
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Snap-shots of cluster growth: structure and properties of a Zintl ion with an Fe3 core, [Fe3Sn18]4−†
Zi-Sheng
Li‡
 a,
Wei-Xing
Chen‡
b,
Harry W. T.
Morgan
a,
Cong-Cong
Shu
b,
John E.
McGrady
*a and
Zhong-Ming
Sun
*b
a,
Wei-Xing
Chen‡
b,
Harry W. T.
Morgan
a,
Cong-Cong
Shu
b,
John E.
McGrady
*a and
Zhong-Ming
Sun
*b
aDepartment of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3QR, UK. E-mail: john.mcgrady@chem.ox.ac.uk
bState Key Laboratory of Elemento-Organic Chemistry, Tianjin Key Lab for Rare Earth Materials and Applications, School of Materials Science and Engineering, Nankai University, Tianjin 300350, China. E-mail: sunlab@nankai.edu.cn
First published on 2nd December 2023
Abstract
The endohedral Zintl-ion cluster [Fe3Sn18]4− contains a linear Fe3 core with short Fe–Fe bond lengths of 2.4300(9) Å. The ground state is a septet, with significant σ and π contributions to the Fe–Fe bonds. The Sn18 cage is made up of two partially fused Sn9 fragments, and is structurally intermediate between [Ni2CdSn18]6−, where the fragments are clearly separated and [Pd2Sn18]4−, where they are completely fused. It therefore represents an intermediate stage in cluster growth. Analysis of the electronic structure suggests that the presence of the linear Fe–Fe–Fe unit is an important factor in directing reactions towards fusion of the two Sn9 units rather than the alternative of oligomerization via exo bond formation.
Introduction
The chemistry of Zintl ions, and in particular those containing endohedral metals, has been the subject of several recent reviews,1–9 and applications in catalysis and materials chemistry are beginning to emerge.10,11 The vast majority of these clusters are relatively small (14 main-group atoms or fewer) and contain a single transition metal ion, often with a closed-shell d10 configuration: classic examples include the icosahedral triad [Ni/Pd/PtPb12]2−,12 but the range of encapsulated metals now includes much of the d block. Larger clusters containing multiple transition metals are much less common but they offer the possibility of unusual magnetic phenomena and/or metal–metal bonding. Amongst the few known examples,13 the Ge18 series [Ni2InGe18]5−,14 [Ni3Ge18]4−,15 and [Pd2Ge18]4− (ref. 16) (Fig. 1) maps out a progressive fusion of the two Ge9 polyhedra which are well separated in [Ni2InGe18]5− but fully coalesced in the Pd cluster. [Ni3Ge18]4− appears to be an intriguing intermediate case, where the fusion is only partially complete. It is far from clear how these clusters are actually formed in situ, but it is certainly plausible that the stepwise fusion of pre-formed polyhedral E9 or ME9 fragments is involved. Indeed, Sevov and Goicoechea proposed the fusion of NiGe9 and Ni2Ge9 units as a possible route to formation of [Ni3Ge18]4−,17 and Dehnen's analysis of fragmentation patterns for [TaGe4As8]3− and [TaGe6As6]3− identified cluster fragments such as [Ge2As2]2− and [Ge3As]3− that may play a role in growth.18 The challenge from a synthetic perspective is that these component polyhedra typically carry high negative charges, and so their close approach incurs a high coulombic penalty. Transition metal ions that can bridge two polyhedral units may, therefore, play an important role in fusion by buffering these repulsions and also, potentially, by removing excess electron density through the extrusion of metal in the elemental form. A further complication is that the oxidative fusion of clusters is, at least in principle, in competition with oxidative oligomerisation via the formation of exo E–E bonds (Fig. 1(b)). This phenomenon is well established in Ge chemistry where linked chains of Ge9 are known.17,19 A deeper understanding of the factors that control cluster growth and the balance between fusion and oligomerisation may provide access to a wider range of element combinations and compositions, and to tailored structural, magnetic and catalytic properties.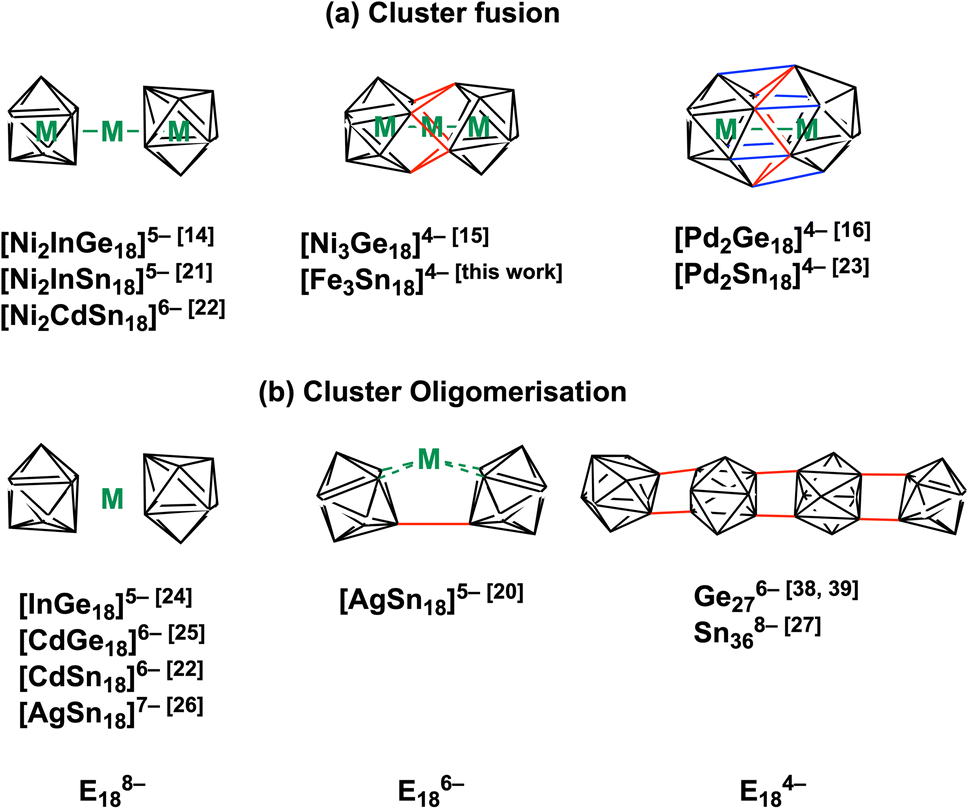 | ||
| Fig. 1 Cluster fusion vs. cluster oligomerisation of E9 polyhedra, E = Ge, Sn.14–16,19–29 | ||
In this paper, we extend our recent work on the Zintl-ion chemistry of tin by reporting the synthesis of a new cluster, [Fe3Sn18]4−, which has a linear Fe3 chain and Fe–Fe bond lengths of 2.4300(9) Å. The Fe3 chain is of significant interest in its own right – there are few examples of metal–metal bonded units encapsulated inside Zintl clusters, and the short Fe–Fe distances are a clear a priori indication of strong bonding. Of equal interest is the structure of the Sn18 cage because the degree of fusion of the two Sn9 polyhedra appears to be mid-way between the completely separated limited, as observed in [Ni2CdSn18]6−, and the completely fused limit in [Pd2Sn18]4. [Fe3Sn18]4− is, therefore, the Sn analogue of the Ge18 unit in [Ni3Ge18]4−. Our analysis of the electronic structure indicates that the E18 cages in [Ni3Ge18]4− and [Fe3Sn18]4− share a common −6 charge state, as does the Sn18 unit in [AgSn18]5−,20 where the two Sn9 units are not fused but rather oligomerised via an exo bond (Fig. 1(b)). A comparison of the different structural chemistry of these isoelectronic species offers a fascinating insight into the factors that control the balance between fusion in [Fe3Sn18]4− and [Ni3Ge18]4− and oligomerisation in [AgSn18]5−.
