
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe allosteric mechanism of mTOR activation can inform bitopic inhibitor optimization†
Yonglan
Liu
 a,
Mingzhen
Zhang
b,
Hyunbum
Jang
b and
Ruth
Nussinov
*bc
a,
Mingzhen
Zhang
b,
Hyunbum
Jang
b and
Ruth
Nussinov
*bc
aCancer Innovation Laboratory, National Cancer Institute, Frederick, MD 21702, USA
bComputational Structural Biology Section, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA. E-mail: NussinoR@mail.nih.gov; Tel: +1-301-846-5579
cDepartment of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel
First published on 7th December 2023
Abstract
mTOR serine/threonine kinase is a cornerstone in the PI3K/AKT/mTOR pathway. Yet, the detailed mechanism of activation of its catalytic core is still unresolved, likely due to mTOR complexes' complexity. Its dysregulation was implicated in cancer and neurodevelopmental disorders. Using extensive molecular dynamics (MD) simulations and compiled published experimental data, we determine exactly how mTOR's inherent motifs can control the conformational changes in the kinase domain, thus kinase activity. We also chronicle the critical regulation by the unstructured negative regulator domain (NRD). When positioned inside the catalytic cleft (NRD IN state), mTOR tends to adopt a deep and closed catalytic cleft. This is primarily due to the direct interaction with the FKBP–rapamycin binding (FRB) domain which restricts it, preventing substrate access. Conversely, when outside the catalytic cleft (NRD OUT state), mTOR favors an open conformation, exposing the substrate-binding site on the FRB domain. We further show how an oncogenic mutation (L2427R) promotes shifting the mTOR ensemble toward the catalysis-favored state. Collectively, we extend mTOR's “active-site restriction” mechanism and clarify mutation action. In particular, our mechanism suggests that RMC-5552 (RMC-6272) bitopic inhibitors may benefit from adjustment of the (PEG8) linker length when targeting certain mTOR variants. In the cryo-EM mTOR/RMC-5552 structure, the distance between the allosteric and orthosteric inhibitors is ∼22.7 Å. With a closed catalytic cleft, this linker bridges the sites. However, in our activation mechanism, in the open cleft it expands to ∼24.7 Å, offering what we believe to be the first direct example of how discovering an activation mechanism can potentially increase the affinity of inhibitors targeting mutants.
Introduction
A structural/functional overview
The mechanistic target of rapamycin (mTOR) plays a crucial role in cell growth and homeostasis.1–3 It belongs to the PI3K-related protein kinase (PIKK) family which shares a complex domain organization.4,5 mTOR consists primarily of four parts: the N-terminal HEAT (N-HEAT) (Huntingtin, EF3A, ATM, TOR) and middle HEAT (M-HEAT) repeats, the FAT domain (coined after FRAP, ATM, and TRRAP), and the kinase domain (Fig. S1a†).6–9 Together with specific proteins, mTOR assembles into two functional complexes (mTORCs), mTORC1 and mTORC2 (Fig. S1b†).10–16 The complexes share an accessory protein, mammalian lethal with SEC13 protein 8 (mLST8), but differ in other components.17–19 Regulatory-associated protein of mTOR (Raptor) is unique to mTORC1, while Raptor-independent companion of mTOR (Rictor) to mTORC2. mTORC1 is a dimer of heterotrimers containing three basic proteins, mTOR, Raptor, and mLST8, while mTORC2 is a dimer of heterotetramers with four conserved subunits, mTOR, Rictor, mLST8, and mammalian stress-activated map kinase interacting protein 1 (mSIN1).20 mTORCs' activation can be triggered through the PI3K/AKT/mTOR pathway.1,21–23 When growth factors such as insulin bind to receptor tyrosine kinases (RTKs), PI3K is activated, converting lipid phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3).24 PIP3-mediated activation of PDK1 stimulates AKT phosphorylation, which subsequently inhibits a negative regulator, the tuberous sclerosis complex (TSC).25–28 In signaling, TSC acts as the GTPase activating protein (GAP) and inhibits GTP-bound Ras homolog enriched in brain protein (RHEB, a member of the Ras superfamily).29,30 Thus, AKT-mediated inhibition of TSC promotes RHEB activation. Active RHEB binds to the FAT domain of mTOR in mTORC1, allosterically changing the alignment of the active-site residues in the kinase domain in favor of catalysis. Full activation of mTORC1 requires the uptake of amino acids, such as Arg, Leu, and Glu, which regulate the recruitment of mTORC1 to lysosomes and endosomes through the Rag GTPases-mediated pathway.1,19,31–33 mTORC2 is predominately mediated by growth factor recruitment. Binding of mSIN1 to PIP3 can release mTORC2 inhibition. mTORC2 activity can be tuned by the negative feedback of mTORC1. Ribosomal protein S6 kinase 1 (S6K1), a substrate of mTORC1, can phosphorylate insulin receptor substrate 1 (IRS1), which then attenuates the PI3K/AKT signaling, thus mTORC2 activity.34The classes and status of mTOR inhibitors
Studies associated constitutive mTORCs' activation with cancer.22,35–40 Alterations of mTOR regulators in the PI3K/AKT/mTOR pathway, as well as mTOR itself, can stimulate signaling, triggering cell growth and proliferation,7,41–47 impacting neurodevelopmental disorders,48 neurodegenerative diseases, and type 2 diabetes.14,34 The activation of the PI3K/AKT/mTOR pathway imparts non-single residue mutational resistance in acute lymphoblastic leukemia (ALL) linked to the oncoprotein Bcr-Abl1, which is encoded by the fusion BCR-ABL1 gene in the Philadelphia chromosome.49 Suppressing aberrant mTOR can extend life and healthspan.3,16,50,51 mTOR inhibitors can be classified into three generations. The first is rapalogs, the variants of rapamycin. Notable successes of rapalogs include temsirolimus (Torisel) by Wyeth, everolimus (Afinitor) by Novartis, and ridaforolimus by Merck, with a bulkier group replacing the hydroxyl group at C40 of rapamycin. The U.S. Food and Drug Administration (FDA) has approved temsirolimus for treating renal cell cancer and everolimus for treating breast cancer, renal cell carcinoma, and neuroendocrine carcinoma. Ridaforolimus' effectiveness is under evaluation in clinical trials. These drugs bind to the interfacial pocket between the FKBP12-rapamycin-binding (FRB) domain of mTOR and the 12 kDa FK506-binding protein (FKBP12), which appears to cause steric hindrance and obstruct the active-site cleft.4,52 The drug prefers binding to mTORC1 over mTORC2.53,54 In mTORC2, the rapamycin/FKBP12 binding site is occluded by Rictor's C-terminal domain, making it insensitive to rapamycin and rapalogs. However, under prolonged treatment, rapamycin can inhibit mTORC2.21 Side effects limit rapalogs' broad clinical applications.55 In addition to rapalogs, there are non-rapalog allosteric inhibitors of mTOR. These inhibitors are specifically engineered to target the same binding site as rapamycin or rapalogs, but they exhibit distinctive interaction patterns.56 Like other kinase inhibitors, the second generation binds to the ATP-binding pocket of the kinase domain in mTORC1 and mTORC2,57 but often lack selectivity. Examples encompass sapanisertib (MLN0128, INK-128 or TAK-128), vistusertib (AZD2014), AZD8055, torkinib (PP 242), torin-1, torin-2, OSI-027 (ASP7486), and PP30. The dual PI3K/mTOR inhibitors effectively inhibit both PI3K and mTOR due to the catalytic domains' structural similarities.58 Illustrative instances include omipalisib (GSK2126458), dactolisib (NVP-BEZ235), gedatolisib (PF-05212384), apitolisib (GDC-0980), bimiralisib (PQR309), voxtalisib (SAR245409), panulisib (P7170), and PI-103. The combination regimen involving both allosteric and orthosteric drugs has been recognized as having increased potential in effectively suppressing mTOR activity, making it a valuable approach for overcoming drug resistance.59–61 The third generation, known as rapalinks, are bitopic inhibitors that combine features of the first and second generations. Rapalinks can simultaneously occupy the cavity between the mTOR FRB domain and FKBP12, and the ATP-binding site.34,61–65 This dual binding mechanism enhances the affinity and selectivity of the inhibitors towards mTOR.mTOR suppresses catalytic activity by restricting substrate access to the active site
The obligate cellular localization and substrate recruitment of mTORCs rely on their components.9,66 X-ray and cryo-EM structures of mTORC1 and mTORC2 provided structural insights into their regulation,2,4,14,53,67–69 and studies largely focused on mTORCs' ability to sense substrate signals. For both complexes, mTOR is the catalytic core. A recent crystal structure featured a complex of N-terminal truncated mTOR (ΔNmTOR) with mLST8, a subunit of both mTORC1 and mTORC2 (ΔNmTOR/mLST8).4 mLST8 is a scaffolding protein for mTORC2 cofactors Rictor and mSIN1 but apparently not critical for mTORC1.70 The crystal structure suggested that mTOR suppressed the catalytic activity by restricting substrate access to the active site.4 This mechanism resembles those of PI3K and SHP2.24,71 Two factors contribute to the restricted access. First, the extensions of the N-lobe by the mTOR FRB domain and the C-lobe by mLST8 generate a deep “V-shape” catalytic cleft. Second, the kα9b-helix (residues 2425–2436) and the negative regulator domain (NRD, residues 2430–2450) block the active site. The static crystal structure could not capture the conformational dynamics required to precisely discern how these domains and motifs orchestrate the conformational changes of mTOR for its kinase activity, in the wild type and the mutant.72,73Here we compiled cellular and medicinal experimental data and performed extensive molecular dynamics (MD) simulations to establish and depict the mechanism of the intrinsic activation of mTOR. We then apply this mechanism to describe how oncogenic mutations of mTOR can promote its activation, and the drug actions. Building upon this mechanism, we suggest a drug strategy that is promising to effectively target the mTOR variants.
