
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConvergent synthesis of thiodiazole dioxides from simple ketones and amines through an unusual nitrogen-migration mechanism†
Kunlayanee
Punjajom‡
 a,
Paul P.
Sinclair‡
a,
Ishika
Saha
b,
Mark
Seierstad
b,
Michael K.
Ameriks
b,
Pablo
García-Reynaga
*b,
Terry P.
Lebold
*b and
Richmond
Sarpong
*a
a,
Paul P.
Sinclair‡
a,
Ishika
Saha
b,
Mark
Seierstad
b,
Michael K.
Ameriks
b,
Pablo
García-Reynaga
*b,
Terry P.
Lebold
*b and
Richmond
Sarpong
*a
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: rsarpong@berkeley.edu
bJanssen Research and Development, San Diego, California 92121, USA. E-mail: pgarciar@its.jnj.com; terry.lebold@gmail.com
First published on 27th November 2023
Abstract
We report the modular preparation of dihydro-1,2,5-thiodiazole dioxide heterocycles starting from methyl ketones and primary amines. This one-pot, three-component coupling employs 2,3-dimethylimidazole-1-sulfonyl azide triflate as a coupling reagent and oxidant. The transformation is scalable and various ketones and amines can be used, yielding thiodiazole dioxide products in up to 89% yield. In addition, 15N- and 13C-labeling studies suggest a mechanism involving a 1,2-nitrogen migration. Together with the mechanistic studies, DFT calculations provide insight into the reaction pathway and set the stage for further exploration of the mechanistic nuances of reactions that use sulfamoyl azides. In combination with the demonstrated modularity of the approach reported herein, the derivatization of the thiodiazole dioxide products highlights the potential of this methodology to rapidly access diverse chemical structures.
Introduction
Sulfamides and their derivatives, including the tetrahydro-1,2,5-thiodiazole dioxide heterocycle, are well represented structural motifs in pharmaceutical and other bioactive compounds (see Fig. 1A).1,2 These heterocycles are typically installed either by reaction of the corresponding diamine (II) with sulfamide (III)3 or by intramolecular C–H amination from the corresponding sulfamoyl azide (IV)4 (Fig. 1B). While useful, these strategies do not allow for late-stage diversification and in some cases rely on starting materials with limited availability (e.g., diamines, II)—two factors which are often important in medicinal chemistry campaigns. To address these limitations, an alternate synthetic entry to tetrahydro-1,2,5-thiodiazole dioxides through functionalization of the closely related dihydro-1,2,5-thiodiazole dioxide (V) would be beneficial. In general, the discovery of direct, yet versatile, synthetic methods for the construction of the dihydro-1,2,5-thiodiazole dioxide structural motif is of value to synthetic and medicinal chemists. One of the most direct approaches to V is the condensation of sulfamides onto carbonyl compounds. The key challenge with this approach is the need for α-oxidation of readily available ketones in order to install the C–N bond at the α-carbon. Historically, the requisite oxidation pattern has been obtained by starting from α-oxygenated ketones such as α-hydroxy ketones (VI)5 or 1,2 diketones (VII).6 However, there is limited commercial availability of these highly oxidized starting materials. A simple method for the synthesis of the thiodiazole dioxide motif from readily available commercial materials (e.g., methyl ketones and aliphatic amines) could accelerate medicinal chemistry efforts to explore the function of diverse thiodiazole dioxides.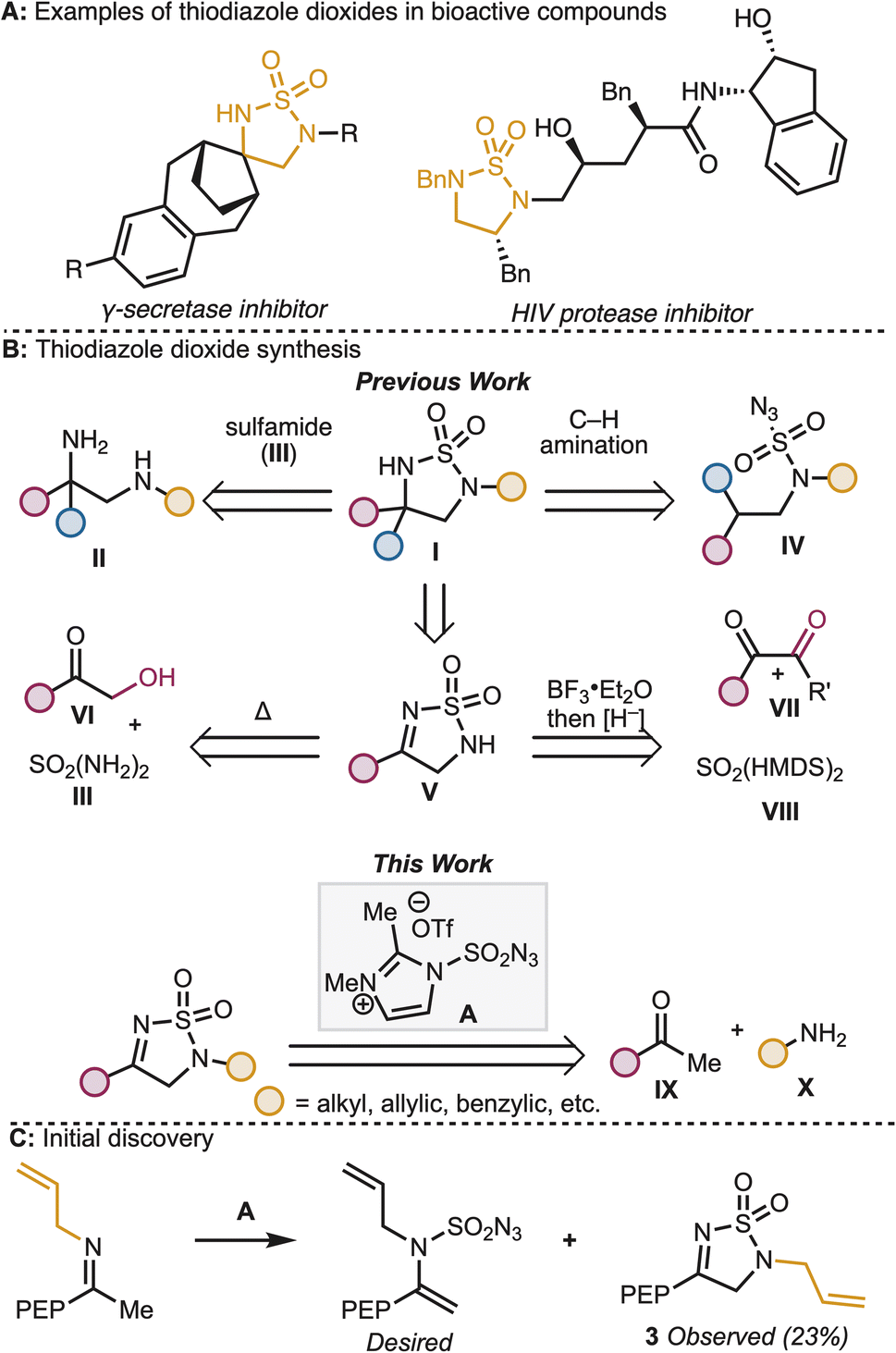 | ||
| Fig. 1 (A) Biologically active molecules containing the thiodiazole dioxide motif. (B) Strategies for the preparation of thiodiazole dioxides. (C) Unexpected formation of thiodiazole dioxide 3. PEP = para-ethoxyphenyl HMDS: hexamethyldisilazide. | ||