Results and discussion
Structure and properties of [Fe3Sn18]4−
The reaction of ethylenediamine (en) solutions of K4Sn9 with [K(thf)Fe(OtBu)3]2 (thf = tetrahydrofuran) results in the formation of the tri-iron cluster [Fe3Sn18]4− in the form of its [K(2.2.2-crypt)]+ salt [K(2.2.2-crypt)]4[Fe3Sn18] (1). Electrospray ionisation mass spectrometry (ESI-MS) of freshly-prepared DMF (DMF = dimethylformamide) solutions of 1 reveals a peak attributable to the dianion [Fe3Sn18]2− (m/z 1152.0323 – note the peak-to-peak separations of 0.5 between isotopologues that confirm the −2 charge, Fig. 2(c)) and also a very weak signal assigned to the cation–dianion pair [K(2.2.2-crypt)Fe3Sn18]− (m/z 2719.2300). It is common to observe only singly charged anions in the ESI-MS of Zintl clusters, but the large size of the Fe3Sn18 unit reduces the coulomb repulsion in the dianion to the extent that it is not ionized under the prevailing conditions. 1 crystallises in the monoclinic space group P21/c and the unit cell contains a single anionic [Fe3Sn18]4− cluster with four [K(2.2.2-crypt)]+ cations (Fig. 2(a) and (b), CCDC 2170116). The Sn18 unit adopts a D3d-symmetric structure based on two Sn9 polyhedra in a staggered, face-to-face arrangement, with a chain of three Fe centers aligned along the principal axis. In this section and the following discussion of the electronic structure, we focus first on the Fe3 chain, where Fe–Fe bonding is the primary interest, before turning to the Sn18 cage which we try to place in the wider context of Zintl-ion chemistry. The Fe–Fe bond lengths of 2.4300(9) Å in 1 are remarkably short, much shorter than those in the other known Fe2-containing Zintl cluster, [Fe2Ge16]4− (2.636(3) Å). Even shorter bonds are known in Fe2 dimers such as the FeIFeI paddlewheel complex30 (2.127 Å) and the (as-yet unknown) Fe2C30 (2.10 Å).31 Direct comparison with other Fe3 chains is restricted to classical coordination compounds such as Guillet's bis[(trimethylsilyl)amido]pyridine complex (Fig. 3(a), referred to henceforth as Fe3L3) where the Fe–Fe bond lengths are 2.4416(5) Å (ref. 32) and to the [Fe3(DpyF)4]2+ complex (DpyF = dipyridylformamide) first synthesised by Cotton and Murrillo33 and subsequently studied by Hillard and co-workers,34 where the Fe–Fe bond lengths are longer, at 2.7838(5) Å. These two FeIIFeIIFeII complexes share a common S = 6 ground state and a common formal σ bond order of 0.25 (per Fe–Fe bond), but differ in the distribution of electrons in the levels of π symmetry, with only Fe3L3 having an additional π component to the Fe–Fe bond. Correlations between bond order and bond length are notoriously difficult when bridging ligands are present, but nevertheless the similar bond lengths in [Fe3Sn18]4− and Fe3L3 offers an initial indication that Fe–Fe π bonding may also be significant in the former. We return to this question in the following discussion of the