
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHollow anatase TiO2 tetrakaidecahedral crystals with an active {001}/{110} redox interface toward high-performance photocatalytic activity†
Liming
Sun
a,
Yaya
Yuan
a,
Xiaoxiao
He
*c,
Wenwen
Zhan
a,
Dong
Li
 c,
Yanli
Zhao
*b,
Xiao-Jun
Wang
a and
Xiguang
Han
*a
c,
Yanli
Zhao
*b,
Xiao-Jun
Wang
a and
Xiguang
Han
*a
aJiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, Department of Chemistry, School of Chemistry and Chemical Engineering, Jiangsu Normal University, Xuzhou, 221116, P. R. China. E-mail: xghan@jsnu.edu.cn
bSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 21 Nanyang Link, Singapore 637371, Singapore
cState Key Laboratory of Precision Spectroscopy, East China Normal University, Shanghai, 200241, P. R. China
First published on 5th December 2023
Abstract
The existence of the oxidation/reduction interface can promote the performance of a photocatalyst, due to its effect on the separation of photogenerated carriers and the surface reactivity. However, it is difficult to construct two sets of oxidation/reduction interfaces in a single crystal and compare their separation efficiency for photogenerated carriers. Introducing a high proportion of active facets into the co-exposed facets is even more challenging. Herein, a hollow anatase TiO2 tetrakaidecahedron (HTT) with two sets of oxidation/reduction interfaces ({001}/{101} and {001}/{110}) is synthesized by directional chemical etching. Theoretical and experimental results indicate that the {001}/{110} interface is a dominant oxidation/reduction interface, showing a better promotion on the separation of photogenerated carriers than the {001}/{101} interface. In the HTT, the ratio of dominant {001}/(110) is increased and the proportion of the active {110} facet is about 40% (generally about 15%). Therefore, the HTT shows excellent catalytic activity for photocatalytic reductive (hydrogen production) and oxidative (selective oxidation of sulfides) reactions. The HTT also demonstrates favorable photocatalytic activity for the cross-dehydrogenative coupling reaction, where both photogenerated electrons and photogenerated holes are involved, further verifying its high separation efficiency of photogenerated carriers and surface reactivity. This work provides an important guideline for developing advanced structures with a predetermined interface toward desired applications.
Introduction
The photocatalytic reaction consists of three main processes, namely, photoinduced generation of carriers, separation and migration of the photogenerated carriers to the surface of the catalyst, and the separated photogenerated electrons and holes participating in surface reductive/oxidative reactions at the active sites.1–3 The effective separation of photogenerated carriers is a prerequisite for a photocatalytic reaction.4–11 While the deposition of noble metals and the construction of a semiconductor heterojunction can promote the separation of photogenerated carriers, the high cost of noble metals and the low effective interface rate in the heterojunction are two main disadvantages limiting their development.12–14 It has been found that the oxidation/reduction interface, with different aggregations of photogenerated holes and electrons on each facet, can be constructed in a single crystal to achieve effective separation of photogenerated carriers by the directional migration of photogenerated elections and holes.15–22 Obviously, the oxidation/reduction interface composed of different facets should have different promotion on the separation of photogenerated carriers. There is a lack of relevant research on this issue,19–22 because it is challenging to construct two sets of oxidation/reduction interfaces in a single crystal and compare their separation efficiency for photogenerated carriers.Although the separation efficiency of photogenerated carriers has a crucial influence on the activity of photocatalysts, the impact of surface reactivity of exposed facets on photocatalytic activity cannot be ignored. Generally, the highly active facets possess more unsaturated surface atoms and high surface energy.23–31 It is very difficult to expose highly active facets in a high proportion in single crystal photocatalysts, as their high surface energy contradicts the principle of crystal growth.32 Therefore, introducing a high proportion of highly active facets into the oxidation/reduction interface is an effective but challenging strategy for fabricating efficient photocatalysts.
Anatase TiO2, as one of the most well-known functional materials, shows promising applications in various photocatalytic fields, such as water splitting, wastewater treatment, and catalytic organic reactions, due to its low cost, non-toxicity, high activity and high photostability.33–36 The sequence of the average surface energy from low index anatase TiO2 crystal facets is γ {110} (1.09 J m−2) > γ {001} (0.90 J m−2) > γ {100} (0.53 J m−2) > γ {101} (0.44 J m−2).25,37–40 The surface energy of the {110} facet is the highest, which means that the {110} facet is a highly active facet of anatase TiO2. The high surface energy of the {110} facet results in its low exposure ratio in the anatase TiO2 single crystal.40–44 At present, the highest exposure ratio of the {110} facet is only about 15%.45 The surface energy of the {101} facet is the lowest, indicating its best stability. From the perspective of reduction and oxidation facets, anatase TiO2 {101} and {110} facets provide the sites for reduction, which benefit the aggregation of photogenerated electrons, while the {001} facet offers the sites for oxidation, which is beneficial to the concentration of photogenerated holes.46–48 Therefore, we first constructed a solid anatase TiO2 tetrakaidecahedron (STT) containing {001}/{101} and {001}/{110} oxidation/reduction interfaces. By introducing the stable {101} facet, the difficulty of synthesizing TiO2 with a highly active {110} facet was effectively solved. In the prepared STT, the exposed proportion of the highly active {110} facet accounts for 20%. From theoretical calculations, it can be found that the {001}/{110} interface has a better effect on promoting charge separation than the {001}/{101} interface. That is, the {001}/{110} interface is the dominant oxidation/reduction interface. Then, the hollow anatase TiO2 tetrakaidecahedron (HTT) was prepared by the directional etching method (Fig. S1†), in which the {001}/{110} interface ratio was increased and the proportion of the highly active {110} facet was further expanded to 40%. The results of photoelectrochemical and transient absorption measurements confirm the dominant position of the {001}/{110} interface in facilitating the photogenerated carrier separation predicted by the theoretical calculation. Benefiting from the synergistic effect of the increased dominant {001}/{110} interface proportion and high proportion of the highly active {110} facet, the HTT shows excellent photocatalytic activity for both reductive (hydrogen production) and oxidative (selective oxidation of sulfides) reactions. Moreover, the prepared HTT also demonstrates favorable photocatalytic activity for the cross-dehydrogenative coupling (CDC) reaction, in which both photogenerated electrons and photogenerated holes are involved, further verifying its high separation efficiency of photogenerated carriers and surface reactivity. This work shows that screening and increasing the proportion of the dominant oxidation/reduction interface with highly active facets would be useful to construct highly efficient photocatalysts.