Results
Description of ΔNmTOR/mLST8, and comparison of mTOR with PI3K
The crystal structure (PDB ID: 4JSP) of ΔNmTOR/mLST8 has a compact architecture (Fig. 1a).4 ΔNmTOR (residues 1376–2549) consists of the FAT domain with most α-helical repeats forming a “C-shape” α-solenoid conformation and the kinase domain with a typical bilobate conformation. The FAT domain winds around half of the kinase domain. The mTOR kinase domain has approximately 300 residues more than those of canonical kinases (Fig. 1b). The kinase domain contains three inserted domains: the N-lobe inserted FRB (residues 2021–2118) domain, the C-lobe inserted Lst8 binding element (LBE, residues 2258–2296) domain, and the C-lobe inserted FAT C-terminal (FATC, residues 2517–2549) domain.9 The FRB domain may operate as a gatekeeper, providing a specific binding site for both rapamycin and substrates (Fig. 1c).2,4 The LBE domain is bound to mLST8. The FATC domain appears to be indispensable for C-lobe integration, packing beside the activation loop (A-loop). In addition to these three inserted domains (FRB, LBE, and FATC), the mTOR kinase domain has other inserted motifs. Within the N-lobe, there are insertions of a kinked helix (kα1) immediately before FRB (Fig. S2a†), and a β-strand (kβ0) and two short helices (kα2 and kα2b) right after FRB (Fig. S2b†). The kα1-helix is a conserved motif in other PIKKs and PI3K.4,74,75 In PI3K, the kα1-helix is followed by the kα2. The position of kα2 in PI3K is taken by kβ0, kα2, and kα2b in mTOR, likely as an anchor for the FRB domain onto the N-lobe. Within the C-lobe of the mTOR kinase domain, kα9b is also embedded, together with the LBE and FATC domains forming an A-loop-centered interaction spine in the vicinity of the active site (Fig. S2c†). Analogous to class III PI3KC, the A-loop displays an ordered helix (kαAL). Unlike the A-loop in the inactive conformation of those canonical kinases, kαAL within the A-loop of mTOR does not block the active site. The four inserted helices, ka3, kα9, kα9b, and kα10, form a compact structure centered on kα9b (Fig. S2d†).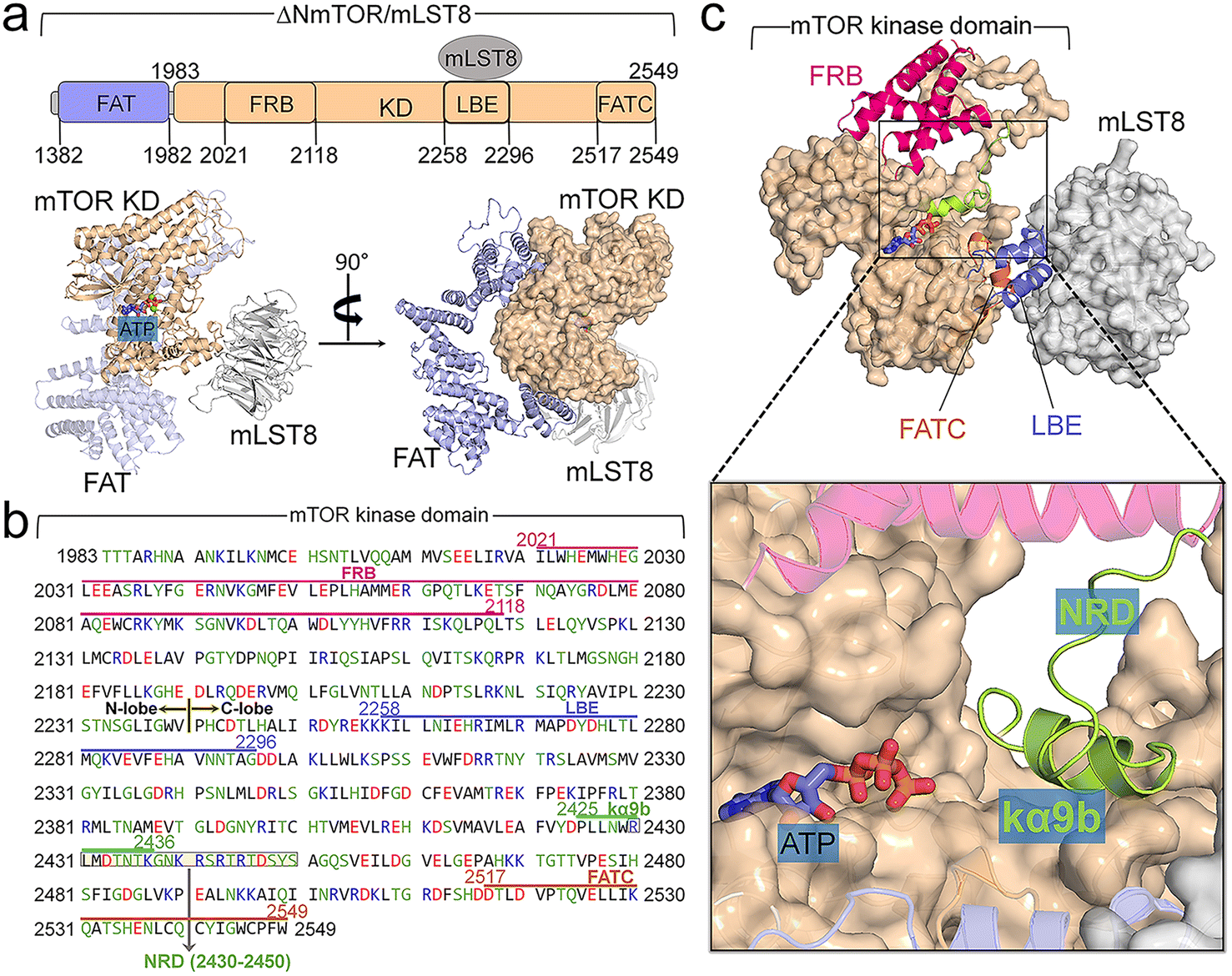 | ||
| Fig. 1 The mTOR domain, sequence, and structure. (a) Domains and structure of ΔNmTOR/mLST8. (b) The sequence of the kinase domain of mTOR. There are several important inserted domains or motifs within the kinase domain of mTOR: the FRB domain (residues 2021–2118) in the N-lobe, and the LBE domain (residues 2258–2296), the kα9b-helix (residues 2425–2436), the NRD (residues 2430–2450), and the FATC domain (residues 2517–2549) in the C-lobe. (c) Conformation of the catalytic cleft of mTOR with the kα9b-helix and NRD inside the catalytic cleft, blocking the active site. | ||
The αC-helix of mTOR in both mTORC1 and mTORC2 is in the “IN” conformation,4,14,69,76 characteristic of active kinases. The crystal structure of ΔNmTOR/mLST8 shows that mTOR inhibition is primarily due to the blocked catalytic cleft,4,76,77 resembling PI3K, whose active site is blocked by the iSH2 domain. PI3K activation is initiated with nSH2 release, followed by the rotation of iSH2 and the exposure of the active site.71 In mTOR, the kα9b-helix and NRD inside the catalytic cleft can obstruct substrate access, suppressing its kinase activity. Experimental data showed that NRD removal greatly enhanced the basal activity of mTOR in vitro and in vivo,78–80 supporting the critical role of NRD in the regulation. The C-terminal of the kα9b-helix partially overlaps the N-terminal of NRD (Fig. 1b). In this work, we aim to unravel the activation mechanism of the mTOR core, not considering mTOR effectors, such as Raptor, Rictor, and mSIN1. Experimental data indicated that the ΔNmTOR/mLST8 complex has kinase activity comparable to mTORC1,4 indicating that it is a functional complex. Thus, we constructed the simulation models based on the crystal structure of the ΔNmTOR/mLST8 complex. Since mTORC1 and mTORC2 share the same binding posture between mTOR and mLST8 (Fig. S3†),14,69 we expect the elucidated mechanism to explain the basic activation of mTOR for both complexes.
Activation mechanism of mTOR by NRD release
To explore how NRD affects mTOR activity, we constructed two ΔNmTOR/mLST8 complex models, ΔNmTORWT/mLST8 with NRD in the catalytic cleft (termed IN NRD) and ΔNmTORΔNRD/mLST8 in the absence of NRD and performed MD simulations (Table S1†). During the simulations, we observed that mTOR in both systems underwent conformational changes in the kinase domain, mainly in the “V-shape” catalytic cleft. However, their changes differed. To characterize these changes, we measured the distance of FRB–LBE (dFRB–LBE) (Fig. S4a†) and the angle of FRB–ATP–LBE (θFRB–ATP–LBE) (Fig. S4b†) along the trajectories for these two systems. In ΔNmTORWT/mLST8, dFRB–LBE and θFRB–ATP–LBE consistently decrease, ultimately populated at ∼23 Å (Fig. 2a) and ∼52° (Fig. 2b), respectively, which are much smaller than those (∼31 Å and ∼67°) in the crystal structure, eventually resulting in a deeper and narrower catalytic cleft (Fig. 2c, left). In sharp contrast, the catalytic cleft of mTOR in the ΔNmTORΔNRD/mLST8 complex was more prone to open (Fig. 2c, right). The removal of NRD resulted in dFRB–LBE and θFRB–ATP–LBE peaking at ∼36 Å and ∼73°, respectively, much larger than for the ΔNmTORWT/mLST8 complex with NRD inside the catalytic cleft. The FRB domain harbors the second substrate binding site.4 This conformational change may favor the recruitment of the substrate at the FRB site (Fig. S5†).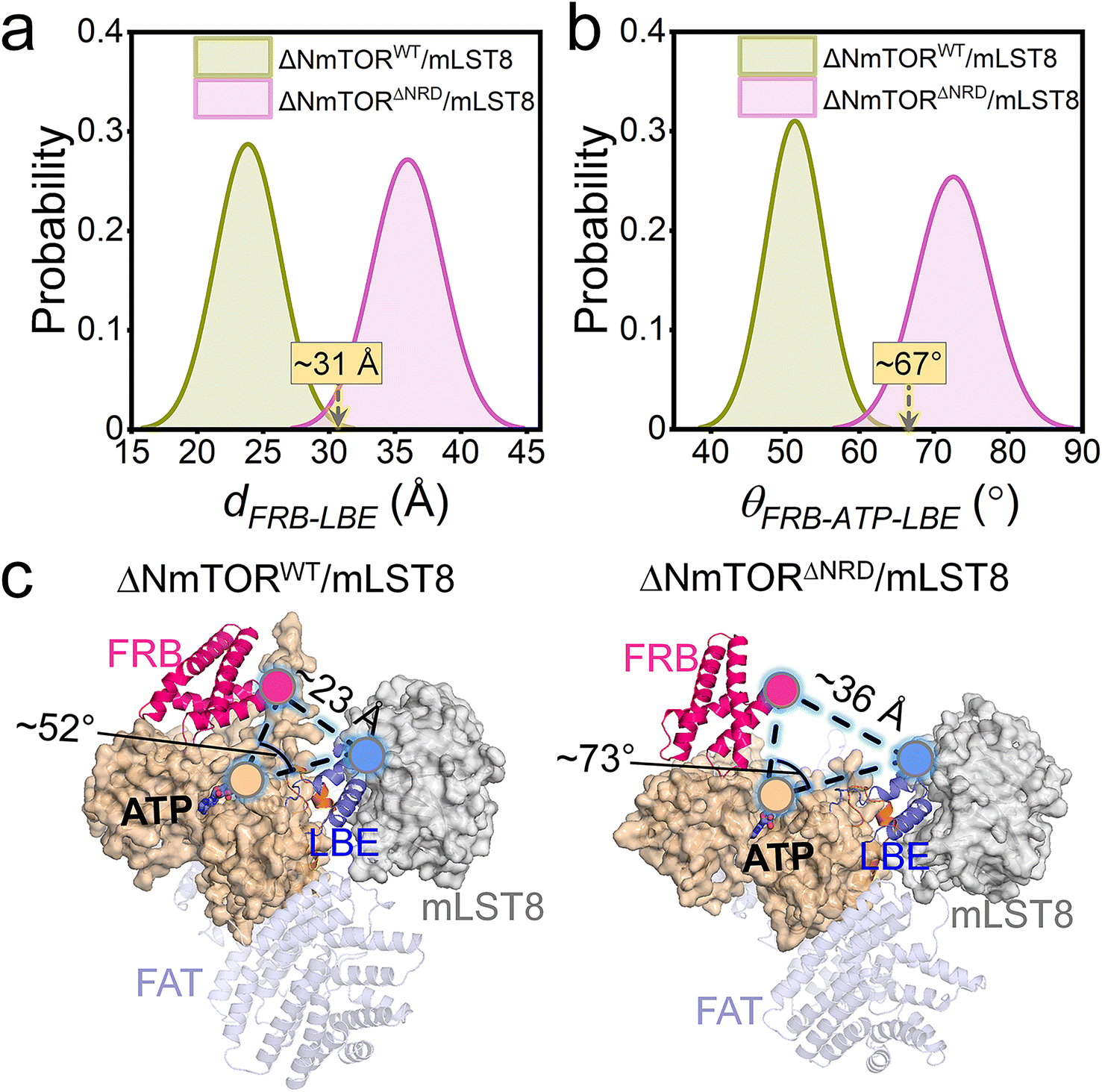 | ||
| Fig. 2 NRD regulation of the mTOR kinase domain. Probability distributions of (a) the distance between the FRB and LBE domains (dFRB–LBE) and (b) the angle of FRB–ATP–LBE (θFRB–ATP–LBE), and (c) final snapshots to represent dFRB–LBE and θFRB–ATP–LBE of mTOR in ΔNmTORWT/mLST8 and ΔNmTORΔNRD/mLST8. dFRB–LBE is the distance between the Cα atom of residue M2039 in the FRB domain and the Cα atom of residue M2277 in the LBE domain. θFRB–ATP–LBE is the angle between the vector from the phosphorus atom of the γ-phosphate of ATP to the Cα atom of residue M2039 in the FRB domain and the vector from the phosphorus atom of the γ-phosphate of ATP to the Cα atom of residue M2277 in the LBE domain. | ||
These observations were consistent with the principal component analysis (PCA). The first two principal components (PCs) of the PCA for the kinase domains of mTOR in both ΔNmTORWT/mLST8 and ΔNmTORΔNRD/mLST8 were clustered according to three timeframes, 0–500 ns, 500–1000 ns, and 1000–1500 ns (Fig. 3a), which reflect the motion patterns of the kinase domains during different periods. Overall, the kinase domain of mTOR in ΔNmTORWT/mLST8 exhibits relatively light variations, as evidenced by the considerable overlap of the three clusters of the first two PCs. For ΔNmTORWT/mLST8, in the first 500 ns, the main change was the loss of half of the helical component of the kα9b-helix, which couples with NRD approaching the FRB domain (Fig. 3b). This leads to the formation of some salt bridges between the positively charged residues (K2440, R2241, R2443) in NRD and the negatively charged residues (E2032 and E2033) in the FRB helix (residues 2022–2040) (Fig. S6†). Such interactions favor the FRB domain getting closer to the LBE domain (or mLST8), giving rise to a closed catalytic cleft. This conformation persisted for 500–1500 ns. In sharp contrast, the ΔNmTORΔNRD/mLST8 complex displayed a remarkably sparse distribution of the first two PCs, indicating that NRD release results in a remarkable variation in the mTOR kinase domain (Fig. 3a). In ΔNmTORΔNRD/mLST8, the FRB domain initially underwent a weak rotation. Without restriction from NRD, the FRB domain moved away from the LBE domain (or mLST8), significantly enlarging the entrance of the catalytic cleft and shallowing it (Fig. 3c). Simultaneously, the helical components of kα9b entirely disappeared, and the region after NRD shifted out and moved away from the catalytic cleft. These observations support the essential role of NRD in regulating mTOR activity. Importantly, the IN NRD not only directly blocks the active site but also serves as the determinant for maintaining a deep and closed catalytic cleft through its restriction of the FRB domain, thereby prohibiting substrate access to the active site. Upon NRD release, the FRB domain will move, giving rise to an open catalytic cleft and making the secondary substrate-binding site in the FRB domain more accessible, increasing the probability of substrate recruitment and access to the active site.