electronic structure of the cluster. Turning our focus now to the structure of the Sn18 cage, we note first that the cluster can be viewed as two FeSn9 units, bridged by a third Fe center. We can make useful comparison to the pair of closely-related clusters identified in Fig. 3(b), [Ni2CdSn18]6−,22 and [Pd2Sn18]4−,23 where we judge the degree of fusion of the two Sn9 units in terms of two distinct Sn–Sn distances identified in Fig. 2, Sn8−Sn9′ and Sn4−Sn9′. The average values of these are 3.46 Å and 4.10 Å, respectively in [Fe3Sn18]4− compared to 5.24 Å and 6.71 Å for [Ni2CdSn18]6− and 3.31 Å and 3.10 Å for [Pd2Sn18]6−. The Sn8−Sn9′ bond lengths in [Fe3Sn18]4− are therefore very similar to those in [Pd2Sn18]4−, but the Sn4−Sn9′ bonds are fully 1 Å longer. On this basis, we argue that the Sn18 cage in [Fe3Sn18]4− represents an intermediate stage in the cluster fusion process that occupies the same central position in Sn cluster chemistry as [Ni3Ge18]4− cluster15 does in the [Ni2InGe18]5−, [Ni3Ge18]4−, [Pd2Ge18]4− series identified in Fig. 1.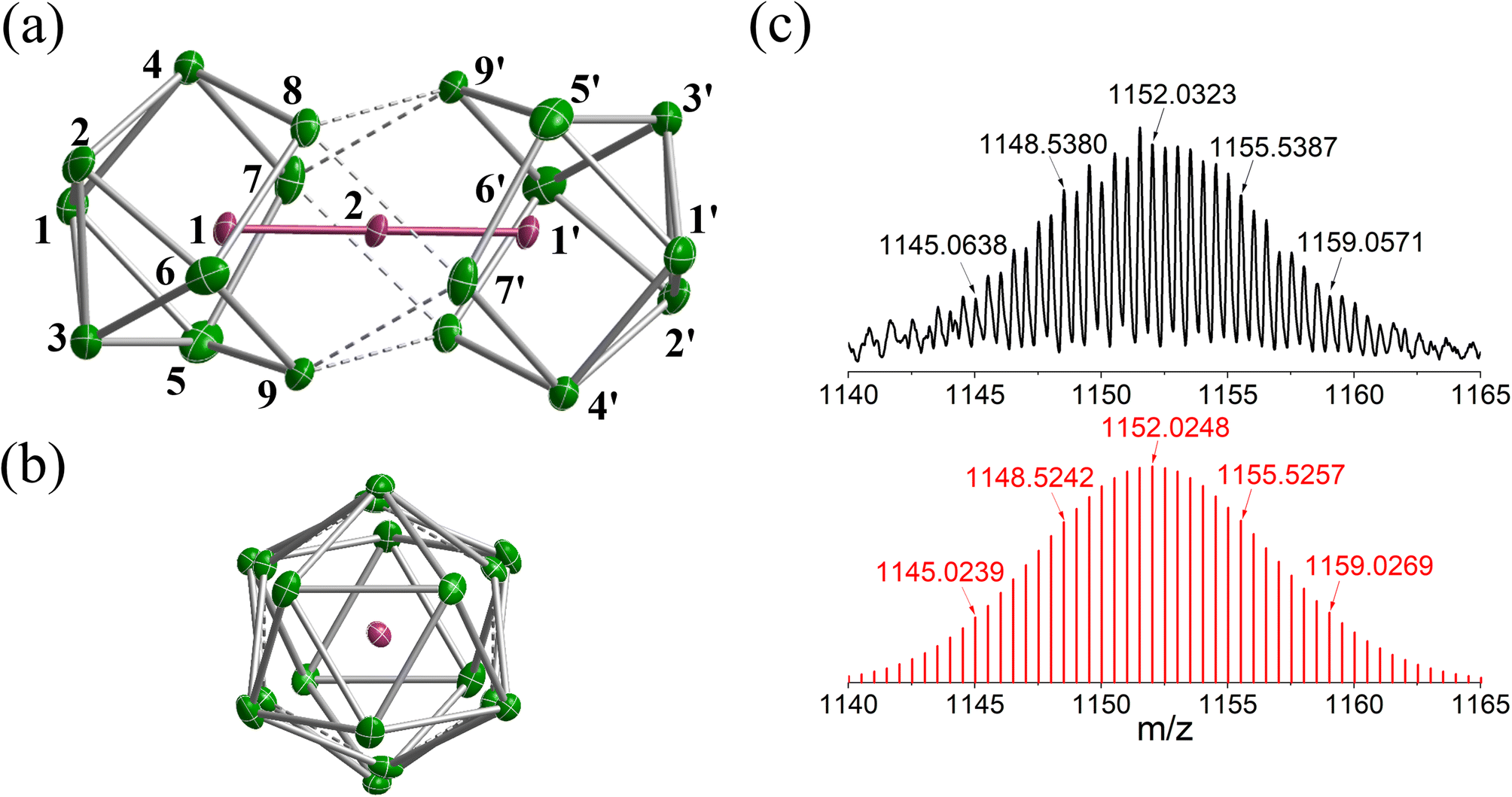 | ||
| Fig. 2 (a) and (b) Structure of anionic component, [Fe3Sn18]4−, of 1 and (c) the ESI-MS of a freshly-prepared solution of 1 in DMF. | ||
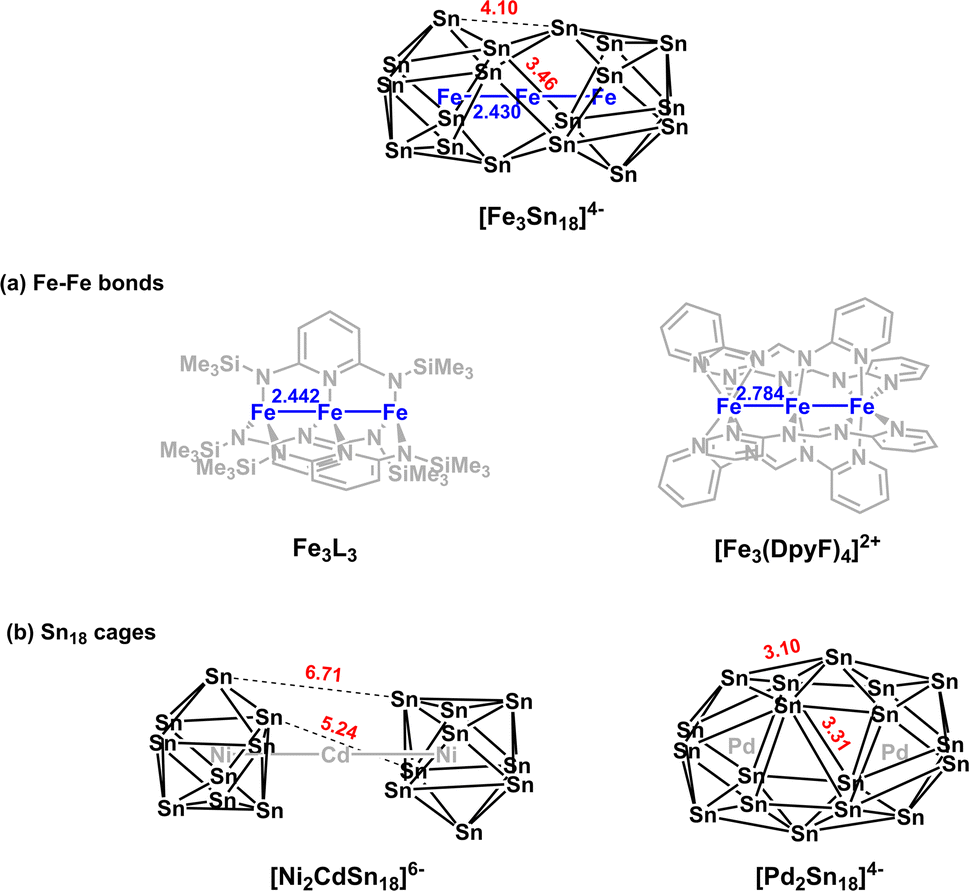 | ||
| Fig. 3 Structural comparisons with [Fe3Sn18]3−: (a) other linear Fe3 coordination compounds and (b) other E18 Zintl clusters. | ||
Electronic structure
Geometry optimisations using the PBE functional indicate that the lowest energy state for [Fe3Sn18]4− is a spin septet (S = 3), 7A2g, with optimised Fe–Fe bond lengths of 2.45 Å, in excellent agreement with the available X-ray data (Table 1). The Fe–Sn and Sn–Sn bond lengths are also fully consistent with experiment. Despite multiple attempts, we have been unable to measure reproducible magnetic susceptibilities to confirm the paramagnetism of [Fe3Sn18]4−: this likely reflects the challenges in producing a homogeneous sample, and in avoiding oxidative degradation during the course of the experiment. The spin-polarised Kohn–Sham eigenvalues and eigenfunctions of the 7A2g ground state are collected in Fig. 4: levels that are localised primarily on the Fe3 chain are shown in green while those localised primarily on Sn are in grey. The same data, in the form of projected density of states (PDOS) and overlap projected density of states (OPDOS), is presented and discussed in the ESI, Fig. S8.† Of the 100 valence electrons of the cluster, we can identify 22, colored green, that are distributed over the 15 linear combinations of Fe 3d orbitals in Fig. 4 (4eg, 5a1g, 4eu, 7a2u, 6eg, 5eu, 7eg, 8eg and 9a1g in the α set). It is notoriously difficult to assign oxidation formal states in endohedral Zintl clusters, where transition- and main-group metal orbitals are typically well mixed, but the presence of 22 valence electrons indicates a Fe23+ chain, and hence a Sn18 cluster in a −6 oxidation state.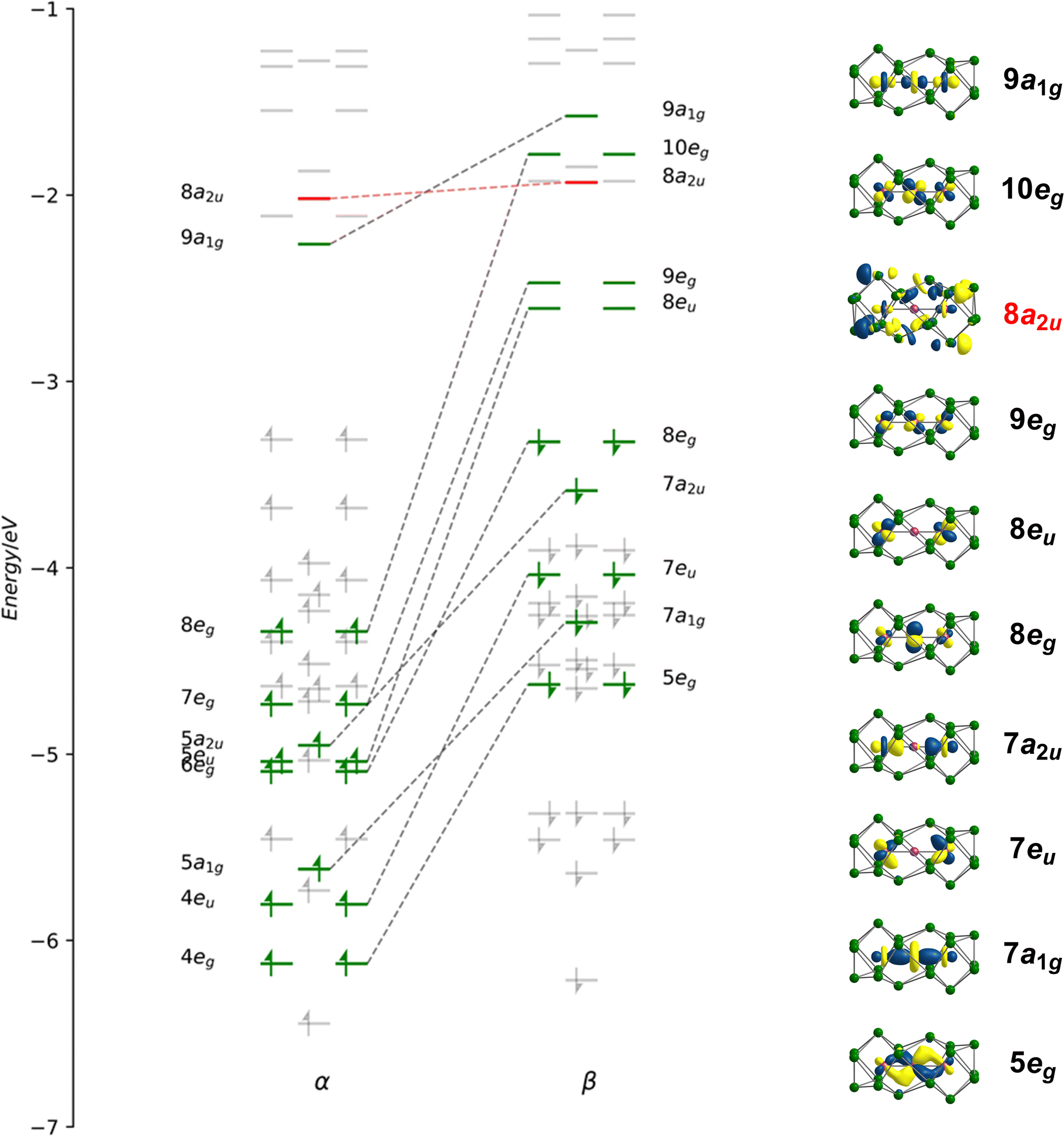 | ||