Results and discussion
Structure and composition of the photocatalysts
Well-defined STT particles were synthesized by a hydrothermal method in the presence of potassium titanate nanowires (KTNWs), H2O2 and HF at 180 °C for 12 h. Field emission scanning electron microscopy (FESEM, Fig. 1a) confirmed the formation of a solid tetrakaidecahedron with high uniformity and a smooth surface. The powder X-ray diffraction (XRD) pattern (Fig. S2†) showed that all the identified peaks could be assigned to anatase TiO2 (JCPDS Card No: 01-071-1166), indicating that the as-prepared STT was pure anatase TiO2. Energy-dispersive X-ray (EDX) spectroscopy mapping (Fig. S3†) showed that the Ti and O elements were evenly dispersed throughout the solid tetrakaidecahedral structure. In order to reveal the exposed crystal facets of the STT, SEM images (Fig. 1b–g) observed at different zone axes were obtained and compared with the corresponding simulated models. There was a good agreement between the SEM images and the geometrical models from different orientations, indicating that the STT was enclosed by the {001}, {101} and {110} facets. According to the ideal model, the proportion of each exposed crystal plane was calculated to be 43% for the {101} facet, 37% for the {001} facet, and 20% for the {110} facet (Fig. S4†). To investigate the photocatalytic oxidation–reduction activity of anatase TiO2 {001}, {101} and {110} facets, the photo-deposition of Pt and MnOx on the surface of the STT was carried out. The SEM images and corresponding energy dispersive spectroscopy (EDS) spectra (Fig. S5†) clearly demonstrate the selective deposition of Pt particles on the {101} and {110} facets, while the MnOx particles are solely deposited on {001} facets. Concurrent photo-deposition of Ag/MnOx on the STT was conducted to further prove the distinct accumulation of electrons or holes, as depicted in Fig. S6.† It is evident that Ag was deposited on the {101} and {110} facets, while MnOx was deposited on the {001} facet, respectively. This finding aligns with those obtained from individual photo-deposition of Pt and MnOx. These observations indicated that the photogenerated electrons were concentrated on the {101} and {110} facets to prepare for the reductive reaction, and the photogenerated holes tended to migrate to the {001} facet for the oxidative reaction. Thus, there are two sets of oxidation/reduction interfaces in the prepared STT, namely {001}/{110} and {001}/{101} interfaces.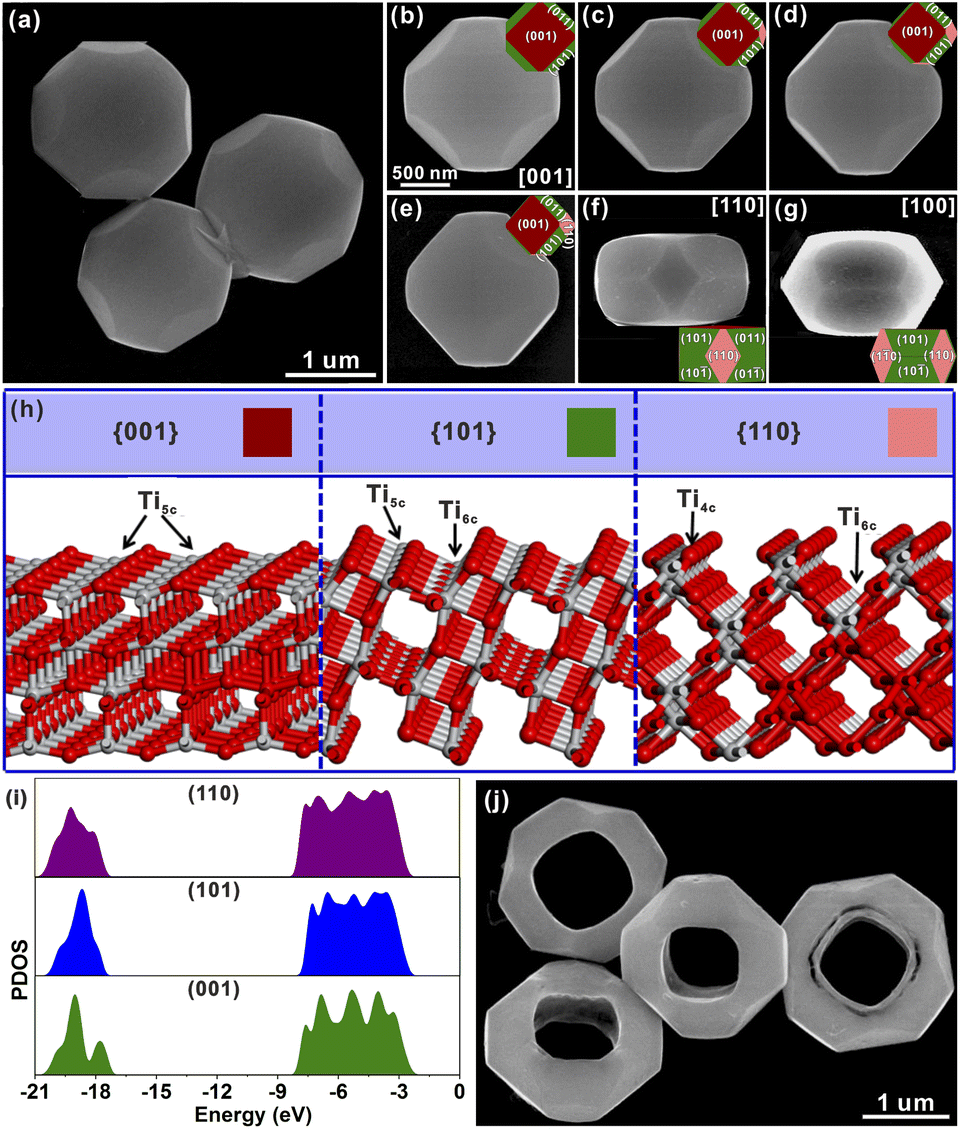 | ||
| Fig. 1 (a) Typical SEM images of the STT. (b) An STT particle viewed from the [001] direction and the corresponding ideal geometrical model. (c–e) The same STT particle rotated at a certain angle and corresponding ideal geometrical model. (f and g) STT particles viewed from the [110] and [100] orientations. (h) Corresponding surface structures of anatase TiO2 {001}, {101} and {110} facets. (i) Density of states for anatase TiO2 {001}, {101} and {110} facets. (j) Typical SEM images of the HTT. | ||
In order to study the origin of this spatial separation and effectively utilize the spatial separation effect, the surface microstructures and the energy levels of the valence band for anatase TiO2 {001}, {101} and {110} facets were evaluated by theoretical calculations. From the perspective of atomic structure (Fig. 1h), the top layers of the {001} and {101} facets were composed of unsaturated five-coordinated Ti (Ti5c) and two-coordinated O (O2c) atoms, while the Ti atom on the top layer of the {110} facet was the four-coordinated Ti (Ti4c) atom. Generally, the more the surface dangling bonds, the better the surface reactivity.23–31 Thus, the {110} facet possessed the highest activity among these three anatase TiO2 facets, which was consistent with the result inferred from the surface energy. In Fig. 1i, the density of states (DOS) was aligned with respect to the Fermi level, which is defined as 0 eV.19 The difference of the energy levels in the valence bands (ΔEVB) between the {001} and {101} facets is ca. 0.25 eV, while the ΔEVB between the {001} and {110} facets is calculated to be ca. 0.36 eV. Although the calculated differences may be much larger than the actual value, the existing difference well demonstrated the feasibility of transferring the photogenerated electrons from the {001} to {101} and {110} facets, and holes from the {101} and {110} to {001} facets, resulting in the formation of reduction and oxidation facets. The higher ΔEVB value between the {001} and {110} facets compared to that between the {001} and {101} facets indicates a stronger ability of the {001}/{110} interface for separating photogenerated electron–hole pairs than the {001}/{101} interface. Based on the above evidence, the goal of promoting the separation of photogenerated carriers can be achieved by increasing the ratio of the {001}/{110} interface.
Starting from such a design objective, directional etching of the prepared STT was carried out using HF solution as the etching agent. The etching process of the STT is schematically depicted in Fig. S1.† STT particles were homogenously dispersed in a solution of HF. During this etching process at 180 °C for 6 h, the product well maintained the tetrakaidecahedral morphology and high monodispersity with a small hollow structure (SHTT), as shown from the FESEM and TEM images (Fig. S7a and b†), while the particles were etched into hollow structures from the top (001) facet to the bottom (00![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) facet. According to the selected-area electron diffraction (SAED) pattern (Fig. S7c†), the obtained SHTT was single-crystalline and could be indexed to the [001] zone axis of tetragonal TiO2. The structural features agreed well with the model of SHTT with exposed {001}, {101} and {110} facets projected along the [001] zone axis (Fig. S7d†). The powder XRD pattern indicated that SHTT was still pure anatase TiO2 phase (Fig. S8†). EDX mapping (Fig. S9†) also showed that the Ti and O elements were uniformly distributed over the small hollow tetrakaidecahedral structure. Upon further etching, the hollow structure of SHTT was gradually enlarged to afford the HTT (Fig. 1j). The phase of the HTT still retained anatase TiO2 (Fig. S10†). EDX mapping showed that the Ti and O elements were still uniformly distributed in the hollow tetrakaidecahedral structure of the HTT (Fig. S11†). In order to explore the etching mechanism, the HF etching solution was replaced by HCl solution, HCl and NaF mixed solution, and NaF solution, respectively. After etching at 180 °C for 12 h, the morphology of the STT sample in HCl solution (Fig. S12a†) did not show any changes, while the STT samples in HCl and NaF mixed solution (Fig. S12b†) and NaF solution (Fig. S12c) exhibited the etched phenomenon on the {001} facet. The results of these comparative experiments indicated that F ions played a key role in this etching process.
