
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew light on the imbroglio surrounding the C8H+6 isomers formed from ionized azulene and naphthalene using ion–molecule reactions†
Corentin
Rossi
 a,
Giel
Muller
b,
Roland
Thissen
cd,
Claire
Romanzin
cd,
Christian
Alcaraz
cd,
Sandesh
Gondarry
e,
Paul M.
Mayer
e and
Ugo
Jacovella
*a
a,
Giel
Muller
b,
Roland
Thissen
cd,
Claire
Romanzin
cd,
Christian
Alcaraz
cd,
Sandesh
Gondarry
e,
Paul M.
Mayer
e and
Ugo
Jacovella
*a
aUniversité Paris-Saclay, CNRS, Institut des Sciences Moléculaires d'Orsay, 91405 Orsay, France. E-mail: ugo.jacovella@universite-paris-saclay.fr
bMolecular Horizons and School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, New South Wales 2522, Australia
cUniversité Paris-Saclay, CNRS, Institut de Chimie Physique, UMR8000, 91405 Orsay, France
dSynchrotron SOLEIL, L'Orme des Merisiers, 91192 Saint Aubin, Gif-sur-Yvette, France
eDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa K1N 6N5, Canada
First published on 24th November 2023
Abstract
Most polycyclic aromatic hydrocarbons (PAHs) can isomerize with internal energies near to or below the dissociation threshold. The C10H+8 group of ions, made up of the naphthalene (Naph+) and the azulene (Azu+) radical cations, is a prototypical example. C8H+6 isomers are important species in the growth kinetics and formation of complex organic molecules, and more generally fragments from larger PAHs, yet information about C8H+6 structures is scarce and contradictory. Here, ion–molecule reactions were carried out and the tunable photoionization chemical monitoring technique was used to probe the C8H+6 isomers formed upon C2H2-loss from Naph+ and Azu+. The experimental findings were interpreted with the support of ab initio and kinetics calculations. To facilitate the interpretation of these data, chemical reactivity starting from phenylacetylene (PA) was studied. It was found that most of the C8H+6 ions formed from C10H8, in a timescale of 40 μs, are PA+ in the vicinity of the dissociation threshold. No evidence of the pentalene radical cation (PE+) was observed and explanations to reconcile previous results are presented.
Introduction
Energetic gaseous polycyclic aromatic hydrocarbons (PAHs) react in environments spanning from plasmas, flames, planetary ionospheres, to stellar and interstellar regions.1–8 The photoprocessing of gaseous PAHs leads to unimolecular reactions such as isomerization and dissociation, which often compete. The major dissociation channels of PAHs involve the loss of atomic or molecular hydrogen and C2H2, with dissociative ionization giving a quasi-balanced branching ratio for small PAHs such as naphthalene (Naph), and in favor of H/H2 loss as the PAH size increases.9 The C2H2-loss reaction, in particular, plays a central role in stirring the composition of species formed in environments where energized PAHs can be found.Better understanding of the unimolecular reactions of small prototypical PAH radical cations such as Naph+ and Azu+ is necessary to predict reactivities of larger PAHs and elucidate their relationship with carbon clusters.10–12 The pioneering works on the fate of Naph and Azu were performed by the groups of Leach13–16 and Lifshitz.17–21 Through use of photoionization spectroscopy, time-resolved photodissociation spectroscopy, ab initio calculations, and Rice–Ramsperger–Kassel–Marcus (RRKM) calculations, these studies revealed H-loss and C2H2-loss as the two primary dissociation channels, occurring with equal branching ratios owing to their similar activation energies and dissociation rate constants. Furthermore, photoionization mass spectra of Naph and Azu have been shown to be similar at photon energy greater than 20 eV.
Structural information about the C8H+6 fragment of Naph+ and Azu+ has been scattered and continually evolving over the last three decades. The structures of the molecular ions of interest are provided in Scheme 1. In the early 1990s, Leach and coworkers proposed the formation of the phenylacetylene radical cation (PA+) as the primary structure resulting from dissociative photoionization (within a few tens of μs).14 They confirmed this proposition one year later by conducting similar experiments with deuterated species.15 In the same year, the Lifshitz group further supported the hypothesis that PA+ is the major product resulting from ionized Naph and Azu, using time-resolved photoionization mass spectrometry (TPIMS), allowing the ions to evolve for 40 μs to 400 ms.17 In the same study, they also corroborated this finding through collision activation (CA) spectra.
 | ||
| Scheme 1 Structures of the molecular ions and neutral targets of interest for this study. | ||
The cards were reshuffled in 1995 with the theoretical work of Granucci et al.,22 which involved structure optimizations for C8H+6 using Møller–Plesset perturbation theory (MP2) and the complete active space self-consistent field (CASSCF) method for energy calculations. The structures they considered included PA+, benzocyclobutadiene radical cation (BCB+), and the benzocyclopropenemethylene radical cation. The latter structure was discarded due to its significantly higher energy, but BCB+ was found to be more stable than PA+. Two years later, Lifshitz and coworkers theoretically confirmed, using a coupled cluster method (CCSD(T)), that BCB+ was the most stable structure among the three considered by Granucci et al.22 The dissociation limit into C8H+6 (BCB+) + C2H2 was then adopted as the lowest one in subsequent works by the Lifshitz group.20,21
In 1999, the Schwarz group employed a variety of complementary mass spectrometric techniques to confirm the structure of the C8H+6 species formed from Naph+.23 They expanded the range of possible structures to include acyclic, eight-membered ring, and were the first to consider the pentalene radical cation (PE+). Acyclic and eight-membered ring structures were considered unlikely products. BCB+ was proposed as the most probable structure, mainly based on charge reversal (CR) experiments (30 μs timescale and ions formed by electron impact), in agreement with the previous theoretical work.19,22 A couple of years later, van der Hart used density-functional theory (DFT) to study the most probable C8H+6 structures formed from Naph+, concluding that both BCB+ and PA+ should be observable in photodissociation experiments.24
A major step forward came from the theoretical work of Mebel and coworkers, who mapped out the potential energy surface of C10H+8 using the G3(MP2,CC)//B3LYP method.25 They then performed RRKM and microcanonical variational state theory calculations to investigate the possible photodissociation mechanisms of Azu+ at different photon energies.25 They also computed the relative branching ratios of C8H+6 structures obtained after C2H2 elimination using the steady-state approach. These branching ratios indicate that below 5.2 eV of internal energy in Azu+, PA+ is the prevailing C8H+6 structure but gets supplanted by PE+ at internal energies greater than 5.2 eV above the Azu+ ionization energy. The contribution of BCB+, although marginal, increases with the internal energy. It is important to note that the total photon energy was assumed to be transferred into internal energy of the ion, neglecting Franck–Condon considerations and any radiative relaxation processes.