In the course of a recent collaborative study between the Sarpong group and Janssen Research and Development to prepare aza-bicyclohexanes and bicyclopentanes,7 we identified 3 as an unexpected product of the reaction between an imine and 2,3-dimethylimidazole-1-sulfonyl azide triflate (A, Fig. 1C). We recognized that this transformation seemingly overcomes the key challenge described above by obviating the need for pre-oxidation of the α-carbon of the ketone coupling partner en route to the thiodiazole dioxide structural motif. This transformation represents a modular, three-component coupling that allows for the rapid diversification of methyl ketones and primary amines, two functional groups that are well-represented in pharmaceutical libraries.
In addition to the practical value of this discovery, we also recognized an opportunity to explore the reactivity of 2,3-dimethylimidazole-1-sulfonyl azide triflate (A).8 Sulfamoyl azides have been used in sulfonyl8 and diazo9 transfer reactions, cycloadditions,10 and even nitrogen deletion reactions.11 Because of their diverse array of reactivity, the study of these reagents with a variety of functional groups may give rise to new mechanistic insights and strategies for chemical diversification.
Results and discussion
We first set out to optimize the yield of 3a (Table 1) by performing the reaction in two steps consisting of imine formation from 4-fluoroacetophenone (1a) and allylamine (2a), followed by reaction of the imine with A. Initial conditions involved stirring 1a and 2a in the presence of 3 Å molecular sieves at ambient temperature in cyclohexane. After 24 h, the solvent was removed, the reaction vessel cooled to 0 °C, and a solution of sulfonyl azide A in dry MeCN was slowly added. The reaction mixture was then allowed to warm to room temperature yielding the desired thiodiazole (3a) in 15% yield along with a 59% yield of recovered ketone (entry 1). The large amount of remaining starting ketone indicated to us that the condensation step (to make the imine) did not reach completion. Increasing the temperature for the imine formation to 60 °C led to a significant increase in overall yield (entry 2). However, running the reaction at 80 °C resulted in mostly decomposition (entry 3). We also speculated that a base additive could facilitate the conversion of the initially formed imine to intermediates such as an enamine en route to the product. Using 2,6-di-tert-butylpyridine as a base led to a pronounced increase in the reaction yield (entry 4), with 2, 6-lutidine proving less effective (entry 5). Lastly, we examined other common solvents, including tetrahydrofuran (THF), and dichloromethane (DCM) (entries 6 and 7), which led to decreased yields of 3a. The optimized conditions (entry 4) also proved viable without the need to remove the cyclohexane solvent needed for imine formation, leading to an efficient one-pot reaction protocol.|
|
|||||
|---|---|---|---|---|---|
| Entry | Temp | Solvent | Additive (1.0 equiv.) | Yielda (%) | RSM (%) |
| a Yield was determined by 1H NMR with dimethyl sulfone as an internal standard. b Decomposition was observed. c Isolated yield. CyH = cyclohexane RSM: recovered starting material MS: molecular sieves. | |||||
| 1 | 23 °C | MeCN | — | 15 | 59 |
| 2 | 60 °C | MeCN | — | 43 | 26 |
| 3 | 80 °C | MeCN | — | N/Ab | N/A |
| 4 | 60 °C | MeCN | Di-tert-butylpyridine | 71c | 18c |
| 5 | 60 °C | MeCN | 2,6-Lutidine | 48 | 47 |
| 6 | 60 °C | THF | Di-tert-butylpyridine | 61 | 15 |
| 7 | 60 °C | DCM | Di-tert-butylpyridine | 38 | 6 |
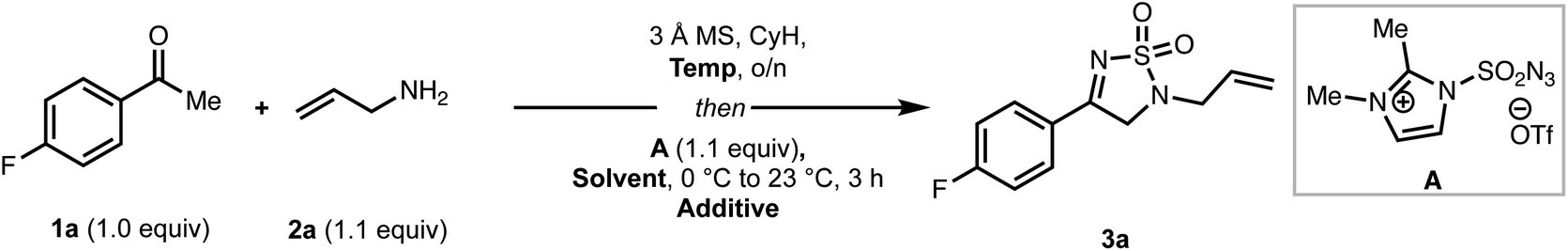
