
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective template-directed synthesis of antibody Fc conjugates with concomitant ligand release†
Viktoriia
Postupalenko‡
 a,
Léo
Marx‡
b,
Mathilde
Pantin
b,
David
Viertl
cd,
Nadège
Gsponer
a,
Gaëlle
Giudice
a,
Natalia
Gasilova
e,
Margret
Schottelius
cf,
Frédéric
Lévy
g,
Patrick
Garrouste
b,
Jean-Manuel
Segura
a and
Origène
Nyanguile
*a
a,
Léo
Marx‡
b,
Mathilde
Pantin
b,
David
Viertl
cd,
Nadège
Gsponer
a,
Gaëlle
Giudice
a,
Natalia
Gasilova
e,
Margret
Schottelius
cf,
Frédéric
Lévy
g,
Patrick
Garrouste
b,
Jean-Manuel
Segura
a and
Origène
Nyanguile
*a
aInstitute of Life Technologies, HES-SO Valais-Wallis, Rue de l'Industrie 23, CH-1950 Sion, Switzerland. E-mail: origene.nyanguile@hevs.ch
bDebiopharm Research & Manufacturing SA, Campus “après-demain”, Rue du Levant 146, 1920 Martigny, Switzerland
cTranslational Radiopharmaceutical Sciences, Departments of Nuclear Medicine and of Oncology, CHUV/UNIL, 1011 Lausanne, Switzerland
dIn Vivo Imaging Facility, Department of Research and Training, University of Lausanne, CH-1011, Lausanne
eEPFL Valais Wallis, MSEAP, ISIC-GE-VS, rue de l'Industrie 17, 1951 Sion, Switzerland
fAgora, pôle de recherche sur le cancer, 1011 Lausanne, Switzerland
gDebiopharm International SA, Forum “après-demain”, Chemin Messidor 5-7, Case postale 5911, 1002 Lausanne, Switzerland
First published on 14th December 2023
Abstract
Template-directed methods are emerging as some of the most effective means to conjugate payloads at selective sites of monoclonal antibodies (mAbs). We have previously reported a method based on an engineered Fc-III reactive peptide to conjugate a radionuclide chelator to K317 of antibodies with the concomitant release of the Fc-III peptide ligand. Here, our method was redesigned to target two lysines proximal to the Fc-III binding site, K248 and K439. Using energy minimization predictions and a semi-combinatorial synthesis approach, we sampled multiple Fc-III amino acid substituents of A3, H5, L6 and E8, which were then converted into Fc-III reactive conjugates. Middle-down MS/MS subunit analysis of the resulting trastuzumab conjugates revealed that K248 and K439 can be selectively targeted using the Fc-III reactive variants L6Dap, L6Orn, L6Y and A3K or A3hK, respectively. Across all variants tested, L6Orn-carbonate appeared to be the best candidate, yielding a degree and yield of conjugation of almost 2 and 100% for a broad array of payloads including radionuclide chelators, fluorescent dyes, click-chemistry reagents, pre-targeted imaging reagents, and some cytotoxic small molecules. Furthermore, L6Orn carbonate appeared to yield similar conjugation results across multiple IgG subtypes. In vivo proof of concept was achieved by conjugation of NODAGA to the PD1/PD-L1 immune checkpoint inhibitor antibody atezolizumab, followed by PET imaging of PD-L1 expression in mice bearing PD-L1 expressing tumor xenograft using radiolabeled [64Cu]Cu-atezolizumab.
Introduction
Site-selective conjugation chemistries have become an important means of building antibody drug conjugates (ADCs). It is widely accepted that site-specific conjugation of a payload in a region of the antibody that is not involved in antigen recognition improves the pharmacodynamic effect and therapeutic window of the product, as illustrated by several studies.2–12 In the ADC field, site-specific technologies of all types now dominate new ADCs entering into clinical trials. However, the recent work of ImmunoGen comparing homogenous and heterogenous ADCs that generate the same metabolites, suggests that site-specific technologies may not always enhance the pharmacokinetics of the drug and may also detrimentally alter its toxicity profile.13–15 In fact, several criteria such as the nature of the payload, the linker, the conjugation chemistry, the drug-antibody ratio (DAR), the hydrophobicity of the ADC may have an impact on the in vivo properties of the conjugate, which are for the time being difficult to predict. The large number of upcoming clinical studies of site-specifically prepared ADCs may help clarifying if there is a single conjugation chemistry that will become of widespread use, or whether other methods will also be applicable. Therefore, developing various technologies is of interest for further progress in the field.Site-specific conjugation to an antibody is challenging due to the large number of solvent-exposed nucleophilic amino acids, in particular lysines. Despite this difficulty, the field has been very prolific through developing a wide array of technologies that can be summarized as engineered cysteines, disulfide rebridging, non-canonical amino acids, C-/N-terminal modifications, transglutaminase, other enzymatic methods and glycan modification (reviewed by Walsh et al.16). However, all these methods require to modify the antibody through mutating, subcloning, reducing or enzymatic reaction that may ultimately alter the pharmacological properties of the ADCs. To address these issues, site-selective lysine conjugation methods are emerging as promising tools.17 Among these, template-directed methods in which a ligand is used to bring a chemical reactive site in proximity to an antibody lysine are of particular interest,18–26 firstly because the ligand is amenable to regioselectivity through binding to the Fc or F(ab′)2 domain, secondly because the chemistry is engineered onto the ligand and not the antibody. In the first-generation technologies, the ligand remained attached to the antibody resulting in possible loss of FcRn binding due to the overlap between the binding site of the ligand and FcRn.19,20 To solve this issue, methods where the ligand is removed following the conjugation step (traceless chemistry) were developed,18,25,26 but they usually require several chemical steps to achieve this goal. We have recently reported a novel template-directed method, which allows to attach a radionuclide chelator to the Fc domain of antibodies with the concomitant release of the ligand, thereby providing an antibody Fc conjugate in a single step.1 Following our publication, another one-pot traceless chemistry was reported.27 Although our approach was regioselective to the Fc domain, we did observe some unspecific conjugation to the F(ab)2′ domain and could not achieve 100% conjugation. To be applicable to ADCs site-specific engineering, achieving near 100% modification at a single lysine residue in the Fc region of the antibody is required. Here, we wish to improve the efficiency of our methodology with the aim of expanding our approach to a wider range of payloads.
Results and discussion
In our original publication describing a ligand-templated approach, Fc-III was equipped with a PEG spacer at its N-terminus to bring a carbonate cleavable reactive site linked to a radionuclide chelator in proximity to a nucleophilic antibody lysine (K317) located 17–26 Å away from the Fc-III ligand binding site.1 Using DOTA-PEG10-Fc-III, we were able to attach DOTA to trastuzumab with a labeling efficiency of 72% and a Fc/Fab selectivity of 4.1. Unspecific conjugation at the Fab domain may alter antigen recognition and achieving 100% modification at single lysine of the Fc region may create ADCs with favorable PK properties. We hypothesized that the non-specific payload conjugation observed at the F(ab′)2 domain (DoC F(ab′)2 of 0.22) may be due to the high flexibility of the PEG spacer, which does not allow the carbonate reactive site to be present at all times in proximity with the antibody targeted lysine (K317). Attaching the chemically cleavable reactive site directly onto the Fc-III ligand without spacer may restrict flexibility and improve regioselectivity toward the IgG Fc region by increasing the local effective concentration of the reactive termini, and thereby increasing the kinetics of the reaction when compared to our previous spacer approach. In that case, the chemically cleavable reactive site should be attached directly to the N- or C-terminus of Fc-III, or to one of its amino-acid side chains. Inspection of the X-ray crystal structure of the Fc-III/IgG Fc bound complex28 revealed that the N- and C-terminus of Fc-III are located at the opposite end of the binding site and are therefore not suitable for this approach. On the other hand, Fc-III His5, Leu6 and Glu8 appear to be located in proximity to Lys248 (Fig. 1A), a conserved residue located in the Fc region of most human IgG antibodies. By substituting His5, Leu6 or Glu8 with a side chain bearing an amine moiety, the chemically cleavable reactive site can be attached to Fc-III via an amide bond linkage using the chemistry concept we devised previously1 (Fig. 1B), thereby providing a Fc-III reactive conjugate which should allow to conjugate the payload selectively to Lys248.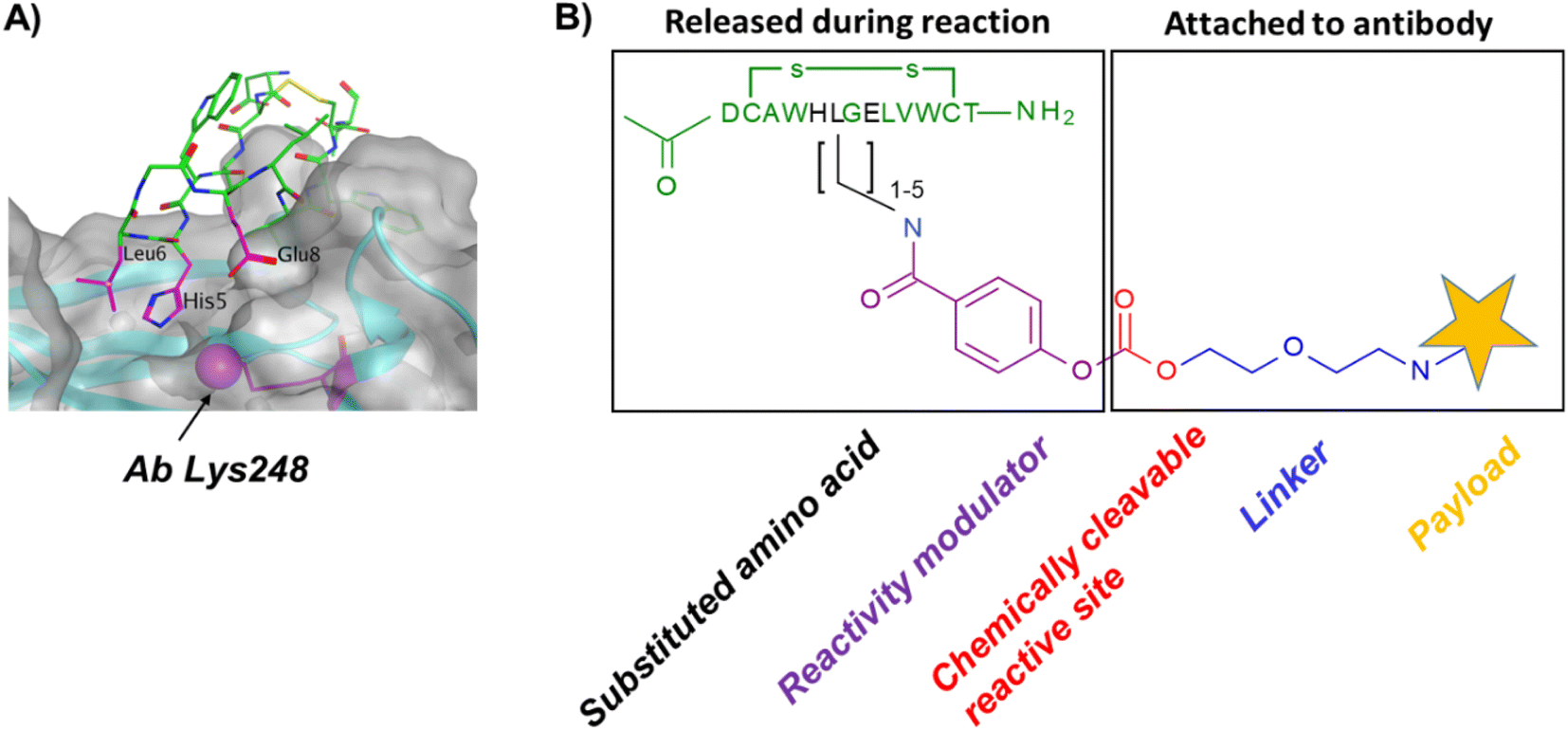 | ||