) facet. According to the selected-area electron diffraction (SAED) pattern (Fig. S7c†), the obtained SHTT was single-crystalline and could be indexed to the [001] zone axis of tetragonal TiO2. The structural features agreed well with the model of SHTT with exposed {001}, {101} and {110} facets projected along the [001] zone axis (Fig. S7d†). The powder XRD pattern indicated that SHTT was still pure anatase TiO2 phase (Fig. S8†). EDX mapping (Fig. S9†) also showed that the Ti and O elements were uniformly distributed over the small hollow tetrakaidecahedral structure. Upon further etching, the hollow structure of SHTT was gradually enlarged to afford the HTT (Fig. 1j). The phase of the HTT still retained anatase TiO2 (Fig. S10†). EDX mapping showed that the Ti and O elements were still uniformly distributed in the hollow tetrakaidecahedral structure of the HTT (Fig. S11†). In order to explore the etching mechanism, the HF etching solution was replaced by HCl solution, HCl and NaF mixed solution, and NaF solution, respectively. After etching at 180 °C for 12 h, the morphology of the STT sample in HCl solution (Fig. S12a†) did not show any changes, while the STT samples in HCl and NaF mixed solution (Fig. S12b†) and NaF solution (Fig. S12c) exhibited the etched phenomenon on the {001} facet. The results of these comparative experiments indicated that F ions played a key role in this etching process.
To further study the structural information of the obtained HTT, the product was characterized by transmission electron microscopy (TEM). Fig. 2a shows a TEM image of an individual HTT projected from the [001] direction, which was in good agreement with the SEM observation. The external appearance of the particle presented an octahedron-like outline and the internal appearance exhibited a square morphology. The corresponding SAED pattern is given in Fig. 2b. This SAED pattern implied that the obtained HTT was single-crystalline and could be indexed to the [001] zone axis of tetragonal TiO2. These structural features agreed well with the model of the HTT with exposed {001}, {101} and {110} facets projected along the [001] zone axis, and the newly exposed internal surfaces corresponded to the {110} facet (Fig. 2c). High resolution transmission electron microscope (HRTEM) observation indicated that the interplanar distance is 0.377 nm (Fig. 2d), corresponding to the lattice spacing of the (010) facet of tetragonal TiO2. In order to confirm the exposed surfaces of the HTT, the same particle was rotated to the other zone axis (Fig. 2e). The SAED pattern (Fig. 2f) indicated that the particle was rotated to the [02![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ] zone axis. The outline and the apex angle of the particle still matched well with that of the ideal HTT model projected along the same direction (Fig. 2g). The HRTEM image (Fig. 2h) also corresponded well with the (010) facet of tetragonal TiO2. This HTT particle from another viewing angle (Fig. 2i) was investigated to verify the correctness of the model. The SAED pattern (Fig. 2j) showed that the viewing angle was in the [010] direction. The profile was still in good agreement with the model of an ideal HTT viewed along the [010] direction (Fig. 2k). Fig. 2l shows that the interplanar distance is 0.377 nm, corresponding to the lattice spacing of the (010) facet. Based on these structural analyses, the exposed surfaces of the as-prepared HTT could be inferred as the {101}, {001} and {110} facets, and the newly exposed internal surfaces could be deduced as the {110} facet. The proportion of each crystal plane was 30% for the {101} facet, 30% for the {001} facet, and 40% for the {110} facet. After etching, the proportion of the {110} facet increases as a result of the exposure of the internal surfaces, while the proportion of the {101} facet decreases, thus achieving the design goal of increasing the {001}/{110} ratio. In addition, the proportion of the highly active {110} facet in the HTT is as high as 40%. The concurrent photo-deposition of Ag/MnOx on thr HTT was conducted to illustrate the separation of photogenerated carriers. As shown in Fig. S13†, the deposition of MnOx is observed on the top surface of the HTT, whereas the inner and outer side surfaces exhibit the presence of Ag particles. This observation suggests that the photogenerated electrons and holes accumulate predominantly on the {001} facet and the {110} and {101} facets, respectively, leading to spatial separation between them.
] zone axis. The outline and the apex angle of the particle still matched well with that of the ideal HTT model projected along the same direction (Fig. 2g). The HRTEM image (Fig. 2h) also corresponded well with the (010) facet of tetragonal TiO2. This HTT particle from another viewing angle (Fig. 2i) was investigated to verify the correctness of the model. The SAED pattern (Fig. 2j) showed that the viewing angle was in the [010] direction. The profile was still in good agreement with the model of an ideal HTT viewed along the [010] direction (Fig. 2k). Fig. 2l shows that the interplanar distance is 0.377 nm, corresponding to the lattice spacing of the (010) facet. Based on these structural analyses, the exposed surfaces of the as-prepared HTT could be inferred as the {101}, {001} and {110} facets, and the newly exposed internal surfaces could be deduced as the {110} facet. The proportion of each crystal plane was 30% for the {101} facet, 30% for the {001} facet, and 40% for the {110} facet. After etching, the proportion of the {110} facet increases as a result of the exposure of the internal surfaces, while the proportion of the {101} facet decreases, thus achieving the design goal of increasing the {001}/{110} ratio. In addition, the proportion of the highly active {110} facet in the HTT is as high as 40%. The concurrent photo-deposition of Ag/MnOx on thr HTT was conducted to illustrate the separation of photogenerated carriers. As shown in Fig. S13†, the deposition of MnOx is observed on the top surface of the HTT, whereas the inner and outer side surfaces exhibit the presence of Ag particles. This observation suggests that the photogenerated electrons and holes accumulate predominantly on the {001} facet and the {110} and {101} facets, respectively, leading to spatial separation between them.
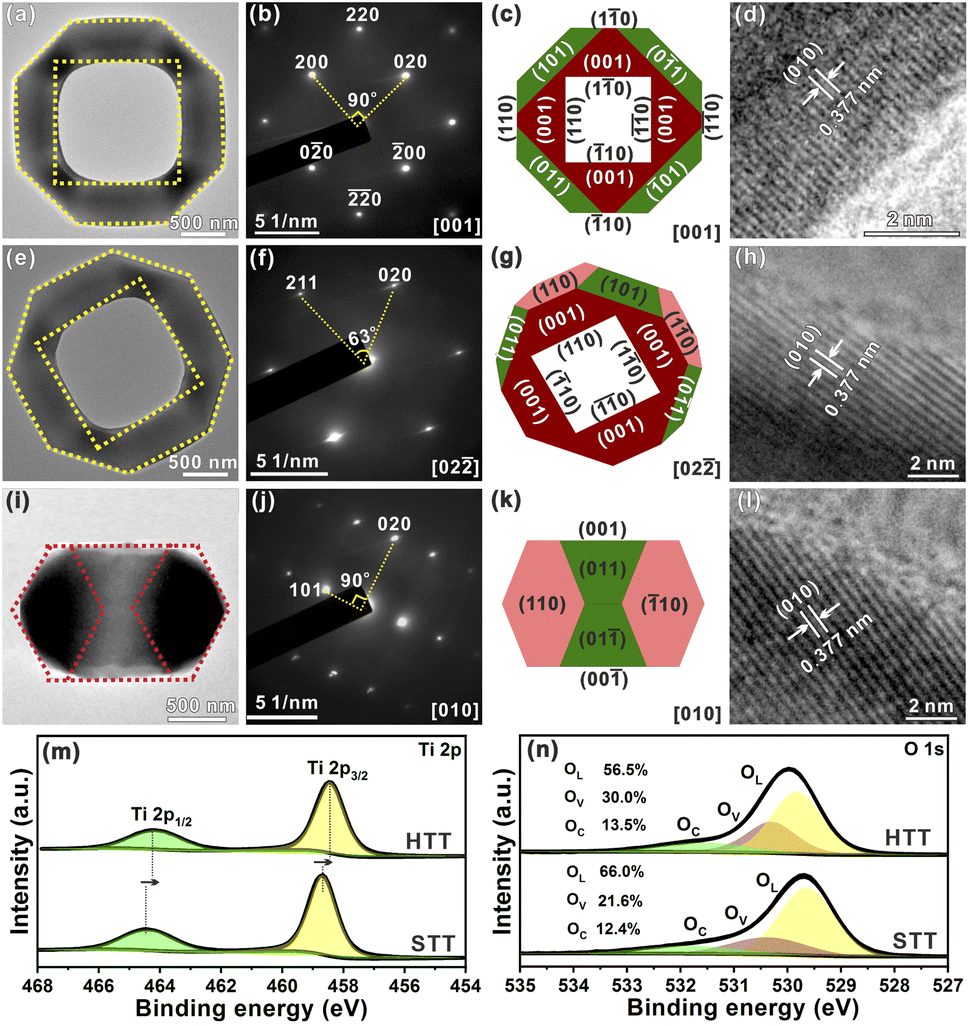 | ||
| Fig. 2 (a, e and i) Typical TEM images of an individual HTT projected from the [001], [02] and [010] directions, respectively. (b, f and j) Corresponding SAED patterns of the HTT projected from the [001], [02] and [010] directions, respectively. (c, g and k) Models of the HTT viewed along the [001], [02] and [010] directions, respectively. (d, h and l) Corresponding HRTEM images. High-resolution XPS spectra of Ti 2p (m) and O 1s (n). | ||
In the high-resolution Ti 2p XPS spectrum (Fig. 2m), the peaks at 464.2 eV for the HTT and 464.5 eV for the STT, are in agreement with Ti 2p1/2, and the peaks at 458.4 eV for the HTT and 458.6 eV for the STT, correspond to Ti 2p3/2. The surface of the {110} facet has lots of unsaturated Ti4c atoms, and is enriched with a large number of electrons. The calculated Mulliken charges of surface Ti atoms on the (110), (110) and (001) surfaces are 1.31e, 1.25e and 1.23e, respectively. The greater the positive charge value, the fewer electrons are lost by the surface Ti atoms. Compared with the STT, the HTT has a higher proportion of the {110} crystal facet. Therefore, the peaks of Ti 2p moved to the low-energy region. The O 1s high-resolution spectrum could be split into three major peaks, corresponding to lattice oxygen (OL), the oxygen-deficient region (OV), and dissociated and chemisorbed oxygen (OC), respectively (Fig. 2n). Compared with the STT, the HTT has a higher proportion of OV, which is consistent with the result of more Ti4c on the {110} facet. The above results show that the HTT has a higher proportion of the {110} facet with more unsaturated coordination Ti4c and more residual electrons, which can act as active sites to improve the catalytic activity of the material.