With optimized conditions in hand, we next investigated the scope of the coupling partners (Scheme 1). Simple allylic amines (see 3a–3b) perform well in this reaction, while cinnamyl amine results in reduced yield of the product (3c). Propargylic and homoallylic amines are also competent coupling partners giving moderate yields of the desired products (3d and 3e, respectively). Diverse benzylic amines, including those bearing electronically-disparate groups (see 3f–h) serve competently as coupling partners in the reaction, although α-substitution (see 3i) leads to decreased yields compared to other benzylic amines. Finally, aliphatic amines can also be employed, giving the products (3j–3k) in moderate to good yields. X-Ray crystallographic analysis of a single crystal of 3b provided support for the assigned structures. To further establish the practicality of this approach, a gram-scale reaction using 1a (1.00 g, 7.23 mmol) and 2a under the standard reaction conditions proceeded without any loss in efficiency to give 3a in 70% yield along with 5% of recovered methyl ketone starting material.
 | ||
| Scheme 1 Substrate scope of amine. aReaction conditions: see Table 1, entry 4. bp-Fluoroacetophenone. c(E)-Cinnamylamine. Isolated yield reported. Yield in parenthesis refer to recovered starting material. CyH: cyclohexane MS: molecular sieves. | ||
We then investigated the substrate scope of the ketone coupling partner. Para- and meta-substitution is well tolerated on the ketone coupling partner (see 3m–3n), while ortho-substitution significantly reduces reaction efficiency (see 3o). Electron-neutral and electron-poor ketones react well (see 3l–3q), whereas decreased yields are observed for an electron-rich ketone (see 3p). Various heteroaryl substituted ketones are competent substrates and deliver the corresponding products (3r–3u) in up to 89% yield. A series of cyclic aromatic ketones, including 5-fluoro-1-indanone, α-tetralone, and β-tetralone were successful substrates as well. Notably, α-tetralone and β-tetralone led to the same product (3w), presumably through diverging pathways (vide infra). Finally, vinyl and aliphatic ketones were competent substrates, giving 3x and 3y–3ab, respectively. Ketones bearing α-branched, cyclic, groups are superior substrates, whereas linear substrates (e.g., 1aa) gave lower yields of the desired products (e.g., 3aa).
There are also several limitations in the scope of this reaction. Specifically, we found that ketones bearing α-substitution were poor substrates. For example, 3ac was not formed from the corresponding ketone even upon extended reaction times. Amino acids and anilines also proved to be unsuccessful amine coupling partners.
We hypothesized that inefficient formation of the intermediate imine could account for the poor reaction outcomes observed in several cases. To probe this possibility, we monitored the imine formation for one successful substrate (i.e., to form 3a) and two unsuccessful substrates (to form 3ac and 3ad) as well as one low yielding product (3o). As shown in Scheme 2, for substrates that reacted poorly or unsuccessfully, only partial conversion of the ketone to the imine was observed. When these ketone/imine mixtures were treated with A, product was only obtained from imines 4a and 4o. The mixtures containing imines 4ac and 4ad returned only starting ketone after treatment with A and workup. We then synthesized pure 4o and subjected it to reaction with A. The product (3o) was isolated in a much improved 43% yield. From these experiments, we conclude that for some ketone substrates, low yields of the product might result due to incomplete imine formation. However, in some cases (e.g., 3ac), even substrates that form the imine intermediate efficiently can lead to low yields of the products due to other incompatibility issues (e.g., difficulty with enamine formation).