Solano and Mayer conducted similar work for Naph+ but used a potential energy surface computed at a lower level of theory (UB3LYP/6-311+G(3df,2p)//UB3LYP/6-31G(d)), and found that PA+ only becomes the major isomer when Naph+ has more than 7.6 eV of internal energy.26 Discrepancies between the results of kinetic calculations and those of Mebel group25 may be due to the fact that kinetic modeling did not fully follow the master equation approach, included simplifying assumptions, and the initial internal energy distribution was different.
The dissociation pathway from C10H+8 leading to PE+ was experimentally evidenced for the first time by recording the infrared spectroscopic fingerprint of PE+.27 C8H+6 ions were generated by multiphoton dissociative ionization from Naph and were allowed to evolve for 200 ms before being ejected from the trap, which could be considered as equilibrium for a molecular system of this size. Other experiments on nitrogen-containing PAHs and larger PAHs then suggested a general trend for the formation of pentagons in the ionic species through the destruction of PAHs containing pure hexagonal geometries.28,29 These mechanisms may help to rationalize the formation of fullerenes from larger energized PAHs10,11
Bull and coworkers30 obtained similar results to Mebel and coworkers25 using a coupled master equation combined with high-level potential energy surface calculations (CCSD(T)/cc-pVTZ).30 At around 5.1 eV of internal energy in Azu+ and Naph+, they calculated a ratio of 3/4 of C8H+6 being PA+ and 1/4 being PE+ at equilibrium. This confirms that PA+ is the dominant one at low internal energy before giving way to PE+, which should dominate the C8H+6 population above its thermodynamic formation threshold, provided sufficient time is granted.
The present study focuses on the reactivity of C8H+6 structures with allene and propyne, where the C8H+6 structures are generated either by direct photoionization of phenylacetylene (PA) or dissociative photoionization of Naph and Azu molecules. We employ the tunable photoionization chemical monitoring technique,31 an approach that has been successful in elucidating the structures of ionic fragments from dissociative ionization32 and uncovering isomerization pathways.33 The data set obtained from the current study provides chemical reference for one of the expected C8H+6 species, specifically PA+, facilitating the data interpretation from dissociative ionization of C10H8 species. The experimental data are complemented by ab initio and kinetics calculations. Finally, results from former experimental works are discussed in the light of the present results and new interpretations are proposed to reconcile all the results.
Methods
Experimental
Parent ions were produced through photoionization using light over the 7.2 to 20 eV photon energy range using the DESIRS undulator-based vacuum ultraviolet (VUV) beamline34 at the SOLEIL synchrotron facility in Saint-Aubin (France). The temperature of the neutral precursors when irradiated by the VUV light was approximately 300 K. Higher-order light was removed from the beam using a gas filter filled with either Kr or Ne depending on the energy range. The reaction products were monitored using the CERISES apparatus consisting of a tandem mass spectrometer (quadrupole–octopole–octopole–quadrupole) coupled to a photoionization source.35 Parent ions produced in the source were mass filtered by the first quadrupole and then guided by