Photoelectrochemical properties of the photocatalysts
In order to verify that increasing the proportion of the {001}/{110} interface can improve the separation of photogenerated electron–hole pairs, the photoelectrochemical properties of a solid TiO2 cuboctahedron composed of {101} and {001} facets (STC),49 the STT and HTT, were tested. The detailed synthesis procedure of the STC is shown in the ESI,† and the corresponding structure and morphology are presented in Fig. S14.† As shown from the UV-vis absorption spectra (Fig. S15†), the light response of the STC, STT and HTT is almost the same, indicating that the ability of these three samples to produce photogenerated carriers is similar. The photocurrent density of the STC, STT and HTT was normalized for the catalyst loading amount and surface area (Fig. 3a). Compared with the STC containing only the {001}/{101} interface, the {001}/{110} interface was introduced in the STT with the appearance of the {110} facet, and the photocurrent density of the STT was twice that of the STC. The photocurrent density continued to increase with the further increased proportion of the {001}/{110} interface, so that the photocurrent density of the HTT was twice that of the STT and four times that of the STC. In order to exclude the influence of the interaction between exposed crystal facets of the catalyst and electrolyte on the photocurrent density, besides NaH2PO4/Na2HPO4 electrolyte, the photocurrent density of the STC, STT and HTT in Na2SO4 electrolyte and NaOH electrolyte was also measured. As shown in Fig. S16,† the photocurrent density of the STC, STT and HTT in Na2SO4 electrolyte and NaOH electrolyte still followed the order of HTT > STT > STC. The photoluminescence (PL) emission spectra of the STC, STT and HTT were then measured. As shown in Fig. 3b, the intensity of the main emission peak (centered at about 398 nm) assigned to the band–band photoluminescence phenomenon presented an obvious sequence of HTT < STT < STC. This meant that the HTT possessed the lowest recombination rate of photogenerated electron–hole pairs, which was consistent with the trend of the photocurrent density. The results of photocurrent density and photoluminescence spectra indicated that the anatase TiO2 {001}/{110} interface had a more significant promoting effect on photogenerated carrier separation than the {001}/{101} interface, which is consistent with the theoretical prediction.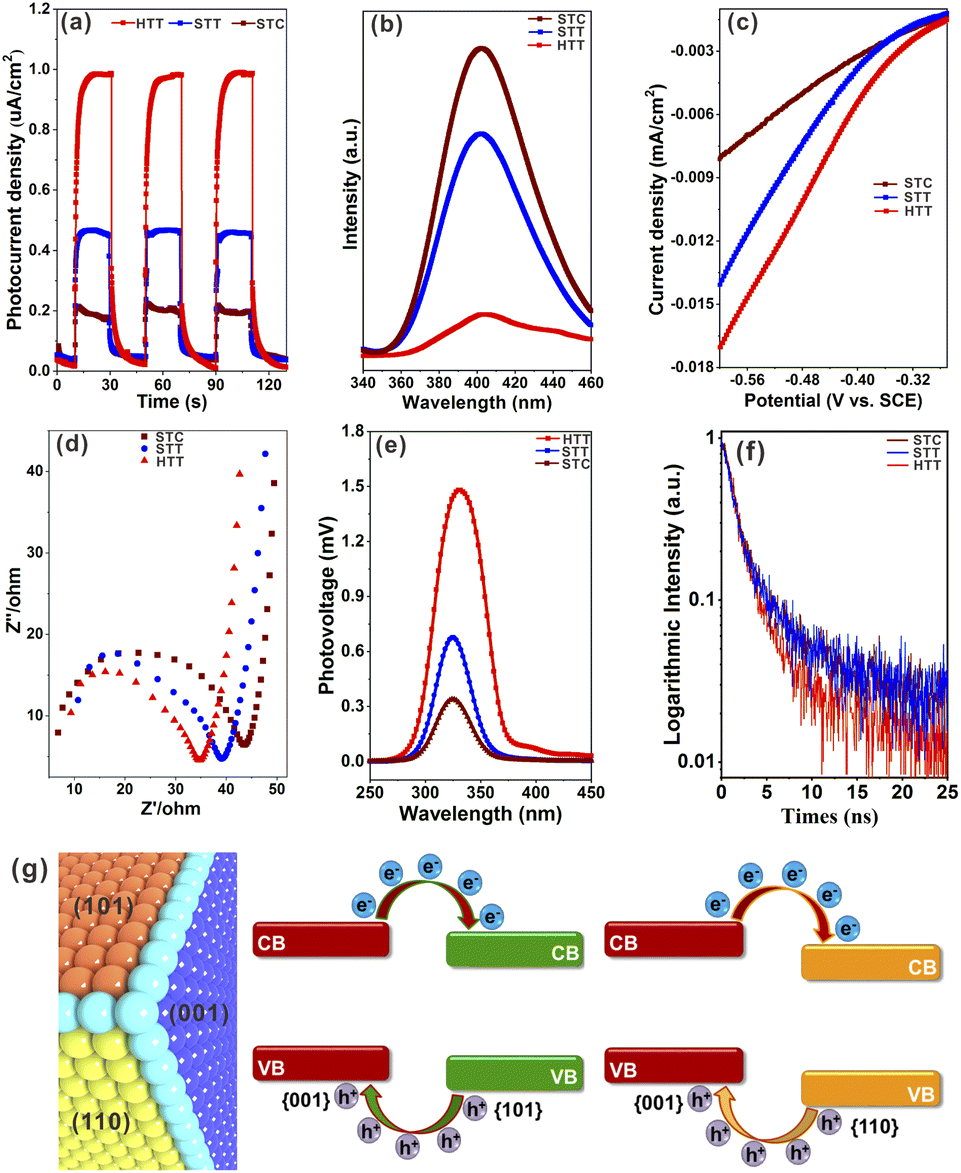 | ||
| Fig. 3 (a) Photocurrent density of the STC, STT and HTT measured at 0.2 V versus Hg/Hg2Cl2 in NaH2PO4/Na2HPO4 electrolyte under illuminated (i.e., light) and non-illuminated (i.e., dark) conditions. (b) Photoluminescence spectra of these three samples. (c) LSV curves of these three samples. (d) EIS spectra of these three samples measured at open-circuit potential versus Hg/Hg2Cl2 under illuminated conditions in the low-frequency range and equivalent circuits. (e) SPV spectrum of the STC, STT and HTT. (f) Time resolved PL spectra. (g) Schematic diagram of energy levels and photogenerated carrier transfer for anatase TiO2 {001}/{101} and {001}/{110} interfaces. | ||
Linear sweep voltammetry (LSV) curves (Fig. 3c) showed that the HTT had higher cathodic current density than the STT and STC, indicating that the HTT had the highest electron transfer efficiency among these three samples. Electrochemical impedance spectroscopy (EIS) of the STC, STT and HTT was carried out to further clarify the kinetics of the charge transfer. Fig. 3d shows the EIS spectra of these samples and equivalent circuits, and the fitting parameters are listed in Table S1.† The order of the transfer resistance (RCT) values among these samples is HTT < STT < STC, revealing that the order of the electron transfer at the interface of anatase TiO2 electrodes and electrolyte is HTT > STT > STC. Quantitatively, the charge transfer efficiency (ηtrans) was determined to be 92.6%, 69.5% and 59.9% for the HTT, STT and STC, respectively, on adding the electron scavenger MV2Cl2 (Fig. S17†).22 The surface charge transfer in the HTT is significantly enhanced, attributed to the increased proportion of the dominant {001}/{110} interface. To further gain direct evidence of the separation efficiency of photogenerated carriers, surface photovoltage spectroscopy (SPV) has been carried out. Compared to the STC and STT, the HTT showed significantly stronger SPV signals, demonstrating the increment of the photogenerated carrier accumulation concentration on the surface of the TiO2 crystal (Fig. 3e). Time-resolved PL spectra (Fig. 3f) had determined the PL lifetimes to be 25.08 ns for the HTT, 5.71 ns for the STT, and 3.99 ns for the STC. A shorter PL lifetime means a faster transfer of charge carriers and a more efficient separation of electrons and holes. Based on the above theoretical calculations (Fig. 1i) and experimental results (Fig. 3), it can be inferred that the {001}/{110} interface of anatase TiO2 has well matched valence band and conduction band positions to separate photogenerated carriers, which can indeed be improved by increasing the proportion of the {001}/{110} interface in the case of the HTT (Fig. 3g). Therefore, the HTT has a suitable crystal surface structure and has great potential for photocatalytic activity as a photocatalyst.
