
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high affinity pan-PI3K binding module supports selective targeted protein degradation of PI3Kα†
Werner Theodor
Jauslin
 a,
Matthias
Schild
a,
Thorsten
Schaefer
b,
Chiara
Borsari‡
b,
Clara
Orbegozo
b,
Lukas
Bissegger
b,
Saule
Zhanybekova
a,
Danilo
Ritz
c,
Alexander
Schmidt
c,
Matthias
Wymann
*b and
Dennis
Gillingham
*a
a,
Matthias
Schild
a,
Thorsten
Schaefer
b,
Chiara
Borsari‡
b,
Clara
Orbegozo
b,
Lukas
Bissegger
b,
Saule
Zhanybekova
a,
Danilo
Ritz
c,
Alexander
Schmidt
c,
Matthias
Wymann
*b and
Dennis
Gillingham
*a
aDepartment of Chemistry, University of Basel, 4056 Basel, Switzerland. E-mail: dennis.gillingham@unibas.ch
bDepartment of Biomedicine, University of Basel, 4031 Basel, Switzerland. E-mail: matthias.wymann@unibas.ch
cProteomics Core Facility, Biozentrum, University of Basel, 4056 Basel, Switzerland
First published on 12th December 2023
Abstract
Class I phosphoinositide 3-kinases (PI3Ks) control cellular growth, but are also essential in insulin signaling and glucose homeostasis. Pan-PI3K inhibitors thus generate substantial adverse effects, a reality that has plagued drug development against this target class. We present here evidence that a high affinity binding module with the capacity to target all class I PI3K isoforms can facilitate selective degradation of the most frequently mutated class I isoform, PI3Kα, when incorporated into a cereblon-targeted (CRBN) degrader. A systematic proteomics study guided the fine tuning of molecular features to optimize degrader selectivity and potency. Our work resulted in the creation of WJ112-14, a PI3Kα-specific nanomolar degrader that should serve as an important research tool for studying PI3K biology. Given the toxicities observed in the clinic with unselective PI3Kα inhibitors, the results here offer a new approach toward selectively targeting this frequently mutated oncogenic driver.
Introduction
The PIK3CA locus, frequently mutated in a diverse range of tumors, encodes the druggable kinase p110α. Despite the apparent promise of this target, drug development against it has been challenging. Most p110α inhibitors have been hampered by a lack of isoform specificity, often affecting other isoforms such as p110β, p110δ, and p110γ, as well as, to a lesser extent, mTOR and DNA-PK, especially at biologically meaningful concentrations. Even the most specific inhibitors, like alpelisib,1 have shown metabolic adverse effects, notably hyperglycemia and hyperinsulinemia,2 primarily due to the inhibition of glucose import upon pan-PI3K inhibition, which, in turn, prompts insulin secretion. This counteractive response not only undermines the drug's efficacy but can even fuel tumor growth.3 These metabolic adverse effects would be mitigated if inhibitors were perfectly isoform selective since there is significant functional redundancy across related PI3Ks. A recent report4 has shown that high affinity engagement of p110α can lead to protein degradation in a limited number of cell lines expressing mutated p110α, while sparing the wild-type. While this pioneering study established that mutant p110α could suffer greater instability than wild-type under certain conditions, the scope of the degradation was limited. Whether isoform selectivity in p110α degradation could be achieved with other protein degradation techniques remains unstudied.Proteolysis targeting chimeras (PROTACs) are bispecific small molecules that can hijack E3 ubiquitin ligases and steer them to novel targets.5 After several processive ubiquitin-transfers to a protein (typically a 4-6-mer ubiquitin chain linked at K48), it is recognized and degraded by the 26S proteasome. The ability of small molecules to behave as effectors that adapt substrate selection of E3 ligases has generated great excitement in drug discovery.6 The approach is fundamentally different in mechanism and consequence than conventional reversible inhibitors, offering catalytic turnover and durable inactivation of the target protein. The durability arises because the target protein must be resynthesized for it to regain its function, meaning the therapeutic effect can endure even after the drug is gone. Indeed, protein degradation can even be optimized to achieve selectivity amongst closely related targets, such as kinase isoforms.5b,7 Despite the importance of p110α in oncology and challenges of targeting it selectively, PROTACs have not been investigated as a means to improve selectivity for specific class I PI3K isoforms. This is primarily because previous efforts could not identify potent PROTACs. One study with a cereblon-directing PROTAC8 and another one with a VHL-directing PROTAC9 have shown only moderate degradation of p110α at high concentrations – isoform selectivity and mechanism of action were not studied. Here, we investigate whether E3 ligase ubiquitination specificity could be exploited to promote isoform-selective degradation of class I PI3Ks (see Fig. 1A for schematic).
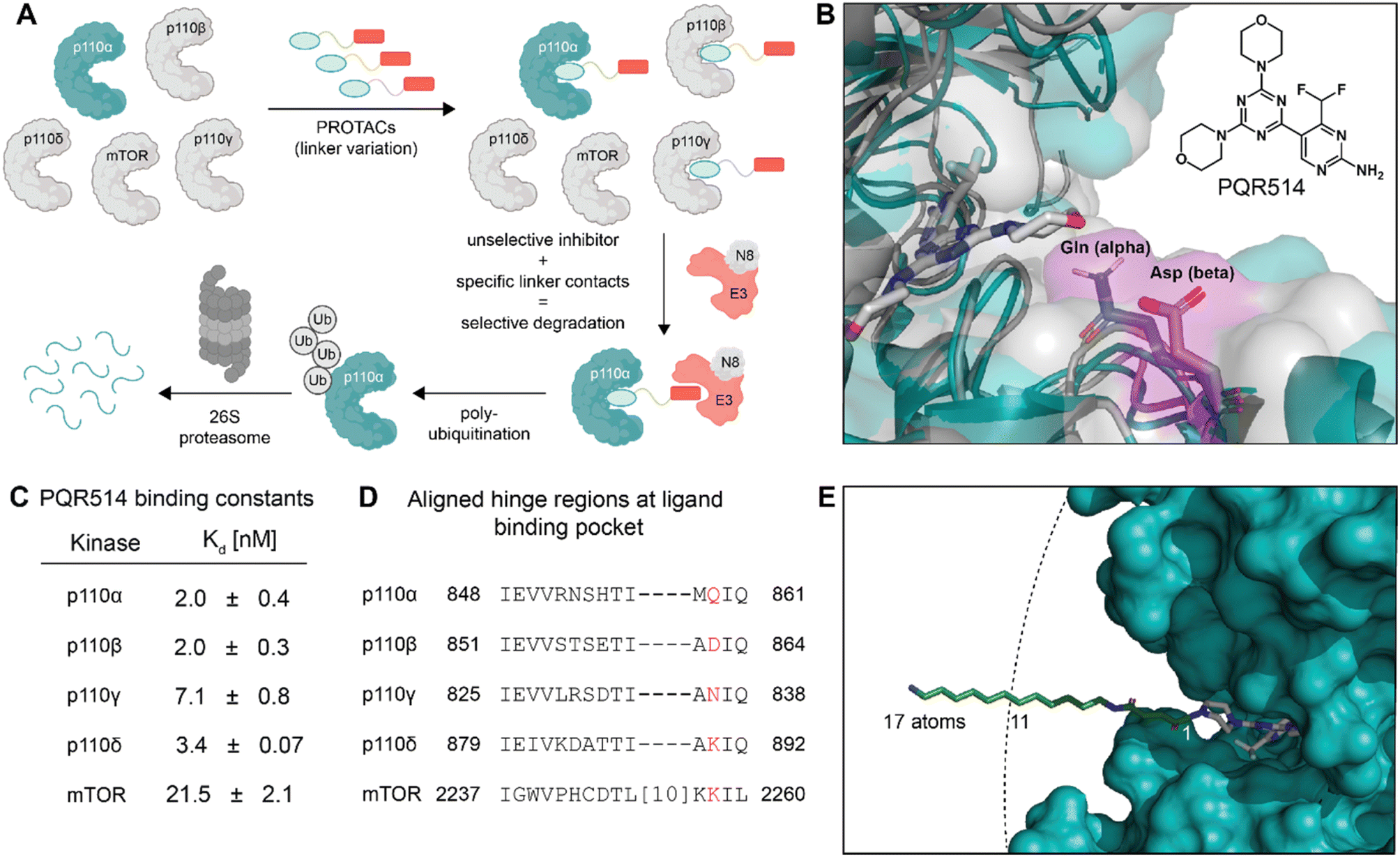 | ||
| Fig. 1 (A) Protein degradation promoted by ubiquitin tagging could be selective if the ternary complex is only active with one isoform. (B) Structural overlay of p110α in blue (PDB: 7R9V, with covalent inhibitor 19 (ref. 10) that we modified for modelling) and p110β in grey (PDB: 4BFR) show strongly conserved pocket, but with differences around the exit channel of the ATP binding site. Molecule in grey represents PQR514. (C) PQR514 binding constants. (D) Sequence alignment of human PI3K isoforms. (E) Modelling the required linker (green) distance to achieve an exit from the p110α pocket suggests at least 11 atoms (PDB: 6OAC, containing PQR530 in grey). | ||
Results & discussion