an octopole to the reaction cell while defining the collision energy. The ionic products of reactions were then radially confined and directed into the second quadruple mass filter. The absolute reaction cross sections were obtained by measuring the absolute pressure of the neutral reactant in the reaction cell and the parent and product ion counts. Measurements were taken across the photon energy range while the collision energy was fixed at 0.11 eV in the center-of-mass frame. The neutral reactants were introduced into the reaction cell at a low pressure (typically below 70 nbar) to ensure the attenuation of parent ion signal was less than 10%, providing a single-collision regime.Computational
Stationary points on the ground state C8H+6 potential energy surface around PA+ were found by conducting relaxed potential energy surface scans of C8H+6 structures along various internal degrees of freedom at the ωB97X-D/aug-cc-pVTZ level of theory using the Gaussian 16 software package.36 The optimized geometry for each saddle point was verified by its single negative vibrational frequency and by intrinsic reaction coordinate (IRC) calculations.Rice–Ramsperger–Kassel–Marcus (RRKM) theory was employed to calculate the k(E) for relevant reactions according to the equation:37
| k(E) = (σN‡(E − E0))/(hρ(E)), | (1) |
Results and discussions
Fig. 1 and 5 show reaction cross sections for the products of C8H+6 ions, formed from PA (Fig. 1), Naph, and Azu (Fig. 5), reacting with propyne and allene. The cross sections are given as a function of photon energy. Below, we will first present results of reactions carried out using the PA precursor and then analyze the results with the support of ab initio calculations and RRKM modeling. Later, a similar scheme is followed for reactions of C8H+6 produced from Naph and Azu. | ||
| Fig. 1 The top panels (black traces) display the C8H+6 photoion yield starting from phenylacetylene precursor (gray region was recorded with Kr in gas filter while dark region was recorded with Ne in gas filter). Bottom panels are reaction cross sections for the products resulting from C8H+6 produced from phenyacetylene reacting, with (left) propyne and (right) allene as a function of photon energy. | ||
C8H+6 from PA
Reactions with Propyne
Reactions with Allene
Discussion
 | ||
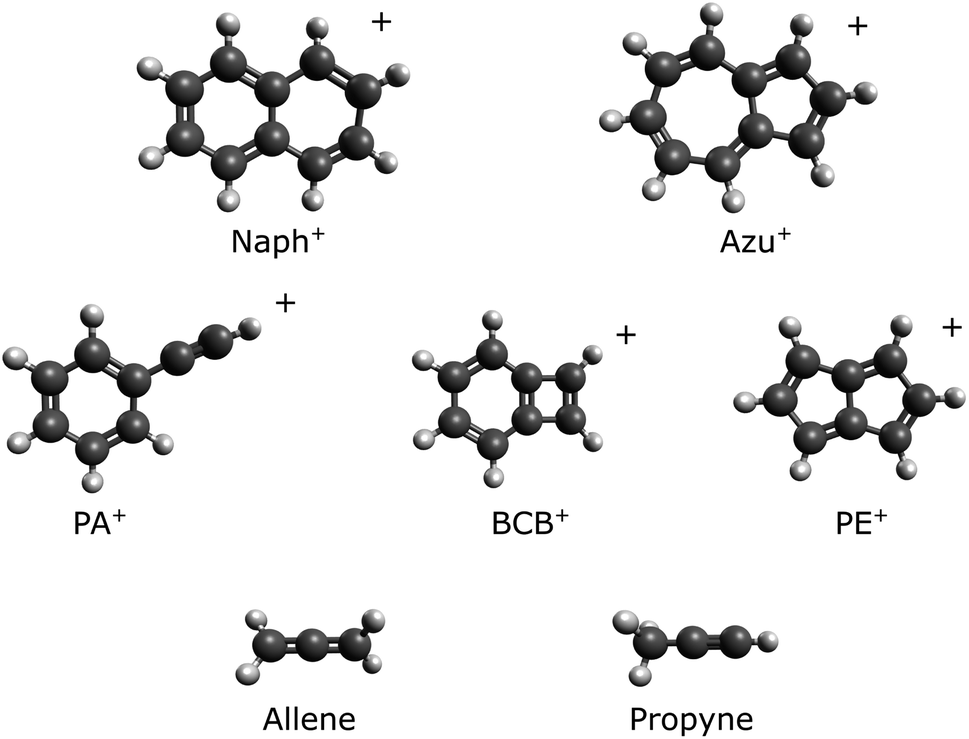
| Fig. 2 C8H+6 potential energy surface calculated at ωB97X-D/aug-cc-pVTZ level of theory. | ||
| Minima | Energies/eV | TSs | Energies/eV |
|---|---|---|---|
| PA+ | 0.00 | PA+-1+ | 2.56 |
| BCB+ | −0.20 | PA+-2+ | 2.57 |
| PE+ | −0.60 | PA+-3+ | 2.64 |
| 1+ | 1.89 | PA+-4+ | 2.61 |
| 2+ | 1.93 | PA+-8+ | 2.88 |
| 3+ | 2.22 | PA+-10+ | 3.26 |
| 4+ | 2.19 | 1+-6+ | 2.86 |
| 5+ | 0.62 | 2+-5+ | 3.08 |
| 6+ | 0.85 | 2+-7+ | 2.75 |
| 7+ | 2.60 | 3+-5+ | 3.19 |
| 8+ | 2.60 | 4+-6+ | 3.13 |
| 9+ | 2.62 | 7+-8+ | 3.13 |
| 10+ | 1.18 | 7+-9+ | 3.41 |
| 11+ | 1.87 | 8+-10+ | 3.49 |
| 12+ | 2.01 | 9+-11+ | 3.15 |
| 13+ | 0.66 | 10+-12+ | 2.86 |
| 14+ | 0.90 | 10+-13+ | 1.94 |
| 10+-14+ | 2.75 | ||
| 11+-BCB+ | 1.96 | ||
| 12+-BCB+ | 2.42 | ||
| BCB+-PE+ | 2.57 |
 | ||
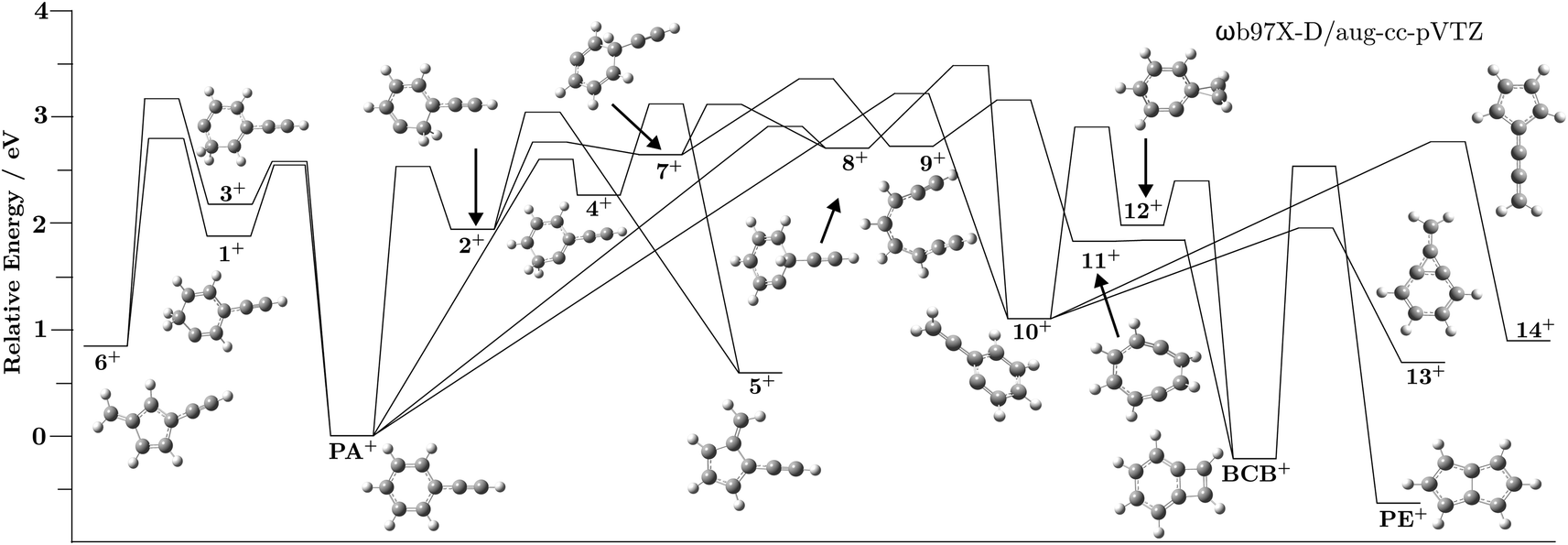
| Fig. 3 Fractional abundances of PA+ and 1+–4+ as a function of the internal energy of PA+. The shaded grey area represents the energies accessible in region 2. Note that these relative populations do not include the reactions out of the PA/1–4 ion well and thus do not reflect the true distribution of structures from Fig. 2. | ||