Photocatalytic activity of the photocatalysts
The effect of the dominant {001}/{110} interface on the photocatalytic activity of the TiO2 system was investigated by photocatalytic hydrogen production and selective oxidation of sulfides into sulfoxides using the STC, STT and HTT as the photocatalysts. The rate of photocatalytic H2 evolution over the STC, STT and HTT was compared, as shown in Fig. 4a. The STC with only the {001}/{101} interface produced almost no H2 in two hours. When the {001}/{110} interface appeared, the H2 yield of the STT showed an enhancement. With a further increase in the proportion of the {001}/{110} interface, the H2 yield of the HTT improved significantly, and was nearly four times that of the STC. This outcome indicates that the {001}/{110} interface plays a crucial role in the high H2 evolution activity of the HTT. The H2 yield rate of the HTT did not decrease significantly after five cycles of testing, indicating that the HTT possessed high cycling stability for photocatalytic H2 evolution. In order to explore the mechanism for the high H2 evolution of the HTT, the adsorption and activation of a H2O molecule on the (101) and (110) surfaces were studied. The adsorption configuration of a H2O molecule on the (101) and (110) surfaces is presented in Fig. 4b and c, and the corresponding adsorption energy was −1.12 and −1.31 eV, respectively, indicating that the (110) surface had better adsorption capacity for the H2O molecule than the (101) surface. The reason for this phenomenon was that the Ti4c on the (110) surface could provide more electrons to the adsorbed H2O molecule than the Ti5c on the (101) surface. The calculated Mulliken charge of O in the H2O molecule adsorbed on the (110) surface was −0.91e, while the charge of O from the H2O molecule adsorbed on the (101) surface was only −0.88e. On account of the stronger interaction between Ti4c on the (110) surface and O in the adsorbed H2O molecule, the bond length of O–H1 was elongated to 1.033 Å, and the charge of the H2 atom was increased to 0.51e. This result means that, in the activated state, the H2 atom of the H2O molecule adsorbed on the (110) surface is more likely to receive photogenerated electrons and be reduced as compared to the H2O molecule adsorbed on the (101) surface. The total electron density and electron density difference of molecular H2O adsorbed on the (101) and (110) surfaces were calculated to further investigate the interaction between the H2O molecule and TiO2 surface. Comparing Fig. 4d and e, it can be seen that the electron density between the H2O molecule and (110) surface was significantly denser than that between the H2O molecule and (101) surface. From the electron density difference of molecular H2O adsorbed on the (101) and (110) surfaces (Fig. 4f and g), it can be clearly observed that the electron interaction between the H2O molecule and (110) surface was stronger than that between the H2O molecule and (101) surface, which was consistent with the result of the total electron density.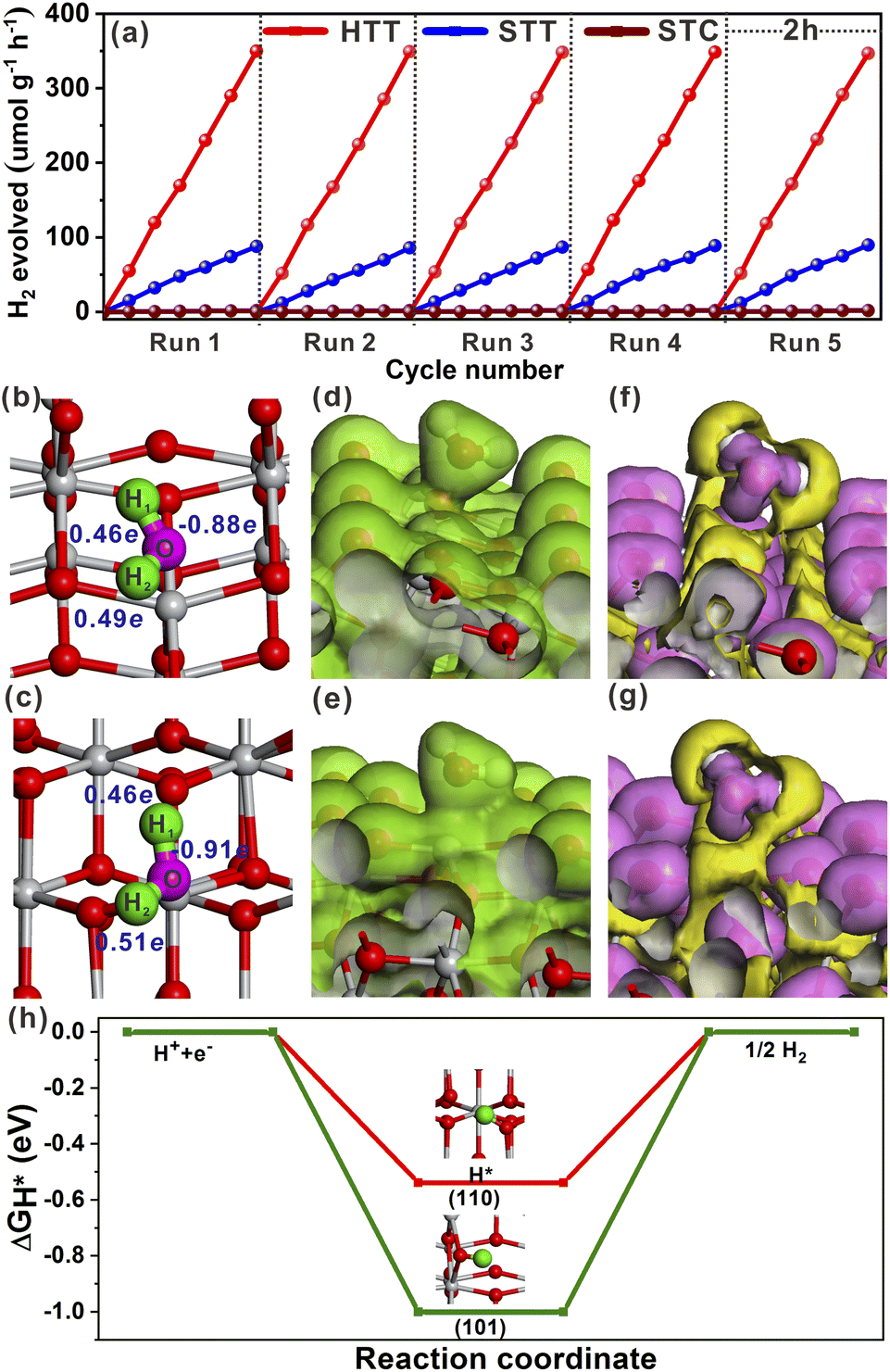 | ||
| Fig. 4 (a) Recyclability of the STC, STT and HTT in H2 production under a Hg lamp. Optimized structural models of molecular H2O adsorbed on the (101) (b) and (110) surfaces (c). Total electron density of molecular H2O adsorbed on the (101) (d) and (110) surfaces (e). Electron density difference of molecular H2O adsorbed on the (101) (f) and (110) surfaces (g). (h) Calculated free-energy diagram of the HER at the equilibrium potential for the (101) and (110) surfaces. | ||
In order to understand the state of the excited H2O molecule on the anatase TiO2 surface, the adsorption of dissociative H2O on (101) and (110) surfaces was also taken into account. On the (101) surface, the adsorption of dissociative H2O ultimately became molecular adsorption (Fig. S18a†), and the corresponding adsorption energy was −1.11 eV, which was close to the adsorption energy of molecular H2O on the (101) surface. The dissociative H2O adsorbed on the (110) surface still remained in the dissociated state, with an adsorption energy of −2.15 eV. The lower adsorption energy of dissociative H2O than that of molecular H2O on the (110) surface indicates that the molecularly adsorbed H2O state is metastable, tending to the dissociated H2O state on the (110) surface. As shown in Fig. S18b†, the O–H bonded with the Ti4c site on the (110) surface, and one of the H atoms is transferred to the adjacent oxygen site. The calculated adsorption of dissociative H2O further verified the better activation effect of the (110) surface on the H2O molecule than the (101) surface. Generally, the Gibbs free-energy of the intermediate adsorbed H* (|ΔGH*|) could be considered as a major descriptor of the activity of catalysts for the hydrogen evolution reaction (HER).50–52 The near-zero value of |ΔGH*| corresponded to the outstanding hydrogen production activity. The |ΔGH*| value of the (101) and (110) surfaces had been calculated and is shown in Fig. 4h. The smaller |ΔGH*| value (ΔGH* = −0.51 eV) of the (110) surface indicated that the (110) surface was more conducive to the HER than the (101) surface, the ΔGH* value of which was −0.96 eV. Therefore, the excellent photocatalytic H2 evolution of the HTT was ascribed not only to the better separation rate of photogenerated electron–hole pairs at the {001}/{110} interface, but also to the higher hydrogen production activity of the {110} facet.