A piperazine-derivative of the potent pan-PI3K inhibitor PQR514 was used as the target binding motif because it has excellent selectivity for class I PI3Ks (Fig. 1C) over a wide range of proteins (>400) and lipid kinases.11 In addition, the exchange of the morpholine pointing to the solvent exposed region by piperazine provides a validated exit vector (having already been used in studies on covalent p110α targeting)10 for the creation of chimeric molecules. We paid particular attention to the linker design as we thought it might provide additional isoform-specific stabilizing interactions that could drive selectivity (see residues with magenta shading in Fig. 1B). In particular, we imagined that residue interactions along the hinge region and first solvent-exposed residues (see Fig. 1D) might provide an additional handle for potency and selectivity in degradation. Indeed, we find key differences in binding and degradation between closely matched lipid kinase isoforms p110α and p110β.CRBN binding PROTACs with hydrophobic linker degrade p110α
The bispecific molecules would need to bridge an E3 ligase receptor and p110α protein. Hence, we began by using publicly available structural data to model the distance required to span this space (Fig. 1E), settling on an 11-atom linker as a minimal starting point. Already at this modelling stage, we compared the pockets of the class I PI3Ks at both the sequence and structural level (Fig. 1B–D). In particular we could see key differences in the cone defining the exit channel of the ATP binding site, potentially offering binding contacts to drive selectivity. The potential of this hinge region (Fig. 1D) for inhibitor selectivity has been widely discussed for the development of the FDA approved PI3Kα drug alpelisib and other isoform selective inhibitors.12 Comparison of PI3Kα and PI3Kβ was our primary focus, since some redundant function between these isoforms suggests that metabolic side-effects would be attenuated if specificity in degradation could be achieved.13Based on the modelling, bifunctional molecules with 11-atom linkers bridging PQR514 with a CRBN-targeting molecule or a VHL-targeting molecule (these two E3 ligases represent the vast majority of successful PROTACs) were built and tested for binding to p110α protein. While the VHL targeting molecule (JS047, Fig. 2B) interrupted p110α binding, the CRBN-targeting example (WJ111-11, Fig. 2A) was as proficient in binding as the parent PQR514, prompting us to explore a more systematic series in the CRBN bifunctionals (Fig. 2B). The full molecular series we prepared included flexible linkers from 11 to 17 atoms and specific additional examples were included to probe hydrophilicity versus hydrophobicity and rigidity versus flexibility (calculated physical–chemical properties are included for all molecules in the ESI Table S1†). The bifunctional compounds were tested for in vitro binding to the catalytic subunit of PI3Kα (p110α) using TR-FRET assays (Fig. 2C), and for inhibition of the PI3K pathway in MCF7 cells (which bear a E545K mutation in p110α) using In-Cell Western technology (ICW, Fig. 2D). While TR-FRET data enabled the calculation of Ki-values, the ICW assays provided insights about their cellular inhibitory activity quantified as IC50-values based on the phosphorylation levels (Ser473) of the first PI3Kα downstream target protein kinase B (PKB/Akt). Comparing the data across this compound series revealed some key trends. First, all molecules with the PQR514 core have Ki-values below 10 nM, many of them even lower than PQR514 itself, suggesting that linker interactions in or around the active site can further stabilize binding. Additionally, the TR-FRET data and the ICWs exhibit the same potency trends, indicating strong PI3Kα inhibition by WJ213-14 and WJ204-12 in both a biochemical and cellular context. Moving forward from target interactions to protein quantification, we used integrated western blot data (Fig. 2E) to report on each molecule's degradation proficiency. This analysis pointed to a sweet spot in hydrophobic linker length between 14 and 16 atoms, where molecules were especially potent degraders (except for VHL targeting molecule WJ203-14, which was inactive). This stands in stark contrast to a hydrophilic PEG-based linker, which was completely inactive at all concentrations (see WJ117-16 in Fig. 2A). A rigid bis-piperidyl linker (WJ204-12), with a different exit vector from the imide scaffold and a 12-atom linker, also led to potent degradation. The tolerance of a shorter linker when connected at the 3-position in the isoindoline (WJ204-12) scaffold is consistent with structural data placing this position further out of the CRBN binding pocket. Hence at the end of the systematic structure–degradation relationship analysis three molecules were selected for further study because they were highly potent degraders (WJ112-14, WJ213-14, and WJ204-12, Fig. 2A), were soluble in delivery vehicle, and were structurally different enough that we might expect differences in activity between isoforms. An additional confirmatory data set for p110α degradation using an orthogonal measurement technique is included in the ESI (Fig. S11 and S12†). In particular, we created an engineered HEK293T cell line stably expressing PI3Kα-EGFP and measured protein degradation (flow cytometry and WB) after 24 hour treatments with WJ112-14 and WJ213-14 at four concentrations (1 μM, 100 nM, 10 nM and 1 nM). Although the increased expression of p110α in this cell line meant that the optimal degradation concentration of the drug was 10-fold higher, the results using this assay are otherwise fully consistent with the data in parental cell lines expressing p110α at their endogenous level.