| Fig. 4 (a) Kohn–Sham orbitals for [Fe3Sn18]4− in its 7A2g ground state. Levels shown in green are the primarily Fe-based orbitals while the remainder, in grey, have dominant Sn character. Orbitals with similar spatial characteristics are joined by a dashed line. | ||
| M1–M2 | M1–E1 | M2–E8 | E2–E8 | E2–E4 | E6–E9 | E8−E9′ | E4−E9′ | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|
| [Fe3Sn18]4− | X-ray (100 K) | 2.4300(9) | 2.882 | 2.689 | 3.690 | 3.001 | 3.004 | 3.460 | 4.10 | This work |
| DFT (7A2g) | 2.45 | 2.98 | 2.70 | 3.64 | 3.07 | 3.04 | 3.50 | 4.22 | ||
| [Ni3Ge18]3− | X-ray (100 K) | 2.395(1) | 2.487 | 2.529 | 3.016 | 2.749 | 2.612 | 3.47 | 4.05 | 15 |
| DFT (1A1g) | 2.43 | 2.50 | 2.52 | 3.09 | 2.76 | 2.66 | 3.29 | 4.01 | ||
| [Ni2CdSn18]6− | X-ray (100 K) | 4.201 | 2.595 | 3.206 | 3.549 | 3.011 | 2.981 | 5.24 | 6.71 | 22 |
| DFT (1A1g) | 4.26 | 2.64 | 3.13 | 3.71 | 3.07 | 3.02 | 5.32 | 6.81 |
Of the 15 metal-based orbitals, only one, the strongly Fe–Fe–Fe σ anti-bonding 9a1g orbital, is vacant in both spin-α and spin-β manifolds, while the complementary σ bonding and non-bonding orbitals, 5a1gα, 7a1gβ and 5a2uα, 7a2uβ, are doubly occupied: the σ2σnb2σ*0 configuration gives a net σ bond order of 0.5 per Fe–Fe bond. Fe–Fe π and δ interactions are mixed in the orbitals of eg and eu symmetry, but the π interactions are primarily contained in 4eg, 4eu and 8eg in the α manifold, 5eg, 7eu and 10eg in β. The prominent positive and negative peaks in the OPDOS shown in ESI, Fig. S8,† corresponding to 5egβ (π bonding) and 10egβ (π antibonding), respectively, confirm the very significant π overlap. The π4πnb4π*2 configuration then defines a formal Fe–Fe π bond order of 0.5 per bond. There are no large peaks in the OPDOS for the orbitals with dominant Fe–Fe δ symmetry (5euα, 7egα, 9egα, 8euβ, 8egβ, 9egβ), so δ bonding can be assumed to be negligible, as might be expected at a distance of 2.4300(9) Å. The overall formal Fe–Fe bond order is therefore 1.0 per Fe–Fe bond made up of 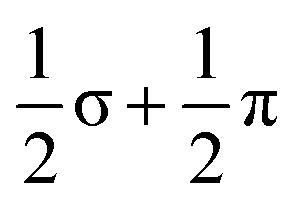 . Returning to the comparison with the coordination complexes [Fe(DpyF)4]2+ (ref. 34) and Fe3L3,32 identified in Fig. 3, the overall oxidation state of the Fe3 unit is lower in [Fe3Sn18]4− (Fe23+vs. Fe63+) and the Fe–Fe σ* orbital is unoccupied, both of which contribute to the lower multiplicity (S = 3 vs. S = 6) and stronger Fe–Fe bonding in the cluster compared to the coordination complexes.
. Returning to the comparison with the coordination complexes [Fe(DpyF)4]2+ (ref. 34) and Fe3L3,32 identified in Fig. 3, the overall oxidation state of the Fe3 unit is lower in [Fe3Sn18]4− (Fe23+vs. Fe63+) and the Fe–Fe σ* orbital is unoccupied, both of which contribute to the lower multiplicity (S = 3 vs. S = 6) and stronger Fe–Fe bonding in the cluster compared to the coordination complexes.