In addition to the reductive reactions such as photocatalytic H2 evolution from water, photocatalytic selective oxidation of organic species was an important type of index reaction to test the activities of photocatalysts. In most photocatalytic selective organic oxidation reactions, the active intermediate is the superoxide radical (O2˙−). Thus, the charge interaction between the two reduction (101) and (110) surfaces and O2 molecule was investigated by using total electron density and electron density difference. The adsorption energies of O2 molecules on the (101) and (110) surfaces were −0.58 eV and 1.59 eV, respectively. The resulting O–O bond lengths of adsorbed O2 molecules on the (101) and (110) surfaces were 1.402 Å and 1.426 Å, respectively. As shown in Fig. 5a and b, the total electron density between the adsorbed O2 molecule and (110) surface was higher than that between the O2 molecule and (101) surface. From the electron density difference (Fig. 5c and d), it could be seen that the charge interaction between the (110) surface and O2 was stronger than that between the (101) surface and O2. The stronger interaction between the adsorbed O2 molecule and (110) surface could be more clearly observed from the electron density difference maps (Fig. 5e and f). The calculated Mulliken charge of O2 adsorbed on the (101) surface was −0.34e, while the charge of O2 adsorbed on the (110) surface was −0.43e. The calculated results of Mulliken charge indicated that O2 adsorbed on the (101) surface gained more charge than that adsorbed on the (101) surface, which was consistent with the analysis result of electron density difference maps. Based on these theoretical calculation results, it could be inferred that increasing the proportion of the {001}/{110} interface was beneficial to the formation of O2˙−. The selective oxidation of sulfides into sulfoxides with the active intermediate of O2˙− by the HTT, STT, and STC photocatalysts was selected to verify the above inference. As shown in Fig. 5g, the sequence of the photocatalytic yield was HTT > STT > STC, indicating that the photocatalytic activity of the HTT was improved with the increase in the {001}/{110} interface ratio. This result was consistent with the theoretical inference. The recycling stability of the HTT was investigated by five cycles of this photocatalytic reaction under the same conditions. It could be found that the activity loss of the HTT was negligible. After catalytic cycle experiments, the composition and morphology of the HTT photocatalyst did not show obvious changes (Fig. S19†), demonstrating that the HTT possessed high catalytic stability. Then, various thioanisole derivatives were used as reactants to evaluate the universality of the HTT as a photocatalyst. As shown in Fig. S20,† the yields and selectivity of the desired oxidized products were 83–98% and 82–94% respectively, indicating that the HTT photocatalyst had good universality for selective oxidation of sulfides into sulfoxides.
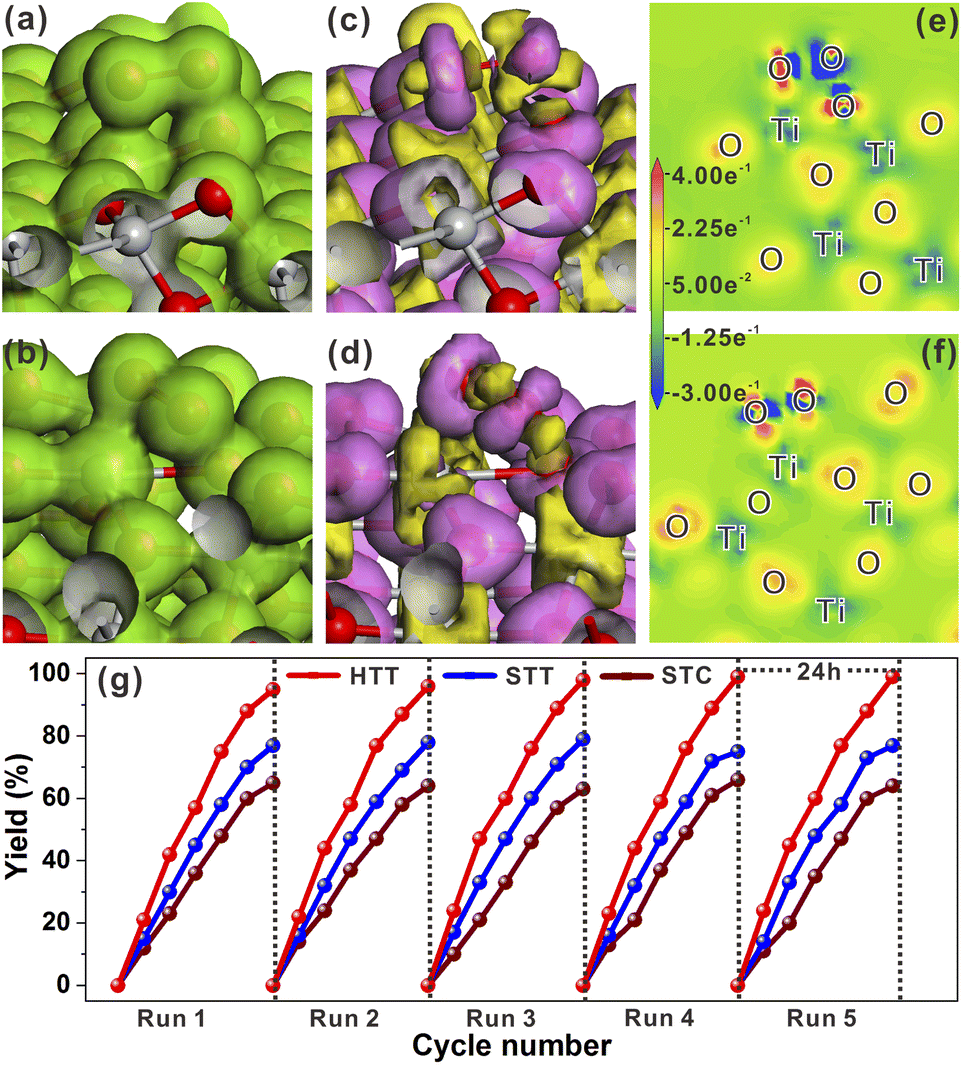 | ||
| Fig. 5 Total electron density of the O2 molecule adsorbed on the (101) (a) and (110) surfaces (b). Electron density difference of the O2 molecule adsorbed on the (101) (c) and (110) surfaces (d). Electron density difference maps of the O2 molecule adsorbed on the (101) (e) and (110) surfaces (f). (g) Yield of sulfoxides by selective oxidation of sulfides over the STC, STT and HTT under a Hg lamp. | ||
Compared with the half reactions with only oxidation or reduction, the photocatalytic reaction with the involvement of both electrons and holes can better reflect the photogenerated carrier separation efficiency and catalytic activity of the catalyst. Thus, the photocatalytic activity of the HTT, STT and STC for CDC reactions (Fig. S21a†) was measured to further verify their separation efficiency of photogenerated carriers and surface reactivity. As shown in Fig. S21b,† with the increasing proportion of the dominant {001}/{110} interface, the yield of CDC reactions was improved sequentially, i.e., HTT > STT > STC. This result further confirmed the efficient separation of photogenerated carriers and high surface reactivity of the HTT.