| Fig. 1 (A) Overview of the IgG Fc-III binding site. The IgG surface is highlighted in grey, and Fc-III (tubular structure) is shown in green, except for the residues in proximity with Ab Lys 248, His5, Leu6 and Glu8, which are color-coded in magenta. (B) Fc-reactive conjugate; His5, Leu6 or Glu8 (shown in black) are substituted with a residue bearing an amino moiety. The chemically cleavable reactive site is stable in aqueous media, yet sufficiently reactive to react with an antibody lysine. The reactivity modulator fine tunes the reactivity of the reactive site and ensures that the ligand is released concomitantly with the conjugation of the payload to the antibody and detached from the antibody after acidic treatment. | ||
Fc-III lysine scan suggests that Leu6, Glu8 and Ala3 are the best residues for site selective conjugation
To identify the best Fc-III residues to attach to the chemically cleavable reactive site, we performed a lysine scanning mutagenesis experiment. Each Fc-III amino acid was substituted with a lysine, except at positions Cys2 and Cys12 which are used for a disulfide bridge. The impact of Lys substitution for binding to trastuzumab was assessed using a biochemical fluorescence polarization (FP) competition assay.1 Briefly this assay is based on the fluorescence polarization signal that can be measured when a fluorescently labeled Fc-III probe (Fc-III-FAM) is bound to trastuzumab. If the binding affinity of the variant peptide is not affected by the substitution, it should be able to compete with binding of Fc-III-FAM to trastuzumab and yield IC50 values. We found that most variants were able to decrease the FP signal in a similar extent than Fc-III, except for W4K, V10K and W11K, which displayed IC50 values higher than 3 μM (Table S1†). Since the addition of the chemically cleavable site and the payload to Fc-III may also result in higher IC50 values, and the antibody concentration used during the conjugation step is of 34 μM, it can be expected that W4K, V10K and W11K will have weak binding affinities to Fc-III.Next, the Fc-III lysine variants were converted into Fc-III DOTA reactive conjugates by coupling the substituted lysine to compound 1 by HATU coupling (Scheme 1A).1 The resulting Fc-III-K-DOTA reactive conjugates were then used to conjugate DOTA to trastuzumab and the antibody-DOTA conjugates were characterized by LC-FTMS analysis as reported previously.1 Briefly, the antibody conjugation efficiency and degree of conjugation (DoC) was determined by LC-FTMS analysis of the intact antibody conjugate following deglycosylation of the trastuzumab-DOTA conjugate with EndoS, and the Fc/F(ab)2′ selectivity was determined by LC-FTMS analysis of the F(ab)2′ and Fc subunits following digestion of trastuzumab-DOTA conjugate with GinghisKHAN. Consistent with the FP competition data, the W4K, V10K and W11K reactive conjugates yielded poor conjugation of DOTA to trastuzumab, resulting in only 20% of labeled antibody and a very low Fc/F(ab′)2 selectivity (Table 1; 0.01, 0.11 and 0.20, respectively). Similarly, the D1K, G7K, L9K and T13K Fc-III reactive conjugates yielded low Fc/Fab selectivity (1.01, 1.44, 1.68 and 0.09, respectively). For simplification, F(ab′)2 is referred to Fab throughout the text. These results are expected given that D1, G7, L9 and T13 are all pointing away from the Fc-III binding site. On the other hand, the antibody labeling efficiency increased significantly for the H5K, L6K and E8K Fc-III-DOTA reactive conjugates (Table 1), consistent with the fact that H5, L6 and E8 are deeply buried in the antibody binding site in close proximity to K248 (Fig. 1A).28 Surprisingly, the conjugation data of Fc-III A3K-DOTA were also excellent, despite that A3 appears to be located far from K248. Inspection of the X-ray structure of Fc-III/IgG Fc bound complex suggests that K439 may be the antibody lysine targeted by this variant. In summary, the best results were observed with A3K, E8K and L6K yielding a DoC of 1.37, 1.70 and 1.24, and a Fc/Fab selectivity of 25.85, 3.97 and 15.52, respectively. These data are a significant improvement in selectivity over our former approach with DOTA-PEG10-Fc-III (DoC = 1.2 and Fc/Fab = 4.1).
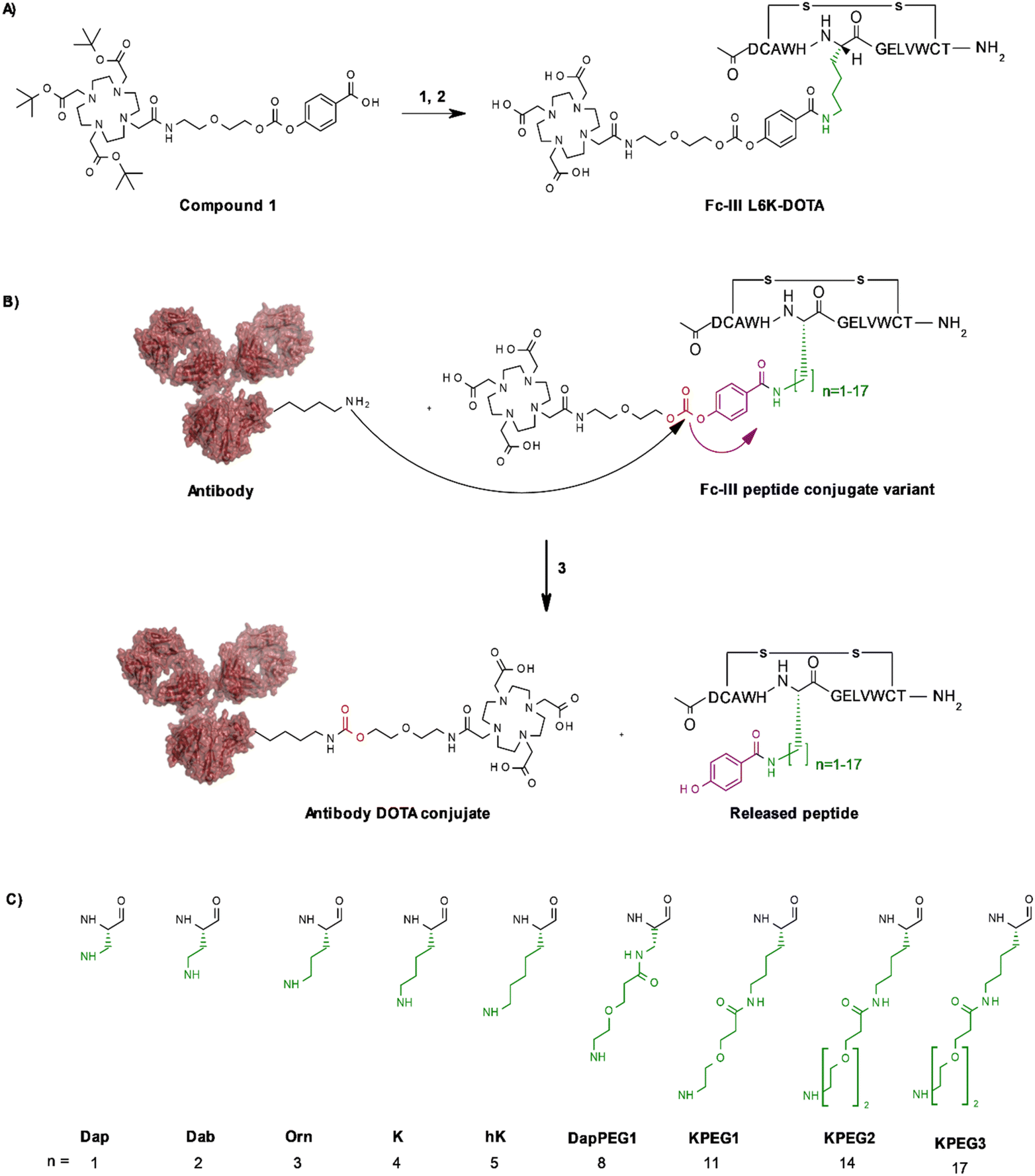 | ||
| Scheme 1 Fc-III lysine and variant scan (A) synthesis of Fc-III-L6K-DOTA reactive conjugate. (1) HATU, DIEA, DMF, Fc-III-L6K peptide, compound 11; (2) TFA/TIS/H2O. (B) Synthesis of antibody-DOTA conjugates; the Fc-III side chains that are varied are color coded in green, the reactivity modulator and the carbonate reactive center in violet and red, respectively; (3) 34 μM of antibody, 2 equivalents of peptide conjugate, pH 9, 2 h incubation at room temperature. (C) Chemical structures of variants used in this study. As the amino acids bearing the hydrocarbon side chains beyond n = 5 are not commercially available, we attached 1, 2 or 3 PEG units via an amide bond to the A3 Dap or L6K variants. The main and side chain of each variant are shown in black and green, respectively. | ||
| Peptide conjugates | DoC mAba | Labeled mAb | Selectivity Fc/Fabb | DoC Fcb | DoC Fabc |
|---|---|---|---|---|---|
| a The DOTA-antibody conjugates were deglycosylated with EndoS enzyme and then characterized by liquid chromatography coupled with orbitrap fourier transform mass spectrometry (LC-FTMS) under denaturing conditions, thereby providing the degree of conjugation (DoC) of DOTA molecules. b For simplification, F(ab′)2 is referred to Fab throughout the text. c To calculate the conjugation selectivity (DoC Fc and DoC F(ab′)2), the DOTA-antibody conjugates were then treated with GingisKHAN™ enzyme, and the digested fragments were analyzed by LC-FTMS. d When the sum of DoC of Fc plus F(ab′)2 was much lower than the DoC of intact mAb, the Fc/F(ab′)2 selectivity was extrapolated based on extrapolated Doc Fc. | |||||
| Fc-III-D1K-DOTAd | 0.52 | 42% | 0.22 (1.01d) | 0.06 | 0.26 |
| Fc-III-A3K-DOTA | 1.37 | 85% | 25.85 (29.40d) | 1.16 | 0.04 |
| Fc-III-W4K-DOTA | 0.23 | 21% | 0.01 | 0 | 0.31 |
| Fc-III-H5K-DOTA | 0.37 | 33% | 3.85 | 0.37 | 0.10 |
| Fc-III-L6K-DOTA | 1.24 | 82% | 15.52 | 1.19 | 0.08 |
| Fc-III-G7K-DOTA | 0.66 | 49% | 0.29 (1.44d) | 0.08 | 0.27 |
| Fc-III-E8K-DOTA | 1.70 | 90% | 3.97 (5.42d) | 1.05 | 0.26 |
| Fc-III-L9K-DOTA | 0.30 | 27% | 0.11 (1.68d) | 0.01 | 0.11 |
| Fc-III-V10K-DOTA | 0.19 | 18% | 0.11 | 0.02 | 0.19 |
| Fc-III-W11K-DOTA | 0.27 | 24% | 0.20 (0.80d) | 0.03 | 0.15 |
| Fc-III-T13K-DOTA | 0.20 | 19% | 0.09 | 0.02 | 0.20 |
Fc-III-L6Orn-DOTA, Fc-III-E8Orn-DOTA and Fc-III-A3hK-DOTA yield excellent antibody labeling efficiency and exquisite Fc selectivity