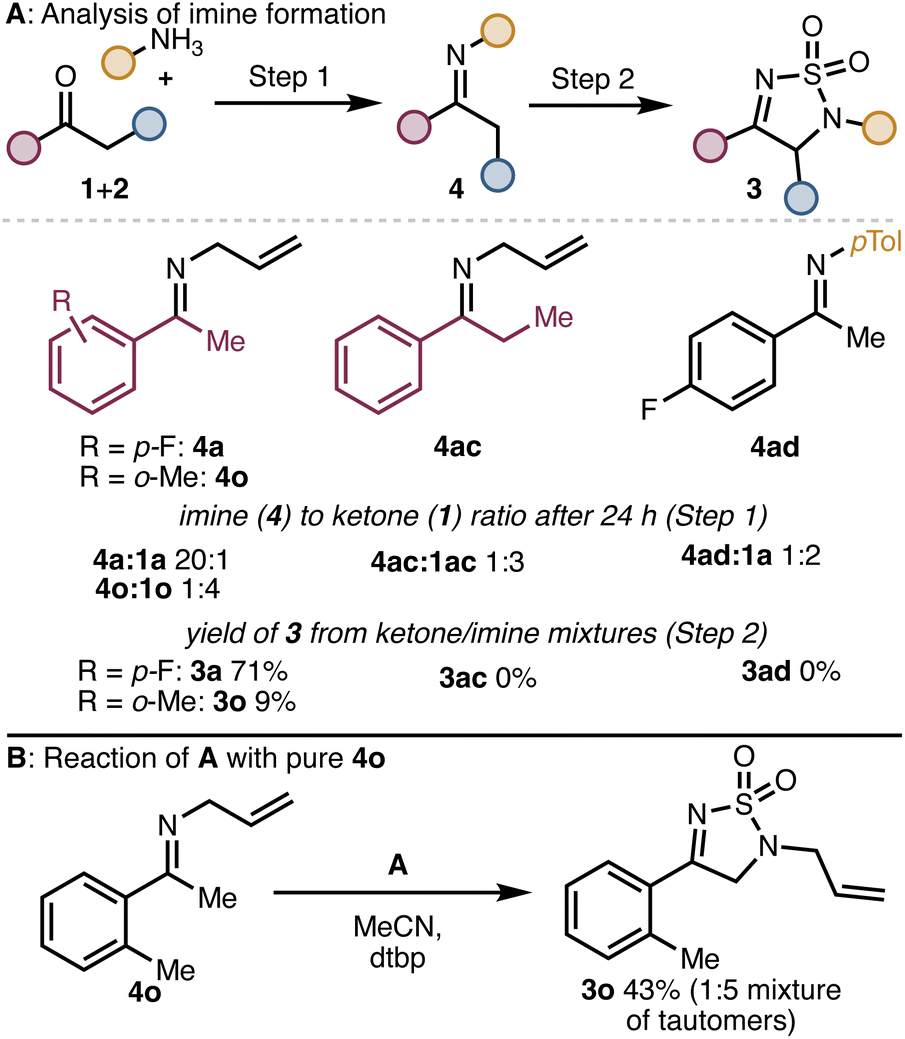 | ||
Scheme 2 (A) Ratios of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 after 24 h (3 Å MS, CyH, 60 °C) and outcome of subsequent reaction of A. (B) Success of reaction with pure 4o step 1: 3 Å MS, cyclohexane, 60 °C. Step 2: A, MeCN, dtbp, 0 °C to r. t. dtbp: 2, 6-ditertbutylpyridine. :3 after 24 h (3 Å MS, CyH, 60 °C) and outcome of subsequent reaction of A. (B) Success of reaction with pure 4o step 1: 3 Å MS, cyclohexane, 60 °C. Step 2: A, MeCN, dtbp, 0 °C to r. t. dtbp: 2, 6-ditertbutylpyridine. | ||
To gain insight into the reaction mechanism, we chose to conduct a series of studies with isotopically labeled substrates. The use of α-13C acetophenone (13C-1l) provided thiodiazole dioxide 13C-3l (Scheme 3A) bearing the 13C label at the imine carbon, which suggests that the carbon chain remains intact (i.e., a phenyl migration does not occur). Next, we conducted the reaction with 15N labelled allyl amine, which gave 15N-3a (Scheme 3B) where the 15N label was incorporated only at the amine position and not at the imine nitrogen. This observation indicates that the imine nitrogen in 4 undergoes a 1,2 migration during the course of the reaction. Notably, the nitrogen also migrates when an aliphatic ketone (1y) is used to give 15N-3y—further evidence that the aromatic system is not involved or critical to the rearrangement.
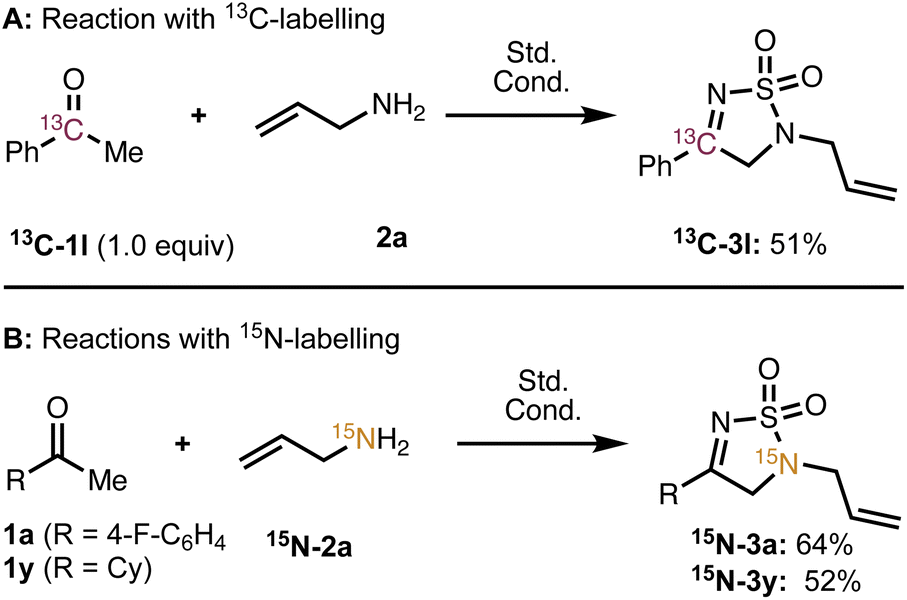 | ||
| Scheme 3 (A) Outcome of reaction with 13C-acetophenone. (B) Outcome of reaction with 15N-allylamine for conditions see Table 1 entry 4. | ||
Nitrogen migrations of this type are known for enamine/azide cycloadditions.12 These cycloadditions can generate amino-triazolines which can in turn generate amino-aziridines (Fig. 2A). Each of these species can further transform in several ways, including through C–C bond cleavage (Type I)13 and migration of a carbon (Type II),13a–d,14 nitrogen (Type III),13e,14b,c,15b or hydride (Type IV)10,13e,14a,15 substituent (Fig. 2A). Product mixtures are often observed, and the selectivity is dictated by the nature of the substituents on both the enamine and the azide components. Importantly, our system differs from those described in the literature in several ways. Most notably, our reaction proceeds through an imine formed from a primary amine (see Scheme 3A) rather than an enamine formed from a secondary amine (see ref. 12–15). Additionally, sulfamoyl azide A is able to react with the imine at sulfur, to generate a sulfamoyl enamine. These key differences, as well as the superb selectivity for the Type III nitrogen migration that we observe, led us to further investigate the reaction pathway with DFT. For ease of computation, calculations were performed with a model enamine (5), where the N-allyl group is modified to N-Me. Structure minima were generated with DFT or UDFT in the gas phase in Jaguar (B3LYP-D3/6-31G+*). Transition structures were identified using coordinate scans, linear synchronous transit, quadratic synchronous transit, or a combination thereof, at the same level of theory.