Using this concept, we can investigate the contrasted unreactive behavior with propyne to provide an explanation. Fig. 4 presents possible entrance channels and adducts for the reactions of 1+ and 2+ with propyne and allene. The minimum reaction energy paths (MREPs) were calculated at the ωB97X-D/aug-cc-pVTZ level of theory and show that the reactions of 1+ and 2+ with allene are strongly exothermic, while those with propyne are either endothermic or thermoneutral. Interestingly, this difference in chemistry is rooted in the propyne/allene structures, leading to an easy H atom transfer through a four member intermediate in allene, impossible in the case of propyne, resulting in a much more stable structure when C2H3 is lost. Note that the loss of C2H3 compared to C2H2 + H is thermodynamically favored.
 | ||
| Fig. 4 Minimum reaction energy paths of isomers 1+ (top panel) and 2+ (bottom panel) with (left) propyne and (right) allene, leading to m/z 115 + C2H3. All calculations were done at the ωB97X-D/aug-cc-pVTZ level of theory. | ||
C8H+6 from Naph and Azu
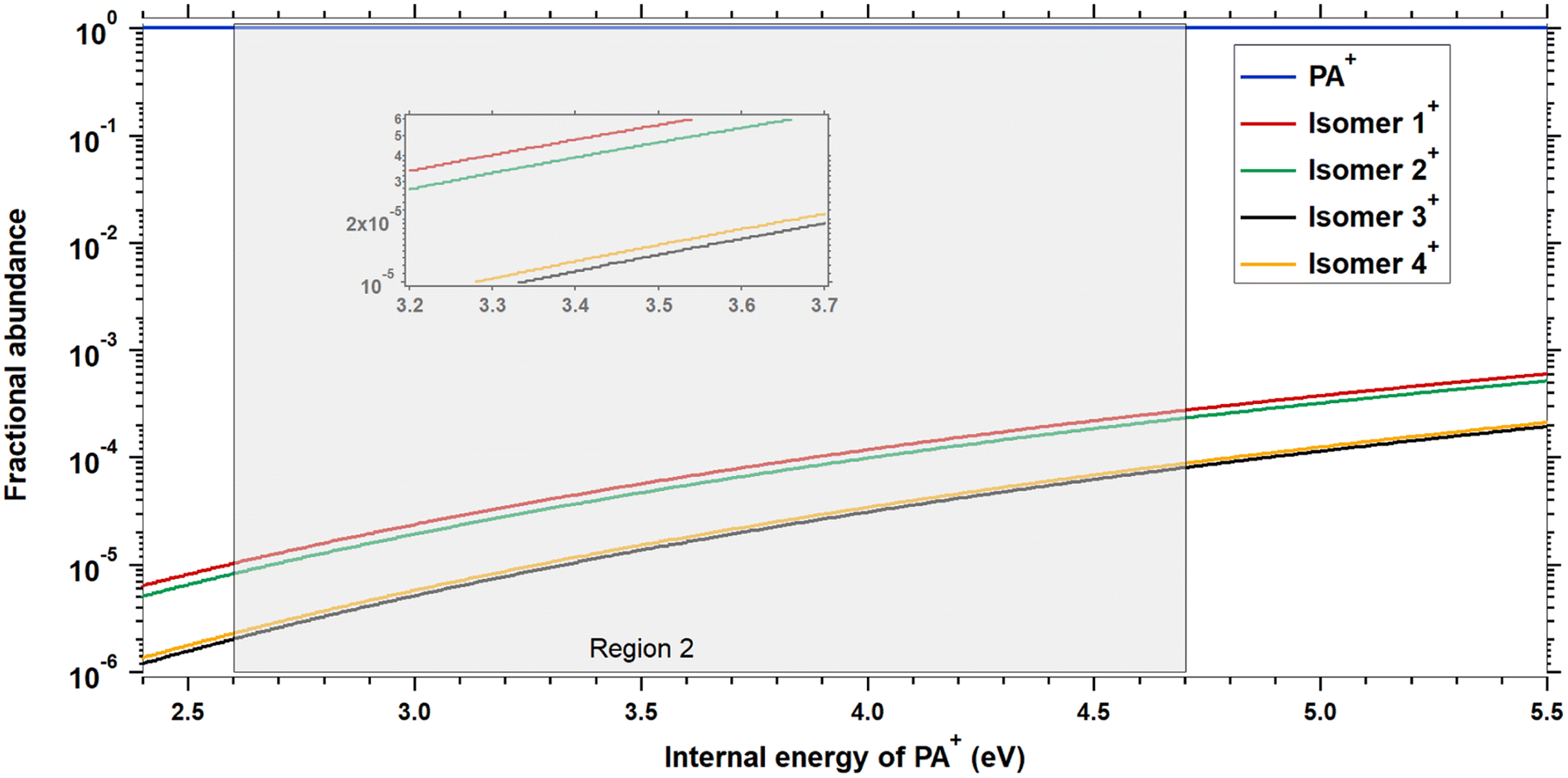
The reaction cross sections of C8H+6, produced by dissociative ionization of Naph and Azu and subsequent reaction with either propyne or allene, can be seen in the panels of Fig. 5. Only reaction channels leading to the formation of products with reaction cross sections greater than 0.1 Å2 are presented. Only two channels meet this criterion, m/z 115 and 141, also observed with PA. The channels at m/z 116, 129, and 139 were not observed or had reaction cross-section below 0.1 Å2. | ||
| Fig. 5 The top panels (black traces), (a and b), display the C8H+6 photoion yield starting from Naph and Azu precursors, respectively. Middle panels, (c and d), are reaction cross sections of C8H+6 with propyne as a function of photon energy starting with Naph and Azu as precursors, respectively. Bottom panels, (e and f), are the reaction cross sections with allene from Naph and from Azu), respectively. We defined the AE as the photon energy where more than 20 C8H+6 ions/s are detected. | ||
Observations
It is striking that there is no isomeric effect, as the data appear to be identical for Naph and Azu, when corrected by their respective appearance energies, as clearly shown by Fig. 6 below. Similarly to previous studies,14,17 the C8H+6 threshold is very elusive, we therefore defined the AE as the photon energy where more than 20 C8H+6 ions/s are detected.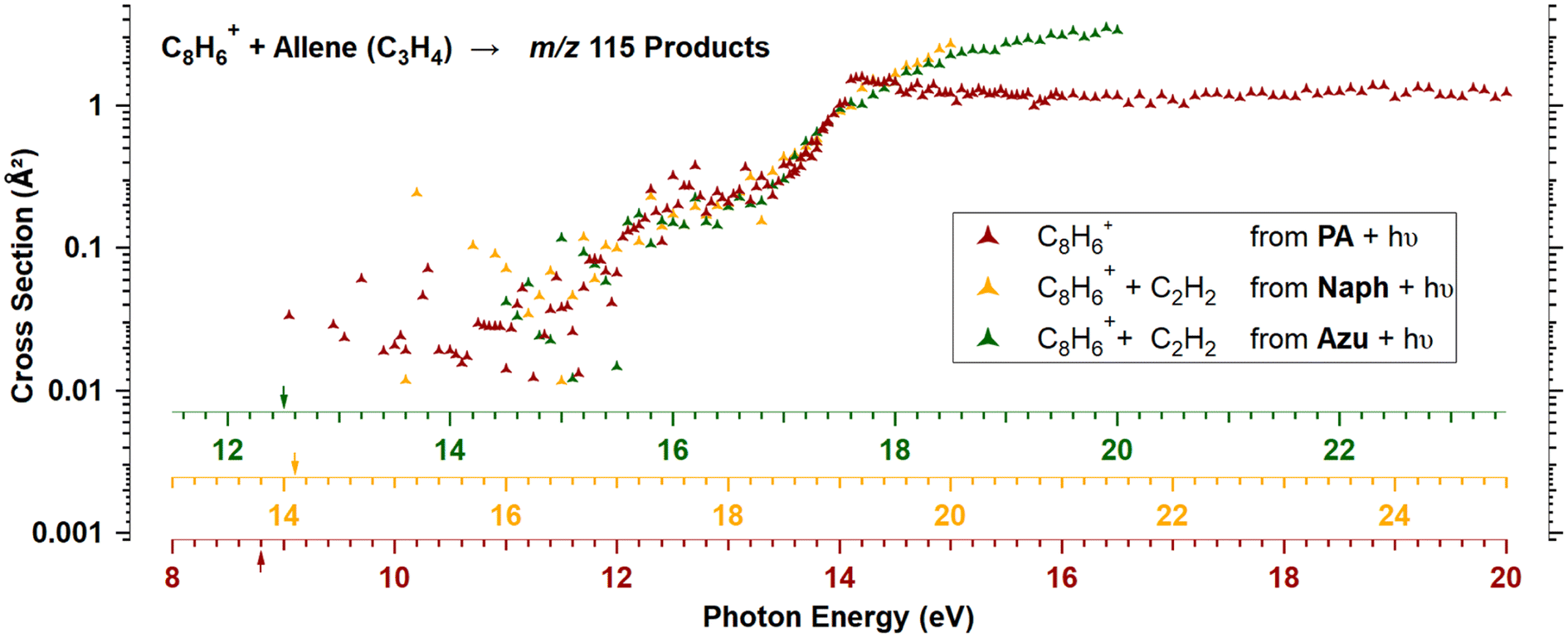 | ||
| Fig. 6 Reaction cross sections of C8H+6 reacting with allene to produce ions with m/z 115 when C8H+6 are formed from PA (red), Naph (orange), and Azu (green). The energy axes have been individually shifted to superimpose together. The arrows represent the IE of PA and the AEs of C8H+6 from Naph (14.1 eV) and Azu (12.5 eV) precursors. | ||
Discussion
Experimental and theoretical studies have disputed the isomeric structures of the major C8H+6 fragment formed upon C2H2-loss but the most recent ones agree that Azu+ should fragment into PE+, while Naph+ should fragment into PA+ and BCB+. Typically starting either from Azu+ or Naph+, it was found that it requires ≈ 5 eV of internal energy for C10H+8 systems to produce PE+ as the main fragment, which underlies the importance of the isomerization kinetics of the C10H+8 ions. In our apparatus, when considering dissociative photoionization, only unimolecular dissociation reactions occurring faster