Microscopic mechanism of charge separation
In order to investigate the microscopic mechanism of charge separation at the {001}/{110} interface, we measured femtosecond transient absorption (TA) to study the excited state dynamics of the HTT and STC. The UV-vis absorption spectra of the HTT and STC were measured (Fig. S15†) to better understand the TA spectral changes. Fig. 6a and b show the 3D contour plots of TA spectra at 430–800 nm. Both samples present a negative TA signal, which reflects the photo excited carrier trapped in surface states.53 The TA of the STC is mainly characterized by the absorption peak near 500 nm. For the HTT, a broad and featureless TA band was observed at 450–800 nm. Carrier dynamics in TiO2 have been extensively studied and reviewed in the 1990s.54–56 Yoshihara and co-workers identified electron and hole TA signals of TiO2 from 400 to 2500 nm.57 They found that the hole in the valence band mainly absorbs in the visible range (400–700 nm) and trapped electrons have a strong absorption peak in the NIR range (∼800 nm). The strong TA signal in the NIR range means the large number of surface trapped electrons for the HTT. However, the marked decrease in TA intensity >700 nm compared with that at ∼500 nm reflects the strong electron trapping ability of deep defect states, which decrease the number of electrons for catalysis in the STC. Considering the high ratio of the {001}/{110} interface in the HTT, the strong TA signal for the surface trapped electron reflects the efficient charge separation of the interface. So that with only the {001}/{101} interface, the STC showed poor charge separation and a low TA signal for the surface trapped electron. As a conclusion, the {001}/{110} interface performs high-efficiency charge separation and the electron dynamic at {001}/{110} interface can reflect the ability of charge separation. The TA signal near 800 nm can efficiently reflect the shallow trapped electron in TiO2. The shallow trapped electrons can not only regain the activation energy for the catalytic reaction, but can also be further captured by the deep defect states and lose the catalytic activity. In this case, we extracted a kinetic trace at 770 nm to analyze the electron dynamics in the TiO2 samples.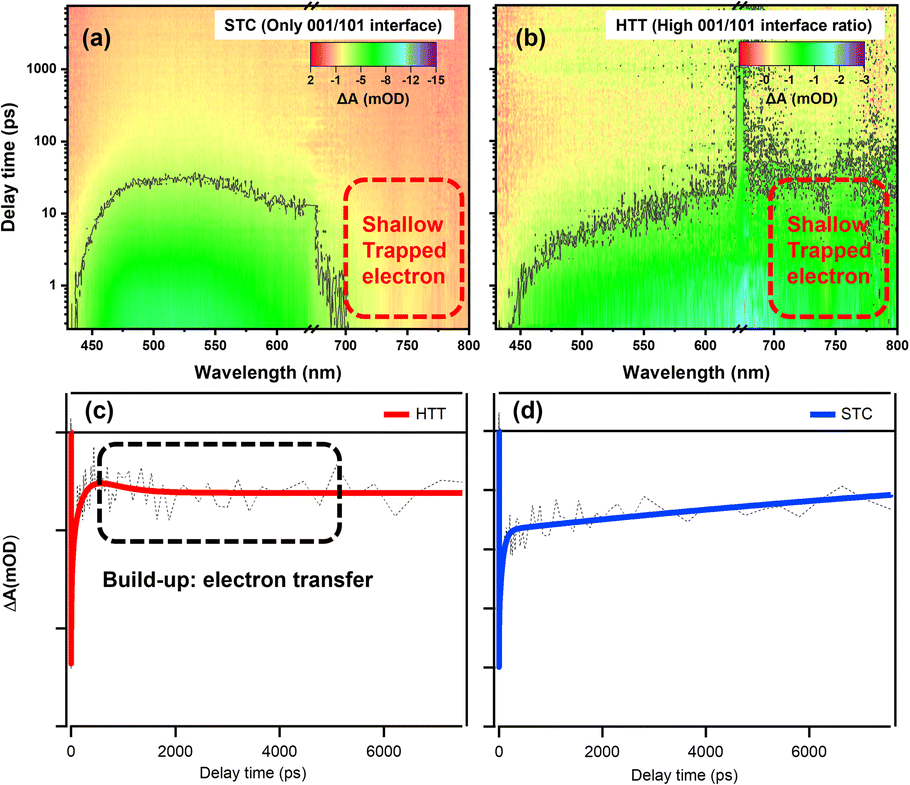 | ||
| Fig. 6 3D contour plots of transient absorption spectra of the (a) STC and (b) HTT and kinetic traces of the HTT (c) and STC (d) at 770 nm. | ||
Fig. 6c and d show the kinetic curves at 770 nm and the data are fitted using multi-exponential decay components as listed in Table S2.† In general, photo-excited electrons first reach the edge of the conduction band and then they can be trapped in surface defects in TiO2. The trapping may occur from a few picoseconds to hundreds of picoseconds. After this, electrons could take part in chemical reactions or recombine with holes in the valence band.58,59 As clearly shown in the decay process for those traces, HTT firstly undergoes a build-up process with a time constant of about 0.4 ns. The formation process reflects the increasing number of free electrons that de-trapped surface defects. Considering the high-efficiency charge separation at {001}/{110} interface, the build-up originates from the electron transfer from {001} facet to {110} facet. The build-up process (0.4 ns) was also observed due to the performance of the {001}/{110} interface in the STT (as shown in Fig. S22†) , and the amplitude variation of the kinetic trace for build-up corresponded with the variation of the {001}/{110} ratio between the HTT and STT. It is further proved that the electron transfer can be attributed to the build-up process. When the filling or the electron transfer is finished, the bleach signal reaches a maximum and remains almost constant at 8 ns, suggesting the long-lived electron for the HTT. The absence of the {001}/{110} interface means the shallow trapped electrons cannot be reactivated, resulting in the electrons being captured by the deeper defects states, and then losing the catalytic activity. An obvious decay tendency was eventually observed. In summary, the electron transfer efficiency between the {001} facet and the {110} facet is the reason why the HTT shows enhanced photocatalytic performance. This result is consistent with the steady optical and electronic measurements discussed above.
Conclusions
In order to obtain excellent photocatalysts by constructing the oxidation/reduction interface with a high proportion of exposed highly active facets, the solid anatase TiO2 tetrakaidecahedron (STT) composed of {001}, {101} and {110} facets has been synthesized. The difficulty of synthesizing TiO2 with a highly active {110} facet has been effectively reduced due to the introduction of the stable {101} facet. In the prepared STT, the exposed proportion of the highly active {110} facet accounts for 20%. The selective photo-deposition of Pt and MnOx on the surface indicated that the STT possessed two sets of oxidation/reduction interfaces, namely the {001}/{101} interface and {001}/{110} interface. Theoretical calculations have demonstrated that the effect of the {001}/{110} oxidation/reduction interface on promoting charge separation is better than that of the {001}/{101} oxidation/reduction interface. Then, the hollow anatase TiO2 tetrakaidecahedron (HTT) has been prepared by the directional etching method to increase the proportion of the {001}/{110} interface. The proportion of the highly active {110} facet in the HTT was further expanded to 40%. The photoelectrochemical properties and transient absorption measurement of the solid TiO2 cuboctahedron (STC) with only the {001}/{101} oxidation/reduction interface, STT and HTT confirmed that the {001}/{110} interface played a dominant role in promoting photogenerated carrier separation as compared to the {001}/{101} interface. The theoretical calculations showed that the Ti4c atoms on the {110} surface served as active sites to provide sufficient electrons for the adsorption-dissociation of the H2O molecule and the generation of O2˙−. The HTT showed excellent photocatalytic activity for both reductive (hydrogen production) and oxidative (selective oxidation of sulfides) reactions, attributed to the synergistic effect of increased dominant {001}/{110} interface proportion and high proportion of the highly active {110} facet. Moreover, the prepared HTT also demonstrated favorable photocatalytic activity for the CDC reactions, in which both photogenerated electrons and photogenerated holes are involved, further verifying its high separation efficiency of photogenerated carriers and high surface reactivity. The present work provides an important guideline for developing advanced structures with a predetermined interface toward desired applications.Data availability
All supporting data is provided in the ESI.†Author contributions
L. Sun and X. Han conceived the study. L. Sun, Y. Yuan and X. He designed and conducted the experiments. W. Zhan, D. Li and X.-J. Wang helped collect the data. L. Sun, X. He and X. Han analysed the data. L. Sun, Y. Zhao and X. Han supervised the work, and drafted and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22005126), the Natural Science Foundation of Jiangsu Province (BK20211549), the Singapore Agency for Science, Technology and Research (A*STAR) Manufacturing, the Trade and Connectivity Individual Research Grant (M21K2c0105), and the Singapore Ministry of Education under its Academic Research Funds (RG2/22 and RG85/22). We thank the Materials Characterization Center of East China Normal University for help with the measurement of femtosecond TA.Notes and references
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- A. Hagfeldt and M. Graetzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341–357 CrossRef CAS.