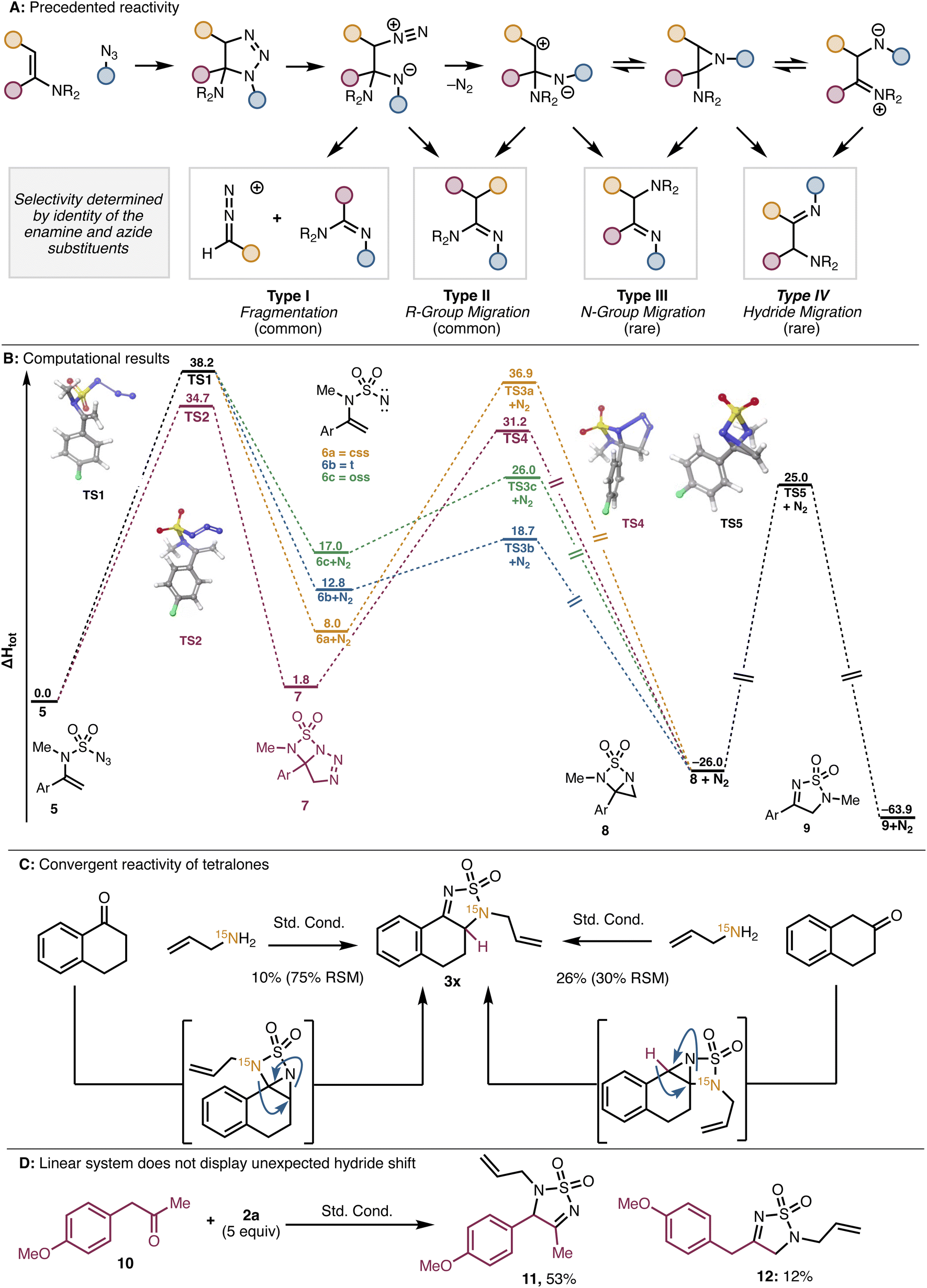 | ||
| Fig. 2 (A) Rearrangements of amino-triazolines. (B) DFT investigation of possible reaction pathways (values given in kcal mol−1). (C) Reactivity of tetralone substrates (css: closed shell singlet. oss: open shell singlet. t: triplet). | ||
We considered two possible pathways to access the amino-aziridine precursor to the Type III product. Pathway 1 involving loss of nitrogen followed by (2 + 1) cycloaddition, and Pathway 2 involving (3 + 2) cycloaddition followed by nitrogen extrusion (Fig. 2B). To interrogate Pathway 1, we first identified a transition state corresponding to loss of nitrogen from 5 (TS1) to form nitrene 6 in one of three spin states. At this stage, (2 + 1) cycloaddition would generate aziridine 8 through the respective transition structure, which subsequently undergoes exothermic rearrangement viaTS5 to give final product 9. For Pathway 2, we were able to identify a transition state for the intramolecular cycloaddition of the azide and enamine (TS2) in 5, which results in the formation of triazoline 7. A transition structure for the direct conversion of 7 to 9 by either a concerted or stepwise mechanism could not be identified. However, TS4 was identified, corresponding to loss of nitrogen from triazoline 7 to give 8. Analogous to the first pathway, 8 could undergo exothermic rearrangement to give the final product (9). The computationally computed barrier for the (3 + 2) cycloaddition at TS2 is 3.5 kcal mol−1 lower than the barrier for loss of nitrogen at TS1, suggesting the former is the favored pathway. Consistent with the isotope labelling experiments discussed above, one of the C–N aziridine bonds is lengthened at TS5, and the imaginary frequency at this transition structure corresponds to atomic motion of the C and N atoms involved in the bond rearrangement.