To place the Fe–Fe bonding into the wider context of Zintl cluster chemistry, we can make a connection to Sevov and Goicoechea's [Ni3Ge18]4−, which is a spin singlet with a total valence electron count of 106. The additional six electrons occupy the three doubly degenerate, metal-based, spin-β orbitals, 8euβ, 9egβ and 10egβ, eliminating the π component of the metal–metal bond but leaving the σ2σnb2σ*0 framework intact. The M–M Mayer bond order35 is reduced from 0.95 in [Fe3Sn18]4− to 0.45 in [Ni3Ge18]4−, and the delocalisation index (DI)36 from 0.69 to 0.44, both metrics pointing to a significant π component to the Fe–Fe bond in [Fe3Sn18]4−. The addition of 6 electrons to the metal chain generates a Ni23+ unit and, hence, a Ge18 cluster in the −6 oxidation level. Switching our focus now to the Sn18 cage, we can identify a single vacant orbital, 8a2u, picked out in red in Fig. 4, that has Sn–Sn σ* character between the Sn3 faces bound to the central Fe atom (Sn8−Sn9′ in Fig. 2). This orbital, along with its doubly-occupied Sn–Sn bonding counterpart, generates a 6-center-2-electron bond that links the two Sn9 units.
Cluster fusion vs. cluster oligomerisation
In the previous section we have established a link between the new cluster [Fe3Sn18]4− and [Ni3Ge18]4− through their common oxidation level of −6 for the E18 cluster unit. In this section, we try to identify broader relationships between the family of clusters with 18 tetrel vertices (E18) but rather different structures. Amongst these, we can pick out the two pairs, [Ni2CdSn18]6− and [Ni2InGe18]5−and [Pd2Sn18]4− and [Pd2Ge18]4− shown in Fig. 1 all of which share a common point symmetry, D3d, with [Fe3Sn18]4− and [Ni3Ge18]4−. The oxidation states of the E18 unit in these four clusters are rather easier to establish: in [Pd2Ge18]4− and [Pd2Sn18]4−, the Pd atoms are diamagnetic and can be assigned straightforwardly a formal oxidation state of 0 (d10), leaving the Sn18 unit in a −4 charge state. In [Ni2InGe18]5− and [Ni2CdSn18]6−, in contrast, the diamagnetism implies Ni0, Cd2+ and In3+ (all d10), defining a charge of −8 on the Sn18 unit. Structurally, the two E9 units are separated by more than 4.0 Å in [Ni2InGe18]5− and [Ni2CdSn18]6− but are very tightly compressed in [Pd2Ge18]4− and [Pd2Sn18]4−, where the E18 unit forms a continuous ellipsoidal cage. From a structural perspective, [Fe3Sn18]4− and [Ni3Ge18]4− appear to be precisely intermediate between the two limits, with two partially but not fully coalesced E9 units, consistent with the formal charge assignment of Sn186−.If we wish to analyse the electronic origins of these structural trends we are faced with the immediate problem that in some cases the clusters contain 3 transition metal ions but in others only 2. In order to circumvent this difficulty, we choose to focus on the electronic structure of the empty cage, Sn18, and explore its dependence on charge state: −8 → −6 → −4. The relationship between structure and charge state can be made explicit by the Walsh diagram for the isolated E18 cluster shown in Fig. 5 (calculated using extended Hückel theory). This figure is constructed by extracting the structures of the Sn18 units from DFT optimisations of [Ni2CdSn18]6−, [Fe3Sn18]4− and [Pd2Sn18]4− and interpolating between these three geometries. A comment on the choice of reaction coordinate is necessary here. The fusion of the two Sn9 units proves to be highly asynchronous: the structural impact of the first 2-electron oxidation is very different from the second 2-electron oxidation. In such circumstances, no single structural parameter can adequately capture the changes occurring across the entire spectrum, from 2 × Sn94− on the left to Sn184− on the right. We therefore choose to identify two distinct Sn–Sn distances that serve as independent measures of structural change. The Sn8−Sn9′ distance is closely related to the distance between the centroids of the two Sn9 units, and it varies rapidly as we go from 2 × Sn184− to Sn186−, and then more slowly from Sn186− to Sn184−. The Sn8−Sn9′ distance, in contrast, varies strongly in the left half of the diagram, but is relatively constant as we move from Sn186− to Sn184−. We can, therefore associate the first 2-electron oxidation with a relative motion of the two Sn9 units towards each other, such that both Sn4−Sn9′ and Sn8−Sn9′ contract. The second 2-electron step is then associated almost exclusively with the formation of the Sn4−Sn9′ bonds, with little further change in Sn8−Sn9′.
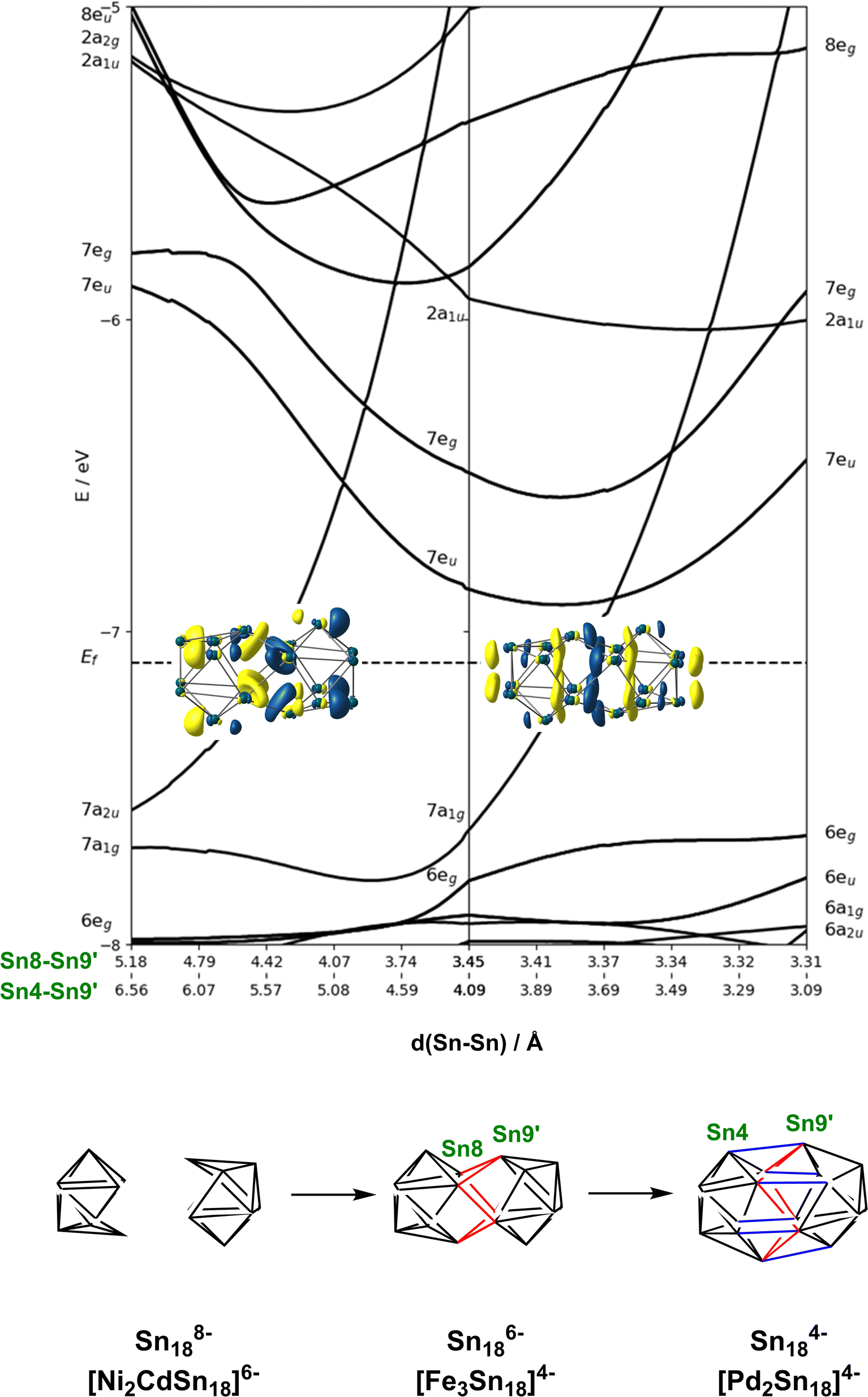 | ||
| Fig. 5 Walsh diagram showing the coalescence of the two Sn9 polyhedra to a single ellipsoidal Sn18 unit. The figure is generated by interpolating between the optimised structures of the Sn18 unit as it is found in the optimised geometries of [Ni2CdSn18]6−, [Fe3Sn18]4− and [Pd2Sn18]4−. | ||