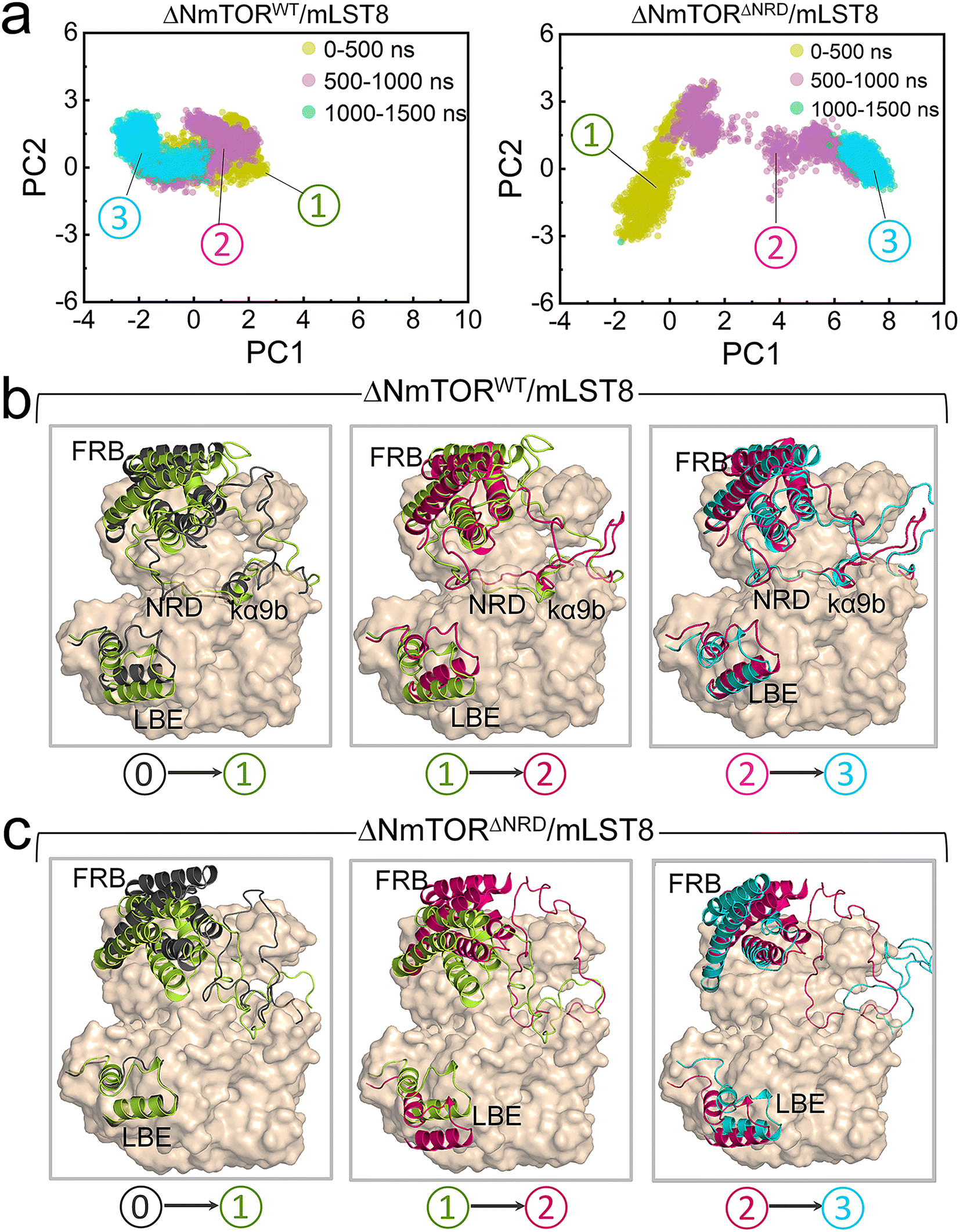 | ||
| Fig. 3 PCA for the mTOR kinase domain in ΔNmTORWT/mLST8 and ΔNmTORΔNRD/mLST8. (a) The projections of the first and second principal components (PC1 and PC2) from the PCA, and (b) structural alignment for the mTOR kinase domain in the three different simulation time frames (0–500 ns, 500–1000 ns, and 1000–1500 ns) for the ΔNmTORWT/mLST8 and ΔNmTORΔNRD/mLST8 systems. In (b) and (c), structure ⓪ represents the initial configuration of the mTOR kinase domain, and structures ①, ②, and ③ are the representative configurations of the mTOR kinase domain in the 0–500 ns, 500–1000 ns, and 1000–1500 ns trajectories, respectively. | ||
Oncogenic activation of mTOR
mTOR mutations are frequently detected in cancer patients. The activating mutations upregulate signaling through the PI3K/AKT/mTOR pathway, driving cancer.81 Most mTOR kinase domain mutations occur in the ka3-, kα9-, kα9b-, and kα10-helices (Fig S2d†),4 disturbing the kα9b-centered packed structure. The top-3 most frequent mutating sites are S2215 at the interface between the N- and C-lobe, I2500 in the kα10-helix, and L2427 in the kα9b-helix (Fig S7†).2 Here, we chose L2427. The three cancer databases, TCGA, GENIE, and COSMIC cataloged 26, 19, and 7 individuals harboring the L2427R, L2427Q, and L2427P mutations, respectively. We verified L2427R pathogenicity,82 modeled an L2427R mutated complex (ΔNmTORL2427R/mLST8) and conducted MD simulations (Table S1†). As expected, this mutation significantly affected the conformation and mobility of the kα9b-helix and NRD, with the helical components of the helix entirely disappearing (Fig. 4a). Compared to ΔNmTORWT/mLST8, the averaged interaction energy between NRD and FRB dramatically increases from ∼−198.0 kcal mol−1 to ∼−85.7 kcal mol−1 (Fig. 4b), indicating that the conformational change weakens their interactions. In turn, this resulted in increasing the mobilities of FRB, kα9b, and NRD, as well as in the unstructured region after NRD (Fig. 4c).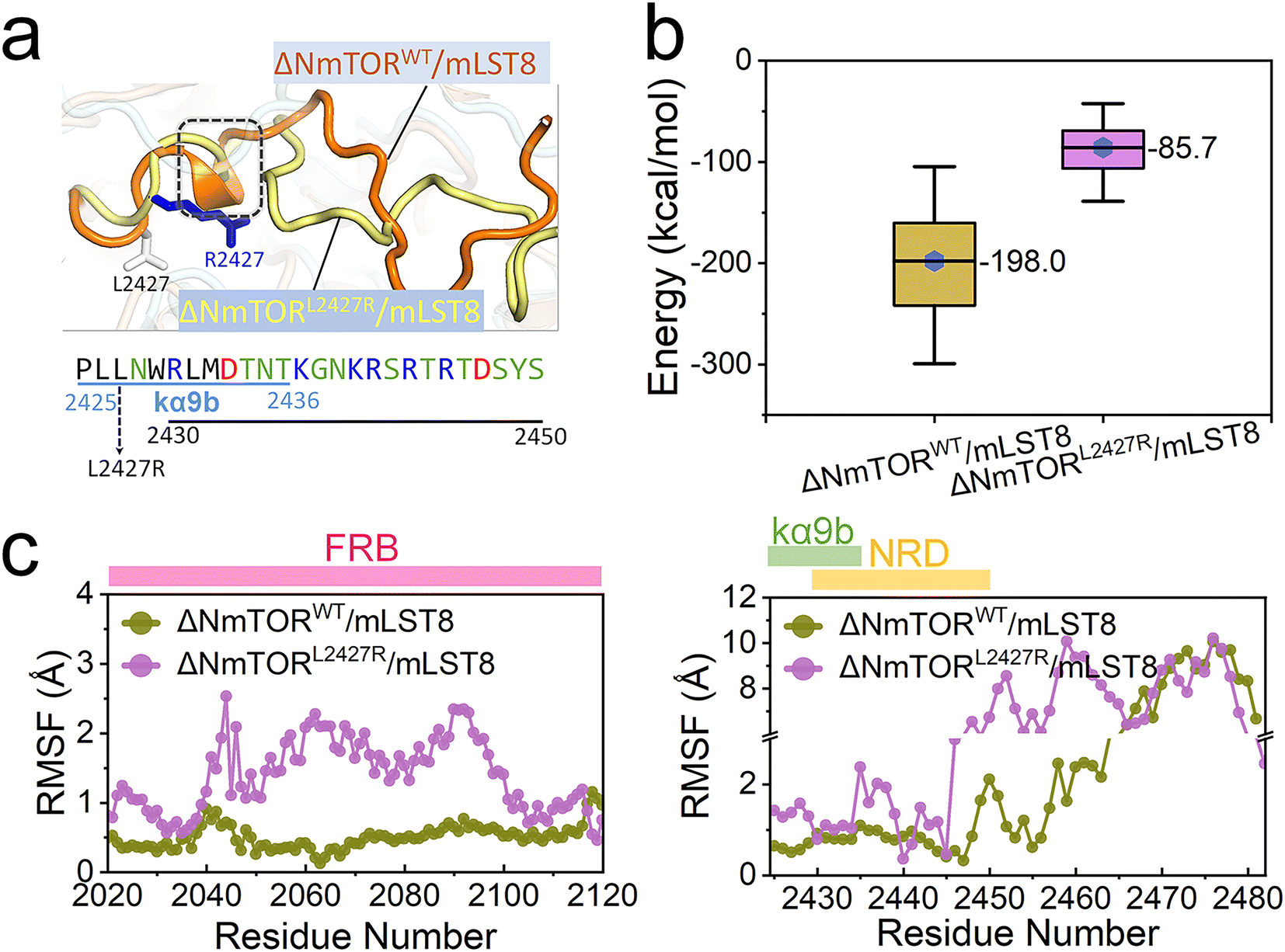 | ||
| Fig. 4 Oncogenic L2427R mutation increases the high dynamics of kα9b, NRD, and FRB. (a) Comparison of the conformations of the kα9b-helix and NRD, (b) interaction energy between NRD and FRB, and (c) root mean square fluctuations (RMSF) of the FRB domain (left), and the kα9b-helix, NRD, and the unstructured region after NRD (right) for the ΔNmTORWT/mLST8 and ΔNmTORΔNRD/mLST8 systems. | ||
The sparse distribution of the first two components observed in the PCA further confirms the significant conformational change in the ΔNmTORL2427R/mLST8 kinase domain (Fig. 5a), with the L2427R mutation driving fast unfolding of the kα9b-helix (Fig. 5b). Without the strong NRD–FRB interaction, NRD and the unstructured region after it move out from the catalytic cleft (termed OUT NRD) and extend. The structural alignment of the mTOR kinase domains in the 0–500 ns, 500–1000 ns, and 1000–1500 ns trajectories further confirms the large movement and large fluctuation of FRB, kα9b, and NRD. Their instabilities facilitate the opening of the catalytic cleft, supporting the premise that some mTOR-activating mutations may loosen the structural framework around the kα9b-helix,4 promoting the shift of the conformational ensemble of mTOR toward the catalysis-favored state.