To further improve the antibody conjugation efficiency and selectivity of our method, we sought to optimize the length of the side chains A3K, L6K and E8K to bring the chemically cleavable reactive site as close as possible to the antibody nucleophilic lysine. Energy minimization was performed§ to estimate the binding affinity of the Fc-III-K-DOTA reactive conjugates to the antibody as well as the distance of the antibody lysine to the carbonate reactive site. As can be seen in Table 2, K248 appeared to be at 9.29 and 4.14 Å of the carbonate reactive site for L6K and E8K, respectively. In the case of Fc-III-A3K-DOTA, K439 appeared to be at 4.97 Å. Taking into account that our model may deviate from the actual Fc-III/IgG Fc interaction seen in the X-ray structure, we devised a limited combinatorial strategy whereby multiple Fc-III amino acid side chain lengths were sampled based on our energy minimization predictions. This was performed by replacing A3, L6 and E8 with Dap, Dab, Orn, hK, DapPEG1, KPEG1, KPEG2 or KPEG3 thereby varying the number (n) of carbon (or heteroatom for PEG containing variants) of the amine bearing side chain to 1, 2, 3, 5, 8, 11, 14 and 17, respectively (Scheme 1B and C, Table 2). The resulting Fc-III variants were then converted into Fc-III-X-DOTA reactive conjugates as described above for the K substituents and used to conjugate DOTA to trastuzumab as described above (Scheme 1B). LC-FTMS analysis of the antibody-DOTA conjugates showed that the A3hK, L6Orn and E8Orn Fc-III DOTA variants yielded excellent results with antibody labeling efficiencies of 92%, 99% and 97%, respectively, and negligeable labeling at the Fab domain with DoC Fab values of 0.03, 0.05 and 0, respectively (highlighted in bold in Table 2). Overall, the conjugation results were consistent with the energy minimization data, where the best results were achieved at the shorter distance of the carbonate center to K248 or K439, and the highest affinity of the Fc-III reactive conjugate-antibody bound complex. Unexpectedly, when the length of the L6 variant was extended beyond the most optimal distance to K248, i.e. L6KPEG1, L6KPEG2 and L6KPEG3, the conjugation results turned out to be also excellent, however with a decrease of Fc/Fab selectivity with L6KPEG3. Altogether, we have discovered 3 variants, A3hK, L6Orn and E8Orn, which allow to conjugate DOTA to trastuzumab with high degree of conjugation and efficiency using our Fc-III template directed conjugation approach.| Peptide conjugate variants
|
n | DoC mAb | Labeled mAb | Selectivity Fc/Fab | DoC Fc | DoC Fab | Distance to Ab K248 or K439b (Å) | Affinity (kcal mol−1) |
|---|---|---|---|---|---|---|---|---|
| a Energy minimization was performed using Molecular Operating Environment (MOE) with the structure depicted in Fig. 1B up to the NH2 moiety of the linker (blue). In all cases the NH2 function pointed away from the Fc-III binding site at the solvent exposed face. Energy minimization with E8Orn showed that the distance to K248 was identical when DOTA was included (data not shown). b The distance of the A3 variants were measured relative to antibody K439. All other variants were measured relative to K248. | ||||||||
| A3Dap | 1 | 0.29 | 26% | 0.48 | 0.06 | 0.12 | 9.62 | −14.39 |
| A3Orn | 3 | 0.64 | 47% | 8.79 | 0.37 | 0.04 | 7.28 | −14.96 |
| A3K | 4 | 1.37 | 85% | 25.85 | 1.16 | 0.04 | 4.52 | −14.69 |
| A3hK | 5 | 1.54 | 92% | 46.26 | 1.50 | 0.03 | 3.96 | −14.85 |
| A3DapPEG1 | 8 | 0.63 | 46% | 4.77 | 0.24 | 0.05 | 13.14 | −14.36 |
| L6Dap | 1 | 1.68 | 96% | 15.49 | 1.74 | 0.11 | 13.17 | −13.56 |
| L6Dab | 2 | 1.11 | 76% | 7.96 | 1.09 | 0.14 | 5.25 | −8.16 |
| L6Orn | 3 | 1.87 | 99% | 41.17 | 1.91 | 0.05 | 5.03 | −14.08 |
| L6K | 4 | 1.24 | 82% | 15.52 | 1.19 | 0.08 | 9.29 | −15.08 |
| L6KPEG1 | 11 | 2.22 | 98% | 33.28 | 1.74 | 0.05 | 15.63 | −13.03 |
| L6KPEG2 | 14 | 1.68 | 95% | 35.22 | 1.91 | 0.05 | 16.92 | −12.77 |
| L6KPEG3 | 17 | 2.29 | 100% | 15.27 | 2.12 | 0.14 | 14.72 | −13.21 |
| E8Dap | 1 | 2.09 | 97% | 5.97 | 1.69 | 0.28 | 3.87 | −11.10 |
| E8Orn | 3 | 1.76 | 97% | Fc selective | 1.77 | 0 | 4.99 | −14.60 |
| E8K | 4 | 1.70 | 90% | 3.97 | 1.05 | 0.26 | 4.14 | −12.75 |
High plasticity of the Fc-III binding site toward various conjugation chemistries
To investigate if other conjugation chemistries can be applied using our approach, we first replaced the carbonate reactive site by a thioester reactive site (Scheme 2A). Similarly, to the native chemical ligation methodology,29 this chemistry has the advantage to result in a native amide bond following the conjugation step of the payload to the antibody in contrast to the carbamate linkage resulting from our original approach. To fine tune the aqueous stability and lysine reactivity of the thioester bond, two electro-donating methylenes were added to the phenyl ring of the reactivity modulator (Scheme 2A). The same combinatorial strategy described above was applied: Fc-III-L6 was replaced by Dap, Dab, Orn or Lys and then linked to compound 004 (ESI†) via amide coupling. As the reactivity modulator contains two additional carbon atoms when compared to the carbonate approach, it would be expected that the best thioester variant would be Dap, given that L6Orn was the best carbonate variant. As expected, when the Fc-III-L6X-thioester-DOTA variants were incubated with trastuzumab, we found that L6Dap-thioester-DOTA gave the conjugation results yielding an excellent antibody labeling efficiency of 94% and a Fc/Fab selectivity of 36.24. In fact, all reactive conjugate variants gave excellent results, except for L6Dab-thioester-DOTA (Table 3). Energy minimization showed that all these variants are positioned quite close to the thioester bond, with excellent binding affinity. This is due to the 2 methylenes that provide the flexibility necessary to nicely fit the phenyl ring in the hydrophobic pocket (Fig. S1†). Based on the energy minimization, it would be expected that the thioester approach yields better results than the carbonate approach. However, despite excellent selectivity, the thioester chemistry provided a lower degree of antibody conjugation efficiency (94% versus 99% for L6Dap-thioester and L6Orn-carbonate, respectively). Additionally, the thioester Fc-III reactive conjugates appeared to be less stable in aqueous media than the corresponding carbonate conjugates.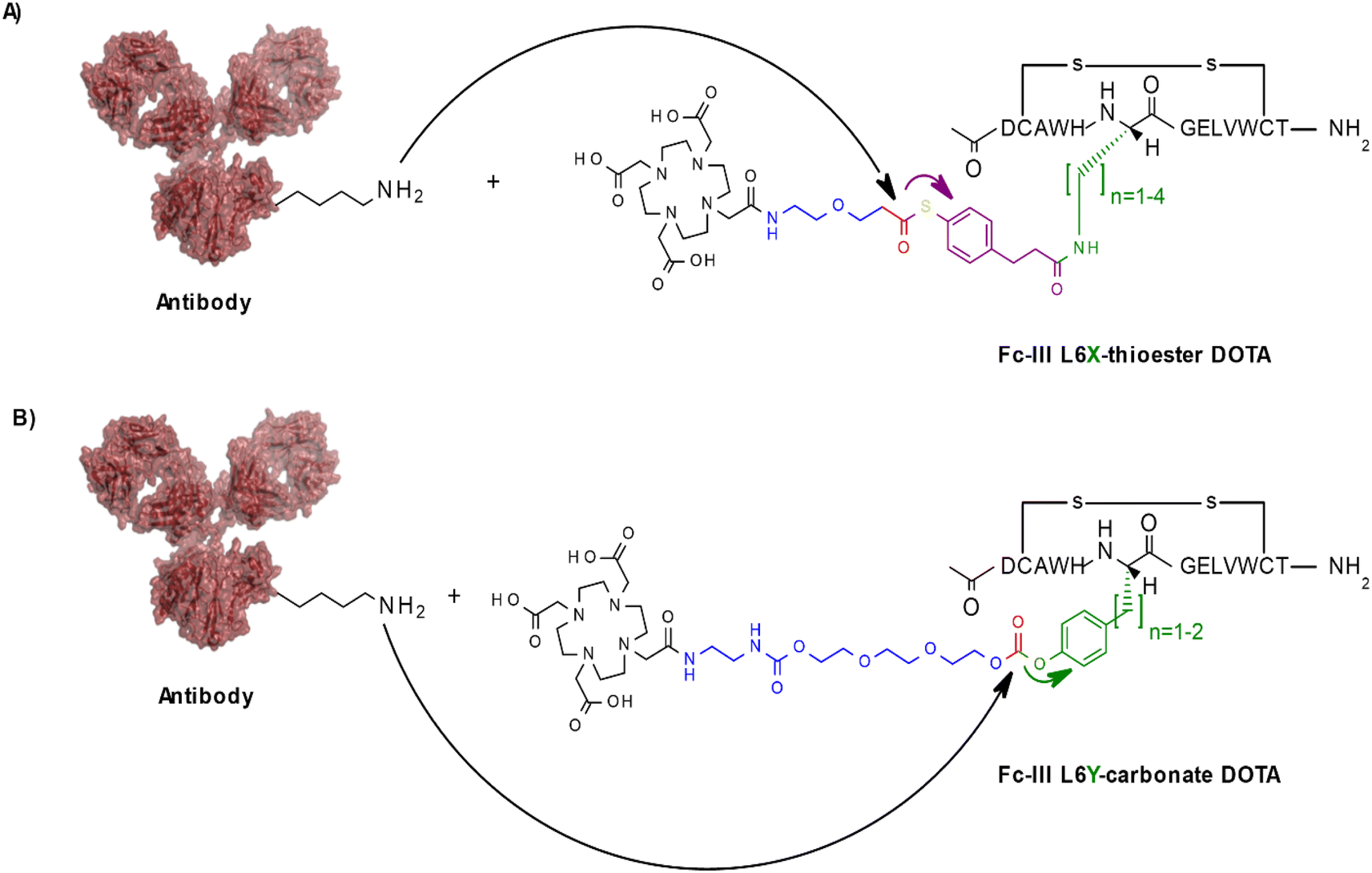 | ||
| Scheme 2 Schematic representation of other conjugation chemistries. The color codes are green for the Fc-III substituted residue, violet for the reactivity modulator (green for the tyrosine approach given that both the substituted side chain becomes the reactivity modulator), red for the chemical reactive center and blue for the linker between the chemical reactive site and the payload (A) Thioester approach; (B) Tyrosine approach. Only the L6 variants are shown here. | ||
| Peptide conjugate variants
|
n | DoC mAb | Labeled mAb | Selectivity Fc/Fab | DoC Fc | DoC Fab | Distance to Ab K248 (Å) | Affinity (kcal mol−1) |
|---|---|---|---|---|---|---|---|---|
| L6Dap | 1 | 1.63 | 94% | 36.24 | 1.52 | 0.04 | 4.53 | −14.14 |
| L6Dab | 2 | 0.70 | 55% | 2.03 | 0.34 | 0.17 | 4.94 | −13.86 |
| L6Orn | 3 | 1.48 | 94% | Fc selective | 1.39 | 0 | 3.36 | −14.44 |
| L6K | 4 | 1.37 | 88% | 24.56 | 1.22 | 0.05 | 4.18 | −16.18 |

| Peptide conjugate variants
|
n | DoC mAb | Labeled mAb | Selectivity Fc/Fab | DoC Fc | DoC Fab | Distance to Ab K248 (Å) | Affinity (kcal mol−1) |
|---|---|---|---|---|---|---|---|---|
| Fc-III-L6Y-DOTA | 1 | 1.42 | 92% | Fc selective | 1.56 | 0 | 4.91 | −15.22 |
| Fc-III-L6hY-DOTA | 2 | 0.32 | 30% | 14.94 | 0.33 | 0.02 | 5.80 | −14.73 |
| Fc-III-E8hY-DOTA | 1 | 0.73 | 60% | Fc selective | 0.78 | 0 | 3.49 | −15.48 |
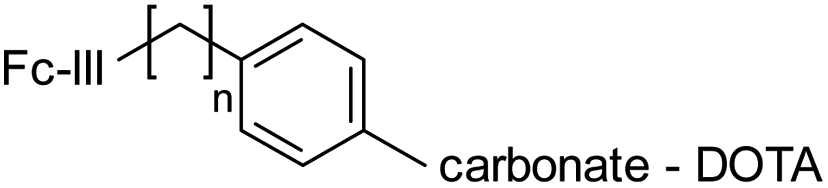
In another approach, rather than coupling the reactivity modulator by amide linkage to a Fc-III variant bearing an amine moiety as described above (Fig. 1B), the Fc-III amino acid side chain was converted into the reactivity modulator by substitution with a tyrosine analog (Scheme 2B). We substituted the positions L6 and E8 with Tyrosine or homo-Tyrosine (hY) to which was added the carbonate reactive site and the linker shown in blue in Scheme 2B. Energy minimization of the resulting peptide reactive conjugates suggested that the L6Y, L6hY and E8hY might be good candidates for the tyrosine approach. The Fc-III-Y variants were synthesized, and their binding affinity to recombinant trastuzumab was assessed using the FP competition assay described above. We found that these variants are all capable of competing with binding of Fc-III-FAM to trastuzumab at concentrations lower than required for the antibody conjugation reaction (34 μM, IC50 values in Table S1†). Similar to the thioester approach, the Fc-III-Y-carbonate-DOTA reactive conjugates improved significantly the selectivity of the conjugation of DOTA to trastuzumab when compared to the results we reported previously with Fc-III-PEG10-DOTA (Table 3). The best candidate Fc-III-L6Y-DOTA was able to label 92% of trastuzumab with a DoC Fc of 1.56 and a DoC Fab of 0. Note that the conjugation efficiency of this approach may be lower in part because of the lower stability of the Fc-III-Y conjugates in aqueous solution as it was observed during the HPLC purification step (20–40% of hydrolyzed form).