than 40 μs can be probed. This is the time for ions to reach the entrance of the first mass filter. We have shown in a previous study considering a similar ionization energy range that within a timescale of 80 μs, only the isomerization from Azu+ to Naph+ is observed.33 This observation is therefore not in favor of the formation of PE+ in our instrument. According to the calculations of Mebel and coworkers25 (specifically Fig. 16 of ref. 25) and Bull and coworkers,30 one could expect to have only PA+ formed in the vicinity of the C8H+6 appearance energies of Naph and Azu at equilibrium. It is crucial to emphasize that in these calculations, it is considered that the entire population has a well defined internal energy value. However, when ions are prepared by photoionization, their internal energy varies between 0 and the photon energy minus IE or AE, depending on whether we consider PA or Naph/Azu, respectively. Consequently, this implies that the calculations provide an upper limit for the population transfer from PA+ to PE+.Fig. 6 shows the reaction cross sections of m/z 115 with the three different precursors. The 3 datasets are only shifted in energy to be superimposed. This leads to values of C8H+6 AEs from Naph and Azu precursors, and IE of PA to coincide within 0.3 eV. Although it is not a surprise that the reaction cross sections from Naph and Azu are similar, the reaction cross section from PA also overlays perfectly. This a decisive observable that indicates that PA+ is the reactive species at all photon energies up to the start of region 4 upon direct ionization of PA or dissociative ionization of Naph and Azu near dissociation thresholds. This result is a strong support for our hypothesis that close to 100% of PA+ is formed by dissociative ionization (for a timescale shorter than 40 μs), whatever the precursor but also informs that all internal energy is conserved by the ionic fragment when PA+ is formed by C2H2 elimination from Naph and Azu. This low kinetic energy release has been observed previously in a similar system, the n-butylbenzene cation (C10H+14) upon loss of C3H6, and was explained by the fact that the rate-limiting step is the isomerization reaction, prior to dissociation.43
In the energy range that would correspond to region 4 for the 3 precursors, it is disconcerting that, although the reaction cross sections are perfectly superimposed at lower energies, the cross section of m/z 115 becomes more important from Naph and Azu compared to PA. An explanation for this different behavior would relate to the previous observation that a strong increase of parent ion yield is observed for PA, at the energy corresponding to region 4, while such increase is absent in C8H+6 yield from Naph and Azu (Fig. 5). The departure from similarity in the m/z 115 channel would indicate that the electronic state of PA+ efficiently dissociates and/or fluoresces and this state is efficiently populated by direct photoionization and not by dissociative ionization.