At the separated limit (left hand side of Fig. 5) there is a total of 40 low-lying valence orbitals (up to 7a2u) that can accommodate 80 valence electrons, the count for Sn188−. The transition from this separated limit to the intermediate structure typical of [Fe3Sn18]4− (or [Ni3Ge18]4−) involves a reduction in the separation between the centroids of the two Sn9 units, resulting in contraction of both the Sn8−Sn9′ and Sn4−Sn9′ distances. The result is the rapid destabilisation of a single orbital, 7a2u, that is antibonding across Sn8−Sn9′ – this is the Sn–Sn antibonding orbital discussed previously in the context of Fig. 4 (where it was labelled 8a2u due to the presence of a lower-lying Fe/Ni-based level of the same symmetry that is obviously absent in the empty cluster). In the second step, from the intermediate structure found in [Fe3Sn18]4− to the fully coalesced one in [Pd2Sn18]4−, the Sn4−Sn9′ distance contracts from 4.22 Å to 3.10 Å, causing a rapid destabilisation of a second cluster-based orbital, 7a1g, which is bonding with respect to the Sn8−Sn9′ contact but strongly anti-bonding with respect to Sn8−Sn9′. We note here that Lin and co-workers have also analysed the fusion of two PdSn9 units from the perspective of the ‘principal interacting orbital’ model,37–39 where they identified a σ-symmetry interaction between ‘principal interacting orbitals' localised on the Sn4 and Sn9′ atoms. To the extent that the structurally characterised clusters illustrated in Fig. 1 can be viewed as snapshots of the oxidative coalescence of two separated Sn9 clusters, it seems that the 4-electron oxidation of Sn188− to Sn184− is a rather asynchronous one, with the two units coming together first via the formation of Sn8−Sn9′ bonds (shown in red in Fig. 5), followed by a distinct second 2-electron oxidation step that leads to formation of the Sn4−Sn9′ bonds (shown in blue in Fig. 5), completing the fusion of the two units.§
In the introduction we noted that there is, in principle, a competing pathway for oxidative coupling of Zintl clusters that leads to oligomerisation via exo bond formation rather than fusion to form a single ellipsoidal cage. This is precisely what is observed in the oxidation of [AgSn18]7− (ref. 26) to [AgSn18]5− (Fig. 1) where the charge on the Sn18 unit (assuming a redox non-innocent Ag+ ion) is reduced from −8 to −6, precisely the same as in the [Ni2CdSn18]6− to [Fe3Sn18]4− comparison. What, then, are the factors that determine the preference for fusion of the two Sn9 units in [Fe3Sn18]4− with retention of three-fold rotational symmetry but oligomerisation in [AgSn18]5−? From an electronic perspective, the Sn18 units in the two clusters both have a 2-electron bond linking the two Sn9 units – the only difference is that in [Fe3Sn18]4− this bond is delocalised over 6 Sn–Sn contacts, each with a formal bond order of  , while in [AgSn18]5− it is localised on one. It is possible that the preference for a fused architecture with a 6-center-2-electron bond in [Fe3Sn18]4− (and also in [Ni3Ge18]4−) is connected to the presence of the underlying Fe–Fe–Fe or Ni–Ni–Ni bonded framework, which provides a rigid ‘strut’ that resists the bending at the central metal necessary to form a localised exo Sn–Sn bond. Where metal–metal bonding is absent, as it necessarily is in [AgSn18]5−, bending to form a localised 2-center-2-electron bond is the preferred outcome: a series of DFT calculations on different isomers of [AgSn18]5− confirms a 0.2 eV preference for the bent structure shown in Fig. 1 over the alternative D3d-symmetric [Fe3Sn18]4−-like alternative. Taking the argument a step further, a second 2-electron oxidation step could, in principle, generate clusters with two exo bonds linking the Sn9 units as an alternative to forming the coalesced cage typical of [Pd2Sn18]4− – doubly bonded E9 units of this kind have been identified in the Ge2738,39 and Sn36 nanorod27 where again there is no underlying metal–metal bonded framework to oppose the bending.
, while in [AgSn18]5− it is localised on one. It is possible that the preference for a fused architecture with a 6-center-2-electron bond in [Fe3Sn18]4− (and also in [Ni3Ge18]4−) is connected to the presence of the underlying Fe–Fe–Fe or Ni–Ni–Ni bonded framework, which provides a rigid ‘strut’ that resists the bending at the central metal necessary to form a localised exo Sn–Sn bond. Where metal–metal bonding is absent, as it necessarily is in [AgSn18]5−, bending to form a localised 2-center-2-electron bond is the preferred outcome: a series of DFT calculations on different isomers of [AgSn18]5− confirms a 0.2 eV preference for the bent structure shown in Fig. 1 over the alternative D3d-symmetric [Fe3Sn18]4−-like alternative. Taking the argument a step further, a second 2-electron oxidation step could, in principle, generate clusters with two exo bonds linking the Sn9 units as an alternative to forming the coalesced cage typical of [Pd2Sn18]4− – doubly bonded E9 units of this kind have been identified in the Ge2738,39 and Sn36 nanorod27 where again there is no underlying metal–metal bonded framework to oppose the bending.