Altogether these data show that the Fc-III binding site is capable of accommodating a wide array of chemical modifications that can be used successfully with our ligand templated approach. For all variants and chemistries that were tested, L6Orn, E8Orn or A3hK carbonate, L6Dap thioester and L6Y carbonate appeared to be the best Fc-III reactive conjugates providing excellent antibody conjugation efficiency and Fc selectivity, with a DOTA to Fc antibody ratio close to 2. In comparison, random conjugation of trastuzumab with DOTA-NHS provided only 73% of labeled mAb and a Fc/Fab selectivity of 1.46 (DoC = 1.34, Table S4†). In addition to these significant improvements, another advantage of our templated conjugation approach is the low amount of Fc-III reactive conjugate that is required for the conjugation step (equimolar amount versus 10-fold excess for the random NHS conjugation methods).30
In regards of lysine selectivity, comparing the conjugation results in terms of DoC and percent antibody labeling can be misleading even if the Fc/Fab ratio is determined, because the degree of conjugation and efficiency are calculated based on the average of conjugated species. In an ideal case where conjugation occurs at a single lysine with 100% conjugation at both Fc domains, only one species should be detected by LC-FTMS analysis.
Fig. 2 shows the complete data set obtained by LC-FTMS analysis and highlighted in bold in Tables 2 and 3. We found that the L6Orn and E8Orn carbonate variants provided the best data with the major species being at D2, that is two DOTA chelators attached to trastuzumab. Conjugates bearing one or three DOTAs were also detected but to a minor extent (D1 and D3, respectively). These data suggest that DOTA is conjugated to at least two lysines using L6Orn- or E8Orn-carbonate. In all other cases, the amount of the D1 species was more important and some unconjugated antibody was also observed (D0). Note that no D3 species was observed when L6Y carbonate was used, suggesting that this may be the only variant resulting in conjugation at a single lysine residue. Because the LC-FTMS analysis obtained using the E8Orn variant showed a slightly higher amount of D1 species, the L6Orn carbonate variant was selected as the best candidate for further studies.
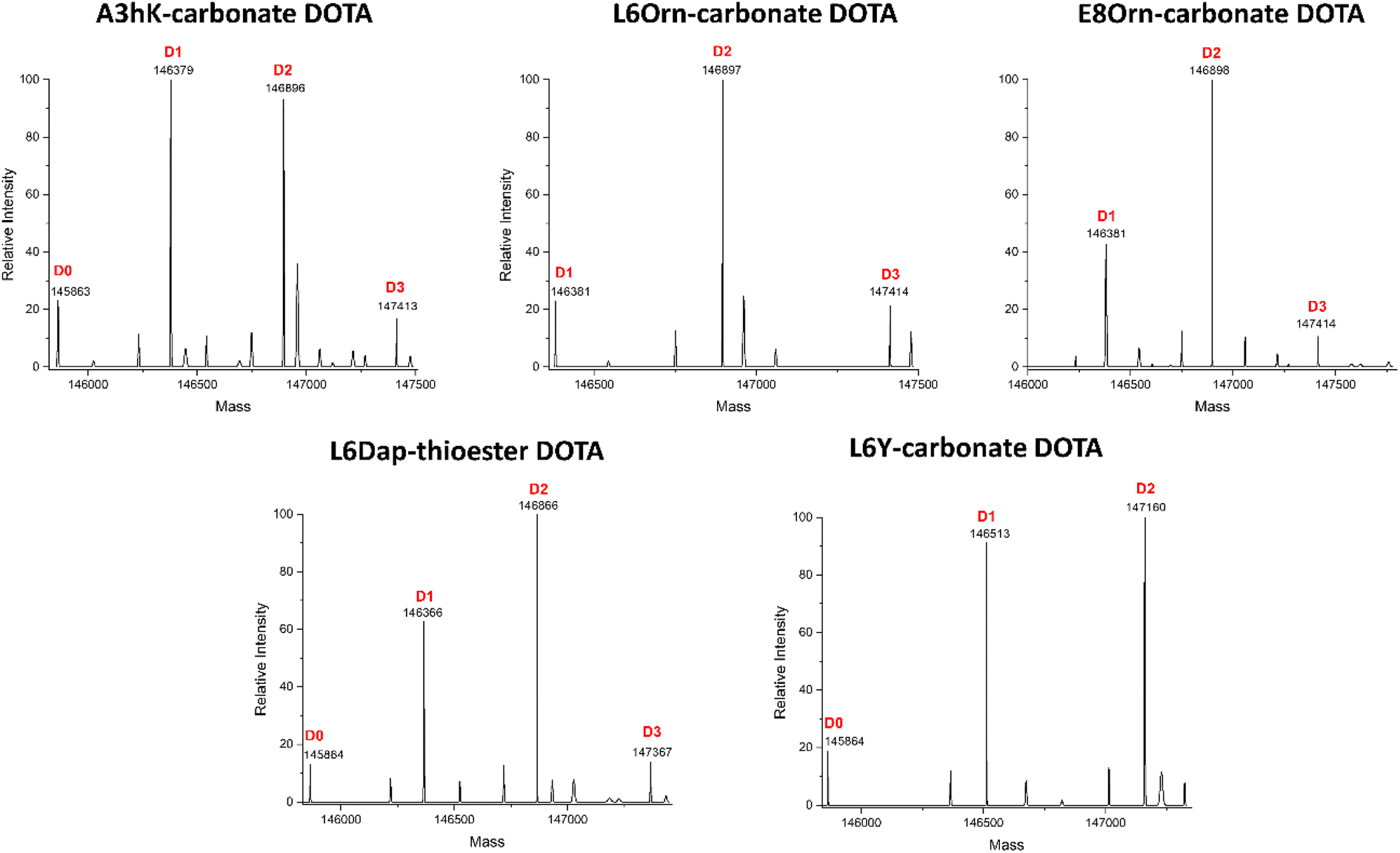 | ||
| Fig. 2 Intact mass analysis of the trastuzumab-DOTA conjugates by LC-FTMS. The Fc-III reactive conjugate variant used to conjugate DOTA to trastuzumab is listed above each spectrum. The deconvolved mass spectra of the intact deglycosylated sample is shown after EndoS treatment. | ||
Middle-down mass spectrometry analysis of trastuzumab DOTA conjugate
Middle-down mass spectrometry (MS/MS) was used to identify the antibody conjugation site(s) of DOTA obtained with our Fc-III carbonate peptide variants as reported previously.1 The trastuzumab-DOTA conjugates were digested with FabRICATOR™ (IdeS) or GinghisKHAN™ proteases, and the disulfide bonds were subsequently reduced with TCEP, yielding 25 kDa antibody fragments, i.e., Lc, Fd′ and Fc/2. The resulting sub-units were then analyzed by LC-FTMS/MS with high-energy collision-induced dissociation (HCD) as a fragmentation technique (more details in ESI†). Using this approach, we found that the antibody DOTA conjugation sites were at K246, K248 or K274 for FabRICATOR™-derived Fc/2 subunit resulting from the L6Orn, L6Y and E8Orn Fc-III carbonate variants (Table 4). Since K246 and K248 could not be discriminated due to the lack of fragmentation in that specific part of Fc/2 sub-unit during middle-down MS/MS analysis, the D3 species observed in Fig. 2 suggest that conjugation occur at K246/248 and K274. As shown above (Tables 2 and 3) L6Orn, L6Y and E8Orn position the carbonate chemically cleavable reactive site in proximity with Ab K248. On the other hand, energy minimization suggests that K246 and K274 are located at least 12 Å and 31 Å away from these variants, respectively. Based on these observations, we conclude that D2 results most likely from conjugation at K248, and the minor D3 species from conjugation at K274. It is noteworthy that DOTA conjugation was observed at K392 for L6K. Compared with L6Orn, the length of L6K seems to be too long to position the carbonate reactive site in proximity to Ab K248 (9.4 Å). Likewise, energy minimization suggests that L6KPEG1, L6KPEG2 and L6PEG3 variants are too far from K248 (15.6 Å, 16.9 Å and 14.7 Å, respectively) for conjugation to occur at this lysine. Despite our peptide mapping and modeling efforts, we were not able to identify the conjugation site of these longer variants. These data corroborate our hypothesis that L6Orn, L6Y and E8Orn mainly result in conjugation at K248, and suggest that it may also be feasible to target a single lysine beyond the Fc-III binding site with longer spacers, in contrary to our former findings when PEG spacers were attached at the Fc-III N-terminus.| Fc-III reactive conjugates | mAb labeled site [distance to carbonate (Å), sequence coverage (%)] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 246a | 248a | 274 | 392 | 317b | ||||||
| a Lysines 246 and 248 cannot be discriminated due to the lack of fragmentation in that specific part of Fc/2 sub-unit during middle-down MS/MS analysis b, therefore the same percentile is listed for both positions. b Lysine 317 could not be discriminated from Lysine326. Lys326 is located at 38.54 Å away of the carbonate cleavable site of Fc-III-L6K-PEG2-DOTA. c Same remark for Lysines 409 and 414. | ||||||||||
| Fc-III-L6Dap-DOTA | 23.1 Å | 13.2 Å | 28% | 45.4 Å | 24% | |||||
| Fc-III-L6Dab-DOTA | 16.1 Å | 26% | 5.3 Å | 26% | 40.6 Å | 28% | ||||
| Fc-III-L6Orn-DOTA | 14.4 Å | 32% | 5.1 Å | 32% | 38.2 Å | 30% | ||||
| Fc-III-L6K-DOTA | 9.4 Å | 41.1 Å | 22% | 8.6 Å | 21% | |||||
| Fc-III-L6KPEG1-DOTA | 28.1 Å | 30% | 15.6 Å | 29% | 49.9 Å | 29% | ||||
| Fc-III-L6KPEG2-DOTA | 35.9 Å | 100% | ||||||||
| Fc-III L6Y-DOTA | 15.5 Å | 32% | 4.9 Å | 32% | 39.9 Å | 28% | ||||
| Fc-III E8Orn-DOTA | 16.4 Å | 27% | 5.0 Å | 41.0 Å | ||||||
| Fc-III-E8hY-DOTA | 12.3 Å | 31% | 3.5 Å | 31% | 34.5 Å | 31% | ||||
Last, middle-down MS/MS analysis of sub-units suggested conjugation at K409 only for A3hK. This data is unexpected first because A3hK positions the carbonate chemically cleavable site at 25 Å from K409, and second because D3 was observed in the intact deglycosylated antibody conjugate, indicating conjugation to at least two lysines (Fig. 2).