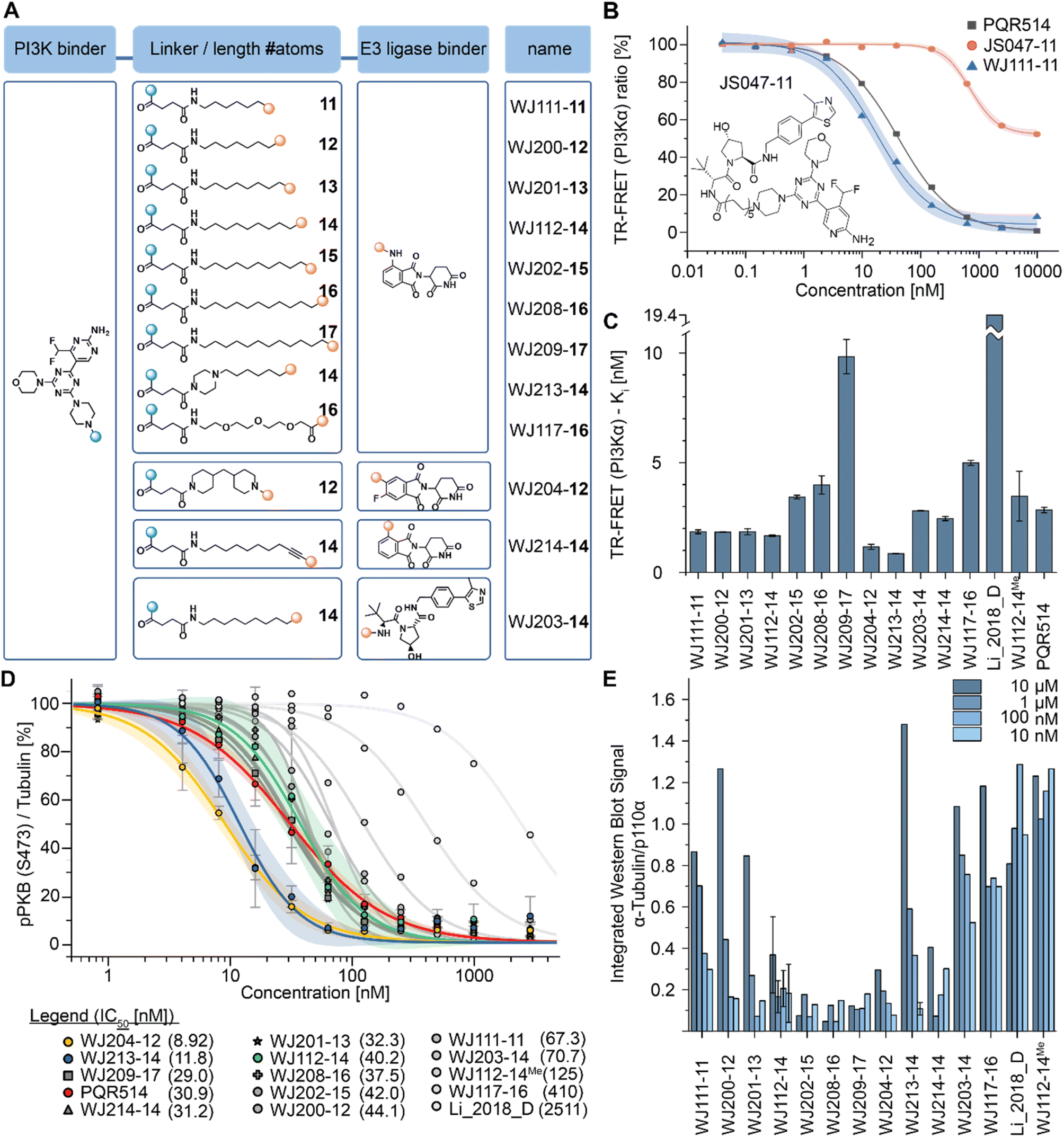 | ||
| Fig. 2 (A) Chemical formulae of bifunctional molecules. (B) To determine which E3 ligase to target, an initial scouting series of bifunctionals was prepared. TR-FRET tracer displacement assay to assess p110α binding was performed (triplicates). (C) TR-FRET curves as in (B) were used for PI3Kα Ki calculations (triplicates). Mean ± SD are reported in Table S2 of ESI.† (D) In-cell western data obtained in MCF7 cells (biological triplicates) incubated for 2 hours with the indicated inhibitors before phosphorylation of Ser473 in PKB/Akt was determined (IC50-values are shown in the panel legend in parenthesis). Mean ± SD are reported in Table S3 of ESI.† (E) Integrated western blot signals of PI3Kα normalized to DMSO and α-tubulin of the full panel of degraders. The MCF7 cells were treated for 6 h. The standard deviation was calculated for WJ112-14 at all 4 concentrations (biological triplicates) and for WJ213-14 at the optimal concentration of 10 nM (biological triplicates). | ||
Mechanistic studies confirm direct protein degradation mediated by CRBN
A series of experiments was performed to examine whether p110α degradation occurred through the ubiquitin-proteasome system (UPS) and particularly through the CRBN E3 ligase complex. A methylated probe in the glutarimide ring of thalidomide derivatives abolishes CRBN binding,7b and indeed this derivative (WJ112-14-Me, Fig. 3A) is incapable of degrading p110α in MCF7 cells (compare first two lanes in Fig. 3B). Pretreatments of cells with the 26S-proteasome inhibitor bortezomib, neddylation inhibitor MLN4924, and ubiquitin activation inhibitor TAK-243 each gave full rescue of p110α (lanes 3, 4, 5 respectively, Fig. 3B). We also saturated CRBN-binding sites with an excess of pomalidomide (Fig. 3C) and observed a rescue of p110α. In summary, the mechanistic studies are fully consistent with a CRBN and UPP-dependent degradation mechanism.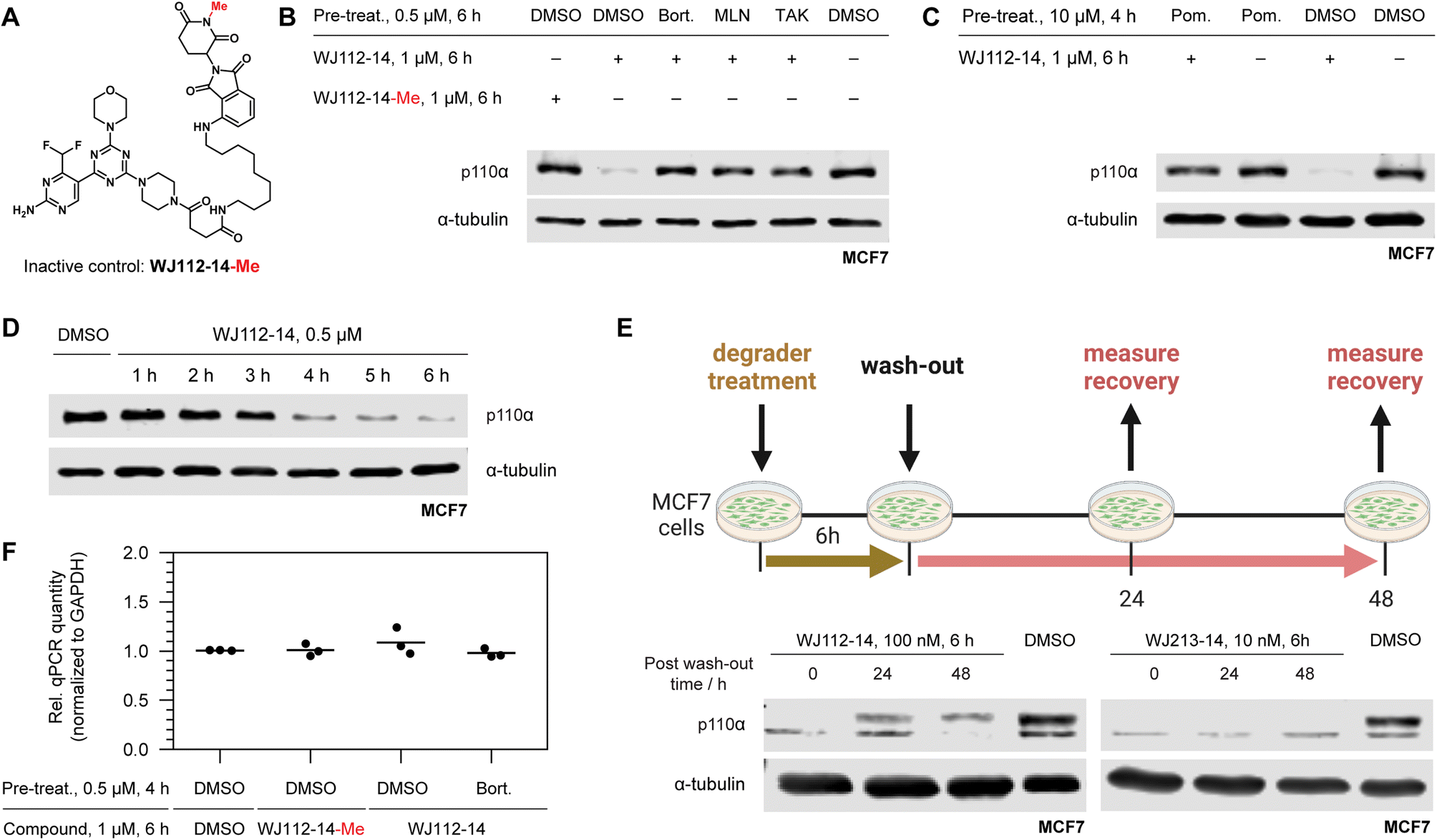 | ||
| Fig. 3 Mechanistic studies of p110α degradation. (A) Structure of methylated CRBN-inactive probe. (B) Chemical rescue experiments implicate WJ112-14 as a CRBN- and UPP-dependent degrader: comparing lanes 1 and 2 demonstrate importance of CRBN-engagement; lanes 3–5 demonstrate the degradation is cullin-ring ligase (lane 4) and UPP dependent (lanes 3 and 5). (C) Saturation of CRBN by pretreating with the tight binder pomalidomide leads to rescue of p110α degradation, further corroborating CRBN involvement. (D) Rapid degradation is consistent with a protein-level effect. (E) Degradation recovery after depletion establishes p110α as a valid degradation target. (F) qPCR of p110α shows no impact on RNA level when normalized to GAPDH further establishing a protein-level effect. | ||
Protein levels and activity of p110α are tightly regulated because it is a powerful cellular growth driver.14 Hence, to assess whether compensatory mechanisms such as transcriptional adaption or rapid protein resynthesis might counteract the effect of degradation, we examined protein degradation rate, resynthesis rate, and monitored transcript levels. Protein degradation is nearly complete at 6 h with WJ112-14 (Fig. 3D), and its recovery after compound washout is slow (Fig. 3E). In particular, at 48 h with WJ112-14, approximately 20–30% recovery occurs – with the more potent WJ213-14, however, p110α levels remained undetectable at 48 h after washout (Fig. 3E). Importantly qPCR data indicates that none of the observed changes in p110α are related to transcription (Fig. 3F).