Summary and conclusions
In this paper, we have reported the synthesis and structure of a new Zintl-ion cluster, [Fe3Sn18]4−, containing a linear Fe3 chain with short Fe–Fe bond lengths of 2.4300(9) Å. Electronic structure analysis indicates the presence of both Fe–Fe σ and π bonding, with a formal net bond order of 1.0 (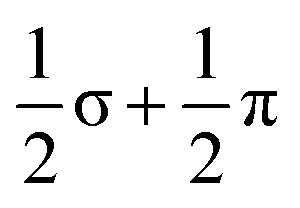 ) per Fe–Fe bond. The cluster is structurally similar to the [Ni3Ge18]4− anion reported previously by Sevov and co-workers, although the Ni–Ni bonding in that case lacks the π component. The structure of the Sn18 unit in [Fe3Sn18]4− is intermediate between that in [Ni2CdSn18]6−, where the two Sn18 units are almost completely separated, and [Pd2Sn18]4−, where they are completely fused to form a continuous ellipsoidal Sn18 unit. These structural differences correlate with the redox level of the cage, with successive 2-electron oxidations starting from Sn188− leading first to partial fusion of the two cages and then to their complete coalescence. One of the obvious challenges in forming ever larger Zintl ions from smaller fragments is that the latter carry high negative charges, so their close approach necessarily involves a substantial coulomb barrier. The identification of [Fe3Sn18]4− as an intermediate stage of cluster fusion presents the intriguing possibility that the central metal cation may act as a buffer, templating the close approach of the anionic components. Transfer of electron density from the main-group cage to the transition metal may then drive the fusion of the two polyhedral fragments, with concomitant reduction of the cations and their expulsion as metal atoms, as is observed, for example, in NaSi, where elevated pressures lead to the formation of Na metal with concomitant amorphisation of the Si4 cluster units through Si–Si bond formation.40 The presence of Fe–Fe bonding appears to play an important role in this process by preventing bending at the central metal atom, directing the reaction towards cluster fusion rather than the competing oxidative oligomerisation observed in the [AgSn18]7−/5− pair.
) per Fe–Fe bond. The cluster is structurally similar to the [Ni3Ge18]4− anion reported previously by Sevov and co-workers, although the Ni–Ni bonding in that case lacks the π component. The structure of the Sn18 unit in [Fe3Sn18]4− is intermediate between that in [Ni2CdSn18]6−, where the two Sn18 units are almost completely separated, and [Pd2Sn18]4−, where they are completely fused to form a continuous ellipsoidal Sn18 unit. These structural differences correlate with the redox level of the cage, with successive 2-electron oxidations starting from Sn188− leading first to partial fusion of the two cages and then to their complete coalescence. One of the obvious challenges in forming ever larger Zintl ions from smaller fragments is that the latter carry high negative charges, so their close approach necessarily involves a substantial coulomb barrier. The identification of [Fe3Sn18]4− as an intermediate stage of cluster fusion presents the intriguing possibility that the central metal cation may act as a buffer, templating the close approach of the anionic components. Transfer of electron density from the main-group cage to the transition metal may then drive the fusion of the two polyhedral fragments, with concomitant reduction of the cations and their expulsion as metal atoms, as is observed, for example, in NaSi, where elevated pressures lead to the formation of Na metal with concomitant amorphisation of the Si4 cluster units through Si–Si bond formation.40 The presence of Fe–Fe bonding appears to play an important role in this process by preventing bending at the central metal atom, directing the reaction towards cluster fusion rather than the competing oxidative oligomerisation observed in the [AgSn18]7−/5− pair.
Experimental section
Materials and reagents
All manipulations and reactions were performed under a nitrogen atmosphere using standard Schlenk or glovebox techniques. Ethylenediamine (en) (Aldrich, 99%) and DMF (Aldrich, 99.8%) were freshly distilled by CaH2 prior to use, and stored in N2 prior to use. Tol (Aldrich, 99.8%) was distilled from sodium/benzophenone under nitrogen and stored under nitrogen. 2.2.2-crypt (4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo (8.8.8) hexacosane, purchased from Sigma-Aldrich, 98%) was dried in vacuum for one day prior to use. K4Sn9 was synthesised by heating a stoichiometric mixture of the elements (K: +99% and Sn: 99.99% all from Aladdin) at 850 °C for 36 h in a niobium tube. [K(thf)Fe(OtBu)3]2 was prepared according to literature methodology.41Synthesis
X-ray crystallography
Crystallographic data for 1 were collected on Rigaku XtalAB Pro MM007 DW diffractometer with graphite monochromated Cu Kα radiation (λ = 1.54184 Å). The crystal structure was solved using direct methods and then refined using SHELXL-2014 (ref. 42) and Olex2,43 with all non-hydrogen atoms refined anisotropically during the final cycles. All hydrogen atoms of the organic molecule were placed by geometrical considerations and were added to the structure factor calculation. The SQUEEZE procedure44 to remove the solvent molecules which could not be modeled properly. A summary of the crystallographic data for the title compounds is presented in the ESI, Tables S1 and S2.† These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.Computational details
All calculations are performed using density functional theory as implemented in the ADF 2021.104 package.45 The Perdew–Burke–Ernzerhof (PBE) functional46 was used and triple-zeta basis sets included with two polarization functions are used for all atoms.47 All electrons are treated as valence in the calculations. Relativistic effects were incorporated using the Zeroth-Order Regular Approximation (ZORA).48 A Conductor-like Screening Model (COSMO) with dielectric constant of 78.39 was used to simulate the confining environment of the ionic lattice.49 A fine numerical grid was used for the integrations (grid setting ‘verygood’),50 and the calculations were considered converged when the commutator of the Fock and density matrices was below 10−6. Optimized structures were confirmed to be minima through the absence of imaginary frequencies.51 The sensitivity of the results to choice of functional was also explored by repeating the calculations using the M06-L and PBE0 functionals.52,53 The Walsh diagram in Fig. 5 was calculated using Extended Hückel theory with the following parameters for the 5s and 5p valence orbitals of Sn: 5s Hii = −16.16 eV, ζ = 2.30, 5p Hii = −7.32 eV, ζ = 2.00. The reaction coordinate was defined by interpolating between the structures of the Sn18 unit as it is found in the optimised structures of [Ni2CdSn18]6−, [Fe3Sn18]4− and [Pd2Sn18]4−. A python script to perform these calculations is available on request.Data availability
Crystallographic data are available from the CCDC. Full details of optimised geometries are summarised in the ESI.† Input files are available from the authors on request.Author contributions
W.-X. C. and C.-C. S. performed the synthesis and characterisation, Z. L. and H. W. T. M. performed the computational analysis. Z.-M. S. and J. E. M. conceived the project and supervised the experimental and computational aspects of the research, respectively. All authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 92161102 and 22371140), the Natural Science Foundation of Tianjin City (no. 21JCZXJC00140) and the PhD Candidate Research Innovation Fund of NKU School of Materials Science and Engineering, and 111 project (B18030) from China (MOE).References
- J. Zhao, Q. Du, S. Zhou and V. Kumar, Chem. Rev., 2020, 120, 9021–9163 CrossRef CAS PubMed.
- B. Weinert and S. Dehnen, in Binary and Ternary Intermetalloid Clusters, ed. S. Dehnen, Springer International Publishing, Cham, 2017, pp. 99–134 Search PubMed.
- B. Oelkers, M. V. Butovskii and R. Kempe, Chem.–Eur. J., 2012, 18, 13566–13579 CrossRef CAS PubMed.
- N. Lichtenberger, R. J. Wilson, A. R. Eulenstein, W. Massa, R. Clérac, F. Weigend and S. Dehnen, J. Am. Chem. Soc., 2016, 138, 9033–9036 CrossRef CAS PubMed.
- X. Min, I. A. Popov, F.-X. Pan, L.-J. Li, E. Matito, Z.-M. Sun, L.-S. Wang and A. I. Boldyrev, Angew. Chem., Int. Ed., 2016, 55, 5531–5535 CrossRef CAS PubMed.