On the other hand, DOTA conjugation was found at K392, K409, K414 and K439 for Fc-III-A3K-DOTA. A3K is only one methylene shorter than A3hK and also positions the carbonate chemically cleavable site in close proximity to A439 (Table 2). Based on the results, we suggest that the A3 variants conjugate DOTA at K439 for D2 and at K392, K409 or K424 for D3. Since the A3 variants expose the carbonate reactive site outside of the hydrophobic Fc-III binding site where a higher structural flexibility is possible, we cannot exclude that another lysine (i.e. K392, K409 or K424) displays great structural flexibility and is capable of approaching the A3 carbonate variants to a greater extent as modeled in the Fc-III/IgG Fc X-ray structure. In conclusion, our middle-down MS/MS experiments with our various Fc-III carbonate variants are summarized in Fig. 3.
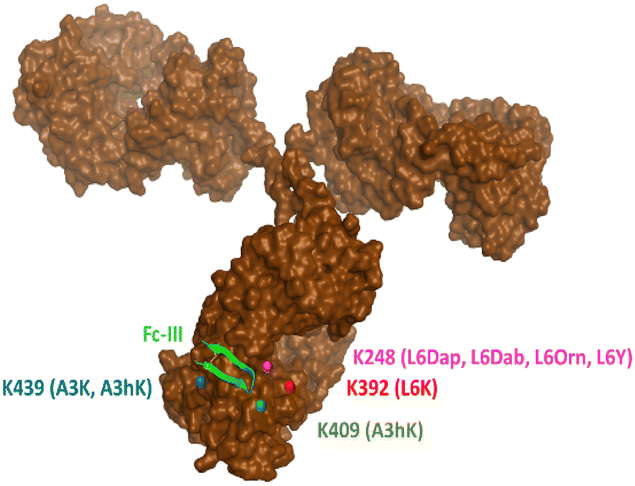 | ||
| Fig. 3 Representation of the antibody conjugation sites identified by middle-down MS/MS sub-unit analysis. The antibody lysines to which conjugation is likely to occur are color coded in red, green, blue and magenta and the respective Fc-III X-DOTA peptide variants used for the conjugation are shown in brackets (abbreviated as the relevant Fc-III amino acid variant). K409 is hidden inside the antibody and was slightly moved at the surface to be visible on this figure. | ||
Application to other antibodies/payloads
To investigate the scope of application of our site-selective templated approach, we tested our conjugation methods on various antibodies and payloads using the Fc-III-L6Orn carbonate variant. The payloads shown in Table 5 were attached via amide coupling to Fc-III-L6Orn-carbonate-NH2 or Fc-III-L6Orn-carbonate-SH (Scheme 3), and the resulting Fc-III reactive conjugates (ESI†) were incubated with several monoclonal antibodies. As can be seen in Table 5, DOTA could be successfully conjugated to human antibodies of all subtypes (IgG1-4) to an extent of almost 100% with exclusive conjugation at IgG Fc, as it was observed with trastuzumab in our previous combinatorial scanning approach (Table 2). Similarly, the conjugation of radionuclide chelators (DTPA, Bn-TCMC, NOGADA), fluorescent dyes (FITC, AF647), click chemistry reagents (N3, DBCO, BCN) and reagents for pre-targeted imaging applications (Tz, TCO)31,32 also yielded excellent results, despite some decrease of conjugation efficiency for Bn-TCMC, DBCO, TCO and Tc (83%, 87%, 79% and 70%, respectively), but with no detrimental effect on the selectivity. In comparison, random conjugation of DOTA-NHS, DTPA-SCN, FITC-SCN and AF647-NHS to trastuzumab provided a much lower selectivity with Fc/Fab values ranging between 0.87 and 2.43 (Table S5†).| Payload | DoC mAb | Labeled mAb | Selectivitya Fc/Fab | DoC Fc | DoC Fab |
|---|---|---|---|---|---|
| a The antibody digestion with GingisKHAN™ or FabRICATOR™ enzymes often caused a cleavage of more hydrophobic payload from Fc subunit, leading to a lower DoC of Fc. In these cases, the DoC of Fc were extrapolated (values in brackets). b 2.8 equivalent of Fc-III reactive conjugate was used. The acetyl group of Fc-III-L6Orn carbonate-NH2 (Scheme 3) was replaced with PEG20 and then conjugated to SPDB-DM4 as described in supporting information. | |||||
| DOTA | |||||
| Trastuzumab (IgG1) | 1.87 | 99% | 41.17 | 1.91 | 0.05 |
| Atezolizumab (IgG1) | 2.12 | 100% | 27.15 | 1.60 | 0.06 |
| Rituximab (IgG1) | 1.67 | 96% | Fc selective | 1.48 | 0 |
| Panitumumab (IgG2) | 2.13 | 99% | 158.60 | 1.92 | 0.01 |
| Pembrolizumab IgG4) | 1.90 | 98% | 85.40 | 1.85 | 0.02 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Chelators | |||||
| CHX-A′′-DTPA | 2.05 | 99% | 34.72 | 1.72 | 0.05 |
| Bn-TCMC | 1.44 | 83% | Fc selective | 1.54 | 0 |
| NODAGA | 1.81 | 97% | Fc selective | 1.77 | 0 |
|
|||||
| Fluorescent dyes | |||||
| FITC | 2.04 | 95% | Fc selective | 0.96 | 0 |
| AF647 | 1.67 | 93% | 56.15 (81.60) | 1.13 | 0.02 |
|
|||||
| Click chemistry | |||||
| N3 | 2.20 | 99% | Fc selective | 2.01 | 0 |
| DBCO | 1.65 | 87% | 19.46 | 1.56 | 0.08 |
| BCN | 1.97 | 92% | 26.47 (64.59) | 0.80 | 0.03 |
| TCO | 1.34 | 79% | Fc selective | 0.64 | 0 |
| Tz | 0.93 | 70% | Fc selective | 0.45 | 0 |
|
|||||
| Cytotoxins | |||||
| VC-PAB-MMAE | 1.87 | 98% | 52.00 (73.20) | 1.31 | 0.03 |
| DM1 | 1.12 | 73% | 5.70 (13.50) | 0.44 | 0.08 |
| PNU-159682 | 1.05 | 46% | 5.40 (24.40) | 0.22 | 0.04 |
| DM4 | 1.05 | 68% | 6.80 (16.20) | 0.41 | 0.06 |
|
|||||
| Pegylated Fc-III conjugateb | |||||
| PEG20-Fc-III-L6Orn-DM4 | 1.27 | 78% | 8.59 (14.00) | 0.73 | 0.08 |
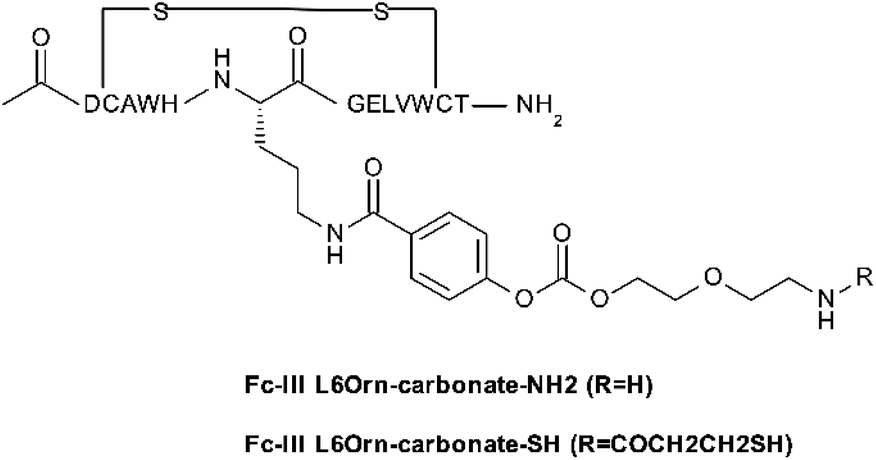 | ||
| Scheme 3 Fc-III-L6Orn carbonate derivative used to prepare the Fc-reactive conjugates with various payloads (mainly through reacting to –NHS or –SCN payloads). | ||
To further evaluate if our method is also suitable for the preparation of antibody drug conjugates (ADCs), Fc-III-L6Orn-carbonate-NH2 was equipped with the cytotoxic agents DM1, DM4, MMAE and PNU-159682 as described above for the other payloads. Maytansinoid (DM1 and DM4) and monomethyl auristatin E (MMAE) are microtubule binding agents that have been successfully used as payloads for clinically approved ADCs,33 and PNU-159682 is a DNA damaging agent, an anthracycline analog with more than thousandfold increased cytotoxicity compared to doxorubicin.34–36 In contrast to the other payloads investigated, we found that the antibody labeling with cytotoxic payloads was less efficient with values ranging from 46% to 98%. Although the Fc/Fab selectivity was still much greater than our original spacer approach, we found that it was not as good as for the other payloads (13 to 73). These data were unexpected and suggest that there must be a unique feature in some cytotoxins not present in other payloads, that appears to affect the efficiency and selectivity of our chemistry.