- L. Wang, D. W. Bahnemann, L. Bian, G. Dong, J. Zhao and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 8103–8108 CrossRef CAS PubMed.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- E. W. McFarland and J. Tang, Nature, 2003, 421, 616–618 CrossRef CAS PubMed.
- M. Murdoch, G. I. N. Waterhouse, M. A. Nadeem, J. B. Metson, M. A. Keane, R. F. Howe, J. Llorca and H. Idriss, Nat. Chem., 2011, 3, 489–492 CrossRef CAS PubMed.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- L. Mu, Y. Zhao, A. Li, S. Wang, Z. Wang, J. Yang, Y. Wang, T. Liu, R. Chen, J. Zhu, F. Fan, R. Li and C. Li, Energy Environ. Sci., 2016, 9, 2463–2469 RSC.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- J. Pan, G. Liu, G. Q. Lu and H.-M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- G. Liu, J. C. Yu, G. Q. Lu and H.-M. Cheng, Chem. Commun., 2011, 47, 6763–6783 RSC.
- J. Li, L. Cai, J. Shang, Y. Yu and L. Zhang, Adv. Mater., 2016, 28, 4059–4064 CrossRef CAS PubMed.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- J. Yu, S. Chang, L. Shi and X. Xu, ACS Catal., 2023, 13, 3854–3863 CrossRef CAS.
- F. Chen, H. Huang, L. Ye, T. Zhang, Y. Zhang, X. Han and T. Ma, Adv. Funct. Mater., 2018, 28, 1804284 CrossRef.
- M. Li, S. Yu, H. Huang, X. Li, Y. Feng, C. Wang, Y. Wang, T. Ma, L. Guo and Y. Zhang, Angew. Chem., Int. Ed., 2019, 58, 9517–9521 CrossRef CAS PubMed.
- H. Yan, R. He, J. Pham and P. Yang, Adv. Mater., 2003, 15, 402–405 CrossRef CAS.
- X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisatos, Nature, 2000, 404, 59–61 CrossRef CAS PubMed.
- W. Jiao, L. Wang, G. Liu, G. Q. Lu and H.-M. Cheng, ACS Catal., 2012, 2, 1854–1859 CrossRef CAS.
- X. Zong, Z. Xing, H. Yu, Z. Chen, F. Tang, J. Zhou, G. Q. Lu and L. Wang, Chem. Commun., 2011, 47, 11742–11744 RSC.
- A. S. Ichimura, B. M. Mack, S. M. Usmani and D. G. Mars, Chem. Mater., 2012, 24, 2324–2329 CrossRef CAS PubMed.
- H. Xu, S. Ouyang, P. Li, T. Kako and J. Ye, ACS Appl. Mater. Interfaces, 2013, 5, 1348–1354 CrossRef CAS PubMed.
- H. Xu, P. Reunchan, S. Ouyang, H. Tong, N. Umezawa, T. Kako and J. Ye, Chem. Mater., 2013, 25, 405–411 CrossRef CAS.
- H. S. Jung, Y. J. Hong, Y. Li, J. Cho, Y.-J. Kim and G.-C. Yi, ACS Nano, 2008, 2, 637–642 CrossRef CAS PubMed.
- F. Wang, L. Sun, Y. Li, W. Zhan, X. Wang and X. Han, Inorg. Chem., 2018, 57, 4550–4555 CrossRef CAS PubMed.
- D. Su, S. Dou and G. Wang, NPG Asia Mater., 2015, 7, e155 CrossRef CAS.
- R. Katal, S. K. Eshkalak, S. Masudy-panah, M. Kosari, M. Saeedikhani, M. Zarinejad and S. Ramakrishna, Nanomaterials, 2019, 9, 163 CrossRef CAS PubMed.
- T. Kawahara, Y. Konishi, H. Tada, N. Tohge, J. Nishii and S. Ito, Angew. Chem., Int. Ed., 2002, 41, 2811–2813 CrossRef CAS PubMed.
- C. P. Sajan, S. Wageh, A. A. Al-Ghamdi, J. Yu and S. Cao, Nano Res., 2016, 9, 3–27 CrossRef CAS.
- R. Katal, M. Salehi, M. H. D. A. Farahani, S. Masudy-Panah, S. L. Ong and J. Hu, ACS Appl. Mater. Interfaces, 2018, 10, 35316–35326 CrossRef CAS PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155409 CrossRef.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 119901 CrossRef.
- M. Liu, L. Piao, L. Zhao, S. Ju, Z. Yan, T. He, C. Zhou and W. Wang, Chem. Commun., 2010, 46, 1664–1666 RSC.
- Q. Wu, Z. Wu, Y. Li, H. Gao, L. Piao, T. Zhang and L. Du, Chin. J. Catal., 2012, 33, 1743–1753 CrossRef CAS.
- L. Pan, J.-J. Zou, S. Wang, X.-Y. Liu, X. Zhang and L. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 1650–1655 CrossRef CAS PubMed.
- R. Katal, S. Masudy-Panah, M. Tanhaei, M. H. D. A. Farahani and J. Hu, Chem. Eng. J., 2020, 384, 123384 CrossRef CAS.
- Q. Hu, J. Huang, Q. Li, C. Wang, G. Li, J. Chen and Y. Cao, Int. J. Hydrogen Energy, 2017, 42, 5478–5484 CrossRef CAS.
- Q. Li, T. Li, S. Chang, Q. Tao, B. Tian and J. Zhang, CrystEngComm, 2016, 18, 5074–5078 RSC.
- W. E. Farneth, R. S. McLean, J. D. Bolt, E. Dokou and M. A. Barteau, Langmuir, 1999, 15, 8569–8573 CrossRef CAS.
- T. Ohno, K. Sarukawa and M. Matsumura, New J. Chem., 2002, 26, 1167–1170 RSC.
- X. Wang, R.-G. Li, Q. Xu, H.-X. Han and C. Li, Acta Phys.-Chim. Sin., 2013, 29, 1566–1571 CAS.
- Z. Lai, F. Peng, Y. Wang, H. Wang, H. Yu, P. Liu and H. Zhao, J. Mater. Chem., 2012, 22, 23906–23912 RSC.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- A. Hagfeldt and M. Graetzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS.
- D. P. Colombo Jr, K. A. Roussel, J. Saeh, D. E. Skinner, J. J. Cavaleri and R. M. Bowman, Chem. Phys. Lett., 1995, 232, 207–214 CrossRef.
- N. Serpone, D. Lawless, R. Khairutdinov and E. Pelizzetti, J. Phys. Chem., 1995, 99, 16655–16661 CrossRef CAS.
- A. Furube, T. Asahi, H. Masuhara, H. Yamashita and M. Anpo, J. Phys. Chem. B, 1999, 103, 3120–3127 CrossRef CAS.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 3817–3823 CrossRef CAS.
- J. J. M. Vequizo, H. Matsunaga, T. Ishiku, S. Kamimura, T. Ohno and A. Yamakata, ACS Catal., 2017, 7, 2644–2651 CrossRef CAS.
- L. Dworak, S. Roth and J. Wachtveitl, J. Phys. Chem. C, 2017, 121, 2613–2619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04328b |
| This journal is © The Royal Society of Chemistry 2024 |