- D. M. P. Mingos, in Electron Counting Rules for Gold Clusters Which Are Stereochemically Non-rigid and Exhibit Skeletal Isomerism, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2021, pp. 1–67 Search PubMed.
- F. Gam, J. Wei, S. Kahlal, J.-Y. Saillard and J.-F. Halet, in Electron Counting in Ligated High Nuclearity Late Transition Metal Clusters, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2021, pp. 69–102 Search PubMed.
- W. Klein, A. Schier and T. F. Fässler, in Molecules Meet Solids: From Wade–Mingos Clusters to Intermetalloid Clusters, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2021, pp. 149–195 Search PubMed.
- F. Pan, B. Weinert and S. Dehnen, in Binary Zintl Anions Involving Group 13–15 (Semi-)Metal Atoms, and the Relationship of Their Structures to Electron Count, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2021, pp. 103–148 Search PubMed.
- L. A. O. P. Braunstein and P. R. Raithby, in Metal Clusters in Chemistry, Wiley-VCH, 1999 Search PubMed.
- C. Dong, Y. Li, D. Cheng, M. Zhang, J. Liu, Y.-G. Wang, D. Xiao and D. Ma, ACS Catal., 2020, 10, 11011–11045 CrossRef CAS.
- E. N. Esenturk, J. Fettinger and B. Eichhorn, J. Am. Chem. Soc., 2006, 128, 9178–9186 CrossRef CAS PubMed.
- A. Spiekermann, S. Hoffmann, T. Fässler, I. Krossing and U. Preiss, Angew. Chem., Int. Ed., 2007, 46, 5310–5313 CrossRef CAS PubMed.
- H.-L. Xu, L. Qiao and Z.-M. Sun, Chin. J. Chem., 2023, 41, 2432–2438 CrossRef CAS.
- J. M. Goicoechea and S. C. Sevov, Angew. Chem., Int. Ed. Engl., 2005, 44, 4026–4028 CrossRef CAS PubMed.
- J. M. Goicoechea and S. C. Sevov, J. Am. Chem. Soc., 2005, 127, 7676–7677 CrossRef CAS PubMed.
- S. C. Sevov and J. M. Goicoechea, Organometallics, 2006, 25, 5678–5692 CrossRef CAS.
- S. Mitzinger, L. Broeckaert, W. Massa, F. Weigend and S. Dehnen, Nat. Commun., 2016, 7, 10480 CrossRef CAS PubMed.
- A. Ugrinov and S. C. Sevov, C. R. Chim., 2005, 8, 1878–1882 CrossRef CAS.
- J.-Q. Wang, B. Wahl and T. Fässler, Angew. Chem., Int. Ed., 2010, 49, 6592–6595 CrossRef CAS PubMed.
- C. Zhang, H. W. T. Morgan, Z.-C. Wang, C. Liu, Z.-M. Sun and J. E. McGrady, Dalton Trans., 2019, 48, 15888–15895 RSC.
- Y.-N. Yang, L. Qiao and Z.-M. Sun, Chin. Chem. Lett., 2023, 34, 107207 CrossRef CAS.
- Z.-M. Sun, H. Xiao, J. Li and L.-S. Wang, J. Am. Chem. Soc., 2007, 129, 9560–9561 CrossRef CAS PubMed.
- D. F. Hansen, B. Zhou and J. M. Goicoechea, J. Organomet. Chem., 2012, 721–722, 53–61 CrossRef CAS.
- L. Lin, Y. Wang and L. Xu, Organometallics, 2022, 41, 450–455 CrossRef CAS.
- F. S. Geitner, W. Klein and T. F. Fässler, Dalton Trans., 2017, 46, 5796–5800 RSC.
- N. V. Tkachenko, W.-X. Chen, H. W. T. Morgan, A. Muñoz-Castro, A. I. Boldyrev and Z.-M. Sun, Chem. Commun., 2022, 58, 6223–6226 RSC.
- A. Ugrinov and S. C. Sevov, J. Am. Chem. Soc., 2002, 124, 10990–10991 CrossRef CAS PubMed.
- L. Yong, S. D. Hoffmann and T. F. Fässler, Z. Anorg. Allg. Chem., 2005, 631, 1149–1153 CrossRef CAS.
- T. Nguyen, W. Merrill, C. Ni, H. Lei, J. Fettinger, B. Ellis, G. Long, M. Brynda and P. Power, Angew. Chem., Int. Ed. Engl., 2008, 47, 9115–9117 CrossRef CAS PubMed.
- N. Fujima and T. Oda, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 115412 CrossRef.
- G. L. Guillet, K. Y. Arpin, A. M. Boltin, J. B. Gordon, J. A. Rave and P. C. Hillesheim, Inorg. Chem., 2020, 59, 11238–11243 CrossRef CAS PubMed.
- F. Cotton, C. A. Murillo and X. Wang, Inorg. Chem. Commun., 1998, 1, 281–283 CrossRef CAS.
- A. Srinivasan, R. A. Musgrave, M. Rouzières, R. Clérac, J. E. McGrady and E. A. Hillard, Chem. Commun., 2021, 57, 13357–13360 RSC.
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS.
- J. Poater, M. Solà, M. Duran and X. Fradera, Theor. Chem. Acc., 2002, 107, 362–371 Search PubMed.
- F. K. Sheong, W.-J. Chen, J.-X. Zhang, Y. Li and Z. Lin, Dalton Trans., 2017, 46, 2214–2219 RSC.
- J.-X. Zhang, F. K. Sheong and Z. Lin, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2020, 10, e1469 CrossRef CAS.
- J.-X. Zhang, F. K. Sheong and Z. Lin, Chem.–Eur. J., 2018, 24, 9639–9650 CrossRef CAS PubMed.
- R. Q. Cabrera, A. Salamat, O. I. Barkalov, O. Leynaud, P. Hutchins, D. Daisenberger, D. Machon, A. Sella, D. W. Lewis and P. F. McMillan, J. Solid State Chem., 2009, 182, 2535–2542 CrossRef CAS.
- Y. Xia, PhD thesis, Soochow University, Suzhou, China, 2012.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- E. Van Lenthe and E.-J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- E. van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 Search PubMed.
- M. Franchini, P. H. T. Philipsen and L. Visscher, J. Comput. Chem., 2013, 34, 1819–1827 CrossRef CAS PubMed.
- L. Versluis and T. Ziegler, J. Chem. Phys., 1988, 88, 322–328 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the X-ray crystallography, further details of the computational experiments including a list of optimised coordinates of all clusters discussed. CCDC 2170116. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04709a |
| ‡ These authors contributed equally |
| § [Fe3Sn18]2− was identified as a prominent peak in the ESI-MS in Fig. 2. This cluster is formed by 2-electron oxidation of the [Fe3Sn18]4− and so, if the oxidation is Sn-based, the cage is valence isoelectronic with that in [Pd2Sn18]4−. However, the optimised structure does not show a completely coalesced cage, but rather one that is similar to [Fe3Sn18]4− despite the 2-electron oxidation. The presence of the Fe3 unit clearly prevents the close approach of the two Sn9 units that is possible for [Pd2Sn18]4−. |
| This journal is © The Royal Society of Chemistry 2024 |