Another aspect that differentiate ions produced by direct ionization or dissociative photoionization is visible in the measured cross section for m/z 141. In Fig. 1, the fact that the channel m/z 141 continuously decreases (as do the channels m/z 116 and 129) indicates that these channels correspond to reaction mechanism where a complex (adduct) is formed. Such process is strongly increasing when energy is reduced. This was verified by conducted studies of the influence of the collision energy (see ESI†), with a rapid increase of cross-section when collision energy is decreased, faster than the Langevin trend. Similarly to collision energy, when considering photon energy, the channel of reaction from PA has a cross section of 10 Å2 at photoionization threshold, and decreases to roughly 1 Å2 when photon energy increase. When C8H+6 is produced by dissociative photoionization from Naph and Azu, a maximum cross section of roughly 1 Å2 is measured and decreases with photon energy. This discrepancy in numbers (apparent lack of 90% of the reaction cross section) is somewhat antagonist with the previous conclusion of 100% PA+ in all the C8H+6 handled in our experiment, whatever the precursor. To reconcile these, one should consider that our experiments are performed without coincidence with energy-selected electrons. This implies that the ion population receives incremental amount of species in excited states as the photon energy increases, and this increment comes in addition to other states with less internal energy, depending on the Franck–Condon parameters leading to the states and the evolution of their relative cross section with photon energy. A more detailed discussion of this effect can be found in ref. 31. It is known that the Franck–Condon factor of the photoionizing origin band of PA is very strong as can be seen in the sharp threshold behavior of PA+ ion in Fig. 1 and in photoelectron spectra, for instance in ref. 42. This will efficiently produce PA+ with very limited internal energy by direct photoionization, which we can expect to exhibit a strong propensity to generate long-lived intermediate and exhibit sizable cross section for m/z 141 production. In contrast, the population of C8H+6 ions produced into their lowest vibronic ground state by dissociative ionization and a mass selection 40 μs after ionization is expected to be very minor, as seen in Fig. 5 with the very receding threshold behavior of the C8H+6 signal. This explains the reduction of the reaction cross section of m/z 141 and the non-detection of the channels m/z 116 and 129.
The latter reasoning can also be invoked to explain the apparent discrepancy between the present results and the conclusion from the group of Schwarz and co-workers,23 who derived an absence (or at max 10%) of PA+ produced from dissociative ionization of Naph. The decisive method that leads to their conclusion is the double charge inversion +CR – where a high velocity collision with O2 leads to the production of anion fragments provided they have a sufficient electron affinity. The formation of C8H−5 was thus used as a proxy of the formation of PA+, as it was the main channel observed when C8H+6 ions were produced by direct ionization from PA precursor. Since at most 10% of total signal was within the C8H−5 channel when C8H+6 ions were produced by dissociative ionization from Naph precursor, the formation of PA+ was considered as negligible. Yet, the 90% reduction in signal might have a similar origin as the one observed for the m/z 141 channel in our experiment: a high sensitivity dependence to energy. Their apparatus allows similar timescale (maximum of 30 μs compared with 40 μs for us) for C8H+6 ions to rearrange before to be probed. The difference in the outcome of the isomeric population compared with our experimental findings can thus be primarily imputed to the much larger amount of energy that they initially deposited into the C10H8 system.