We hypothesize that the preference for Type III reactivity over either Type I or Type II arises from the sulfur dioxide group connecting the two nitrogens. This tether would make amidine formation difficult due to developing strain en route to a four-membered heterocycle. Although we cannot rule out an intermolecular enamine/azide cycloaddition, the product selectivity suggests that the S–N bond forms prior to rearrangement. Additionally, we propose that the nitrogen rearrangement accounts for the convergent reactivity of α- and β-tetralone. In the case of α-tetralone, the expected Type III nitrogen migration is observed (as confirmed by 15N NMR; see Fig. 2C). In the β-tetralone system, however, the nitrogen does not migrate. Instead, the observed product results from Type IV hydride migration (see Fig. 2A and C). We posit that in this case, the nitrogen shift is disfavored by developing “peri-strain” between the ortho hydrogen on the aromatic ring and the N-allyl substituent. In all other cases, the migrating group is moving away from the aromatic ring, leading instead to a lessening of any unfavorable interactions between the nitrogen substituent and the aromatic protons. On the basis of this hypothesis, we anticipated that a linear analog of β-tetralone (e.g., phenylacetone) should give only the expected nitrogen shift product because of the absence of “peri-strain” in this conformationally flexible system. Indeed, using 10 and 2a in the thiadiazole dioxide forming reaction, we isolated only the two expected products that involve a nitrogen shift (i.e., 11, arising from an intermediate benzylic enamine and 12, arising from a terminal enamine intermediate). This observation supports our hypothesis that the conformational rigidity of the α-tetralone system leads to the unique reactivity that is observed in that case.
With empirical and computational support for our proposed mechanism secured, we then set out to demonstrate the synthetic utility of our dihydro-1,2,5-thiodiazole 1,1-dioxides (Scheme 4). For example, tetrahydro-1,2,5-thiodiazole dioxide 13 is obtained through either reduction with sodium borohydride or enantioselective transfer hydrogenation using ruthenium catalyst B to give 13 in high yield and, in the case of the transfer hydrogenation, good enantiomeric excess (91% ee).6 In addition to hydrides, 3a is a competent electrophile for carbon nucleophiles, exemplified by the addition of ethyl magnesium bromide to form 14. The cyclic sulfamide moiety can also be opened to 1,2 diamine 15 by heating with hydrazine. This process allows for easy and modular access to diamines which are precursors to a variety of biologically relevant structural motifs (imidazolidinones, etc.).16
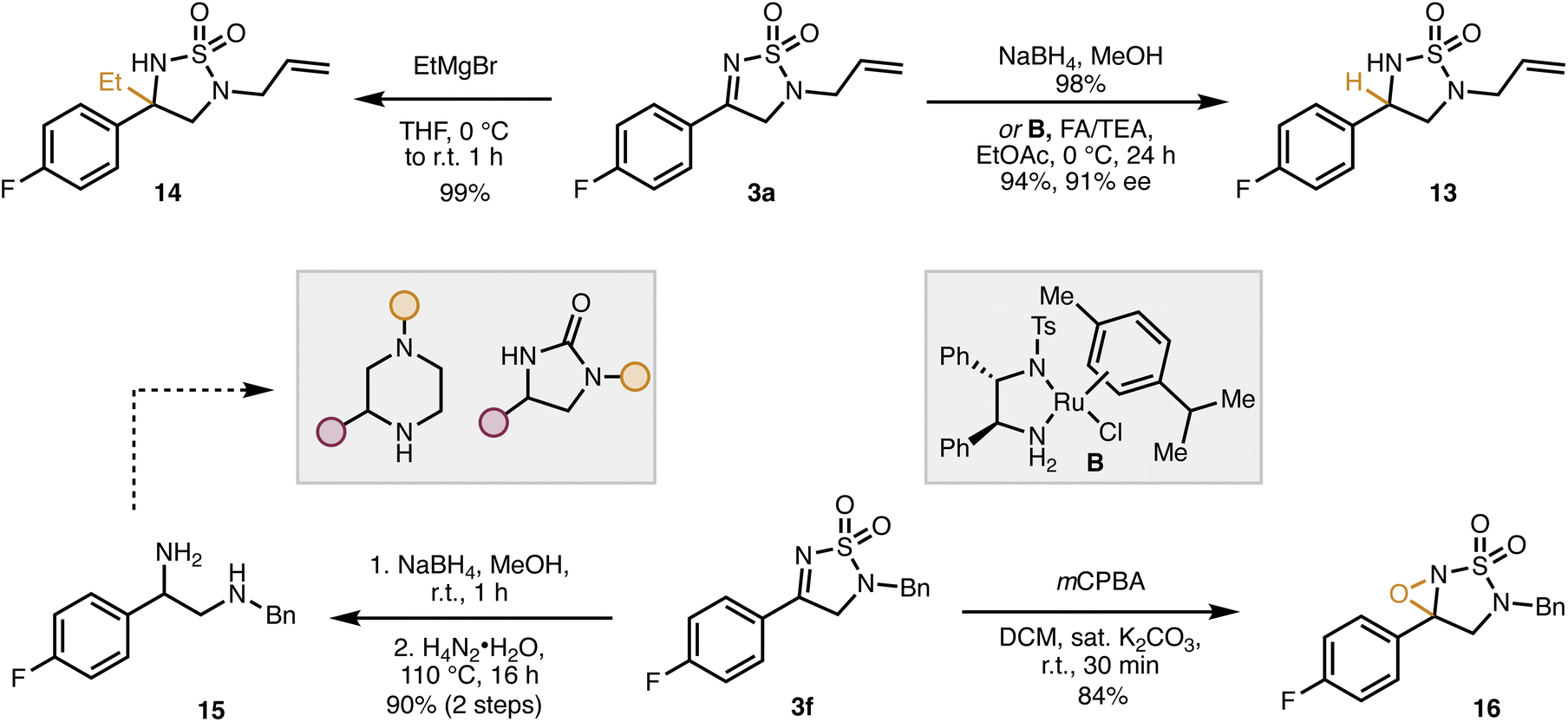 | ||
| Scheme 4 Synthetic transformations of dihydro-1,2,5-thiodiazole dioxide. FA/TEA: formic acid/triethylamine (∼5:2). | ||
Sulfonyl imines are well-established precursors to N-sulfonyl oxaziridines, a very useful class of oxygen-transfer reagents.17 Accordingly, the dihydro-1,2,5-thiodiazole dioxide 3f was readily converted to the corresponding oxaziridine (16) by oxidation with meta-chloroperoxybenzoic acid (mCPBA). The modular nature of our method should allow for the synthesis of a diverse array of oxaziridines which could aid in the discovery of new reagents for oxygen transfer.