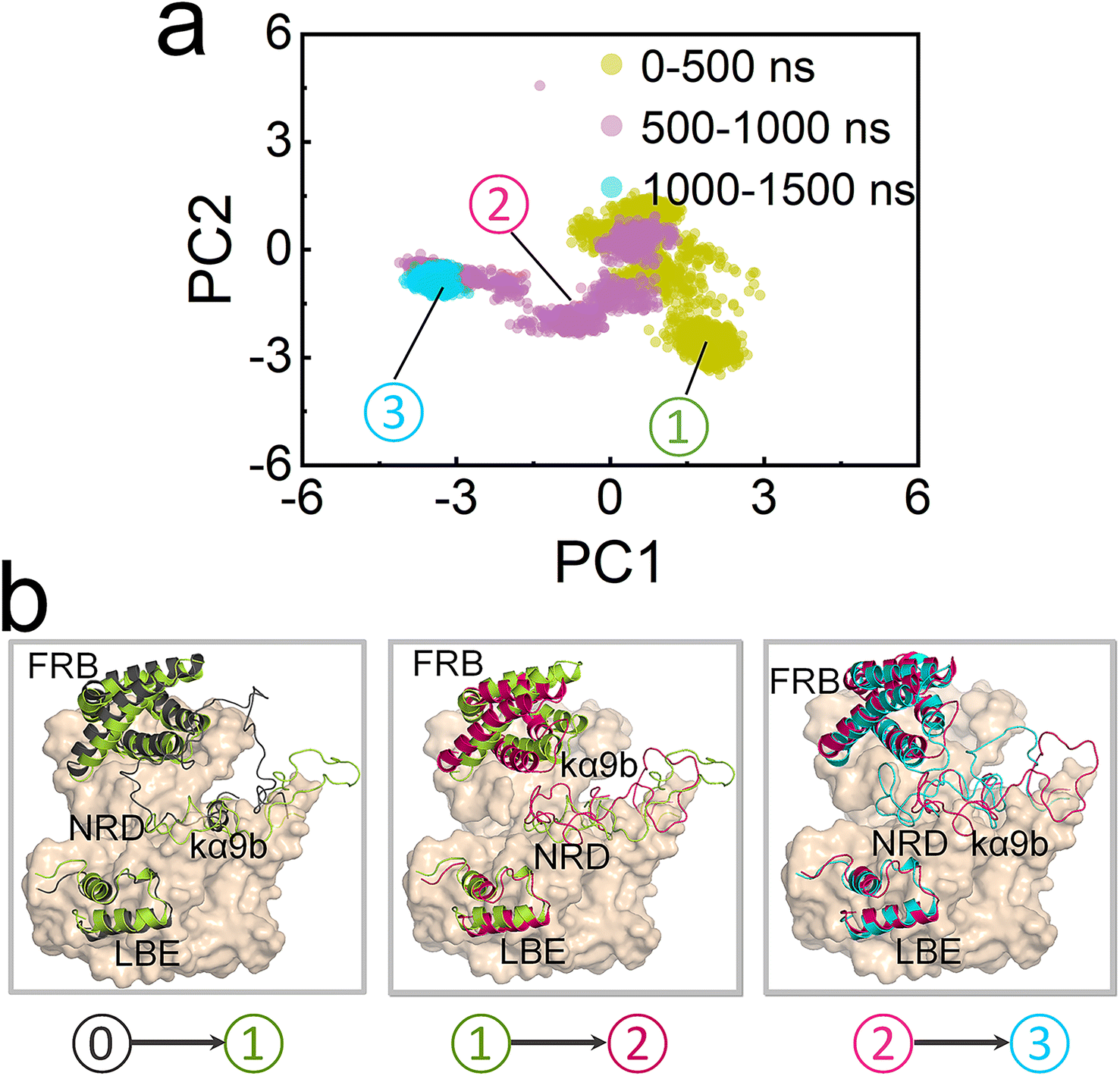 | ||
| Fig. 5 PCA for the mTOR kinase domain in ΔNmTORL2427R/mLST8. (a) The projections of the first and second principal components (PC1 and PC2) from the PCA, and (b) structural alignment for the mTOR kinase domain in the three different simulation time frames (0–500 ns, 500–1000 ns, and 1000–1500 ns) for the ΔNmTORL2427R/mLST8 system. In (b), structure ⓪ represents the initial configuration of the mTOR kinase domain, and structures ①, ②, and ③ are the representative configurations of the mTOR kinase domain in the 0–500 ns, 500–1000 ns, and 1000–1500 ns trajectories, respectively. | ||
mTOR drug discovery
mTOR is a compelling drug target, with recent advances garnering increasing attention.34,64,83–86 Beyond fundamental therapeutic indices such as solubility, affinity, and broad kinase toxicity with off-target effects, ensuring high selectivity for mTORC1 over mTORC2 is critical. mTORC2 inhibition has been associated with compromised insulin sensitivity and glucose intolerance.55 A noteworthy advancement in mTOR drug discovery is the emergence of novel bitopic inhibitors. Shokat et al. devised Rapalink-1, a compound connecting an allosteric inhibitor (rapamycin) and an orthosteric inhibitor (MLN0128) using a PEG-rich linker (Fig. 6a).62,63 The linked chemotypes in the bitopic inhibitor trigger a mutually beneficial interaction, which can cooperatively enhance the binding of both components, augmenting the affinity and selectivity for the target protein. Some residues in the FRB and kinase domains contribute to the allosteric and orthosteric drug binding. Their mutations promote resistance to the monovalent drug interventions. Patients harboring the A2034V and F2108L mutations within the mTOR FRB domain demonstrate resistance to the allosteric drug (everolimus) and the M2327I mutation is able to disrupt the binding of the orthosteric drug (AZD8055).61,87 With the cooperation of rapamycin and MLN0128, Rapalink-1 can effectively overcome the resistance.61 Recently, Revolution Medicines Inc. created two exceptionally potent bitopic inhibitors, RMC-5552 and RMC-6272 with a linker comprised of eight EG units (PEG8) to connect a rapalog with “rigid MLN” and XL388, respectively. These inhibitors exhibited elevated selectivity for mTORC1 and potent anti-tumor effects, positioning them as promising contenders for targeted therapeutic approaches.65 RMC-5552 is currently undergoing clinical trials (NCT ID: NCT05557292 and NCT04774952) to evaluate its effectiveness in treating patients with recurrent glioblastoma and relapsed/refractory solid tumors.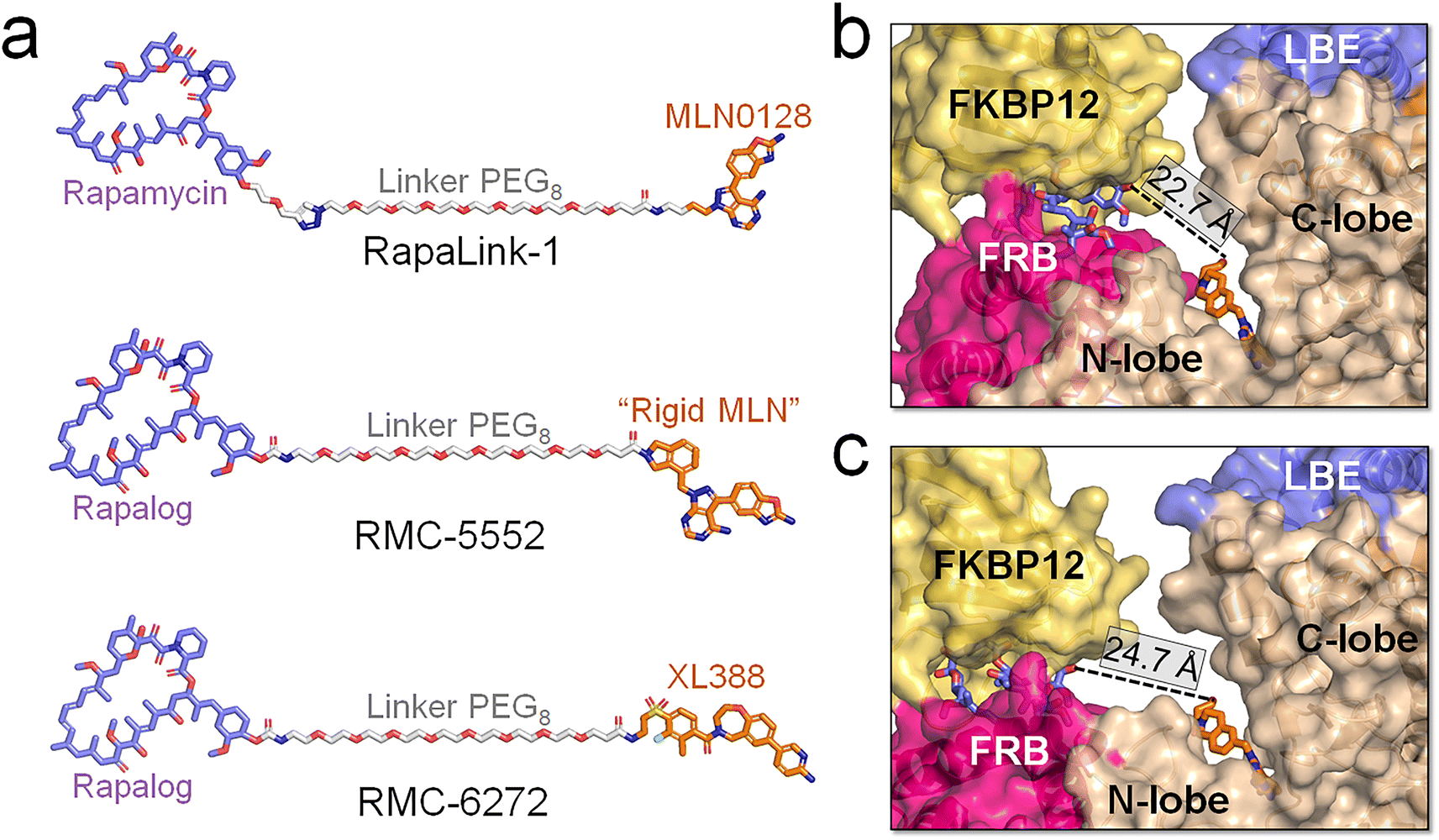 | ||
| Fig. 6 Structural insights into the binding of the bitopic inhibitors and mTOR. (a) Structures of mTOR's bitopic inhibitors, RapaLink-1, RMC-5552, and RMC-6272. Binding modes between RMC-5552 and mTOR with the (b) closed and (c) open catalytic cleft. The distances between allosteric (blue sticks) and orthosteric (orange sticks) inhibitors in (b) and (c) were measured. The RMC-5552/mTOR complex in (b) is extracted from the cryo-EM structure (PDB ID: 8ERA), in which the mTOR adopts the closed catalytic cleft. In (c), the RMC-5552/mTOR complex was modeled by integrating RMC-5552 with a representative configuration of mTOR extracted from the trajectories of the ΔNmTORΔNRD/mLST8 system, in which mTOR populates the open catalytic cleft. | ||
Our simulations indicate that mTOR adopts an open catalytic cleft when activated. In its wild-type form, mTOR maintains a closed catalytic cleft. However, oncogenic mutations, such as L2427R, result in a high population of the open catalytic cleft, accompanied by the FRB domain displacement. A cryo-EM structure of the mTOR/RMC-5552 complex (PDB ID: 8ERA) reveals a closed catalytic cleft of mTOR (Fig. 6b), suggesting that RMC-5552 exhibits potency against wild-type mTOR. However, its effectiveness against oncogenic mTOR remains uncertain. The distance between the allosteric and orthosteric inhibitors is ∼22.7 Å in the cryo-EM mTOR/RMC-5552 structure. The length of the linker in bitopic inhibitors is critical for high binding affinity.62,63,88,89 In mTOR with a closed catalytic cleft, RMC-5552 and RMC-6272, with the PEG8 linker, aptly occupy the space between mTOR's allosteric and orthosteric sites. In our modeled RMC-5552/mTOR complex with an open catalytic cleft the distance between the allosteric and orthosteric inhibitors expands to ∼24.7 Å (Fig. 6c). This indicates that bitopic inhibitors with PEG8 linker may encounter reduced affinity to mTOR with open cleft, possibly diminishing their effectiveness against oncogenic mTOR variants. At the same time, long linkers are more flexible, and depending on their chemistry, can be allergenic. Collectively, our analysis suggests that it might be advantageous to consider adjusting the linker length and/or chemistry of the bitopic inhibitors when targeting a certain mTOR variant. In experiments, it would be valuable to have the compounds with shorter and longer linkers to determine if the linker length is indeed responsible for the observed changes in distance.
Discussion
The allosteric mechanism underlying mTOR activation
Here we provide a detailed account of the allosteric mechanisms underlying the activation of the wild-type and mutated mTOR at the atomic level. Our findings are consistent with a broad range of experimental data and support the previously proposed concept of “active-site restriction”.4,78–80 We demonstrate that two partially overlapping motifs, the kα9b-helix and NRD, play a crucial role in regulating the conformation and activity of the mTOR kinase domain. The FRB domain and mLST8 extending from the N- and C-lobes of the mTOR kinase domain, respectively, create a deep “V-shape” catalytic cleft that partially restricts substrate access to the active site. The NRD can switch between two states, inside (IN) and outside (OUT) the catalytic cleft (Fig. 7 & Movie S1†). As proposed earlier,4 when the NRD and the kα9b-helix are in the IN state, they can obstruct the catalytic cleft, effectively preventing substrate access. Our simulations reveal that the NRD can also directly interact with the FRB domain, further restricting its movement and stabilizing the deep and closed “V-shape” cleft. This interaction strengthens the restriction imposed on substrate access. Conversely, when the NRD is in the OUT state, these restrictions are relieved, leading to increased mobility of the FRB domain. The relaxed FRB domain becomes more likely to move away from the mTOR LBE domain (or mLST8), resulting in a shallow and open catalytic cleft, with substrates able to more easily access the active site. The movement of the FRB domain also exposes its secondary substrate-binding site, facilitating substrate recruitment. These structural changes create a seemingly optimal environment for efficient catalysis. Our results also shed light on why the deletion of NRD (residues 2430–2450), rather than the partially overlapping/adjoining unstructured segment of 2443–2486, can significantly increase the kinase activity of mTOR.76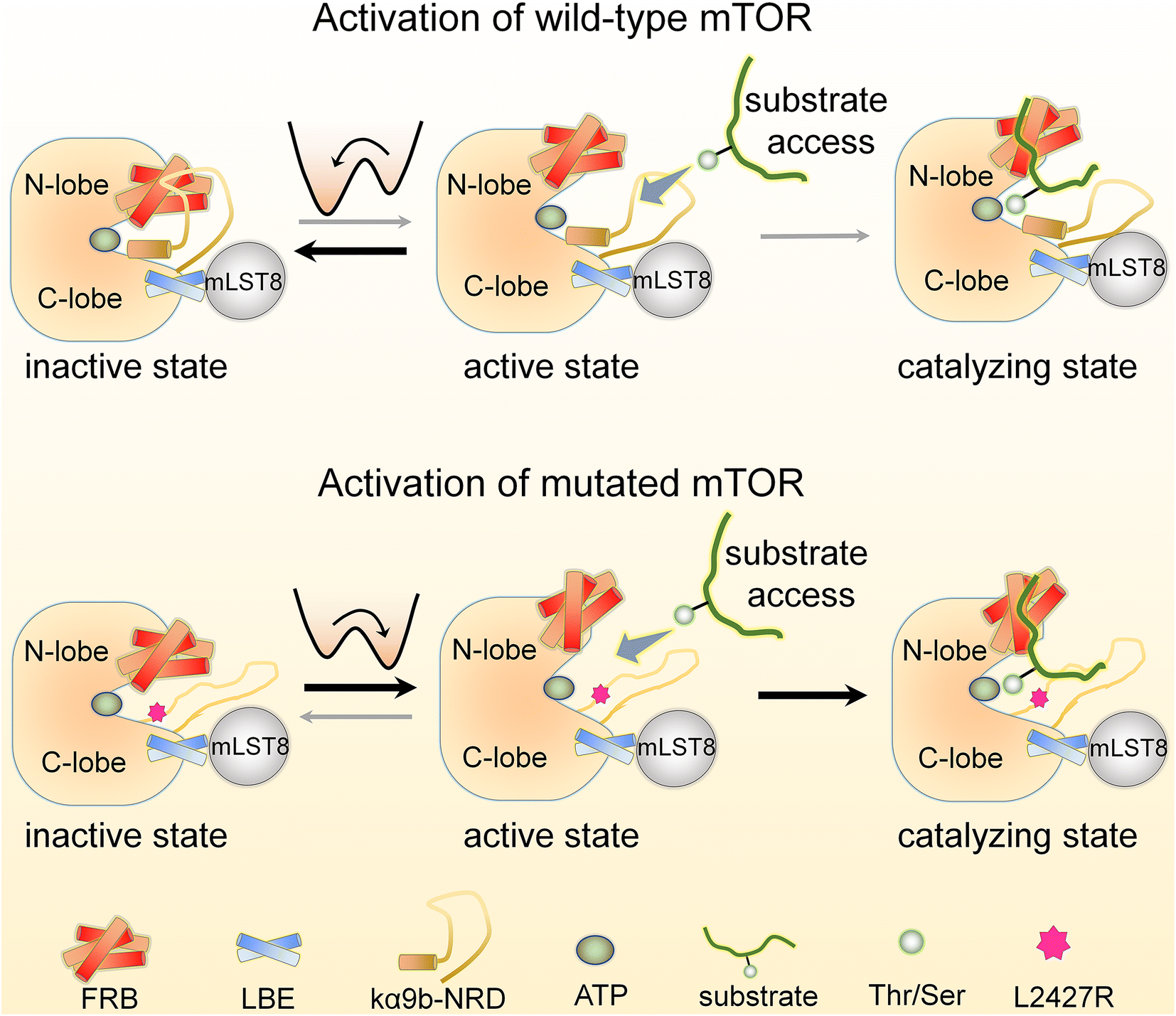 | ||
| Fig. 7 A schematic illustration of mTOR's activation mechanism. The release of NRD from the catalytic cleft of mTOR leads to the opening of the catalytic cleft and the substrate-binding site on FRB making them more accessible, which allows for catalysis. Oncogenic mutations, such as L2427R, can promote this activation process, shifting the conformational ensemble of the mTOR kinase domain toward the catalysis-preferred state. | ||
mTOR's autoinhibition and its release
Wild-type mTOR predominantly adopts an autoinhibited state characterized by a closed catalytic cleft (Fig. 7). Activating physiological interactions allosterically release mTOR from this state to assume an active conformation. In mTORC1, these include the interaction between RHEB and the mTOR FAT domain. For mTORC2, they include the binding of the mSIN1 PH domain to the membrane. The interaction between RHEB and the mTOR FAT domain is known to allosterically promote activation of mTORC1 by releasing the FAT domain autoinhibition,2 with RHEB's C-terminal farnesylation promoting its lysosomal membrane interactions and activation,90 while the binding of the pleckstrin homology (PH) domain of mSIN1 to the membrane facilitates activation of mTORC2. RHEB also stimulates a conformational change that allosterically rearranges mTORC1 active-site residues, accelerating catalysis.2The kα9b-helix plays a critical role in regulating the activity of mTOR. The movement of this helix is closely linked to the mobility of the NRD. When the NRD exhibits higher mobility, it is more likely to shift out from the catalytic cleft (Fig. 7). Several oncogenic mutations were identified within the kα3-, kα9-, and kα10-helices of mTOR, which are neighboring motifs that surround the kα9b-helix. These mutations have the potential to disrupt packing around kα9b, leading to its, and the NRD's, increased mobility, ultimately promoting a shift of the mTOR ensemble towards a catalytically favored state. The L2427R mutation, which stimulated unfolding of the kα9b-helix, provides an example. The mutation enhanced the mobility of both the kα9b-helix and the associated NRD and FRB. Our mTORL2427R simulations observed that NRD tends to adopt an OUT conformation, favoring catalytic activity. We expect that other oncogenic mutations at the 2427-site, such as L2427R and L2427Q, or mutations at positions adjacent to the 2427-site, like S2215 and I2500, may similarly enhance mTOR activity through such a mechanism. Increased dynamics of the FRB domain can also destabilize the IN conformation of the NRD, leading to a preference for the OUT state. Oncogenic mutations in the FRB domain (e.g., E2029K, E2032A, E2033G) may weaken the interactions with the NRD, resulting in a shallower and more open catalytic cleft, which, in conjunction with the OUT orientation of the NRD, further promotes mTOR activity. This also suggests the presence of allosteric communication between these mutations and the orthosteric ATP-binding site.91,92 As recorded in the ClinVar database, pathogenic mTOR variants manifest as somatic and germline mutations. Somatic and germline alterations can drive cancer, while germline mutations are also commonly detected in disorder diseases. These allele origins of mTOR variants can share identical mutations, suggesting that individuals with disorder diseases may have an elevated risk of developing cancer.93,94
Different than other protein kinases, mTOR may regulate its activity through dynamic opening and closing of its deep catalytic cleft
mTOR is a protein kinase, but its mechanism differs. Kinases' catalytic activity is governed by the energy barrier of the conformational change toward the active state (Ka) and substrate accessibility to the active site (Km).95 Typically, activation involves an OUT-to-IN movement of the αC-helix and collapsed-to-extended transition of the A-loop. These features can be used in kinase drug discovery strategies.96 In contrast, the mTOR αC-helix maintains the IN conformation, suggesting that it is constitutively active. mTOR appears to have evolved to regulate its activity through the dynamic opening and closing of its catalytic cleft, as observed in our simulations. The depth of mTOR's active site may restrict phosphorylation sites adjacent to bulky substrate structures. Common mTORC1 and mTORC2 substrates include the AGC kinase family, e.g., AKT, S6K1, SGK1, PKN1, and PKCs. They primarily phosphorylate serine or threonine within hydrophobic motifs (HM), TOR interacting motifs (TIM), and turn motifs (TM), rather than those within the A-loop of the bulk kinase domain.20 mTOR protein kinase populates heteromeric mTOR complexes, unlike most protein kinases in human cells that exist as monomers or homomeric assemblies. Other mTORCs' subunits play obligatory roles in cellular localization and regulation, primarily through substrate recruitment (Km).9 Raptor is responsible for recruiting mTORC1 substrates through direct binding to the TOR signaling (TOS) motifs within the substrate, whereas for mTORC2, mSIN1, rather than Rictor, appears to be the substrate recruiter. Anchoring mSIN1 onto the complex appears the only explored function of Rictor.20 Experimental data have also suggested mSIN1 inhibition of mTORC277 through interaction of mSIN1's PH domain with mTOR kinase domain. Competitive binding of mSIN1 PH domain to PIP3 relieves kinase domain inhibition. Binding of mSIN1 Ras binding domain (RBD) to H-Ras and K-Ras can dampen MAPK signaling.66,97,98 DEP domain-containing mTOR interacting protein (DEPTOR) operates as an endogenous mTOR inhibitor. DEPTOR binding to the mTOR FAT domain induces an inactive conformation, partially inhibiting mTORC1 and mTORC2.99 Proline-rich AKT substrate 40 kDa (PRAS40) inhibits mTORC1 by blocking the first substrate-binding site on Raptor and the second on the mTOR FRB domain.2 These interactions can compete with substrates, such as S6K1 and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), binding to mTORC1. RHEB's regulation of mTORC1 does not relate to substrate recruitment. It can associate with the FAT domain of mTORC1 promoting allosteric changes that favor catalysis by altering the alignment of active site residues.2 mLST8 is a common component in both mTOR complexes but functions differently in mTORC1 and mTORC2.70 Since mLST8 is indispensable for maintaining the Rictor–mTOR interaction but apparently not Raptor–mTOR, it is required for phosphorylation of AKT and protein kinase C alpha (PKCα) by mTORC2 but not S6K1 by mTORC1.50,69Our work elucidates exactly how the domains and inherent motifs of mTOR regulate conformational changes in the mTOR kinase domain, thereby controlling kinase activity. Our mechanism focuses on conformational sampling, thereby elucidating activation principles of both mTORCs and helps explain how mutations can drive mTOR transition to the active state. In particular, our proposed allosteric activation mechanism can suggest how to optimize bitopic inhibitors informing selective targeting of the mTOR mutants. Finally, it connects protein conformational ensembles to function, mechanisms of activation and drug discovery, in line with theory.100,101
Conclusions
In this work, we decipher the mechanism of intrinsic and oncogenic activation of mTOR at the atomic level. Our findings highlight the significance of the unstructured N-terminal regulatory domain (NRD) (residues 2430–2450) as a key regulator and show that NRD dynamics are closely associated with conformational changes in the kinase domain. When NRD is located inside the catalytic cleft along with kα9b, it obstructs the active site, effectively blocking mTOR activity. NRD interacts directly with the mTOR FRB domain, thereby constraining its motion. This interaction contributes to maintaining a deep and closed catalytic cleft, which restricts substrate access. Upon NRD release from the catalytic cleft, the mTOR FRB domain moves away from the mTOR LBE domain (or mLST8), resulting in an open conformation of the catalytic cleft and a more accessible substrate-binding site on FRB, which is favorable for substrate access and recruitment. The motion of NRD is affected by the dynamics of the adjacent kα9b-helix. The oncogenic mutation L2427R promotes unfolding of the kα9b-helix. This results in increasing the mobilities of NRD, kα9b, and FRB, which reduces the interaction between NRD and FRB, accelerating mTOR activation by shifting the equilibrium toward the catalysis-favored state. These scenarios lead us to expect that mutations that can break the kα9b-helix-centered packed structure to promote mTOR activation follow this activation principle. Our proposed mechanism aligns with previous experimental observations, expands the concept of the active-site restriction mechanism of mTOR, and clarifies how oncogenic mutations can drive mTOR activation. Our simulations may help in more potent bitopic inhibitors that could effectively target mTOR mutants. This study advances our understanding of mTOR regulation and offers insights for therapeutic intervention.Materials and methods
Modeling of different ΔNmTOR/mLST8 complex systems
We modeled three different ΔNmTOR/mLST8 complex systems based on the crystal structure of N-terminal truncated mTOR complexing with mLST8 (PDB ID: 4JSP). They are ΔNmTORWT/mLST8, ΔNmTORΔNRD/mLST8, and ΔNmTORL2427R/mLST8 (Table S1†). The missing regions in the crystal structure were modeled using the AlphaFold2 package.102,103 Based on the active-site restriction mechanism that the kα9b-helix and NRD block the active site of mTOR, we manually adjusted NRD, leading it to approach the active site. The resulting complex was used as ΔNmTORWT/mLST8. Based on ΔNmTORWT/mLST8, the ΔNmTORΔNRD/mLST8 and ΔNmTORL2427R/mLST8 systems were constructed by deleting the 2430–2450 segment, and substituting L2427 with Arg, respectively. An ATP molecule and two magnesium ions (Mg2+) were placed in the ATP-binding pocket of mTOR in the three complexes. The resulting systems were solvated using the TIP3P solvent model. The Na+ and Cl− ions were added into the solvated systems to neutralize the systems and to generate a physiological salt concentration of ∼0.15 mol L−1.MD simulation protocol
The all-atom MD simulations were performed for the three systems using the NAMD 2.13 package104 with the CHARMM105 all-atom additive force field (version C36).106,107 The simulation protocol closely followed the methodology employed in our previous studies.71,108–111 Prior to MD simulation, we performed several cycles of minimization and dynamics to eliminate atom contacts within the systems. Each system underwent 1.5 μs MD simulations under the NPT ensemble with 3D periodic boundary conditions. To assess the consistency of outcomes, we executed three parallel trajectories concurrently for each individual system. The results derived from these three parallel trajectories exhibited similarities and were found to be comparable. Throughout the simulations, pressure was maintained at 1 atm using the Langevin piston control algorithm, while the temperature was kept at 310 K using the Langevin thermostat method with a damping coefficient of 1 ps−1. The SHAKE algorithm was used to constrain covalent bonds, including hydrogen atoms. A 2 fs time-step was used in the simulations. The long-range electrostatic and short-range van der Waals (vdW) potential energies were calculated using the particle mesh Ewald method and switching functions, respectively. The analysis used the FORTRAN script in the CHARMM package (version c45b1) and the TCL script in the VMD package.Data availability
Data is available upon request.Author contributions
YL, MZ, HJ, and RN conceived and designed the study. YL and MZ conducted the simulations. YL also analyzed the results and drafted the manuscript. YL, HJ, and RN validated, reviewed, and edited the manuscript. RN supervised the project.Conflicts of interest
The authors declare no potential conflicts of interest.Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261201500003I. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the US Government. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, CCR. The calculations had been performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (https://hpc.nih.gov/).References
- D. Mossmann, S. Park and M. N. Hall, mTOR signalling and cellular metabolism are mutual determinants in cancer, Nat. Rev. Cancer, 2018, 18(12), 744–757 CrossRef CAS PubMed.
- H. Yang, X. Jiang, B. Li, H. J. Yang, M. Miller and A. Yang, et al., Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40, Nature, 2017, 552(7685), 368–373 CrossRef CAS PubMed.
- D. M. Sabatini, Twenty-five years of mTOR: Uncovering the link from nutrients to growth, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(45), 11818–11825 CrossRef CAS PubMed.
- H. Yang, D. G. Rudge, J. D. Koos, B. Vaidialingam, H. J. Yang and N. P. Pavletich, mTOR kinase structure, mechanism and regulation, Nature, 2013, 497(7448), 217–223 CrossRef CAS PubMed.
- C. T. Keith and S. L. Schreiber, PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints, Science, 1995, 270(5233), 50–51 CrossRef CAS PubMed.
- K. Switon, K. Kotulska, A. Janusz-Kaminska, J. Zmorzynska and J. Jaworski, Molecular neurobiology of mTOR, Neuroscience, 2017, 341, 112–153 CrossRef CAS PubMed.
- A. K. Murugan, mTOR: Role in cancer, metastasis and drug resistance, Semin. Cancer Biol., 2019, 59, 92–111 CrossRef CAS PubMed.
- A. Gonzalez, M. N. Hall, S. C. Lin and D. G. Hardie, AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control, Cell Metab., 2020, 31(3), 472–492 CrossRef CAS PubMed.
- L. Tafur, J. Kefauver and R. Loewith, Structural Insights into TOR Signaling, Genes, 2020, 11(8), 885 CrossRef CAS PubMed.
- R. Loewith, E. Jacinto, S. Wullschleger, A. Lorberg, J. L. Crespo and D. Bonenfant, et al., Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control, Mol. Cell, 2002, 10(3), 457–468 CrossRef CAS PubMed.
- K. Hara, Y. Maruki, X. Long, K. Yoshino, N. Oshiro and S. Hidayat, et al., Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action, Cell, 2002, 110(2), 177–189 CrossRef CAS PubMed.
- D. H. Kim, D. D. Sarbassov, S. M. Ali, J. E. King, R. R. Latek and H. Erdjument-Bromage, et al., mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery, Cell, 2002, 110(2), 163–175 CrossRef CAS PubMed.
- D. D. Sarbassov, S. M. Ali, D. H. Kim, D. A. Guertin, R. R. Latek and H. Erdjument-Bromage, et al., Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton, Curr. Biol., 2004, 14(14), 1296–1302 CrossRef CAS PubMed.
- C. H. Aylett, E. Sauer, S. Imseng, D. Boehringer, M. N. Hall, N. Ban and T. Maier, Architecture of human mTOR complex 1, Science, 2016, 351(6268), 48–52 CrossRef CAS PubMed.
- C. C. Thoreen, L. Chantranupong, H. R. Keys, T. Wang, N. S. Gray and D. M. Sabatini, A unifying model for mTORC1-mediated regulation of mRNA translation, Nature, 2012, 485(7396), 109–113 CrossRef CAS PubMed.
- P. P. Hsu, S. A. Kang, J. Rameseder, Y. Zhang, K. A. Ottina and D. Lim, et al., The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling, Science, 2011, 332(6035), 1317–1322 CrossRef CAS PubMed.
- E. J. Chen and C. A. Kaiser, LST8 negatively regulates amino acid biosynthesis as a component of the TOR pathway, J. Cell Biol., 2003, 161(2), 333–347 CrossRef CAS PubMed.
- D. H. Kim, D. D. Sarbassov, S. M. Ali, R. R. Latek, K. V. Guntur and H. Erdjument-Bromage, et al., GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR, Mol. Cell, 2003, 11(4), 895–904 CrossRef CAS PubMed.
- Y. Sancak, T. R. Peterson, Y. D. Shaul, R. A. Lindquist, C. C. Thoreen, L. Bar-Peled and D. M. Sabatini, The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1, Science, 2008, 320(5882), 1496–1501 CrossRef CAS PubMed.
- S. Battaglioni, D. Benjamin, M. Walchli, T. Maier and M. N. Hall, mTOR substrate phosphorylation in growth control, Cell, 2022, 185(11), 1814–1836 CrossRef CAS PubMed.
- D. D. Sarbassov, S. M. Ali, S. Sengupta, J. H. Sheen, P. P. Hsu and A. F. Bagley, et al., Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB, Mol. Cell, 2006, 22(2), 159–168 CrossRef CAS PubMed.
- D. A. Guertin, D. M. Stevens, M. Saitoh, S. Kinkel, K. Crosby and J. H. Sheen, et al., mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice, Cancer Cell, 2009, 15(2), 148–159 CrossRef CAS PubMed.
- R. Nussinov, C. J. Tsai and H. Jang, Allostery, and how to define and measure signal transduction, Biophys. Chem., 2022, 283, 106766 CrossRef CAS PubMed.
- M. Zhang, H. Jang and R. Nussinov, PI3K inhibitors: review and new strategies, Chem. Sci., 2020, 11(23), 5855–5865 RSC.
- M. Sacerdoti, L. Z. F. Gross, A. M. Riley, K. Zehnder, A. Ghode and S. Klinke, et al., Modulation of the substrate specificity of the kinase PDK1 by distinct conformations of the full-length protein, Sci. Signaling, 2023, 16(789), eadd3184 CrossRef CAS PubMed.
- K. Busschots, L. A. Lopez-Garcia, C. Lammi, A. Stroba, S. Zeuzem and A. Piiper, et al., Substrate-Selective Inhibition of Protein Kinase PDK1 by Small Compounds that Bind to the PIF-Pocket Allosteric Docking Site, Chem. Biol., 2012, 19(9), 1152–1163 CrossRef CAS PubMed.
- A. Ghode, L. Z. F. Gross, W. V. Tee, E. Guarnera, I. N. Berezovsky, R. M. Biondi and G. S. Anand, Synergistic Allostery in Multiligand-Protein Interactions, Biophys. J., 2020, 119(9), 1833–1848 CrossRef CAS PubMed.
- S. S. Taylor, C. Kim, D. Vigil, N. M. Haste, J. Yang, J. Wu and G. S. Anand, Dynamics of signaling by PKA, Biochim. Biophys. Acta, 2005, 1754(1–2), 25–37 CrossRef CAS PubMed.
- K. Inoki, Y. Li, T. Xu and K. L. Guan, Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling, Genes Dev., 2003, 17(15), 1829–1834 CrossRef CAS PubMed.
- K. Inoki, Y. Li, T. Zhu, J. Wu and K. L. Guan, TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling, Nat. Cell Biol., 2002, 4(9), 648–657 CrossRef CAS PubMed.
- E. Kim, P. Goraksha-Hicks, L. Li, T. P. Neufeld and K. L. Guan, Regulation of TORC1 by Rag GTPases in nutrient response, Nat. Cell Biol., 2008, 10(8), 935–945 CrossRef CAS PubMed.
- Y. Sancak, L. Bar-Peled, R. Zoncu, A. L. Markhard, S. Nada and D. M. Sabatini, Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids, Cell, 2010, 141(2), 290–303 CrossRef CAS PubMed.
- A. L. Cangelosi, A. M. Puszynska, J. M. Roberts, A. Armani, T. P. Nguyen and J. B. Spinelli, et al., Zonated leucine sensing by Sestrin-mTORC1 in the liver controls the response to dietary leucine, Science, 2022, 377(6601), 47–56 CrossRef CAS PubMed.
- P. Oleksak, E. Nepovimova, Z. Chrienova, K. Musilek, J. Patocka and K. Kuca, Contemporary mTOR inhibitor scaffolds to diseases breakdown: A patent review (2015-2021), Eur. J. Med. Chem., 2022, 238, 114498 CrossRef CAS PubMed.
- K. Xu, P. Liu and W. Wei, mTOR signaling in tumorigenesis, Biochim. Biophys. Acta, 2014, 1846(2), 638–654 CAS.
- T. Tian, X. Li and J. Zhang, mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy, Int. J. Mol. Sci., 2019, 20(3), 755 CrossRef CAS PubMed.
- N. Nathan, K. M. Keppler-Noreuil, L. G. Biesecker, J. Moss and T. N. Darling, Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway, Dermatol. Clin., 2017, 35(1), 51–60 CrossRef CAS PubMed.
- H. Populo, J. M. Lopes and P. Soares, The mTOR signalling pathway in human cancer, Int. J. Mol. Sci., 2012, 13(2), 1886–1918 CrossRef CAS PubMed.
- I. Vivanco and C. L. Sawyers, The phosphatidylinositol 3-Kinase AKT pathway in human cancer, Nat. Rev. Cancer, 2002, 2(7), 489–501 CrossRef CAS PubMed.
- A. C. Hsieh, Y. Liu, M. P. Edlind, N. T. Ingolia, M. R. Janes and A. Sher, et al., The translational landscape of mTOR signalling steers cancer initiation and metastasis, Nature, 2012, 485(7396), 55–61 CrossRef CAS PubMed.
- C. M. Robbins, W. A. Tembe, A. Baker, S. Sinari, T. Y. Moses and S. Beckstrom-Sternberg, et al., Copy number and targeted mutational analysis reveals novel somatic events in metastatic prostate tumors, Genome Res., 2011, 21(1), 47–55 CrossRef CAS PubMed.
- J. Urano, T. Sato, T. Matsuo, Y. Otsubo, M. Yamamoto and F. Tamanoi, Point mutations in TOR confer Rheb-independent growth in fission yeast and nutrient-independent mammalian TOR signaling in mammalian cells, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(9), 3514–3519 CrossRef CAS PubMed.
- T. Sato, A. Nakashima, L. Guo, K. Coffman and F. Tamanoi, Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer, Oncogene, 2010, 29(18), 2746–2752 CrossRef CAS PubMed.
- M. Hager, H. Haufe, R. Kemmerling, G. Mikuz, C. Kolbitsch and P. L. Moser, PTEN expression in renal cell carcinoma and oncocytoma and prognosis, Pathology, 2007, 39(5), 482–485 CrossRef CAS PubMed.
- S. V. Madhunapantula and G. P. Robertson, The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma, Pigm. Cell Melanoma Res., 2009, 22(4), 400–419 CrossRef CAS PubMed.
- J. M. Han and M. Sahin, TSC1/TSC2 signaling in the CNS, FEBS Lett., 2011, 585(7), 973–980 CrossRef CAS PubMed.
- D. J. Kwiatkowski, T. K. Choueiri, A. P. Fay, B. I. Rini, A. R. Thorner and G. de Velasco, et al., Mutations in TSC1, TSC2, and MTOR Are Associated with Response to Rapalogs in Patients with Metastatic Renal Cell Carcinoma, Clin. Cancer Res., 2016, 22(10), 2445–2452 CrossRef CAS PubMed.
- R. Nussinov, B. R. Yavuz, M. K. Arici, H. C. Demirel, M. Zhang and Y. Liu, et al., Neurodevelopmental disorders, like cancer, are connected to impaired chromatin remodelers, PI3K/mTOR, and PAK1-regulated MAPK, Biophys. Rev., 2023, 15(2), 163–181 CrossRef CAS PubMed.
- A. A. Mian, U. Zafar, S. M. A. Ahmed, O. G. Ottmann and E. N. M. A. Lalani, Oncogene-independent resistance in Philadelphia chromosome - positive (Ph) acute lymphoblastic leukemia (ALL) is mediated by activation of AKT/mTOR pathway, Neoplasia, 2021, 23(9), 1016–1027 CrossRef CAS PubMed.
- D. A. Guertin, D. M. Stevens, C. C. Thoreen, A. A. Burds, N. Y. Kalaany and J. Moffat, et al., Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1, Dev. Cell, 2006, 11(6), 859–871 CrossRef CAS PubMed.
- S. Sengupta, T. R. Peterson, M. Laplante, S. Oh and D. M. Sabatini, mTORC1 controls fasting-induced ketogenesis and its modulation by ageing, Nature, 2010, 468(7327), 1100–1104 CrossRef CAS PubMed.
- A. Y. Choo and J. Blenis, Not all substrates are treated equally: Implications for mTOR, rapamycin-resistance, and cancer therapy, Cell Cycle, 2009, 8(4), 567–572 CrossRef CAS PubMed.
- C. Gaubitz, T. M. Oliveira, M. Prouteau, A. Leitner, M. Karuppasamy and G. Konstantinidou, et al., Molecular Basis of the Rapamycin Insensitivity of Target Of Rapamycin Complex 2, Mol. Cell, 2015, 58(6), 977–988 CrossRef CAS PubMed.
- C. C. Thoreen, S. A. Kang, J. W. Chang, Q. Liu, J. Zhang and Y. Gao, et al., An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1, J. Biol. Chem., 2009, 284(12), 8023–8032 CrossRef CAS PubMed.
- D. W. Lamming, L. Ye, P. Katajisto, M. D. Goncalves, M. Saitoh and D. M. Stevens, et al., Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity, Science, 2012, 335(6076), 1638–1643 CrossRef CAS PubMed.
- R. C. Deutscher, C. Meyners, S. C. Schäfer, M. L. Repity, W. O. Sugiarto and J. Kolos, Discovery of fully synthetic FKBP12-mTOR molecular glues, ChemRxiv, 2023 DOI:10.26434/chemrxiv-2023-4vb0m.
- Q. Liu, C. Thoreen, J. Wang, D. Sabatini and N. S. Gray, mTOR Mediated Anti-Cancer Drug Discovery, Drug Discovery Today: Ther. Strategies, 2009, 6(2), 47–55 CAS.
- R. Roskoski Jr, Properties of FDA-approved small molecule phosphatidylinositol 3-kinase inhibitors prescribed for the treatment of malignancies, Pharmacol. Res., 2021, 168, 105579 CrossRef PubMed.
- L. Feng, S. Y. Lu, Z. Zheng, Y. Y. Chen, Y. Y. Zhao and K. Song, et al., Identification of an allosteric hotspot for additive activation of PPARγ in antidiabetic effects, Sci. Bull., 2021, 66(15), 1559–1570 CrossRef CAS PubMed.
- D. Ni, Y. Li, Y. R. Qiu, J. Pu, S. Y. Lu and J. Zhang, Combining Allosteric and Orthosteric Drugs to Overcome Drug Resistance, Trends Pharmacol. Sci., 2020, 41(5), 336–348 CrossRef CAS PubMed.
- V. S. Rodrik-Outmezguine, M. Okaniwa, Z. Yao, C. J. Novotny, C. McWhirter and A. Banaji, et al., Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor, Nature, 2016, 534(7606), 272–276 CrossRef CAS PubMed.
- Z. Zhang, Q. Fan, X. Luo, K. Lou, W. A. Weiss and K. M. Shokat, Brain-restricted mTOR inhibition with binary pharmacology, Nature, 2022, 609(7928), 822–828 CrossRef CAS PubMed.
- K. Lou, D. R. Wassarman, T. Yang, Y. Paung, Z. Zhang and T. A. O'Loughlin, et al., IFITM proteins assist cellular uptake of diverse linked chemotypes, Science, 2022, 378(6624), 1097–1104 CrossRef CAS PubMed.
- Y. Chen and X. Zhou, Research progress of mTOR inhibitors, Eur. J. Med. Chem., 2020, 208, 112820 CrossRef CAS PubMed.
- G. L. Burnett, Y. C. Yang, J. B. Aggen, J. Pitzen, M. K. Gliedt and C. M. Semko, et al., Discovery of RMC-5552, a Selective Bi-Steric Inhibitor of mTORC1, for the Treatment of mTORC1-Activated Tumors, J. Med. Chem., 2023, 66(1), 149–169 CrossRef CAS PubMed.
- S. Pudewell, J. Lissy, H. Nakhaeizadeh, N. Mosaddeghzadeh, S. Nakhaei-Rad, R. Dvorsky and M. R. Ahmadian, New mechanistic insights into the RAS-SIN1 interaction at the membrane, Front. Cell Dev. Biol., 2022, 10, 987754 CrossRef PubMed.
- D. Baretic, A. Berndt, Y. Ohashi, C. M. Johnson and R. L. Williams, Tor forms a dimer through an N-terminal helical solenoid with a complex topology, Nat. Commun., 2016, 7, 11016 CrossRef CAS PubMed.
- C. K. Yip, K. Murata, T. Walz, D. M. Sabatini and S. A. Kang, Structure of the human mTOR complex I and its implications for rapamycin inhibition, Mol. Cell, 2010, 38(5), 768–774 CrossRef CAS PubMed.
- A. Scaiola, F. Mangia, S. Imseng, D. Boehringer, K. Berneiser and M. Shimobayashi, et al., The 3.2-A resolution structure of human mTORC2, Sci. Adv., 2020, 6(45), eabc1251 CrossRef CAS PubMed.
- Y. Hwang, L. C. Kim, W. Song, D. N. Edwards, R. S. Cook and J. Chen, Disruption of the Scaffolding Function of mLST8 Selectively Inhibits mTORC2 Assembly and Function and Suppresses mTORC2-Dependent Tumor Growth In Vivo, Cancer Res., 2019, 79(13), 3178–3184 CrossRef CAS PubMed.
- M. Zhang, H. Jang and R. Nussinov, The mechanism of PI3Kα activation at the atomic level, Chem. Sci., 2019, 10(12), 3671–3680 RSC.
- P. Grudzien, H. Jang, N. Leschinsky, R. Nussinov and V. Gaponenko, Conformational Dynamics Allows Sampling of an “Active-like” State by Oncogenic K-Ras-GDP, J. Mol. Biol., 2022, 434(17), 167695 CrossRef CAS PubMed.
- R. Nussinov, C. J. Tsai and H. Jang, Protein ensembles link genotype to phenotype, PLoS Comput. Biol., 2019, 15(6), e1006648 CrossRef CAS PubMed.
- B. L. Sibanda, D. Y. Chirgadze and T. L. Blundell, Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats, Nature, 2010, 463(7277), 118–121 CrossRef CAS PubMed.
- M. Zhang, H. Jang and R. Nussinov, Structural Features that Distinguish Inactive and Active PI3K Lipid Kinases, J. Mol. Biol., 2020, 432(22), 5849–5859 CrossRef CAS PubMed.
- D. R. Alessi and Y. Kulathu, Structural biology: Security measures of a master regulator, Nature, 2013, 497(7448), 193–194 CrossRef CAS PubMed.
- P. Liu, W. Gan, Y. R. Chin, K. Ogura, J. Guo and J. Zhang, et al., PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex, Cancer Discovery, 2015, 5(11), 1194–1209 CrossRef CAS PubMed.
- L. P. McMahon, K. M. Choi, T. A. Lin, R. T. Abraham and J. C. Lawrence Jr, The rapamycin-binding domain governs substrate selectivity by the mammalian target of rapamycin, Mol. Cell. Biol., 2002, 22(21), 7428–7438 CrossRef CAS PubMed.
- A. Sekulic, C. C. Hudson, J. L. Homme, P. Yin, D. M. Otterness, L. M. Karnitz and R. T. Abraham, A direct linkage between the phosphoinositide 3-Kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells, Cancer Res., 2000, 60(13), 3504–3513 CAS.
- A. L. Edinger and C. B. Thompson, An activated mTOR mutant supports growth factor-independent, nutrient-dependent cell survival, Oncogene, 2004, 23(33), 5654–5663 CrossRef CAS PubMed.
- R. Nussinov, C. J. Tsai and H. Jang, A New View of Activating Mutations in Cancer, Cancer Res., 2022, 82(22), 4114–4123 CrossRef CAS PubMed.
- Y. B. Chen, J. Xu, A. J. Skanderup, Y. Dong, A. R. Brannon and L. Wang, et al., Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets, Nat. Commun., 2016, 7(1), 13131 CrossRef CAS PubMed.
- C. Borsari, M. De Pascale and M. P. Wymann, Chemical and Structural Strategies to Selectively Target mTOR Kinase, ChemMedChem, 2021, 16(18), 2744–2759 CrossRef CAS PubMed.
- Y. J. Zhang, Y. Duan and X. F. Zheng, Targeting the mTOR kinase domain: the second generation of mTOR inhibitors, Drug Discovery Today, 2011, 16(7–8), 325–331 CrossRef CAS PubMed.
- B. B. Mao, Q. Zhang, L. Ma, D. S. Zhao, P. Zhao and P. Z. Yan, Overview of Research into mTOR Inhibitors, Molecules, 2022, 27(16), 5295 CrossRef CAS PubMed.
- R. Roskoski Jr, Rule of five violations among the FDA-approved small molecule protein kinase inhibitors, Pharmacol. Res., 2023, 191, 106774 CrossRef PubMed.
- N. Wagle, B. C. Grabiner, E. M. Van Allen, A. Amin-Mansour, A. Taylor-Weiner and M. Rosenberg, et al., Response and acquired resistance to everolimus in anaplastic thyroid cancer, N. Engl. J. Med., 2014, 371(15), 1426–1433 CrossRef PubMed.
- S. Lu, S. Li and J. Zhang, Harnessing allostery: a novel approach to drug discovery, Med. Res. Rev., 2014, 34(6), 1242–1285 CrossRef CAS PubMed.
- C. Zhu, X. Lan, Z. Wei, J. Yu and J. Zhang, Allosteric modulation of G protein-coupled receptors as a novel therapeutic strategy in neuropathic pain, Acta Pharm. Sin. B, 2023 DOI:10.1016/j.apsb.2023.07.020.
- B. Angarola and S. M. Ferguson, Coordination of Rheb lysosomal membrane interactions with mTORC1 activation, F1000Research, 2020, 9(F1000 Faculty Rev), 450, DOI:10.12688/f1000research.22367.1.
- S. Y. Lu, X. H. He, Z. Yang, Z. T. Chai, S. H. Zhou and J. Y. Wang, et al., Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design, Nat. Commun., 2021, 12(1), 4721 CrossRef CAS PubMed.
- D. Ni, J. C. Wei, X. H. He, A. U. Rehman, X. Y. Li and Y. R. Qiu, et al., Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy, Chem. Sci., 2021, 12(1), 464–476 RSC.
- B. R. Yavuz, M. K. Arici, H. C. Demirel, C. J. Tsai, H. Jang, R. Nussinov and N. Tuncbag, Neurodevelopmental disorders and cancer networks share pathways, but differ in mechanisms, signaling strength, and outcome, npj Genomic Med., 2023, 8(1), 37 CrossRef PubMed.
- R. Nussinov, C.-J. Tsai and H. Jang, How can same-gene mutations promote both cancer and developmental disorders?, Sci. Adv., 2022, 8(2), eabm2059 CrossRef CAS PubMed.
- M. Zhang, H. Jang and R. Nussinov, PI3K Driver Mutations: A Biophysical Membrane-Centric Perspective, Cancer Res., 2021, 81(2), 237–247 CrossRef CAS PubMed.
- M. Zhang, Y. Liu, H. Jang and R. Nussinov, Strategy toward Kinase-Selective Drug Discovery, J. Chem. Theory Comput., 2023, 19(5), 1615–1628 CrossRef CAS PubMed.
- Y. Zheng, L. Ding, X. Meng, M. Potter, A. L. Kearney and J. Zhang, et al., Structural insights into Ras regulation by SIN1, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(19), e2119990119 CrossRef CAS PubMed.
- P. Castel, S. Dharmaiah, M. J. Sale, S. Messing, G. Rizzuto and A. Cuevas-Navarro, et al., RAS interaction with Sin1 is dispensable for mTORC2 assembly and activity, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(33), e2103261118 CrossRef CAS PubMed.
- M. Walchli, K. Berneiser, F. Mangia, S. Imseng, L. M. Craigie and E. Stuttfeld, et al., Regulation of human mTOR complexes by DEPTOR, eLife, 2021, 10, e70871 CrossRef PubMed.
- R. Nussinov, Y. Liu, W. Zhang and H. Jang, Cell phenotypes can be predicted from propensities of protein conformations, Curr. Opin. Struct. Biol., 2023, 83, 102722 CrossRef CAS PubMed.
- R. Nussinov, Y. L. Liu, W. A. Zhang and H. Jang, Protein conformational ensembles in function: roles and mechanisms, RSC Chem. Biol., 2023, 4(11), 850–864 RSC.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov and O. Ronneberger, et al., Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596(7873), 583–589 CrossRef CAS PubMed.
- M. Varadi, S. Anyango, M. Deshpande, S. Nair, C. Natassia and G. Yordanova, et al., AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models, Nucleic Acids Res., 2022, 50(D1), D439–D444 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid and E. Villa, et al., Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26(16), 1781–1802 CrossRef CAS.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr, L. Nilsson, R. J. Petrella and B. Roux, et al., CHARMM: the biomolecular simulation program, J. Comput. Chem., 2009, 30(10), 1545–1614 CrossRef CAS PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig and B. L. de Groot, et al., CHARMM36m: an improved force field for folded and intrinsically disordered proteins, Nat. Methods, 2017, 14(1), 71–73 CrossRef CAS PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias and C. Mondragon-Ramirez, et al., Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types, J. Phys. Chem. B, 2010, 114(23), 7830–7843 CrossRef CAS.
- M. Zhang, H. Jang, Z. Li, D. B. Sacks and R. Nussinov, B-Raf autoinhibition in the presence and absence of 14-3-3, Structure, 2021, 29(7), 768–777 CrossRef CAS e2..
- R. C. Maloney, M. Zhang, H. Jang and R. Nussinov, The mechanism of activation of monomeric B-Raf V600E, Comput. Struct. Biotechnol. J., 2021, 19, 3349–3363 CrossRef CAS PubMed.
- H. Jang, I. N. Smith, C. Eng and R. Nussinov, The mechanism of full activation of tumor suppressor PTEN at the phosphoinositide-enriched membrane, iScience, 2021, 24(5), 102438 CrossRef CAS PubMed.
- Y. Liu, H. Jang, M. Zhang, C. J. Tsai, R. Maloney and R. Nussinov, The structural basis of BCR-ABL recruitment of GRB2 in chronic myelogenous leukemia, Biophys. J., 2022, 121(12), 2251–2265 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04690g |
| This journal is © The Royal Society of Chemistry 2024 |