Another experimental finding that, at first sight, is contradictory with our results comes from the work of Bouwman et al.27 An infrared spectrum of the C8H+6 fragments resulting from the dissociative ionization of Naph after multi photon excitation at 193 nm indicates unambiguously that the main structure formed is PE+. This conclusion is conflicting with ours. However, no power dependency studies were conducted to ensure that the ionization process was restricted to only two photons. Hence, two 193 nm photons correspond to an energy of ≈12.8 eV, barely above the thermodynamic dissociation threshold, and should mainly lead to PA+, but three or four photons will bring 5 to 11 eV above the IE of PA, a region where PE+ is the major structure of C8H+6 ions according to Fig. 16 from ref. 25. Another experimental factor that plays a role in these conflicting results is the timescale of the experiment. Indeed, in Bouwman et al. experiment, the time given to the ions to explore the PES is extremely long (200 ms, with buffer gas (He) pressure of 2 × 10−5 mbar), so that if thermodynamically accessible, the most stable isomer, e.g.PE+ should be formed.
The conclusions drawn by the work of Leach and coworkers using an experimental setup with a similar timescale as ours (a few tens of μs) were primarily based on the available heats of formation in the literature at that time for PA+, PE+, and one acyclic structure. They assumed that the most stable cation would be the preferred structure formed upon dissociation, which at that time was PA+.14,15 In the experimental studies performed by the Lifshitz group, they fit the dissociation curves as a function of time based on RRKM calculations. They first used the energy associated with the dissociation limit to PA+ + C2H2,17 and then to BCB+ + C2H2,21 once this structure was calculated to be more stable.19,22 It seems that the experimental data against which the RRKM calculations were fit did not have the resolution to differentiate between the close-lying dissociation limits of the three main C8H+6 isomers.
Conclusion
Recent theoretical studies converge on PA+ as the main isomer resulting from the loss of acetylene from the C10H+8 ions with less than 5.2 eV internal energy, whereas the results of experimental studies are more scattered. The origin of these experimental discrepancies can be twofold: (i) the way to generate C8H+6 from C10H+8 was different (electron ionization, one-photon dissociation, multi-photon dissociation) and (ii) the time integral for the chain of unimolecular reactions to proceed was very different. These two factors can greatly influence the isomeric distribution obtained. We found that PA+ is the only isomer formed from single VUV photon dissociative ionization from Naph and Azu at least up to 4.7 eV above the C8H+6 AEs within 40 μs. This is based on the exact same behavior of the reactivity leading to the product m/z 115 in region 1, 2, and 3 from PA with the results from Naph and Azu. The apparent underestimation of the branching ratio PE+/PA+ up to 4.7 eV above the AE of Naph and Azu in our experiment compared with Dyakov et al.‘s calculations25 (215 kcal mol−1 in Fig. 16 of their paper) lies in the fact that the two cannot be directly compared. The calculations assume that the equilibrium is reached and that all the ion population has a unique internal energy, but in our experiment there is a kinetic restriction and all ions are produced with a distribution of internal energy based on Franck–Condon considerations.This study has highlighted several crucial factors that should be consistently taken into account when comparing experimental studies conducted with varying apparatus and theoretical investigations:
• Consideration of measurement times and RRKM times.
• Consideration of the amount of internal energy depending on the ionization method, multiphoton absorption and their impact on reactivity and spectroscopic observables.
• Recognition of stimulated or assisted reactivity within reaction complexes, which can reveal ion structures kinetically absent when the object is isolated.
As VUV photon are ubiquitous in space, the present finding highlights the importance of PA+ into the interstellar chemical network. The complementary study starting from PA also informed on the kinetics of the C2H2 elimination to form PA+ from Naph+ and Azu+. It was observed that PA+ conserves all the energy put in the C10H8 systems, leaving the ejected C2H2 unit with nearly no internal or kinetic energy. We believe that this study sheds new light on the imbroglio concerning the C8H+6 structures from dissociation of Naph+ and Azu+. Furthermore, it also paves the way to challenging experiments where single and well-defined internal energy ions are generated by coincidence. Such experiments will allow direct comparisons with kinetic calculations restricted to 40 μs and should therefore end this imbroglio.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
U. J. conceived the project. U. J., C. Rossi, R. T., C. R., and C. A. performed the experiments and collected the data. R. T. and C. Rossi performed the data processing. C. Rossi generated the figures. U. J. wrote the manuscript. G. M. calculated the PES. P. M. and S. G. performed the RRKM and the MREP calculations. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the referees for their critical and constructive comments, which enabled us to rectify part of the manuscript and thus strengthen it significantly. U. J. is grateful to the SOLEIL facility for providing beamtime under project 20201123. C. A and C. R. acknowledge the synchrotron SOLEIL for the support to the associated CERISES setup of ICP since 2008 and subsistence expenses during beamtime periods. This research/project was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), which is supported by the Australian Government. PMM thanks the Natural Science and Engineering Research Council of Canada for continuing financial support and the Digital Research Alliance of Canada for computational resources.References
- A. Léger and J. L. Puget, Astron. Astrophys., 1984, 137, 5–8 Search PubMed.
- L. Allamandola, A. Tielens and J. Barker, Astrophys. J., Suppl. Ser., 1989, 71, 733–775 CrossRef CAS PubMed.
- H. Richter, S. Granata, W. H. Green and J. B. Howard, Proc. Combust. Inst., 2005, 30, 1397–1405 CrossRef.
- C. S. McEnally, L. D. Pfefferle, B. Atakan and K. Kohse-Höinghaus, Prog. Energy Combust. Sci., 2006, 32, 247–294 CrossRef CAS.
- A. G. Tielens, Annu. Rev. Astron. Astrophys., 2008, 46, 289–337 CrossRef CAS.
- Y. Wang, A. Raj and S. H. Chung, Combust. Flame, 2013, 160, 1667–1676 CrossRef CAS.
- M. López-Puertas, B. M. Dinelli, A. Adriani, B. Funke, M. García-Comas, M. Moriconi, E. D'Aversa, C. Boersma and L. J. Allamandola, Astrophys. J., 2013, 770, 132 CrossRef.
- L. Zhao, R. Kaiser, W. Lu, B. Xu, M. Ahmed, A. N. Morozov, A. M. Mebel, A. H. Howlader and S. F. Wnuk, Nat. Commun., 2019, 10, 1–7 CrossRef PubMed.