Conclusions
In conclusion, we have developed an efficient method for the conversion of commercially available methyl ketones and primary amines to dihydro-1,2,5-thiodiazole dioxides in a one-pot protocol using 2,3-dimethylimidazole-1-sulfonyl azide. The overall transformation constitutes a highly modular three-component coupling and provides a general and practical approach to diversified drug-like thiodiazole dioxides. The transformation displays a reasonably broad functional group tolerance. A proposed reaction mechanism is supported by isotopic labelling of nitrogen and carbon atoms in the amine and ketone coupling partners, respectively. The subsequent transformation of thiodiazole dioxides into other compounds of medicinal relevance was also demonstrated.Data availability
Data supporting the work in this publication are available via the ESI and associated crystallographic data.†Author contributions
This manuscript was written by P. S. and K. P. with input from I. S., R. S., T. P. L., P. G. R. and M. A. P. P. G. R. first characterized the unexpected thiodiazole dioxide product. The majority of the synthetic work was carried out by K. P. and P. S. Experiments were designed by P. S. and R. S. with input from M. A., T. P. L. and P. G. R. All calculations were carried out by I. S. under the supervision of M. S., T. P. L, P. G. R., M. A., and R. S. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. S. is grateful to Janssen Research and Development for support (Agreement No. 1501907). Partial support of the work conducted at Berkeley was supported by the NIGMS (R35 GM130345). K. P. was supported by a fellowship from the National Research Council of Thailand (NRCT): NRCT5-RGJ63023-175. We thank Dr Hasan Celik and UC Berkeley's NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance. Instruments in CoC-NMR are supported in part by NIH S10OD024998. We thank Dr Nicholas Settineri (UC Berkeley) for X-ray crystallographic studies of 3b. We thank Drs Ulla Andersen and Zongrui Zhou at the UC Berkeley QB3 Mass Spectrometry Facility for mass spectrometry analysis. Special thanks to Deszra Shariff, Dr Mona Sharar and Jonathan Malone at Janssen Research and Development for NMR analysis and acquisition of HRMS. We thank Dr Michael Black (formerly from the Sarpong Group), who was the first to synthesize the thiodiazole dioxide product. We are grateful to Dr Ian Bakanas for insightful discussions regarding the nitrogen migration mechanism.Notes and references
- For reviews, see: (a) J. Y. Winum, A. Scozzafava, J. L. Montero and C. T. Supuran, Med. Res. Rev., 2006, 26, 767–792 CrossRef CAS; (b) A. B. Reitz, G. R. Smith and M. H. Parker, Expert Opin. Ther. Pat., 2009, 19, 1449–1453 CrossRef CAS PubMed.
- (a) T. Sparey, D. Beher, J. Best, M. Biba, J. L. Castro, E. Clarke, J. Hannam, T. Harrison, H. Lewis, A. Madin, M. Shearman, B. Sohal, N. Tsou, C. Welch and J. Wrigley, Bioorg. Med. Chem. Lett., 2005, 15, 4212–4216 CrossRef CAS PubMed; (b) A. F. Kreft, R. Martone and A. Porte, J. Med. Chem., 2009, 52, 6169–6188 CrossRef CAS PubMed; (c) L. E. Keown, I. Collins, L. C. Cooper, T. Harrison, A. Madin, J. Mistry, M. Reilly, M. Shaimi, C. J. Welch, E. E. Clarke, H. D. Lewis, J. D. J. Wrigley, J. D. Best, F. Murray and M. S. Shearman, J. Med. Chem., 2009, 52, 3441–3444 CrossRef CAS PubMed; (d) J. Zhong, X. Gan, K. R. Alliston and W. C. Groutas, Bioorg. Med. Chem., 2004, 12, 589–593 CrossRef CAS PubMed; (e) A. Spaltenstein, M. R. Almond, W. J. Bock, D. G. Cleary, E. S. Furfine, R. J. Hazen, W. M. Kazmierski, L. L. Wright, F. G. Salituro and R. D. Tung, Bioorg. Med. Chem. Lett., 2000, 10, 1159–1162 CrossRef CAS; (f) M. Benltifa, M. I. García Moreno, C. Ortiz Mellet, J. M. García Fernández and A. Wadouachi, Bioorg. Med. Chem. Lett., 2008, 18, 2805–2808 CrossRef CAS.