Increasing the hydrophilicity of Fc-III-L6Orn-cytotoxin reactive conjugates increases the efficiency of antibody modification
Most of the challenges of ADCs are linked to the hydrophobicity of the cytotoxic drugs used, which degrade the pharmacological properties of the antibody when attached to it.37 We hypothesized that the lower labeling efficiency and selectivity observed for the cytotoxins may be due to the high hydrophobicity of these payloads. Additionally, given that Fc-III is a rather hydrophobic peptide ligand by itself, the addition of highly lipophilic cytotoxins may further decrease the solubility of the reactive conjugates, and thereby may not be well soluble in aqueous media conditions used for the conjugation step. PEGylation of biological molecules is known to improve the solubility and other properties of drugs.38 We PEGylated the Fc-III-L6Orn-DM4 reactive conjugate, where PEG20 was attached to the N-terminus of the Fc-III prior to activating the peptide ligand with the carbonate cleavable reactive site and DM4. As it can be seen in Table 5, we were able to increase the yield of DM4 conjugation to the antibody (68% to 78%) and maintain similar Fc/Fab selectivity. Other combinations were tested where PEGs varying in length were attached to the N- or C-terminus of the Fc-III-L6Orn-DM4 reactive conjugate, and they all yielded similar conjugation results (data not shown). These data suggest that it might also be feasible to achieve near 100% conjugation for the preparation of ADCs using our site-selective templated approach pending some improvement of aqueous solubility.In vitro toxicity studies of ADCs
To compare the in vitro potency of our ADCs to ADCs produced using other methods, we compared Tmab-VC-PAB-MMAE to Tmab-Vedotin and T-DM1. Tmab-Vedotin consists of the same valine-citrulline-payload linked antibody, whose native interchain disulfide bonds were partially reduced and reacted with the maleimide containing auristatin derivative MMAE. As expected, we found that Tmab-VC-PAB-MMAE, Tmab-Vedotin and T-DM1 maintained a similar level of toxicity against human breast carcinoma cell line SK-BR-3 (HER-2 positive) regardless of the method used to conjugate MMAE or DM1 (Fig. S65,† IC50 = 0.258, 0.126 and 0.145 μM, respectively). However, note that the cytotoxin distribution per antibody was lower using our approach (DAR = 1.91 versus 3.28 and 3.5, for Tmab-Vedotin and T-DM1, respectively).In vivo biodistribution and PET imaging of PD-L1 expression with 64Cu-NODAGA-atezolizumab
Immune check-point inhibitors such as PD1/PD-L1 are widely recognized as a promising approach to counter immune escape of tumor cells.39 Despite the remarkable clinical responses observed with three recently approved antibodies (Atezolizumab, Avelumab, and Durvalumab) not all patients respond to PD1/PD-L1 blockade.Position emission tomography (PET) of PD-L1 expression in patients with radiolabeled atezolizumab has been proposed as a non-invasive method to predict clinical response.40 To evaluate our template site-specific conjugation method on a clinically validated target, [64Cu]Cu-NODAGA-atezolizumab was prepared using Fc-III-L6Orn-carbonate-NOGADA, hereby referred as [64Cu]Cu-atezolizumab. The radiochemical purity of [64Cu]Cu-atezolizumab was tested by instant thin layer chromatography (iTLC) and found to be higher than 97% (Fig. S43†).
The in vitro stability of the radiotracer was assessed by incubation of [64Cu]Cu-atezolizumab in human serum at 37 °C and at different time points (3, 24, and 48 h). iTLC and radio-SEC analysis confirmed that the radiolabeled antibody conjugate was stable under these conditions (>95% for the different time points). To verify that [64Cu]Cu-atezolizumab retained its affinity to PD-L1 after NOGADA conjugation and radiolabeling, the immunoreactive fraction of the radioconjugate was quantified by cell-binding assays in MDA-MB-231 (PD-L1hi) breast cancer. A constant amount of [64Cu]Cu-atezolizumab was incubated with increasing numbers of human breast adenocarcinoma PD-L1 positive MDA-MB-231 (PD-L1hi) and MDA-MB-157 PD-L1 negative cells. The resulting radioactivity of atezolizumab bound to the cells was then plotted as a function of the number of cells. As it can be seen in Fig. 4, the immune-reactive fraction of [64Cu]Cu-atezolizumab was 99.5% for PD-L1 positive MDA-MB-231 and 2.1% for the PD-L1 negative cells. Together with radiochemical purity, these data suggest that the integrity of the atezolizumab conjugate and its affinity to PD-L1 is preserved.
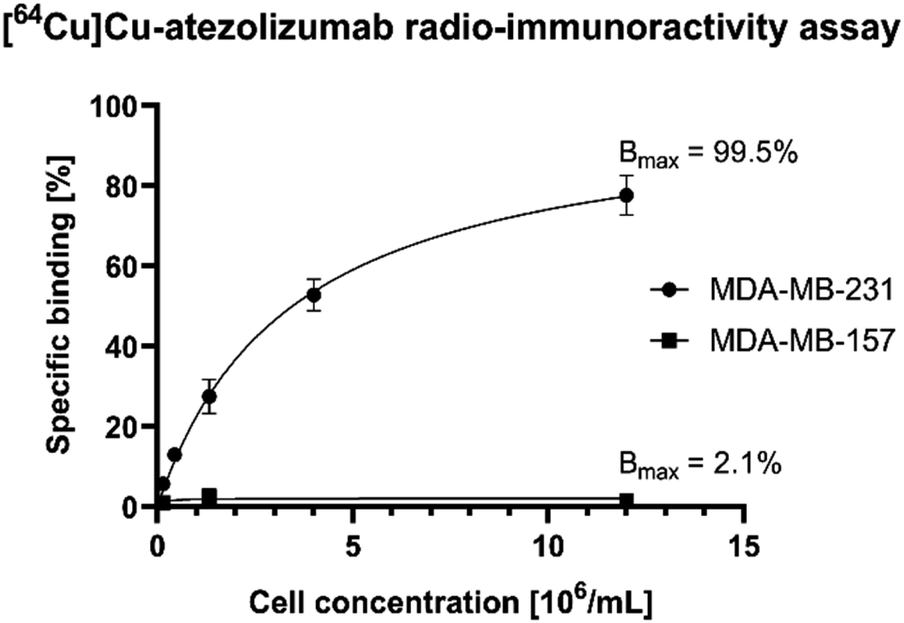 | ||
| Fig. 4 Radio-immunoreactivity assay with [64Cu]Cu-atezolizumab against the MDA-MB-231 (PD-L1hi-PD-L1 positive) and MDA-MB-157 (PD-L1-negative) cell lines. The specific cell binding is expressed as a function of the number of cells. The binding curves were fitted by nonlinear regression and the immunoreactive fraction (Bmax) corresponding to the extrapolation to infinite antigen excess was calculated to be equal to 99.5% for MDA-MB-231 and to 2.1% for MDA-MB-157. | ||
Next, [64Cu]Cu-atezolizumab (0.25 MBq, corresponding to 0.05 μg [64Cu]Cu-atezolizumab plus 49.95 μg cold atezolizumab to saturate nonspecific Fc receptors involved in the turnover of antibodies) was administered by intravenous injection to mice bearing MDA-MB-231 (PD-L1hi) tumor xenograft implanted subcutaneously. Mice were sacrificed at different time points (2, 24 and 48 hours) post-injection, selected organs were collected, and the radioactivity and weight of organs was measured to further study the level of antibody biodistribution (Table 6). The specificity of [64Cu]Cu-atezolizumab for its antigen was established at 24 hours by co-injecting an excess of “cold” atezolizumab (2 mg per mouse) in combination with [64Cu]Cu-atezolizumab (0.25 MBq; 0.05 μg).
| Organ | [64Cu]Cu-atezolizumab | |||
|---|---|---|---|---|
| 2 h [% ID g−1] | 24 h [% ID g−1] | 48 h [% ID g−1] | + Blocking (24 h) [% ID g−1] | |
| Tumo (MDA-MB-231) | 5 ± 1.2 | 11.4 ± 1.8 | 5.5 ± 2.1 | 4.3 ± 1 |
| Liver | 8.2 ± 0.1 | 11.8 ± 1.9 | 8.5 ± 1.6 | 5.3 ± 0.6 |
| Kidney | 11.8 ± 1.3 | 11.9 ± 1.3 | 8 ± 1.8 | 4.2 ± 0.1 |
| Lung | 14.3 ± 1 | 14.2 ± 1.9 | 6.6 ± 0.7 | 6.4 ± 0.7 |
| Spleen | 28.1 ± 4 | 42.8 ± 10.7 | 21.3 ± 6.5 | 17.9 ± 0.1 |
| Heart | 7.8 ± 0.6 | 6.8 ± 0.5 | 3.8 ± 0.6 | 4 ± 0.3 |
| Muscle | 1.3 ± 0.1 | 2.8 ± 0.6 | 1.1 ± 0.1 | 1 ± 0.2 |
| Bone | 2.5 ± 0.4 | 4.2 ± 1.7 | 2.2 ± 0.4 | 1.7 ± 0.1 |
| Skin | 1.1 ± 0.3 | 2.7 ± 0.7 | 2 ± 0.8 | 2.5 ± 0.4 |
| Stomach | 1.6 ± 0.6 | 4.5 ± 2.3 | 2.1 ± 0.5 | 1.5 ± 0.3 |
| Small intestine | 3.8 ± 0.9 | 8.7 ± 0.9 | 4.8 ± 1.3 | 2.6 ± 0.1 |
| Large intestine | 1.2 ± 0.5 | 6.1 ± 0.9 | 3.7 ± 1 | 2.7 ± 0.2 |
| Pancreas | 1.8 ± 1.6 | 9.6 ± 3.6 | 3.1 ± 0.2 | 3.6 ± 0.2 |
| Blood | 6.3 ± 3.8 | 3.1 ± 1.7 | 2 ± 1.2 | 10.9 ± 0.9 |
MDA-MB-231 tumor uptake was of 11.4 ± 1.9 % ID g−1 at 24 hours post injection and was specific, as confirmed by a reduction of tumor uptake to 4.3 ± 1.0 % ID g−1 upon coinjection of an excess of cold atezolizumab). Similar levels of activity were observed at 24 hours post injection in liver (11.8 ± 1.9 % ID g−1), kidney (11.9 ± 1.3 % ID g−1), lungs (14.2 ± 1.9 % ID g−1) and pancreas (9.6 ± 3.6 % ID g−1), while activity levels in blood and heart quickly decreased after 2 hours. Tumor to muscle ratio is equal to 4.0 at 24 hours. The spleen displayed the largest uptake of [64Cu]Cu-atezolizumab at 24 hours with 42.8 ± 10.7 % ID g−1. The detailed results are reported in Table 6 and are expressed in percentage of injected dose per gram of tissue (% ID g−1) (n = 4 animals/group).
Last, in vivo PET/CT imaging of MDA-MB-231 xenograft bearing mice was performed using an Albira Si PEC/SPECT/CT small animal scanner following i.v. injection of [64Cu]Cu-atezolizumab (9 MBq; 50 μg total dose of atezolizumab). PET/CT scans were acquired at 2, 24, and 48 hours post-injection. The specificity to PD-L1 was demonstrated by accumulation of the tracer in the tumor following co-injection of 2 mg of unlabeled atezolizumab. After acquisitions, PET/CT images were analyzed using PMOD 4.3. MDA-MB-231 tumors were delineated, and the activity in the corresponding volumes of interest (VOI) were expressed in % ID g−1 (Fig. 5). The specific tumor uptake of the antibodies increased over time, whereas the background activity decreased, resulting in an increased contrast of the tumor over time with an optimum at 24 h consistent with the biodistribution experiments. As already found in the biodistribution study, an important uptake of the radiotracer was observed in liver and in spleen.
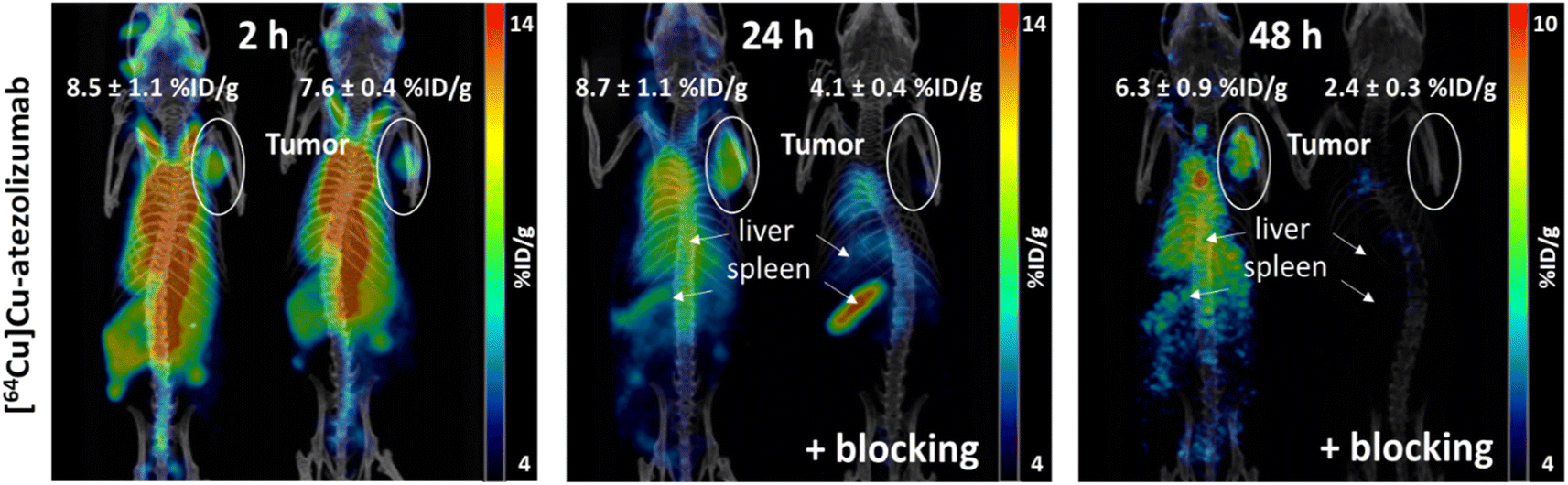 | ||
| Fig. 5 Coronal volume rendered (MIP) microPET/CT images of [64Cu]Cu-atezolizumab in mice bearing MDA-MB-231 xenograft at 2, 24 and 48 hours post injection. Mice were injected with 9 MBq of [64Cu]Cu-atezolizumab. At 24 and 48 hours mice blocked for PD-L1-specific tracer accumulation by co injection of 2 mg cold atezolizumab are shown (+blocking). Tumors are delineated in white ovals, liver and spleen are located by white arrows. The % ID g−1 for the tumors are reported. | ||
Site-selective conjugation of NODAGA to atezolizumab with Fc-III-L6Orn-NODAGA provides an excellent means for the robust and high-yield preparation of [64Cu]Cu-atezolizumab. The resulting radiotracer displays an unbiased immunoreactivity and enables to visualize PD-L1 expressing tumors in vivo using PET/CT imaging. The biodistribution and PET imaging studies reported here using [64Cu]Cu-atezolizumab are consistent with previous reports using similar models.41,42
Conclusions
Progress in drug linkers and site-selective conjugation technologies may result in better clinical efficacy of antibody drug conjugates. The recent approval of Enhertu, an ADC targeting HER2, shows that conjugating Deruxtecan homogenously to Trastuzumab (DAR = 7.5) improves patient survival significantly compared to its predecessor Kadcyla.43 This is mainly due to a better therapeutic index of Enhertu, as the ratio between the dose required for efficacy versus the dose resulting in toxicity is increased in comparison to Kadcyla.44 In this work, we have demonstrated that our template-based approach can be used to conjugate payloads with high efficiency and selectivity at a single lysine of the antibody Fc region. Through sampling various Fc-III variants of A3, H5, L6 and E8, we have shown that the Fc-III DOTA reactive carbonate of A3hK, L6Orn, L6Y and E8Orn results in site selective conjugation of DOTA to trastuzumab. We have also shown the flexibility of our method by replacing the carbonate chemically cleavable site with a thioester moiety, thereby resulting in the formation of a native amide bond following the conjugation step. Using middle-down mass spectrometry analysis of the antibody conjugates, we have demonstrated that the Fc-III L6Orn carbonate reactive peptide provides the best conjugation results with almost all payload conjugated to K248 of each Fc domain. Last, L6Orn carbonate was exemplified with a broad array of payloads including cytotoxic small molecules. In comparison with other technologies available, the method is quite convenient as the conjugation is performed in a single step simply by mixing the antibody with the Fc-III reactive conjugate and does not require prior modification of the antibody. For a successful ADC application, the selected drug linker may need to be optimized in order to reach 100% conjugation and a DAR of 2.Data availability
All experimental data are described in ESI.†Author contributions
V. P., N. G., N. G., D. V., M. P. and G. D. devised and performed the experiments. D. V. and O. N. wrote the paper. V. P. contributed partly to the writing of the paper. L. M. and F. L. thoroughly reviewed the paper. O. N. and L. M. devised the concept. P. G., F. L., D. V., L. M., V. P., M. S. and O. N. managed the project. M. S. devised and supervised the in vivo experiments. O. N. and J. M. S. prepared the Innosuisse grant application. All authors revised and approved the last version of the paper.Conflicts of interest
There are no conflicts to declare. V. P., L. M., F. L., P. G., J.-M. S., O. N. are coinventors of application WO 2022/078566 A1 filed on 12 October 2020, entitled “Reactive conjugates”.Acknowledgements
This work was co-financed by Innosuisse (project number 26695.2 PFLS-LS) and Debiopharm International SA, Switzerland, whose support we gratefully acknowledge. We thank Genochem (Grasse, France) and Concept Life Sciences for the synthesis of the key building blocks, and Gael Coppey for his contribution to the synthesis of peptide-reactive conjugates. We also thank the ARRONAX team and in particular F. Haddad and C. Hérault for the production and delivery of Cu-64. We are most grateful to Stéphane Baeriswyl for his support in preparing the figures and formatting the manuscript and to Dr Michel A Cuendet from the Swiss Institute of Bioinformatics for the molecular dynamics simulation of some tyrosine variants. We thank Lorianne Rey-Bellet, Selena Vigano and Catherine Salvat-Ortis (Debiopharm Bioresearch group) for the in vitro cytotoxicity studies of ADCs.References
- V. Postupalenko, L. Marx, D. Viertl, N. Gsponer, N. Gasilova, T. Denoel, N. Schaefer, J. O. Prior, G. Hagens, F. Levy, P. Garrouste, J. M. Segura and O. Nyanguile, Template directed synthesis of antibody Fc conjugates with concomitant ligand release, Chem. Sci., 2022, 13, 3965–3976 RSC.
- F. Lhospice, D. Bregeon, C. Belmant, P. Dennler, A. Chiotellis, E. Fischer, L. Gauthier, A. Boedec, H. Rispaud, S. Savard-Chambard, A. Represa, N. Schneider, C. Paturel, M. Sapet, C. Delcambre, S. Ingoure, N. Viaud, C. Bonnafous, R. Schibli and F. Romagne, Site-Specific Conjugation of Monomethyl Auristatin E to Anti-CD30 Antibodies Improves Their Pharmacokinetics and Therapeutic Index in Rodent Models, Mol. Pharm., 2015, 12, 1863–1871 CrossRef CAS PubMed.
- J. R. Junutula, K. M. Flagella, R. A. Graham, K. L. Parsons, E. Ha, H. Raab, S. Bhakta, T. Nguyen, D. L. Dugger, G. Li, E. Mai, G. D. Lewis Phillips, H. Hiraragi, R. N. Fuji, J. Tibbitts, R. Vandlen, S. D. Spencer, R. H. Scheller, P. Polakis and M. X. Sliwkowski, Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer, Clin. Cancer Res., 2010, 16, 4769–4778 CrossRef CAS.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- R. M. Barfield, Y. C. Kim, S. Chuprakov, F. Zhang, M. Bauzon, A. O. Ogunkoya, D. Yeo, C. Hickle, M. D. Pegram, D. Rabuka and P. M. Drake, A Novel HER2-targeted Antibody-drug Conjugate Offers the Possibility of Clinical Dosing at Trastuzumab-equivalent Exposure Levels, Mol. Cancer Ther., 2020, 19, 1866–1874 CrossRef CAS PubMed.
- B. Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S. F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- T. H. Pillow, J. Tien, K. L. Parsons-Reponte, S. Bhakta, H. Li, L. R. Staben, G. Li, J. Chuh, A. Fourie-O'Donohue, M. Darwish, V. Yip, L. Liu, D. D. Leipold, D. Su, E. Wu, S. D. Spencer, B. Q. Shen, K. Xu, K. R. Kozak, H. Raab, R. Vandlen, G. D. Lewis Phillips, R. H. Scheller, P. Polakis, M. X. Sliwkowski, J. A. Flygare and J. R. Junutula, Site-specific trastuzumab maytansinoid antibody-drug conjugates with improved therapeutic activity through linker and antibody engineering, J. Med. Chem., 2014, 57, 7890–7899 CrossRef CAS.
- F. Tian, Y. Lu, A. Manibusan, A. Sellers, H. Tran, Y. Sun, T. Phuong, R. Barnett, B. Hehli, F. Song, M. J. DeGuzman, S. Ensari, J. K. Pinkstaff, L. M. Sullivan, S. L. Biroc, H. Cho, P. G. Schultz, J. DiJoseph, M. Dougher, D. Ma, R. Dushin, M. Leal, L. Tchistiakova, E. Feyfant, H. P. Gerber and P. Sapra, A general approach to site-specific antibody drug conjugates, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1766–1771 CrossRef CAS PubMed.
- D. Jackson, J. Atkinson, C. I. Guevara, C. Zhang, V. Kery, S. J. Moon, C. Virata, P. Yang, C. Lowe, J. Pinkstaff, H. Cho, N. Knudsen, A. Manibusan, F. Tian, Y. Sun, Y. Lu, A. Sellers, X. C. Jia, I. Joseph, B. Anand, K. Morrison, D. S. Pereira and D. Stover, In vitro and in vivo evaluation of cysteine and site specific conjugated herceptin antibody-drug conjugates, PLoS One, 2014, 9, e83865 CrossRef PubMed.
- P. Strop, K. Delaria, D. Foletti, J. M. Witt, A. Hasa-Moreno, K. Poulsen, M. G. Casas, M. Dorywalska, S. Farias, A. Pios, V. Lui, R. Dushin, D. Zhou, T. Navaratnam, T. T. Tran, J. Sutton, K. C. Lindquist, B. Han, S. H. Liu, D. L. Shelton, J. Pons and A. Rajpal, Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading, Nat. Biotechnol., 2015, 33, 694–696 CrossRef CAS PubMed.
- C. R. Behrens, E. H. Ha, L. L. Chinn, S. Bowers, G. Probst, M. Fitch-Bruhns, J. Monteon, A. Valdiosera, A. Bermudez, S. Liao-Chan, T. Wong, J. Melnick, J. W. Theunissen, M. R. Flory, D. Houser, K. Venstrom, Z. Levashova, P. Sauer, T. S. Migone, E. H. van der Horst, R. L. Halcomb and D. Y. Jackson, Antibody-Drug Conjugates (ADCs) Derived from Interchain Cysteine Cross-Linking Demonstrate Improved Homogeneity and Other Pharmacological Properties over Conventional Heterogeneous ADCs, Mol. Pharm., 2015, 12, 3986–3998 CrossRef CAS PubMed.
- K. J. Hamblett, T. Le, B. M. Rock, D. A. Rock, S. Siu, J. N. Huard, K. P. Conner, R. R. Milburn, J. W. O'Neill, M. E. Tometsko and W. C. Fanslow, Altering Antibody-Drug Conjugate Binding to the Neonatal Fc Receptor Impacts Efficacy and Tolerability, Mol. Pharm., 2016, 13, 2387–2396 CrossRef CAS PubMed.
- C. Bai, E. E. Reid, A. Wilhelm, M. Shizuka, E. K. Maloney, R. Laleau, L. Harvey, K. E. Archer, D. Vitharana, S. Adams, Y. Kovtun, M. L. Miller, R. Chari, T. A. Keating and N. C. Yoder, Site-Specific Conjugation of the Indolinobenzodiazepine DGN549 to Antibodies Affords Antibody-Drug Conjugates with an Improved Therapeutic Index as Compared with Lysine Conjugation, Bioconjug. Chem., 2020, 31, 93–103 CrossRef CAS PubMed.
- N. C. Yoder, C. Bai, D. Tavares, W. C. Widdison, K. R. Whiteman, A. Wilhelm, S. D. Wilhelm, M. A. McShea, E. K. Maloney, O. Ab, L. Wang, S. Jin, H. K. Erickson, T. A. Keating and J. M. Lambert, A Case Study Comparing Heterogeneous Lysine- and Site-Specific Cysteine-Conjugated Maytansinoid Antibody-Drug Conjugates (ADCs) Illustrates the Benefits of Lysine Conjugation, Mol. Pharm., 2019, 16, 3926–3937 CrossRef CAS PubMed.
- Y. Matsuda, T. Seki, K. Yamada, Y. Ooba, K. Takahashi, T. Fujii, S. Kawaguchi, T. Narita, A. Nakayama, Y. Kitahara, B. A. Mendelsohn and T. Okuzumi, Chemical Site-Specific Conjugation Platform to Improve the Pharmacokinetics and Therapeutic Index of Antibody-Drug Conjugates, Mol. Pharm., 2021, 18, 4058–4066 CrossRef CAS PubMed.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Site-selective modification strategies in antibody-drug conjugates, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- M. Haque, N. Forte and J. R. Baker, Site-selective lysine conjugation methods and applications towards antibody-drug conjugates, Chem. Commun., 2021, 57, 10689–10702 RSC.
- K. Yamada, N. Shikida, K. Shimbo, Y. Ito, Z. Khedri, Y. Matsuda and B. A. Mendelsohn, AJICAP: Affinity Peptide Mediated Regiodivergent Functionalization of Native Antibodies, Angew Chem. Int. Ed. Engl., 2019, 58, 5592–5597 CrossRef CAS PubMed.
- J. Park, Y. Lee, B. J. Ko and T. H. Yoo, Peptide-Directed Photo-Cross-Linking for Site-Specific Conjugation of IgG, Bioconjug. Chem., 2018, 29, 3240–3244 CrossRef CAS PubMed.
- S. Kishimoto, Y. Nakashimada, R. Yokota, T. Hatanaka, M. Adachi and Y. Ito, Site-Specific Chemical Conjugation of Antibodies by Using Affinity Peptide for the Development of Therapeutic Antibody Format, Bioconjug. Chem., 2019, 30, 698–702 CrossRef CAS PubMed.
- C. Yu, J. Tang, A. Loredo, Y. Chen, S. Y. Jung, A. Jain, A. Gordon and H. Xiao, Proximity-Induced Site-Specific Antibody Conjugation, Bioconjug. Chem., 2018, 29, 3522–3526 CrossRef CAS PubMed.
- J. Ohata and Z. T. Ball, A Hexa-rhodium Metallopeptide Catalyst for Site-Specific Functionalization of Natural Antibodies, J. Am. Chem. Soc., 2017, 139, 12617–12622 CrossRef CAS PubMed.
- N. Gupta, A. Ansari, G. V. Dhoke, M. Chilamari, J. Sivaccumar, S. Kumari, S. Chatterjee, R. Goyal, P. K. Dutta, M. Samarla, M. Mukherjee, A. Sarkar, S. K. Mandal, V. Rai, G. Biswas, A. Sengupta, S. Roy, M. Roy and S. Sengupta, Computationally designed antibody-drug conjugates self-assembled via affinity ligands, Nat. Biomed. Eng., 2019, 3, 917–929 CrossRef CAS PubMed.
- T. Lee, J. H. Kim, S. J. Kwon, J. W. Seo, S. H. Park, J. Kim, J. Jin, J. H. Hong, H. J. Kang, C. Sharma, J. H. Choi and S. J. Chung, Site-Selective Antibody-Drug Conjugation by a Proximity-Driven S to N Acyl Transfer Reaction on a Therapeutic Antibody, J. Med. Chem., 2022, 65, 5751–5759 CrossRef CAS PubMed.
- T. Fujii, Y. Matsuda, T. Seki, N. Shikida, Y. Iwai, Y. Ooba, K. Takahashi, M. Isokawa, S. Kawaguchi, N. Hatada, T. Watanabe, R. Takasugi, A. Nakayama, K. Shimbo, B. A. Mendelsohn, T. Okuzumi and K. Yamada, AJICAP Second Generation: Improved Chemical Site-Specific Conjugation Technology for Antibody-Drug Conjugate Production, Bioconjug. Chem., 2023, 34, 728–738 CAS.
- M. M. Tanriver, D. Richards, S. Majima, A. DeMello and J. W. Bode, Peptide-Directed Attachment of Hydroxylamines to Specific Lysines of IgG Antibodies for Bioconjugations with acylboronates, ChemRxiv, preprint, 2023, DOI:10.26434/chemrxiv-2023-4f6kr.
- Y. Zeng, W. Shi, Q. Dong, W. Li, J. Zhang, X. Ren, C. Tang, B. Liu, Y. Song, Y. Wu, X. Diao, H. Zhou, H. Huang, F. Tang and W. Huang, A Traceless Site-Specific Conjugation on Native Antibodies Enables Efficient One-Step Payload Assembly, Angew Chem. Int. Ed. Engl., 2022, 61, e202204132 CrossRef CAS PubMed.
- W. L. DeLano, M. H. Ultsch, A. M. de Vos and J. A. Wells, Convergent solutions to binding at a protein-protein interface, Science, 2000, 287, 1279–1283 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Synthesis of proteins by native chemical ligation, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- M. Acchione, H. Kwon, C. M. Jochheim and W. M. Atkins, Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates, MAbs, 2012, 4, 362–372 CrossRef PubMed.
- V. F. C. Ferreira, B. L. Oliveira, A. D'Onofrio, C. M. Farinha, L. Gano, A. Paulo, G. J. L. Bernardes and F. Mendes, In Vivo Pretargeting Based on Cysteine-Selective Antibody Modification with IEDDA Bioorthogonal Handles for Click Chemistry, Bioconjug. Chem., 2021, 32, 121–132 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- R. V. Chari, M. L. Miller and W. C. Widdison, Antibody-drug conjugates: an emerging concept in cancer therapy, Angew Chem. Int. Ed. Engl., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- L. Quintieri, C. Geroni, M. Fantin, R. Battaglia, A. Rosato, W. Speed, P. Zanovello and M. Floreani, Formation and antitumor activity of PNU-159682, a major metabolite of nemorubicin in human liver microsomes, Clin. Cancer Res., 2005, 11, 1608–1617 CrossRef CAS PubMed.
- S. F. Yu, B. Zheng, M. Go, J. Lau, S. Spencer, H. Raab, R. Soriano, S. Jhunjhunwala, R. Cohen, M. Caruso, P. Polakis, J. Flygare and A. G. Polson, A Novel Anti-CD22 Anthracycline-Based Antibody-Drug Conjugate (ADC) That Overcomes Resistance to Auristatin-Based ADCs, Clin. Cancer Res., 2015, 21, 3298–3306 CrossRef CAS PubMed.
- M. Scalabrin, L. Quintieri, M. Palumbo, F. Riccardi Sirtori and B. Gatto, Virtual Cross-Linking of the Active Nemorubicin Metabolite PNU-159682 to Double-Stranded DNA, Chem. Res. Toxicol., 2017, 30, 614–624 Search PubMed.
- L. Conilh, G. Fournet, E. Fourmaux, A. Murcia, E. L. Matera, B. Joseph, C. Dumontet and W. Viricel, Exatecan Antibody Drug Conjugates Based on a Hydrophilic Polysarcosine Drug-Linker Platform, Pharmaceuticals, 2021, 14(3), 247 Search PubMed.
- J. S. Kang, P. P. Deluca and K. C. Lee, Emerging PEGylated drugs, Expert. Opin. Emerg. Drugs, 2009, 14, 363–380 Search PubMed.
- J. Gong, A. Chehrazi-Raffle, S. Reddi and R. Salgia, Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations, J. Immunother. Cancer, 2018, 6, 8 Search PubMed.
- F. Bensch, E. L. van der Veen, M. N. Lub-de Hooge, A. Jorritsma-Smit, R. Boellaard, I. C. Kok, S. F. Oosting, C. P. Schroder, T. J. N. Hiltermann, A. J. van der Wekken, H. J. M. Groen, T. C. Kwee, S. G. Elias, J. A. Gietema, S. S. Bohorquez, A. de Crespigny, S. P. Williams, C. Mancao, A. H. Brouwers, B. M. Fine and E. G. E. de Vries, (89)Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer, Nat. Med., 2018, 24, 1852–1858 CrossRef CAS PubMed.
- W. G. Lesniak, S. Chatterjee, M. Gabrielson, A. Lisok, B. Wharram, M. G. Pomper and S. Nimmagadda, PD-L1 Detection in Tumors Using [(64)Cu]Atezolizumab with PET, Bioconjug. Chem., 2016, 27, 2103–2110 CrossRef CAS PubMed.
- S. P. Vinod Nagarajan, Deok Il Kim, Sang-Keun Woo and ILHAN LIM, PET Imaging and Biodistribution of 64Cu-DOTA-Atezolizumab in Mice Bearing MDA-MB231 Xenograft, J. Nucl. Med., 2021, 62, 1490 Search PubMed.
- S. J. Keam, Trastuzumab Deruxtecan: First Approval, Drugs, 2020, 80, 501–508 CrossRef CAS PubMed.
- E. Tarcsa, M. R. Guffroy, H. Falahatpisheh, C. Phipps and J. C. Kalvass, Antibody-drug conjugates as targeted therapies: Are we there yet? A critical review of the current clinical landscape, Drug Discov. Today Technol., 2020, 37, 13–22 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04324j |
| ‡ These authors contributed equally to this work. |
| § Peptide modeling was performed with Molecular Operating Environment (Chemical computer group ULC) based on the previously published structure of Fc-III that was used to guide our design (PDB ID 1DN2).28 |
| This journal is © The Royal Society of Chemistry 2024 |