- B. West, S. R. Castillo, A. Sit, S. Mohamad, B. Lowe, C. Joblin, A. Bodi and P. M. Mayer, Phys. Chem. Chem. Phys., 2018, 20, 7195–7205 RSC.
- J. Zhen, P. Castellanos, D. M. Paardekooper, H. Linnartz and A. G. Tielens, Astrophys. J., Lett., 2014, 797, L30 CrossRef CAS.
- O. Berné, J. Montillaud and C. Joblin, Astron. Astrophys., 2015, 577, A133 CrossRef.
- H. R. Hrodmarsson, J. Bouwman, A. G. Tielens and H. Linnartz, Int. J. Mass Spectrom., 2022, 116834 CrossRef CAS.
- E. Ruehl, S. D. Price and S. Leach, J. Phys. Chem., 1989, 93, 6312–6321 CrossRef CAS.
- H. Jochims, H. Rasekh, E. Rühl, H. Baumgärtel and S. Leach, Chem. Phys., 1992, 168, 159–184 CrossRef CAS.
- H. Jochims, H. Rasekh, E. Rühl, H. Baumgärtel and S. Leach, J. Phys. Chem., 1993, 97, 1312–1317 CrossRef CAS.
- H. Jochims, E. Ruhl, H. Baumgartel, S. Tobita and S. Leach, Astrophys. J., 1994, 420, 307–317 CrossRef CAS.
- Y. Gotkis, M. Oleinikova, M. Naor and C. Lifshitz, J. Phys. Chem., 1993, 97, 12282–12290 CrossRef CAS.
- Y.-P. Ho, R. C. Dunbar and C. Lifshitz, J. Am. Chem. Soc., 1995, 117, 6504–6508 CrossRef CAS.
- Y. Ling, J. M. Martin and C. Lifshitz, J. Phys. Chem. A, 1997, 101, 219–226 CrossRef CAS.
- G. Koster, J. M. Martin and C. Lifshitz, J. Chem. Soc., Perkin Trans. 2, 1999, 2383–2387 RSC.
- W. Cui, B. Hadas, B. Cao and C. Lifshitz, J. Phys. Chem. A, 2000, 104, 6339–6344 CrossRef CAS.
- G. Granucci, Y. Ellinger and P. Boissel, Chem. Phys., 1995, 191, 165–175 CrossRef CAS.
- K. Schroeter, D. Schröder and H. Schwarz, J. Phys. Chem. A, 1999, 103, 4174–4181 CrossRef CAS.
- W. Van der Hart, Int. J. Mass Spectrom., 2002, 214, 269–275 CrossRef CAS.
- Y. A. Dyakov, C.-K. Ni, S. Lin, Y. Lee and A. Mebel, Phys. Chem. Chem. Phys., 2006, 8, 1404–1415 RSC.
- E. A. Solano and P. M. Mayer, J. Chem. Phys., 2015, 143, 104305 CrossRef PubMed.
- J. Bouwman, A. J. de Haas and J. Oomens, ChemComm, 2016, 52, 2636–2638 RSC.
- A. J. de Haas, J. Oomens and J. Bouwman, Phys. Chem. Chem. Phys., 2017, 19, 2974–2980 RSC.
- B. J. West, L. Lesniak and P. M. Mayer, J. Phys. Chem. A, 2019, 123, 3569–3574 CrossRef CAS PubMed.
- J. W. Lee, M. H. Stockett, E. K. Ashworth, J. E. Navarro Navarrete, E. Gougoula, D. Garg, M. Ji, B. Zhu, S. Indrajith and H. Zettergren, et al. , J. Chem. Phys., 2023, 158, 174305 CrossRef CAS PubMed.
- C. Rossi, C. Alcaraz, R. Thissen and U. Jacovella, J. Phys. Org. Chem., 2023, e4489 CrossRef CAS.
- U. Jacovella, C. Rossi, C. Romanzin, C. Alcaraz and R. Thissen, ChemPhysChem, 2022, 23, e202100871 CrossRef CAS.
- U. Jacovella, C. Rossi, C. Romanzin, C. Alcaraz and R. Thissen, ChemPhysChem, 2023, 24, e202200474 CrossRef CAS.
- L. Nahon, N. de Oliveira, G. A. Garcia, J.-F. Gil, B. Pilette, O. Marcouillé, B. Lagarde and F. Polack, J. Synchrotron Radiat., 2012, 19, 508–520 CrossRef CAS PubMed.
- B. Cunha de Miranda, C. Romanzin, S. Chefdeville, V. Vuitton, J. Žabka, M. Polášek and C. Alcaraz, J. Phys. Chem. A, 2015, 119, 6082–6098 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian Inc, Wallingford CT, 2016 Search PubMed.
- T. Baer and W. L. Hase, Unimolecular reaction dynamics: theory and experiments, Oxford University Press on Demand, 1996, vol. 31 Search PubMed.
- T. Beyer and D. Swinehart, Commun. ACM, 1973, 16, 379 CrossRef.
- T. Baer and P. M. Mayer, J. Am. Soc. Mass Spectrom., 1997, 8, 103–115 CrossRef CAS.
- J. Dyke, H. Ozeki, M. Takahashi, M. Cockett and K. Kimura, J. Chem. Phys., 1992, 97, 8926–8933 CrossRef CAS.
- U. Jacovella, C. Rossi, C. Romanzin, C. Alcaraz and R. Thissen, ChemPhysChem, 2022, 23, e202100871 CrossRef CAS.
- J. Maier and D. Turner, J. Chem. Soc., Faraday Trans., 1973, 69, 521–531 RSC.
- T. Baer, O. Dutuit, H. Mestdagh and C. Rolando, J. Phys. Chem., 1988, 92, 5674–5679 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03015f |
| This journal is © The Royal Society of Chemistry 2024 |