Comparison to other p110α degraders
The structurally related p110α inhibitors taselisib (GDC-0032, see Fig. 4A) and inavolisib (GDC-0077 – currently undergoing several late-stage clinical trials)4a,15 have been reported to induce degradation of mutated p110α in a limited number of cell lines and circumstances, while sparing the p110β isoform.4a Although taselisib and inavolisib show nearly identical degradation in side-by-side experiments, we selected taselisib as a benchmark compound for comparing with our PROTACs as its PI3Kα degradation activity has been more extensively characterized.4a We compared degradation behavior of WJ112-14 and taselisib in several PIK3CA-mutant cell lines as well as one PTEN loss cell line (precise genetic changes found in Fig. 4B). The data indicates that even in taselisib-responsive cell lines the PROTAC degrader outperforms taselisib (MDA-MB453 and HCC-1954, Fig. 4B); in addition, WJ112-14 is active in cells where taselisib is inactive, such as with MCF7 and T47D cell lines (Fig. 4B). The only cell line where WJ112-14 failed to deliver strong degradation is the A2058 cell line (Fig. 4B). A closer look at p110α and CRBN levels in the A2058 cell line suggest an unfavorable ratio of the two proteins (i.e. high p110α and low CRBN) as a possible explanation for the poor degradation in this case (see Fig. S10† for WB).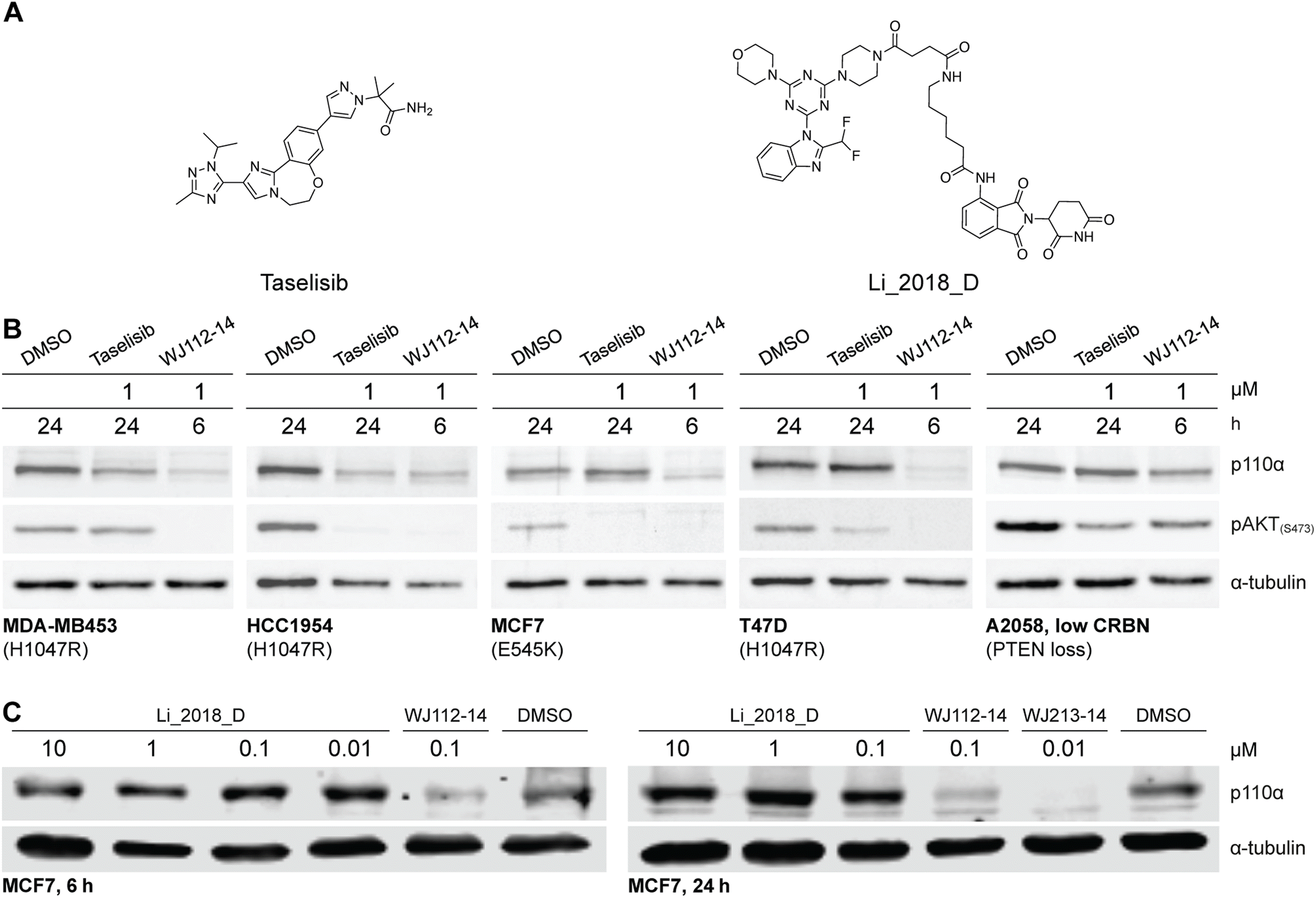 | ||
| Fig. 4 Comparison to state-of-the-art degraders. (A) Structures of taselisib and previously reported CRBN-targeted p110α degrader Li_2018_D. (B) In cell lines where taselisib degrades p110α, WJ112-14 delivers similar levels of degradation in shorter times (6 h versus 24 h). WJ112-14 can also degrade p110α in cell lines resistant to taselisib-mediated degradation. Measurements in cell line A2058 suggest that WJ112-14 is poorly active when levels of CRBN are low relative to p110α (see ESI Fig. S10† for measurements of CRBN levels) (C) WJ112-14 and the related WJ213-14 are more active than CRBN-based bifunctional degrader Li_2018_D. | ||
A previous report on CRBN-based bifunctional degraders also reported molecules with degradation activity for p110α.8 Hence we synthesized the best performer from that study, Li_2018_D (see Fig. 4A for structure) and compared it to WJ112-14 and WJ213-14 (see Fig. 4C). While Li_2018_D was reported to give some degradation at high concentration (50 μM) in HepG2 cells, we see little detectable degradation in MCF7 cells (see Fig. 4C) at 6 h or 24 h (Fig. S14† with targeted proteomics data corroborating these findings). In summary, WJ112-14 is a best-in-class, highly potent and isoform selective degrader of p110α. While WJ213-14 is also an extremely potent degrader, our later studies (vide infra) reveal that it is less isoform selective than WJ112-14 in its degradation profile.
Bifunctional molecules lead to potent and tunable degradation
To characterize more deeply the potency and selectivity of the best degraders identified in Fig. 2, we performed a series of quantifications including targeted and global proteomics, as well as a KINOMEscan focused on lipid kinases and mTOR. Previous work had shown that PQR514 is highly selective for lipid kinases.11 Interestingly, we found that WJ112-14 and WJ213-14, while still generally strong inhibitors of lipid kinases, had some key differences. For example, while both are strong binders of class I PI3K kinases, WJ112-14 exhibits improved selectivity as it is one order of magnitude less effective at binding PIK3C2B, PIK3C2G and MTOR (see Fig. 5D; tabulated data, see Table S4†). Importantly, although the difference in binding efficiency of WJ112-14 for PI3Kα and PI3Kβ seems subtle, it translates to surprising levels of degradation selectivity. Testing WJ112-14 across a panel of cell lines (Fig. 5B), we observed dose-dependent degradation of p110α (10 nM to 1 μM) over short treatment times (6 h). In contrast, the closely related p110β shows no detectable degradation at any concentration. To glean a quantitative picture of degradation selectivity we turned to targeted proteomics with parallel reaction monitoring (PRM) and global proteome analysis using Tandem Mass Tags (TMT). Heavy peptides for all isoforms and regulatory subunits of p110α and p110β as well as mTOR were obtained and used to monitor protein changes when MCF7 cells were treated for 6 h. All compounds from Fig. 2A were analyzed using PRM and representative data for compounds WJ112-14 and WJ213-14 are shown in Fig. 5B. The full data set is shown in the ESI Fig. S14–S18.† The quantitative results closely parallel the previously presented qualitative data from western blots for p110α/β, while adding additional information on the regulatory subunits and mTOR. An immediate and important observation is that none of the compounds degrade mTOR at any of the measured concentrations (see lower panel of Fig. 5B). Another finding is that the protein levels of the regulatory subunits of alpha (PIK3R1) and beta (PIK3R2) follow the general trends of their corresponding catalytic subunits, albeit with a small absolute reduction in protein levels. Consistent with the western blotting and Kd data (see Fig. S13†), we see distinct differences in degradation selectivity between WJ112-14 and WJ213-14. In particular, WJ213-14, which showed strong binding to p110α and p110β according to TR-FRET and the Kd binding assays (Fig. S13†), degraded both isoforms almost completely at 10 nM. There is, however, selectivity at 1 nM, where 80% of p110α and 20% of p110β is degraded. In contrast, WJ112-14 shows strong α/β selectivity, giving no measurable p110β degradation at concentrations that fully deplete p110α. The most notable data point from the extended data is that WJ204-12 shows strong degradation for p110α and around 50% degradation of p110β (see ESI Fig. S14 and S15†) at 100 nM concentration. In summary, the PRM data corroborated earlier western blot observations in terms of optimal linker length, observation of hook effects for active degraders, and degradation efficacies that match binding preferences.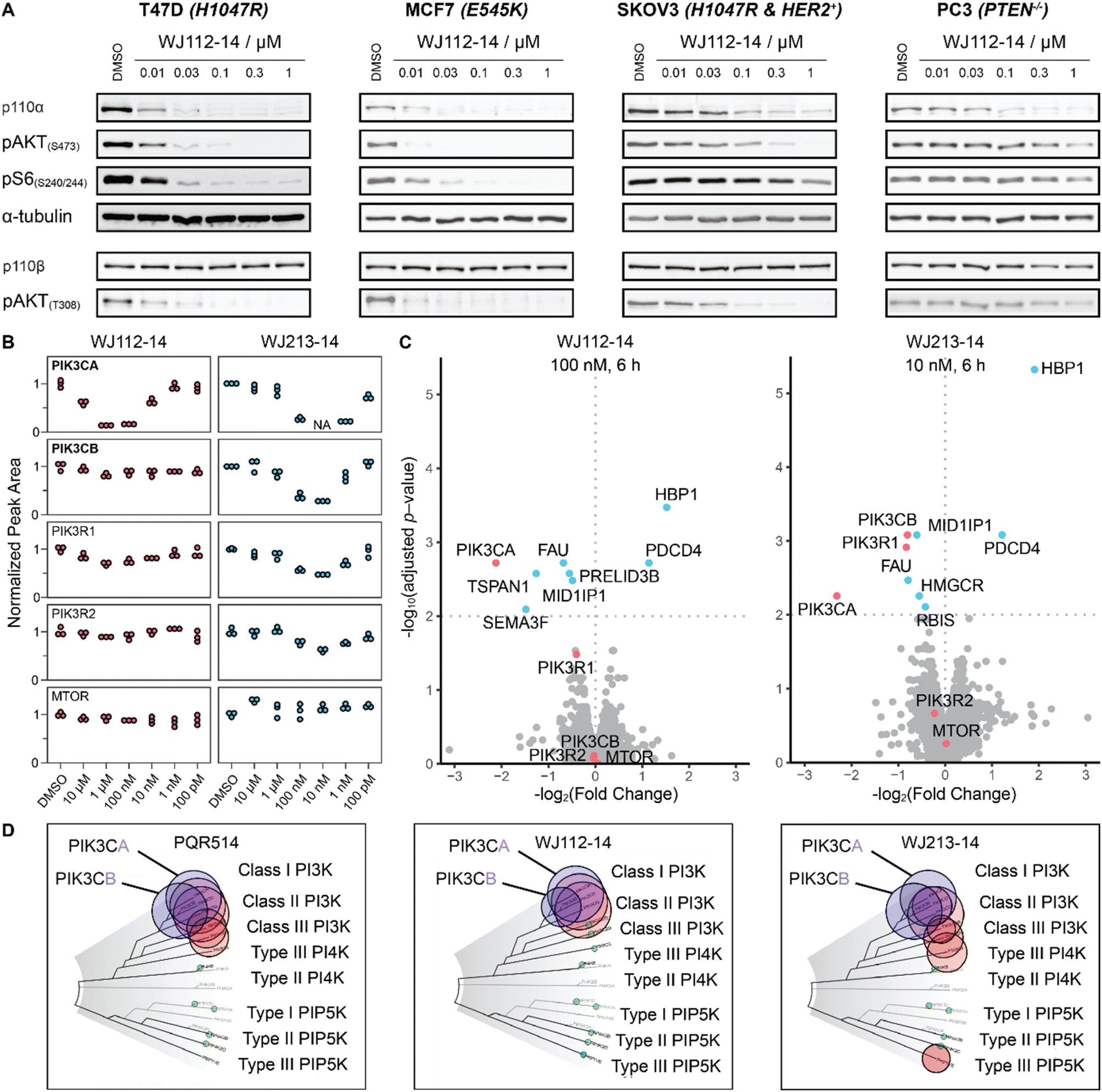 | ||
| Fig. 5 Selectivity profile of degraders. (A) p110α degradation with WJ112-14 in a panel of cell lines shows selective degradation and strong downstream impact upon 6 h treatments. (B) PRM data for WJ112-14 and WJ213-14 at 6 concentrations each upon 6 h treatments. Peptides for PIK3CA, PIK3CB, PIK3R1, PIK3R2 and mTOR measured. (C) TMT global proteomics for WJ112-14 (100 nM, 6 h) and WJ213-14 (10 nM, 6 h). (D) Lipid Kinome scan data for PQR514,11WJ112-14 and WJ213-14 at 10 μM concentration. | ||
While targeted proteomics gave us valuable insights into isoform selectivity, we were also interested in the proteome-wide impact of the most potent PROTACs. Therefore, we performed a global proteome analysis using three compounds (WJ112-14, WJ204-12 and WJ213-14) at treatment concentrations that gave maximum degradation of p110α. These experiments support the findings from PRM and western blot, specifically that WJ112-14 has selectivity for PIK3CA over PIK3CB while WJ213-14 shows potent degradation of both PIK3CA and PIK3CB (see Fig. 5C). WJ204-12 showed strong degradation of PIK3CA while the data point for PIK3CB was below the significance threshold (see Fig. S19†). Further, we could see upregulation of HBP1 and PDCD4 for all three compounds, which agrees with reports that these proteins are repressed upon activated PI3K signaling.16
Rationale for changes in degradation selectivity between WJ112-14 and WJ213-14
We performed computational studies to explain the different profile for the two optimal degraders WJ112-14 and WJ213-14. In terms of structure only the piperazine ring attached to the succinyl-diamide in the linker changes. Otherwise, the binding units for the protein of interest and the E3 ligase stay the same, the linker length is the same, as are the functional groups immediate to the binding site. Hence, we suggest as the origin of increased degradation potency of p110α for WJ213-14 that the piperazine reinforces coordination of the two carbonyls to the Q859 (in human; PDB: 7R9V) residue due to enhanced rigidity, thus improving binding to PI3Kα (see Fig. 6A and B). WJ112-14 does not degrade p110β. The linker does not contain other functional groups that could lead to specific linker interactions with the mutated residue (D856 in mouse; PDB: 4BFR) in p110β. The origin of the lower p110α/p110β selectivity for WJ213-14 likely stems from potential linker interactions of WJ213-14 with an aspartate (D856 in mouse; PDB: 4BFR) in the beta isoform. Specifically, protonation of the piperazine nitrogen provides a potential salt bridge with D856 in p110β (see Fig. 6B). Whatever the rationale, the data clearly show that the linker and its functional groups influence the selectivity of our PROTACs.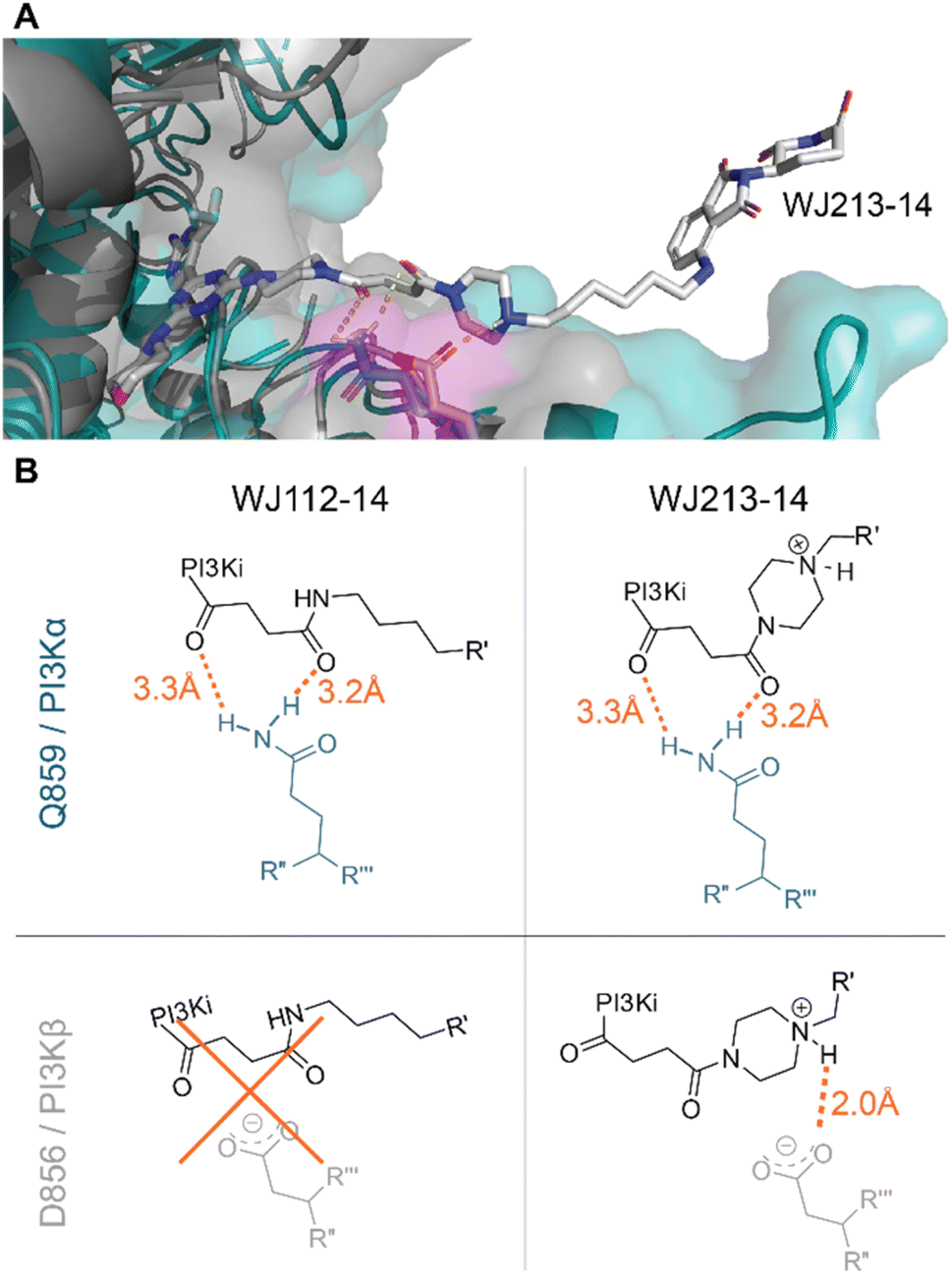 | ||
| Fig. 6 Model for isoform selectivity. (A) Structural overlay of p110α (human; PDB: 7R9V) from ref. 10 in slate blue and p110β (mouse; PDB: 4BFR) in grey with modelling of compound WJ213-14 in white. Magenta shaded residues indicate potential interacting residues with the linker. (B) Measured distances in PyMOL (orange) for suggested compound–protein interactions. | ||
Conclusion
We present a set of new molecules that target the PI3K class I protein family for degradation by recruiting it to the CRBN-bound CRL4 E3 ligase. Computed models based on published structural data were carefully analyzed to investigate sensible linker lengths and to study potential linker-specific interactions with residues in the hinge and solvent-exposed region proximal to the ATP pocket. This approach led us to discover WJ112-14, a first in class highly potent, selective degrader of PI3Kα as well as WJ213-14, which is a low-nanomolar degrader that depletes both PI3Kα and PI3Kβ. The degraders have been tested in eight different cell lines, giving activity in all except for A2058 – which is low in CRBN. Degradation potency, selectivity, and mechanism have been confirmed with several orthogonal measurement methods.Studies to uncover the biological roles of PI3K isoforms have historically relied on genetic methods since small molecule probes were unselective. Although selective chemical probes would be far easier to use experimentally, and could also be used in medical applications, it has proven difficult to precisely target each isoform selectivity. Our work here opens the door to an alternative approach where each isoform could be selectively degraded with a specific chemical probe. Our studies have focused on PI3Kα because of its importance in oncology, but looking forward from our findings we expect that judicious linker optimization could also deliver selective probes for other PI3K isoforms.
Data availability
Proteomic data have been deposited to the ProteomeXchange Consortium (https://www.proteomexchange.org/) via the MassIVE partner repository with MassIVE data set identifier MSV000093594 and ProteomeXchange identifier PXD047599.Author contributions
W. J., M. S., M. W., and D. G. collaborated in all aspects of project design and conception. W. J. performed chemical synthesis and initial cellular testing of all molecules, including mechanistic experiments. W. J. and M. S. designed proteomics experiments and performed initial experimental setup in collaboration with D. R. and A. S., D. R. and M. S. performed the global and targeted proteomics in collaboration under the supervision of D. G. and A. S. Cellular engineering was by S. Z. and testing of engineered cells was done by W. J. Measurement of binding data and in-cell westerns was performed by L. B. and C. O. under the supervision of M. W. The first draft of the manuscript was prepared by W. J., M. S., M. W. and D. G. Refinement was done with contributions from all authors.Conflicts of interest
WJ, MS, MW, and DG have filed for patent protection of the work described herein.Acknowledgements
We thank Pedro Mota, Thanos Halazonetis and Nicolas Thomä for enlightening discussions and Basilius Sauter for his help with data presentation. DG has received funding from the Swiss National Science Foundation (CRSII5_186230) and the European Research Council (Horizon 2020, ExploDProteins, 866345) for the work herein. MPW is supported by the Swiss National Science Foundation grants 200021_204602 and 310030_189065, and the Swiss Cancer League (KFS-5442-08-2021).References
- F. André, E. Ciruelos, G. Rubovszky, M. Campone, S. Loibl, H. S. Rugo, H. Iwata, P. Conte, I. A. Mayer, B. Kaufman, T. Yamashita, Y.-S. Lu, K. Inoue, M. Takahashi, Z. Pápai, A.-S. Longin, D. Mills, C. Wilke, S. Hirawat, D. Juric and N. Engl, J. Med., 2019, 380, 1929–1940 Search PubMed.
- (a) H. S. Rugo, M. E. Lacouture, M. D. Goncalves, U. Masharani, M. S. Aapro and J. A. O'Shaughnessy, Breast, 2022, 61, 156–167 CrossRef PubMed; (b) B. Pla Peris, A. Arranz Martin, A. Ballesteros García, F. Sebastián-Valles and M. Marazuela Azpiroz, Front. Endocrinol., 2022, 13, 802612 CrossRef PubMed.
- B. D. Hopkins, C. Pauli, X. Du, D. G. Wang, X. Li, D. Wu, S. C. Amadiume, M. D. Goncalves, C. Hodakoski, M. R. Lundquist, R. Bareja, Y. Ma, E. M. Harris, A. Sboner, H. Beltran, M. A. Rubin, S. Mukherjee and L. C. Cantley, Nature, 2018, 560, 499–503 CrossRef CAS PubMed.
- (a) K. W. Song, K. A. Edgar, E. J. Hanan, M. Hafner, J. Oeh, M. Merchant, D. Sampath, M. A. Nannini, R. Hong, L. Phu, W. F. Forrest, E. Stawiski, S. Schmidt, N. Endres, J. Guan, J. J. Wallin, J. Cheong, E. G. Plise, G. D. Lewis Phillips, L. Salphati, T. P. Heffron, A. G. Olivero, S. Malek, S. T. Staben, D. S. Kirkpatrick, A. Dey and L. S. Friedman, Cancer Discovery, 2022, 12, 204–219 CrossRef CAS PubMed; (b) E. J. Hanan, M.-G. Braun, R. A. Heald, C. MacLeod, C. Chan, S. Clausen, K. A. Edgar, C. Eigenbrot, R. Elliott, N. Endres, L. S. Friedman, E. Gogol, X.-H. Gu, R. H. Thibodeau, P. S. Jackson, J. R. Kiefer, J. D. Knight, M. Nannini, R. Narukulla, A. Pace, J. Pang, H. E. Purkey, L. Salphati, D. Sampath, S. Schmidt, S. Sideris, K. Song, S. Sujatha-Bhaskar, M. Ultsch, H. Wallweber, J. Xin, S. Yeap, A. Young, Y. Zhong and S. T. Staben, J. Med. Chem., 2022, 65, 16589–16621 CrossRef CAS PubMed.
- (a) M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed; (b) D. A. Nalawansha and C. M. Crews, Cell Chem. Biol., 2020, 27, 998–1014 CrossRef CAS PubMed; (c) D. P. Bondeson, A. Mares, I. E. D. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Nat. Chem. Biol., 2015, 11, 611–617 CrossRef CAS PubMed; (d) J. Lu, Y. Qian, M. Altieri, H. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew, K. Coleman and C. M. Crews, Chem. Biol., 2015, 22, 755–763 CrossRef CAS PubMed; (e) K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed; (f) G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS PubMed; (g) M. Zengerle, K. H. Chan and A. Ciulli, ACS Chem. Biol., 2015, 10, 1770–1777 CrossRef CAS PubMed.
- A. C. Lai and C. M. Crews, Nat. Rev. Drug Discovery, 2017, 16, 101–114 CrossRef CAS PubMed.
- (a) G. M. Burslem, B. E. Smith, A. C. Lai, S. Jaime-Figueroa, D. C. McQuaid, D. P. Bondeson, M. Toure, H. Dong, Y. Qian, J. Wang, A. P. Crew, J. Hines and C. M. Crews, Cell Chem. Biol., 2018, 25, 67–77 CrossRef CAS PubMed; (b) D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Cell Chem. Biol., 2018, 25, 78–87 CrossRef CAS PubMed; (c) B. E. Smith, S. L. Wang, S. Jaime-Figueroa, A. Harbin, J. Wang, B. D. Hamman and C. M. Crews, Nat. Commun., 2019, 10, 131 CrossRef PubMed; (d) M. Brand, B. Jiang, S. Bauer, K. A. Donovan, Y. Liang, E. S. Wang, R. P. Nowak, J. C. Yuan, T. Zhang, N. Kwiatkowski, A. C. Müller, E. S. Fischer, N. S. Gray and G. E. Winter, Cell Chem. Biol., 2019, 26, 300–306 CrossRef CAS PubMed; (e) S. Rana, M. Bendjennat, S. Kour, H. M. King, S. Kizhake, M. Zahid and A. Natarajan, Bioorg. Med. Chem. Lett., 2019, 29, 1375–1379 CrossRef CAS PubMed.
- W. Li, C. Gao, L. Zhao, Z. Yuan, Y. Chen and Y. Jiang, Eur. J. Med. Chem., 2018, 151, 237–247 CrossRef CAS PubMed.
- H. Wang, C. Li, X. Liu and M. Ma, Bioorg. Med. Chem., 2022, 61, 116707 CrossRef CAS PubMed.
- C. Borsari, E. Keles, J. A. McPhail, A. Schaefer, R. Sriramaratnam, W. Goch, T. Schaefer, M. De Pascale, W. Bal, M. Gstaiger, J. E. Burke and M. P. Wymann, J. Am. Chem. Soc., 2022, 144, 6326–6342 CrossRef CAS PubMed.
- C. Borsari, D. Rageot, F. Beaufils, T. Bohnacker, E. Keles, I. Buslov, A. Melone, A. M. Sele, P. Hebeisen, D. Fabbro, P. Hillmann and M. P. Wymann, ACS Med. Chem. Lett., 2019, 10, 1473–1479 CrossRef CAS PubMed.
- M. Zhang, H. Jang and R. Nussinov, Chem. Sci., 2020, 11, 5855–5865 RSC.
- A. Molinaro, B. Becattini, A. Mazzoli, A. Bleve, L. Radici, I. Maxvall, V. R. Sopasakis, A. Molinaro, F. Bäckhed and G. Solinas, Cell Metab., 2019, 29, 1400–1409 CrossRef CAS PubMed.
- B. Vanhaesebroeck, L. Stephens and P. Hawkins, Nat. Rev. Mol. Cell Biol., 2012, 13, 195–203 CrossRef CAS PubMed.
- P. L. Bedard, M. K. Accordino, A. Cervantes, V. Gambardella, E. P. Hamilton, A. Italiano, D. Juric, K. Kalinsky, I. E. Krop, M. Oliveira, C. Saura, P. Schmid, N. C. Turner, A. Varga, N. Shankar, J. Schutzman, S. Royer-Joo, M. V. Martin and K. L. Jhaveri, J. Clin. Oncol., 2022, 40, 1052 CrossRef.
- (a) A. Coomans de Brachène, E. Bollaert, A. Eijkelenboom, A. de Rocca Serra, K. E. van der Vos, B. M. T. Burgering, P. J. Coffer, A. Essaghir and J.-B. Demoulin, Biochem. J., 2014, 460, 25–36 CrossRef PubMed; (b) S. Matsuhashi, M. Manirujjaman, H. Hamajima and I. Ozaki, Int. J. Mol. Sci., 2019, 20, 2304 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04629j |
| ‡ Present address: Department of Pharmaceutical Sciences, University of Milan, Via Mangiagalli 25, 20133, Milano, Italy. |
| This journal is © The Royal Society of Chemistry 2024 |