- (a) J. Hannam, T. Harrison, F. Heath, A. Madin and K. Merchant, Synlett, 2006, 833–836 CAS; (b) J. Liu, P. P. Shao, D. Guiadeen, A. Krikorian, W. Sun, Q. Deng, A. M. Cumiskey, R. A. Duffy, B. A. Murphy, K. Mitra, D. G. Johns, J. L. Duffy and P. Vachal, Bioorg. Med. Chem. Lett., 2021, 32, 127668–127677 CrossRef CAS.
- (a) K. Lang, C. Li, I. Kim and X. P. Zhang, J. Am. Chem. Soc., 2020, 142, 20902–20911 CrossRef CAS PubMed; (b) K. Lang, S. Torker, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2019, 141, 12388–12396 CrossRef CAS PubMed; (c) H. Jiang, K. Lang, H. Lu, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2016, 55, 11604–11608 CrossRef CAS; (d) H. Lu, K. Lang, H. Jiang, L. Wojtas and X. P. Zhang, Chem. Sci., 2016, 7, 6934–6939 RSC; (e) Y. Yang, I. Cho, X. Qi, P. Liu and F. H. Arnold, Nat. Chem., 2019, 11, 987–993 CrossRef CAS PubMed; (f) P. Dydio, H. M. Key, H. Hayashi, D. S. Clark and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 1750–1753 CrossRef CAS PubMed; (g) H. Lu, H. Jiang, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192–10196 CrossRef CAS; (h) X. Nie, Z. Yan, S. Ivlev and E. Meggers, J. Org. Chem., 2021, 86, 750–761 CrossRef CAS.
- S. A. Lee, S. H. Kwak and K. I. Lee, Chem. Commun., 2011, 47, 2372–2374 RSC.
- C. Schüttler, Z. Li-Böhmer, K. Harms and P. Von Zezschwitz, Org. Lett., 2013, 15, 800–803 CrossRef PubMed.
- B. A. Wright, A. Matviitsuk, M. J. Black, P. García-Reynaga, L. E. Hanna, A. T. Herrmann, M. K. Ameriks, R. Sarpong and T. P. Lebold, J. Am. Chem. Soc., 2023, 145, 10960–10966 CrossRef CAS PubMed.
- J. C. Culhane and V. V. Fokin, Org. Lett., 2011, 13, 4578–4580 CrossRef CAS PubMed.
- (a) M. Y. Stevens, R. T. Sawant and L. R. Odell, J. Org. Chem., 2014, 79, 4826–4831 CrossRef CAS PubMed; (b) E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797–3800 CrossRef CAS PubMed.
- (a) M. H. Shen, K. Xu, C. H. Sun and H. D. Xu, Org. Lett., 2015, 17, 3654–3657 CrossRef CAS PubMed; (b) M. H. Shen, K. Xu, C. H. Sun and H. D. Xu, Org. Biomol. Chem., 2016, 14, 1272–1276 RSC.
- (a) X. Zou, J. Zou, L. Yang, G. Li and H. Lu, J. Org. Chem., 2017, 82, 4677–4688 CrossRef CAS PubMed; (b) H. Qin, W. Cai, S. Wang, T. Guo, G. Li and H. Lu, Angew. Chem., Int. Ed., 2021, 60, 20678–20683 CrossRef CAS.
- For a review, see: V. A. Bakulev, T. Beryozkina, J. Thomas and W. Dehaen, Eur. J. Org. Chem., 2018, 262–294 CrossRef CAS.
- (a) D. Pocar and P. Trimarco, J. Chem. Soc., Perkin Trans. 1, 1976, 622–624 RSC; (b) P. D. Croce and R. Stradi, Tetrahedron, 1977, 33, 865–867 CrossRef; (c) F. Cassani, G. Celentano, E. Erba and D. Pocar, Synthesis, 2004, 1041–1046 CAS; (d) I. Efimov, V. Bakulev, N. Beliaev, T. Beryozkina, U. Knippschild, J. Leban, F. Zhi-Jin, O. Eltsov, P. Slepukhin, M. Ezhikova and W. Dehaen, Eur. J. Org. Chem., 2014, 2014, 3684–3689 CrossRef CAS; (e) T. Gao, M. Zhao, X. Meng, C. Li and B. Chen, Synlett, 2011, 1281–1284 CAS.
- (a) L. Citerio, M. L. Saccarello and P. Trimarco, J. Heterocycl. Chem., 1979, 16, 289–292 CrossRef CAS; (b) A. Contini and E. Erba, RSC Adv., 2012, 2, 10652–10660 RSC; (c) S. Pellegrino, A. Contini, M. L. Gelmi, L. Lo Presti, R. Soave and E. Erba, J. Org. Chem., 2014, 79, 3094–3102 CrossRef CAS PubMed; (d) G. Audran, C. Adiche, P. Brémond, D. El Abed, M. Hamadouche, D. Siri and M. Santelli, Tetrahedron Lett., 2017, 58, 945–948 CrossRef CAS.
- (a) L. Sitario, M. L. Saccarello and R. Stradi, Synthesis, 1979, 4, 305–308 Search PubMed; (b) J. F. Stephen and E. Marcus, J. Heterocycl. Chem., 1969, 6, 969–974 CrossRef CAS.
- (a) K. E. Gettys, Z. Ye and M. Dai, Synth, 2017, 49, 2589–2604 CrossRef CAS; (b) S. P. Swain and S. Mohanty, ChemMedChem, 2019, 14, 291–302 CrossRef CAS PubMed; (c) D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580–2627 CrossRef CAS.
- (a) K. S. Williamson, D. J. Michaelis and T. P. Yoon, Chem. Rev., 2014, 114, 8016–8036 CrossRef CAS PubMed; (b) S. Ravindra, C. P. Irfana Jesin, A. Shabashini and G. C. Nandi, Adv. Synth. Catal., 2021, 363, 1756–1781 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2284144 for 3b. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04478e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |