. Catal., 1983, 79, 359–377 CrossRef CAS.
- P. Zhai, C. Xu, R. Gao, X. Liu, M. Li, W. Li, X. Fu, C. Jia, J. Xie, M. Zhao, X. Wang, Y.-W. Li, Q. Zhang, X.-D. Wen and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 9902–9907 CrossRef CAS PubMed.
- Z. Li, L. Zhong, F. Yu, Y. An, Y. Dai, Y. Yang, T. Lin, S. Li, H. Wang, P. Gao, Y. Sun and M. He, ACS Catal., 2017, 7, 3622–3631 CrossRef CAS.
- N. Lang, S. Holloway and J. Nørskov, Surf. Sci., 1985, 150, 24–38 CrossRef CAS.
- G. Ertl, S. Lee and M. Weiss, Surf. Sci., 1982, 114, 527–545 CrossRef CAS.
- D. Le and T. S. Rahman, Nat. Catal., 2022, 5, 977–978 CrossRef.
- J. Xie, P. P. Paalanen, T. W. van Deelen, B. M. Weckhuysen, M. J. Louwerse and K. P. de Jong, Nat. Commun., 2019, 10, 167 CrossRef PubMed.
- Y. Zhai, D. Pierre, R. Si, W. Deng, P. Ferrin, A. U. Nilekar, G. Peng, J. A. Herron, D. C. Bell and H. Saltsburg, Science, 2010, 329, 1633–1636 CrossRef CAS PubMed.
- M. Yang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2013, 135, 3768–3771 CrossRef CAS PubMed.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498–1501 CrossRef CAS PubMed.
- M. Yang, J. Liu, S. Lee, B. Zugic, J. Huang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2015, 137, 3470–3473 CrossRef CAS PubMed.
- S. Chen, Z.-J. Zhao, R. Mu, X. Chang, J. Luo, S. C. Purdy, A. J. Kropf, G. Sun, C. Pei and J. T. Miller, Chem, 2021, 7, 387–405 CAS.
- W. Wang, J. Xu and F. Deng, Natl. Sci. Rev., 2022, 9, nwac155 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- I. S. Nam and J. Kittrell, Ind. Eng. Chem. Process Des. Dev., 1984, 23, 237–242 CrossRef CAS.
- Z. Lian, C. Si, F. Jan, S. Zhi and B. Li, ACS Catal., 2021, 11, 9279–9292 CrossRef CAS.
- O. Pozdnyakova, D. Teschner, A. Wootsch, J. Kröhnert, B. Steinhauer, H. Sauer, L. Toth, F. C. Jentoft, A. Knop-Gericke, Z. Paál and R. Schlögl, J. Catal., 2006, 237, 1–16 CrossRef CAS.
- J. Wang, X. Chang, S. Chen, G. Sun, X. Zhou, E. Vovk, Y. Yang, W. Deng, Z.-J. Zhao and R. Mu, ACS Catal., 2021, 11, 4401–4410 CrossRef CAS.
- G. Sun, Z.-J. Zhao, R. Mu, S. Zha, L. Li, S. Chen, K. Zang, J. Luo, Z. Li, S. C. Purdy, A. J. Kropf, J. T. Miller, L. Zeng and J. Gong, Nat. Commun., 2018, 9, 4454 CrossRef PubMed.
- L. Liu, M. Lopez-Haro, C. W. Lopes, S. Rojas-Buzo, P. Concepcion, R. Manzorro, L. Simonelli, A. Sattler, P. Serna, J. J. Calvino and A. Corma, Nat. Catal., 2020, 3, 628–638 CrossRef CAS.
- V. J. Cybulskis, B. C. Bukowski, H.-T. Tseng, J. R. Gallagher, Z. Wu, E. Wegener, A. J. Kropf, B. Ravel, F. H. Ribeiro and J. Greeley, ACS Catal., 2017, 7, 4173–4181 CrossRef CAS.
- H. Jiang and D. A. Stewart, ACS Appl. Mater. Interfaces, 2017, 9, 16296–16304 CrossRef CAS PubMed.
- F. Jiang, L. Zeng, S. Li, G. Liu, S. Wang and J. Gong, ACS Catal., 2015, 5, 438–447 CrossRef CAS.
- S. Sun, G. Sun, C. Pei, Z.-J. Zhao and J. Gong, J. Phys. Chem. C, 2021, 125, 18708–18716 CrossRef CAS.
- J. Zhu, M.-L. Yang, Y. Yu, Y.-A. Zhu, Z.-J. Sui, X.-G. Zhou, A. Holmen and D. Chen, ACS Catal., 2015, 5, 6310–6319 CrossRef CAS.
- K. Chen, E. Iglesia and A. T. Bell, J. Catal., 2000, 192, 197–203 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04310j |
| This journal is © The Royal Society of Chemistry 2024 |